
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical low valent cobalt-catalyzed addition of aryl and vinyl chlorides to α-ketoamides via C–Cl bond activation†
Prashant S.
Shinde
a,
Valmik S.
Shinde
*ab and
Magnus
Rueping
 *a
*a
aKing Abdullah University of Science and Technology (KAUST), KAUST Catalysis Center (KCC), Thuwal 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
bMedicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, Lucknow 226031, Uttar Pradesh, India. E-mail: valmik.shinde@cdri.res.in
First published on 18th March 2024
Abstract
The development of an electrochemical cobalt catalyzed C–Cl bond activation at room temperature for the nucleophilic addition of aryl and vinyl chlorides to α-ketoamides is described. The overall method operates through an electrochemically induced low valent cobalt catalyst that oxidatively adds to aryl or vinyl chlorides affording medicinally important 3-hydroxy oxindole and 3-hydroxypyrrolidinone scaffolds. The development of an enantioselective version using a chiral pyrox ligand is also demonstrated.
The activation of carbon–halogen bonds via oxidative addition with transition metals is one of the fundamental steps in organometallic chemistry. Particularly, the development of the direct catalytic nucleophilic reaction of aryl or vinyl halides with electrophilic reaction partners is an attractive method as no preceding preparation of stoichiometric organometallic reagents, such as Grignard-, organo zinc, organoborane, or organolithium-reagents is required, allowing for a high tolerance toward sensitive functional groups. In recent times, earth-abundant first-row transition metals have gained particular attention owing to their relatively lower cost and toxicity as well as greater sustainability.1 The organometallic chemistry of cobalt, in particular, cobalt(II) complexes has proven useful, especially in several catalytic reactions ranging from hydrogen evolution,2 and cross-couplings3 to C–H functionalizations.4 The versatility of Co(II) complexes in electrocatalysis can be attributed to the reactive nature of their corresponding electrochemically generated Co(I) or Co(0) intermediates.5 The oxidative addition process is highly dependent on both the cobalt complex and the substrate involved and can proceed through a variety of polar and/or radical intermediates. Mechanistic investigations involving such complexes have illustrated that Co(I) intermediates are highly reactive and also prone to a multitude of decomposition pathways. The pioneering study by Périchon's research group proved that the predominant decomposition pathway of electrochemically generated Co(I) species proceeds via disproportionation to Co(II) and Co(0), which subsequently results in precipitation of metallic Co(0), removing cobalt from the electro-catalytic cycle.6 To evade these deleterious pathways, cobalt electrolysis catalysts have been traditionally stabilized through polydentate ligands such as salen complexes7 and cobalamin derivatives.8
There have been several literature examples for direct nucleophilic addition of carbon–halogen bonds under relatively forcing reaction conditions; however, most of the substrates were more reactive aryl bromides and iodides. Aryl chlorides have not been extensively explored in this field despite their great diversity, affordability, and ease of access. This is likely because of their low reactivity, which is generally attributed to their higher bond energy and thus, making the activation by transition-metal catalysts more challenging9 [bond dissociation energies (kcal mol−1) for PhX: Cl (95); Br (80); I (65)]. We envisioned that highly reactive in situ generated cobalt (I) and cobalt (0) species obtained by electrochemical reduction of Co(II) complexes may activate aryl or vinyl chlorides through oxidative addition processes at relatively less forcing conditions or even at room temperature to avoid catalyst deactivation and decomposition pathways.10 Furthermore, an additional hetero-atom in substrate could assist in stabilizing the insitu generated low valent cobalt intermediate via cyclic intermediate. Consequently, this stabilized low-dentate ligands–metal complex, would harbor additional free coordination sites to bind organic substrates and facilitate alternative organometallic steps. The corresponding “aryl or vinyl cobalt” species would then undergo an intramolecular addition with the carbonyl group to produce addition products. This hypothesis was also supported by the pioneering work of Gosmini10 and also Minteer11 who extensively studied the role of ligand electronics on stabilizing the electrocatalytically generated low valent Co(I) intermediates.
Herein, we report the electrochemical Co-catalyzed intramolecular nucleophilic addition of aryl and vinyl chlorides to α-ketoamides through C–Cl bond activation at room temperature to generate 3-hydroxy oxindoles scaffolds. These oxindoles containing a tetra substituted carbon at the benzylic position constitute a common structural motif that shows a broad range of biological activities (Fig. 1).12 Structure activity studies have also shown that the biological activities of these compounds are significantly affected by both the configuration of the C3 and its substitution pattern.13,15
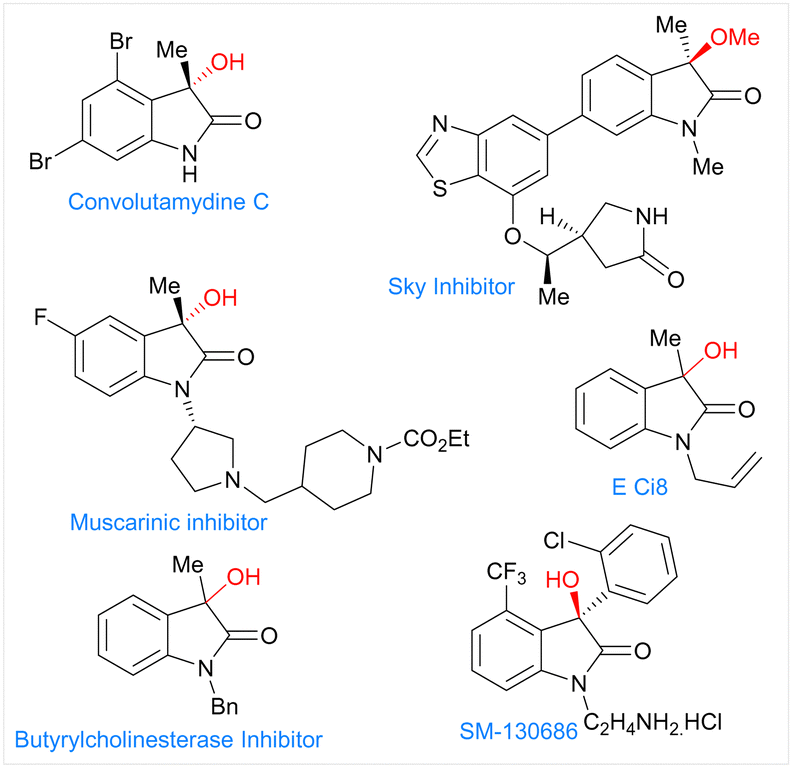 | ||
| Fig. 1 Bioactive 3-hydroxy oxindoles scaffolds. | ||
In order to realize the proof of concept, we initiated our study using CoBr2(2,2′-bipyridine) complex14 as it appeared to be a particularly suitable catalyst for the activation of a variety of aromatic substrates ranging from halides to triflates. We began our study by examining the reaction using a α-keto 2-chloroanilide 1a as substrate in the presence of CoBr2(bpy) (10 mol%) as the catalyst and NaI as supporting electrolyte in DMA as solvent (Table 1). The reaction was performed in an undivided cell equipped with a stainless steel as anode and cathode that yielded 81% of the expected product 2a. The further development concentrated on optimizing the reaction parameters, including variation of the electrodes (cathodes and anodes), electrolytes, current densities, solvents, and cobalt complexes (for detailed optimization studies see ESI†). After extensive efforts, the desired product 2a was obtained in 92% isolated yield by using CoBr2(bpy) as the catalyst, NaI as an electrolyte, in DMA solvent (entry 1, Table 1). Use of different cobalt catalysts resulted in slightly lower yields of product along with some dehalogenation of 1a as a side reaction (entries 3 and 4, Table 1). Altering the anode or cathode resulted in decreased yields (entries 5 and 7). The use of a sacrificial donor such as Et3N yielded no product (entry 8). Subsequent examination revealed that the reaction outcome was significantly dependent on the solvent. The reaction was less efficient in solvents such as ACN, DMF and NMP (entries 9–11). Noticeably, use of other supporting electrolytes such as quaternary ammonium salt and KBr also proved to be efficient in this reaction (entries 12 and 13). Finally, the control experiments verified that the electricity and cobalt catalyst were both indispensable for this transformation (entries 14 and 15).
|
|
||
|---|---|---|
| Entry | Variation from standard conditionsa,b | 2a (%) |
| a Standard conditions. 1a (0.4 mmol), CoBr2(bpy) (10 mol%), NaI (50 mol%) DMA (dimethylacetamide 4.0 mL), rt, 12 h. b Yields were determined by GC analysis, calibrated using dodecane as an internal stand. c Isolated yield. | ||
| 1 | Standard conditions | 92 |
| 2 | 10 mol% CoBr2(dtbbpy) | 87c |
| 3 | 10 mol% CoCl2(PPh3)2 | 74c |
| 4 | 10 mol% CoCl2(dppe) | 78 |
| 5 | (+) SS/(−) Ni foam | 81 |
| 6 | (+) Fe/(−) Ni foam | 86c |
| 7 | (+) Zn/(−) SS | 89c |
| 8 | (+) RVC/(−) SS, Et3N | nr |
| 9 | ACN as solvent | 42 |
| 10 | DMF as solvent | 78 |
| 11 | NMP as solvent | 72 |
| 12 | (nBu)4NBr instead of NaI | 81 |
| 13 | KBr insted of NaI | 84 |
| 14 | w/o CoBr2(bpy) | nr |
| 15 | w/o electricity | nr |
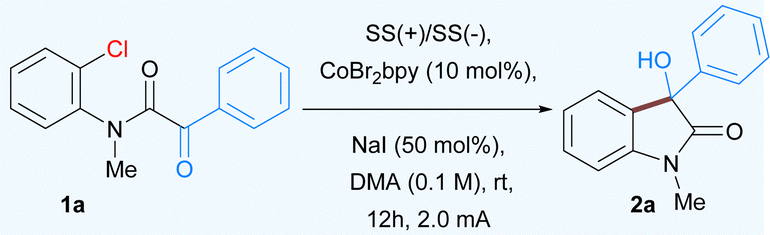
With the optimized conditions in hand, we next explored the scope of this nucleophilic addition of aryl chlorides to α-ketoamides. As shown in Table 2, good to excellent yields of the corresponding 3-hydroxyoxindoles 2a–2q were observed for the substrates bearing either electron-withdrawing groups or electron-donating groups on the phenyl rings. The replacement of methyl group to easily removable benzyl protected α-ketoamide afforded the product 2b in 81% yield. In general, methyl groups at various positions such as ortho to keto group or ortho and meta-substituted with respect to the amide group is well tolerated under the optimized reaction conditions (2d–2f). A variety of substituents, both electron-rich and electron-poor groups on either of the aromatic rings as well as methoxy, halogens, or naphthyl groups resulted in the corresponding products 2g–2l. In addition, heteroaromatic α-ketoamides with groups such as thiophene (2m), furan (2n), and indole (2o), also provided corresponding 3-hydroxyoxindoles in good to moderate yield. Notably, the current methodology is also suitable for aliphatic α-keto amides, affording butyryl cholinesterase inhibitor 2p as well as 2q in moderate yields. Surprisingly, the α-ketoester did not undergo cyclization to give 3-hydroxy-3-phenylbenzofuranone 2r.
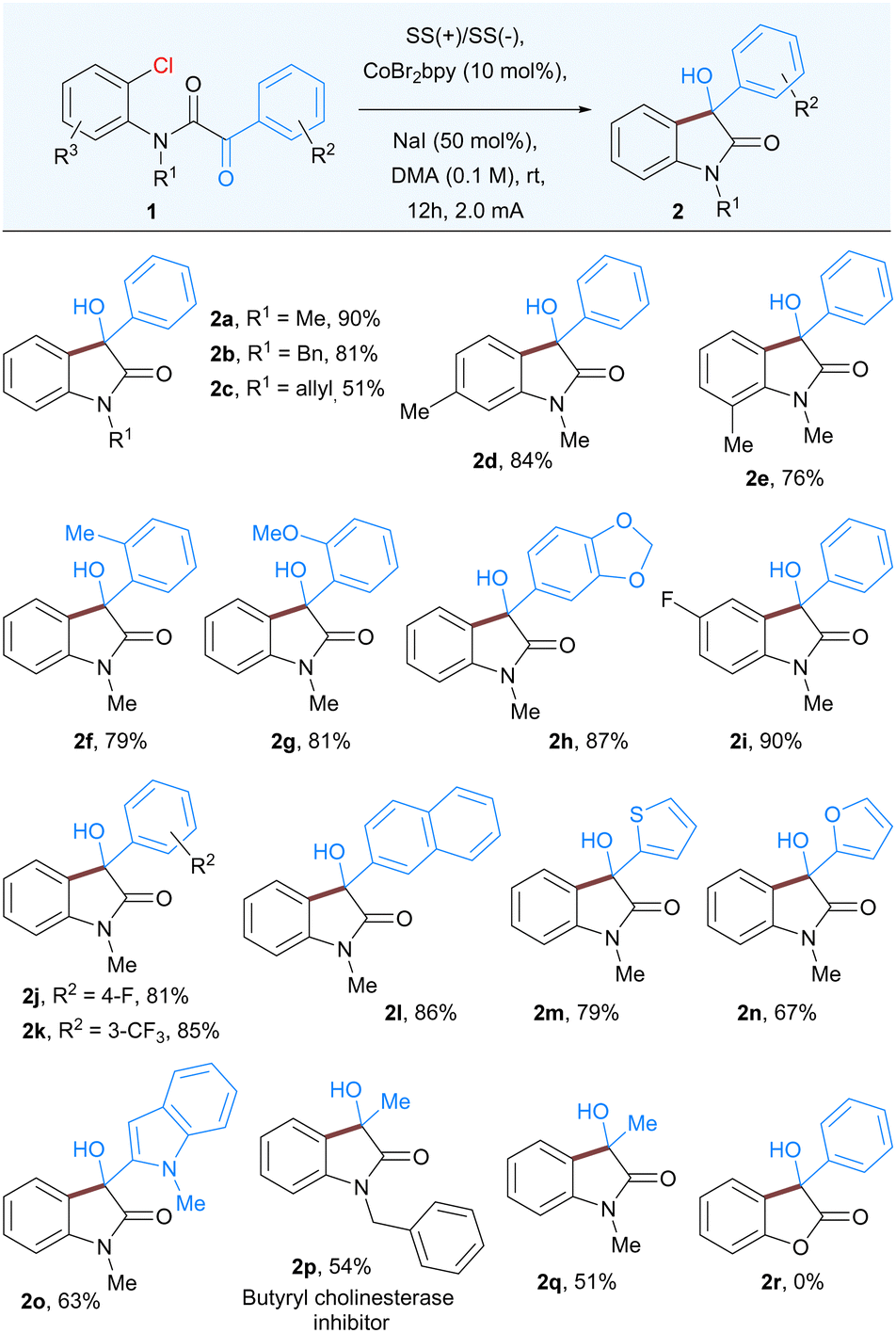
|
In order to further expand the scope and generality of the developed protocol we studied the Co-catalyzed intramolecular nucleophilic addition to α-keto amide 3 having a vinyl chloride group. As anticipated the reaction of 3a proceeded well to afford 3-hydroxy-4-methylene-1,3-diphenylpyrrolidin-2-ones 4a, albeit in lower yield (68%). Further increase in current from 2 mA to 3 mA proved beneficial for complete consumption of starting material to afford corresponding product 4a in 87% isolated yield. As expected, a variety of substituents having various steric and electronic properties at different positions of both aryl rings were well tolerated under the standard reaction conditions. Substituents, including methoxy (4b), fluoro (4c), styrenyl (4d), and thiophenyl (4e) groups were well tolerated under the optimized reaction conditions affording 3-hydroxypyrrolidinones. The electron-donating p-methoxy (4f-a), as well as electron-withdrawing substituents on the aromatic ring of aniline part such as cyano (4f-b), CF3 (4g), and a keto group (4h-a), ester (4h-b), resulted good to moderate yields. Further to compare and confirm the reactivity between aryl and vinyl chloride we prepared the substrate 3j having both chloro (aryl and vinyl) groups and subjected them to optimized reaction conditions. The reaction outcome confirmed that the vinyl chloride underwent the nucleophilic addition on α-ketoamide that led to the formation 4j-a in 30% yield as well as dehydro-chlorinated product 4j-b in 32% yield (Table 3).
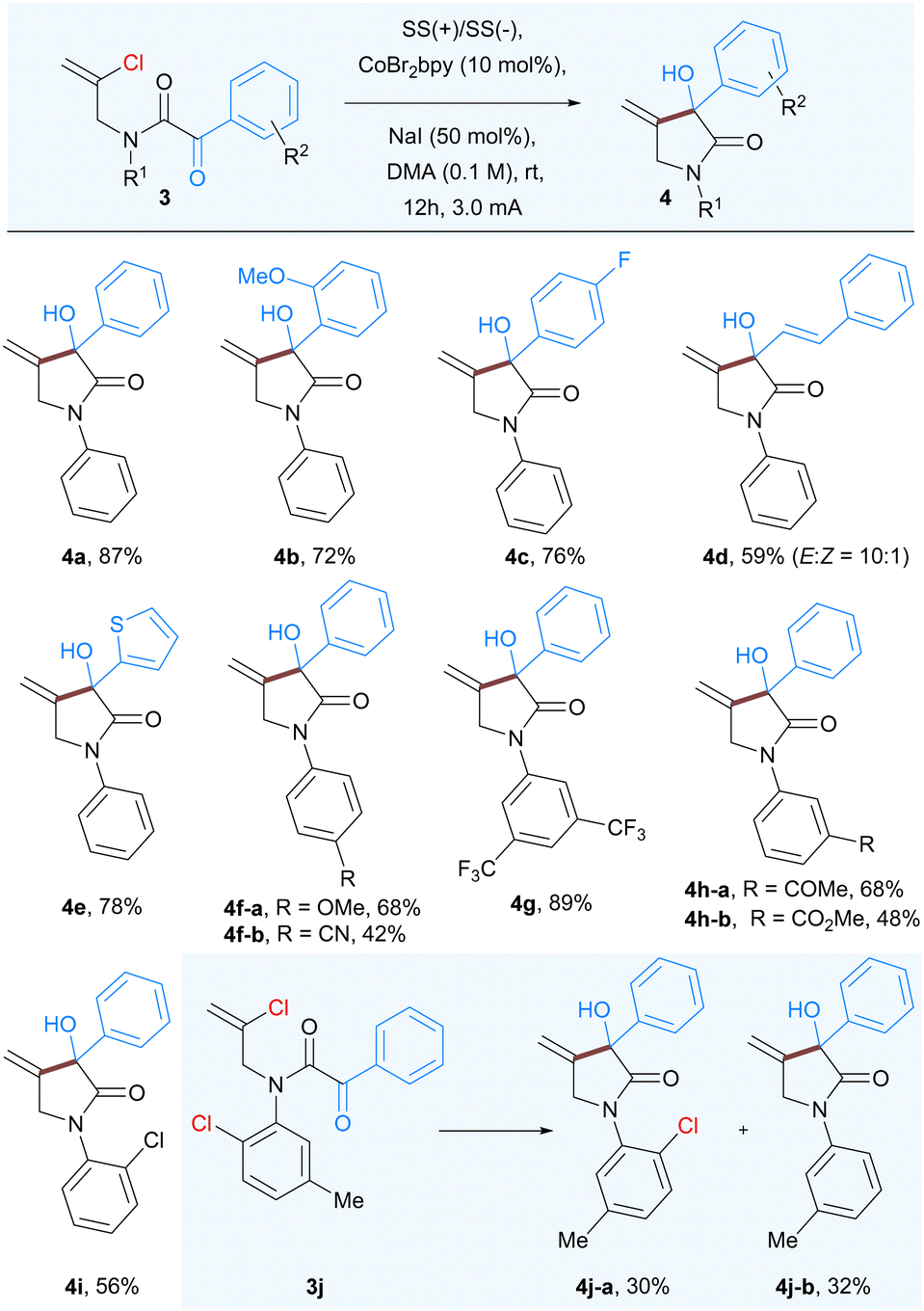
|
Furthermore, the scalability of the developed protocol is exemplified with the use of α-keto 2-chloroanilide 1a as a substrate on a 1.2-gram scale affording 90% of 3-hydroxyoxindoles 2a. Moreover, we also aimed to develop an enantioselective version of this electrochemical cobalt-catalyzed reaction. To realize the proof of concept, we tested the reaction of intramolecular nucleophilic addition of aryl chloride to α-ketoamides 1a in the presence of cobalt catalyst along with a variety of chiral ligands (for detailed optimization study see ESI†). Under electrochemical conditions, enantioenriched 3-hydroxyoxindoles 2a bearing a quaternary stereogenic center was obtained in 86% yield with 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 er (Scheme 1).15
:20 er (Scheme 1).15
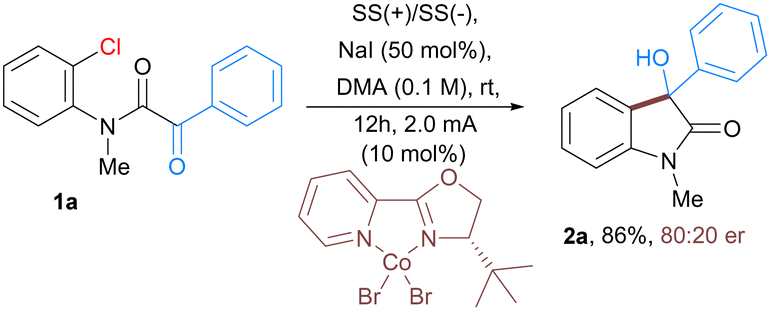 | ||
| Scheme 1 Enantioselective version of electrochemical Co-catalyzed intramolecular addition of aryl chlorides to α-ketoamide. | ||
Based on the literature reports, experimental results, and cyclic voltammetry study (for a detailed study see ESI†) we propose a reaction mechanism that accounts for the formation of the observed product (Scheme 2). The reaction is initiated by the reductive generation of the low valent cobalt species at the cathode surface from pre-catalyst CoBr2bpy via the electron transfer process. The low valent cobalt then undergoes oxidative addition with aryl chloride of 1a to provide 7-membered organometallic Co(II) intermediate B which subsequently undergoes nucleophilic addition to the electrophilic carbonyl group resulting in the formation of intermediate C. The Co(II) intermediate C then undergoes transmetalation with iron generated at the anode to give the Co(II) species that again participates in the next catalytic cycle. Another feasible pathway involves the generation of a low-valent Co(I) which undergoes oxidative addition to 1a to give a Co(III) intermediate B′. Subsequent addition and reduction leads to C which is hydrolysed during the acidic workup to afford the 3-hydroxy indole 2a.
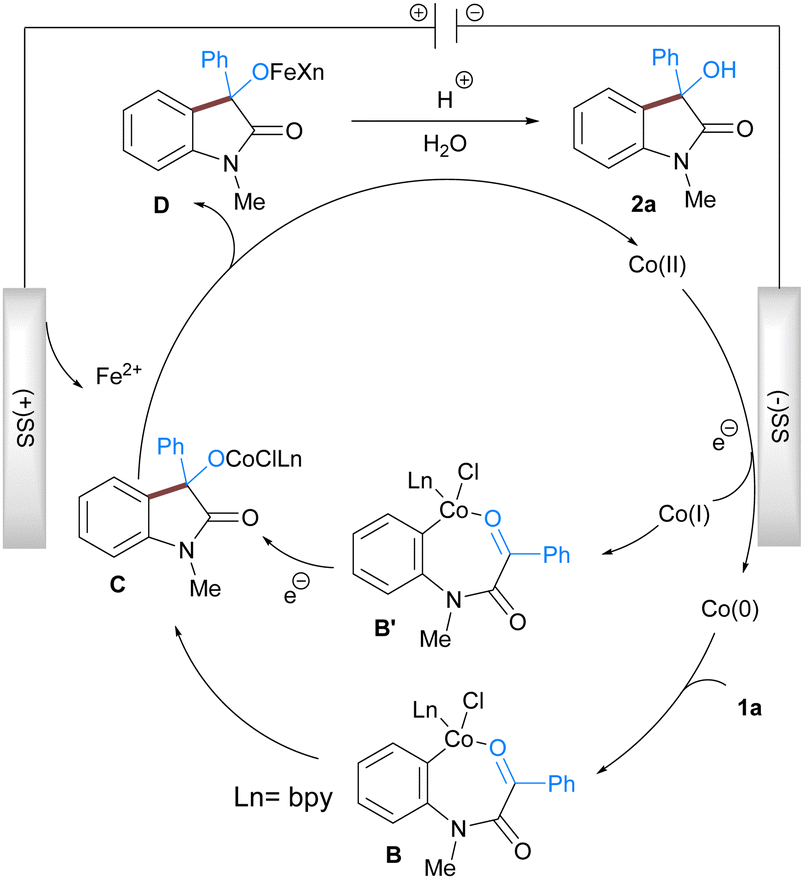 | ||
| Scheme 2 Plausible reaction mechanism. | ||
In summary, we have developed a cobalt-catalyzed nucleophilic addition of aryl and vinyl chlorides to α-ketoamides via C–Cl bond activation at room temperature under electrochemical conditions. The overall method operates via an electrochemically induced low valent cobalt catalyst that first oxidatively adds to the aryl or vinyl chlorides followed by Grignard type addition to the carbonyl group affording medicinally important 3-hydroxy oxindole and 3-hydroxypyrrolidinone scaffolds with a quaternary carbon center. Of note, the use of stoichiometric Zn, Mn, alkyl zinc reagent,16 or expensive silanes in such reductive couplings can thus be avoided. An enantioselective version of the established protocol demonstrates the feasibility under electrochemical conditions,17 thereby affording valuable enantioenriched products and paving the way for numerous prospective asymmetric electrochemical cobalt-catalyzed reactions.
We gratefully acknowledge the funding support from the King Abdullah University of Science and Technology (KAUST), Saudi Arabia, Office of Sponsored Research (URF/1/4405). VSS also gratefully acknowledged generous financial support through a Core Research Grant (CRG/2022/004970) by the Science and Engineering Research Board (SERB), DST, Government of India.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. C. Haibach, S. Shekhar, T. S. Ahmed and A. R. Ickes, Org. Process Res. Dev., 2023, 27, 423 CrossRef CAS; (b) R. A. Singer, S. Monfette, D. Bernhardson, S. Tcyrulnikov, A. K. Hubbell and E. C. Hansen, Org. Process Res. Dev., 2022, 26, 3204 CrossRef CAS; (c) H. Wu, B. Qu, T. Nguyen, J. C. Lorenz, F. Buono and N. Haddad, Org. Process Res. Dev., 2022, 26, 2281 CrossRef CAS; (d) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245 CrossRef CAS PubMed; (e) G. Pototschnig, N. Maulide and M. Schnürch, Chem. – Eur. J., 2017, 23, 9206 CrossRef CAS PubMed; (f) P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed.
- (a) C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164 CrossRef CAS PubMed; (b) X. Hu, B. M. Cossairt, B. S. Brunschwig, N. S. Lewis and J. C. Peters, Chem. Commun., 2005, 4723 RSC.
- (a) P. Gomes, C. Gosmini, J.-Y. Nedélec and J. Périchon, Tetrahedron Lett., 2000, 41, 3385 CrossRef CAS; (b) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115 RSC.
- (a) J. C. A. Oliveira, U. Dhawa and L. Ackermann, ACS Catal., 2021, 11, 1505 CrossRef CAS; (b) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452 CrossRef CAS PubMed; (c) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef CAS; (d) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed.
- (a) H. Chen, C. Zhu, H. Yue and M. Rueping, ACS Catal., 2023, 13, 6773 CrossRef CAS; (b) A. Guérinot and J. Cossy, Acc. Chem. Res., 2020, 53, 1351 CrossRef PubMed; (c) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435 CrossRef CAS PubMed; (d) C. Gosmini and A. Moncomble, Isr. J. Chem., 2010, 50, 568 CrossRef CAS; (e) C. Gosmini, J.-M. Begouin and A. Moncomble, Chem. Commun., 2008, 3221 RSC.
- (a) O. Buriez, E. Labbe and J. Périchon, J. Electroanal. Chem., 2006, 593, 99 CrossRef CAS; (b) O. Buriez, C. Cannes, J. Y. Nédélec and J. Périchon, J. Electroanal. Chem., 2000, 495, 57 CrossRef CAS.
- (a) A. J. Fry and U. N. Sirisoma, J. Org. Chem., 1993, 58, 4919 CrossRef CAS; (b) A. J. Fry, U. N. Sirisoma and A. S. Lee, Tetrahedron Lett., 1993, 34, 809 CrossRef CAS; (c) J.-C. Folest, J.-M. Duprilot, J. Périchon, Y. Robin and J. Devynck, Tetrahedron Lett., 1985, 26, 2633 CrossRef CAS.
- (a) B. P. Branchaud and W. D. Detlefsen, Tetrahedron Lett., 1991, 32, 6273 CrossRef CAS; (b) S. Torii, T. Inokuchi and T. Yukawa, J. Org. Chem., 1985, 50, 5875 CrossRef CAS; (c) R. Scheffold, M. Dike, S. Dike, T. Herold and L. Walder, J. Am. Chem. Soc., 1980, 102, 3642 CrossRef CAS.
- (a) Y.-X. Jia, D. Katayev and E. P. Kundig, Chem. Commun., 2010, 46, 130 RSC; (b) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS PubMed; (c) M. Durandetti, J. Périchon and J.-Y. Nedelec, Org. Lett., 2001, 3, 2073 CrossRef CAS PubMed; (d) M. Durandetti, J. Périchon and J.-Y. Nedelec, Tetrahedron Lett., 1999, 40, 9009 CrossRef CAS.
- (a) C. Dorval and C. Gosmini, Cobalt Catal. Org. Synth., 2020, 163 CAS; (b) C. Dorval, M. Tricoire, J.-M. Begouin, V. Gandon and C. Gosmini, ACS Catal., 2020, 10, 12819 CrossRef CAS; (c) M. Amatore and C. Gosmini, Chem. – Eur. J., 2010, 16, 5848 CrossRef CAS PubMed; (d) Y. Cai, A. D. Benischke, P. Knochel and C. Gosmini, Chem. – Eur. J., 2017, 23, 250 CrossRef CAS PubMed.
- D. P. Hickey, C. Sandford, Z. Rhodes, T. Gensch, L. R. Fries, M. S. Sigman and S. D. Minteer, J. Am. Chem. Soc., 2019, 141, 1382 CrossRef CAS PubMed.
- (a) A. Kumar and S. S. Chimni, RSC Adv., 2012, 2, 9748 RSC; (b) S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS; (c) H. V. Erkizan, Y. Kong, M. Merchant, S. Schlottmann, J. S. BarberRotenberg, L. Yuan, O. D. Abaan, T.-H. Chou, S. Dakshanamurthy, M. L. Brown, A. Üren and J. A. Toretsky, Nat. Med., 2009, 15, 750 CrossRef CAS PubMed; (d) D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6946 CrossRef CAS PubMed; (e) T. Tokunaga, W. E. Hume, T. Umezome, K. Okazaki, Y. Ueki, K. Kumagai, S. Hourai, J. Nagamine, H. Seki, M. Taiji, H. Noguchi and R. Nagata, J. Med. Chem., 2001, 44, 4641 CrossRef CAS PubMed.
- (a) S. Denoyelle, T. Chen, H. Yang, L. Chen, Y. Zhang, J. A. Halperin, B. H. Aktas and M. Chorev, Eur. J. Med. Chem., 2013, 69, 537 CrossRef CAS PubMed; (b) J. Totobenazara, P. Bacalhau, A. A. San Juan, C. S. Marques, L. Fernandes, A. Goth, A. T. Caldeira, R. Martins and A. J. Burke, ChemistrySelect, 2016, 1, 3580 CrossRef CAS.
- L. Polleux, E. Labbé, O. Buriez and J. Périchon, Chem. – Eur. J., 2005, 11, 4678 CrossRef CAS PubMed.
- (a) Y. Li and S. Xu, Chem. – Eur. J., 2018, 24, 16218 CrossRef CAS PubMed; (b) B. Yu, H. Xing, D.-Q. Yu and H.-M. Liu, Beilstein J. Org. Chem., 2016, 12, 1000 CrossRef CAS PubMed.
- J.-X. Hu, H. Wu, C.-Y. Li, W.-J. Sheng, Y.-X. Jia and J.-R. Gao, Chem. – Eur. J., 2011, 17, 5234 CrossRef CAS PubMed.
- M. Gosh, V. S. Shinde and M. Rueping, Beilstein J. Org. Chem., 2019, 15, 2710 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00309h |
| This journal is © The Royal Society of Chemistry 2024 |