Magnetic Fe–N–C nanoparticles as a dual nanozyme for label-free colorimetric detection of antibiotics†
Received
15th December 2022
, Accepted 22nd March 2023
First published on 23rd March 2023
Abstract
In this study, we report a simple pyrolytic strategy for N–C co-doped Fe-based nanoparticles (Fe–N–C) and exhibit their oxidase-mimicking and peroxidase-mimicking activity in a chromogenic reaction with 3,3′,5,5′-tetramethylbenzidine dihydrochloride (TMB) as a substrate. Structural analysis revealed that Fe–NX and Fe3C structures were formed with Fe-Imace coordination compounds as precursors and melamine as the nitrogen source, which provided abundant active sites for the Fe–N–C nanozyme. Interestingly, owing to the blocking effect triggered by π–π stacking between the tetracycline antibiotics (TCs) tetraphenyl skeleton and Fe–N–C, the substrate affinity of the Fe–N–C was significantly blocked, resulting in the solution fading. Under the optimum conditions, the UV absorption intensity versus the concentration of TCs was found to be linear over the range of 0.08–90 μM for tetracycline (TC), 0.1–80 μM for chlortetracycline (CTC) and 0.09–100 μM for oxytetracycline (OTC). The detection limits were 62 nM for TC, 85 nM for CTC and 88 nM for OTC. The Fe–N–C displayed good storage and long-term stability when stored at 4 °C for 30 days. In addition, based on its superparamagnetism, Fe–N–C can be effectively recycled and reused through an external magnetic field, avoiding secondary pollution.
Environmental significance
Today, the presence of tetracycline antibiotics (TCs) in aquatic environments has drawn great attention from environmental scientists. The establishment of fast and convenient detection methods is one of the first problems to be solved in environmental analysis. Therefore, nanozymes are deemed ideal materials for constructing colorimetric sensing platforms for detection and analysis of antibiotics and have important environmental significance.
|
1 Introduction
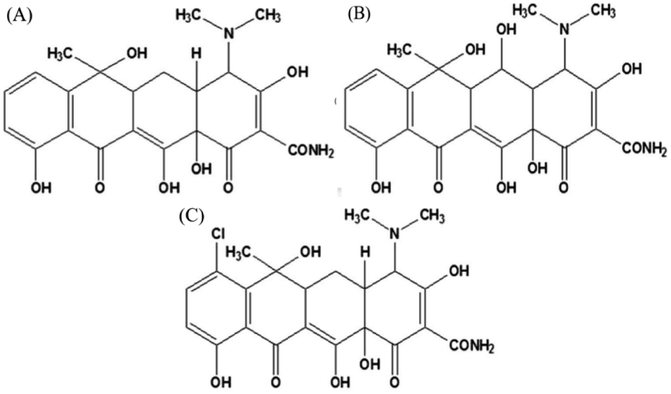
Tetracycline antibiotics (TCs) are a class of broad-spectrum antibiotics that include tetracycline (TC), chlortetracycline (CTC), oxytetracycline (OTC), etc. (Fig. 1). Due to their good bactericidal effect, they are typically used to treat human diseases or in livestock husbandry. However, improper or excessive use of TCs can lead to high levels of drug residues, posing a serious threat to the environment and human health. To date, various convenient and accurate techniques have been applied to TCs monitoring. Instrumental analysis such as liquid chromatography-mass spectrometry (LC/MS),1 high performance liquid chromatography (HPLC)2,3 and capillary electrophoresis (CE)4 showed a high degree of automation, as well as satisfactory sensitivity and accuracy. However, the above techniques require long analysis time and expensive instruments, which make it difficult to achieve real-time analysis of large-scale samples. In addition, electrochemical,5 fluorescent6 and chemiluminescent (CL)7 sensors have been developed for the detection of TCs due to their advantages of low detection limit and fast detection speed, but they still require complex electrode processing procedures or rely on toxic organic compounds as fluorescent sources. Colorimetry, by contrast, is one of the simplest detection methods that depends on the variation of color during the detection program. In addition, colorimetry does not require sophisticated or expensive instruments, and color variation can be seen directly by the naked eye to facilitate field and real-time analysis. Among them, nanozymes are deemed ideal materials for constructing colorimetric sensing platforms due to their ability to produce reactive oxygen species (ROS) and thus cause color variation in substrates such as 3,3′,5,5′-tetramethylbenzidine dihydrochloride (TMB), 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulphonate) (ABTS) and o-phenylenediamine (OPD).
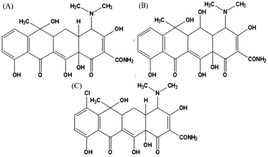 |
| | Fig. 1 Chemical structure of (A) tetracycline, (B) oxytetracycline, and (C) chlortetracycline. | |
Due to their excellent specificity and high catalytic activity, natural enzymes are widely used in biosensors, the food industry and pharmaceutical processes.8 However, their broad-spectrum applications are still limited due to their environmental sensitivity and the high cost of large-scale preparation. Recent reports in the field of nanotechnology, such as those on quantum dots,9 metal–organic frameworks,10 metal nanoparticles11 and metal oxide nanoparticles,12 indicated that nanomaterials with enzyme-mimicking properties are expected to be viable substitutes for natural enzymes due to their high catalytic activity, recoverability and stability.13–15 Notably, nanozymes have been proved to possess the properties needed for producing ROS, such as having oxidase (OXD)-mimicking or peroxidase (POD)-mimicking activity with the ability to catalyze O2 and H2O2 to produce highly oxidizing ROS, respectively.16 Among them, researchers have confirmed that the mechanism of OXD-mimicking activity is similar to the oxygen reduction reaction (ORR).17 Nevertheless, the practical application of nanozymes still faces some challenges. For example, non-precious metal catalysts (NPMCs) with high enzyme-mimicking activity need to replace precious metal group (PMG) materials; it is necessary to improve the activity and stability of nanozymes by regulating the material's morphology and constructing recyclable nanozymes to ensure the economic benefit and environmental performance. Studies have revealed that metal ion centers are beneficial for the initial adsorption of oxygen molecules by reaction intermediates, thus enhancing the oxygen reduction activity.18 It has been reported that the specificity and catalytic activity of nano-enzyme materials can be effectively improved by the introduction of nitrogen heteroatoms.19 Therefore, transition metals containing nitrogen coordination sites (M–NX, M = Cu, Fe, Co, etc.) are generally considered to be the most promising alternatives to PMG. Furthermore, carbon-based nanomaterials have unique advantages in simulating the active sites and specific geometries of natural enzymes.20 In particular, N–C co-doped Fe material (Fe–N–C) is expected to be an efficient nanozyme and a suitable investigation platform. Wang et al. synthesized and compared M–N–C (M = Fe, Co, Ni) catalysts with similar coordination, and found that Fe–N–C had the highest enzyme-mimicking catalytic activity.21 Although many researchers have reported that nanomaterials mimic the enzyme properties of either oxidase or peroxidase separately to detect antibiotics, our study proposes a novel Fe–N–C nanozyme with dual enzyme activities without binding aptamers for TCs detection.
Here, Fe–N–C was successfully synthesized by a one-step pyrolysis method as a functional nanozyme with dual enzyme-mimicking catalytic activities. In a weakly acidic environment, the Fe–N–C nanozyme could not only act as an OXD-mimic to generate ROS through the reduction of molecular oxygen in the absence of H2O2, but also showed significant POD-mimicking catalytic performance to generate a large amount of ˙OH in the presence of H2O2. The synergistic enhancement mechanism of the above dual enzyme-mimicking catalytic activities was used for the catalytic oxidation of substrates TMB, OPD and ABTS, and the sensing solution showed typical color variation. In the presence of TCs, by contrast, the oxidation of substrates was inhibited, and the color depth of the sensing solution negative correlated with the concentration of TCs. More importantly, the potential mechanisms of Fe–N–C mimicking enzyme activity and TC inhibiting enzyme activity of Fe–N–C were systematically studied by steady-state kinetics, ROS detection and scavenging experiments, and density functional theory (DFT) calculations. Finally, the specific colorimetric detection of TCs was realized based on the prepared nano-enzyme, and the validity of the sensor was finally verified by real samples.
2 Experimental section
2.1 Materials and apparatus
Iron(II) acetate ((CH3COO)2Fe, >90%), 1-imidazoleacetic acid (Imace, 98 wt%) and melamine (99 wt%) were purchased from Shanghai Aladdin Technology Co., Ltd (Shanghai, China). Tetracycline hydrochloride, oxytetracycline dihydrate, chlortetracycline hydrochloride, doxorubicin (DOX), amoxicillin (AML), nitrofurazone (NFZ), kanamycin (KAN), streptomycin (STR), and chloramphenicol (CPL) were purchased from Shanghai Macklin Co., Ltd (Shanghai, China). Acetic acid (HAc, 99.5 wt%), sodium acetate (NaAc, 99 wt%), 3,3′,5,5′-tetramethylbenzidine dihydrochloride (TMB, >99 wt%), potassium thiocyanate (99 wt%), p-benzoquinone (p-BQ) (99 wt%), L-histidine (99 wt%) and isopropanol were purchased from Shanghai Macklin Co., Ltd (Shanghai, China). Hydrogen peroxide solution (30 wt%) was purchased from Shanghai Aladdin Technology Co., Ltd (Shanghai, China). All of the chemicals were of analytical reagent grade and deionised water was used throughout the whole experiment.
The pyrolysis treatment was carried out in a tube electric resistance furnace (SK-G05123K, Tianjin Zhonghuan Electric Furnace Co., Ltd). The microscopic structure of the samples was characterized by scanning electron microscopy (SEM, FEI QUANTA FEG250, USA) and high resolution transmission electron microscopy (HRTEM, JEM-2100 F, Japan). The surface chemical composition was tested using X-ray photoelectron spectroscopy (XPS, Thermo Fisher, ESCALAB 250Xi, USA). The X-ray diffraction (XRD) patterns of the samples were examined with a Rigaku MiniFlex600 X-ray diffractometer (Japan) with radiation under the 2θ range of 10° to 80°. Ultraviolet spectra were recorded using a UV-vis spectrometer (Thermo Fisher, Lambda 365, USA).
2.2 Synthesis of Fe–N–C
Fe–N–C was synthesized based on Li's work with minor modifications.22 Briefly, 3.36 g Imace and 2.32 g (CH3COO)2Fe were added to 40 mL deionised water and stirred continuously for 6 h at 70 °C. Subsequently, 67 mL deionised water and 5.33 g melamine were added for ultrasonic treatment. After being stirred at room temperature for 6 h, the above solution was dried at 70 °C for 15 h, and a brown powder was obtained. To synthesize Fe–N–C, the product was first placed in a ceramic vessel and the tube furnace was heated to 550 °C at a programmed rate of 3 °C min−1, and kept for 2 h at 550 °C. Then, the temperature was raised to 700, 800 and 900 °C and maintained for 2 h with a ramp rate of 3 °C min−1, respectively. All the above pyrolysis processes were carried out in a nitrogen atmosphere. After cooling to room temperature, the black products were leached in 1 M HCl solution and deionised water three times to remove the unstable iron nanoparticles, then dried overnight at 70 °C. The obtained black materials were denoted as Fe–N–C-700, Fe–N–C-800 and Fe–N–C-900 respectively to represent pyrolysis temperature. The synthesis process of N–C was similar to that of the Fe–N–C, except that no (CH3COO)2Fe was added. The black powders obtained were named Fe–N–C-X, where “X” indicates the pyrolysis temperature.
2.3 Dual-response enzyme mimicking activity of Fe–N–C
The OXD-mimicking and POD-mimicking activities of Fe–N–C were assessed by OPD, ABTS and TMB as reaction substrates in a colorimetric assay. In a typical experiment, 80 μL of 10 mM TMB, 80 μL of 0.1 M H2O2 (present or absent) and 9 μL of 1 mg mL−1 Fe–N–C nanozyme aqueous dispersion were mixed with 1.5 mL HAc-NaAc buffer solution (pH = 4). The color variation of the mixed solution and the absorbance change at 652 nm in a UV-vis spectrophotometer was monitored at room temperature over 17 minutes.
2.4 Steady-state kinetic analysis
The steady-state kinetic measurements were carried out in time-course mode, and all measurements were based on the absorbance change of ox-TMB at 652 nm. The absorbance coefficient of the product (ox-TMB) at 652 nm is 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1. To determine the initial reaction rate, different concentrations of H2O2 were reacted with Fe–N–C at a certain concentration of TMB, and vice versa. The rate of reaction is the slope of the absorbance changes with time. The kinetic parameters of enzyme activity were calculated using the Lineweaver–Burk plot of the double reciprocal of the Michaelis Menten equation: 1/v = Km/Vmax(1/[S] + 1/Km), where V, Vmax, Km and [S] represent the initial velocity of the reaction, the maximal reaction velocity, the Michaelis–Menten constant and the substrate concentration, respectively.
000 M−1 cm−1. To determine the initial reaction rate, different concentrations of H2O2 were reacted with Fe–N–C at a certain concentration of TMB, and vice versa. The rate of reaction is the slope of the absorbance changes with time. The kinetic parameters of enzyme activity were calculated using the Lineweaver–Burk plot of the double reciprocal of the Michaelis Menten equation: 1/v = Km/Vmax(1/[S] + 1/Km), where V, Vmax, Km and [S] represent the initial velocity of the reaction, the maximal reaction velocity, the Michaelis–Menten constant and the substrate concentration, respectively.
2.5 Mechanism analysis
Different active species, including O2−, ˙OH and 1O2, are captured through various scavengers. Fe–N–C, H2O2, TMB, p-BQ, isopropanol and L-histidine were put into HAc-NaAc buffer (pH = 4) with a total volume of 1.5 mL under the optimum experimental conditions. The absorbance (652 nm) of the system was recorded at ambient temperature after 17 min.
All simulations were performed using first principles calculations based on density functional theory (DFT) within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) formulation with Grimme's energy method in the Dmol3 module. All the geometry optimizations were performed using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm. The k-points were set at gamma (1 × 1 × 1) and the convergence tolerance was set at 1.0 × 10−5 Ha, 2.0 × 10−3 Ha Å, and 5.0 × 10−3 Å for energy, maximum force, and maximum displacement, respectively.
2.6 TCs detection
In a typical assay of antibiotics, 9 μL of 1 mg mL−1 Fe–N–C aqueous dispersion, 80 μL of 10 mM TMB, 80 μL of 0.1 M H2O2 and gradient concentrations of antibiotics were mixed in 1.5 mL HAc-NaAc buffer solution (pH = 4) for 17 minutes at room temperature, and the absorbance of the mixed solution was recorded at 652 nm.
2.7 Detection of antibiotics in actual samples
Lake water samples were collected from Shanxi University and filtered through a 0.22 μm microporous filter. The subsequent detection procedures were similar to those described previously.
3 Results and discussion
3.1 Characterization of Fe–N–C
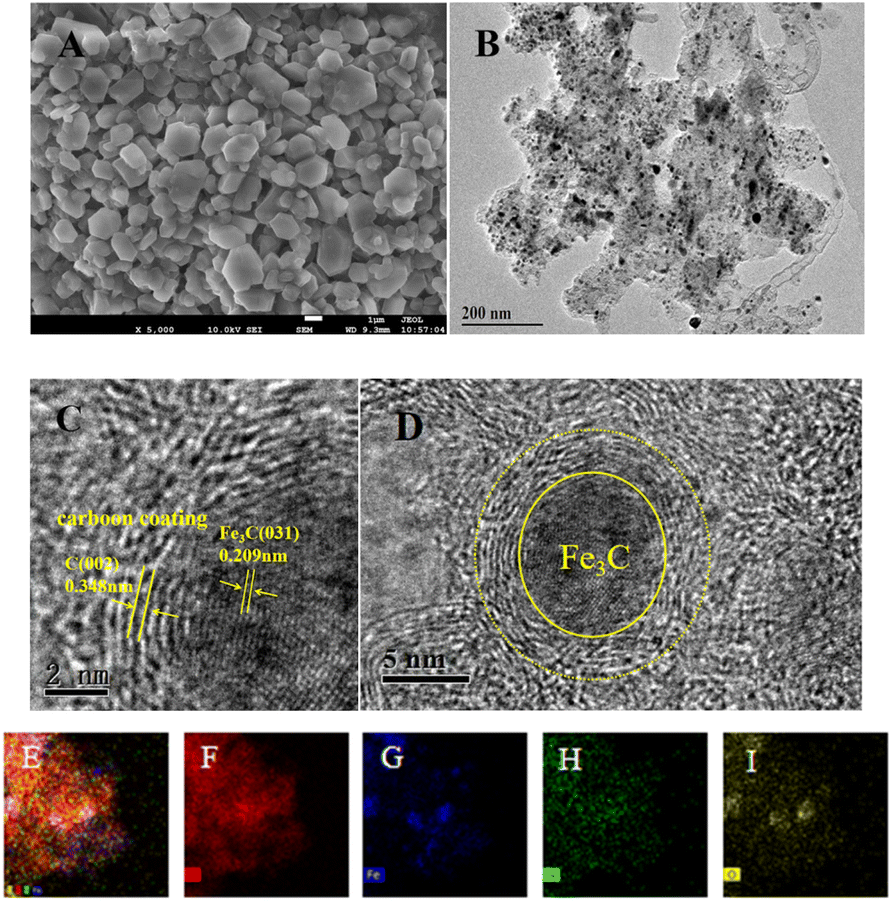
The micromorphology and microstructure of the prepared Fe–N–C were characterized by SEM and TEM. As depicted in Fig. 2A, the SEM micrograph clearly shows that the Fe–N–C has uniform polyhedral morphology and is stacked up to some extent. Fig. S1† shows the micromorphology of Fe–N–C-X at different calcination temperatures. It can be seen that the particle size of Fe–N–C-X decreased with the increase of calcination temperature, and the smaller particle size indicates a higher catalytic efficiency to some extent. More importantly, higher calcination temperatures lead to a higher degree of graphitization. As seen from Fig. 2B–D, the TEM images show that lots of tiny Fe based nanoparticles in the form of hyperfine Fe3C nanoparticles are evenly distributed on or in the carbon layers, which is due to the fact that Fe3C nanoparticles can effectively promote the graphitization of the surrounding C atoms. The Fe3C nanoparticles coated by graphitic carbon layers can not only enrich the electron density on the carbon surface, but also avoid the dissolution and agglomeration caused by direct contact between Fe3C and the external medium, ensuring favorable catalytic and stability activity of Fe–N–C. Furthermore, the elemental mapping images validate the uniform dispersion of C, Fe, N and O elements in the carbon matrix (Fig. 2E–I). These results indicate that Fe–N co-doped carbon structure catalysts can be easily synthesized by pyrolysis.
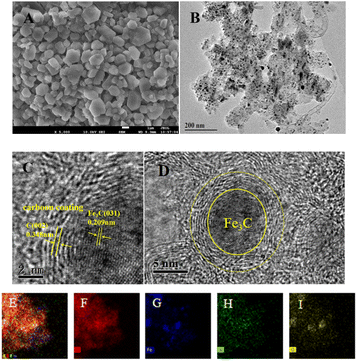 |
| | Fig. 2 (A) SEM image of Fe–N–C-900; (B) TEM and (C and D) HR-TEM images of Fe–N–C-900; (E–I) the TEM-EDX mapping images of C, Fe, N, and O elements. | |
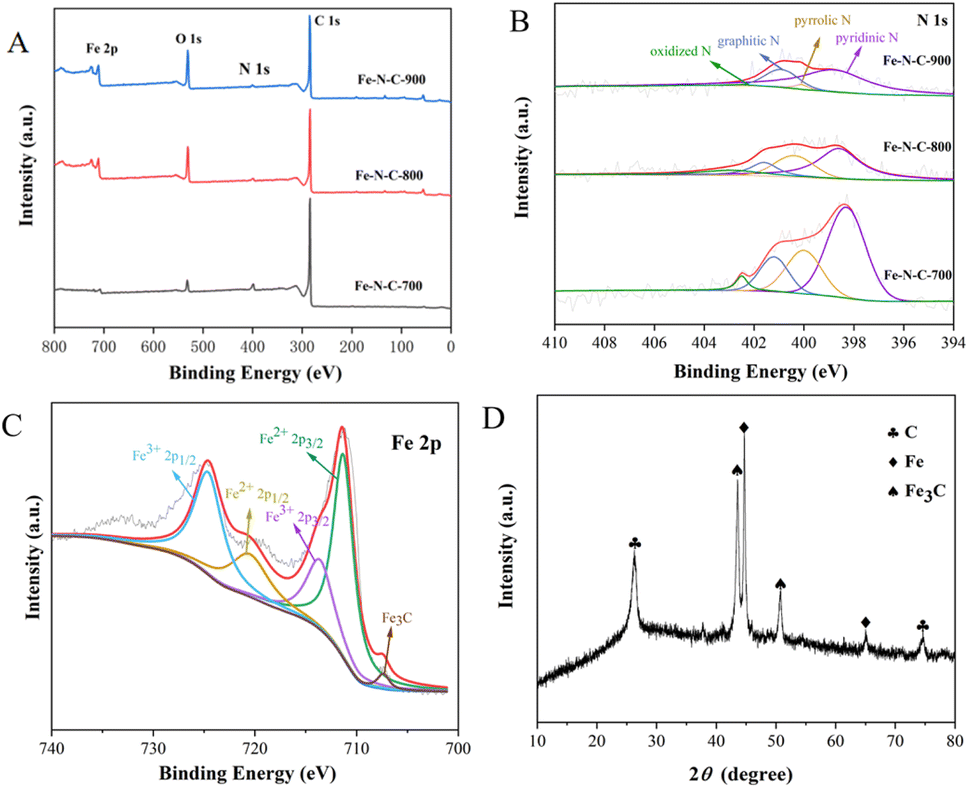
The chemical composition and elemental binding states of Fe–N–C-X were further characterized by XPS. As illustrated in Fig. 3A, Fe, C, N and O can be clearly identified from XPS spectra. This result matched well with the mapping result. According to the integrated peak areas, the atomic ratios of Fe, C, N and O on the surfaces of Fe–N–C-X are listed in Table S1.† The results showed that the atomic percentage of Fe increased gradually with increasing calcination temperatures. However, the N atomic percentage showed the opposite trend. Among them, the low Fe content may be due to the fact that Fe3C nanoparticles are wrapped in the graphitized carbon layer and cannot be detected. The N 1s emission spectra (Fig. 3B) of Fe–N–C can be fitted as four peaks at 398.4 eV, 399.7 eV, 401.2 eV and 402.5 eV, corresponding to pyridinic N, pyrrolic N, graphitic N and oxidized N, respectively. Moreover, although the N atomic percentage decreased with the increase of calcination temperature, the ratio of graphitic N varied from 14.12% to 23.30%, indicating that the graphitization degree of Fe–N–C catalysts was enhanced by increasing the temperature. The results are in good agreement with the TEM results. It has been proved that the pyridinic N and graphitic N are rich in active sites and thus accelerate the ORR process by facilitating the adsorption of O2 and the breaking of O–O bonds.23 Besides, pyridinic N could link with Fe atoms to form Fe–NX active centers, which further improves oxygen reduction performance because they are conducive to the initial adsorption of oxygen.24 Furthermore, the high resolution Fe 2p spectra of Fe–N–C-X are shown in Fig. 3C, which can be fitted to two pairs of peaks (Fe 2p3/2 and Fe 2p1/2). Among them, the peaks at the binding energies of 711.3 eV and 713.6 eV are attributed to the Fe 2p3/2 orbitals of Fe(II) and Fe(III), and the peaks at the binding energies of 720.5 eV and 724.6 eV are attributed to the Fe 2p1/2 orbitals of Fe(II) and Fe(III), indicating the co-existence of Fe(II) and Fe(III). Abundant Fe(II) species are beneficial to the generation of the Fe–NX configuration with high catalytic activity. Meanwhile, the peak of Fe3C is observed at the binding energy of 706.8 eV, which is consistent with TEM results. According to the above XPS analysis, the prepared Fe–N–C is rich in active sites due to the coexistence of Fe3C, pyridinic N (Fe–NX) and graphitic N, which plays a positive role in improving the catalytic activity of the catalyst.
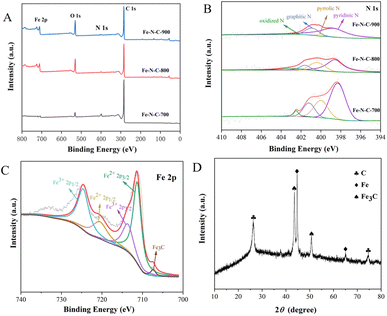 |
| | Fig. 3 (A) XPS survey spectra of Fe–N–C-X; (B and C) XPS spectra of N 1s and Fe 2p; (D) XRD pattern of Fe–N–C. | |
To determine the structure of the Fe–N–C, the XRD spectrum was measured and is presented in Fig. 3D. In the XRD pattern, the strong peak near 26.0° and the weak peak around 75.0° both correspond to the typical (002) lattice plane of graphitic carbon, while the diffraction peaks at 43.7° and 51.0° are attributed to Fe3C, and the remaining diffraction peaks are attributed to metallic iron. The XRD results show that the Fe–N–C is crystalline, and demonstrate the co-existence of graphitic carbon and iron carbide in the Fe–N–C, which confirmed the TEM results again. Previous studies have suggested that the most significant parameters affecting the catalytic efficiency and stability are crystallinity and structure.25 SEM micrographs have revealed that Fe–N–C has a clear boundary, neat arrangement and sharp edges. At the same time, the good crystal structure of Fe–N–C is conducive to the improvement of catalytic activity.
3.2 Dual response enzyme-mimicking activity
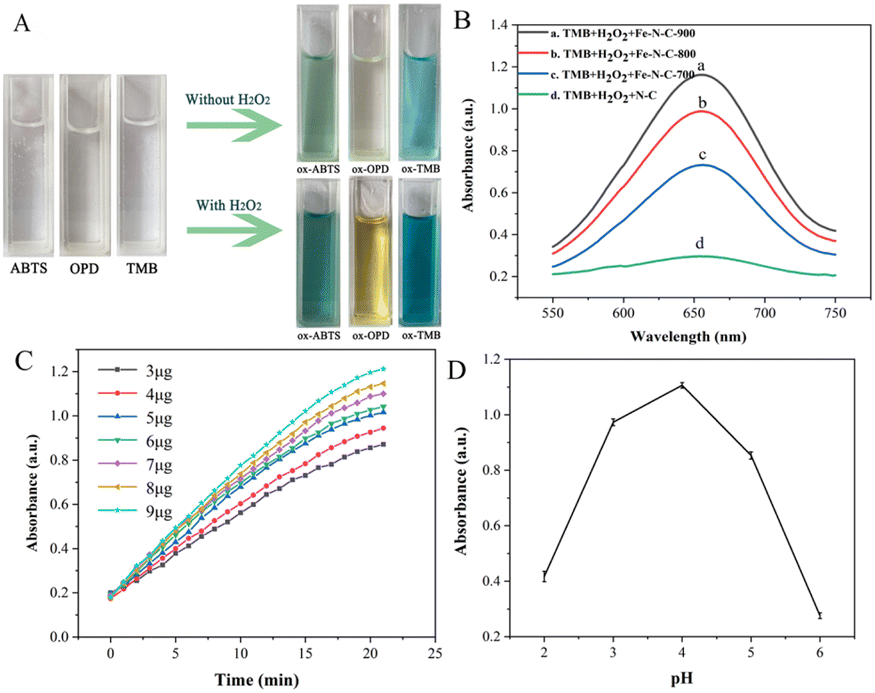
OPD, ABTS and TMB are commonly used as chromogenic substrates to demonstrate oxidase and peroxidase activities in nanozyme. To assess the catalytic activities of Fe–N–C, the experiments of catalytic substrate oxidation were carried out in the presence and absence of H2O2, respectively. We found that the Fe–N–C can catalyze the oxidation of OPD, ABTS and TMB by dissolved oxygen in the absence of H2O2, and the above solutions eventually changed from colorless to typical yellow, green and blue (Fig. 4A). Fig. S2† displays the UV absorption spectrum of TMB catalyzed oxidation, with the maximum absorption peak at approximately 652 nm. The experimental results support our initial presumption that the Fe–N–C has OXD-mimicking activity. In contrast, with the participation of H2O2, the color variations were much faster and showed darker colors at the same time intervals. The results confirmed that the Fe–N–C exhibits intrinsic POD-mimicking properties. An additional control experiment without Fe–N–C showed no color variation, which further proved the bifunctional enzyme-like catalytic activities of Fe–N–C.
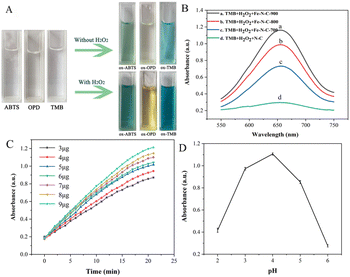 |
| | Fig. 4 (A) Dual response enzyme-mimicking activity; (B) effect of materials on the catalytic efficiency of Fe–N–C; (C) time-dependent absorbance changes of the reaction system for different Fe–N–C concentrations; (D) effect of pH on the catalytic efficiency of Fe–N–C. | |
The effect of materials on the catalytic activity of TMB was investigated. Fig. 4B shows that the Fe–N–C/TMB/H2O2 system exhibits stronger absorption at 652 nm than the N–C/TMB/H2O2 system, indicating the excellent enzyme-mimicking activity of Fe–N–C and the importance of Fe active sites. By comparison, we also evaluated the catalytic activity of Fe–N–C synthesized at different temperatures for TMB, respectively. It can be observed that with the increase of pyrolysis temperature from 700 °C to 900 °C, the catalytic activity of Fe–N–C for TMB is enhanced. This is because high temperature increases the value of the specific surface area and more active sites are exposed after pickling, which is favorable for efficient oxygen transport and rapid catalytic reaction. In addition, it has been confirmed that high temperature is conducive to nucleation of nanoparticles, while low temperature is conducive to nuclei growth. An increase of synthesis temperature results in small-sized nanoparticles. Taking this into account as well as the results of both TEM and XPS, Fe–N–C-900 was selected as an optimal material for subsequent analyses.
Similar to the natural enzyme, the catalytic activity of Fe–N–C is influenced by many external factors in practical measurements. Therefore, we optimized the experimental conditions, including the reaction time, the amount of Fe–N–C added and the pH of the HAc–NaAc buffer solution. The reaction time ranged from 0 to 20 minutes, and the dark blue solution appeared at 17 minutes (Fig. 4C). After 17 minutes, the intensity of the characteristic peak remained almost constant, demonstrating that the TMB molecules were basically completely oxidized. Considering the practicability and operability, the addition amount of Fe–N–C was set at 9 μg for subsequent analysis. Similar to natural enzymes, Fe–N–C exhibited highly pH-dependent properties. For the tests to determine the pH effect, the pH value of HAc–NaAc buffer was adjusted from 2 to 6, and the results showed that pH = 4 was the optimal pH for this catalytic reaction, which was similar to the results of previous studies (Fig. 4D).26 Some publications have confirmed that a slightly acidic solution is conducive to the decomposition of H2O2 that generates the ˙OH radical, but H2O2 becomes unstable at high pH and easily decomposes into H2O and O2.27
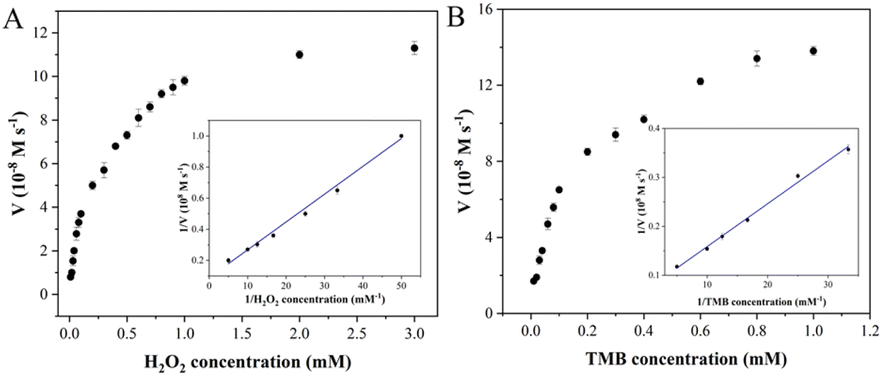
3.3 Kinetic analysis of Fe–N–C
Steady-state kinetic analysis of Fe–N–C with enzyme-like activity was performed by adjusting one substrate concentration while keeping the other substrate concentration fixed. The experimental data were obtained by detecting absorbance at 652 nm, and the Michaelis–Menten curves (Fig. 5) were obtained. The molar extinction coefficient of ox-TMB in the Beer–Lambert law equation was used to calculate the variation of absorbance per unit time.
where A652 represents the absorbance of ox-TMB at 652 nm, εox-TMB represents the molar extinction coefficient (39000 M−1 cm−1) and l represents the path length of the sample in the cuvette (1 cm). Subsequently, the data were fitted to the Lineweaver–Burk plot (insets in Fig. 5A and B) to obtain the kinetic parameters such as Michaelis–Menten constant (Km) and maximum initial velocity (Vmax). Among them, Km is an indicator of enzyme affinity to the substrate, and the smaller the Km value is, the higher the affinity of the enzyme to the substrate is. Horseradish peroxidase (HRP) with high peroxidase–mimicking activity was used as the reference. As shown in Table S2,† the Km values of Fe–N–C with H2O2 and TMB as the substrate are 0.2 and 0.124 respectively, both lower than that of HRP, illustrating Fe–N–C had a higher affinity and activity for H2O2 and TMB. That is to say, H2O2 is easily adsorbed on the surface of Fe–N–C and then rapidly decomposed by Fe–N–C. Meanwhile, the Vmax value of Fe–N–C was significantly higher than that of HRP. These results suggest that the high enzyme-like activity of Fe–N–C is derived from the superior affinity for the substrates.
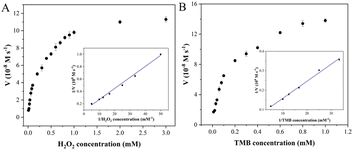 |
| | Fig. 5 Steady-state kinetic analysis utilizing the Michaelis–Menten model (A and B) and Lineweaver–Burk double-reciprocal model for Fe–N–C at pH 4.0. | |
3.4 Colorimetric detection of TCs with Fe–N–C
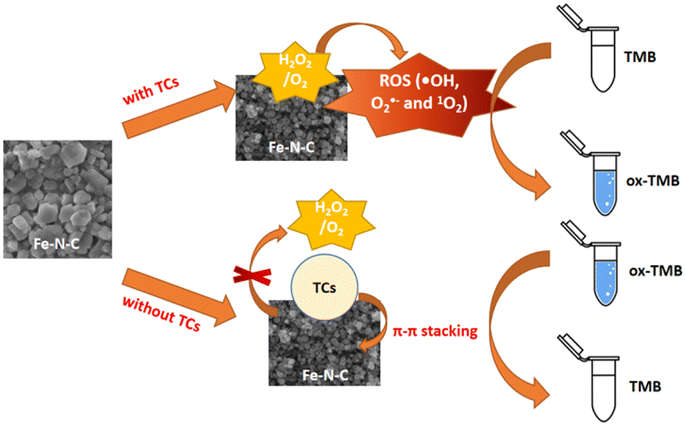
On the basis of the excellent enzyme-mimicking activity of the Fe–N–C, a colorimetric method for TCs detection was established. As illustrated in Scheme 1, the Fe–N–C is first added to an HAc-NaAc buffer containing TMB and H2O2, and the solution gradually changed from colorless to a typical blue. Next, in the presence of TCs, π–π stacking was formed between the TCs tetraphenyl skeleton and Fe–N–C, resulting in the catalytic blocking effect of the material on the substrate. Eventually, the solution faded and the absorption intensity of ox-TMB decreased at 652 nm.
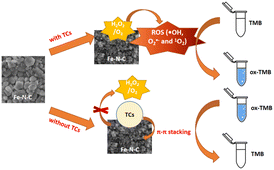 |
| | Scheme 1 Schematic illustration of the colorimetric biosensor for TCs detection using Fe–N–C as a nanozyme. | |
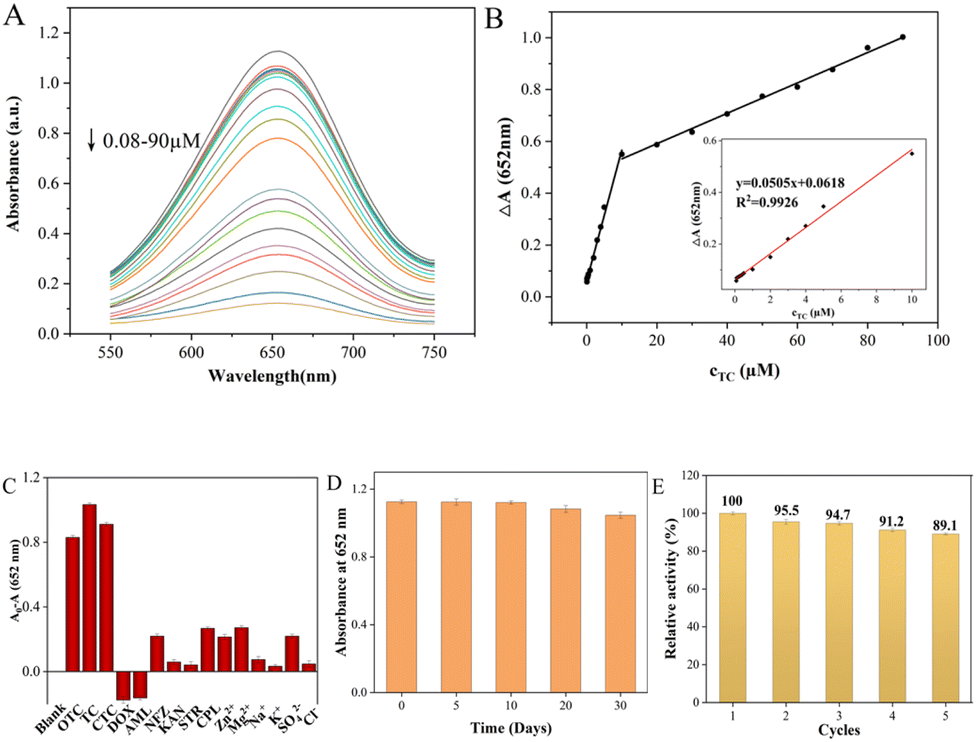
To evaluate the performance of the colorimetric method for TCs detection, the UV-vis absorbance of ox-TMB at 652 nm was measured with different concentrations of TCs. As shown in Fig. 6 and S3,† the colorimetric biosensor had similar responses to the three TCs due to their similar chemical structures, and the UV absorbance at 652 nm decreased gradually as TCs concentration increased. There was a linear relationship between the ΔA value (ΔA = A0 − A, where A0 and A represent the absorbance intensities of ox-TMB at 652 nm in the absence and presence of TC, respectively) and TC concentrations in the range of 0.08–10 μM and 10–90 μM (Fig. 6A). Color variation with the increase of TC dosage could also be visualized by the naked eye, gradually going from blue to colorless (Fig. S4†). And the limit of detection (LOD) for TC calculated as 3σ/slope was 62 nM. The linear range of UV absorbance measured by CTC was found to be 0.1–20 μM and 20–80 μM while the LOD was 85 nM. And the OTC detection as a function of concentration was 0.09–10 μM and 10–100 μM while the LOD was 88 nM. The above results strongly proved that the colorimetric biosensor has a satisfactory and wide linear range in the case of low TCs concentration, which provides a possibility for its practical application. Compared with other detection methods, the performance of the Fe–N–C biosensor has comparable or better features. Although some reported methods showed higher sensitivity or a wider linear range, our proposed method has a simpler analytical process, lower cost and reliable analytical results (Table S3†).
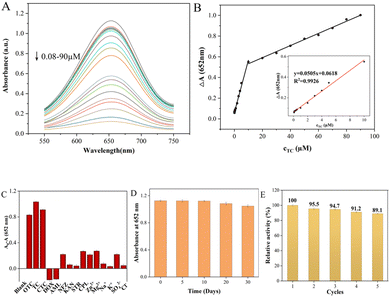 |
| | Fig. 6 (A) UV-vis absorption spectra of this Fe–N–C nanozyme sensing system following incubation with TC in the range from 0.08 to 90.0 μM. (B) Calibration curve of absorbance variations versus quantities of TC added. (C) Selectivity of the Fe–N–C nanozyme sensing system for TC over ions and other antibiotics. (D) Long-time stability of the Fe–N–C nanozyme sensing system. (E) Five consecutive catalysis cycles of Fe–N–C. | |
In actual sample detection, the high selectivity is a key factor for assessing the feasibility of the analytical detection platform. The selectivity of TCs colorimetric sensors was evaluated using cations (Zn2+, Mg2+, K+ and Na+), anions (SO42−, Cl−), common antibiotics (DOX, AML, NFZ, KAN, STR and CPL) and TCs (TC, CTC and OTC) under the same conditions. The error bar represents the standard deviation of three measurements. As shown in Fig. 6C, the A0–A values of the TCs/Fe–N–C/TMB/H2O2 system were all as high as expected due to the similar structure of TCs. However, they are rarely added to the same drug simultaneously, so that they do not interfere with each other when they are tested separately. In addition, the absorbance variation of the Fe–N–C/TMB/H2O2 system containing other competitive antibiotics was much lower than that of the sensor solution containing TCs, indicating that the proposed system can detect the three TCs with high specificity. Furthermore, the long-term performance stability of Fe–N–C nanomaterials for the catalytic oxidation of TMB substrates was also tested to evaluate the efficiency of the sensor system for a long duration. Based on their absorbance at 652 nm, the stability of the nanoparticles did not significantly decrease within 20 days of storage at 4 °C (Fig. 6D). On the 30th day, the catalytic activity of the Fe–N–C decreased slightly, possibly due to its increased hydrodynamic size, but its catalytic activity remained at 92.98%. In addition, even after 631 days had passed since we synthesized this material, its activity was still considerable. Based on colorimetric experiments and UV measurements, we demonstrate that Fe–N–C remains enzyme active after 631 days at 85.07% of its original activity (Fig. S5†). It is worth mentioning that the nanomaterials prepared in this paper exhibited superparamagnetic features, which creates a great possibility for the regeneration and reutilization of the catalyst. As can be seen from Fig. S6,† a magnet is placed outside a glass container containing the Fe–N–C dispersion, and the catalyst is magnetically drawn to one side of the container's inner wall. In the enzyme-mimicking cyclic experiment, the Fe–N–C was collected after each measurement and repeatedly cleaned with deionized water and ethanol, then dried in a vacuum oven at 60 °C for later use. The Fe–N–C was then stored in a sealed dark bottle in a refrigerator at 4 °C and the catalytic oxidation to TMB substrates was monitored at 5 day intervals. As shown in Fig. 6E, the catalytic properties of Fe–N–C remained at 93.2% after 5 cycles under the same reaction conditions, confirming the satisfactory recycling stability of the catalyst. Furthermore, it is easy to collect in the actual application of TCs detection due to Fe–N–C's superparamagnetism, thus minimizing the secondary pollution caused by the detection process.
3.5 Investigation of the catalysis mechanism of Fe–N–C
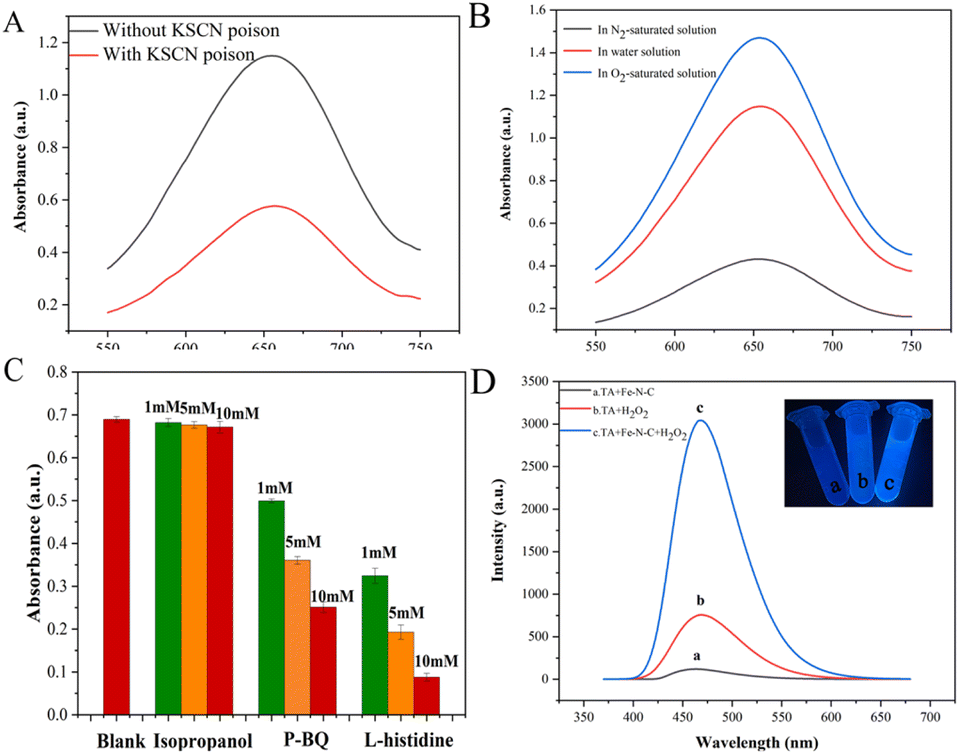
To further determine the active sites of Fe–N–C, a 10 mM KSCN poison treatment blocking experiment was performed. As shown in Fig. 7A, the absorbance of the solution after KSCN poison treatment was significantly decreased, indicating that Fe sites played the key role of the active site in the catalytic process. In addition, we further evaluated the role of Fe(II) and Fe(III) on the catalytic activity of Fe–N–C. Fig. S7† shows the XPS spectra of Fe 2p before and after the catalytic reaction. By calculating the peak area, the ratio of Fe(II)/Fe(III) dropped from 1.25 to 0.50, which confirmed the dominant activity of Fe(II). Meanwhile, we observed that the catalytic activity of Fe–N–C was severely inhibited in N2-saturated solution but enhanced obviously in O2-saturated solution, confirming that O2 is indispensable in this research (Fig. 7B).
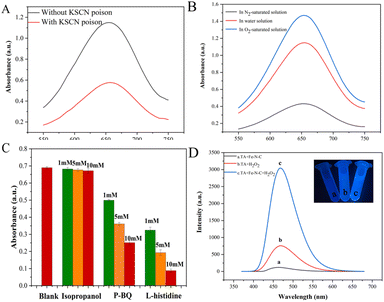 |
| | Fig. 7 (A) The absorbance changes of ox-TMB poisoned with 10 mM KSCN in HAc–NaAc buffer (pH = 4). (B) UV absorbance spectra of the colorimetric reaction in a saturated N2/O2 atmosphere. (C) Absorbances after inhibition of the reaction by different scavengers. (D) The effect of the Fe–N–C on the formation of the hydroxyl radical with TA as a fluorescence probe. | |
Previous reports have indicated that the OXD-mimicking or POD-mimicking activity of metal-based nanomaterials was mainly attributed to an electron transfer mechanism caused by ROS production or the valence variations of metal ions.28 To verify the above conjecture, we firstly conducted radical scavenging experiments without H2O2 participation. As shown in Fig. 7C, the contribution of the ˙OH radical was explored through the inhibition of ˙OH by isopropanol. The results showed that the absorbance of ox-TMB was almost unchanged with the increase of isopropanol concentration, indicating that almost no ˙OH was produced during the catalytic reaction. In addition, the absorbance at 652 nm decreased obviously when the concentration of O2˙− radical scavenger p-BQ increased from 1 mM to 10 mM, which proved that Fe–N–C effectively catalyzed the formation of O2˙− from O2 in solution. This hypothesis was further investigated by adding L-histidine as an 1O2 radical scavenger. Fig. 7C shows a strong inhibitory effect in the presence of L-histidine, and it can be deduced that 1O2 was involved in a significant inhibitory effect. To sum up, the main active substance in the reaction system was 1O2 and the O2˙− radical rather than the ˙OH radical. Apart from mimicking oxidase, the prepared Fe–N–C can also act simultaneously as a POD-mimicking nanozyme in the presence of H2O2. It has been reported that a POD-mimicking nanozyme can decompose the O–O bond of H2O2 into strong oxidizing double hydroxyl radicals (˙OH).29 Simultaneously, reactions involving ˙OH are triggered to perform one or two electron oxidations of substrates. Fe–N–C acts as the catalyst, organic substrates act as electron donors, molecular oxygen or H2O2 acts as the electron acceptor and is reduced according to the nature of enzyme catalysis.30 In this system, Fe(II) on the surface of Fe–N–C plays a leading role in reacting with H2O2 to produce ˙OH. The process can be described in the following reactions:31
| | | FeIII + H2O2 → FeII + ˙OOH + H+ | (1) |
| | | FeIII + ˙OOH → FeII + O2 + H+ | (2) |
| | | FeII + H2O2 → FeIII + ˙OH + OH− | (3) |
| | | ˙OH + TMB → ox-TMB + H+2O | (4) |
To confirm this assumption, we chose terephthalic acid (TA) as a fluorescent probe to evaluate the formation of ˙OH. TA hardly produces fluorescence on its own, but easily reacts with the ˙OH radical to form highly fluorescent 2-hydroxyterephthalic acid (TAOH). As exhibited in Fig. 7D, TA is catalyzed by Fe–N–C with a higher fluorescence signal, indicating that more ˙OH was generated. The above results proved that the prepared Fe–N–C possessed the ability to catalyze the decomposition of H2O2 to generate the ˙OH radical, which could oxidize the peroxidase substrate TMB to ox-TMB. This result is consistent with some early reports.32
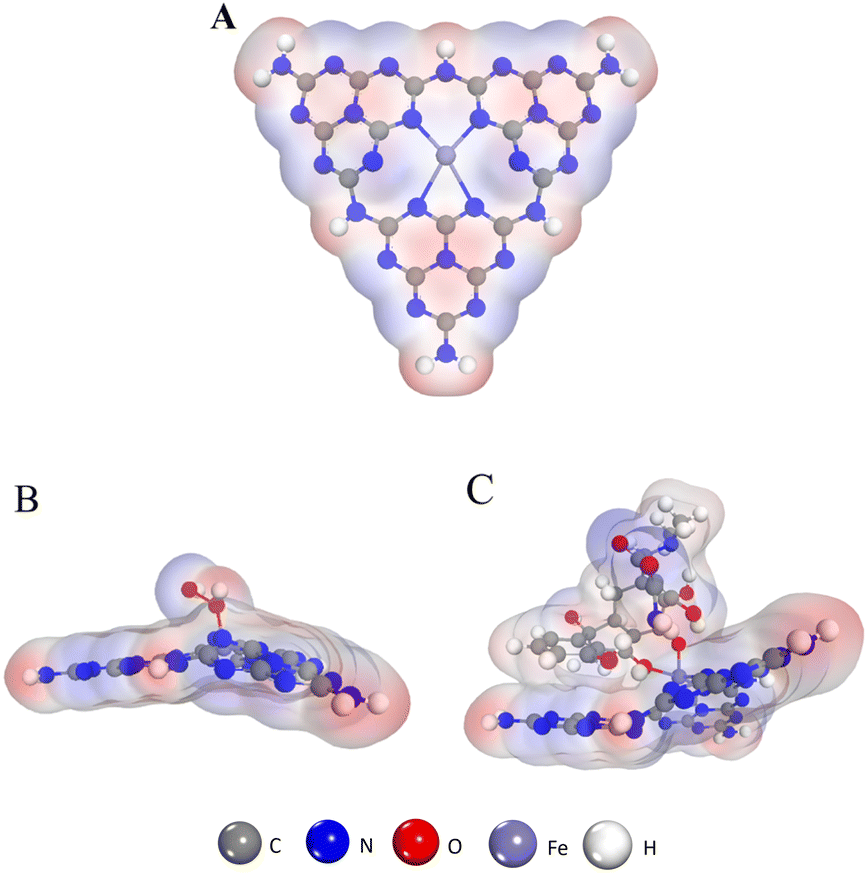
DFT calculations were performed to elucidate possible inhibitory catalytic pathways. The configuration of Fe–N–C (Fig. 8A) and structures of H2O2 and TC adsorption configurations on the Fe–N–C surface are presented in Fig. 8. In all the configurations, in accordance with what is observed in Fig. 8B, H2O2 is chemically adsorbed on the top surface of Fe–N–C with an O atom of H2O2 being bonded with an Fe atom. And the DFT calculation signifies that H2O2 adsorbs on Fe–N–C with an adsorption energy of −0.97 eV. In comparison, the interaction between TC and Fe–N–C is more intense, with the two O atoms of TC being bonded by one Fe atom through a similar coordination. More importantly, the unsaturated (poly)cyclic structures of TC and Fe–N–C are tightly adsorbed by π–π stacking (Fig. 8C). In the TC_Fe–N–C configuration, the adsorption energy of TC on Fe–N–C is −2.24 eV, which inhibits the decomposition of H2O2 into ˙OH. Hence, according to the above calculations, the binding interaction between TC and Fe–N–C is strong enough to compete with H2O2 for Fe–N–C and fade the solution.
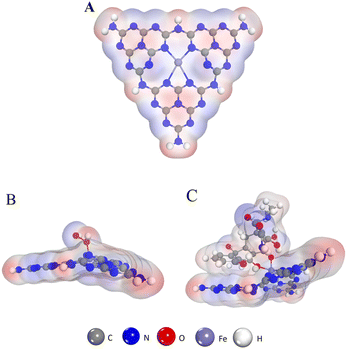 |
| | Fig. 8 (A) The structure of Fe–N–C. (B) The structure of the H2O2 absorption process on the Fe–N–C. (C) The structure of the TC absorption process on the Fe–N–C. | |
3.6 Application of the Fe–N–C in the determination of TCs in water samples
Due to their high hydrophilicity and low volatility characteristics, TCs can exhibit continuous persistence in aqueous environments.33 Therefore, we have evaluated the practical application of the colorimetric biosensor for TCs in lake water samples. The recoveries of TCs ranged between 90.2% and 112.8%, and the RSDs ranged from 2.94% to 4.3% (Table S4†), indicating that the colorimetric sensor based on Fe–N–C is reliable for TCs detection in lake water.
4 Conclusions
In summary, dual enzyme mimicking Fe–N–C was synthesized through a one-step pyrolytic process. The established colorimetric platform showed excellent catalytic performance in terms of high selectivity to TCs, and is a promising sensor in environmental analysis. In addition, this assay displayed a recoverability and stable sensing performance, and its practical application in water samples has been verified.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project was supported by the Shanxi Scholarship Council of China (2022-017), Natural Science Foundation of Shanxi Province (20210302123494 and 20210302124396) and Program of the Ministry of Science and Technology of China (G2022004012L).
Notes and references
- K. Bousova, H. Senyuva and K. Mittendorf, J. Chromatogr. A, 2013, 1274, 19–27 CrossRef CAS PubMed.
- J. Zhou, X. Xue, Y. Li, J. Zhang, F. Chen, L. Wu, L. Chen and J. Zhao, Food Chem., 2009, 115, 1074–1080 CrossRef CAS.
- K. Phomai, S. A. Supharoek, J. Vichapong, K. Grudpan and K. Ponhong, Talanta, 2023, 252, 123852 CrossRef CAS.
- C. Zhou, J. Deng, G. Shi and T. Zhou, Electrophoresis, 2017, 38, 1060–1067 CrossRef CAS PubMed.
- X. Liu, S. Zheng, Y. Hu, Z. Li, F. Luo and Z. He, Food Anal. Methods, 2016, 9, 2972–2978 CrossRef.
- J. Zhang, Z. Bao, J. Qian, H. Zhou and K. Zhang, Anal. Chim. Acta, 2022, 1216, 339991 CrossRef CAS.
- M. Kaczmarek, K. Staninski and M. Stodolny, J. Lumin., 2021, 36, 1945–1952 CrossRef CAS.
- W. Kong, X. Guo, M. Jing, F. Qu and L. Lu, Biosens. Bioelectron., 2020, 150, 111875 CrossRef CAS PubMed.
- W. Xia, P. Zhang, W. Fu, L. Hu and Y. Wang, Chem. Commun., 2019, 55, 2039–2042 RSC.
- C. Song, W. Ding, H. Liu, W. Zhao, Y. Yao and C. Yao, New J. Chem., 2019, 43, 12776–12784 RSC.
- D. Singh, D. Sillu, A. Kumar and S. Agnihotri, Environ. Sci.: Nano, 2021, 8, 1308–1325 RSC.
- W. Li, G. C. Fan, F. Gao, Y. Cui, W. Wang and X. Luo, Biosens. Bioelectron., 2019, 127, 64–71 CrossRef CAS.
- B. Xu, Y. Cui, W. Wang, S. Li, C. Lyu, S. Wang, W. Bao, H. Wang, M. Qin, Z. Liu, W. Wei and H. Liu, Adv. Mater., 2020, 32, e2003563 CrossRef PubMed.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- P. Zhang, D. Sun, A. Cho, S. Weon, S. Lee, J. Lee, J. W. Han, D. P. Kim and W. Choi, Nat. Commun., 2019, 10, 940 CrossRef PubMed.
- K. Fan, J. Xi, L. Fan, P. Wang, C. Zhu, Y. Tang, X. Xu, M. Liang, B. Jiang, X. Yan and L. Gao, Nat. Commun., 2018, 9, 1440 CrossRef PubMed.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- T. Zhang, H. Li, M. Liu, H. Zhou, Z. Zhang, C. Yu, C. Wang and G. Wang, Sens. Actuators, B, 2021, 341, 129987 CrossRef CAS.
- X. Zhou, M. Wang and X. Su, Sens. Actuators, B, 2021, 338, 129777 CrossRef CAS.
- Y. Wang, Z. Zhang, G. Jia, L. Zheng, J. Zhao and X. Cui, Chem. Commun., 2019, 55, 5271–5274 RSC.
- D. Li, Y. Qu, X. Liu, C. Zhai and Y. Liu, Int. J. Hydrogen Energy, 2021, 46, 18364–18375 CrossRef CAS.
- M. Liu, L. Wang, K. Zhao, S. Shi, Q. Shao, L. Zhang, X. Sun, Y. Zhao and J. Zhang, Energy Environ. Sci., 2019, 12, 2890–2923 RSC.
- L. Wang, K. Liang, L. Deng and Y.-N. Liu, Appl. Catal., B, 2019, 246, 89–99 CrossRef CAS.
- Y. Xiong, S. Chen, F. Ye, L. Su, C. Zhang, S. Shen and S. Zhao, Chem. Commun., 2015, 51, 4635–4638 RSC.
- A. C. Razavi, T. N. Kelly, M. J. Budoff, L. A. Bazzano, J. He, C. Fernandez, J. Lima, K. Nasir, R. S. Blumenthal, M. J. Blaha and S. P. Whelton, Atherosclerosis, 2021, 326, 56–62 CrossRef CAS PubMed.
- H. Q. Zheng, Y. N. Zeng, J. Chen, R. G. Lin, W. E. Zhuang, R. Cao and Z. J. Lin, Inorg. Chem., 2019, 58, 6983–6992 CrossRef CAS.
- H. Wang, K. Wan and X. Shi, Adv. Mater., 2019, 31, e1805368 CrossRef.
- P. Ni, H. Dai, Y. Wang, Y. Sun, Y. Shi, J. Hu and Z. Li, Biosens. Bioelectron., 2014, 60, 286–291 CrossRef CAS PubMed.
- Y. Zhang, C. Dai, W. Liu, Y. Wang, F. Ding, P. Zou, X. Wang, Q. Zhao and H. Rao, Mikrochim. Acta, 2019, 186, 340 CrossRef PubMed.
- B. Liu, H. Zhu, R. Feng, M. Wang, P. Hu, J. Pan and X. Niu, Sens. Actuators, B, 2022, 370, 132451 CrossRef CAS.
- P. Chen, H. Zhong, X.-r. Li, M. Li and S. Zhou, Appl. Clay Sci., 2021, 209, 106109 CrossRef CAS.
- R. Daghrir and P. Drogui, Environ. Chem. Lett., 2013, 11, 209–227 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Guangming
Wen
*b,
Hung-Wing
Li
*a,
Guangming
Wen
*b,
Hung-Wing
Li