
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron nitroprusside as a chemodynamic agent and inducer of ferroptosis for ovarian cancer therapy†
Kanwal
Asif
 ab,
Muhammad
Adeel
*ab,
Md. Mahbubur
Rahman
*c,
Isabella
Caligiuri
b,
Tiziana
Perin
b,
Maja
Cemazar
d,
Vincenzo
Canzonieri
be and
Flavio
Rizzolio
*ab
ab,
Muhammad
Adeel
*ab,
Md. Mahbubur
Rahman
*c,
Isabella
Caligiuri
b,
Tiziana
Perin
b,
Maja
Cemazar
d,
Vincenzo
Canzonieri
be and
Flavio
Rizzolio
*ab
aDepartment of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venice, 30172, Venice, Italy. E-mail: muhammad.adeel@unive.it; flavio.rizzolio@unive.it
bPathology Unit, Centro di Riferimento Oncologico di Aviano (C.R.O.) IRCCS, 33081, Aviano, Italy
cDepartment of Applied Chemistry, Konkuk University, Chungju 27478, Republic of Korea. E-mail: mahbub1982@kku.ac.kr
dDepartment of Experimental Oncology, Institute of Oncology Ljubljana, Zaloska cesta 2, SI-1000 Ljubljana, Slovenia
eDepartment of Medical, Surgical and Health Sciences, University of Trieste, 34149, Trieste, Italy
First published on 8th March 2023
Abstract
ChemoDynamic Therapy (CDT) is a powerful therapeutic modality using Fenton/Fenton-like reactions to produce oxidative stress for cancer treatment. However, the insufficient amount of catalyst ions and ROS scavenging activity of glutathione peroxidase (GPX4) limit the application of this approach. Therefore, a tailored strategy to regulate the Fenton reaction more efficiently (utilizing dual metal cations) and inhibit the GPX4 activity, is in great demand. Herein, a CDT system is based on dual (Fe2+ metals) iron pentacyanonitrosylferrate or iron nitroprusside (FeNP) having efficient ability to catalyze the reaction of endogenous H2O2 to form highly toxic ˙OH species in cells. Additionally, FeNP is involved in ferroptosis via GPX4 inhibition. In particular, FeNP was structurally characterized, and it is noted that a minimum dose of FeNP is required to kill cancer cells while a comparable dose shows negligible toxicity on normal cells. Detailed in vitro studies confirmed that FeNP participates in sustaining apoptosis, as determined using the annexin V marker. Cellular uptake results showed that in a short time period, FeNP enters lysosomes and, due to the acidic lysosomal pH, releases Fe2+ ions, which are involved in ROS generation (˙OH species). Western blot analyses confirmed the suppression of GPX4 activity over time. Importantly, FeNP has a therapeutic effect on ovarian cancer organoids derived from High-Grade Serous Ovarian Cancer (HGSOC). Furthermore, FeNP showed biocompatible nature towards normal mouse liver organoids and in vivo. This work presents the effective therapeutic application of FeNP as an efficient Fenton agent along with ferroptosis inducer activity to improve CDT, through disturbing redox homeostasis.
1. Introduction
To cope with the continuing threat of cancer, ChemoDynamic Therapy (CDT) is considered one of the most promising cancer therapeutic approaches with minimal toxicity through a Fenton reaction.1 In particular, besides iron, other metals including Cu, Mn, Co, Ti, V, Pd, and Ag act as CDT agents via Fenton/Fenton-like reactions.2–10 CDT induces severe oxidative damage via the catalysed conversion of hydrogen peroxide (H2O2) into a lethal hydroxyl radical (˙OH), resulting in an increased intracellular oxidative stress and lipid oxidation, which lead to cell death.2,3,11–14 It has been reported that CDT has low toxicity towards normal cells compared to the cancerous cells, because metals have poor catalytic activity above pH 415 while they are more effective towards tumor cells due to their low acidic tumor microenvironment compared to traditional chemotherapeutics.16–18 However, there is no external stimulation required to initiate the Fenton reaction via CDT,11,19,20 compared to other cancer therapeutic approaches.21 Other than the Fenton reaction to produce reactive oxygen species (ROS),22–24 iron-based materials promote ferroptosis-mediated cell death. Ferroptosis is an iron-dependent form of cell death, which is related to the intracellular iron concentration25,26 and plays an important role in tumor growth suppression.27 Ferroptosis requires the inactivation of glutathione peroxidase (GPX4) or glutathione (GSH) depletion in order to produce an abundant amount of ROS28–30 to induce cell death.To date, several types of iron-based nanomaterials have been explored for the Fenton reaction as anticancer agents.31–36 However, there are not enough ions to catalyse the Fenton reaction and the remaining ROS are readily scavenged by the antioxidant glutathione peroxidase (GPX4), allowing the survival of tumoral cells.37 Therefore, the regulation of a Fenton reaction by increasing nanocatalysts and the induction of ferroptosis by inhibiting the GPX4 enzyme is an effective strategy in cancer treatment. For example, Chen et al. reported the use of ultrasmall poly(ethylene glycol)-modified polydopamine (UPDA-PEG-Fe) nanoparticles to induce ferroptosis through the inactivation of GPX4 activity to induce cell death. This system improves the metal-ion-loading strategy in a pH-controllable manner for the Fenton reaction.38 Li et al. designed tannic acid (TA) and Fe2+ coated zeolite imidazole ester skeleton-8 (ZIF-8) self-assembly, as a carrier of artemisinin (ART) to catalyze the degradation of ferritin and increase the amounts of Fe2+ in cells. This system induced ROS with decreasing GSH and GPX4 activity to promote ferroptosis.39 In another study, Dong et al. developed a liposomal-based BSO/GA–Fe(II) nanoformulation to deliver GSH inhibitor L-buthionine sulfoximine (BSO) to amplify the tumor oxidative stress by targeting H2O2 and GSH within the tumor microenvironment to generate ROS species.40 Liu et al. developed Fe–metal–organic framework nanoparticles conjugated with an iRGD peptide multifunctional nanocomposite, MIL-101(Fe)@sor, as a carrier for sorafenib to inhibit GSH. This proposed nanocomposite increases the concentration of iron and reduces GSH simultaneously to effectively induce tumor ferroptosis.41 However, a previously developed system is based on the delivery of a GSH inhibitor with other therapeutic approaches (e.g., peptide, surface modification) but is far from a satisfactory clinical outcome.42,43 Therefore, it is necessary to design such a system that has efficient catalytic activity (dual metal ions) for ROS production and the capability to inactivate the GPX4 function without any external drug and loading system for a better therapeutic approach.
In this study, for the first time, we reported such a system that shows (dual Fe metal) efficient catalytic activity on H2O2 for ˙OH generation as well as simultaneous involvement in the depletion of GPX4 activity to induce ferroptosis without any delivery system or external stimuli. In our design, a biocompatible Fenton catalyst iron pentacyanonitrosylferrate or iron nitroprusside (FeNP) was prepared using a simple solution mixture-based procedure using FDA-approved sodium nitroprusside as a precursor. Iron nitroprusside compound has naturally NO species together with dual iron metal cations (Fe2+) in its structure to improve CDT together with GPX4 depletion-based therapeutic systems. Additionally, this system presents a novel strategy to amplify tumor oxidative stress with GPX4 depletion at the same time without the requirement of any extra stimuli or delivery system to improve CDT for therapeutic effects. The results were validated using a number of different in vitro, ex vivo and in vivo experiments, showing the biocompatible nature of FeNP towards normal liver organoids and in vivo while being toxic towards tumoral cells and Patient derived tumor organoids (PDTO). Additionally, FeNP decomposed more endogenous H2O2 than its precursors (iron chloride and sodium nitroprusside) and inactivated GPX4 over time to induce ferroptosis.
2. Experimental section
2.1 Materials and methods
Iron(II) chloride (FeCl2), sodium nitroprusside dihydrate (Na2[Fe(CN)5NO]·2H2O, SNP, ≥99%), and anhydrous methanol (99.8%) were purchased from Sigma-Aldrich St. Louis, MO, USA. A Millipore Milli-Q Biocell A10 water purifying system was employed to prepare ultrahigh purity water to use throughout the experiments. A2780, A2780cis, U-87 MG (Sigma-Aldrich, St. Louis, MO, USA), MDA-MB-231 (Cell Biolabs, CA, USA), SK-OV-3, MCF-7, and MRC-5 (ATCC, Manassas, VA, USA) cells were cultured according to the manufacturers’ instructions. LysoTracker Green DND-26, Hoechst 33342, and rhodamine B were acquired from ThermoFisher Scientific, Waltham, MA, USA. CellTiter-Glo® from Promega, Madison, WI, USA and PE-Annexin V Apoptosis Detection Kit from Becton-Dickinson, Franklin Lakes, NJ, USA were used. An ROS-Glo™ H2O2 luminescence assay kit bought from Promega, Madison, WI, USA and FluorSave™ reagent (Catalog no: 345789; Millipore: Burlington, MA, USA) were used.2.2 Instrumentation
The morphologies of the FeNP samples were observed under a field-emission scanning electron microscope (FE-SEM, Carl Zeiss Sigma VP, Jena, Germany) and a transmission electron microscope (TEM, JEM-2100, JEOL, Tokyo, Japan). An energy-dispersive X-ray spectroscopy (EDS) analyzer (INCAx-sight7421, Oxford Instruments, UK) equipped with a FE-SEM instrument was employed to obtain the atomic percentage of the elements present in FeNP. The as-prepared FeNP crystal structure was characterized with an X-ray powder diffractometer (XRD, Philips, X’pert, Netherland) with Cu Kα radiation (λ = 1.5406 Å). A Fourier transform infrared (FTIR) spectrophotometer (MIDAC, M4000, Westfield, MA, USA) was utilized to obtain the chemical functional groups in FeNP. The Raman shift was observed using a Raman spectrophotometer (Horiba Scientific, Xplora Plus, France) operated at 532 nm. X-ray photoelectron spectroscopy (XPS, Thermo Scientific™ K-Alpha, ThermoFisher Scientific, Waltham, MA, USA) was utilized to observe the chemical composition, nature of bonding, and oxidation states of FeNP. A UV-Visible spectrophotometer (UV-VIS-31 Scan, ONDA, Modena, Italy) was used to obtain the optical absorption spectra of the samples. To evaluate IC50 values, a Tecan infinite M1000 (Tecan, Mannedorf, Switzerland) luminescence measurement system was used. AnnexinV was evaluated with a BD FACS CantoII instrument (BD Biosciences, San Jose, CA, USA). Fluorescence images were captured on a Leica DM5500B with an X-Cite 120 PC Q lamp. The chemiluminescent signal was obtained with a chemiluminescent gel imaging system (ChemiDoc™ Touch Imaging System, Bio-Rad, Hercules, CA, USA) and analysed with Image Lab Software (Bio-Rad).2.3 Synthesis of FeNP
A simple solution mixing procedure was employed to synthesize FeNP. Briefly, 40 mL of 0.2 M FeCl2 solution in methanol was mixed with 40 mL of 0.4 M SNP solution in methanol and stirred for 12 h at 70 °C. After the completion of the reaction, the reaction mixture was cooled to RT. Subsequently, the precipitates of FeNP were collected by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RPM for 30 min, washed several times with methanol, and dried at 40 °C overnight in a vacuum oven at 40 °C. The chemical reaction for the synthesis of FeNP can be written as follows:
000 RPM for 30 min, washed several times with methanol, and dried at 40 °C overnight in a vacuum oven at 40 °C. The chemical reaction for the synthesis of FeNP can be written as follows:| Fe2+ + [Fe(CN)5NO]2− → Fe[Fe(CN)5NO] |
2.4 Cell viability
For cell viability measurements, SK-OV-3, MDA-MB-231, A2780, A2780 cis, U-87 MG and MCF-7 cells were seeded in 96-well plates at densities of 1 × 103 and, for MRC-5, 8 × 103 per well and treated with six concentrations (0.001, 0.01, 0.1, 1, 10 and 100 μg) of iron nitroprusside (FeNP) and its precursors iron chloride (FeCl2) and sodium nitroprusside (SNP). As a positive control, we treated cells with six concentrations of CisPt starting from 30 μg ml−1 (100 μM) to 0.001 μg ml−1 (0.03 μM). After 96 hours, cell viability was measured using the CellTiter-Glo® assay system according to the manufacturer's instructions (Promega, Madison, WI, USA). Luminescence was recorded using a Tecan M1000 instrument. Experiments were performed in triplicate and IC50 values were analysed using a non-linear regression method using GraphPad Prism software.2.5 Stability of FeNP
The stability of FeNP was measured by developing an artificial in vivo system through mixing FeNP in physiological solutions with two different pH values (pH 5.5 and pH 7.4). Next, the solutions were kept in an incubator at 37 °C. Then samples were taken at different day intervals (0–7; D0, D1, D2, D3, D5, and D7), centrifuged, and collected. After that, samples were measured by UV-vis absorption spectroscopy by mixing them in DI water.2.6 Intracellular localization
SK-OV-3 cells were plated at a density of 2 × 105 cells per mL in 6 well plates containing a glass coverslip for 24 h to allow cell attachment. Afterward, 50 μg ml−1 of FeNP labelled with 30 μg ml−1 of rhodamine B was added for 6, 24 and 48 h. Cells were washed twice with PBS and then incubated with 200 nM Lysotracker™Green DND-26 (ThermoFisher Scientific, Waltham, MA, USA) for lysosomes and 200 ng ml−1 Hoechst 33342 for nuclear staining, respectively. Next, to fix cells, 4% paraformaldehyde was used for 20 minutes and then the coverslip was mounted using FluorSave mounting media (Merck Millipore, Burlington, MA, USA) for cellular imaging. The cellular uptake of FeNP was observed under a fluorescence microscope with appropriate filters. Images were analysed using ImageJ and JacoP plugins.2.7 Flow cytometry analysis
SK-OV-3 cells were cultured at a density of 2 × 105 in 6 multi-well plates as suggested by the supplier for indicated time points. Following treatment, cells were washed with PBS, then harvested and labelled with a PE-Annexin V Apoptosis Detection Kit from Becton-Dickinson, Franklin Lakes, NJ, USA for 15 min in darkness. After, samples were examined using a BD Canto II flow cytometer and BD FACS DIVA software.2.8 H2O2 level measurement
To quantify the H2O2 level, SK-OV-3 and MRC-5 were plated in 96 multi-well plates at a density of 1 × 104 per well and incubated at 37°C for 24h. Then, cells were treated with 50 μg ml−1 of FeNP, FeCl2, SNP and cisplatin (CisPt) (3 μg ml−1: 10 μM) for different time points (6, 24 and 48 h). The intracellular ROS H2O2 level was measured by ROS-Glo™ H2O2 luminescence assay (Promega, Madison, WI, USA) corresponding to the manufacturer's guidelines on a Tecan M1000 instrument.
2.9 Cytochrome c release
SK-OV-3 cells were grown on coverslips at a density of 2 × 105 cells per mL followed by the treatment of FeNP (50 μg ml−1) and CisPt (3 μg ml−1: 10 μM) for 6, 24 and 48 h. After each time point, 4% paraformaldehyde was used to fix the cells (20 min, RT). Furthermore, cells were permeabilized with 0.3% Triton X-100/PBS (15 min, RT) and blocked with 8% BSA/PBS (1 h, RT). Next, cells were stained with a mouse monoclonal anti-cytochrome c (6H2·B4) antibody (1:100 dilution in 1% BSA/PBS, at 4 °C, overnight) purchased from Cell Signaling Technology, Catalog no: 12963; Danvers, MA, USA, followed by secondary antibody staining (1:1000 dilution, Alexa Fluor® 488 dye) for 2 h at room temperature. For nuclear visualization, cells were stained with 1 mg mL−1 of DAPI (1:10000 dilution in PBS, RT, 1 min). The cells were washed three times with PBS after all incubation steps. The coverslips were mounted on glass slides with the FluorSave™ reagent (Catalog no: 345789; Millipore: Burlington, MA, USA) and images were acquired using a Leica DM5500B fluorescence microscope with an X-Cite 120 PC Q lamp and analysed with Image J software.
2.10 Western blot analysis
SK-OV-3 cells were treated with FeNP, FeCl2, SNP (10 μg ml−1) and CisPt (3 μg ml−1: 10 μM) for 6, 24, and 48 h, then harvested and lysed using a radioimmunoprecipitation assay buffer containing protease and phosphatase (sodium fluoride (NaF) and sodium orthovanadate (Na3VO4)) inhibitors via vigorous sonication on an ice bath and the protein concentration was determined using the Bradford protein assay. 20 μg of proteins was used for SDS-PAGE and transferred to a nitrocellulose membrane. After protein transfer, the membranes were blocked in 5% (wt/v) non-fat milk and incubated with primary antibodies overnight: GPX4 (Catalog no: ab125066, dilution 1:500, Abcam, Cambridge, UK) and Vinculin (Catalog no: sc7649, dilution 1:5000, Santacruz Biotechnology, Dallas, TX, USA), followed by incubation with secondary antibodies conjugated with horseradish peroxidase (goat anti-rabbit immunoglobulin G, Catalog no: 31462, ThermoFisher Scientific, Waltham, MA, USA, and mouse anti-goat immunoglobulin G, Catalog no: 31400, ThermoFisher Scientific, Waltham, MA, USA) with dilution 1:10000. The immunoblot signals were generated using a Liteablot Chemiluminescent Substrate Kit (Catalog no: EMP013001, Euroclone, Milan, Italy). The chemiluminescent signal was captured in a chemiluminescent gel imaging system (ChemiDoc™ Touch Imaging System, Bio-Rad, Hercules, CA, USA) and analyzed with Image Lab Software (Bio-Rad).
2.11 Organoid isolation and culture
Mouse liver organoids were produced from 8-week-old C57/BL6 mice following the protocol described by Stappenbeck.44 Organoids were generated from mouse liver post-mortem (Italian Ministry of Health, 148/2016-PR). Briefly, liver tissue was digested for 30’ with 2 mg ml−1 collagenase, the tissue suspension was centrifuged at 1500 rpm for 5min, and then the pellet was resuspended in a Cultrex growth factor-reduced Basement Membrane Extract (BME), Type2 (R&D Systems Catalog no: 3533-001-02, Milan, Italy) and cultured in 24 well plates. After hydrogel solidification, 450 μl of the organoid medium was added.
Patient-derived tumour organoids (PDTO) were obtained from ovarian cancer patients. Tissues were handled and cultured using the protocol described by Scattolin et al.45 For research purposes, biobank informed consent was available to collect the samples at the National Cancer Institute (CRO) of Aviano.
2.12 Toxicity assay on mouse liver organoids and PDTO
To determine the toxicity, organoids were cultured in 96 multi-well plates and treated with FeNP, FeCl2, and SNP ranging from 100 μg ml−1 to 0.034 μg ml−1 for 96 hours. As a positive control, we used CisPt starting from 30 μg ml−1 (100 μM) to 0.001 μg ml−1 (0.03 μM). After 96 hours, organoid viability was quantified using the CellTiter-Glo® 3D Luminescence assay (Promega, Madison, WI, USA) with a Tecan M1000 instrument (Tecan, Mannedorf, Switzerland).2.13 Histopathological analysis of HGSOC PDTO
For the histopathological study, formalin-fixed paraffin-embedded sections of tumour organoids were used. After culturing, organoids were harvested and fixed in phosphate-buffered 10% formalin and embedded in paraffin using a Micro NextGen Cell Blocking™ Kit (Cat no: M20; AV Bioinnovation) following the manufacturer's instructions. Using a Leica ST5020 multistainer, 5 μm sections were stained with haematoxylin and eosin (H&E) and 2 μm sections were cut for immunohistochemistry (IHC) staining. A heat-induced antigen retrieval method and an UltraVision LP Detection System HRP DAB kit (ThermoFisher Scientific, Waltham, MA, USA) were utilized to perform IHC. The following antibodies were used to characterize PDTO and parental tumour: PAX8 (Catalog no: 10336-1-AP, dilution 1:400, ProteinTech Group, Planegg-Martinsried, Germany), Ca125 (Catalog no: sc-52095, dilution 1:100, Santa Cruz Biotechnology, Dallas, TX, USA) and WT1 (Catalog no: ab89901, dilution 1:300, Abcam, Cambridge, UK). Immunohistochemistry images were captured using a light microscope with different magnifications.
2.14 Animal studies
Animal experiments (EU directive (2010/63/EU)) were performed under the authorization of the National Ethical Committee and the Administration of the Republic of Slovenia for Food Safety, Veterinary and Plant Protection.Tumor-bearing mice were randomly distributed in 3 groups (n = 6 tumors per group). Mice per group were treated intraperitoneally (i.p.) with a vehicle, 8 mg kg−1 of FeNP and 2 mg kg−1 of CisPt. In subsequent treatments, mice were examined every day for toxicity signs for 30 days. Data are reported as the mean and standard error. The dosages of CisPt and FeNP were derived from literature analysis.38,46,47
2.15 Statistical analysis
The data represented the means of at least two independent experiments performed in triplicate. Statistical analysis was performed with a two-tailed t test using GraphPad Prism 8.0 and Origin 9.0 software. For all data, p < 0.05 (*p), p < 0.01 (**p) and p < 0.001 (***p) were considered statistically significant.3. Results and discussion
3.1 Structural and morphological analyses
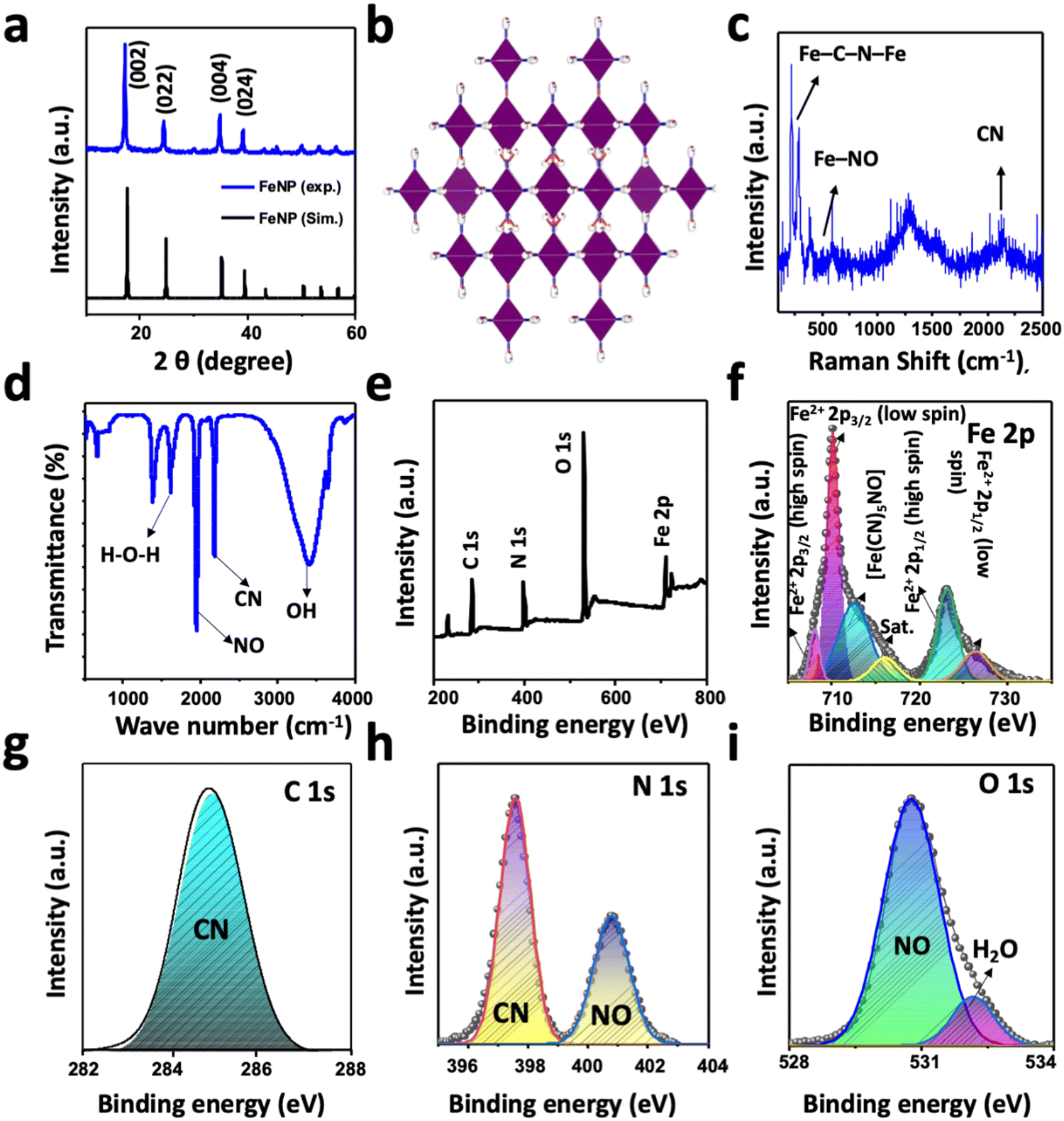
Fig. 1(a) shows the powder XRD pattern of FeNP. To elucidate the exact pattern, the XRD spectra of FeNP are simulated using the pattern of nickel(II) nitroprusside (NiNP).48 This is because the preparation of a single-crystal of FeNP and the corresponding XRD analysis are beyond the scope of this research. The simulated XRD pattern is well-matched with the experimental pattern, and it is isostructural to NiNP.48 The sharp and intense XRD peaks of FeNP are well-matched with the computed XRD pattern. The high intensity and sharp major peaks at 2θ = 17.25°, 24.40°, 34.70°, 39.0°, 50.0°, 53.40°, and 56.40° can be ascribed to the hkl reflections of the (002), (022), (004), (024), (044), (244) and (026) planes, respectively, of the cubic crystal system with the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, as shown in Fig. 1(b).48,49 The absence of additional peaks and the high intensity of the peaks suggest the high crystallinity of the as-prepared FeNP. The crystal structure and the composition of FeNP were examined further by Raman spectroscopy. The high-intensity Raman peaks of FeNP (Fig. 1(c)) at 220, 280, and 383 cm−1 can be assigned to the Fe–C–N–Fe bands, while the peak at 590 cm−1 can be ascribed to the Fe–NO band.48,50 The isolated bands of CN appear in the range of 2050–2250 cm−1 and the intense band at 2150 cm−1 is possibly due to the stretching of the CN group in FeNP.51
m, as shown in Fig. 1(b).48,49 The absence of additional peaks and the high intensity of the peaks suggest the high crystallinity of the as-prepared FeNP. The crystal structure and the composition of FeNP were examined further by Raman spectroscopy. The high-intensity Raman peaks of FeNP (Fig. 1(c)) at 220, 280, and 383 cm−1 can be assigned to the Fe–C–N–Fe bands, while the peak at 590 cm−1 can be ascribed to the Fe–NO band.48,50 The isolated bands of CN appear in the range of 2050–2250 cm−1 and the intense band at 2150 cm−1 is possibly due to the stretching of the CN group in FeNP.51
 | ||
| Fig. 1 (a) Powder XRD pattern of the as-prepared FeNP together with the simulated XRD pattern. (b) The crystal structure of FeNP. (c) Raman and (d) FTIR spectra of FeNP. (e) XPS survey spectra and high-resolution XPS spectra of (f) Fe 2p, (g) C 1s, (h) N 1s, and (i) O 1s. The line with the circle symbol and the shaded region represents the experimental and fitted data, respectively. | ||
The nature of the chemical species and the functional groups present in FeNP was analyzed by FTIR spectroscopy. Fig. 1(d) shows the FTIR spectrum of the as-prepared FeNP, which revealed the presence of high-intensity bands of NO and CN at 1940 cm−1 and 2170 cm−1, respectively.48 The additional high-intensity absorption bands at 3402 cm−1 and 1609 cm−1 can be assigned to the O–H absorption band originating from the adsorbed or crystalline water and the deformational scissor vibrations of water, respectively.48,52 The elements, functional groups, and elemental oxidation states in FeNP were analyzed with XPS measurements. Fig. 1(e) displays the survey XPS spectra of FeNP, which exhibited the existence of C 1s, N 1s, O 1s, and Fe 2p peaks in their reported binding energy values.53 The experimental high-resolution XPS spectra of Fe 2p, C 1s, N 1s, and O 1s are presented in Fig. 1(f)–(i), respectively, along with the fitted results. The high-resolution Fe 2p spectrum of FeNP showed spin–orbit doublet peaks of Fe 2p3/2 and Fe 2p1/2. Both the peaks exhibited Fe2+ low spin states at 710.0 and 726.0 eV, respectively, and Fe2+ high spin states at 708.0 and 723.0 eV, respectively, originated from the Fe2+ species.48,54 The additional peak at 712.0 can be ascribed to the presence of [Fe(CN)5NO] in FeNP.49 The appearance of these high-spin Fe2+ peaks in FeNP can be attributed to the partial decomposition of FeNP induced by the high energy XPS beam that can be further confirmed by the presence of a satellite peak at 716.25 eV. The decomposition of FeNP induced by high energy XPS beam during measurement possibly altering of the Fe–CN bonds to Fe–NC bonds or breaking of the Fe–CN bond.48,54 The deconvoluted core-level C 1s spectrum of FeNP showed only a CN peak at 284.85 eV, while the high-resolution N 1s spectrum showed the CN and NO peaks at the binding energies of 397.58 and 400.80 eV, respectively. These binding energy values of chemical species of C 1s and N 1s spectra of FeNP are close to the reported results of other metal-nitroprussides.48,49,54 The fitted O 1s spectra exhibited two-component peaks at 530.80 and 532.30 eV. The former peak can be assigned to the NO peak in FeNP, while the latter peak can be ascribed to the oxygen peak, originating from the adsorbed or crystalline water in FeNP.48 The stability of FeNP was observed by establishing the artificial in vivo conditions in two different pHs (7.4 and 5.5) at 37 °C. After the treatment samples were taken at different time intervals and investigated by UV-vis absorption measurements (Fig. S1, ESI†). The results showed the presence of the FeNP absorption band at 270 nm without any visible shifting in the absorbance maxima at both pHs for up to 1 week, indicating the high stability of FeNP.
Fig. 2(a) and (b) show the FE-SEM images of FeNP under two-different magnifications. The surface morphology of the as-synthesized FeNP exhibited the formation of mixed morphologies with a wide size distribution range. This was further confirmed with the HR-TEM images of the samples, which are presented in Fig. 2(c) and (d). The appearance of the smaller and larger particles in the sample can be further evidenced by the varying transparency of the electron beam in the HR-TEM images. To further confirm the characteristics of elements, present in FeNP, the EDS spectrum was measured, as shown in Fig. 2(e). The spectrum clearly revealed the presence of Fe, C, N, and O elemental peaks without the presence of other impurity peaks in the sample, and the corresponding elemental weight (%) was 32.51, 24.94, 31.45, and 11.10%, respectively.
 | ||
| Fig. 2 (a) and (b) FE-SEM images, (c) and (d) HR-TEM images, and (e) EDS spectra of FeNP (inset table shows the elemental wt%). | ||
3.2 FeNP induces cytotoxicity in cancer cells
To evaluate the cytotoxicity of FeNP, different cancerous cell lines (SK-OV-3, A2780, A2780cis, U87 MG, MDA-MB-231 and MCF-7) and a normal human fibroblast cell line (MRC-5) were treated with FeNP for 96 h. The potency of FeNP was shown mainly on ovarian cancer cell lines (SK-OV-3, A2780, and A2780cis) as well as other types of cancers including glioblastoma (U-87MG) and breast cancer (MDA-MB-231 and MCF-7). The cytotoxicity of the proposed FeNP is also significantly higher in the CisPt resistant ovarian cancer cell line (A2780cis) compared to the other treated groups (Fig. 3(c)).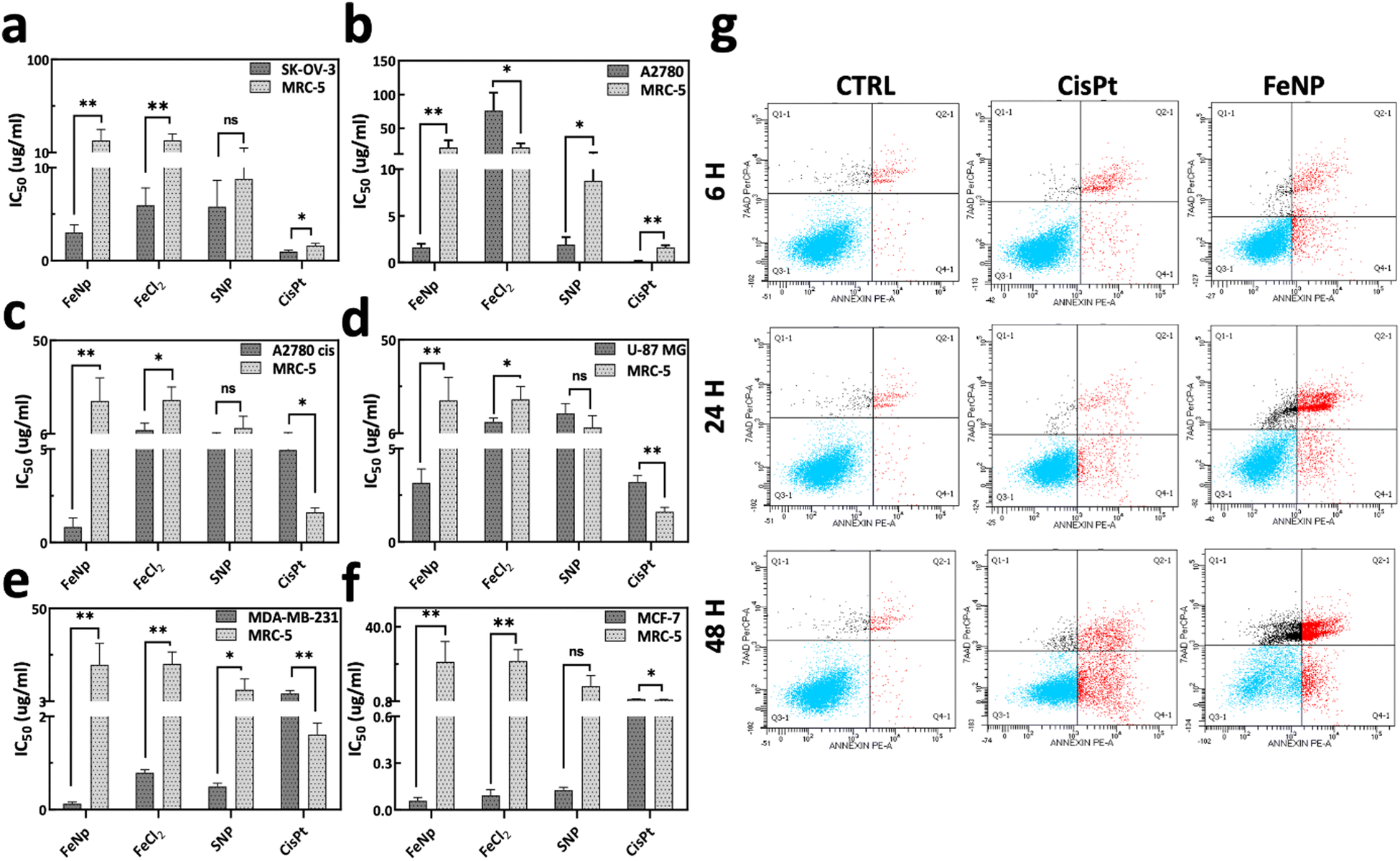 | ||
| Fig. 3 Cytotoxic evaluation. (a)–(f) IC50 values of FeNP toward normal and cancer cells. Values are expressed in μg ml−1. Data represent mean ± SD (n = 3). (g) Apoptosis analysis. The SK-OV-3 cells were analyzed by FACS using Annexin V/7AAD double staining after treatment of FeNP (50 μg ml−1) and CisPt (3 μg ml−1; 10 μM) for 6, 24 and 48 h (Q3: viable cells, Q4: early apoptosis, Q2: late apoptosis, and Q1: necrosis). | ||
Briefly, the IC50 values of FeNP and its precursor (FeCl2) against cancer cell lines are in the range of 0.05–3.16 μg ml−1 and 0.09–76.3 μg ml−1, respectively. In comparison with CisPt on cancer cells, FeNP is more effective up to 54 fold. However, as depicted in Fig. 3(a)–(f), FeNP caused low or negligible toxicity to normal cells (0.07-fold). These data demonstrate that FeNP could release Fe+ ions inside the cells, which induce ROS species production and exert their toxicity. Complete data of IC50 values are listed in Table S1, ESI.† Hence, our results suggest that FeNP exhibits considerable toxicity towards all cancer cell lines while having less toxicity towards normal cells.
To validate the cell death mechanism caused by FeNP, annexin V/7AAD staining was performed following the treatment with FeNP, FeCl2 (10 μg ml−1) and CisPt (6 μg ml−1; 20 μM) of the SK-OV-3 cell line for 6, 24, and 48 h. Our data showed that the FeNP treatment leads to apoptosis, which increased over time as shown in Fig. 3(g) and Fig. S2c (ESI†). We observed a percentage of viable cells similar to control cells (92%) after 6 h of treatment which reduced to 71.3% and 29.0% after 24 h and 48 h of FeNP treatment. Additionally, there is an increase in early apoptosis from 1.2% in the control cells to 2.7%, 4.2% and 13.3% after 6, 24 and 48 h of FeNP treatment. A significant increase in the proportion of late apoptosis was observed, from 2.2% in control samples to 2.7%, 15.7% and 44.0% after 6, 24 and 48 h of FeNP treatment, respectively. Comparatively, apoptosis in precursors and the CisPt group is lower than that due to FeNP (Fig. S2(a) and (b), ESI†). These results suggested that FeNP induces cell death via the apoptotic pathway.
3.3 Cytochrome c release
The release of cytochrome c is the major event during apoptosis.55,56 For this reason, next we examine the release of cytochrome c into the cytosol from mitochondria. Based on the IC50 data, we incubated SK-OV-3 cells with FeNP, FeCl2, SNP (50 μg ml−1) and CisPt (3 μg ml−1; 10 μM) for 6, 24, and 48 h and then compared them with untreated cells. As illustrated in Fig. 4 and Fig. S3 (ESI†) there is an obvious release of cyt c after 24 h of FeNP treatment compared to cisplatin and FeCl2-treated cells. Our data suggest that there is a cyt c release in the cytosol leading to cell death.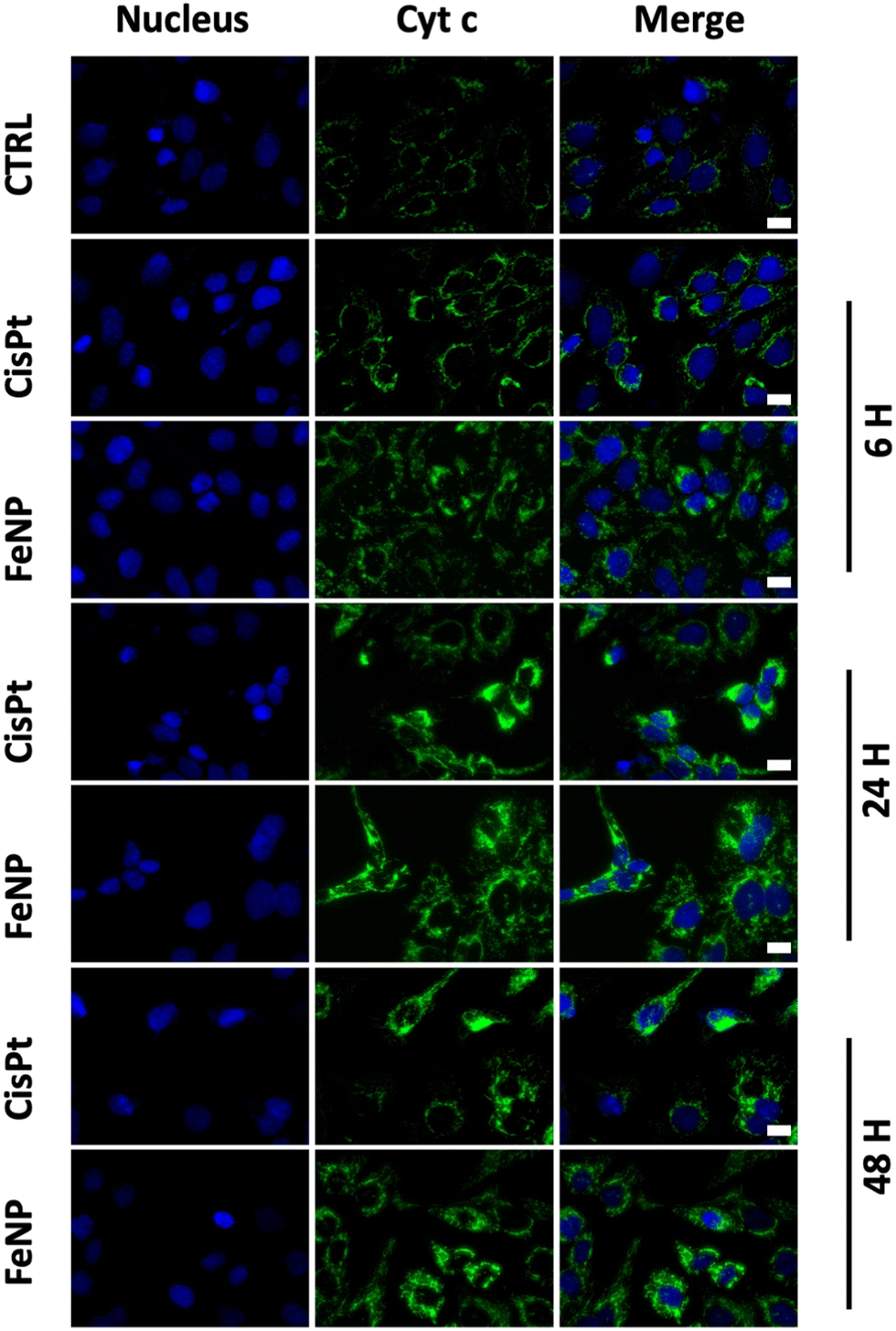 | ||
| Fig. 4 Immunofluorescence analysis of cytochrome c release of SK-OV-3 cells incubated with FeNP (50 μg ml−1) and CisPt (3 μg ml−1; 10 μM) for 6, 24 and 48 h. Bar, 10 μm. | ||
3.4 Cellular uptake analysis
One of the challenges in cancer therapy is the lysosomal degradation of chemotherapeutic agents.57 Colocalization experiments were performed to investigate the amount of FeNP internalized in lysosomes. To evaluate the FeNP accumulation, we incubated the SK-OV-3 cells for 6, 24 and 48 h at 37 °C with FeNP labelled with rhodamine (red), the nucleus with Hoechst 33342 (blue) and lysosomes with Lysotracker™Green DND-26 (green). Fig. 5 shows the internalization of FeNP at each time point. The colocalization of FeNP and lysosomes was evaluated with Pearson's correlation coefficient (R) after each time point. Interestingly, “R” values after 6 and 24 h of treatment were 0.64 and 0.709 but after 48 h the values decreased to 0.59 for FeNP. These results suggest that FeNP initially goes partially inside the lysosomes in agreement with previous studies for iron nanoparticles58–60 demonstrating the endocytotic uptake mechanism of FeNP, but with time, FeNP escapes lysosomal degradation and relocalized to the cytoplasm.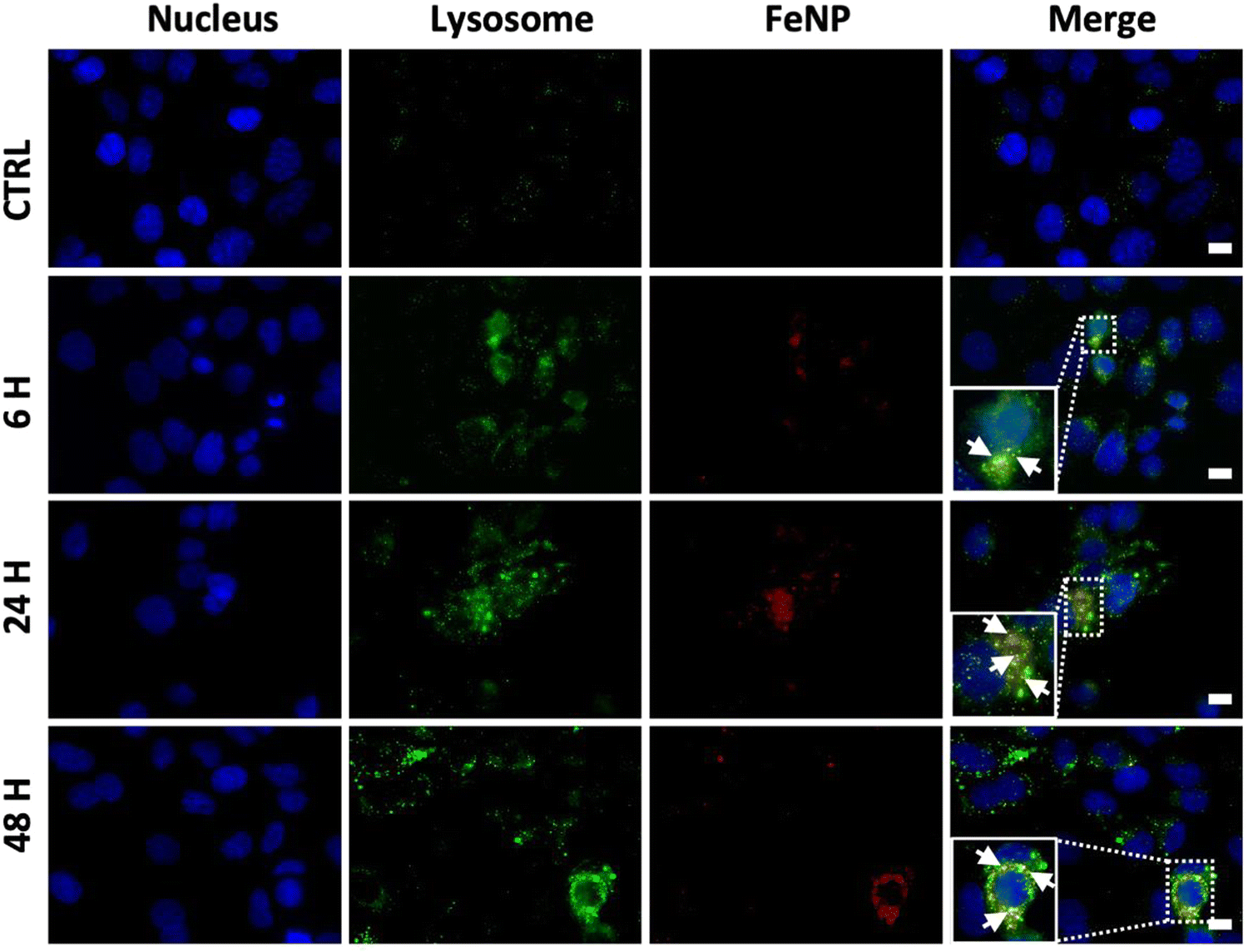 | ||
| Fig. 5 Lysotracker analysis of an FeNP for the cell internalization study. Fluorescence microscopy images of a rhodamine-labeled FeNP of SK-OV-3 ovarian cancer cell line after 6, 24 and 48 h, from the left to the right, respectively. Negative control (only nuclei and lysosome staining, rhodamine) was used to set up the fluorescence intensity and avoid the background signal. Bar, 10 μm. Cells were incubated with FeNP (50 μg ml−1) for 6, 24 and 48 h. | ||
We next examined the cell death mechanism through oxidative stress via ROS production. SK-OV-3 and MRC-5 cell lines were incubated with FeNP (50 μg ml−1) and CisPt (3 μg ml−1, 10 μM) for 6, 24 and 48 h. As shown in Fig. 6(a), in SK-OV-3 cells the H2O2 levels increase after 48 h. This effect could be due to the reduction of the glutathione peroxidase 4 (GPX4) activity. Comparatively, in fibroblasts, the overall H2O2 levels are higher than in SK-OV-3 as shown in Fig. 6(b) but are reduced with time due to the antioxidant activity. In comparison, between cancerous and normal cells, the overall H2O2 levels are lower in cancerous cells because of the FeNP catalytic activity for efficient ˙OH generation as a Fenton agent for subsequent damage of cancer cells.61,62 Our results are in accordance with previous reports, which demonstrate that ˙OH radical production depends on H2O2 and the catalytic activity of iron nanoparticles for efficient ˙OH generation to destroy cancer cells.61
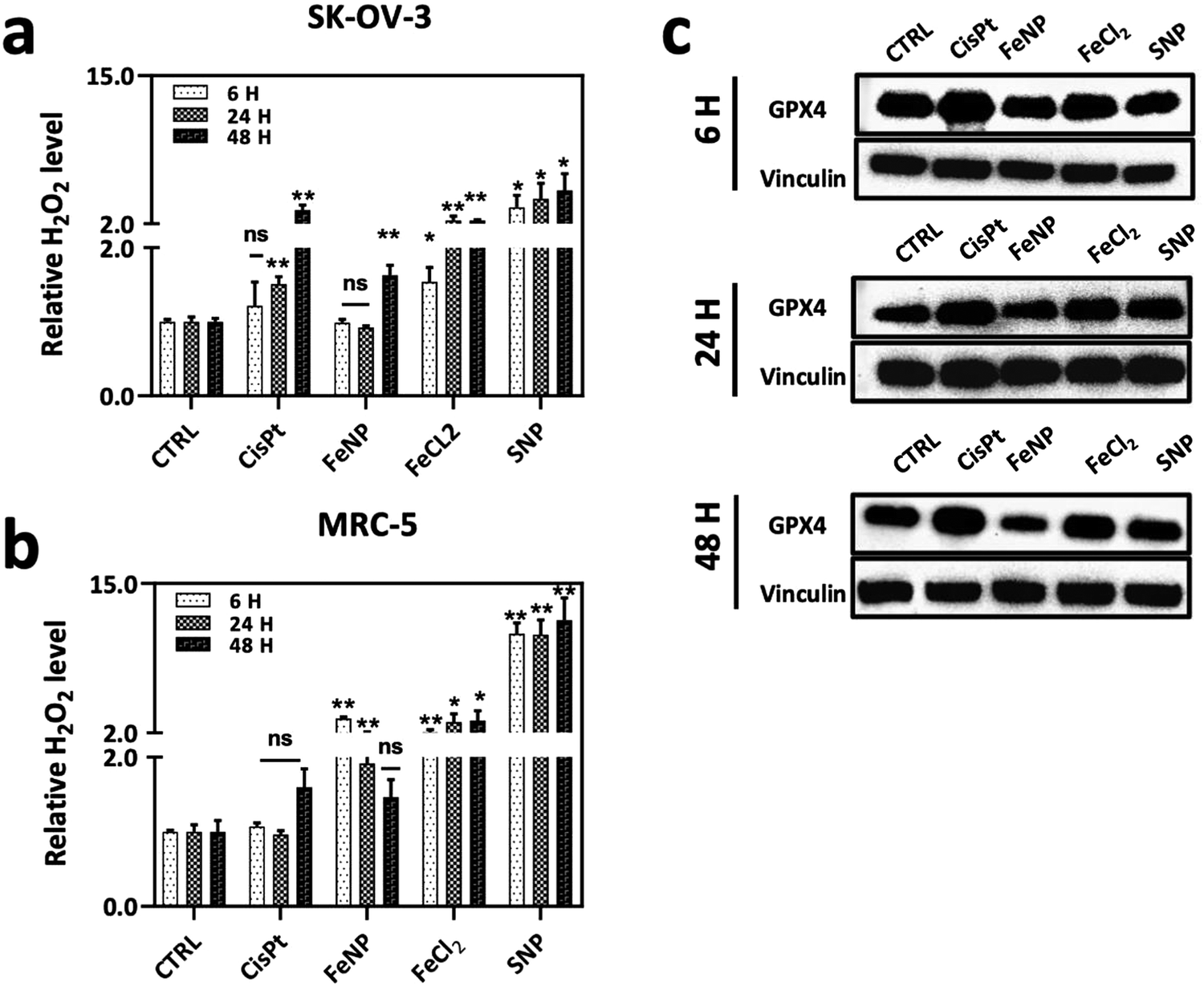 | ||
| Fig. 6 (a) and (b) Relative H2O2 level. (a) SK-OV-3 and (b) MRC-5 incubated with FeNP, FeCl2, SNP (50 μg ml−1) and CisPt (3 μg ml−1; 10 μM) for 6, 24 and 48 h. (c) Effect of FeNP on the GPX4 activity on the SK-OV-3 cell line, assessed by western blot after 6, 24 and 48 h of treatment. Cells were incubated with FeNP (50 μg ml−1) and CisPt (3 μg ml−1; 10 μM). Error bars indicate the SEM, and data shown are for two independent experiments of three replicates. Groups were considered statistically significant if p < 0.05 (*), p < 0.01 (**) and non-significant (ns). | ||
Recently, it has been reported that iron nanoparticles induce cell death via a lysosomal degradation pathway due to an acidic lysosomal environment, which releases iron ions involved in the production of ROS leading to ferroptosis cell death.63,64 There are a number of studies that demonstrated the role of ROS species in the ferroptotic cell death mechanism.65,66 Ferroptosis is an iron-dependent programmed cell death, which is characterized by higher intracellular iron ions and ROS production.67 Ferroptosis is activated by the depletion of the GPX4 activity. GPX4 is an antioxidant enzyme involved in repairing oxidative damage to lipids and acts as an inhibitor of ferroptosis. Thus, GPX4 is an important regulator of ferroptosis.68–70 Therefore, next we determined the GPX4-mediated ferroptosis after FeNP treatment. As shown in Fig. 6(c), the GpX4 activity is similar after 6 and 24 h of treatment, but eventually, after 48 h the GPX4 activity is downregulated in SK-OV-3, which demonstrates the involvement of FeNP in the ferroptosis-mediated cell death pathway with apoptosis. Previously, it has been reported that the reduced glutathione peroxidase activity leads to increased H2O2 levels ultimately leading to ˙OH production.71 CisPt treatment showed upregulation of the GPX4 activity, which indicates a CisPt resistance mechanism via ferroptosis inhibition.72 These data suggest that FeNP is involved in ROS production via a ferroptosis cell death mediated pathway.
3.5 Anticancer activity of FeNP on human ovarian cancer organoids
Normal mouse liver organoids were used to evaluate the toxicity effect of FeNP.73 The IC50 value of FeNP (>200 μg ml−1) was 21-fold higher than that of CisPt (9.2 ± 1.9 μg ml−1). This suggests the biocompatibility of FeNP towards liver organoids compared to its precursors and CisPt (Fig. 7(a) and Table S2, ESI†). | ||
| Fig. 7 Cytotoxicity of FeNP and CisPt (a) in normal mouse liver organoids. (b)–(d) PDTO obtained from a HGSOC patient. Error bars indicate the SEM. | ||
Afterward, we examined the toxicity of FeNP on HGSOC patient-derived tumor organoids (PDTO), the most common type of ovarian cancer with the worst survival rate.74,75 Patient-derived tumor organoids represent a useful ex vivo model to evaluate the therapeutic drug efficacy45,76,77 as they mimic the patients’ response in the clinic78 compared to xenografts.79 To better mimic the treatment option, we generated organoids from HGSOC patients to determine the efficacy of FeNP. In ovarian cancer, organoids could predict drug response in human patients.78,79 As shown in Fig. 7(b)–(d) the IC50 values are in the range of 0.3–2.7 μg ml−1 (Table S2, ESI†).
To investigate the molecular similarity of organoids with the parental tumor, immunohistochemistry analysis was performed. We observed immunopositivity for CA125 (cancer antigen 125), WT1 (Wilms Tumor 1) and PAX8 (paired box gene 8) which are markers of ovarian carcinoma (Fig, S4, ESI†).
3.6 In vivo biocompatibility of FeNP
Since FeNP was less toxic towards normal liver organoids, the biocompatibility of FeNP was investigated in mice. The mice were treated intraperitoneally twice per week with 2 mg kg−1 of CisPt and up to 8 mg kg−1 of FeNP. After treatment, mice were examined on a daily basis for signs of toxicity (physical distresses). As shown in Fig. 8, after FeNP treatment (8 mg kg−1) no obvious body weight changes are observed. These results demonstrate the biocompatible nature of FeNP.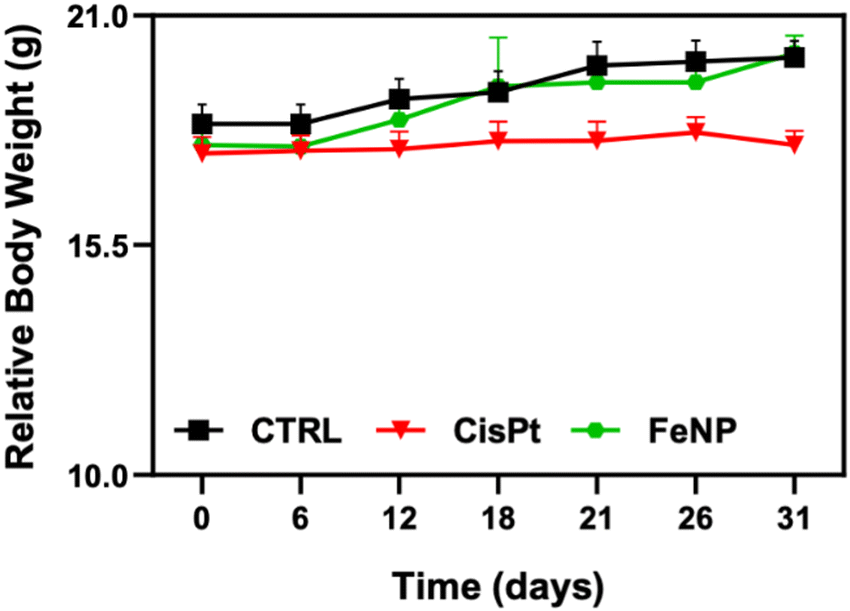 | ||
| Fig. 8 Body weight change of tumor-bearing mice after treatment with FeNP and CisPt. Error bars indicate the SEM. | ||
4. Conclusion
In summary, we demonstrated a iron-based self-therapeutic nanosystem with dual ROS generation through the Fenton reaction for cancer therapy. Our results showed that FeNP decomposed H2O2via the Fenton reaction to produce more hydroxyl radical species in cancer cells due to the lysosomal acidic environment, thereby inducing more cancer cell destruction than in normal cells as demonstrated by flow cytometric analysis and the cytochrome c release test, which showed an increase in apoptosis over time. Furthermore, cell internalization analysis indicated that FeNP escapes the lysosomal degradation pathway and is localized in the cytoplasm. While at 6 and 24 h, FeNP decomposed more intracellular H2O2 for hydroxyl radical species production, after 48 h there is an increase in the H2O2 level due to the downregulation of the glutathione peroxidase gene (GPX4) demonstrating a ferroptosis mediated cell death mechanism. Furthermore, a low toxicity towards normal liver organoids and a high therapeutic efficacy towards cancer organoids indicate the potential of FeNP for future therapeutic applications.Author contributions
M. A. and K. A.: conceptualization, experimental data, and writing; M.d MR and I.C.: data analysis, review & editing, and writing; M. C.: animal data; T. P. and V. C.: biological samples, review & editing, and funding acquisition; F.R.: conceptualization, data analysis, and review & editing.Funding
This research was funded by the Ministry of Health, Ricerca Corrente.Conflicts of interest
The authors declare no conflict of interest.References
- C. Jia, Y. Guo and F. G. Wu, Small, 2022, 18, 2103868 CrossRef CAS PubMed.
- Z. Tang, Y. Liu, M. He and W. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed.
- Z. Tang, P. Zhao, H. Wang, Y. Liu and W. Bu, Chem. Rev., 2021, 121, 1981–2019 CrossRef CAS PubMed.
- L. H. Fu, Y. Wan, C. Qi, J. He, C. Li, C. Yang, H. Xu, J. Lin and P. Huang, Adv. Mater., 2021, 33, 2006892 CrossRef CAS PubMed.
- S. Jin, L. Weng, Z. Li, Z. Yang, L. Zhu, J. Shi, W. Tang, W. Ma, H. Zong and W. Jiang, J. Mater. Chem. B, 2020, 8, 4620–4626 RSC.
- Y. Su, X. Zhang, G. Ren, Z. Zhang, Y. Liang, S. Wu and J. Shen, Chem. Eng. J., 2020, 400, 125949 CrossRef CAS.
- G. Liu, J. Zhu, H. Guo, A. Sun, P. Chen, L. Xi, W. Huang, X. Song and X. Dong, Angew. Chem., Int. Ed., 2019, 58, 18641–18646 CrossRef CAS PubMed.
- W. L. Wang, Z. Guo, Y. Lu, X. C. Shen, T. Chen, R. T. Huang, B. Zhou, C. Wen, H. Liang and B. P. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 17294–17305 CrossRef CAS PubMed.
- P. Liu, Y. Wang, L. An, Q. Tian, J. Lin and S. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 38833–38844 CrossRef CAS PubMed.
- K. Xu, Y. Cheng, J. Yan, Y. Feng, R. Zheng, X. Wu, Y. Wang, P. Song and H. Zhang, Nano Res., 2019, 12, 2947–2953 CrossRef CAS.
- S. L. Li, P. Jiang, F. L. Jiang and Y. Liu, Adv. Funct. Mater., 2021, 31, 2100243 CrossRef CAS.
- X. Chen, Y. Liu and W. Bu, Sci. Sin.: Chim., 2020, 50, 159–172 Search PubMed.
- Q. Tian, F. Xue, Y. Wang, Y. Cheng, L. An, S. Yang, X. Chen and G. Huang, Nano Today, 2021, 39, 101162 CrossRef CAS.
- K. Ni, T. Luo, G. T. Nash and W. Lin, Acc. Chem. Res., 2020, 53, 1739–1748 CrossRef CAS PubMed.
- D. Wu, Y. Chen, Z. Zhang, Y. Feng, Y. Liu, J. Fan and Y. Zhang, Chem. Eng. J., 2016, 294, 49–57 CrossRef CAS.
- J. Dinić, T. Efferth, A. T. García-Sosa, J. Grahovac, J. M. Padrón, I. Pajeva, F. Rizzolio, S. Saponara, G. Spengler and I. Tsakovska, Drug Resist. Updates, 2020, 52, 100713 CrossRef PubMed.
- T. Scattolin, I. Caligiuri, L. Canovese, N. Demitri, R. Gambari, I. Lampronti, F. Rizzolio, C. Santo and F. Visentin, Dalton Trans., 2018, 47, 13616–13630 RSC.
- M. Aghazadeh Tabrizi, P. G. Baraldi, S. Baraldi, E. Ruggiero, L. De Stefano, F. Rizzolio, L. Di Cesare Mannelli, C. Ghelardini, A. Chicca, M. Lapillo, J. Gertsch, C. Manera, M. Macchia, A. Martinelli, C. Granchi, F. Minutolo and T. Tuccinardi, J. Med. Chem., 2018, 61, 1340–1354 CrossRef CAS PubMed.
- X. Liu, Y. Jin, T. Liu, S. Yang, M. Zhou, W. Wang and H. Yu, ACS Biomater. Sci. Eng., 2020, 6, 4834–4845 CrossRef CAS PubMed.
- R. Cao, W. Sun, Z. Zhang, X. Li, J. Du, J. Fan and X. Peng, Chin. Chem. Lett., 2020, 31, 3127–3130 CrossRef CAS.
- P. Liu, Y. Peng, J. Ding and W. Zhou, Asian J. Pharm. Sci., 2022, 17, 177–192 CrossRef PubMed.
- J. Liu, R. Kang and D. Tang, FEBS J., 2021, 289, 7038–7050 CrossRef PubMed.
- D. Vitalakumar, A. Sharma and S. J. S. Flora, J. Biochem. Mol. Toxicol., 2021, 35, e22830 CrossRef CAS PubMed.
- B. Xie and Y. Guo, Cell Death Discovery, 2021, 7, 1–10 Search PubMed.
- L. M. Bystrom, M. L. Guzman and S. Rivella, Antioxid. Redox Signaling, 2014, 20, 1917–1924 CrossRef CAS PubMed.
- S. Doll and M. Conrad, IUBMB Life, 2017, 69, 423–434 CrossRef CAS PubMed.
- Z. Xu, B. Peng, Q. Liang, X. Chen, Y. Cai, S. Zeng, K. Gao, X. Wang, Q. Yi, Z. Gong and Y. Yan, Front. Immunol., 2021, 12, 3728 Search PubMed.
- Y. Xie, W. Hou, X. Song, Y. Yu, J. Huang, X. Sun, R. Kang and D. Tang, Cell Death Differ., 2016, 23, 369–379 CrossRef CAS PubMed.
- A. Banjac, T. Perisic, H. Sato, A. Seiler, S. Bannai, N. Weiss, P. Kölle, K. Tschoep, R. D. Issels, P. T. Daniel, M. Conrad and G. W. Bornkamm, Oncogene, 2007, 27, 1618–1628 CrossRef PubMed.
- S. J. Dixon, D. Patel, M. Welsch, R. Skouta, E. Lee, M. Hayano, A. G. Thomas, C. Gleason, N. Tatonetti, B. S. Slusher and B. R. Stockwell, eLife, 2014, 3, e02523 CrossRef PubMed.
- L. Ying Guo, Q. Sheng Xia, J. Lei Qin, M. Yang, T. Yu Yang, F. Tian You, Z. Hua Chen, B. Liu and H. Shang Peng, J. Colloid Interface Sci., 2022, 616, 81–92 CrossRef PubMed.
- X. Wang, P. Hua, C. He and M. Chen, Acta Pharm. Sin. B, 2022, 12, 3567–3593 CrossRef CAS PubMed.
- Z. Shi, J. Zheng, W. Tang, Y. Bai, L. Zhang, Z. Xuan, H. Sun and C. Shao, Front. Chem., 2022, 10, 269 Search PubMed.
- L. Zhao, X. Zhou, F. Xie, L. Zhang, H. Yan, J. Huang, C. Zhang, F. Zhou, J. Chen and L. Zhang, Cancer Commun., 2022, 42, 88–116 CrossRef PubMed.
- M. Zhang, X. Qin, Z. Zhao, Q. Du, Q. Li, Y. Jiang and Y. Luan, Nanoscale Horiz., 2022, 7, 198–210 RSC.
- F. Zhao, J. Yao, Y. Tong, D. Su, Q. Xu, Y. Ying, W. Li, J. Li, J. Zheng, L. Qiao, W. Cai, X. Mou, S. Che, J. Yu and Y. Hou, Mater. Adv., 2022, 3, 1191–1199 RSC.
- X. Wang, X. Zhong, Z. Liu and L. Cheng, Nano Today, 2020, 35, 100946 CrossRef CAS.
- L. Chen, Z. Lin, L. Liu, X. Zhang, W. Shi, D. Ge and Y. Sun, ACS Biomater. Sci. Eng., 2019, 5, 4861–4869 CrossRef CAS PubMed.
- Z. Li, X. Wu, W. Wang, C. Gai, W. Zhang, W. Li and D. Ding, Nanoscale Res. Lett., 2021, 16, 1–11 CrossRef PubMed.
- Z. Dong, L. Feng, Y. Chao, Y. Hao, M. Chen, F. Gong, X. Han, R. Zhang, L. Cheng and Z. Liu, Nano Lett., 2019, 19, 805–815 CrossRef CAS PubMed.
- X. Liu, X. Zhu, X. Qi, X. Meng and K. Xu, Int. J. Nanomed., 2021, 16, 1037–1050 CrossRef PubMed.
- X. Wang, X. Zhong, H. Lei, Y. Geng, Q. Zhao, F. Gong, Z. Yang, Z. Dong, Z. Liu and L. Cheng, Chem. Mater., 2019, 31, 6174–6186 CrossRef CAS.
- B. Ma, S. Wang, F. Liu, S. Zhang, J. Duan, Z. Li, Y. Kong, Y. Sang, H. Liu, W. Bu and L. Li, J. Am. Chem. Soc., 2019, 141, 849–857 CrossRef CAS PubMed.
- H. Miyoshi and T. S. Stappenbeck, Nat. Protoc., 2013, 8, 2471–2482 CrossRef CAS PubMed.
- T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem. – Eur. J., 2020, 26, 11868–11876 CrossRef CAS PubMed.
- N. K. Chaturvedi, M. J. Kling, D. W. Coulter, T. R. McGuire, S. Ray, V. Kesherwani, S. S. Joshi and J. G. Sharp, Oncotarget, 2018, 9, 16619–16633 CrossRef PubMed.
- E. I. Hassanen, R. M. S. Korany and A. M. Bakeer, J. Biochem. Mol. Toxicol., 2021, 35, e22722 CrossRef CAS PubMed.
- M. M. Rahman, Materials, 2021, 14, 6563 CrossRef CAS PubMed.
- L. Reguera, Y. Avila and E. Reguera, Coord. Chem. Rev., 2021, 434, 213764 CrossRef CAS.
- A. Djebli, A. Boudjemaa, H. Bendjeffal, H. Mamine, T. Metidji, H. Bekakria and Y. Bouhedja, Materials, 2020, 50, 1115–1122 CAS.
- M. E. Chacón Villalba, E. L. Varetti and P. J. Aymonino, Vib. Spectrosc., 2000, 23, 31–38 CrossRef.
- H. Razmi and H. Heidari, Anal. Biochem., 2009, 388, 15–22 CrossRef CAS PubMed.
- A. Mullaliu, G. Aquilanti, J. R. Plaisier, M. Giorgetti, B. Barbiellini, J. Kuriplach and R. Saniz, Condens. Matter, 2021, 6, 27 CrossRef CAS.
- A. Cano, J. Rodríguez-Hernández, A. Shchukarev and E. Reguera, J. Solid State Chem., 2019, 273, 1–10 CrossRef CAS.
- D. R. Green and J. C. Reed, Science, 1998, 31, 118 Search PubMed.
- A. Matapurkar and Y. Lazebnik, Cell Death Differ., 2006, 13, 2062–2067 CrossRef CAS PubMed.
- J. Hraběta, M. Belhajová, H. Šubrtová, M. A. M. Rodrigo, Z. Heger and T. Eckschlager, Int. J. Mol. Sci., 2020, 21, 1–18 Search PubMed.
- O. Lunov, T. Syrovets, B. Büchele, X. Jiang, C. Röcker, K. Tron, G. U. Nienhaus, P. Walther, V. Mailänder, K. Landfester and T. Simmet, Biomaterials, 2010, 31, 5063–5071 CrossRef CAS PubMed.
- N. Kohler, C. Sun, J. Wang and M. Zhang, Langmuir, 2005, 21, 8858–8864 CrossRef CAS PubMed.
- C. Wilhelm and F. Gazeau, Biomaterials, 2008, 29, 3161–3174 CrossRef CAS PubMed.
- C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang and J. Shi, Angew. Chem., Int. Ed., 2016, 55, 2101–2106 CrossRef CAS PubMed.
- H. Ranji-Burachaloo, P. A. Gurr, D. E. Dunstan and G. G. Qiao, ACS Nano, 2018, 12, 11819–11837 CrossRef CAS PubMed.
- S. Wang, J. Luo, Z. Zhang, D. Dong, Y. Shen, Y. Fang, L. Hu, M. Liu, C. Dai, S. Peng, Z. Fang and P. Shang, Am. J. Cancer Res., 2018, 8, 1933 CAS.
- A. Viktorinova, Arch. Biochem. Biophys., 2018, 654, 185–193 CrossRef CAS PubMed.
- M. Gao, P. Monian, Q. Pan, W. Zhang, J. Xiang and X. Jiang, Cell Res., 2016, 26, 1021–1032 CrossRef CAS PubMed.
- L. Jiang, N. Kon, T. Li, S. J. Wang, T. Su, H. Hibshoosh, R. Baer and W. Gu, Nature, 2015, 520, 57–62 CrossRef CAS PubMed.
- A. Weiland, Y. Wang, W. Wu, X. Lan, X. Han, Q. Li and J. Wang, Mol. Neurobiol., 2019, 56, 4880–4893 CrossRef CAS PubMed.
- X. Song, X. Wang, Z. Liu and Z. Yu, Front. Oncol., 2020, 10, 597434 CrossRef PubMed.
- T. M. Seibt, B. Proneth and M. Conrad, Free Radical Biol. Med., 2019, 133, 144–152 CrossRef CAS PubMed.
- A. Seiler, M. Schneider, H. Förster, S. Roth, E. K. Wirth, C. Culmsee, N. Plesnila, E. Kremmer, O. Rådmark, W. Wurst, G. W. Bornkamm, U. Schweizer and M. Conrad, Cell Metab., 2008, 8, 237–248 CrossRef CAS PubMed.
- J. Čapek and T. Roušar, Molecules, 2021, 26, 4710 CrossRef PubMed.
- X. Zhang, G. Gu, X. Li and C. Zhang, Cell Cycle, 2020, 19, 2530–2537, DOI:10.1080/15384101.2020.1809914.
- S. Palazzolo, M. Hadla, C. R. Spena, I. Caligiuri, R. Rotondo, M. Adeel, V. Kumar, G. Corona, V. Canzonieri, G. Toffoli and F. Rizzolio, Cancers, 2019, 11, 1997 CrossRef CAS PubMed.
- D. D. Bowtell, S. Böhm, A. A. Ahmed, P. J. Aspuria, R. C. Bast, V. Beral, J. S. Berek, M. J. Birrer, S. Blagden, M. A. Bookman, J. D. Brenton, K. B. Chiappinelli, F. C. Martins, G. Coukos, R. Drapkin, R. Edmondson, C. Fotopoulou, H. Gabra, J. Galon, C. Gourley, V. Heong, D. G. Huntsman, M. Iwanicki, B. Y. Karlan, A. Kaye, E. Lengyel, D. A. Levine, K. H. Lu, I. A. McNeish, U. Menon, S. A. Narod, B. H. Nelson, K. P. Nephew, P. Pharoah, D. J. Powell, P. Ramos, I. L. Romero, C. L. Scott, A. K. Sood, E. A. Stronach and F. R. Balkwill, Nat. Rev. Cancer, 2015, 15, 668–679 CrossRef CAS PubMed.
- M. Govindarajan, C. Wohlmuth, M. Waas, M. Q. Bernardini and T. Kislinger, J. Hematol. Oncol., 2020, 13, 1–20 CrossRef PubMed.
- T. Scattolin, I. Pessotto, E. Cavarzerani, V. Canzonieri, L. Orian, N. Demitri, C. Schmidt, A. Casini, E. Bortolamiol, F. Visentin, F. Rizzolio and S. P. Nolan, Eur. J. Inorg. Chem., 2022, e202200103 CAS.
- J. Kim, B. K. Koo and J. A. Knoblich, Nat. Rev. Mol. Cell Biol., 2020, 21, 571–584 CrossRef CAS PubMed.
- S. J. Hill, B. Decker, E. A. Roberts, N. S. Horowitz, M. G. Muto, M. J. Worley, C. M. Feltmate, M. R. Nucci, E. M. Swisher, H. Nguyen, C. Yang, R. Morizane, B. S. Kochupurakkal, K. T. Do, P. A. Konstantinopoulos, J. F. Liu, J. V. Bonventre, U. A. Matulonis, G. I. Shapiro, R. S. Berkowitz, C. P. Crum and A. D. D’Andrea, Cancer Discovery, 2018, 8, 1404–1421 CrossRef CAS PubMed.
- O. Kopper, C. J. de Witte, K. Lõhmussaar, J. E. Valle-Inclan, N. Hami, L. Kester, A. V. Balgobind, J. Korving, N. Proost, H. Begthel, L. M. van Wijk, S. A. Revilla, R. Theeuwsen, M. van de Ven, M. J. van Roosmalen, B. Ponsioen, V. W. H. Ho, B. G. Neel, T. Bosse, K. N. Gaarenstroom, H. Vrieling, M. P. G. Vreeswijk, P. J. van Diest, P. O. Witteveen, T. Jonges, J. L. Bos, A. van Oudenaarden, R. P. Zweemer, H. J. G. Snippert, W. P. Kloosterman and H. Clevers, Nat. Med., 2019, 25, 838–849 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb02691k |
| This journal is © The Royal Society of Chemistry 2023 |