
Ligand-regulated photoinduced electron transfer within metal–organic frameworks for efficient photocatalysis†
Yang
Tang
a,
Guanfeng
Ji
a,
Hanning
Li
a,
Hui
Gao
a,
Cheng
He
 a,
Liang
Zhao
*a and
Chunying
Duan
b
a,
Liang
Zhao
*a and
Chunying
Duan
b
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, People's Republic of China. E-mail: zhaol@dlut.edu.cn
bState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210093, P. R. China
First published on 4th August 2023
Abstract
Dye-based metal–organic frameworks (MOFs) have received great attention due to their excellent photochemical stability in photocatalysis. However, the control of photoinduced electron transfer (PET) from dye to substrates rather than to metal nodes is still a challenging task for modern chemistry and organic synthesis, limiting the application of dye-based MOFs in photocatalytic transformations. Herein, we report a new method by introducing a carbonyl group into triphenylamine to form a planar conjugate acridone to compulsively generate a greater degree of conjugation torsion between the dye-based ligands and the metal–carboxylate nodes for controlling the direction of electron flow in the MOF. A new heterogeneous three-dimensional (3D) acridone-based Co-MOF was fabricated by the solvothermal reaction. In comparison with the similar triphenylamine-based Co-MOF, extensive control experiments in combination with density functional theory (DFT) calculations demonstrated that the acridone-based Co-MOF successfully inhibits the intramolecular PET process between the acridone-based ligands and the metal nodes for efficient photocatalytic organic transformations. This new heterogeneous platform exhibits a promising approach to modulating the direction of PET in MOFs by fine modifications of the ligands, thus providing a new avenue to develop multipurpose and flexible catalytic systems.
Introduction
Metal–organic frameworks (MOFs) are widely recognized as porous coordination polymers (PCPs), which are highly regulated, long-range ordered crystalline materials constructed through the self-assembly of multifunctional organic ligands and metal ions or clusters. Over the past few decades, MOFs have garnered significant attention for their immense potential in various photocatalytic transformations.1–3 Incorporating organic dyes into MOFs provides high-density photoactive sites, enabling the dyes to be linked together and well isolated by metal ions. This strategy overcomes the challenges posed by non-recyclable catalysts and the strong aggregation and polymerization phenomena of organic dyes, which greatly limit the development and utilization of various homogeneous dye-based photocatalysts.4–6 Moreover, dye-based MOFs can enhance specific photophysical properties, such as efficient exciton hopping, amplified stimulated emission and higher light harvesting efficiency,7–9 which provides a versatile and adjustable platform for photocatalytic transformations. Photoinduced electron transfer (PET) is a general phenomenon whereby the absorption of light by photocatalysts provides an energetic driving force for electron-transfer reactions.10 Photoredox catalysis provides unique modes for chemical bond activation, cleavage and formation through the PET process, thereby enabling previously inaccessible chemical transformations.11,12 The promise of dye-based MOFs as photoredox catalysts hinges on their electron transfer ability upon photoexcitation.13 However, the direct connection of dyes with the metal nodes leads to electrons being preferentially transferred to the metal nodes from the excited states of the dyes through the coordination bonds under irradiation.14–16 As a consequence, organic dyes in dye-based MOFs are often used as photosensitizers for photocatalytic hydrogen production and CO2 reduction, which does not make sufficient use of the redox properties of organic dyes, thereby limiting the application of MOFs as photocatalysts.17–22Taking advantage of the easy modification features of MOFs, introducing the functional groups within MOFs is a promising and effective strategy for tailoring the physical environment of cavities, pores and electronic structures for targeted applications.23–25 Recent research has indicated that the functionalization of dye-based ligands can regulate the twisted conjugative junction between the ligands and metal nodes, which enables MOFs to markedly enhance the separation efficiency of photogenerated carriers in MOFs and hamper the futile intramolecular fluorescence quenching, thus enhancing the photocatalytic efficiency.26 Our group has also dedicated a lot of effort to realizing the targeted PET process in MOFs by fine-tuning the structures of dye-based ligands. Upon incorporating sterically bulky hindrance groups into the backbone of dye-based ligands or controlling the geometric configuration of dye-based ligands to induce twisted conjugation between ligands and functional metal coordination carboxylate nodes, futile fluorescence quenching between metal sites and dyes can be effectively prevented, which rectifies unidirectional electron transfer routes and enables directional electron transfer.27,28 Note that the direction of the electron transfer was able to be directly regulated for controlling the PET process by adjusting the geometric configuration of the photosensitive ligands. Triphenylamine (TPA) as a common organic photosensitizer has been successfully decorated into MOFs for photocatalytic hydrogen production and CO2 reduction by photoreduction.29,30 We speculate that decorating the TPA-based ligand to form a π-conjugate plane might significantly generate a twist of conjugation between dye-based ligands and metal–carboxylate nodes which avoids unwanted intramolecular PET processes under photoexcitation, thus providing a new strategy to efficiently facilitate a variety of photocatalytic transformations by directly using the outstanding redox properties of dyes.
Herein, by introducing a carbonyl group into a fan-shaped triphenylamine-based ligand to connect two benzene rings of triphenylamine to form a remarkable π-conjugate plane chromophore (acridone) that possesses excellent light absorption capacity31 and the potential to become an excellent photocatalyst, we have developed a new heterogeneous approach for regulating the electron transfer path in a dye-based cobalt–organic framework. The introduction of the carbonyl group resulted in twisted conjugation between the dye moieties and the coordinated carboxylates to inhibit the intramolecular PET process between the dye-based ligands and the metal nodes, which was conducive for electron transfer to the substrate to improve the photocatalytic transformation efficiency (Scheme 1). It is worth noting that the use of acridone as a photocatalyst has been little explored until now. To our knowledge, this is the first instance of an acridone-based MOF in the field of photocatalysis, allowing for precise control over electron transfer pathways. This opens up opportunities for designing photoredox and catalytic systems from an intrinsic perspective, while also facilitating the development of scalable and sustainable synthetic strategies.
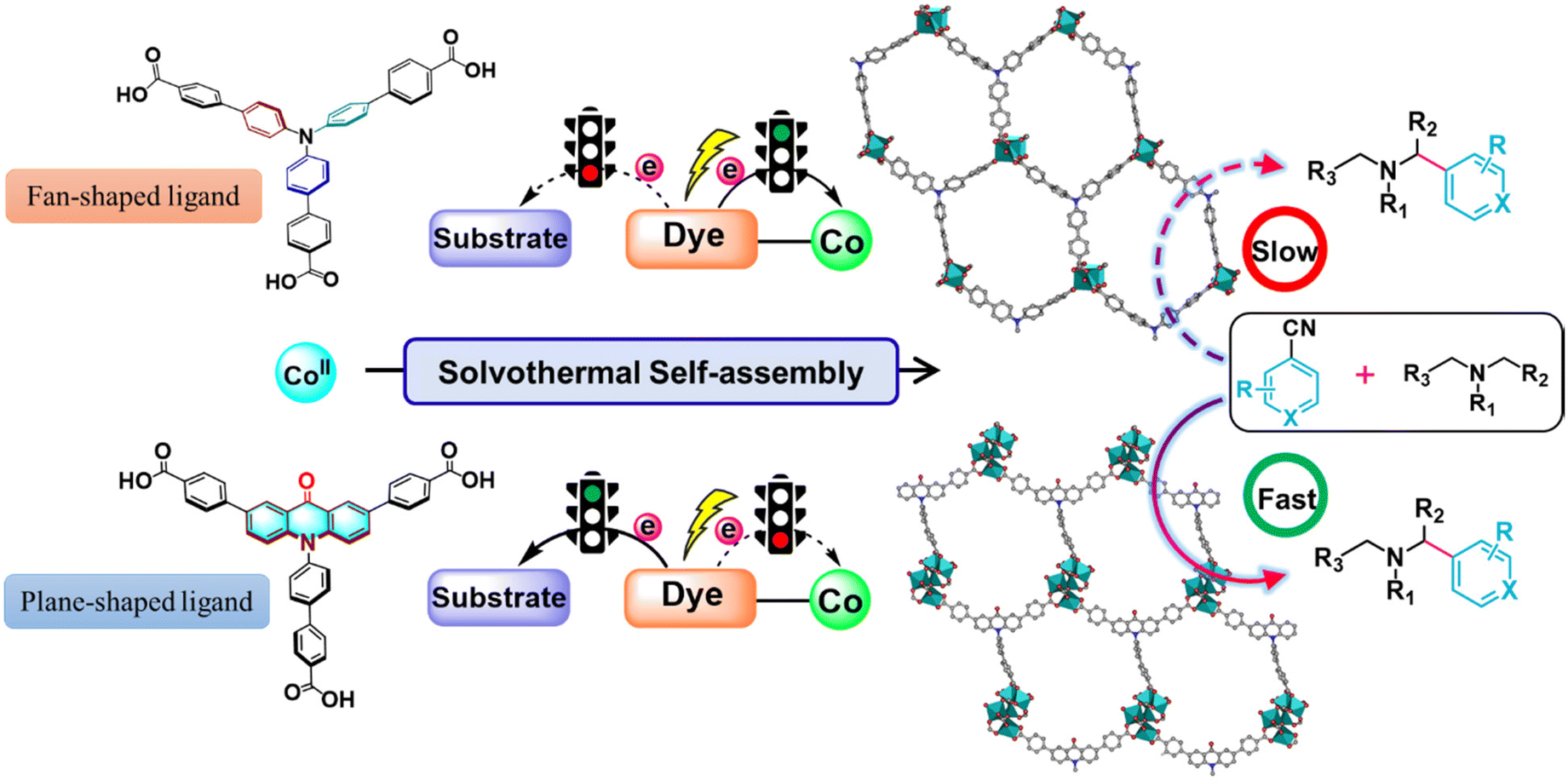 | ||
| Scheme 1 Schematic illustration of the acridone-functionalized Co-based MOF for the photocatalytic coupling reaction. | ||
Experimental
Chemicals and materials
All the chemicals and solvents were of reagent-grade quality, obtained from commercial sources and used without further purification. The synthetic process for N-((phenyl-D5)methyl-D2)pyrrolidine was referenced from the existing literature.32 The synthetic process for 10-(4-bromophenyl)acridin-9(10H)-one was also referenced from the existing literature.33Synthesis of 2,7-dibromo-10-(4-bromophenyl)acridin-9(10H)-one
Compounds 10-(4-bromophenyl)acridin-9(10H)-one (1.00 g, 2.87 mmol) and N-bromosuccinimide (NBS) (1.53 g, 8.61 mmol) were dissolved in DMF (50 mL) and stirred at 80 °C for 12 hours. Then, the mixture was quenched with water and CH2Cl2. After the separation of phases, the organic layer was washed with water (3 × 100 mL) and brine (1 × 100 mL) and dried over anhydrous Na2SO4. After filtration, the filtrate was evaporated to dryness under reduced pressure and the crude product was purified by chromatography on silica gel using CH2Cl2 as the eluent to yield compound 2,7-dibromo-10-(4-bromophenyl)acridin-9(10H)-one. Yield: 89%. 1H NMR (500 MHz, CDCl3) δ 8.58 (d, J = 2.4 Hz, 2H), 7.80 (d, J = 4.7 Hz, 2H), 7.51 (dd, J = 9.1, 2.4 Hz, 2H), 7.17 (d, J = 1.9 Hz, 2H), 6.57 (d, J = 9.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 175.66, 141.51, 137.23, 136.59, 134.80, 131.48, 129.89, 124.39, 123.01, 118.60, 115.69. HRMS (ESI+): calcd for [C19H10Br3NO + H]+: m/z = 507.8365, found: 507.8356. [C19H10Br3NO + Na]+: m/z = 529.8185, found: 529.8172.Synthesis of dimethyl 4,4′-(10-(4′-(methoxycarbonyl)-[1,1′-biphenyl]-4-yl)-9-oxo-9,10-dihydroacridine-2,7-diyl)dibenzoate
Compounds 2,7-dibromo-10-(4-bromophenyl)acridin-9(10H)-one (1.00 g, 1.98 mmol), K2CO3 (1.64 g, 11.88 mmol), [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) chloride (0.15 g, 0.20 mmol) and 4-(methoxycarbonyl)benzeneboronic acid (1.60 g, 7.92 mmol) were dissolved in a mixture containing 100 mL THF and 30 mL H2O. The reaction mixture was refluxed with stirring for 24 hours under an argon atmosphere and then it was cooled to room temperature. The resulting mixture was poured into 100 mL of water and extracted from dichloromethane (3 × 100 mL). The organic phase was washed with water (2 × 100 mL) and dried over anhydrous Na2SO4. After filtration, the filtrate was evaporated to dryness under reduced pressure. The crude product was purified by chromatography on silica gel using dichloromethane as the eluent to yield the compound dimethyl 4,4′-(10-(4′-(methoxycarbonyl)-[1,1′-biphenyl]-4-yl)-9-oxo-9,10-dihydroacridine-2,7-diyl)dibenzoate. Yield: 72%. 1H NMR (500 MHz, CDCl3): δ 8.78 (d, J = 2.2 Hz, 2H), 8.14 (d, J = 8.3 Hz, 2H), 8.04 (d, J = 8.3 Hz, 4H), 7.92 (d, J = 8.3 Hz, 2H), 7.77–7.71 (m, 4H), 7.67 (t, J = 7.1 Hz, 2H), 7.46 (d, J = 8.3 Hz, 1H), 6.90–6.86 (m, 1H), 3.91 (s, 1H), 3.86 (s, 1H). 13C NMR (126 MHz, CDCl3): δ 177.89, 166.88, 166.73, 143.92, 143.82, 142.69, 141.75, 138.47, 133.46, 132.18, 130.45, 130.43, 130.27, 130.05, 129.95, 129.01, 127.24, 126.74, 125.71, 122.12, 117.74, 52.27, 52.11. HRMS (ESI+): calcd for [C43H31NO7 + H]+: m/z = 674.2173, found: 674.2176. [C43H31NO7 + Na]+: m/z = 696.1993, found: 696.1988.Synthesis of 4,4′-(10-(4′-carboxy-[1,1′-biphenyl]-4-yl)-9-oxo-9,10-dihydroacridine-2,7-diyl)dibenzoic acid (H3CDDA)
Compounds dimethyl 4,4′-(10-(4′-(methoxycarbonyl)-[1,1′-biphenyl]-4-yl)-9-oxo-9,10-dihydroacridine-2,7-diyl)dibenzoate (5.00 g, 7.43 mmol) and NaOH (4.4 g, 0.11 mol) were dissolved in a mixture containing 50 mL ethanol and 50 mL H2O. The mixture was then refluxed for 24 hours. After cooling to room temperature, the mixture was acidified with HCl. The pH of the solution was adjusted to 3 to obtain a yellow precipitate. Finally, the precipitate was filtered and washed with plenty of water and dried in an oven to obtain the desired product H3CDDA. Yield 90%. 1H NMR (400 MHz, DMSO-d6): δ 13.02 (s, 3H), 8.70 (d, J = 2.3 Hz, 2H), 8.19 (d, J = 8.4 Hz, 2H), 8.12 (d, J = 8.4 Hz, 1H), 8.09–8.04 (m, 3H), 8.02 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 8.4 Hz, 4H), 7.77 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 8.9 Hz, 2H). 13C NMR (126 MHz, DMSO-d6): δ 176.60, 167.06, 142.91, 142.86, 142.39, 140.49, 138.01, 132.45, 132.40, 130.52, 130.47, 130.33, 130.12, 130.08, 129.81, 129.63, 127.19, 126.50, 124.18, 121.36, 118.12. HRMS (ESI+): calcd for [C40H24NO7]−: m/z = 630.1547, found: 630.1554. Elemental analysis calcd for C40H25NO7: C 76.05, H 3.99, N 2.22%; found: C 76.07, H 4.04, N 2.18%.Preparation of Co-CDDA
A mixture of H3CDDA (31.56 mg, 0.05 mmol) and Co(OAc)2·4H2O (10.45 mg, 0.055 mmol) was dissolved in water (0.5 mL) and N,N-dimethylformamide (DMF) (3 mL), which were placed in a 10 mL Teflon vessel within the autoclave. The mixture was heated to 120 °C for 72 hours and then cooled to room temperature at a rate of 3 °C h−1. Red block crystals of Co-CDDA were obtained and collected from the mixture system above, washed with DMF and air-dried. Yield: 40% (based on the crystal dried under room temperature). Elemental analysis calcd for C127.34H83.49Co5.10N5.45O27.44: C 62.91 H 3.44, N 3.14%; found: C 63.28, H 3.26, N 3.32%. IR (KBr): 3400 (br, vs), 1606 (v), 1545 (w), 1520 (w), 1480 (m), 1400 (s), 1331 (m), 1300 (m), 1255 (m), 1106 (w), 1033 (w), 1006 (m), 991 (m), 1006 (m), 957 (w), 921 (m), 864 (m), 837 (w), 821 (m), 783 (s), 748 (m), 734 (m), 712 (m), 691 (m), 664 (m) cm−1 (Fig. S9†).Crystallography
The intensities were collected on a Bruker SMART APEX CCD diffractometer equipped with a graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation source; the data were acquired using the SMART and SAINT programs.34,35 The structure was solved by direct methods and refined by full matrix least-squares methods using the program SHELXL-2014.36 In the structural refinement of Co-CDDA, all non-hydrogen atoms were refined anisotropically. Hydrogen atoms within the ligand backbones, DMF and the coordinate water molecules were fixed geometrically at calculated distances and allowed to ride on the parent non-hydrogen atoms. The three disordered benzene rings were refined in two parts while constraining the sum of the occupancies to unity. The relative occupancies of the two parts converged to values of 0.732![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.268; 0.816:0.174 and 0.626:0.374, respectively, after refinement. To assist the stability of the refinements, the disordered benzene rings were restrained as idealized regular polygons. The thermal parameters of adjacent atoms in benzene rings were restrained to be similar, respectively. The SQUEEZE subroutine in PLATON was used.37
:0.268; 0.816:0.174 and 0.626:0.374, respectively, after refinement. To assist the stability of the refinements, the disordered benzene rings were restrained as idealized regular polygons. The thermal parameters of adjacent atoms in benzene rings were restrained to be similar, respectively. The SQUEEZE subroutine in PLATON was used.37
General procedure for the photocatalytic α-amino C(sp3)–H arylation of amines with 1,4-dicyanobenzene
A 10 mL flame-dried Schlenk quartz flask was filled with 1,4-dicyanobenzene (1.0 mmol), Co-CDDA (20.0 μmol), K2CO3 (1.0 mmol), amine (2.0 mmol), a mini-stirrer and DMAc (4.0 mL). Then the reaction mixture was degassed by bubbling argon for 20 minutes under atmospheric pressure. After that, the flask containing the reaction mixture was then irradiated by a 420 nm LED irradiation for 14 hours with fan cooling to maintain the flask at room temperature. After the reaction, the mixture was centrifuged at 9000 rpm for 5 min, and the supernatant was concentrated under vacuum distillation. The residues were separated on a silica gel column (EtOAc/n-hexane) to obtain the isolated yields.General procedure for photocatalytic benzylamine oxidation
A 10 mL flame-dried Schlenk quartz flask was filled with benzylamine (0.2 mmol), nitrobenzene (0.4 mmol), Co-CDDA (10.0 μmol), a mini-stirrer and CH3CN (4.0 mL). Then the reaction mixture was degassed by bubbling argon for 20 minutes under atmospheric pressure. After that, the flask containing the reaction mixture was then irradiated by a 420 nm LED irradiation for 6 hours with fan cooling to maintain the flask at room temperature. After the reaction, the products were detected by GC analyses.Results and discussion
Preparation and characterization of metal–organic frameworks
The introduction of a carbonyl group at the ortho-position of the N-atom in the tris(4′-carboxybiphenyl)amine ligand, H3TBPA,38 resulted in the formation of an acridone dye group, which extended to a 4,4′-(10-(4′-carboxy-[1,1′-biphenyl]-4-yl)-9-oxo-9,10-dihydroacridine-2,7-diyl)dibenzoic acid (named H3CDDA). Co-CDDA, an acridone-containing cobalt-based metal–organic framework, was obtained in a yield of approximately 40% via a solvothermal reaction of ligand H3CDDA and Co(OAc)2·4H2O in a mixed solvent of N,N-dimethylformamide (DMF) and H2O (v:v = 6:1) at 120 °C for 72 hours. Single-crystal X-ray structural analysis revealed that Co-CDDA adopts a 3D framework and crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (CCDC number: 2263257†), indicating its highly ordered structure. Five independent Co ions are connected by two μ3-OH and coordinated with sixteen oxygen atoms from eight carboxyl groups of different H3CDDA ligands, two H2O molecules and three DMF molecules to form a unique Co5 cluster (Fig. S1†). Interestingly, there are three types of Co coordination modes. Co1, Co2 and Co4 have a distorted octahedral geometry, while Co3 is four-coordinated, showing a pyramidal coordination environment, and Co5 is five-coordinated. Each deprotonated H3CDDA ligand serves as a three-point connected node to connect three pentanuclear Co5 clusters to form a 3D framework with opening channels of 15.8 × 19.4 Å2 (Fig. 1a, S2 and S3†). The void volume of Co-CDDA was calculated to be approximately 4600 Å3 per unit cell (approximately 49.1% of the cell volume) using the PLATON program.37 The nitrogen adsorption–desorption isotherm of Co-CDDA at 77 K showed a reversible type-II isotherm with a Brunauer–Emmett–Teller (BET) surface area of 252.1 m2 g−1, confirming the architectural stability and permanent porosity of Co-CDDA (Fig. S4†).39 The pore size distributions of Co-CDDA were approximately 16 Å and 20 Å (Fig. S5†), which matched well with those of the single-crystal X-ray structural analysis, indicating that the pores were large enough and accessible for smooth substrate diffusion and catalyst–substrate interactions.40 Moreover, the elemental mapping images indicated that each element was uniformly distributed inside the Co-CDDA sample (Fig. S6†). Importantly, the introduction of the carbonyl group promoted the N-atom and two benzene rings of triphenylamine to form a conjugate plane of the acridone group,31 resulting in a twisted dihedral angle of 47° between the conjugated carboxylic coordination group and the acridone group (Fig. 1b). This twisted connection with the CoII-carboxylate nodes provided the probability for regulating the direction of electronic communication between metal centers and dye fragments and restraining unwanted inter- and intramolecular fluorescence quenching of CoII sites towards dye-based ligands.
(CCDC number: 2263257†), indicating its highly ordered structure. Five independent Co ions are connected by two μ3-OH and coordinated with sixteen oxygen atoms from eight carboxyl groups of different H3CDDA ligands, two H2O molecules and three DMF molecules to form a unique Co5 cluster (Fig. S1†). Interestingly, there are three types of Co coordination modes. Co1, Co2 and Co4 have a distorted octahedral geometry, while Co3 is four-coordinated, showing a pyramidal coordination environment, and Co5 is five-coordinated. Each deprotonated H3CDDA ligand serves as a three-point connected node to connect three pentanuclear Co5 clusters to form a 3D framework with opening channels of 15.8 × 19.4 Å2 (Fig. 1a, S2 and S3†). The void volume of Co-CDDA was calculated to be approximately 4600 Å3 per unit cell (approximately 49.1% of the cell volume) using the PLATON program.37 The nitrogen adsorption–desorption isotherm of Co-CDDA at 77 K showed a reversible type-II isotherm with a Brunauer–Emmett–Teller (BET) surface area of 252.1 m2 g−1, confirming the architectural stability and permanent porosity of Co-CDDA (Fig. S4†).39 The pore size distributions of Co-CDDA were approximately 16 Å and 20 Å (Fig. S5†), which matched well with those of the single-crystal X-ray structural analysis, indicating that the pores were large enough and accessible for smooth substrate diffusion and catalyst–substrate interactions.40 Moreover, the elemental mapping images indicated that each element was uniformly distributed inside the Co-CDDA sample (Fig. S6†). Importantly, the introduction of the carbonyl group promoted the N-atom and two benzene rings of triphenylamine to form a conjugate plane of the acridone group,31 resulting in a twisted dihedral angle of 47° between the conjugated carboxylic coordination group and the acridone group (Fig. 1b). This twisted connection with the CoII-carboxylate nodes provided the probability for regulating the direction of electronic communication between metal centers and dye fragments and restraining unwanted inter- and intramolecular fluorescence quenching of CoII sites towards dye-based ligands.
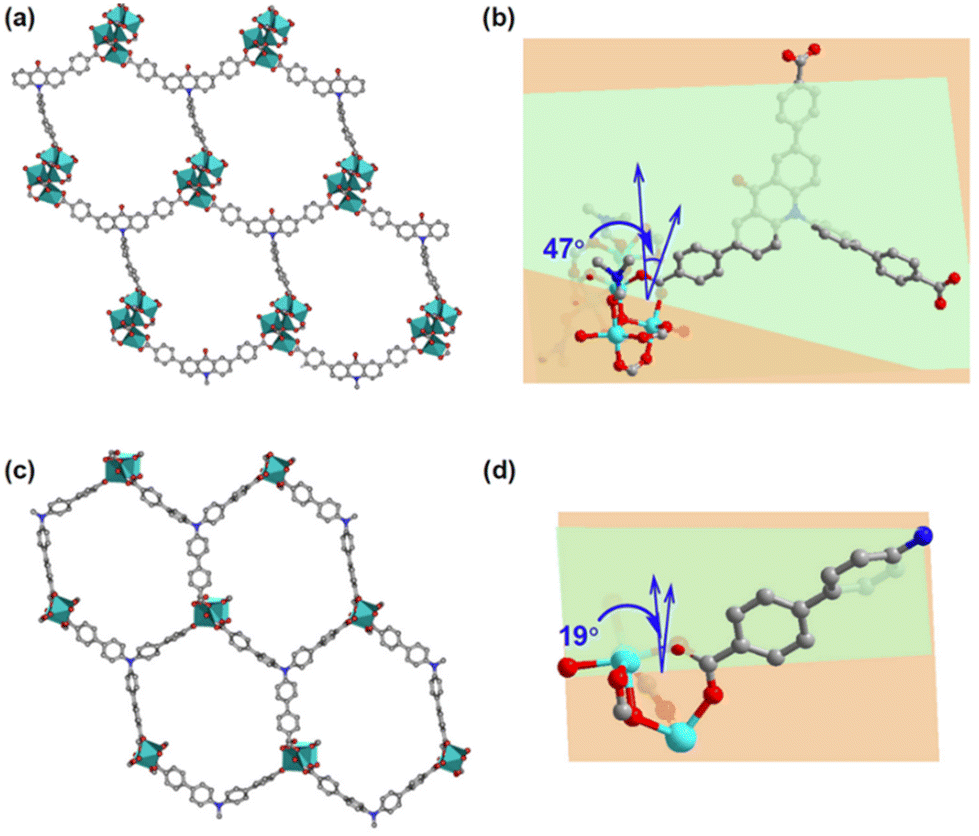 | ||
| Fig. 1 (a) Hexagonal pores in Co-CDDA. (b) Twisted dihedral angle between the CoII-carboxylate coordination plane and the dye plane of Co-CDDA. (c) Similar hexagonal pores in Co-TBPA. (d) Twisted dihedral angle between the CoII-carboxylate coordination plane and the dye plane of Co-TBPA. | ||
In order to investigate the influence of introducing the carbonyl group into triphenylamine on the electron-transport path under photoexcitation, Co-TBPA was prepared according to the reported literature.38 Single-crystal X-ray structural analysis revealed that Co-TBPA possesses a similar internal cavity and open window to Co-CDDA (Fig. 1a and c), facilitating rapid mass transfer during catalytic applications. Moreover, the identical distances between dyes and metal nodes in both Co-CDDA and Co-TBPA and the non-interpenetrated nature of both MOFs excluded intermolecular luminescence quenching of CoII nodes to the excited state. It should be noted that the absence of the carbonyl group from the acridone resulted in a maximum twisted dihedral angle of only 19° between the CoII-carboxylate node and the adjacent phenyl moiety of the TPA-based ligand (Fig. 1d). Therefore, Co-TBPA was an ideal comparison compound to Co-CDDA for exploring the effects of ligands in regulating electron transfer within MOFs in photocatalysis.
The powder X-ray diffraction (PXRD) patterns of the MOF samples matched well with those of Co-CDDA and Co-TBPA, respectively, based on their single crystal structural data, indicating the high phase purity of the samples (Fig. S7†). Thermogravimetric analysis (TGA) revealed that Co-CDDA and Co-TBPA38 exhibited structural stability at room temperature, as evidenced by their lack of decomposition below 400 °C (Fig. S8†), ensuring the maintenance of the framework structure at the reaction temperature. Malachite green was chosen as the indicator for the dye-uptake study. A quantum uptake equivalent to 16.1% of the MOFs’ weight was obtained by soaking Co-CDDA in a methanol solution containing malachite green (Fig. S10†).41,42 Strong fluorescence emission was observed using the confocal laser scanning microscope, which was attributed to the malachite green dye adsorbed by Co-CDDA (Fig. S12†). The image showed that malachite green molecules were uniformly distributed throughout the Co-CDDA crystals, indicating that the dye molecules had penetrated deeply into the crystals through the channels rather than being surface adsorbed on the crystals.43 An identical phenomenon was also observed in Co-TBPA (Fig. S11 and S13†), demonstrating that both Co-CDDA and Co-TBPA allowed organic substrate molecules to access their open pores.44
The solid-state ultraviolet–visible (UV-Vis) absorption spectrum of Co-CDDA exhibited a full range of absorption. The strong absorption of Co-CDDA in the range of 400–450 nm was attributed to the π–π* transition of the π electron in the molecule, which was caused by the charge transfer of the acridone main body (Fig. 2a).45 This indicated that the absorption behavior in Co-CDDA mainly benefited from the absorption of the ligand H3CDDA. Another strong broad visible absorption peak ranging from 450 nm to 700 nm was reasonably assigned to the typical d–d transition peak of cobalt nodes.46 The absorption performance of Co-TBPA was similar to that of Co-CDDA, showing good absorption under visible light. To elucidate the photocatalytic abilities of Co-CDDA and Co-TBPA, Mott–Schottky measurements were performed at frequencies of 500, 1000 and 1500 Hz (Fig. 2b and c).47,48 The LUMO positions determined from the intersection for Co-CDDA and Co-TBPA were −2.01 V and −1.92 V (vs. Ag/AgCl), respectively. The band gaps (Eg) of Co-CDDA and Co-TBPA were estimated to be 2.66 eV and 2.83 eV, respectively, by the Kubelka–Munk method from Tauc plots (Fig. 2d).49,50 The HOMO positions were then calculated to be 0.65 V and 0.91 V (vs. Ag/AgCl), respectively. These electrochemical measurement results demonstrated that both Co-CDDA and Co-TBPA with a sufficient potential of the HOMO and LUMO could be widely applied in the activation of substrates such as 1,4-dicyanobenzene (−1.65 V vs. SCE),51,52 for various photoinduced organic synthesis reactions. To further explore the charge separation ability of these two heterogeneous photocatalysts, electrochemical impedance spectroscopy (EIS) and photocurrent analysis were carried out for comparison. The results revealed that Co-TBPA (Rct, 1.9 kΩ) had a smaller radius of the semicircle in comparison with Co-CDDA (Rct, 2.6 kΩ), which represented a lower charge transfer resistance (Fig. 2e).53 Meanwhile, both Co-MOFs showed well-reproducible photocurrents during on/off cycles of light irradiation. Co-TBPA presented a more effective charge separation efficiency than Co-CDDA under the same irradiation conditions (Fig. 2f), indicating a more obvious intramolecular PET process. Clearly, a more powerful twisted conjunction mode did not affect the photoreduction abilities of the MOFs but remarkably altered the kinetic features of intramolecular electronic communications.27
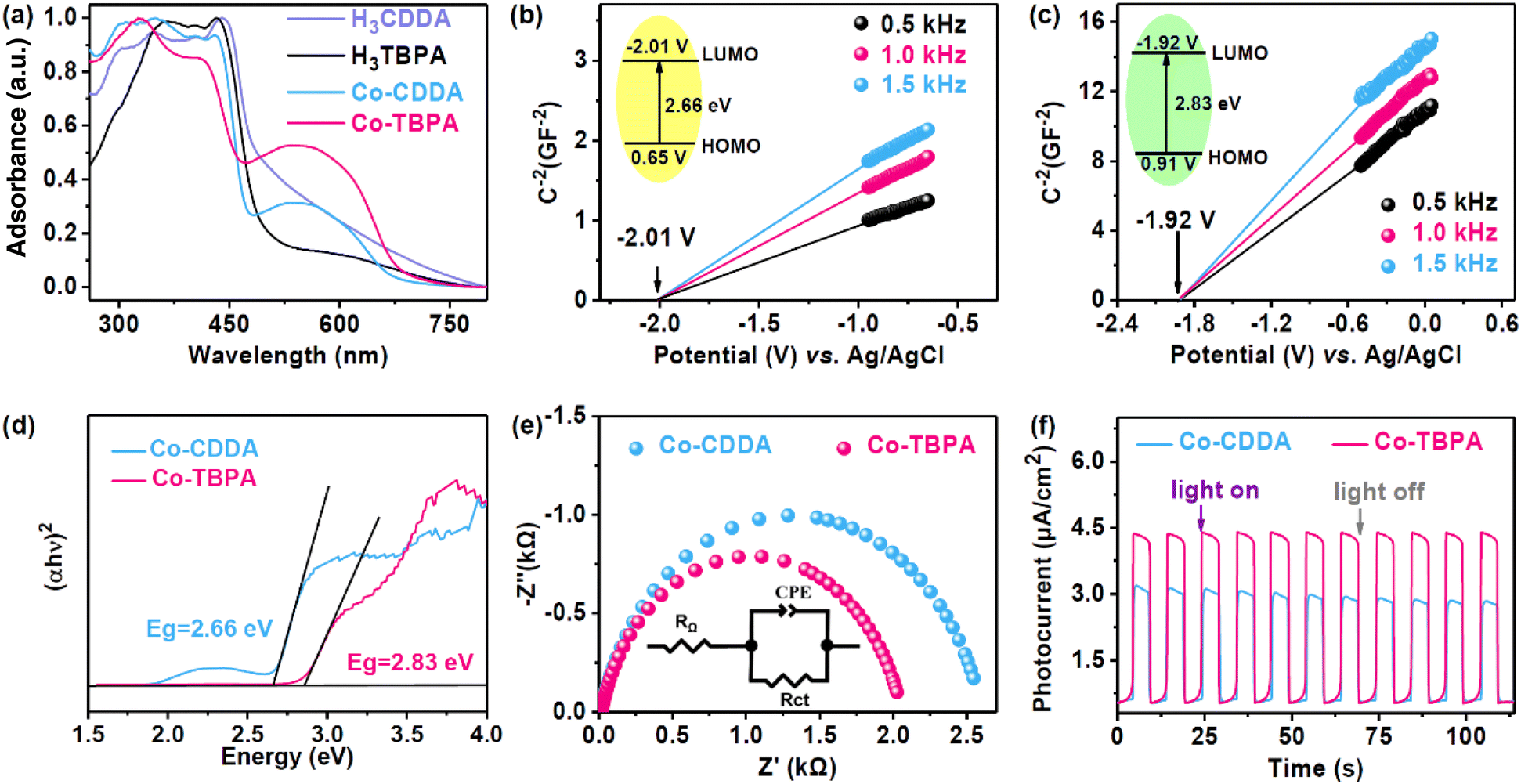 | ||
| Fig. 2 (a) The solid UV-vis spectrum of H3CDDA, H3TBPA, Co-CDDA and Co-TBPA measured at room temperature. (b) and (c) Mott–Schottky plots for Co-CDDA and Co-TBPA, respectively, in 0.1 M Bu4NPF6 CH3CN solution (the insets are the energy diagrams of HOMO and LUMO levels). (d) Tauc plots of Co-CDDA and Co-TBPA. (e) EIS plots and (f) photocurrent responses of Co-CDDA and Co-TBPA. | ||
The fluorescence spectra of Co-CDDA and Co-TBPA were recorded for exploring the behaviors of the intramolecular PET process between Co nodes and dye-functionalized ligands under irradiation (Fig. 3a). Under the same conditions, the fluorescence intensity of Co-CDDA was approximately 50 times that of Co-TBPA, suggesting that Co-CDDA significantly restrains fluorescence quenching behaviors from the Co nodes towards the acridone-based ligands compared with Co-TBPA. Moreover, the fluorescence lifetime of the Co-CDDA suspension was longer (τ = 2.9 ns) than that of Co-TBPA (τ = 2.2 ns) (Fig. 3b) under the same conditions, which matched well with the fluorescence measurement. These results provide evidence that the introduction of the carbonyl group made acridone a more rigid structure than triphenylamine, which was conducive to avoiding the unwanted intramolecular PET processes and regulating the direction of electronic communication within the MOFs, providing more opportunities for directly applying the excellent redox properties of the dyes to engage with the reaction substrates and participate in the subsequent reaction.54
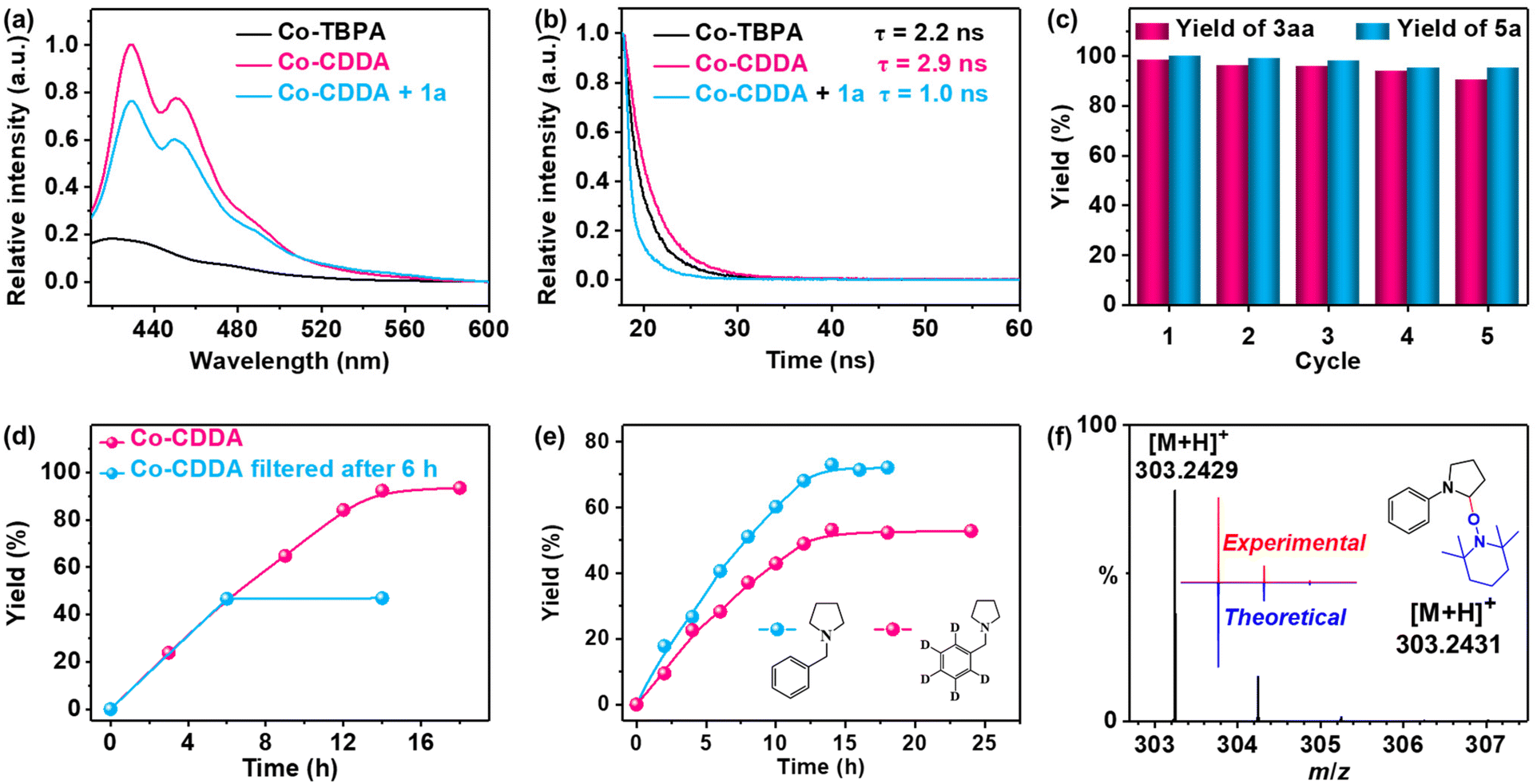 | ||
| Fig. 3 (a) Luminescence spectra and (b) time-dependent luminescence decay of Co-TBPA, Co-CDDA and Co-CDDA with 1,4-dicyanobenzene 1a in DMAc. (c) Recycling catalytic experiments of C–H arylation of N-phenylpyrrolidine 2a and the oxidation of benzylamine 4a with Co-CDDA as the photocatalyst, respectively. (d) Time-dependent catalytic traces of 2a C–H arylation within 14 hours and with Co-CDDA filtered after 6 hours. (e) Kinetics curves of the C–H arylation using N-benzylpyrrolidine 2k or N-((phenyl-D5)methyl-D2)pyrrolidine 2kD with 1a under standard conditions (kH/kD = 1.72). (f) Radical trapping experiments by TEMPO under standard conditions. ESI-MS calcd for [C19H30N2O + H]+: 303.2431; found for [C19H30N2O + H]+: 303.2429. | ||
α-Amino C(sp3)–H arylation reactions
The α-arylated amines are ubiquitous structural motifs among various kinds of organic compounds, including many natural products, pharmaceutical agents and agrochemicals.55 However, α-amino C(sp3)–H bond activation by hydrogen atom transfer (HAT) agents is one of the effective methods for the synthesis of α-arylated amines.56 Encouraged by the efficiency of HAT agents, the high-efficiency formation of HAT agents in the framework materials by inhibiting intramolecular PET prompted us to first evaluate their catalytic activities in the functionalization of α-amino C(sp3)–H. Direct C(sp3)–H arylation of 1,4-dicyanobenzene (1a) with N-phenylpyrrolidine (2a) was chosen as the model reaction to investigate our strategy. The standard reaction was conducted in a flame-dried Schlenk quartz flask containing 1a (1.0 mmol), 2a (2.0 mmol), K2CO3 (1.0 mmol) and Co-CDDA (20 μmol), N,N-dimethylacetamide (DMAc, 4.0 mL) under argon at room temperature. The photocatalytic transformation gave the target product α-amino cyanobenzene 3aa with a yield of 92.3% upon irradiation with a 420 nm LED within 14 hours (Table 1, entry 1). Control experiments revealed that light, a base and a photocatalyst were necessary for the α-amino C(sp3)–H arylation reaction (Table 1, entries 2–4). Using only ligand H3CDDA as a photocatalyst resulted in approximately 62.8% yield (Table 1, entry 5), whereas using only cobalt salt exhibited no activity (Table 1, entry 6). These results suggested that the catalytic activity of the MOF originated from the photosensitive ligand and the efficiency of the reaction was also improved. It is worth noting that simply physically mixing the ligand H3CDDA with the cobalt salt instead of using Co-CDDA as the photocatalyst resulted in only approximately 14.2% yield (Table 1, entry 7), suggesting that the intense intermolecular PET process between the cobalt salt and the ligand significantly decreased the yield of the target product. When the TPA-based ligand H3TBPA was used as the photocatalyst, the yield was similar to that of H3CDDA (Table 1, entry 8). However, when Co-TBPA was used as a photocatalyst, the yield only reached about 37.9% under identical reaction conditions (Table 1, entry 10). Although the LUMO position of Co-TBPA was similar to that of Co-CDDA and was sufficient to reduce the substrate, the reaction efficiency of Co-CDDA was obviously higher than that of Co-TBPA. This indicated that forcing a twist between the CoII-carboxylate node and the adjacent phenyl moiety is beneficial to inhibit the PET process between the ligands and metal nodes.|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield (%) |
| Standard conditions: Co-CDDA (20 μmol), 1,4-dicyanobenzene (1.0 mmol), K2CO3 (1.0 mmol), N-phenylpyrrolidine (2.0 mmol) and DMAc (4.0 mL) for 14 hours under an Ar atmosphere at room temperature. The yields were determined by 1H NMR, using 1,3,5-trimethoxybenzene as the internal standard. N.R. = No reaction. | ||
| 1 | None | 92.3 |
| 2 | No light | N.R. |
| 3 | No K2CO3 | 40.2 |
| 4 | No Co-CDDA | 14.5 |
| 5 | H3CDDA | 62.8 |
| 6 | Co(OAc)2·4H2O | N.R. |
| 7 | H3CDDA/Co(OAc)2·4H2O | 14.2 |
| 8 | H3TBPA | 56.8 |
| 9 | H3TBPA/CoCl2·6H2O | 9.8 |
| 10 | Co-TBPA instead of Co-CDDA | 37.9 |
| 11 | TEMPO | Trace |
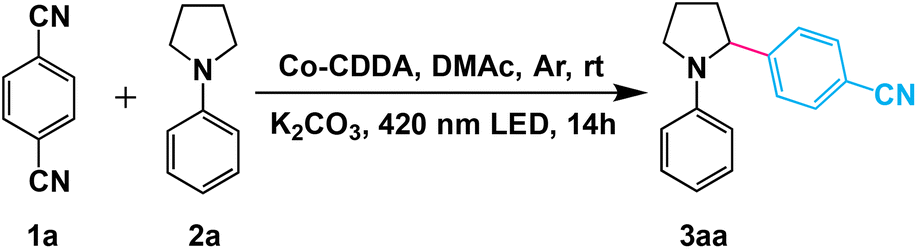
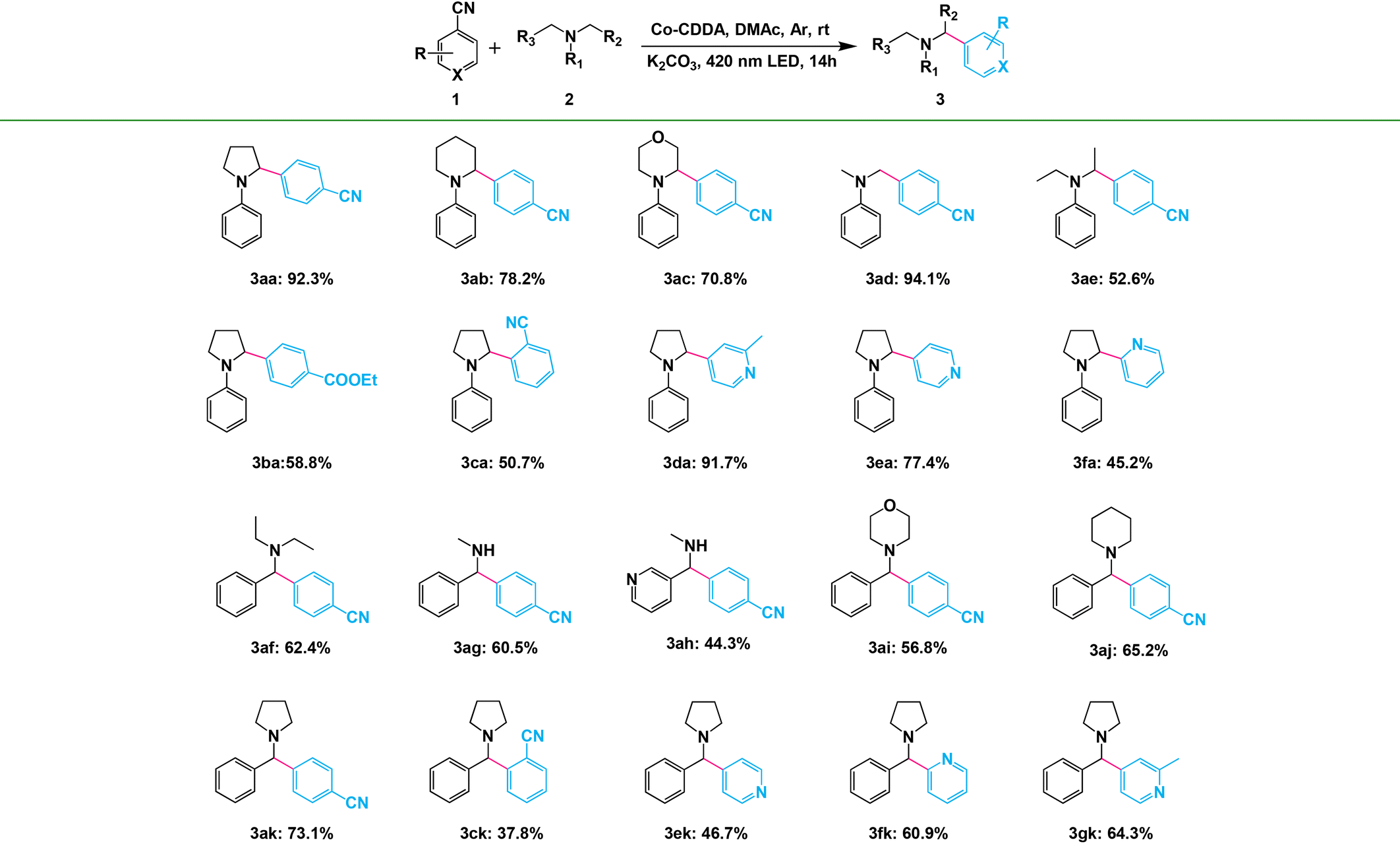
Amines with phenyl groups were successfully converted into the desired products with moderate conversions to N-phenylic arylation products. Furthermore, functional group tolerance was examined by coupling nitrile compounds (1) with various amines (2) (Table 2). Satisfyingly, a broad variety of functional groups such as piperidines (3ab), morpholines (3ac) and acyclic amines (3ad and 3ae) provided desirable results. Electron-rich derivatives yielded the highest yields, which were consistent with the polarity effects. Electron-deficient arenes (3ba and 3ca) or heteroaromatic benzonitriles (3da–3fa) as coupling substrates were also compatible with our method and no over-reaction of the products was observed. Compared to the para-substituted derivative 3aa, the ortho-substituted derivatives produced 3ca with a lower yield (50.7%), probably due to steric effects. To further explore the universality of the substrates, amines with benzyl groups were selected as reactants. Owing to the large difference in C–H bond dissociation energy (BDE) between N-methyl C–H and N-benzyl C–H, N-benzylic position C(sp3)–H arylation products were obtained as the main products under standard conditions. Aliphatic substituents (3af–3ah) and heteroatom-bearing cyclic substituents (3ai–3ak) performed equally well under the same reaction conditions, suggesting that different substituted groups on the nitrogen atom did not affect the reaction. Moreover, apart from 1,4-dicyanobenzene, cyanopyridine derivatives provided 3ck–3gk in good to excellent yields. These results indicated that Co-CDDA as a highly active heterogeneous photocatalyst achieved efficient α-amino C(sp3)–H arylation reactions. Besides the advantages of mild reaction conditions, high catalytic efficiency and a wide range of substrates, Co-CDDA exhibited high stability in our heterogeneous visible light-mediated approach.57,58 No significant decrease in conversion was observed after five cycles (Fig. 3c). The PXRD patterns of Co-CDDA before and after catalysis were basically the same (Fig. S14†), indicating that the configuration of Co-CDDA was maintained during the photocatalytic reaction. The emergence of new peaks at higher angles in the PXRD of Co-CDDA after the reaction was believed to be caused by gradual exposure of some crystal faces during the continuous stirring process.59–61 SEM images showed that the crystals after catalysis were broken into small pieces and no obvious formation of amorphous substances was observed (Fig. S15†). Moreover, after 6 h of the photocatalytic reaction, Co-CDDA was removed from the reaction mixture by filtering and the filtrate was allowed to sequentially react for an additional 8 h (Fig. 3d). The results showed that the reaction stopped immediately when the photocatalyst was removed, indicating that Co-CDDA was a truly heterogeneous photocatalyst.62
| Standard conditions: Co-CDDA (20 μmol), benzonitriles (1.0 mmol), K2CO3 (1.0 mmol), amines (2.0 mmol) and 4.0 mL DMAc for 14 hours under an Ar atmosphere at room temperature. Isolated yields were obtained by silica gel flash chromatography (EtOAc/n-hexane). |
|---|
|
To explore the possible mechanism of the reaction above, the fluorescence titration tests were conducted first. 1a was demonstrated to be an efficient quencher of the emission of Co-CDDA in DMAc suspension. The emission intensity of the Co-CDDA suspension was quenched by about 20% with a Stern–Volmer constant of about 6.94 × 103 M−1 (Fig. S16†), which was reasonably ascribed to the typical single electron transfer (SET) process from the excited state of Co-CDDA to 1a.63,64 Upon the addition of nucleophiles 2a or 2k, no change was observed in the emission intensity of the Co-CDDA suspension (Fig. S17 and S18†). Moreover, time-dependent emission decay experiments also matched well with the fluorescence titration tests, which demonstrated an obvious decline in the emission lifetime from the initial Co-CDDA suspension (τ = 2.9 ns) to the addition of 1a into the Co-CDDA suspension (τ = 1.0 ns) (Fig. 3b). The combination of the fluorescence measurement data above and the LUMO position of Co-CDDA indicated the occurrence of swift electron transfer from Co-CDDA to 1a after photoexcitation.65–67 The reaction kinetics studies on 2k and N-((phenyl-D5)methyl-D2)pyrrolidine (2kD) were then carried out in two different flasks under standard conditions (Fig. 3e). The results showed that the reaction yields grew rapidly over time and the reaction rate of 2k was a little faster than that of 2kD, resulting in a kinetic isotope effect (KIE) of 1.72 calculated by kH/kD. This suggested that the cleavage of C–H might not be the rate-determining step but played an important role in the photocatalytic selectivity.68,69 We speculated that the good regioselectivity of the N-benzylic position could be attributed to the HAT catalysis process. Moreover, radical quenching experiments and radical trapping experiments were performed. Upon addition of the radical scavenger 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) into the catalytic system, the reaction was shut down completely under identical reaction conditions (Table 1, entry 11), indicating that it was a reaction process involving free radicals.70 Importantly, the expected adduct of aryl radical and TEMPO was detected by ESI-MS (Fig. 3f). According to the above experimental results and relevant literature,51 the acridone radical cation formed in situ by the SET process acted as a HAT agent for regioselective C(sp3)–H arylation. The mechanistic details of heterogeneous photocatalysis are outlined in Fig. S16.†
Photocatalytic oxidation of benzylamine
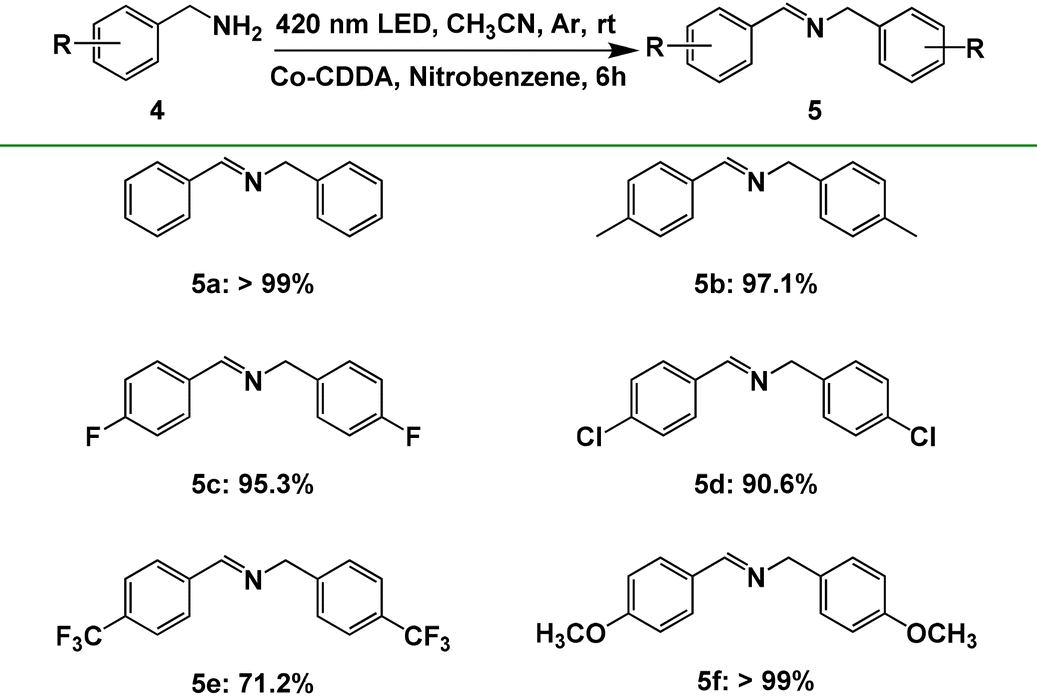
To further explore the impact of the formation of rigid ligands on the electron transfer direction, another typical reaction, benzylamine oxidation, was executed as the model reaction. Our group has developed the combination of hydrogen production and oxidation of benzylamine through the intramolecular PET process with a series of triphenylamine-based Co-MOFs as photocatalysts.71 Co-TBPA is no exception; it also realizes photocatalysis with the oxidation of benzylamine of >99% yield and hydrogen production of 127 μmol g−1 h−1 under the same conditions (Table S4, entry 1†), indicating that electrons are preferentially transferred from the photosensitive ligand triphenylamine to cobalt metal under irradiation. However, when Co-CDDA was used as the photocatalyst, the reaction mentioned above proceeded with difficulty (Table S4, entry 2†), suggesting that the electrons could not transfer efficiently from the photosensitizer acridone to the CoII nodes for hydrogen production. These controlled experimental results indicated that the introduction of a carbonyl group to form a planar acridone dye group caused the intramolecular PET process to be effectively inhibited.27,28 Moreover, we noticed that nitrobenzene was an effective fluorescence quenching agent for Co-CDDA (Fig. S20 and S21†); therefore, we speculated that the introduction of nitrobenzene as an electron acceptor into our photocatalytic reaction system could effectively coordinate with the oxidation of benzylamine. The standard reaction was conducted in a flame-dried Schlenk quartz flask containing the substrate benzylamine 4a (0.2 mmol), nitrobenzene (0.4 mmol), Co-CDDA (10 μmol) and CH3CN (4 mL) under argon at room temperature. The photocatalytic transformation gave the target product N-benzylbenzaldimine product 5a with a yield of >99% upon irradiation with a 420 nm LED within 6 hours (Table S4, entry 3†).The reaction was further extended to different para-substituted benzylamines (p-CH3, p-F, p-Cl, p-CF3, p-OCH3) under standard conditions and all the reactants achieved satisfactory yields (Table 3). Furthermore, Co-CDDA exhibited excellent recyclability and stability in the closed redox cycle involving nitrobenzene reduction and benzylamine oxidation (Fig. 3c). These results indicated that the introduction of the carbonyl group into the TPA-based ligand to form a π-conjugate plane resulted in an increased twisted dihedral angle between the conjugated carboxylic coordination group and the acridone group, which successfully regulated the intramolecular PET process and facilitated the direct use of the dye.
| Standard conditions: Co-CDDA (10 μmol), nitrobenzene (0.4 mmol), benzylamine (0.2 mmol) and 4.0 mL CH3CN for 6 h under an Ar atmosphere at room temperature. Yields were determined by GC analyses using 1,3,5-trimethoxybenzene as the internal standard. |
|---|
|
To further investigate the impact of the π-conjugate torsion formation on the electron transfer process of the excited state MOFs, density of states calculations were performed based on density functional theory (DFT) to assess the band structure of Co-CDDA and Co-TBPA, respectively. The obtained results of the calculations were used to construct density of states (DOS) and partial DOS plots (Fig. 4 and S22†). The plots showed that the HOMO in Co-CDDA was contributed majorly by aromatic carbons and carboxylate oxygen atoms, cobalt played a minor role and other elements in Co-CDDA offered almost negligible contributions to the HOMO. Likewise, the LUMO in Co-CDDA was also mainly contributed by aromatic carbon and oxygen centers. Hence, DOS plots showed that electronic transitions in Co-CDDA were more likely to be ligand-to-ligand type, indicating that the electron transfer from ligands to metal nodes under photoexcitation was inhibited successfully.72,73 For Co-TBPA, the positions of the HOMO and LUMO were consistent when compared with the DOS of cobalt, which meant that electrons could transfer between the ligands and the metal nodes without hindrance. The theoretical calculation and experimental results aligned well, further confirming that the introduction of the carbonyl group into the freely rotating triphenylamine-based ligand to form a conjugate rigid plane caused a much larger torsion angle between the cobalt nodes and the linked benzene ring that inhibited preferential electron transfer from ligands to metal nodes under photoexcitation, which provided an opportunity to transfer electrons from the ligands to the substrates for photocatalytic transformations.
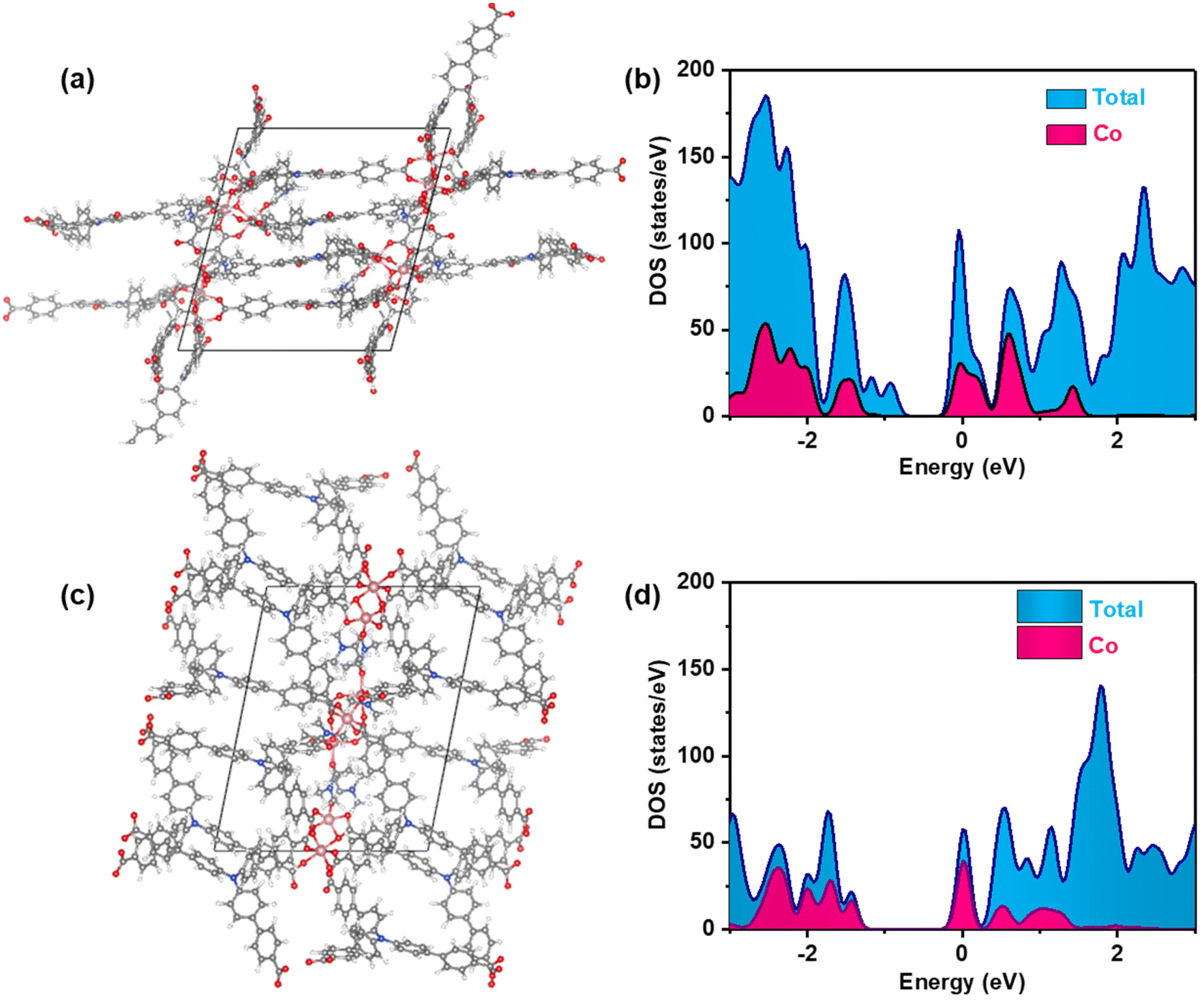 | ||
| Fig. 4 (a) Geometric structure models of Co-CDDA for the DFT calculations. (b) DOS and partial DOS plots for Co-CDDA. (c) Geometric structure models of Co-TBPA for DFT calculations. (d) DOS and partial DOS plots for Co-TBPA. | ||
Conclusions
By elaborately designing the configuration of the ligand, we achieved directional PET processes in MOFs for efficient photocatalysis. By introducing a carbonyl group at the ortho-position of the N atom of a TPA-based ligand to form an acridone-based ligand with a conjugate plane, both MOFs Co-TBPA and Co-CDDA were obtained by coordinating with cobalt, respectively. Single-crystal X-ray structural analysis of Co-CDDA revealed that a larger angle of torsion was compulsively generated between the acridone and the CoII-carboxylate nodes compared with Co-TBPA, which effectively impeded the intramolecular PET process from the ligands to the cobalt nodes. The higher photocatalytic activities of Co-CDDA for α-amino C–H arylation and the combination of benzylamine oxidation with nitrobenzene reduction compared to those of Co-TBPA supported our design strategy. This was further confirmed by DFT theory calculations, which provided a basis for effectively suppressing the extremely widespread photoinduced electron transfer from the dye-based photocatalysts to the metal centers under irradiation and was conducive to realizing better utilization of the redox properties of organic dyes and broadening the application of MOFs as photocatalysts. We believe that this new method has paved a promising way to design novel MOF-targeted catalytic systems, which may trigger a new blueprint in artificial photosynthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22171034, 21890381 and 21820102001), the Natural Science Foundation of Jiangsu Province (Grant No. BK20220033), the Natural Science Foundation of Liaoning Province (Grant No. 2023MS116) and the Fundamental Research Funds for the Central Universities (Grant No. DUT22LAB606).References
- Q. Xia, J. Yang, S. Zhang, J. Zhang, Z. Li, J. Wang and X. Chen, Bodipy-based metal–organic frameworks transformed in solid states from 1D chains to 2D layer structures as efficient visible light heterogeneous photocatalysts for forging C–B and C–C bonds, J. Am. Chem. Soc., 2023, 145, 6123–6134 CrossRef CAS PubMed.
- M. Mian, X. Wang, X. Wang, K. Kirlikovali, H. Xie, K. Ma, K. Fahy, H. Chen, T. Islamoglu, R. Snurr and O. Farha, Structure–activity relationship insights for organophosphonate hydrolysis at Ti(IV) active sites in metal–organic frameworks, J. Am. Chem. Soc., 2023, 145, 7435–7445 CrossRef CAS PubMed.
- C. Dong, J. Yang, L. Xie, G. Cui, W. Fang and J. Li, Catalytic ozone decomposition and adsorptive VOCs removal in bimetallic metal-organic frameworks, Nat. Commun., 2022, 13, 4991–5000 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Catalysis and photocatalysis by metal organic frameworks, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- X. Kong, T. He, J. Zhou, C. Zhao, T. Li, X. Wu, K. Wang and J. Li, In Situ Porphyrin Substitution in a Zr(IV)-MOF for stability enhancement and photocatalytic CO2 reduction, Small, 2021, 17, 2005357–2005366 CrossRef CAS PubMed.
- X. Song, L. Zhao, N. Zhang, L. Liu, X. Ren, H. Ma, C. Luo, Y. Li and Q. Wei, Zinc-based metal–organic framework with MLCT properties as an efficient electrochemiluminescence probe for trace detection of trenbolone, Anal. Chem., 2022, 94, 14054–14060 CrossRef CAS PubMed.
- R. Medishetty, L. Nemec, V. Nalla, S. Henke, M. Samoc, K. Reuter and R. Fischer, Multi-photon absorption in metal–organic frameworks, Angew. Chem., Int. Ed., 2017, 56, 14743–14748 CrossRef CAS PubMed.
- D. Mayer, A. Manzi, R. Medishetty, B. Winkler, C. Schneider, G. Kieslich, A. Pöthig, J. Feldmann and R. Fischer, Controlling multiphoton absorption efficiency by chromophore packing in metal–organic frameworks, J. Am. Chem. Soc., 2019, 141, 11594–11602 CrossRef CAS PubMed.
- R. Medishetty, V. Nalla, L. Nemec, S. Henke, D. Mayer, H. Sun, K. Reuter and R. Fischer, A new class of lasing materials: Intrinsic stimulated emission from nonlinear optically active metal–organic frameworks, Adv. Mater., 2017, 29, 1605637–1605643 CrossRef PubMed.
- I. MacKenzie, L. Wang, N. Onuska, O. Williams, K. Begam, A. Moran, B. Dunietz and D. Nicewicz, Discovery and characterization of an acridine radical photoreductant, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- N. Cowper, C. Chernowsky, O. Williams and Z. Wickens, Potent reductants via electron-primed photoredox catalysis: Unlocking aryl chlorides for radical coupling, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- J. Cole, D. Chen, M. Kudisch, R. Pearson, C. Lim and G. Miyake, Organocatalyzed birch reduction driven by visible light, J. Am. Chem. Soc., 2020, 142, 13573–13581 CrossRef CAS PubMed.
- L. Hou, X. Jing, H. Huang and C. Duan, Merging charge transfer into metal–organic frameworks to achieve high reduction potentials via multiphoton excitation, ACS Appl. Mater. Interfaces, 2022, 14, 15307–15316 CrossRef CAS PubMed.
- S. Guo, L. Kong, P. Wang, S. Yao, T. Lu and Z. Zhang, Switching excited state distribution of metal–organic framework for dramatically boosting photocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202206193 CAS.
- Y. Abderrazak, A. Bhattacharyya and O. Reiser, Visible-light-induced homolysis of earth-abundant metal–substrate complexes: A complementary activation strategy in photoredox catalysis, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS PubMed.
- B. Gui, Y. Meng, Y. Xie, J. Tian, G. Yu, W. Zeng, G. Zhang, S. Gong, C. Yang, D. Zhang and C. Wang, Tuning the photoinduced electron transfer in a Zr-MOF: Toward solid-state fluorescent molecular switch and turn-on sensor, Adv. Mater., 2018, 30, 1802329–1802334 CrossRef PubMed.
- J. Liang, H. Yu, J. Shi, B. Li, L. Wu and M. Wang, Dislocated bilayer MOF enables high-selectivity photocatalytic reduction of CO2 to CO, Adv. Mater., 2023, 35, 2209814–2209825 CrossRef CAS PubMed.
- S. Li, F. Wu, R. Lin, J. Wang, C. Li, Z. Li, J. Jiang and Y. Xiong, Enabling photocatalytic hydrogen production over Fe-based MOFs by refining band structure with dye sensitization, Chem. Eng. J., 2022, 429, 132217–132223 CrossRef CAS.
- Y. Zhao, Y. Cui, L. Xie, K. Geng, J. Wu, X. Meng and H. Hou, Rational construction of metal organic framework hybrid assemblies for visible light-driven CO2 conversion, Inorg. Chem., 2023, 62, 1240–1249 CrossRef CAS PubMed.
- Q. Wu, H. Ma, Y. Wang, J. Chen, J. Dai, X. Xu and X. Wu, Surface electron localization in Cu-MOF-bonded double heterojunction Cu2O induces highly efficient photocatalytic CO2 reduction, ACS Appl. Mater. Interfaces, 2022, 14, 54328–54337 CrossRef CAS PubMed.
- W. Fan and M. Tahir, Recent advances on cobalt metal organic frameworks (MOFs) for photocatalytic CO2 reduction to renewable energy and fuels: A review on current progress and future directions, Energy Convers. Manage., 2022, 253, 115180–115206 CrossRef CAS.
- Y. Song, Y. Pi, X. Feng, K. Ni, Z. Xu, J. Chen, Z. Li and W. Lin, Cerium-based metal–organic layers catalyze hydrogen evolution reaction through dual photoexcitation, J. Am. Chem. Soc., 2020, 142, 6866–6871 CrossRef CAS PubMed.
- W. Lu, Z. Wei, Z. Gu, T. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Boscha and H. Zhou, Tuning the structure and function of metal–organic frameworks via linker design, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- X. Kong, H. Deng, F. Yan, J. Kim, J. Swisher, B. Smit, O. Yaghi and J. Reimer, Mapping of functional groups in metal–organic frameworks, Science, 2013, 341, 882–885 CrossRef CAS PubMed.
- H. Deng, C. Doonan, H. Furukawa, R. Ferreira, J. Towne, C. Knobler, B. Wang and O. Yaghi, Multiple functional groups of varying ratios in metal–organic frameworks, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- Z. Gao, Y. Lai, Y. Tao, L. Xiao, Z. Li, L. Zhang, L. Sun and F. Luo, Creating and tailoring ultrathin two-dimensional uranyl-organic framework nanosheets for boosting photocatalytic oxidation reactions, Appl. Catal., B, 2021, 297, 120485–120493 CrossRef CAS.
- Y. Shi, T. Zhang, X. Jiang, G. Xu, C. He and C. Duan, Synergistic photoredox and copper catalysis by diode-like coordination polymer with twisted and polar copper–dye conjugation, Nat. Commun., 2020, 11, 5384–5394 CrossRef CAS PubMed.
- Y. Tang, L. Zhao, G. Ji, Y. Zhang, C. He, Y. Wang, J. Wei and C. Duan, Ligand-regulated metal–organic frameworks for synergistic photoredox and nickel catalysis, Inorg. Chem. Front., 2022, 9, 3116–3129 RSC.
- L. Dong, L. Zhang, J. Liu, Q. Huang, M. Lu, W. Ji and Y. Lan, Stable heterometallic cluster-based organic framework catalysts for artificial photosynthesis, Angew. Chem., Int. Ed., 2020, 59, 2659–2663 CrossRef CAS PubMed.
- C. Li, H. Xu, J. Gao, W. Du, L. Shangguan, X. Zhang, R. Lin, H. Wu, W. Zhou, X. Liu, J. Yao and B. Chen, Tunable titanium metal–organic frameworks with infinite 1D Ti–O rods for efficient visible-light-driven photocatalytic H2 evolution, J. Mater. Chem. A, 2019, 7, 11928–11933 RSC.
- R. Liu, H. Gao, L. Zhou, Y. Ji and G. Zhang, Effects of N-substitution on the property of acridone, ChemistrySelect, 2019, 4, 7797–7804 CrossRef CAS.
- K. Muralirajan, R. Kancherla and M. Rueping, Dehydrogenative aromatization and sulfonylation of pyrrolidines: Orthogonal reactivityin photoredox catalysis, Angew. Chem., Int. Ed., 2018, 57, 14787–14791 CrossRef CAS PubMed.
- Y. Matsuo, Y. Wang, H. Ueno, T. Nakagawa and H. Okada, Mechanochromism, twisted/folded structure determination, and derivatization of (N-phenylfluorenylidene)acridane, Angew. Chem., Int. Ed., 2019, 58, 8762–8767 CrossRef CAS PubMed.
- SMART, Data collection software (version 5.629), Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- SAINT, Data reduction software (version 6.45), Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- G. Sheldrick, SHELXTL97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- G. Chen, Y. He, S. Zhang and J. Zhang, Tuning a layer to a three-dimensional cobalt-tris(4′-carboxybiphenyl)amine framework by introducing potassium ions, Inorg. Chem. Commun., 2018, 90, 65–68 CrossRef CAS.
- Y. Zhu, H. Long and W. Zhang, Imine-linked porous polymer frameworks with high small gas (H2, CO2, CH4, C2H2) uptake and CO2/N2 selectivity, Chem. Mater., 2013, 25, 1630–1635 CrossRef CAS.
- X. Gong, Y. Shu, Z. Jiang, L. Lu, X. Xu, C. Wang and H. Deng, Metal–organic frameworks for the exploitation of distance between active sites in efficient photocatalysis, Angew. Chem., Int. Ed., 2020, 59, 5326–5331 CrossRef CAS PubMed.
- W. Gong, W. Zhang, F. Son, K. Yang, Z. Chen, X. Chen, J. Jiang, Y. Liu, O. Farha and Y. Cui, Topological strain-induced regioselective linker elimination in a chiral Zr(IV)-based metal–organic framework, Chem., 2021, 7, 190–201 CAS.
- X. Chen, H. Jiang, B. Hou, W. Gong, Y. Liu and Y. Cui, Boosting chemical stability, catalytic activity, and enantioselectivity of metal–organic frameworks for batch and flow reactions, J. Am. Chem. Soc., 2017, 139, 13476–13482 CrossRef CAS PubMed.
- G. Chen, S. Huang, X. Kou, F. Zhu and G. Ouyang, Embedding functional biomacromolecules within peptide-directed metal–organic framework (MOF) nanoarchitectures enables activity enhancement, Angew. Chem., Int. Ed., 2020, 59, 13947–13954 CrossRef CAS PubMed.
- H. Han, X. Zheng, C. Qiao, Z. Xia, Q. Yang, L. Di, Y. Xing, G. Xie, C. Zhou, W. Wang and S. Chen, A stable Zn-MOF for photocatalytic Csp3–H oxidation: Vinyl double bonds boosting electron transfer and enhanced oxygen activation, ACS Catal., 2022, 12, 10668–10679 CrossRef CAS.
- J. Jia, J. Wang, M. Li, C. Gong, G. Liang, Y. Song and Y. She, Phenothiazine metal-organic framework materials with excellent third-order nonlinear properties, Dyes Pigm., 2022, 205, 110398–110406 CrossRef CAS.
- A. Goswami, D. Ghosh, V. Chernyshev, A. Dey, D. Pradhan and K. Biradha, 2D MOFs with Ni(II), Cu(II), and Co(II) as efficient oxygen evolution electrocatalysts: rationalization of catalytic performance vs structure of the MOFs and potential of the redox couples, ACS Appl. Mater. Interfaces, 2020, 12, 33679–33689 CrossRef CAS PubMed.
- C. Zhang, C. Xie, Y. Gap, X. Tao, C. Ding, F. Fan and H. Jiang, Charge separation by creating band bending in metal–organic frameworks for improved photocatalytic hydrogen evolution, Angew. Chem., Int. Ed., 2022, 61, e202204108 CAS.
- N. Li, J. Liu, J. Liu, L. Dong, Z. Xin, Y. Teng and Y. Lan, Adenine components in biomimetic metal–organic frameworks for efficient CO2 photoconversion, Angew. Chem., Int. Ed., 2019, 58, 5226–5231 CrossRef CAS PubMed.
- X. Wang, J. Liu, L. Zhang, L. Dong, S. Li, Y. Kan, D. Li and Y. Lan, Monometallic catalytic models hosted in stable metal–organic frameworks for tunable CO2 photoreduction, ACS Catal., 2019, 9, 1726–1732 CrossRef CAS.
- H. Liu, C. Xu, D. Li and H. Jiang, Photocatalytic hydrogen production coupled with selective benzylamine oxidation over MOF composites, Angew. Chem., Int. Ed., 2018, 57, 5379–5383 CrossRef CAS PubMed.
- Y. Ma, X. Yao, L. Zhang, P. Ni, R. Cheng and J. Ye, Direct arylation of α-amino C(sp3)–H bonds by convergent paired electrolysis, Angew. Chem., Int. Ed., 2019, 58, 16548–16552 CrossRef CAS PubMed.
- T. Ide, J. Barham, M. Fujita, Y. Kawato, H. Egami and Y. Hamashima, Regio- and chemoselective Csp3–H arylation of benzylamines by single electron transfer/hydrogen atom transfer synergistic catalysis, Chem. Sci., 2018, 9, 8453–8460 RSC.
- W. Qiao, I. Waseem, G. Shang, D. Wang, Y. Li, F. Besenbacher, H. Niemantsverdriet, C. Yan and R. Su, Paired electrochemical N–N coupling employing a surface hydroxylated Ni3Fe-MOF-OH bifunctional electrocatalyst with enhanced adsorption of nitroarenes and anilines, ACS Catal., 2021, 11, 13510–13518 CrossRef CAS.
- D. Tan, X. Cheng, J. Zhang, Y. Sha, X. Shen, Z. Su, B. Han and L. Zheng, Photocatalytic carbon dioxide reduction coupled with benzylamine oxidation over Zn-Bi2WO6 microflowers, Green Chem., 2021, 23, 2913–2917 RSC.
- X. Shu, D. Zhong, Y. Lin, X. Qin and H. Huo, Modular access to chiral α-(hetero)aryl amines via Ni/photoredox catalyzed enantioselective cross-coupling, J. Am. Chem. Soc., 2022, 144, 8797–8806 CrossRef CAS PubMed.
- J. Xie, M. Rudolph, F. Rominger and A. Hashmi, Photoredox-controlled mono- and di-multifluoroarylation of C(sp3)–H bonds with aryl fluorides, Angew. Chem., Int. Ed., 2017, 56, 7266–7270 CrossRef CAS PubMed.
- J. Jin, K. Wu, X. Liu, G. Huang, Y. Huang, D. Luo, M. Xie, Y. Zhao, W. Lu, X. Zhou, J. He and D. Li, Building a pyrazole–benzothiadiazole–pyrazole photosensitizer into metal–organic frameworks for photocatalytic aerobic oxidation, J. Am. Chem. Soc., 2021, 143, 21340–21349 CrossRef CAS PubMed.
- N. Huang, H. He, S. Liu, H. Zhu, Y. Li, J. Xu, J. Huang, X. Wang, P. Liao and X. Chen, Electrostatic attraction-driven assembly of a metal–organic framework with a photosensitizer boosts photocatalytic CO2 reduction to CO, J. Am. Chem. Soc., 2021, 143, 17424–17430 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, A scalable general synthetic approach toward ultrathin imine-linked two-dimensional covalent organic framework nanosheets for photocatalytic CO2 reduction, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- Y. Liu, L. Liu, X. Chen, Y. Liu, Y. Han and Y. Cui, Single-crystalline ultrathin 2D porous nanosheets of chiral metal–organic frameworks, J. Am. Chem. Soc., 2021, 143, 3509–3518 CrossRef CAS PubMed.
- L. Li, X. Pan, D. Lan, H. Xu, J. Ge, H. Zhang, Z. Zheng, J. Liu, Z. Xu and J. Liu, Etching of cubic Pd@Pt in UiO-66 to obtain nanocages for enhancing CO2 hydrogenation, Mater. Today Energy, 2021, 19, 100585–100591 CrossRef CAS.
- T. Luo, L. Li, Y. Chen, J. An, C. Liu, Z. Yan, J. Carter, X. Han, M. Alena, A. Sheveleva, F. Tuna, E. McInnes, C. Tang, M. Schröder and S. Yang, Construction of C–C bonds via photoreductive coupling of ketones and aldehydes in the metalorganic-framework MFM-300(Cr), Nat. Commun., 2021, 12, 3583–3592 CrossRef CAS PubMed.
- Z. Dong and D. MacMillan, Metallaphotoredox-enabled deoxygenative arylation of alcohols, Nature, 2021, 598, 451–456 CrossRef CAS PubMed.
- Y. Zhu, G. Lan, Y. Fan, S. Veroneau, Y. Song, D. Micheroni and W. Lin, Merging photoredox and organometallic catalysts in a metal–organic framework significantly boosts photocatalytic activities, Angew. Chem., Int. Ed., 2018, 57, 14090–14094 CrossRef CAS PubMed.
- H. Zheng, S. Huang, M. Luo, Q. Wei, E. Chen, L. He and Q. Lin, Photochemical in situ exfoliation of metal–organic frameworks for enhanced visible-light-driven CO2 reduction, Angew. Chem., Int. Ed., 2020, 59, 23588–23592 CrossRef CAS PubMed.
- Y. Su, Z. Song, W. Zhu, Q. Mu, X. Yuan, Y. Lian, H. Cheng, Z. Deng, M. Chen, W. Yin and Y. Peng, Visible-light photocatalytic CO2 reduction using metal–organic framework derived Ni(OH)2 nanocages: A synergy from multiple light reflection, static charge transfer, and oxygen vacancies, ACS Catal., 2021, 11, 345–354 CrossRef CAS.
- L. Wu, Y. Mu, X. Guo, W. Zhang, Z. Zhang, M. Zhang and T. Lu, Encapsulating perovskite quantum dots in iron-based metal–organic frameworks (MOFs) for efficient photocatalytic CO2 reduction, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- Z. Wang, Q. Liu, X. Ji, G. Deng and H. Huang, Bromide-promoted visible-light-induced reductive minisci reaction with aldehydes, ACS Catal., 2020, 10, 154–159 CrossRef CAS.
- C. Huang, J. Li and C. Li, A cross-dehydrogenative C(sp3)–H heteroarylation via photo-induced catalytic chlorine radical generation, Nat. Commun., 2021, 12, 4010–4018 CrossRef CAS PubMed.
- W. Yu, Y. Luo, L. Yan, D. Liu, Z. Wang and P. Xu, Dehydrogenative silylation of alkenes for the synthesis of substituted allylsilanes by photoredox, hydrogen-atom transfer, and cobalt catalysis, Angew. Chem., Int. Ed., 2019, 58, 10941–10945 CrossRef CAS PubMed.
- H. Li, Y. Yang, X. Jing, C. He and C. Duan, Multi-component metal–organic frameworks significantly boost visible-light-driven hydrogen production coupled with selective organic oxidation, Chem. – Asian J., 2021, 16, 1237–1244 CrossRef CAS PubMed.
- F. Yuan, C. Yuan, B. Cao, Y. Di, S. Wang, M. Liu, A. Kumar, C. Shi and M. Muddassir, Two Ln-based metal–organic frameworks based on the 5-(1H-1,2,4-triazol-1-yl)-1,3-benzenedicarboxylic acid ligand: syntheses, structures, and photocatalytic properties, RSC Adv., 2020, 10, 39771–39778 RSC.
- Q. Ding, Y. Pan, Y. Luo, M. Zhou, Y. Guan, B. Li, M. Trivedi, A. Kumar and J. Liu, Photocatalytic and ferric ion sensing properties of a new three-dimensional metal–organic framework based on cuboctahedral secondary building units, ACS Omega, 2019, 4, 10775–11078 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystal structure and additional spectroscopic data. CCDC 2263257. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01120h |
| This journal is © the Partner Organisations 2023 |