
Functional micro-RNA drugs acting as a fate manipulator in the regulation of osteoblastic death
Zhengwen
Cai
 ab,
Fengshuo
Liu
a,
Yong
Li
ab,
Long
Bai
c,
Maogeng
Feng
c,
Songhang
Li
ab,
Wenjuan
Ma
ab and
Sirong
Shi
*ab
ab,
Fengshuo
Liu
a,
Yong
Li
ab,
Long
Bai
c,
Maogeng
Feng
c,
Songhang
Li
ab,
Wenjuan
Ma
ab and
Sirong
Shi
*ab
aState Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, Sichuan 610041, China. E-mail: sirongshi@scu.edu.cn
bSichuan Provincial Engineering Research Center of Oral Biomaterials, Chengdu, Sichuan 610041, China
cThe Affiliated Hospital of Southwest Medical University, Luzhou, 646000, China
First published on 24th July 2023
Abstract
Bone loss is prevalent in clinical pathological phenomena such as osteoporosis, which is characterized by decreased osteoblast function and number, increased osteoclast activity, and imbalanced bone homeostasis. However, current treatment strategies for bone diseases are limited. Regulated cell death (RCD) is a programmed cell death pattern activated by the expression of specific genes in response to environmental changes. Various studies have shown that RCD is closely associated with bone diseases, and manipulating the death fate of osteoblasts could contribute to effective bone treatment. Recently, microRNA-targeting therapy drugs have emerged as a potential solution because of their precise targeting, powerful curative effect, and limited side effects. Nevertheless, their clinical application is limited by their inherent instability, easy enzymatic degradation, and poor membrane penetrability. To address this challenge, a self-assembling tetrahedral DNA nanostructure (TDN)-based microRNA (Tmi) delivery system has been proposed. TDN features excellent biocompatibility, cell membrane penetrability, serum stability, and modification versatility, making it an ideal nucleic acid carrier for miRNA protection and intracellular transport. Once inside cells, Tmi can dissociate and release miRNAs to manipulate key molecules in the RCD signaling pathway, thereby regulating bone homeostasis and curing diseases caused by abnormal RCD activation. In this paper, we discuss the impact of the miRNA network on the initiation and termination of four critical RCD programs in bone tissues: apoptosis, autophagy, pyroptosis, and ferroptosis. Furthermore, we present the Tmi delivery system as a miRNA drug vector. This provides insight into the clinical translation of miRNA nucleic acid drugs and the application of miRNA drugs in bone diseases.
1. Introduction
With an aging global population, the prevalence of bone diseases, such as osteoporosis (OP), fractures, and osteoarthritis (OA), continues to increase year by year.1–3 For instance, in the United Kingdom, the annual medical costs of osteoporosis and its complications have exceeded £4 billion, placing a huge economic burden on families and society.4 At the cellular level, abnormal bone homeostasis is characterized by a substantial loss of osteoblast cells such as bone marrow mesenchymal stem cells (BMSC), pre-osteoblasts, and chondroblasts, which weakens osteogenic activity.5 Risk factors, such as infection, inflammation, drugs, mechanical forces, metabolic disorders, and immune dysregulation, can disrupt bone homeostasis, resulting in widespread osteoblast death during proliferation and differentiation, ultimately damaging the bone cortex and trabeculae.2 Therefore, the inhibition of aberrant osteoblast death processes and the maintenance of bone homeostasis are crucial research priorities.Death marks the end of cells. Regulated cell death (RCD), a term introduced by the Nomenclature Committee on Cell Death, describes the complex signaling pathways involving the initiation, execution, and propagation of cell death.6 It provides a classification system based on intracellular molecules and specific signaling cascades, moving beyond simplistic assessments of dying or dead cells based solely on macroscopic morphological features.7,8 Therefore, understanding the distinct signaling cascades underlying RCD enables the utilization of specific molecularly targeted drugs to regulate the onset and cessation of cell death.9
Micro-RNAs (miRNAs), a type of non-coding RNA comprising 18–25 nucleotides, serve as crucial mediators for epigenetic regulation during organism growth and development.10 In the cytoplasm, mature double-stranded miRNAs bind to AGO proteins to form the RNA-induced silencing complex (RISC). This complex binds to the 3′-untranslated region (3′-UTR) of target mRNAs, leading to translation repression or mRNA degradation.10–12 Intracellular miRNA expression can be altered by environmental stress, enabling the epigenetic regulation of functional genes.13 Therefore, the delivery of miRNAs into specific cells allows for post-transcriptional gene regulation, facilitating the overexpression of beneficial genes and silencing of detrimental genes. This approach holds potential as a gene therapy strategy to modulate the RCD process in cells.14,15 However, the inherent limitations of miRNAs, including poor targeting, structural instability, a short half-life, susceptibility to enzymatic degradation, and limited membrane penetration, hinder their application as nucleic acid drugs and impede their clinical translation.16 Consequently, improving miRNA delivery strategies and enhancing their bioavailability are crucial for realizing the potential of gene therapy, representing significant research focuses and challenges in recent years.
Tetrahedral DNA nanostructures (TDNs) are emerging nano-scale nucleic acid drugs.17 They possess remarkable features such as stability, precise programmability, simple synthesis, and excellent biocompatibility.18–20 Previous studies have shown that TDNs could remain stable for up to 24 hours in 10% serum at 37 °C without complete degradation.21In vivo studies also demonstrated that TDNs can rapidly disperse in most organs after either caudal vein or intraperitoneal injection, and their presence can still be detectable 24 hours after intraperitoneal injection.22,23 These findings indicate the strong serum stability of TDNs. Moreover, TDNs have shown great potential in the field of tissue repair and regeneration engineering.18,24,25 Furthermore, TDNs can serve as an excellent delivery vehicle to address the transport and storage deficiencies of miRNA, enabling the delivery of miRNA nucleic acid drugs.24,26 To date, several types of TDN-based small RNA delivery systems have been established (Fig. 1A–F), including:
1. A TDN-based vertex-directed connected system,27
2. A TDN-based vertex-single-sticky end system,26,28–30
3. A TDN-based vertex-multi-sticky end system,24,31
4. A TDN-based edge-sticky end system,32
5. A TDN-based RNA-embedded system,33
6. A TDN-based RNA-frame-surrounded system.34
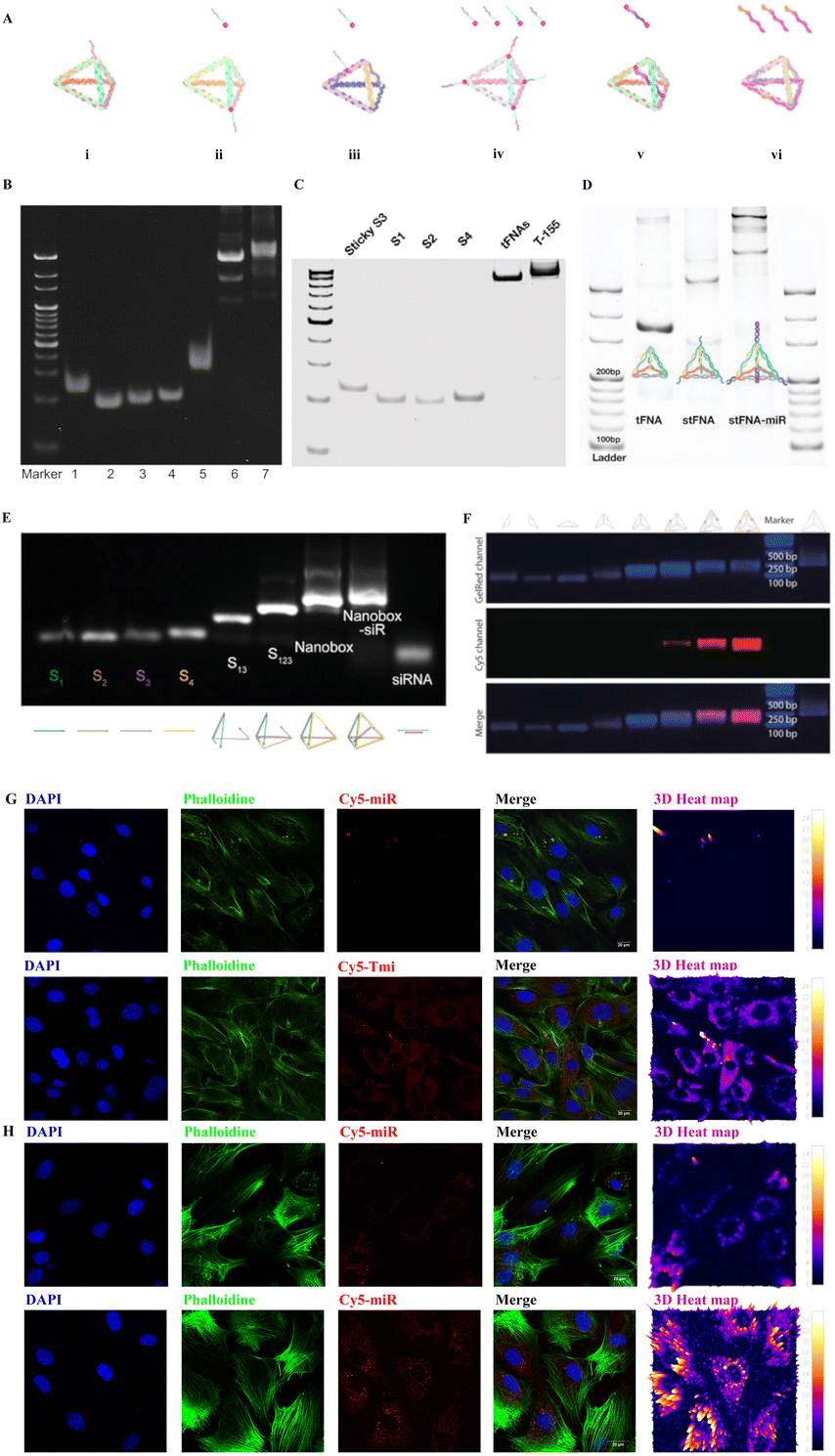 | ||
| Fig. 1 Illustration of the microRNA-TDN delivery system (Tmi), polyacrylamide gel electrophoresis (PAGE) of Tmi synthesis, and fluorescence for the cellular uptake of Tmi. (A) Diagram of six types of Tmi delivery systems. (i) a TDN-based vertex-directed connected system,27 (ii) a TDN-based vertex-single-sticky end system,26,28–30 (iii) a TDN-based vertex-multi-sticky end system;24,31 (iv) a TDN-based edge-sticky end system;32 (v) a TDN-based RNA-embedded system,33 and (vi) a TDN-based RNA-frame-surrounded system.34 (B) 8% PAGE demonstrating a TDN-based vertex-directed connected system (lanes 1: S1; 2: S2; 3: S3; 4: S4; 5: S3-miR; 6: TDN; 7: TDN-miR).27 (C) PAGE showing a TDN-based vertex-single-sticky end system (from left to right: sticky S3, S1, S2, and S4, TDN, and TDN equipped with mir-155).28 (D) PAGE analysis of a TDN-based vertex-multi-sticky end system (from left to right: TDN, TDN-stickers, and TDN-miRs).24 (E) 2% agarose gel electrophoresis confirming the synthesis of a TDN-based RNA-embedded system (from left to right lane: S1; S2; S3; S4; S1 + S3; S1 + S2 + S3; nanobox; nanobox-siR).33 (F) Agarose gel electrophoresis proving Cy5-labeled miRNA inhibitors successfully loaded on the TDN-based RNA-frame-surrounded system.34 (G) Fluorescence images demonstrating that the Tmi treatment group is able to deliver a large quantities of miRNA drugs into MC3T3-E1 cells compared to the miRNA treatment group. Red: Cy5–miRNA; blue: nuclei; green: cytoskeleton. Scale bars: 20 μm. (H) Fluorescence images demonstrating that the Tmi treatment group is able to deliver a large quantities of miRNA drugs into BMSCs compared to the miRNA treatment group. Red: Cy5–miRNA; blue: nuclei; green: cytoskeleton. Scale bars: 20 μm. The Fig. 1B has been reproduced from ref. 27. with permission from Wiley, copyright 2020. The Fig. 1C has been reproduced from ref. 28. with permission from Elsevier, copyright 2022. The Fig. 1D has been reproduced from ref. 24. with permission from Wiley, copyright 2022. The Fig. 1E has been reproduced from ref. 33. with permission from Wiley, copyright 2022. The Fig. 1F has been reproduced from ref. 34. with permission from Wiley, copyright 2022. | ||
More excitingly, all six TDN-based systems have exhibited superior cell membrane penetration and almost no cytotoxicity. Here, we loaded Cy5-labeled miRNAs onto TDNs and incubated them with osteoblasts (MC3T3-E1). As anticipated, the TDN-based miRNA (Tmi) system delivered the miRNAs into the osteoblasts effectively (Fig. 1G). An increased amount of red fluorescence was observed in the cytoplasm of MC3T3-E1 cells in the Tmi treatment group, and the distribution of red fluorescence was colocalized with green fluorescence (Phalloidine), which suggests that Tmi is able to effectively carry miRNA drugs into the cytoplasm for functions. In contrast, only faint red fluorescence was observed in the cytoplasm in the miRNA treatment group, indicating the limited entry capacity of miRNA alone. Similarly, we demonstrated the miRNA drug delivery function of Tmi in bone marrow mesenchymal stem cells (BMSCs) (Fig. 1H). As anticipated, the Tmi treatment group showed a wider range of red fluorescence distribution and a stronger fluorescence intensity. This suggests that Tmi was able to carry a larger number of Cy5-labeled miRNAs across the cell membrane barrier into the cytoplasm of BMSCs compared to miRNA alone. It is clear that TDNs hold great promise in the field of nucleic acid drug delivery, with improvements in stability, drug delivery efficiency, and membrane penetration paving the way for the development of safe and effective nucleic acid drugs in the future.24,26
This review aims to provide a comprehensive overview of the potential of miRNAs in regulating different subtypes of cell death in osteoblasts for the treatment of bone disease. We discuss various candidate miRNAs and their regulatory mechanisms in treating bone diseases, specifically those associated with regulated cell death (RCD), such as apoptosis, autophagy, pyroptosis and ferroptosis (Fig. 2–4). Special attention is given to the utilization of Tmi systems as efficient and precise vehicles for delivering miRNAs. The objective is to offer valuable insights into the development of safe and effective miRNA nucleic acid drugs that can modulate bone diseases.
 | ||
| Fig. 2 Schematic diagram illustrates the regulatory effects of miRNA drugs on apoptotic osteoblasts by targeting the apoptosis-related signaling pathway. TDNs can easily penetrate the phospholipid bilayer without damage to the cell membrane. Based on base pair editing technology, Tmi carries miRNA into cells to achieve gene therapy. When bone tissue is damaged, caspase-3,6,7 can be activated through intrinsic and extrinsic signaling pathways, leading to apoptosis. Other signaling pathways, such as MAPK/JNK, Wnt and JAK/STAT, also influence cell proliferation and apoptosis. By targeting the corresponding mRNA, miRNA drugs can obstruct apoptosis and encourage proliferation. | ||
 | ||
| Fig. 3 Schematic showing the application of miRNA drugs in treating abnormal osteoblastic regulated cell death (RCD) by targeting the autophagy-related signaling pathway (on the left) and the pyroptosis-related signaling pathway (on the right). Tmi systems act as pivotal mediators in the transport of miRNA drugs into cells, delivering miRNAs into the cytoplasm and releasing them to form a RNA-induced silencing complex. mTOR is a key protein in the regulation of autophagy, contributing to the inhibition of autophagosomes. MiRNAs can regulate the disorder of autophagy in pathological states by targeting key PI3K/AKT/mTOR signaling pathways to achieve bone disease therapy. Additionally, the classic NLRP3/caspase-1/GSDMD pathway or AIM2, NLRP1 and other signaling pathways are activated when cells are invaded by a virus, infected by bacteria, or fatally damaged, ultimately causing osteoblasts pyroptosis and releasing IL-1β, IL-18 inflammatory factors. MiRNA drugs can target mRNA and inhibit the translation of key inflammasome proteins, thus impeding the process of osteoblastic pyroptosis. | ||
 | ||
| Fig. 4 Schematic illustrating the application of miRNA drugs in targeting the ferroptosis-related signaling pathway. Excess intracellular iron accumulation and lipid peroxidation promote ferroptosis in osteoblasts. Tmi, as a miRNA drug, can penetrate the cell membrane and unload functional miRNAs to exert gene silencing. They can inhibit ferroptosis by inhibiting the production of PL-PUFA, a key component of the lipid peroxidation system, or enhancing the expression level of the antioxidant reduction system. | ||
2. Fabrication and characteristics of Tmi
In order to provide a clearer picture of the internal structure of a TDN, Table 1 lists the base composition of single-stranded DNA (ssDNA) and ssDNA with sticky ends (ssDNA-st). A TDN nucleic acid deliver system was formed based on the Watson–Crick's complementary base pairing principle. The synthesis methods of the TDN have been described in previous studies. Briefly, four specifically designed ssDNA strands were mixed in equal proportions in TM buffer (MgCl2·6H2O: 50 mmol L−1, Tris-HCL: 20 mmol L−1, PH = 8.0). The mixture was then heated at 95 °C for 10 minutes, followed by rapid annealing at 4 °C for 20 minutes. Next, the TDN was mixed with miRNAs in equal proportions and incubated at room temperature for 30 minutes, allowing for loading of the miRNAs onto the TDN, and generating Tmi. As depicted in Fig. 1A i–iv, the major differences among the four types of Tmi delivery systems are the position and number of sticky ends, which serve as binding sites for carrying miRNA drugs and ultimately forming Tmi drugs. Polyacrylamide gel electrophoresis further confirmed the stepwise synthesis of the TDN using ssDNA (or ssDNA-sticker) and the successful loading of miRNAs (Fig. 1B–D). To optimize the transportation system of the TDN, we developed the 5th and 6th types of Tmi delivery systems, which respond to changes in environmental pH and intelligently release the miRNA/siRNA embedded in the TDN framework (Fig. 1A v–vi). The successful synthesis of these systems was confirmed using 2% agarose gel electrophoresis (Fig. 1E and F). Subsequently, we evaluated the efficiency of Tmi in delivering miRNAs. Specifically, we investigated its effect on MC3T3-E1 cells, an osteoblast progenitor cell, and BMSCs. It was observed that Tmi efficiently transported large quantities of Cy5-labeled miRNA into cells in large quantities, as evidenced by the abundant red fluorescence in the cytoplasm of osteoblasts (Fig. 1G and H). In contrast, the simple Cy5-labeled miRNA treated group exhibited minimal cellular uptake at an equivalent drug concentration.| ssDNA | Sequence (5′-3′) |
|---|---|
| S1 | ATTTATCACCCGCCATAGTAGACGTATCACCAGGCAGTTGAGACGAACATTCCTAAGTCTGAA |
| S2 | ACATGCGAGGGTCCAATACCGACGATTACAGCTTGCTACACGATTCAGACTTAGGAATGTTCG |
| S3 | ACTACTATGGCGGGTGATAAAACGTGTAGCAAGCTGTAATCGACGGGAAGAGCATGCCCATCC |
| S4 | ACGGTATTGGACCCTCGCATGACTCAACTGCCTGGTGATACGAGGATGGGCATGCTCTTCCCG |
| Sticker | TTAAGACCTGTGAA |
| S1-sticker | ATTTATCACCCGCCATAGTAGACGTATCACCAGGCAGTTGAGACGAACATTCCTAAGTCTGAA- |
| TTAAGACCTGTGAA |
3. MiRNA-mediated regulation of distinct death patterns of osteoblasts
MiRNAs play a crucial role in the regulation of osteoblastic biology and functions. Evidence from Dicer and Argonaute 2 gene knockout experiments demonstrates that the deletion of miRNA can lead to abnormal expression of osteogenic makers in osteoblasts.35,36 Furthermore, miRNAs have been identified as crucial players during two pivotal phases of bone formation: (1) the embryonic phase, during which miRNAs promote the proliferation and osteogenic differentiation of osteoblasts, and (2) the growth phase, where they precisely regulate overall bone mass and maintain homeostasis.35 For example, alteration of miR-22 and miR-125b can modulate the occurrence of apoptosis through mitogen-activated protein kinase (MAPK) pathways, resulting in an increased or decreased apoptosis mortality in osteoblasts.37 Additionally, miRNAs can also regulate the life course of osteoblasts by influencing other subtypes of RCD, including autophagic death, pyroptosis, and ferroptosis.37–40 It is worth noting that the death modes in osteoblast cell lines extend beyond the aforementioned styles and may include necroptosis, NETotic cell death and entotic cell death.6 However, due to either the absence of triggering mechanisms in osteoblasts or limited supporting evidence, this paper does not delve into these specific subroutines.3.1. MiRNAs in regulating osteoblast apoptosis
Apoptosis, also known as type I cell death, is an extensively studied and the earliest identified form of RCD in organisms. It involves the activation of the caspase protein family through two signaling pathways: intrinsic and extrinsic, which are in response to cellular damage or stress.6,41 The intrinsic signaling pathway can be initiated by an imbalance in the ratio of B-cell lymphoma 2 (Bcl-2)/Bcl2-associated X (Bax) proteins, leading to increased mitochondrial membrane permeability. Consequently, cytochrome C is released into the cytoplasm, activating downstream caspase-9 and caspase-3 and ultimately inducing apoptosis.42 On the other hand, the extrinsic pathway is triggered by the death-inducing signaling complex (DISC), involving Fas death domain-associated protein (FADD) and the caspase-8 cascade, culminating in the activation of caspase-3 and the induction of apoptosis.41 Therefore, targeting crucial components of the signaling pathway, such as caspase-3, can block the cascade response and inhibit osteoblast apoptosis (Fig. 2).Several studies have reported on miRNAs that promote osteoblast apoptosis. Wang et al. discovered that miR-let-7b can target cyclin D and its overexpression can limit the cellular viability of MC3T3-E1 cells and facilitate apoptosis by blocking the Wnt/β-catenin pathway.43 Similarly, Chen et al. found that miR-22 negatively affects osteoblastic proliferation and osteogenic expression. It binds to ESR1 and indirectly inactivates the MAPK/JNK and p38 pathways, leading to apoptosis in MC3T3-E1-E1 cells.44 Wang et al. also showed that miR-34a could inhibit fibroblast growth factor receptor 1 (FGFR1) and promote osteoblast apoptosis.45 Additionally, other studies found that miR-107 can downregulate calcium binding protein 39 (CAB39), thereby inhibiting the activation of the nuclear factor E2-related factor 2 (Nrf2) cascade of the AMP-activated protein kinase (AMPK) pathway and facilitating oxidative injury and cellular apoptosis.46 MiR-133a plays an important role in bone metabolism, which can significantly promote apoptosis of BMSC and inhibit osteogenesis by jamming MAPK/ERK signaling.47 MiR-135b can inhibit the Janus kinase 2/Signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway, leading to a decreased alkaline phosphatase (ALP) activity and osteoblast-specific proteins and ultimately to osteoblast apoptosis.48 Overexpression of miR-139-3p promotes the apoptosis of MC3T3-E1-E1 cells and inhibits differentiation via targeting the ETS transcription factor ELK1. However, Wang et al. were able to reverse the negative effects of miR-139-3p on osteoblasts by utilizing the sponge effect of long non-coding RNA ODSM.49 In osteoporosis, miR-140-3p promotes osteoblastic apoptosis and inhibits cellular proliferation by blocking the phosphatidylinositol 3-kinase (PI3K)/AKT pathway. Furthermore, it also stimulates osteoclastic differentiation, exacerbating the imbalance of bone homeostasis.50 Tian et al. reported that miR-141 can target TGF-β to promote osteoblast apoptosis, while inhibiting miR-141 can up-regulate the expression of runt related transcription factor 2 (Runx2), secreted phosphoprotein 1 (Spp1), Bcl-2 and bone morphogenetic protein 2 (BMP-2), and reduce the expression of Bax and RANK in rat femoral head tissues, thus protecting against osteonecrosis.51 Similarly, another study indicated that miR-141 could promote osteoblast apoptosis and lead to osteoporosis.52 Activation of the PI3K pathway is positively associated with osteoblast proliferation and osteogenic differentiation. Mi et al. showed that PI3KR1 is one of the targets of miR-7223-5p. Thus, overexpressing miR-7223-5p could inhibit the PI3K/AKT pathway, reduce the activity of osteoblasts, and induce apoptosis.53 Sun et al. found that the upregulation of miR-211 significantly inhibited the PI3K/AKT pathway, which also promoted the activation of the caspase-9/3 apoptotic pathway.54 Recent studies have shown that miR-223-3p and miR-701-3p can downregulate fibroblast growth factor receptor 2 (FGFR2) and FGFR3, respectively, resulting in decreased levels of these receptors and promoting osteoblast apoptosis.55,56 Moreover, miR-3074-5p has been found to target Smad4 protein, leading to inhibition of the TGF-β/Smad signaling pathway and initiation of the apoptotic program in MC3T3-E1-E1 cells. Meanwhile, downregulation of Smad4 synergistically blocked ERK, PI3K/AKT and JAK/STAT signaling pathways and exacerbated apoptosis.57
Researchers also found that some miRNAs inhibit osteoblast apoptosis. Dong et al. reported that miR-23a could downregulate the expression of Fas receptor protein, leading to a reduction in apoptosis in TNF-α-stimulated MC3T3-E1-E1 cells.58 Li et al. reported that miR-26a, which decreases the expression of zeste homologue 2 (EZH2), slows down the apoptosis of osteoblasts and reduces osteoclasts to ameliorate the progression of steroid-induced osteonecrosis.59 Ren et al. found that miR-27a not only inhibits osteoblast apoptosis but also promotes osteogenic differentiation by activating the cryptochrome circadian regulator 2 (CRY2)/ERK1/2 axis.60 Similarly, Qiu et al. found that BMSC-derived miR-150-3p inhibits apoptosis and promotes the expression of osteogenic-related proteins such as Runx2 and Ostrix.61 Other miRNAs, such as miR-193a-3p and miR-708, target PTEN to improve the activity of MC3T3-E1-E1 cells, reduce reactive oxygen species (ROS) levels, and inhibit cell apoptosis to promote fracture repair.58,59,62,63 MiR-206 and miR-214 act as silencers to inhibit E74-like factor 3 (Elf3) and Sox4, which increases the levels of ALP and calcium deposition and ameliorates osteoblastic apoptosis.64 Additionally, miR-214 mediates the phosphorylation of PI3K/AKT or MAPK/JNK signaling pathways to assist fracture healing.65,66 MiR-497, a down-regulator of leucine-rich alpha-2-glycoprotein-1 (LRG1), was corroborated to indirectly engage in activating the TGF-β1/Smads pathway, via which the oxidative stress can be suppressed and restore osteoblastic viability.67 Similarly, MiR-655-3p also engaged in activating the Smad signaling pathway to inhibit osteoblast apoptosis. It has been demonstrated that lysine-specific histone demethylase 1 (LSD1) is the downstream target gene of miR-655-3p, which further effected the BMP-2/Smad signaling to promote osteogenesis.68 Caspase-3 is one of the most critical components in the activation of the apoptosis cascade. Therefore, downregulation of caspase-3 can block the process of apoptosis. MiR-758-3p could target caspase-3 and significantly reduce osteoblast apoptosis in osteoporosis patients.69 MiR-3175 and miR-4523 silence DDB1 and CUL4-associated factor 1 (DCAF1) or phosphoglycerate kinase 1 (PGK1), respectively, efficiently activating the nuclear-factor-E2-related factor 2 (Nrf2) cascade to protect osteoblasts.70,71
Notably, several miRNAs were reported to display disputed impacts in regulating the apoptotic fate of osteoblasts. For instance, Luo et al. demonstrated that miR-142 mimics significantly increase the expression of Bax and caspase-3, while decreasing the expression of Bcl-2.72 In addition, BMP-2 was identified as one of the targets of miR-142, suggesting that miR-142 could facilitate apoptosis and suppress osteogenic expression by inhibiting the BMP/Smad signaling pathway.72 Controversially, Zheng et al. reported that miR-142 has a protective effect on osteoblasts in a high-glucose environment.73 They showed that miR-142 can block the Wnt signaling pathway by targeting β-catenin to suppress the onset of high glucose-induced apoptosis. Another debated one is miR-146a, which was reported to be engaged in both promoting and inhibiting osteoblastic apoptosis.74,75 Cao et al. demonstrated that the expression of caspase-3 and Bax could be inhibited by downregulation of miR-146a. Thus, making use of the competitive endogenous RNA (ce-RNA) mechanism of circRNA–Rtn4 to sponge miR-146a can significantly attenuate apoptosis in osteoblasts.75 Differently, Nan et al. reported that miR-146a could protect against steroid-induced osteonecrosis.74 They showed that miR-146a down-regulated the ROS levels and protected bone trabecular structures via down-mediating the Wnt/FOXO signaling pathway.
In summary, various miRNAs can regulate different apoptotic pathways in osteoblasts under pathological conditions, and the use of effective transmembrane transporters of miRNAs is a prerequisite for regulation. TDNs can carry functional miRNAs or inhibitors to overexpress or suppress the post-transcription levels of apoptosis-related mRNAs, thereby achieving targeted therapy.76 More importantly, previous studies have demonstrated the excellent biological properties of the TDN vector itself in regulating apoptosis activation.77–79 It has an effect on Bcl-2/Bax regulation, caspase-3 inhibition, and cellular viability activation. For instance, Zhou et al. reported that TDNs can significantly upregulate the expression of Bcl-2 while decreasing the expression of caspases-3. Moreover, the expression of caspase-3 was negatively correlated with the concentration gradient of TDNs. Flow cytometry also showed that 250 nmol L−1 TDN could significantly reduce the percentage of apoptosis cells.80 Shi et al. observed that TDN treatment could upregulate the expression of Bcl2 and decrease the expression of Bax and caspase-3 to block chondrocytes apoptosis.77 Flow cytometry also revealed that the number of chondrocytes in the early and late phase apoptosis stages decreased by half in the TDN treatment group. In general, when the intrinsic and extrinsic pathways are triggered in a pathological state, multiple signaling pathways eventually lead to apoptosis. Therefore, combining the anti-apoptotic effect of TDN itself with specific miRNA nucleic acid drugs enables a powerful miRNA-targeting drug therapy (Fig. 2).
3.2. MiRNA in regulating the autophagy of osteoblasts and chondrocytes
Autophagy, also referred to as type II cell death, is a cellular process by which cells degrade intracellular damaged, degenerated, and senescent macromolecules or organelles in response to adversity, enabling nutrient recycling and the maintenance of cellular homeostasis. Therefore, autophagy is generally considered as a self-protective measure of organisms.81 For example, impaired chondrocyte autophagy is considered a significant contributor to cartilage degradation in OA.82 Likewise, the absence of autophagy in osteoblasts results in accumulation of harmful substances within cells, ultimately leading to apoptosis and disease exacerbation. However, excessive activation of autophagy can also lead to cell death, known as “autophagy-dependent cell death” or autosis.83 Hence, comprehending the mechanisms of autophagy is crucial for regulating its activity. Mammalian rapamycin complex 1 (mTORC1) serves as a key regulator of autophagy, and the inhibition of mTORC1 activity triggers the formation of autophagosomes. Under normal physiological conditions, mTOR exists in an activated state and phosphorylates ULK1, suppressing the formation of the ULK complex and downstream Beclin1–Vps34, thereby impeding autophagic vesicle formation.84 Thus, miRNAs can directly or indirectly regulate key mTORC1 by targeting signaling pathways, such as PI3K/AKT, to regulate osteoblast autophagy85 (Fig. 3A).In terms of miRNAs promoting chondrocyte autophagy, Feng et al. found that upregulation of let-7e could promote chondrocyte autophagy and inhibit apoptosis in knee osteoarthritis.86 PI3K is one of the target genes of miR-27, whose over-expression can block the PI3K/AKT/mTOR axis and induce chondrocyte autophagy.85 Similarly, miR-34a-5p promotes chondrocyte autophagy, inhibits cell proliferation, and exacerbates OA disease.87 MiR-100-5p targets the 3′-UTR of mTOR to inhibit translation and significantly upregulates OA chondrocyte autophagy levels.88 The upregulation of miR-335-5p in OA chondrocytes can promote chondrocyte autophagy, improving cell survival and alleviating inflammation.89 On the other hand, some miRNAs can inhibit chondrocyte autophagy. For instance, miR-7 can exacerbate OA by inhibiting autophagy and activating the PI3K/AKT/mTOR pathway. Herein, the use of the siRNA silencing or ce-RNA mechanism to reduce the expression of miR-7 is a potential therapeutic measure.90 MiR-375 acts as a catalyst in the exacerbation of OA, which targets ATG2B to inhibit autophagy while stimulating endoplasmic reticulum.91 MiR-411 was identified to block chondrocyte autophagy in OA by targeting hypoxia-inducible factor 1 alpha (HIF-1α). When transfected with miR-411 mimics, chondrocytes were detected to express decreased levels of autophagy-related molecular including LC3, ULK-1, p62 and Beclin-1.92
In terms of miRNAs promoting osteoblast autophagy, miR-199a-3p has been found to induce autophagy by targeting IGF-1 and mTOR, aggravating osteoporosis under estrogen-deficient conditions.93 According to Huang's investigation, the miR-1252-5p/GNB1 axis can promote osteoporosis by activating osteoblast autophagy and suppressing viability, thus reversing this pathway may be a potential therapeutic strategy for osteoporosis.94 On the other hand, some miRNAs have been found to inhibit osteoblast autophagy. Lu et al. observed that silencing miR-15b in osteoporotic mice resulted in increased bone mineral density. Further investigation revealed that miR-15b regulates KDM6B by targeting USP7, which ultimately inhibits osteoblast autophagy and aggravates osteoporosis.95 Feng et al. reported that miR-204 can negatively regulate autophagy and osteogenic differentiation by targeting microtubule-associated light chain 3B (LC3B), a key component in autophagosome assembly.96 Conversely, miR-27a-5p plays a protective role in bone tissue. Li et al. discovered that exosome-derived miR-27a-5p could target ATG4B to mediate autophagy and stimulate osteogenesis in osteoblasts.97 Overall, it is evident that even autophagy, which is typically considered “protective”, can inhibit the survival of chondrocytes/osteoblasts and negatively impact osteogenic differentiation. Therefore, modulating intracellular miRNAs to maintain appropriate levels of autophagy may be an ideal therapeutic strategy for the treatment of both osteoarthritis and osteoporosis.
Our previous studies have demonstrated the positive regulatory effect of TDN on cellular autophagy.77,98 Specifically, Shao et al. showed that TDNs have pro-chondrogenic regenerative potential.99 Shi et al. observed that TDNs can increase the number of LC3-positive autophagosomes in chondrocytes using immunofluorescence. Western blotting also confirmed an increase in the amount of LC3 protein.98 In an IL-1β-induced OA model, the amount of autophagy markers, LC3-II, Beclin1 and Atg7, was decreased, while the TDN was found to enhance chondrocyte autophagy by upregulating the expression of LC3-II, Beclin1 and Atg7, which exhibited a protective effect against OA.77 Therefore, based on the protective function of the TDN itself, carrying miRNAs to mediate the osteoblast autophagy function at the same time, a more optimal therapeutic effect may be achieved.
3.3. MiRNAs in regulating osteoblast pyroptosis
Pyroptosis is a form of regulated cell death (RCD) characterized by the formation of pores in the cell membrane through the action of gasdermin proteins.6 In response to harsh conditions such as viral or bacterial infections or exposure to lethal hazards, the inflammasome complex within cells can be activated. The classical inflammasome complex consists of pattern recognition receptors, such as NLR family pyrin domain containing 3 (NLRP3), or absent in melanoma 2 (AIM2), along with apoptosis-associated speck-like protein containing a CARD (ASC) and pro-caspase-1.100 Activation of the inflammasome complex leads to the activation of caspase-1, which in turn cleaves the gasdermin D (GSDMD) protein to generate the effector “GSDMD-N domain” that ultimately forms pores in the cell membrane.101 Pyroptotic cells exhibit swelling, rupturing and release of cytokines IL-1β and IL-18102 (Fig. 3B).Numerous studies have established a close relationship between chondrocyte pyroptosis and the progression of OA. MiRNAs were identified to regulate chondrocyte pyroptosis and play a significant role in the disease process.103 For instance, miR-107, miR-140-5p and miR-326 have been shown to inhibit the activation of pyroptosis pathways in chondrocytes, thereby mitigating OA progression.104–106 Qian et al. found that miR-107 can directly target caspase-1, inhibiting the activation of GSDMD proteins and suppressing the release of inflammatory cytokines, thereby promoting cartilage matrix secretion.104 MiR-140-5p downregulates the binding level of histone B to NLRP3 by binding to the 3′-UTR of histone B, thereby inhibiting the activation of inflammasome.105 Moreover, Xu et al. revealed that miR-326 in BMSCs exosomes regulates the STAT1/NF-κB/p65 pathway by targeting HDAC3 to significantly downregulate the expression levels of IL-1β, IL-18, ASC, NLRP3, and GSDMD and inhibit pyroptosis.106 Additionally, miR-155 alleviates OA by inhibiting chondrocyte pyroptosis through targeting SMAD2 to inhibit NLRP3/caspase-1 activation.107 Zhang et al. also found that upregulating the expression level of miR-200-3p in BMSCs can suppress pyroptosis in inflammatory states, promoting the survival of more BMSCs and alleviating bone loss.108
Theoretically, miRNAs that can target the key molecules in the pyroptosis signaling pathway may be potential therapeutic nucleic acid agents. Since the published research studies on osteoblastic pyroptosis are limited, below we additionally list some miRNAs which can target key mRNAs of pyroptosis, aiming at providing ideas for inhibiting pathological pyroptosis in osteoblast cell lines. Duan et al. showed that miR-133a could target NLRP3 and mitigate the progression of acute aortic coarctation by inhibiting endothelial pyroptosis.109 Similarly, miR-223, miR-590-3p, and miR-672-5p downregulate NLRP3, NLRP1, and AIM2, respectively, to inhibit inflammasome activation and prevent pyroptosis.110,111 Zhang et al. reported that miR-223 is able to downregulate NLRP3 expression and inhibit inflammasome.110 NLRP1 is a homologous protein of NLRP3 and forms a NLRP1/ASC/caspase-1 inflammasome complex to trigger cellular pyroptosis. Gu et al. found that miR-590-3p simultaneously interrupts the translation level of NLRP1 and caspase-1 with superior inhibition.111 MiR-214-3p also targets caspase-1 and reduces the level of GSDMD-N domain products, and therefore decreases the level of pyroptosis.38 In addition, miR-672-5p targets AIM2 and inhibits the activation of AIM2/ASC/caspase-1 inflammasome, which plays an important role in preventing virus-induced and intracellular bacteria-induced pyroptosis.112 At the downstream of the pyroptosis pathway, GSDMD is the most important executor to induce membrane perforation.113 Therefore, GSDMD-targeted miRNA may be the most promising agent for pyroptosis therapy. Both miR-193a and miR-204 have been reported to directly target GSDMD, while inhibiting caspase-1 and caspase-11 to protect cells from pyroptosis.114,115
Based on these candidate miRNAs, special Tmi can be fabricated, which has the potential to inhibit pyroptosis gene expression. Additionally, Tmi has the ability to promote various cellular activities and protect cells during pathological states.76,116,117 Studies have identified the regulatory effect of TDN on pyroptosis both in vitro and in vivo. For example, Jiang et al. found that TDNs can significantly inhibit the activated production of cleaved caspase-1 and GSDMD-N domains, and suppress the release of inflammatory factors such as IL-1β and IL-18, thereby inhibiting cell pyroptosis.118 Similarly, Chen et al. reported that TDNs have a therapeutic effect on the systemic inflammatory state of sepsis by inhibiting macrophage pyroptosis.119 In an LPS-stimulated inflammation model, macrophages showed a significant increase in the expression of NLRP3, cleaved caspase-1, and GSDMD-N. However, after TDN treatment, the expression of these pyroptosis-related proteins was significantly decreased, and the inflammatory factors, IL-18 and IL-1β, were suppressed. Nevertheless, given that the subjects in these studies were not osteoblasts, further investigation is required to determine the effect of TDN on osteoblastic pyroptosis. By combining the TDN with specific miRNAs to target pyroptosis-related genes, it is expected that the translation of the therapeutic nucleic acid drug from zero to one can be achieved.
3.4. MiRNAs in regulating osteoblast ferroptosis
In recent years, ferroptosis has garnered attention as a form of RCD. It is associated with excess intracellular iron (Fe2+) concentration and accumulation of lipid peroxidation.120 Ferroptotic cells exhibit reduced mitochondrial cristae and atrophied or ruptured mitochondrial membranes.121 At the molecular level, ferroptosis is primarily mediated by two key components: (1) a lipid peroxidation system, which generates multiple lipid peroxidation products via Fenton or Haber Weiss reactions when cells are under iron overload, ultimately triggering ferroptosis; and (2) an antioxidant system, which primarily consists of the cystine glutamate transporter system XC- and phospholipid peroxidase glutathione peroxidase 4 (GPX4). The antioxidant system utilizes glutathione (GSH) to reduce membranal peroxide polyunsaturated fatty acids (PL-PUFAs) and intracellular ROS to inhibit ferroptosis.122 Additionally, cells can scavenge lipid peroxidation by generating ubiquinol and tetrahydrobiopterin (BH4) through FSP1-coenzyme Q10 and the GTP cyclohydrolase 1 (GCH1) axis.123–125 Therefore, a standoff exists between oxidation and antioxidation. The protection of the antioxidant system eliminates the peroxidised lipids generated intracellularly in a timely manner, preventing cellular ferroptosis (Fig. 4).MiRNAs, as the smallest non-coding RNA, play a crucial role in regulating various physiological processes of cells. However, their functions in ferroptosis remain largely unexplored. In theory, blocking the pathway of PL-PUFA can impede the occurrence of ferroptosis by reducing the concentration of phospholipid substrates or suppressing the activity of phospholipid peroxidase.126 Acyl–CoA synthase long chain family member 4 (ACSL4) is an enzyme that facilitates the integration of free PUFAs into long-chain PL-PUFA, which promotes the production of lipid peroxidation substrates. To this end, Ou et al. found that miR-4291 can target ACSL4, suppressing the synthesis of substrates and inhibiting ferroptosis.127 Similarly, miR-124-3p.1 can target lysophosphatidylcholine acyltransferase 3 (LPCAT3), another crucial enzyme in producing PL-PUFAs, to prevent ferroptosis.128 Additionally, Bin et al. demonstrated that miR-10a-5p can inhibit chondrocyte ferroptosis by targeting IL-6R.129 Li et al. found that miR-874-3p could inhibit ferroptosis by targeting activation transcription factor 3 (ATF3).130 Iron response element-binding protein 2 (IREB2) is a significant regulator of cellular iron homeostasis, and Li et al. reported that miR-29-3p could reduce the level of ferroptosis through the targeted downregulation of (IREB2) expression.131 The transferrin receptor protein (TFRC) encodes a cell surface receptor necessary for cellular iron uptake. Wang et al. reported that miR-370 targets TFRC and inhibits ferroptosis.132
Inhibiting ferroptosis can protect cells, while promoting ferroptosis can damage cells. In cancer research, miRNAs have been utilized in cancer research to induce ferroptosis and kill tumour. Ou et al. reported that circRNA cirLMO1 can decrease miR-4291 to promote ferroptosis in cervical cancer.127 Similarly, in osteosarcoma, Xu et al. found that miR-1287-5p directly targets GPX4 to promote ferroptosis and improve sensitivity to cisplatin chemotherapy.133 SLC7A11, which plays a crucial role in regulating ferroptosis through the transport of cystine and glutamate. Cai et al. showed that miR-587 can target SLC7A11 to promote ferroptosis in ovarian cancer cells.134
Unfortunately, direct evidence regarding the effects of TDN on ferroptosis in osteoblasts is currently lacking. Nevertheless, existing studies have demonstrated the anti-inflammatory and antioxidant properties of TDN, particularly its ability to inhibit cellular reactive oxygen species (ROS) production.135–137 Therefore, it is reasonable to speculate that the TDN has an inhibitory function on lipid peroxide production, subsequently regulating the process of ferroptosis.138–140 Additionally, it is certain that the TDN as a carrier enables delivery of functional miRNA cargos such as miR-4291 (targeting ACSL4) and miR-124-3p (targeting LPCAT3) to realize ferroptosis therapy.
4. Conclusion and prospects
Maintaining bone homeostasis is crucial for preventing bone diseases, as an adequate number of osteoblasts is essential for bone regeneration and treatment. However, during the differentiation of stem cells into osteoblasts or chondrocytes, abnormal environments and pathological conditions can trigger the RCD process, resulting in increased mortality of pre-osteoblasts and mature differentiated osteoblasts.141,142 This disturbance of the osteogenic homeostasis macroscopically leads to bone diseases.143,144MiRNAs are post-transcriptional regulators that play a pivotal role in gene regulation. By assembling into the RISC, miRNAs can silence gene expression through inhibiting the translation of the corresponding mRNAs.145 Therefore, miRNAs can modulate the expression of key molecules in the RCD signaling pathway, thus manipulating the initiation or inhibition of the osteoblastic death program and treating bone diseases. Research studies have demonstrated that screening effective miRNAs to block apoptosis, autosis, pyroptosis and ferroptosis in osteoblasts is a promising therapeutic approach.70,97,105,146
However, miRNAs have inherent limitations including instability, susceptibility to enzymatic degradation, and poor cellular membrane penetrability. Although current transfection reagents can facilitate miRNA delivery to the cytoplasm, they are unsuitable for therapeutic applications in humans due to their cytotoxicity. Fortunately, a potential solution to this challenge is presented by Tmi, which offers advantages such as affordability, ease of synthesis, programmable editing, excellent biocompatibility, efficient cellular penetration, and minimal cytotoxicity.19,20 Building upon the Tmi delivery system that carries miRNAs into cells and targets key genes involved in RCD, it is feasible to regulate osteoblastic RCD. Notably, while this paper focuses on the regulatory role of miRNAs in the RCD pathway, siRNAs, which share a similar structure and function, can also exert silencing effects through the RISC mechanism.29,32,33 Moreover, siRNAs can be designed to match the base sequences completely, providing stronger specificity and more effective silencing compared to the partial base pairing of miRNAs’ seed region (positions 2–7), and all of the key molecules involved in the RCD pathway can be designed for siRNA-targeting sites.147
In this review, the role of miRNAs as molecular targeted agents in four modes of regulated cell death in osteoblasts is summarized. The use of Tmi as a miRNA delivery system is proposed, considering its excellent biocompatibility and cell membrane penetration. Notably, the Tmi delivery system not only transports miRNA but also protects it, which may play an important role in the field of bone diseases. Although the Tmi system has several advantages, there is still room for improvement. First, enzyme resistance needs to be enhanced, as its retention time in serum is currently about 24 hours.22,23 Second, the specific tissue targeting of Tmi needs improvement. The current administration methods of Tmi are mainly intravenous injection and local injection, which implies a broad distribution of TDN drugs and limited tissue specificity. Although aptamer modification and biofilm coating can enhance the targeting ability, more optimization techniques are needed.31,148 Third, the delivery efficiency of TDN needs to be optimized. Different ssDNA editing methods result in varying amounts of miRNA carried by 1 unit of the Tmi delivery system (Fig. 1A). Thus, exploring Tmi delivery systems with higher unit delivery efficiency, stronger miRNA protection efficiency, good cell membrane permeability, and biocompatibility is necessary. With the help of highly membrane-penetrating and excellent biocompatible Tmi, miRNAs are able to overcome their own defects and effectively deliver intracellularly to exert gene silence,16 ultimately realizing the clinical application of RNA nucleic acid drugs.
Author contributions
ZC, SS, and FL conceptualized the paper; ZC, LB, MF, and YL performed the literature collection and wrote the paper; ZC, YL, WM and SL completed the material synthesis and evaluation process; ZC, FL, SL, WM and LB conceived and completed the schematic diagram; ZC and SS edited and revised the paper. All authors discussed and approved the final paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (82101077), the Postdoctoral Science Foundation of China (Grant 2021M692271), and the West China School/Hospital of Stomatology Sichuan University, No. RCDWJS2023-(5). We thank Yang Gao and Xin Qin for their support in the polyacrylamide gel electrophoresis of the Tmi system.References
- D. Prieto-Alhambra, A. Judge, M. K. Javaid, C. Cooper, A. Diez-Perez and N. K. Arden, Ann. Rheum. Dis., 2014, 73, 1659–1664 CrossRef PubMed.
- J. E. Compston, M. R. McClung and W. D. Leslie, Lancet, 2019, 393, 364–376 CrossRef CAS PubMed.
- P. R. Ebeling, H. H. Nguyen, J. Aleksova, A. J. Vincent, P. Wong and F. Milat, Endocr. Rev., 2022, 43, 240–313 CrossRef PubMed.
- M. A. Clynes, N. C. Harvey, E. M. Curtis, N. R. Fuggle, E. M. Dennison and C. Cooper, Br. Med. Bull., 2020, 133, 105–117 Search PubMed.
- T. Komori, Int. J. Mol. Sci., 2016, 17, 2045 CrossRef PubMed.
- L. Galluzzi, I. Vitale, S. A. Aaronson, J. M. Abrams, D. Adam, P. Agostinis, E. S. Alnemri, L. Altucci, I. Amelio, D. W. Andrews, M. Annicchiarico-Petruzzelli, A. V. Antonov, E. Arama, E. H. Baehrecke, N. A. Barlev, N. G. Bazan, F. Bernassola, M. J. M. Bertrand, K. Bianchi, M. V. Blagosklonny, K. Blomgren, C. Borner, P. Boya, C. Brenner, M. Campanella, E. Candi, D. Carmona-Gutierrez, F. Cecconi, F. K. Chan, N. S. Chandel, E. H. Cheng, J. E. Chipuk, J. A. Cidlowski, A. Ciechanover, G. M. Cohen, M. Conrad, J. R. Cubillos-Ruiz, P. E. Czabotar, V. D'Angiolella, T. M. Dawson, V. L. Dawson, V. De Laurenzi, R. De Maria, K. M. Debatin, R. J. DeBerardinis, M. Deshmukh, N. Di Daniele, F. Di Virgilio, V. M. Dixit, S. J. Dixon, C. S. Duckett, B. D. Dynlacht, W. S. El-Deiry, J. W. Elrod, G. M. Fimia, S. Fulda, A. J. García-Sáez, A. D. Garg, C. Garrido, E. Gavathiotis, P. Golstein, E. Gottlieb, D. R. Green, L. A. Greene, H. Gronemeyer, A. Gross, G. Hajnoczky, J. M. Hardwick, I. S. Harris, M. O. Hengartner, C. Hetz, H. Ichijo, M. Jäättelä, B. Joseph, P. J. Jost, P. P. Juin, W. J. Kaiser, M. Karin, T. Kaufmann, O. Kepp, A. Kimchi, R. N. Kitsis, D. J. Klionsky, R. A. Knight, S. Kumar, S. W. Lee, J. J. Lemasters, B. Levine, A. Linkermann, S. A. Lipton, R. A. Lockshin, C. López-Otín, S. W. Lowe, T. Luedde, E. Lugli, M. MacFarlane, F. Madeo, M. Malewicz, W. Malorni, G. Manic, J. C. Marine, S. J. Martin, J. C. Martinou, J. P. Medema, P. Mehlen, P. Meier, S. Melino, E. A. Miao, J. D. Molkentin, U. M. Moll, C. Muñoz-Pinedo, S. Nagata, G. Nuñez, A. Oberst, M. Oren, M. Overholtzer, M. Pagano, T. Panaretakis, M. Pasparakis, J. M. Penninger, D. M. Pereira, S. Pervaiz, M. E. Peter, M. Piacentini, P. Pinton, J. H. M. Prehn, H. Puthalakath, G. A. Rabinovich, M. Rehm, R. Rizzuto, C. M. P. Rodrigues, D. C. Rubinsztein, T. Rudel, K. M. Ryan, E. Sayan, L. Scorrano, F. Shao, Y. Shi, J. Silke, H. U. Simon, A. Sistigu, B. R. Stockwell, A. Strasser, G. Szabadkai, S. W. G. Tait, D. Tang, N. Tavernarakis, A. Thorburn, Y. Tsujimoto, B. Turk, T. Vanden Berghe, P. Vandenabeele, M. G. Vander Heiden, A. Villunger, H. W. Virgin, K. H. Vousden, D. Vucic, E. F. Wagner, H. Walczak, D. Wallach, Y. Wang, J. A. Wells, W. Wood, J. Yuan, Z. Zakeri, B. Zhivotovsky, L. Zitvogel, G. Melino and G. Kroemer, Cell Death Differ., 2018, 25, 486–541 CrossRef PubMed.
- L. Galluzzi, M. C. Maiuri, I. Vitale, H. Zischka, M. Castedo, L. Zitvogel and G. Kroemer, Cell Death Differ., 2007, 14, 1237–1243 CrossRef CAS PubMed.
- J. U. Schweichel and H. J. Merker, Teratology, 1973, 7, 253–266 CrossRef CAS PubMed.
- D. Tang, R. Kang, T. V. Berghe, P. Vandenabeele and G. Kroemer, Cell Res., 2019, 29, 347–364 CrossRef CAS PubMed.
- M. Ha and V. N. Kim, Nat. Rev. Mol. Cell Biol., 2014, 15, 509–524 CrossRef CAS PubMed.
- D. S. Schwarz, G. Hutvágner, T. Du, Z. Xu, N. Aronin and P. D. Zamore, Cell, 2003, 115, 199–208 CrossRef CAS PubMed.
- M. R. Fabian and N. Sonenberg, Nat. Struct. Mol. Biol., 2012, 19, 586–593 CrossRef CAS PubMed.
- J. Du, M. Li, Q. Huang, W. Liu, W. Q. Li, Y. J. Li and Z. C. Gong, Pharmacol. Res., 2019, 142, 294–302 CrossRef CAS PubMed.
- S. W. L. Lee, C. Paoletti, M. Campisi, T. Osaki, G. Adriani, R. D. Kamm, C. Mattu and V. Chiono, J. Controlled Release, 2019, 313, 80–95 CrossRef CAS PubMed.
- R. Rupaimoole and F. J. Slack, Nat. Rev. Drug Discovery, 2017, 16, 203–222 CrossRef CAS PubMed.
- Y. Zhang, Z. Wang and R. A. Gemeinhart, J. Controlled Release, 2013, 172, 962–974 CrossRef CAS PubMed.
- T. Tian, T. Zhang, S. Shi, Y. Gao, X. Cai and Y. Lin, Nat. Protoc., 2023, 18, 1028–1055 CrossRef CAS PubMed.
- T. Tian, Y. Li and Y. Lin, Bone Res., 2022, 10, 40 CrossRef CAS PubMed.
- T. Zhang, T. Tian, R. Zhou, S. Li, W. Ma, Y. Zhang, N. Liu, S. Shi, Q. Li, X. Xie, Y. Ge, M. Liu, Q. Zhang, S. Lin, X. Cai and Y. Lin, Nat. Protoc., 2020, 15, 2728–2757 CrossRef CAS PubMed.
- T. Zhang, T. Tian and Y. Lin, Adv. Mater., 2021, e2107820, DOI:10.1002/adma.202107820.
- T. Zhang, H. S. Ma, X. L. Zhang, S. R. Shi and Y. F. Lin, Adv. Funct. Mater., 2023, 33, 14 Search PubMed.
- T. Zhang, M. Zhou, D. Xiao, Z. Liu, Y. Jiang, M. Feng, Y. Lin and X. Cai, Adv. Sci., 2022, 9, e2202058 CrossRef PubMed.
- Z. Q. Liu, X. Y. Chen, W. J. Ma, Y. Gao, Y. X. Yao, J. J. Li, T. X. Zhang, X. Qin, Y. C. Ge, Y. Y. Jiang and Y. F. Lin, Adv. Funct. Mater., 2022, 32, 14 Search PubMed.
- S. Li, Y. Liu, T. Tian, T. Zhang, S. Lin, M. Zhou, X. Zhang, Y. Lin and X. Cai, Small, 2021, e2104359, DOI:10.1002/smll.202104359.
- X. R. Shao, S. Y. Lin, Q. Peng, S. R. Shi, X. L. Li, T. Zhang and Y. F. Lin, Nanomedicine, 2017, 13, 1809–1819 CrossRef CAS PubMed.
- J. Li, Y. Yao, Y. Wang, J. Xu, D. Zhao, M. Liu, S. Shi and Y. Lin, Adv. Mater., 2022, e2202513, DOI:10.1002/adma.202202513.
- S. Li, Y. Sun, T. Tian, X. Qin, S. Lin, T. Zhang, Q. Zhang, M. Zhou, X. Zhang, Y. Zhou, H. Zhao, B. Zhu and X. Cai, Cell Proliferation, 2020, 53, e12708 CrossRef PubMed.
- X. Qin, L. Xiao, N. Li, C. Hou, W. Li, J. Li, N. Yan and Y. Lin, Bioact. Mater., 2022, 14, 134–144 CrossRef CAS PubMed.
- W. Fu, L. Ma, Y. Ju, J. G. Xu, H. Li, S. R. Shi, T. Zhang, R. H. Zhou, J. W. Zhu, R. X. Xu, C. You and Y. F. Lin, Adv. Funct. Mater., 2021, 31, 12 Search PubMed.
- Y. Ge, Q. Wang, X. Qin, S. Li, Z. Liu, Y. Lin, X. Li and X. Cai, ACS Appl. Mater. Interfaces, 2022, 14, 19091–19093 CrossRef CAS PubMed.
- D. Xiao, Y. Li, T. Tian, T. Zhang, S. Shi, B. Lu, Y. Gao, X. Qin, M. Zhang, W. Wei and Y. Lin, ACS Appl. Mater. Interfaces, 2021, 13, 6109–6118 CrossRef CAS PubMed.
- X. Zhang, M. Zhang, M. Zhou, T. Zhang, Y. Gao, S. Li, Y. Lin and X. Cai, ACS Appl. Mater. Interfaces, 2022, 14, 6442–6452 CrossRef CAS PubMed.
- Y. Gao, X. Chen, T. Tian, T. Zhang, S. Gao, X. Zhang, Y. Yao, Y. Lin and X. Cai, Adv. Mater., 2022, e2201731, DOI:10.1002/adma.202201731.
- S. Li, Y. Liu, T. Zhang, S. Lin, S. Shi, J. He, Y. Xie, X. Cai, T. Tian and Y. Lin, Adv. Mater., 2022, 34, e2204287 CrossRef PubMed.
- T. Gaur, S. Hussain, R. Mudhasani, I. Parulkar, J. L. Colby, D. Frederick, B. E. Kream, A. J. van Wijnen, J. L. Stein, G. S. Stein, S. N. Jones and J. B. Lian, Dev. Biol., 2010, 340, 10–21 CrossRef CAS PubMed.
- S. Shirazi, C. C. Huang, M. Kang, Y. Lu, S. Ravindran and L. F. Cooper, Sci. Rep., 2021, 11, 5953 CrossRef CAS PubMed.
- S. Jiang, Y. Liu, B. Xu, Y. Zhang and M. Yang, Wiley Interdiscip. Rev.: RNA, 2020, 11, e1584 CAS.
- F. Yang, Y. Qin, J. Lv, Y. Wang, H. Che, X. Chen, Y. Jiang, A. Li, X. Sun, E. Yue, L. Ren, Y. Li, Y. Bai and L. Wang, Cell Death Dis., 2018, 9, 1000 CrossRef PubMed.
- J. Yang, S. Hu, Y. Bian, J. Yao, D. Wang, X. Liu, Z. Guo, S. Zhang and L. Peng, Front. Cell Dev. Biol., 2021, 9, 789948 CrossRef PubMed.
- K. Sun, Z. Guo, L. Hou, J. Xu, T. Du, T. Xu and F. Guo, Ageing Res. Rev., 2021, 72, 101481 CrossRef CAS PubMed.
- I. Verbrugge, R. W. Johnstone and M. J. Smyth, Cell, 2010, 143, 1192 CrossRef CAS PubMed.
- G. S. Salvesen and A. Ashkenazi, Cell, 2011, 147, 476–476 CrossRef CAS PubMed.
- L. J. Wang and H. Q. Cai, Kaohsiung J. Med. Sci., 2020, 36, 775–785 CrossRef CAS PubMed.
- G. H. Chen, X. L. Zhang, H. Chen, H. Lin, H. J. Wu, H. Lin and G. Z. Huang, J. Gene Med., 2020, 22, e3174 CAS.
- X. Wang, J. He, H. Wang, D. Zhao, B. Geng, S. Wang, J. An, C. Wang, H. Han and Y. Xia, J. Cell. Mol. Med., 2021, 25, 8734–8747 CrossRef CAS PubMed.
- Y. Zhuang, S. Wang, H. Fei, F. Ji and P. Sun, Aging, 2020, 12, 11754–11767 CrossRef CAS PubMed.
- G. Wang, F. Wang, L. Zhang, C. Yan and Y. Zhang, Stem Cell Res. Ther., 2021, 12, 215 CrossRef CAS PubMed.
- X. T. Zhang, M. Sun, L. Zhang, Y. K. Dai and F. Wang, Kaohsiung J. Med. Sci., 2020, 36, 673–681 CrossRef CAS PubMed.
- Y. Wang, K. Wang, L. Zhang, Y. Tan, Z. Hu, L. Dang, H. Zhou, G. Li, H. Wang, S. Zhang, F. Shi, X. Cao and G. Zhang, Cell Death Dis., 2020, 11, 133 CrossRef CAS PubMed.
- R. Yin, J. Jiang, H. Deng, Z. Wang, R. Gu and F. Wang, Hum. Cell, 2020, 33, 569–581 CrossRef PubMed.
- L. Tian, S. Sun, W. Li, L. Yuan and X. Wang, Cell Cycle, 2020, 19, 772–786 CrossRef CAS PubMed.
- S. Z. Wang, J. Jia and C. H. Chen, Stem Cells Int., 2021, 2021, 7690006 Search PubMed.
- B. Mi, Y. Xiong, L. Chen, C. Yan, Y. Endo, Y. Liu, J. Liu, L. Hu, Y. Hu, Y. Sun, F. Cao, W. Zhou and G. Liu, Aging, 2019, 11, 11988–12001 CrossRef CAS PubMed.
- T. Sun, D. Yang, Y. Wu and Q. Sheng, J. Int. Med. Res., 2020, 48, 300060520926353 CAS.
- B. Wang, W. Wu, K. Xu and H. Wu, Bioengineered, 2021, 12, 12040–12048 CrossRef CAS PubMed.
- L. Chen, Y. Xiong, C. Yan, W. Zhou, Y. Endo, H. Xue, Y. Hu, L. Hu, X. Leng, J. Liu, Z. Lin, B. Mi and G. Liu, FASEB J., 2020, 34, 5208–5222 CrossRef CAS PubMed.
- Y. Feng, P. Y. He, W. D. Kong, W. J. Cen, P. L. Wang, C. Liu, W. Zhang, S. S. Li and J. W. Jiang, Cell. Mol. Biol. Lett., 2021, 26, 37 CrossRef CAS PubMed.
- J. Dong, X. Cui, Z. Jiang and J. Sun, J. Cell. Biochem., 2013, 114, 2738–2745 CrossRef CAS PubMed.
- G. Li, H. Liu, X. Zhang, X. Liu, G. Zhang and Q. Liu, Cell Cycle, 2020, 19, 551–566 CrossRef CAS PubMed.
- L. R. Ren, R. B. Yao, S. Y. Wang, X. D. Gong, J. T. Xu and K. S. Yang, Mol. Med., 2021, 27, 43 CAS.
- M. Qiu, S. Zhai, Q. Fu and D. Liu, Hum. Gene Ther., 2021, 32, 717–729 CrossRef CAS PubMed.
- K. Zheng and Y. Wang, Mol. Biotechnol., 2021, 63, 605–612 CrossRef CAS PubMed.
- W. Zhang, S. Y. Cui, H. Yi, X. H. Zhu, W. Liu and Y. J. Xu, J. Orthop. Surg. Res., 2020, 15, 255 CrossRef PubMed.
- Y. Huang, X. Zhang, J. Zhan, Z. Yan, D. Chen, X. Xue and X. Pan, J. Cell. Mol. Med., 2021, 25, 7734–7745 CrossRef CAS PubMed.
- Z. Xin, D. Cai, J. Wang, L. Ma, F. Shen, C. Tang, L. Hu and W. Sun, J. Musculoskeletal Neuronal Interact., 2020, 20, 429–436 CAS.
- X. Zhu, Z. Zhao, C. Zeng, B. Chen, H. Huang, Y. Chen, Q. Zhou, L. Yang, J. Lv, J. Zhang, D. Pan, J. Shen, G. Duque and D. Cai, Calcif. Tissue Int., 2020, 106, 518–532 CrossRef CAS PubMed.
- Z. Gu, D. Xie, C. Huang, R. Ding, R. Zhang, Q. Li, C. Lin and Y. Qiu, J. Cell. Mol. Med., 2020, 24, 12619–12632 CrossRef CAS PubMed.
- X. J. Wang, J. W. Liu and J. Liu, Hum. Exp. Toxicol., 2020, 39, 1390–1404 CrossRef CAS PubMed.
- H. Liu, Y. W. Wang, W. D. Chen, H. H. Dong and Y. J. Xu, IUBMB Life, 2021, 73, 432–443 CrossRef CAS PubMed.
- J. Chen, J. Q. Liang, Y. F. Zhen, L. Chang, Z. T. Zhou and X. J. Shen, Cell Death Dis., 2021, 12, 1024 CrossRef CAS PubMed.
- J. Q. Liang, Z. T. Zhou, L. Bo, H. N. Tan, J. H. Hu and M. S. Tan, Cell Death Dis., 2021, 12, 964 CrossRef CAS PubMed.
- B. Luo, J. Yang, Y. Yuan, P. Hao and X. Cheng, FEBS Open Bio, 2020, 10, 1793–1801 CrossRef CAS PubMed.
- T. Zheng, G. Ji, J. Chen, J. Lai, T. Liu, J. Mo and Q. Jin, Exp. Ther. Med., 2020, 20, 125 CAS.
- K. Nan, J. P. Pei, L. H. Fan, Y. K. Zhang, X. Zhang, K. Liu, Z. B. Shi, X. Q. Dang and K. Z. Wang, Ann. N. Y. Acad. Sci., 2021, 1503, 23–37 CrossRef CAS PubMed.
- G. Cao, X. Meng, X. Han and J. Li, Biosci. Rep., 2020, 40, BSR20193436 CrossRef CAS PubMed.
- J. J. Li, L. R. Xiao, N. H. Yan, Y. J. Li, Y. Wang, X. Qin, D. Zhao, M. T. Liu, N. Li and Y. F. Lin, Adv. Funct. Mater., 2021, 31, 13 Search PubMed.
- S. Shi, T. Tian, Y. Li, D. Xiao, T. Zhang, P. Gong and Y. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 56782–56791 CrossRef CAS PubMed.
- M. Zhou, T. Zhang, B. Zhang, X. Zhang, S. Gao, T. Zhang, S. Li, X. Cai and Y. Lin, ACS Nano, 2021, 16, 1456–1470 CrossRef PubMed.
- Y. Wang, Y. Li, S. Gao, X. Yu, Y. Chen and Y. Lin, Nano Lett., 2022, 22, 1759–1768 CrossRef CAS PubMed.
- M. Zhou, T. Zhang, X. Zhang, M. Zhang, S. Gao, T. Zhang, S. Li, X. Cai, J. Li and Y. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 37478–37492 CrossRef CAS PubMed.
- N. Mizushima and M. Komatsu, Cell, 2011, 147, 728–741 CrossRef CAS PubMed.
- R. F. Loeser, J. A. Collins and B. O. Diekman, Nat. Rev. Rheumatol., 2016, 12, 412–420 CrossRef CAS PubMed.
- C. Samara, P. Syntichaki and N. Tavernarakis, Cell Death Differ., 2008, 15, 105–112 CrossRef CAS PubMed.
- R. A. Saxton and D. M. Sabatini, Cell, 2017, 168, 960–976 CrossRef CAS PubMed.
- C. Cai, S. Min, B. Yan, W. Liu, X. Yang, L. Li, T. Wang and A. Jin, Aging, 2019, 11, 6371–6384 CrossRef CAS PubMed.
- L. Feng, C. Feng, C. X. Wang, D. Y. Xu, J. J. Chen, J. F. Huang, P. L. Tan and J. M. Shen, Int. J. Mol. Med., 2020, 45, 1464–1476 CAS.
- F. Tian, J. Wang, Z. Zhang and J. Yang, Biol. Res., 2020, 53, 9 CrossRef CAS PubMed.
- J. Wu, L. Kuang, C. Chen, J. Yang, W. N. Zeng, T. Li, H. Chen, S. Huang, Z. Fu, J. Li, R. Liu, Z. Ni, L. Chen and L. Yang, Biomaterials, 2019, 206, 87–100 CrossRef CAS PubMed.
- G. Zhong, H. Long, S. Ma, Y. Shunhan, J. Li and J. Yao, Life Sci., 2019, 226, 164–172 CrossRef CAS PubMed.
- X. Zhou, J. Li, Y. Zhou, Z. Yang, H. Yang, D. Li, J. Zhang, Y. Zhang, N. Xu, Y. Huang and L. Jiang, Aging, 2020, 12, 20163–20183 CAS.
- H. Li, Z. Li, Y. Pi, Y. Chen, L. Mei, Y. Luo, J. Xie and X. Mao, Aging, 2020, 12, 7248–7261 CrossRef CAS PubMed.
- F. Yang, R. Huang, H. Ma, X. Zhao and G. Wang, Med. Sci. Monit., 2020, 26, e921155 CAS.
- J. Fu, L. Hao, Y. Tian, Y. Liu, Y. Gu and J. Wu, J. Cell. Physiol., 2018, 233, 2292–2303 CrossRef CAS PubMed.
- C. Huang, R. Li, C. Yang, R. Ding, Q. Li, D. Xie, R. Zhang and Y. Qiu, Exp. Mol. Med., 2021, 53, 894–906 CrossRef CAS PubMed.
- X. Lu, Y. Zhang, Y. Zheng and B. Chen, J. Cell. Mol. Med., 2021, 25, 2069–2081 CrossRef CAS PubMed.
- Q. Feng, S. Y. Cheng, R. Yang, X. W. Zeng, F. M. Zhao and X. Q. Zhan, Exp. Ther. Med., 2020, 19, 883–890 CAS.
- X. Li, R. Chen, Y. Li, P. Wang, Y. Cui, L. Yang, X. Zhu and R. Zhang, Front. Cell Dev. Biol., 2021, 9, 642646 CrossRef PubMed.
- S. Shi, S. Lin, Y. Li, T. Zhang, X. Shao, T. Tian, T. Zhou, Q. Li and Y. Lin, Chem. Commun., 2018, 54, 1327–1330 RSC.
- X. Shao, S. Lin, Q. Peng, S. Shi, X. Wei, T. Zhang and Y. Lin, Small, 2017, 13, 1602770 CrossRef PubMed.
- K. Schroder and J. Tschopp, Cell, 2010, 140, 821–832 CrossRef CAS PubMed.
- J. Shi, Y. Zhao, K. Wang, X. Shi, Y. Wang, H. Huang, Y. Zhuang, T. Cai, F. Wang and F. Shao, Nature, 2015, 526, 660–665 CrossRef CAS PubMed.
- C. L. Evavold, J. Ruan, Y. Tan, S. Xia, H. Wu and J. C. Kagan, Immunity, 2018, 48, 35–44 CrossRef CAS PubMed.
- S. An, H. Hu, Y. Li and Y. Hu, Aging Dis., 2020, 11, 1146–1157 CrossRef PubMed.
- J. Qian, P. Fu, S. Li, X. Li, Y. Chen and Z. Lin, J. Orthop. Surg. Res., 2021, 16, 40 CrossRef PubMed.
- L. Zhang, J. Qiu, J. Shi, S. Liu and H. Zou, Bioengineered, 2021, 12, 9949–9964 Search PubMed.
- H. Xu and B. Xu, Mediators Inflammation, 2021, 2021, 9972805 Search PubMed.
- G. Li, L. Xiu, X. Li, L. Ma and J. Zhou, J. Orthop. Surg. Res., 2022, 17, 48 CrossRef PubMed.
- L. Zhang, S. Li, J. Li and Y. Li, Stem Cell Rev. Rep., 2021, 17, 2262–2275 CrossRef CAS PubMed.
- H. Duan, X. Zhang, R. Song, T. Liu, Y. Zhang and A. Yu, Acta Biochim. Biophys. Sin., 2020, 52, 988–997 CrossRef CAS PubMed.
- Y. Zhang, X. Liu, X. Bai, Y. Lin, Z. Li, J. Fu, M. Li, T. Zhao, H. Yang, R. Xu, J. Li, J. Ju, B. Cai, C. Xu and B. Yang, J. Pineal Res., 2018, 64, e12449 CrossRef PubMed.
- C. Gu, D. Draga, C. Zhou, T. Su, C. Zou, Q. Gu, T. Lahm, Z. Zheng and Q. Qiu, Invest. Ophthalmol. Visual Sci., 2019, 60, 4215–4223 CrossRef CAS PubMed.
- Z. Zhou, C. Li, T. Bao, X. Zhao, W. Xiong, C. Luo, G. Yin and J. Fan, J. Neurotrauma, 2022, 39, 1057–1074 CrossRef PubMed.
- J. Shi, Y. Zhao, Y. Wang, W. Gao, J. Ding, P. Li, L. Hu and F. Shao, Nature, 2014, 514, 187–192 CrossRef CAS PubMed.
- J. Wang, X. Li, Y. Liu, C. Peng, H. Zhu, G. Tu, X. Yu and Z. Li, Front. Med., 2020, 7, 88 CrossRef PubMed.
- G. Gu, Y. Huo, G. Xu, L. Li, J. Yu, L. Sheng and Z. Yin, Int. Immunopharmacol., 2021, 91, 107227 CrossRef CAS PubMed.
- D. Zhao, M. Liu, J. Li, D. Xiao, S. Peng, Q. He, Y. Sun, Q. Li and Y. Lin, ACS Appl. Mater. Interfaces, 2021, 13, 29439–29449 CrossRef CAS PubMed.
- J. Zhu, M. Zhang, Y. Gao, X. Qin, T. Zhang, W. Cui, C. Mao, D. Xiao and Y. Lin, Signal Transduction Targeted Ther., 2020, 5, 120 CrossRef CAS PubMed.
- Y. Jiang, S. Li, T. Zhang, M. Zhang, Y. Chen, Y. Wu, Y. Liu, Z. Liu and Y. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 15069–15079 CrossRef CAS PubMed.
- X. Chen, J. He, Y. Xie, T. Zhang, S. Li, Y. Zhao, N. Hu and X. Cai, Cell Proliferation, 2023, e13424, DOI:10.1111/cpr.13424.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison 3rd and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- S. J. Dixon and B. R. Stockwell, Nat. Chem. Biol., 2014, 10, 9–17 CrossRef CAS PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascón, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS PubMed.
- S. Doll, F. P. Freitas, R. Shah, M. Aldrovandi, M. C. da Silva, I. Ingold, A. Goya Grocin, T. N. Xavier da Silva, E. Panzilius, C. H. Scheel, A. Mourão, K. Buday, M. Sato, J. Wanninger, T. Vignane, V. Mohana, M. Rehberg, A. Flatley, A. Schepers, A. Kurz, D. White, M. Sauer, M. Sattler, E. W. Tate, W. Schmitz, A. Schulze, V. O'Donnell, B. Proneth, G. M. Popowicz, D. A. Pratt, J. P. F. Angeli and M. Conrad, Nature, 2019, 575, 693–698 CrossRef CAS PubMed.
- V. A. N. Kraft, C. T. Bezjian, S. Pfeiffer, L. Ringelstetter, C. Müller, F. Zandkarimi, J. Merl-Pham, X. Bao, N. Anastasov, J. Kössl, S. Brandner, J. D. Daniels, P. Schmitt-Kopplin, S. M. Hauck, B. R. Stockwell, K. Hadian and J. A. Schick, ACS Cent. Sci., 2020, 6, 41–53 CrossRef CAS PubMed.
- K. Bersuker, J. M. Hendricks, Z. Li, L. Magtanong, B. Ford, P. H. Tang, M. A. Roberts, B. Tong, T. J. Maimone, R. Zoncu, M. C. Bassik, D. K. Nomura, S. J. Dixon and J. A. Olzmann, Nature, 2019, 575, 688–692 CrossRef CAS PubMed.
- K. Hadian and B. R. Stockwell, Cell, 2020, 181, 1188–1188 CrossRef CAS PubMed.
- R. Ou, S. Lu, L. Wang, Y. Wang, M. Lv, T. Li, Y. Xu, J. Lu and R. S. Ge, Front. Oncol., 2022, 12, 858598 CrossRef CAS PubMed.
- H. Zhang, H. Wu, J. Qian, L. Sun, L. Sang, P. Wang, B. Yuan and J. Zhang, Biochem. Biophys. Res. Commun., 2022, 604, 37–42 CrossRef CAS PubMed.
- S. Bin, L. Xin, Z. Lin, Z. Jinhua, G. Rui and Z. Xiang, Exp. Mol. Pathol., 2021, 118, 104570 CrossRef CAS PubMed.
- Y. Li, D. Pan, X. Wang, Z. Huo, X. Wu, J. Li, J. Cao, H. Xu, L. Du and B. Xu, Oxid. Med. Cell. Longevity, 2022, 2022, 4235126 Search PubMed.
- X. Li, L. Wu, X. Tian, W. Zheng, M. Yuan, X. Tian, H. Zuo, H. Song and Z. Shen, Oxid. Med. Cell. Longevity, 2022, 2022, 6520789 Search PubMed.
- H. Wang, X. Qiao, C. Zhang, J. Hou and S. Qi, Bioengineered, 2022, 13, 13070–13081 CrossRef CAS PubMed.
- Z. Xu, L. Chen, C. Wang, L. Zhang and W. Xu, Free Radical Res., 2022, 1–11, DOI:10.1080/10715762.2021.2024816.
- L. Cai, X. Hu, L. Ye, P. Bai, Y. Jie and K. Shu, Bioengineered, 2022, 13, 8226–8239 CrossRef CAS PubMed.
- S. Lin, Q. Zhang, S. Li, T. Zhang, L. Wang, X. Qin, M. Zhang, S. Shi and X. Cai, ACS Appl. Mater. Interfaces, 2020, 12, 11397–11408 CrossRef CAS PubMed.
- M. Zhang, J. Zhu, X. Qin, M. Zhou, X. Zhang, Y. Gao, T. Zhang, D. Xiao, W. Cui and X. Cai, ACS Appl. Mater. Interfaces, 2019, 11, 30631–30639 CrossRef CAS PubMed.
- Q. Zhang, S. Lin, S. Shi, T. Zhang, Q. Ma, T. Tian, T. Zhou, X. Cai and Y. Lin, ACS Appl. Mater. Interfaces, 2018, 10, 3421–3430 CrossRef CAS PubMed.
- W. S. Yang and B. R. Stockwell, Trends Cell Biol., 2016, 26, 165–176 CrossRef CAS PubMed.
- H. Zhang, T. Deng, R. Liu, T. Ning, H. Yang, D. Liu, Q. Zhang, D. Lin, S. Ge, M. Bai, X. Wang, L. Zhang, H. Li, Y. Yang, Z. Ji, H. Wang, G. Ying and Y. Ba, Mol. Cancer, 2020, 19, 43 CrossRef CAS PubMed.
- K. Tomita, T. Nagasawa, Y. Kuwahara, S. Torii, K. Igarashi, M. H. Roudkenar, A. M. Roushandeh, A. Kurimasa and T. Sato, Int. J. Mol. Sci., 2021, 22, 8300 CrossRef CAS PubMed.
- H. Li, D. Li, Z. Ma, Z. Qian, X. Kang, X. Jin, F. Li, X. Wang, Q. Chen, H. Sun and S. Wu, Autophagy, 2018, 14, 1726–1741 CrossRef CAS PubMed.
- S. C. Manolagas, Endocr. Rev., 2000, 21, 115–137 CAS.
- Y. Gao, S. Patil and J. Jia, Int. J. Mol. Sci., 2021, 22, 8182 CrossRef CAS PubMed.
- Y. J. Moon, C. Y. Yun, H. Choi, S. O. Ka, J. R. Kim, B. H. Park and E. S. Cho, Exp. Mol. Med., 2016, 48, e256 CrossRef CAS PubMed.
- M. Ponzetti and N. Rucci, Int. J. Mol. Sci., 2021, 22, 6651 CrossRef CAS PubMed.
- H. Li, H. Xie, W. Liu, R. Hu, B. Huang, Y. F. Tan, K. Xu, Z. F. Sheng, H. D. Zhou, X. P. Wu and X. H. Luo, J. Clin. Invest., 2009, 119, 3666–3677 CrossRef CAS PubMed.
- J. K. Lam, M. Y. Chow, Y. Zhang and S. W. Leung, Mol. Ther.–Nucleic Acids, 2015, 4, e252 CrossRef CAS PubMed.
- W. Ma, Y. Yang, J. Zhu, W. Jia, T. Zhang, Z. Liu, X. Chen and Y. Lin, Adv. Mater., 2022, 34, e2109609 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |