
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComposites based on nitroprusside cyano-bridged coordination polymer particles and chitosan for NO delivery†
Melvyn
Gorra
,
Frantz Ndebulia
Watchou
,
Maria A.
Palacios‡
 ,
Jérôme
Long
,
Saad
Sene
,
Gautier
Félix
,
Nathalie
Tanchoux
,
Françoise
Quignard
,
Joulia
Larionova
* and
Yannick
Guari
*
,
Jérôme
Long
,
Saad
Sene
,
Gautier
Félix
,
Nathalie
Tanchoux
,
Françoise
Quignard
,
Joulia
Larionova
* and
Yannick
Guari
*
ICGM, University Montpellier, CNRS, ENSCM, Montpellier, 34000, France. E-mail: joulia.larionova@umontpellier.fr; yannick.guari@umontpellier.fr
First published on 28th November 2022
Abstract
We report the design of new composite films {Fe[Fe(CN)5(NO)]}0.55@C6H11NO4 and {Ag2[Fe(CN)5(NO)]}0.28@C6H11NO4 based on cyano-bridged coordination polymers FeII[FeII(CN)5(NO)] or Ag2I[FeII(CN)5(NO)] grown into a chitosan matrix that are able to provide a slow NO radical release under white light irradiation. The characterisation methods such as X-ray diffraction, TGA, SEM-EDX, IR and UV-vis spectroscopies show that these composites are made of ca. 50 wt% of the cyano-bridged coordination polymer as microcrystalline particles embedded into chitosan films. Irradiation for 7 days by using white light results in initiating a 6.5 and 10.6% NO radical delivery for {Fe[Fe(CN)5(NO)]}0.55@C6H11NO4 and {Ag2[Fe(CN)5(NO)]}0.28@C6H11NO4, respectively. The rearrangement of the cyano-bridged coordination compounds after the NO departure leads to the formation of Prussian blue or silver Prussian blue analogues, which limits the unwanted release of cyanide as a side effect.
Introduction
Nitric oxide (NO) is a gaseous radical molecule possessing numerous biological functions including gene regulation, vasorelaxation, vascular permeability, bronchodilatation, platelet aggregation, angiogenesis, neuronal communication, hormone secretion, immune system regulation, inflammation and wound healing. Therefore, delivery of a controlled, quantifiable and biologically significant amount of NO is attractive for many in vitro and in vivo applications, such as curative,1–4 antithrombotic,5 anti-inflammatory, and antibiotic-based therapies.6–9 However, its short half-life (between 0.1 and 5 s in aqueous solutions), high chemical reactivity, rapid systemic clearance, instability under physiological conditions, lipophilicity and cytotoxicity make the direct delivery of nitric oxide using classical methods very difficult.10 Furthermore, for certain applications, the kinetics of NO delivery must be slow. This is particularly the case for the treatment of chronic wounds for which the inflammatory phase lasts too long to allow proper healing.11The employment of composite materials allowing the generation of NO in situ and therefore providing its controlled and local release (not systemic) has emerged as a unique strategy for the safe nitric oxide delivery reducing indeed unwanted side effects.10 These materials usually encapsulate organic NO-donor molecules, such as S-nitrosothiols12 and N-diazeniumdiolates, or nitrosylmetal complexes13 into the host matrixes.14 We can cite micro- and nano-scale materials, including functionalized polymers or dendrimers,15–17 silica nanoparticles,18,19 zeolites,20 gold clusters,21 or metal–organic frameworks,22,23 developed in the recent years, capable of producing and releasing NO at different rates and in different amounts. However, many of these employed organic NO donors suffer from carcinogenic or pro-inflammatory side products, which may limit their applicability.
An alternative nitric oxide source is well-known under the name of sodium nitroprusside with the chemical formula Na2[Fe(CN)5NO]·2H2O, for which the hypotensive action linked with the NO release has been demonstrated first in 1929. Indeed, the aqueous solution of this compound is widely used in the clinical practice to induce hypotension during surgery through a fine control of its infusion rate, which allows adjustment of the blood pressure in a very effective and fast response without overshoot.24 Similarly, nitroprusside is used in the treatment of chronic hypertension, in the management of myocardial infarction and other cardiac failures. It is capable of releasing NO enzymatically and non-enzymatically when in the presence of vascular tissue and reducing agents or activated by light irradiation with the quantity of 1 mole of NO per mole of nitroprusside.25 However, the employment of nitroprusside is restricted because the NO delivery is accompanied by compound decomposition inducing the liberation of free cyanides. Moreover, another important disadvantage of employing nitroprusside is its instability in aqueous solutions when exposed to light and therefore the impossibility of the slow and controlled NO delivery. In order to overcome this problem, several works have reported the encapsulation of nitroprusside into silica nanoparticles in order to protect this fragile compound and provide the in situ NO delivery in aqueous solutions.18,19
Another option to render nitroprusside more stable is via its integration as a building block in cyano-bridged coordination polymers of general formula Mx[FeII(CN)5(NO)] (where M = mono- (x = 2) or bivalent (x = 1) metal ions). These bulk compounds present a cubic structure (Fm3m space group) for M = bivalent transition metal ions26 and a monoclinic one (Pc space group) for M = monovalent ions.27 Recently, the photodegradation of these compounds in water under irradiation by white light has been investigated and a structural re-organisation to Prussian blue or its analogues occurred after irradiation and the NO departure has been demonstrated by means of infrared and Mössbauer spectroscopy.28 However, to the best of our knowledge, these coordination polymers have never been proposed as a source of NO and the kinetic monitoring of the NO release has never been investigated. Also note that the design of composite materials involving Mx[FeII(CN)5(NO)] coordination polymers has never been reported till date.
In this article, we describe the synthesis and characterisation of new composite materials based on coordination polymer particles, Mx[FeII(CN)5(NO)] (where M = Fe2+ or Ag+), embedded into chitosan polysaccharide films and their ability to slowly deliver NO under white light irradiation. Chitosan was chosen here as a matrix because it is a non-toxic, biocompatible, biodegradable and a bioactive polymer derived from the partial deacetylation of chitin, which is approved for use on human beings.29,30 Furthermore, it can be shaped in the form of beads or films, still exhibiting a porous structure when dried under supercritical CO2 conditions31 or as an alcogel.32 Thanks to the accessible free amino groups that are able to coordinate with metal ions, it was successfully used in the covalent anchoring of cyano-bridged coordination networks.33–38 We focus here on the growth of FeII[FeII(CN)5(NO)] and AgI2 [FeII(CN)5(NO)] particles into chitosan films because: (i) we expect that these particles will in situ produce the NO radicals under appropriate irradiation and (ii) we expect that NO departure will induce structural reorganisation and formation of biocompatible Prussian blue or Ag+-based Prussian blue analogue particles avoiding a huge CN− release unsuitable for biomedical applications. Indeed, in comparison with M[FeII(CN)5(NO)] analogous with M = Mn2+, Zn2+, Cd2+ and alkali metal ions, the cyano-bridged networks of M = Fe2+ and Ag+ are robust, insoluble in water and thus more resistant to degradation.28 Particular attention is paid to the kinetics of NO delivery during the light exposure in the solid state and in solution, as well as to the resulting rearrangement of the cyano-bridged coordination networks affording the formation of the corresponding Prussian blue or Ag+ Prussian blue analogues without significant leaching of cyanide.
Experimental
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 × g (20000 rpm) for 10 min. The supernatant was removed and the brown solid was washed twice with water and dried under vacuum.
700 × g (20000 rpm) for 10 min. The supernatant was removed and the brown solid was washed twice with water and dried under vacuum.
IR (cm−1): 3844 overtone ν(NO), ν(OH): 3659 and 3387, ν(CN):2182 and 2147, ν(NO): 1941, δ(OH): 1616, δ(Fe–NO): 666, ν(Fe–CN): 520, δ(Fe–CN): 443. TGA analysis: 30–150 °C (23.4 wt%), 240 °C (11.3 wt%), 301 °C (11.9 wt%), 343 °C (6.1 wt%), 520 °C (3 wt%); residual mass 55 wt%. SEM-EDS (at%): Na/Fe = 2/98, estimated formula Na0.04Fe[Fe(CN)5(NO)]. UV-Vis spectroscopy: wide absorption 230–700 nm, maximums at 260, 338, 465 nm. XRD (2θ, hkl): 17.25° (002), 24.40° (022), 30.01° (222), 34.78° (004), 39.01° (024), 42.89° (224), 49.92° (044), 53.20° (006) 56.36° (026); cubic structure, estimated cell parameter a = 10.35 Å.
IR (cm−1): 3854 overtone ν(NO),39ν(OH): 3443, ν(CN):2177 and 2163, ν(NO): 1937, δ(OH): 1625, δ(Fe–NO): 662, ν(Fe–CN): 516, δ(Fe–CN): 418. TGA (weight loss): 30–238 °C (15.1 wt%), 270 °C (4.5 wt%), 320 °C (8.1 wt%), 450 °C (10.3 wt%); residual mass 62 wt%. SEM-EDS (at%): Na/Ag/Fe = 0/67/33, estimated formula Ag2[Fe(CN)5(NO)]. UV-Vis spectroscopy: wide absorption 230–650 nm, maximums at 262, 300, 401, 534 nm. XRD (2θ, hkl): 13.23° (100), 13.84° (010), 16.18° (011), 18.50° (−1–11), 19.17° (110), 21.35° (−112), 21.61° (012), 24.51° (−202), 25.27° (102), 26.57° (200), 27.84° (020), 28.57° (013), 29.05° (021), 30.01° (−104), 30.95° (120), monoclinic structure, estimated cell parameters a = 7.4412(6) Å, b = 6.4141(1) Å, c = 11.8732(2) Å, β = 115.297(2)°.
IR (cm−1): 3838 overtone ν(NO),39ν(OH): 3431, ν(CH): 2917, ν(CN): 2182 and 2145, ν(NO): 1941, ν(CO): 1654, δ(NH): 1525, ν(CO): 1080, δ(Fe–NO): 664, ν(Fe–CN): 522, δ(Fe–CN): 431. SEM-EDX (at%): Na/Fe = 0/100. TGA (weight loss): 30–170 °C (9.48 wt%), 283 °C (47.4 wt%), 356 °C (18.1 wt%), 600 °C (3.6 wt%); estimated amount of particles in the composite film is 48 wt%. Estimated formula {Fe[Fe(CN)5(NO)]}0.55@C6H11NO4. UV-Vis spectroscopy: wide absorption 230–750 nm, maximums at 264, 376, 573 nm. XRD (2θ): 17.13°, 24.28°, 29.88°, 34.65°, 38.95°, 42.89°, 49.92°, 53.14°, 56.18°; cubic structure, estimated cell parameter a = 10.35 Å.
{Ag2[Fe(CN)5(NO)]}0.28@C6H11NO4 composite film 2@chit was obtained as a light pink film following the same method as described above for {Fe[Fe(CN)5(NO)]}0.55@C6H11NO4 by replacing FeCl2·4H2O/ascorbic acid by AgNO3 (1 × 10−2 M).
IR (cm−1): 3844 overtone ν(NO), ν(OH): 3410, ν(CH): 2920, ν(CN) = 2177 and 2141, ν(NO): 1924, ν(CO): 1644, δ(NH): 1543, ν(CO): 1053, δ(Fe–NO): 659, ν(Fe–CN): 507, δ(Fe–CN): 414. SEM-EDX (at%): Na/Ag/Fe = 0/66/34. TGA (weight loss): 30–228 °C (15.4 wt%), 285 °C (15.2 wt%), 358 °C (10.3 wt%), 412 °C (36.4 wt%); estimated amount of particles in the composite film 41 wt%.
Estimated formula {Ag2[Fe(CN)5(NO)]}0.28@C6H11NO4. UV-Vis spectroscopy: wide absorption 230–650 nm, maximums at 260, 379 nm. XRD (2θ): 13.82°, 19.13°, 20.88°, 24.09°, 26.72°, monoclinic structure, estimated cell parameters a = 7.483(6) Å, b = 6.299(2) Å, c = 12.669(6) Å, β = 114.77(3).
Materials and methods
All of the chemical reagents were used as received: chitosan (low molecular weight, deacetylation ≥75%, viscosity: 20–300 cps with 1% in 1% acetic acid, Aldrich), sodium nitroprusside dehydrate, SNP (Na2[Fe(CN)5NO]·2H2O 99%, Aldrich), silver nitrate (AgNO3, 99 + %, Alfa Aesar), sodium hydroxide (NaOH 97%, Honeywell), FeCl2·4H2O (98%, Alfa Aesar), ascorbic acid (99%, Aldrich), acetic acid (Aldrich), methanol (Carlo Erba), N-(1-naphthyl)ethylenediamine dehydrochloride (98%, Aldrich), and sulfanilamide (99%, Aldrich).Physico-chemical characterisation
Infrared spectra were recorded as KBr disks on a PerkinElmer Spectrum two spectrophotometer. UV-Vis spectra were collected on a JASCO V-650 spectrometer. Powder X-ray diffraction (PXRD) patterns were recorded in the 2θ interval 10–60° at room temperature using a PANalytical X’Pert Powder analytical diffractometer mounted in a Debye–Scherrer configuration and equipped with Cu radiation (λ = 1.5418 Å). Thermogravimetric analysis (TGA) was performed with a thermal analyser STA 409 Luxx (Netzsch) in the range of 25–800 °C at a heating speed of 2 °C min−1 under air flow (∼60 mL min−1). Scanning electronic microscopy coupled with energy dispersive X-ray spectroscopy (SEM-EDX) analyses were performed on a FEI Quanta FEG 200 instrument. The films were deposited on an adhesive carbon film and analysed under vacuum. The quantification of the heavy elements was carried out with the INCA software, with a dwell time of 3 μs.700 × g (12000 rpm, 10 min) and replaced with the same volume of fresh distilled water. The NO release from supernatants was then quantified using the Griess assay.40 A volume of supernatant was mixed with sulfanilamide (1 mM) and N-(1-naphthyl)ethylenediamine (1 mM) before being measured via UV-vis spectroscopy. The amount of the released NO was then determined using a calibration curve previously obtained using NaNO2 at concentrations ranging from 10 to 50 μM. All experimental releases were assessed over three experimental replicates and the error bars represent the mean standard error.
Results and discussion
Fig. 1 presents the step-by-step approach for the synthesis of nanocomposites {Mx[Fe(CN)5(NO)]}y@C6H11NO4 (with M = Ag+ or Fe2+) via the growth of the corresponding cyano-bridged coordination polymers by a sequential addition of the nitroprusside and the corresponding metal ion precursors in the presence of the chitosan film. Such a strategy was previously successfully used to design chitosan-based nanocomposite beads with Prussian blue analogue nanoparticles.33–38 As described in more details in the Experimental section, the elaboration of the cyano-bridged coordination polymer within the chitosan is performed via the addition of the metal ion Mn+ (Ag+ or Fe2+) to the –NH2 functionalities of chitosan and then Na2[Fe(CN)5NO]·2H2O (5 × 10−2 M) using methanol solutions. These successive additions were repeated 3 times with intermediate washings leading after a final air drying step to composites 1@chit and 2@chit as brown or light pink films, respectively, while the pristine chitosan film is white.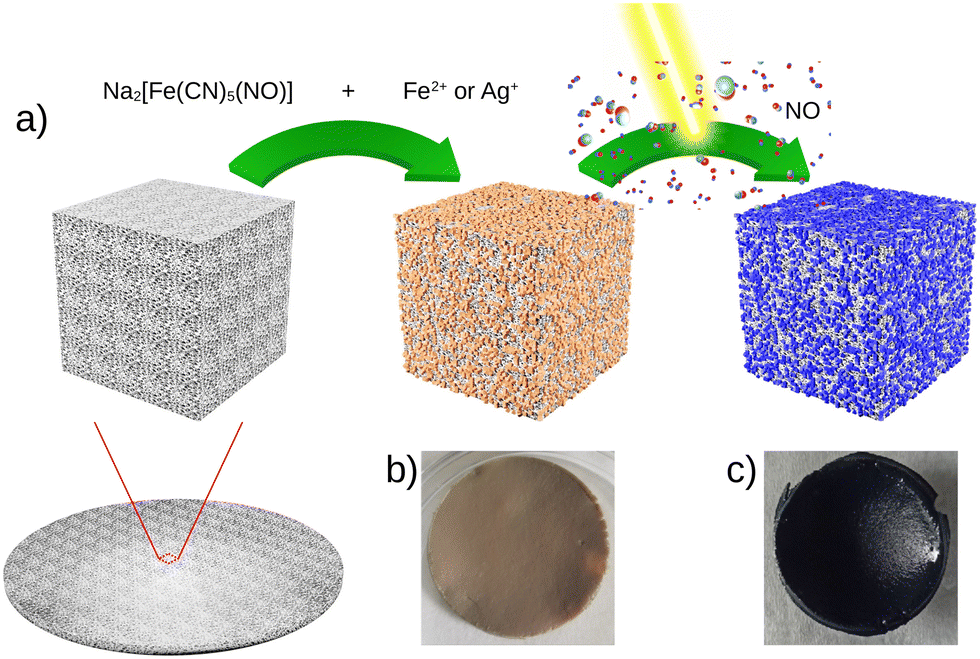 | ||
| Fig. 1 (a) Synthesis of the composite materials {Mx[Fe(CN)5(NO)]}y@C6H11NO4 with M = Fe2+1@chit or Ag+2@chit followed by light irradiation and NO release, and the corresponding photographs of the composite film 1@chit before (b) and after irradiation (c). | ||
The IR spectra were recorded for these composites especially in the spectral window of 1800–2400 cm−1, i.e. in the vicinity of the CN− and NO stretching modes, which are fingerprints of the structural and electronic changes occurring in cyano-bridged coordination polymers and nitroprusside. The IR spectrum of 1@chit shows a strong band at 2182 cm−1 ascribed to the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) stretching vibrations for the bridging (FeII–CN–FeII) mode of Fe[Fe(CN)5NO] with a shoulder at 2145 cm−1, which can be ascribed to the non-bridging cyanides (Fig. 2 and 7). This band can be also seen in the IR spectrum of the bulk analogue 1.41 Sample 2@chit exhibits a strong band at 2141 cm−1 with a shoulder at 2177 cm−1 ascribed to ν(CN) for the non-bridging and AgI–NC–FeII bridging modes, respectively (Fig. 2 and Fig. S1, ESI†).27 Note that the presence of bands ascribed to the non-bridging cyanides has already been observed in the case of Prussian blue analogue nanoparticles embedded in the chitosan matrix.33,36 The spectra of both composites show the characteristic ν(NO) band at 1937 and 1924 cm−1, respectively, for 1@chit and 2@chit (Fig. 2 and 7, Fig. S1, ESI†).39 The characteristic bands of the chitosan matrix are also present in these IR spectra beside the described bands of the coordination polymer particles, as expected.42
N) stretching vibrations for the bridging (FeII–CN–FeII) mode of Fe[Fe(CN)5NO] with a shoulder at 2145 cm−1, which can be ascribed to the non-bridging cyanides (Fig. 2 and 7). This band can be also seen in the IR spectrum of the bulk analogue 1.41 Sample 2@chit exhibits a strong band at 2141 cm−1 with a shoulder at 2177 cm−1 ascribed to ν(CN) for the non-bridging and AgI–NC–FeII bridging modes, respectively (Fig. 2 and Fig. S1, ESI†).27 Note that the presence of bands ascribed to the non-bridging cyanides has already been observed in the case of Prussian blue analogue nanoparticles embedded in the chitosan matrix.33,36 The spectra of both composites show the characteristic ν(NO) band at 1937 and 1924 cm−1, respectively, for 1@chit and 2@chit (Fig. 2 and 7, Fig. S1, ESI†).39 The characteristic bands of the chitosan matrix are also present in these IR spectra beside the described bands of the coordination polymer particles, as expected.42
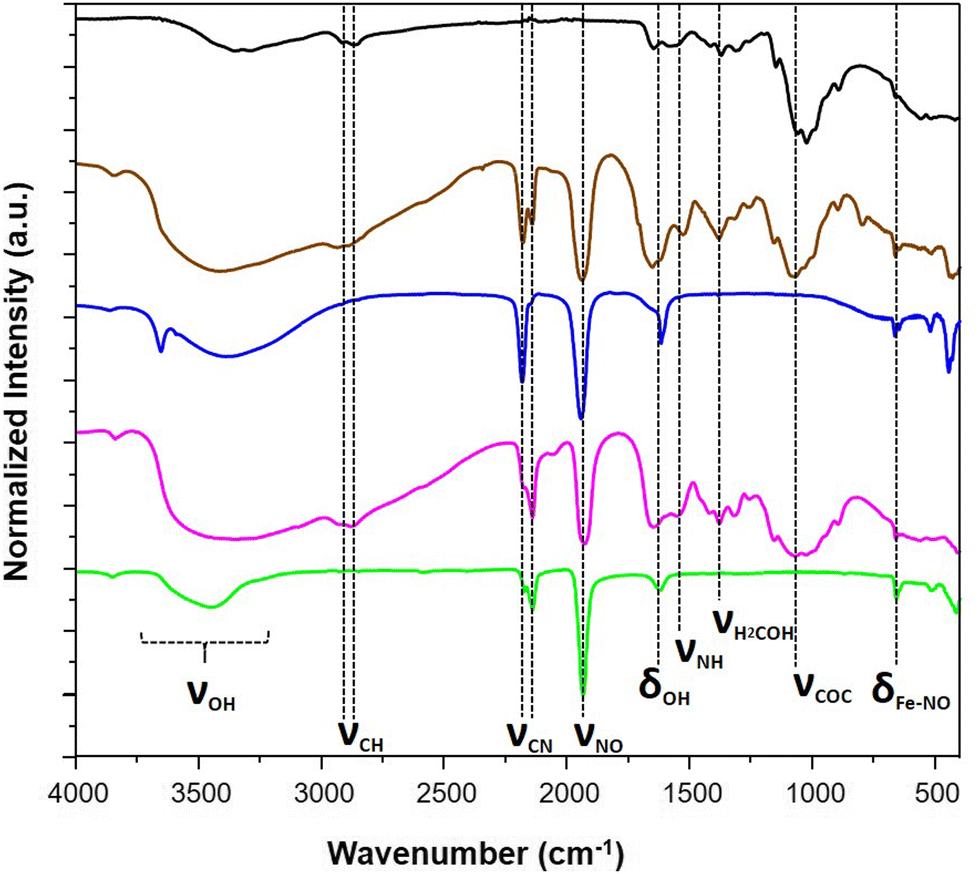 | ||
| Fig. 2 IR spectra of the pristine chitosan (black), 1@chit (brown), 1 (blue), 2@chit (purple) and 2 (green). | ||
The calculated coordination polymers loading from the elemental analysis are equal to 48 and 41 wt% for 1@chit and 2@chit, respectively. The resulting formulas obtained from an ensemble of elemental, TGA (Fig. S2 and S3, ESI†) and SEM-EDX analyses are {Fe[Fe(CN)5(NO)]}0.55@C6H11NO4 for 1@chit and {Ag2[Fe(CN)5(NO)]}0.28@C6H11NO4 for 2@chit. The powder X-ray diffraction (PXRD) pattern for 1@chit shows a cubic structure with an estimated cell parameter of a = 10.35 Å in accordance with the PXRD pattern of 1 (CCDC 1730892).43 Nanocomposite 2@chit shows a monoclinic structure with estimated cell parameters a = 7.483(6) Å, b = 6.299(2) Å, c = 12.669(6) Å, β = 114.77(3) in accordance with the PXRD pattern of 2 (CCDC 1056385) even though some discrepancies can be observed (Fig. 3).27 Such discrepancies can be attributed to the kinetics of formation of 2 which is much faster than that of 1 and to the fact that its synthesis is carried out on chitosan that can explain an imperfect crystallization of 2 within the chitosan matrix.
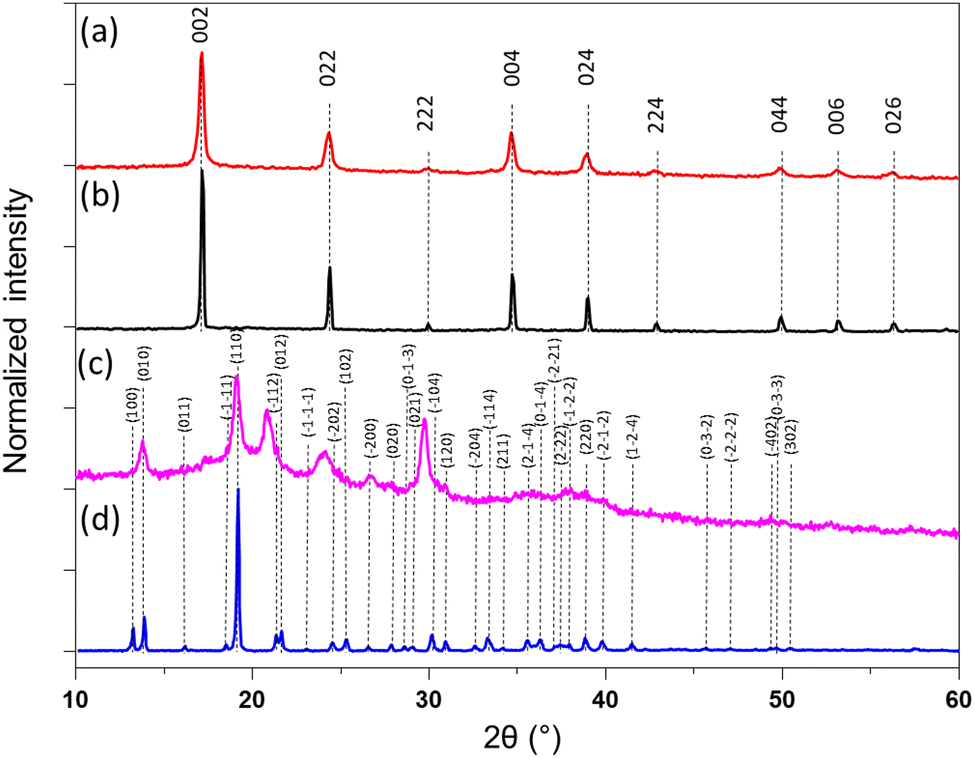 | ||
| Fig. 3 (a) PXRD pattern of 1@chit; (b) 1; (c) 2@chit; (d) 2. | ||
To obtain information about the morphological characteristics, SEM was performed on the obtained composite and the pristine chitosan films (Fig. 4, Fig. S4, ESI†). The images demonstrate the formation of the crystalline particles of a few μm in the chitosan. The homogeneity of the composite films 1@chit and 2@chit can be clearly seen using EDS mapping images of iron and silver atoms (Fig. 5) both on the surface and on the transversal cuts of the films. Both of them present a homogenous distribution of Fe or Fe and Ag atoms on the surface (Fig. 5a and b). Transversal cuts for 2@chit (Fig. 5d) indicate that the occurrence of silver and iron atoms is more evidenced in the first layer (ca. 0.15 mm) of the film's surface, indicating that the Ag2[Fe(CN)5NO] coordination polymer particles are more dense in that location probably due to the very fast formation kinetics of this compound. On the other hand, the homogenous distribution of iron and then the Fe[Fe(CN)5NO] particles in 1@chit can be observed in the whole width of the film (Fig. 5c).
 | ||
| Fig. 4 SEM top film images of the chitosan film surface (a) and its transversal cut (b); (b) the 1@chit film surface (c) and its transversal cut (d); the 2@chit film surface (e) and its transversal cut (f). | ||
 | ||
| Fig. 5 SEM and SEM-EDS atomic mapping for the 1@chit surface (a), its transversal cut (c) and the 2@chit surface (b), its transversal cut (d). Iron atoms are shown in purple and silver one in yellow. | ||
Previously, it has been demonstrated that the NO release for aqueous solutions of nitroprusside can be triggered either using endogenous (such as pH, glutathione or H2O2) or exogenous stimuli (such as light, X-ray or ultrasound).14 In the present study, the NO release was triggered by light irradiation in the visible region using white light from a simple desk lamp. The measurement of NO release was performed using 1@chit and 2@chit dispersed in water under irradiation using a white lamp (60 W) for a period of 7 days. At each time point, the amount of the formed NO was detected by the Griess assay in water at room temperature. This analysis has previously been used for detection of NO in other NO donor systems and relies on the aerobic fast conversion of NO to NO2−.44–46 The amount of cumulative NO release as a function of time (Fig. 6) indicates that the liberation of NO in water is rapid and almost linear within 1 day for both samples. After 1 day, samples continue to release NO, but much slowly. After 7 days, the saturation is not reached for 2@chit, suggesting that the sample will still slowly produce nitric oxide beyond 7 days.
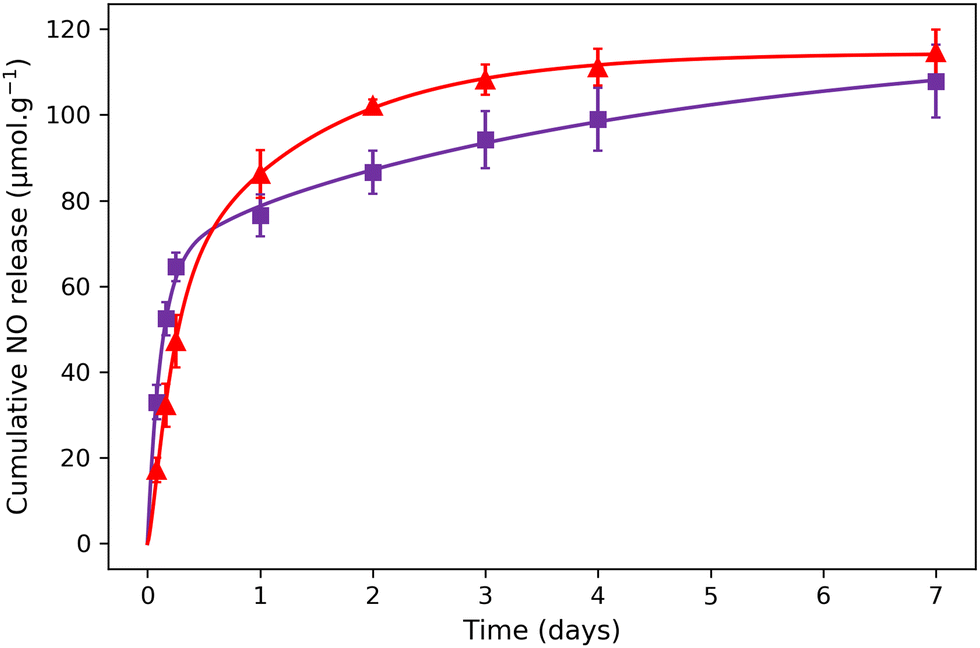 | ||
| Fig. 6 NO release experimental points for (▲) 1@chit and (■) 2@chit under white light irradiation. The solid lines represent the theoretical fit with the model described in the ESI.† | ||
The total cumulated amounts of nitrite of 114.5 ± 5.4 and 107.9 ± 8.5 μmol g−1 detected after 7 days correspond to 6.5% and 10.6% of the released NO for 1@chit and 2@chit, respectively (Fig. 6). The maximal calculated amounts of the available NO in these samples are 1770 μmol g−1 (for 1@chit) and 1021 μmol g−1 (for 2@chit) (Table 1).
| 1@chit | 2@chit | |
|---|---|---|
| NO release (μmol g−1) | 114.5 ± 5.4 | 107.9 ± 8.5 |
| Relative % of NO release | 6.5% | 10.6% |
| CN− release (μmol g−1) | 5.5 ± 1.4 | 202 ± 50.5 |
| Relative % of CN− release | 6.2 × 10−2% | 4.0% |
We show in Table 2 different parameters for different materials reported in the literature able to deliver NO. Entries 1–3 (entry 3 relates to the present work) are related to materials using nitroprusside as the NO source, while entries 4–7 are related to materials using N-diazoniumdiolate and entry 8 is related to a material using the ruthenium tetraammine nitrosyl complex. The NO release is triggered by light for materials containing nitroprusside and ruthenium tetraammine nitrosyl complex, while it is triggered through pH for materials containing N-diazoniumdiolate. Depending on the different materials and their compositions, the quantity of NO released varies strongly from 0.14 to 920 μmol g−1 with maximum delivery times varying from few hours (entries 2, 4–8) to almost 1 day (entries 1, 6). The results obtained in the present study (entry 3) are in the same order of magnitude in terms of the total quantity delivered to other studies carried out for the most efficient but with much slower (by a factor of ten) kinetic release (up to 7 days), which is particularly suited to issues related to the healing of chronic wounds (Table 2).18,19,21,44,47–49 In this perspective, it is also particularly interesting to note that for the materials containing nitroprusside, the amount of cyanide released is considerably lower in the present study (entry 3) compared with other nitroprusside-containing materials (entry 1). It should be noted that carboxymethyl chitosan/sodium alginate films loaded with S-nitrosoglutathione were shown to in vitro release NO for 168 h.50
| Entry | NO-donor | Matrix | Trigger | Total NO released, μmol g−1 | Relative % of NO release | Relative % of CN− release | Maximum delivery time (h) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The release of nitroprusside and not NO was measured in this study precluding straight comparison. b NO-release half-life. | ||||||||
| 1 | Nitroprusside | Mesoporous SiO2 nanoparticles bearing amine functions | Blue LED | 335 | 100 | 36 | 18 | 44 |
| 2 | Nitroprusside | Mesoporous SiO2 nanoparticles | —a | —a | —a | n.d. | 3 | 19 |
| 3 | Nitroprusside | Chitosan | White light | 107–114 | 6.5–10.6 | 0.062–4 | 168 | This work |
| 4 | N-Diazoniumdiolate | Mesoporous SiO2 nanoparticles bearing amine functions | pH | 2.78–920 | 5.8–51.7 | n.a. | 0.11–0.18 | 18 |
| 5 | N-Diazoniumdiolate | Au nanoparticles/tiopronin | pH | 0.14–40.4 | 2–33.4 | n.a. | 1.5–16 | 21 |
| 6 | N-Diazoniumdiolate | SiO2 nanoparticles/quaternary ammonium epoxides | pH | ca. 300 | 100 | n.a. | 24< | 48 |
| 7 | N-Diazoniumdiolate | Alkylamine-modified alginates | pH | 80–550 | 24–100 | n.a. | 4b | 47 |
| 8 | Ruthenium tetraammine nitrosyl complex | Silica gel/isonicotinamide | White light | 11.3 | 87 | n.a. | 1 | 49 |
The experimental NO release has been fitted using a diffusion model based on the Higuchi equation, which takes into account the formation of NO gas from the nitroprusside-containing cyano-bridged coordination polymers and then models the diffusion of NO molecules inside the chitosan matrix and their release in the solution (see the ESI† for details).51 In the model, the NO gas formation depends on the chemical coefficient rate kNO and the total number per unit of NO gas molecules, which can be released from the nitroprusside-containing cyano-bridged coordination polymers per mass unit of the latter CTOT-NO. The diffusion time of NO gas in the chitosan matrix depends on the diffusion coefficient D, which is a constant rate per surface unit depending on the sample physical-chemical properties. From D, we can define the diffusion flux J, which represents the quantity of NO molecules which passes through a surface unit (see the ESI†). For sample 1@chit, the obtained parameters gave: D = 12553 ± 687 μm2 days−1, CTOT-NO = 114.6 ± 0.8 μmol g−1 and kNO = 0.331 ± 0.014 hour−1. The experimental data for NO release for sample 2@chit present a different behaviour compared to sample 1@chit (see Fig. 6). As Fig. 5d shows a higher concentration of the cyano-bridged material on the surfaces for 2@chit, we adapted the model taking into account a difference of NO concentrations between the surface (CS-NO) and the volume (CTOT-NO–CS-NO). The obtained parameters gave: D = 9670 ± 5285 μm2 days−1, CTOT-NO = 119.4 ± 11.8 μmol g−1, CS-NO = 60.7 ± 4.4 μmol g−1 and kNO = 0.390 ± 0.053 hour−1. It is important to note that the release of NO at the surface is almost instantaneous and not linked to the diffusion coefficient D, which induces a high error in the D parameter obtained from the fit. These results indicate that the diffusion coefficient D and the coefficient rate kNO are in the same order of magnitude for both samples due to a similar matrix and the close physicochemical properties of the two nitroprusside-containing cyano-brigded coordination polymers. However, the release behaviour of NO within the composites is significantly different between both composites. The fitting of the NO release curve obtained from 1@chit does not imply to consider a surface source term, however this term is necessary to fit the NO release curve obtained from 2@chit. This is in agreement with the results obtained from SEM-EDS that shows a homogeneous distribution of the cyano-bridged coordination polymer in the chitosan film for 1@chit, while a preferential location of the cyano-bridged coordination polymer in the first layer near the surface is observed for 2@chit.
The measurement of CN− release for samples 1@chit and 2@chit in aqueous solutions after irradiation for 7 days (same conditions, as for NO release experiments) was realised by photometric titration. The cumulative quantities of the released cyanides are equal to 5.5 μmol g−1 for 1@chit and 202 μmol g−1 for 2@chit, while the expected CN− amounts in the hypothesis of their full release are 8850 μmol g−1 and 5105 μmol g−1. This corresponds to 6.2 × 10−2% and 4.0% of CN− leaching in the solution for 1@chit and 2@chit, respectively (Table 1). Note that in comparison with previously published works,44 such particularly low amount of released cyanides after NO departure in the case of composite film 1@chit indicates the mobilisation of cyanide ligands for the rearrangement to the more stable Prussian blue networks. This fact signifies that NO may be delivered without important side cyanide release, which is promising for future biomedical applications, such as wound healing.
After being exposed to white light for 7 days, the IR spectra of composites 1@chit and 2@chit were recorded in order to gather the information about the fate of the coordination polymers after irradiation and a partial NO departure. A new large band at 2070 cm−1 clearly appeared for 1@chit after irradiation (Fig. 7), as it can also be observed for 1 (Fig. S5, ESI†). It can be attributed to the stretching vibration of the bridging cyanides in the FeII–CN–FeIII linkage mode attesting the formation of the Prussian blue compound.52 Indeed, the release of NO is accompanied by a partial oxidation of Fe2+ to Fe3+ ions affording very stable Prussian blue. Note also that the relative intensity of the ν(NO) band at 1939 cm−1 notably decreases for 1 after irradiation. This is perfectly coherent with the results obtained by others after a white light irradiation of the respective bulk coordination polymer 1.28 Moreover, the absorption spectrum in the visible region of 1@chit after irradiation demonstrates the appearance of a broad band typical for Prussian blue at ca. 730 nm occurring due to the Fe3+ to Fe2+ electron transfer (Fig. S6, ESI†).53
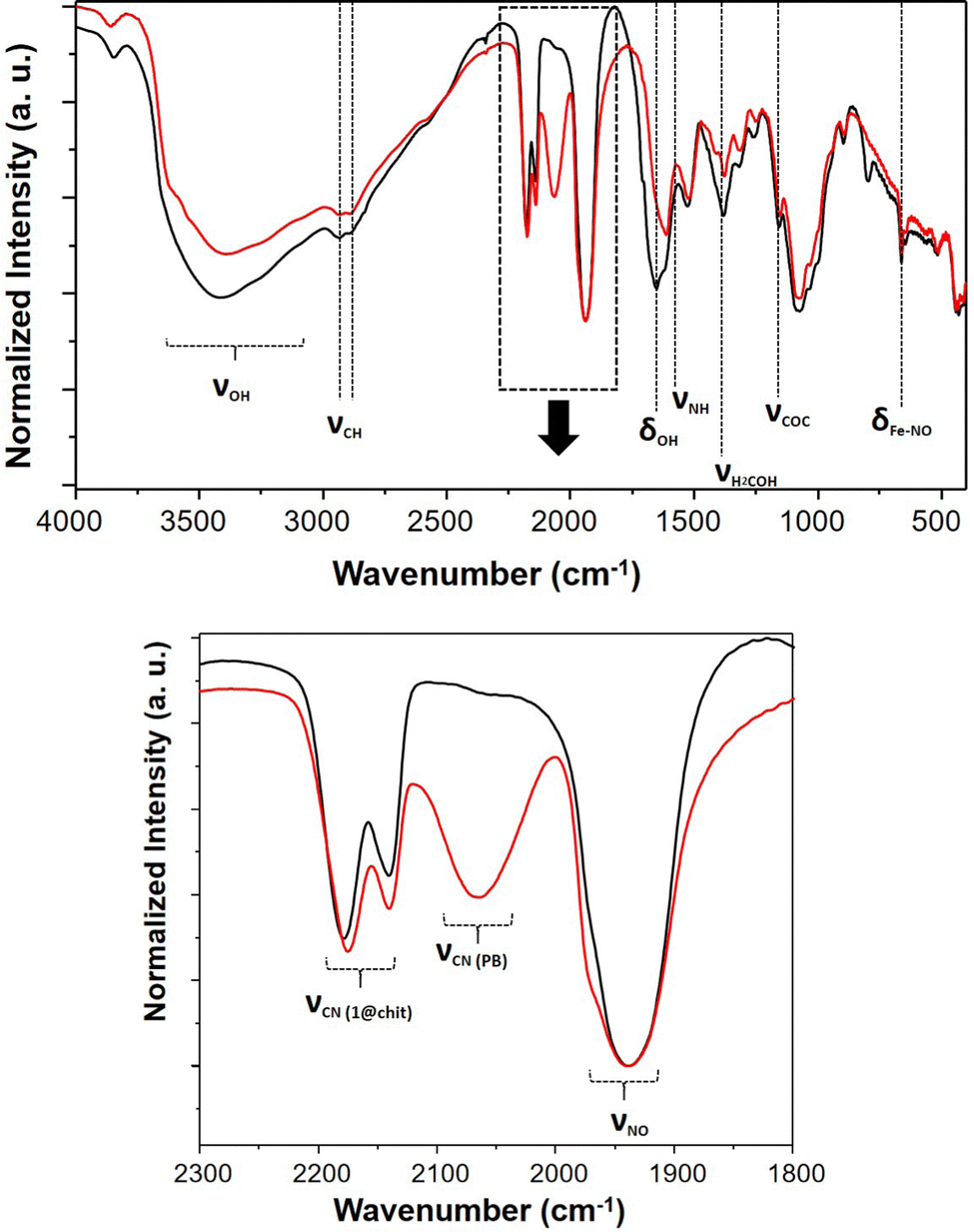 | ||
| Fig. 7 IR spectra of 1@chit before (black) and after (red) NO release; (up) spectral window 4000–400 cm−1, (down) spectral window 2300–1800 cm−1. | ||
For the composite film 2@chit, a new band appears at ca. 2055 cm−1 in the IR spectrum, which corresponds to the stretching vibration of the cyanide groups in the bridging mode FeII–CN–AgI due to the formation of the Prussian blue analogue Ag4[Fe(CN)6] (Fig. S1, ESI†).54 Note that in this case, no Fe2+ to Fe3+ oxidation occurs since ν(CN) for the FeIII–CN–AgI mode is expected at 2092 cm−1.55 Interestingly, the related bulk compound 2 does not release NO upon irradiation and no new bands have been observed in its IR spectrum taken after irradiation under similar conditions. Thus, for both nanocomposites, the NO departure is accompanied with a self-restructuration of the material leading to the formation of the corresponding cyano-bridged coordination polymers limiting the concomitant release of cyanide. However, this reaction seems more efficient for 1@chit than 2@chit, as the amount of cyanide release is strongly limited for the former.
Conclusions
To summarize, in this work, we showed that cyano-bridged coordination polymer particles FeII[FeII(CN)5(NO)] or Ag2I[FeII(CN)5(NO)] can be grown by the step-by-step coordination of Fe2+ or Ag+ and [Fe(CN)5(NO)]2− precursors inside the chitosan film as a matrix. Their morphological and structural characterisation demonstrated that those composite materials are made of ca. 50–50 wt% of the cyano-bridged coordination polymer and chitosan, the formers being present as microcrystalline components. Note that while FeII[FeII(CN)5(NO)] is shown to be homogeneously distributed in the chitosan film, the AgI2 [FeII(CN)5(NO)]-containing film exhibits a preferential location of the particles in the first layer near the surface of the chitosan film. Continuous irradiation of both nanocomposites using a desk lamp in aqueous media initiates the slow NO release, which was measured during the experimentation time. It indicates a maximum amount of 6.5 and 10.6% released 7 days after irradiation for 1@chit and 2@chit, respectively. Modelling of the NO release for 1@chit can be obtained satisfactorily without considering a specific contribution of the surface, while for 2@chit, the surface contribution has to be taken into account. This is in agreement with a higher amount of the cyano-bridged coordination material on the surfaces of the chitosan film for 2@chit. The infrared and absorption spectroscopies performed before and after irradiation demonstrated that a structural rearrangement of the cyano-bridged coordination polymers leading to the formation of Prussian blue or the Prussian blue analogue Ag2[Fe(CN)6] particles has occurred after NO departure. Such a self-structuring of the composite associated with chitosan allows the concomitant release of cyanides to be limited, which is shown to be particularly efficient in the case of 1@chit. Such a strategy allowing the successful release of NO from nitroprusside-based composites while limiting the release of cyanide opens interesting opportunities for different potential applications where delivery of small quantities of NO for a long period of time is needed without side effects linked to cyanide.Author contributions
Conceptualization: N. T., F. Q., J. La., and Y. G.; data curation: M. G., N. W. M. A. P., and G. F.; formal analysis: G. F.; funding acquisition: Y. G.; investigation: M. G., N. W. M. A. P., S. S., and G. F.; methodology: G. F.; project administration: J. La. and Y. G.; resources: S. S.; software: G. F. supervision: N. T., F. Q., J. La., and Y. G.; validation: M. G., N. W. M. A. P., and S. S.; writing – original draft: S. S., G. F., and Y. G.; writing – review & editing: N. T., J. La., J. Lo, and Y. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Institut Carnot Chimie Balard Cirimat and SATT AxLR for financial support.Notes and references
- S. P. Nichols, et al. , Adv. Drug Delivery Rev., 2012, 64, 1177–1188 CrossRef PubMed.
- C. Szabo, Nat. Rev. Drug Discovery, 2016, 15, 185–203 CrossRef PubMed.
- S. Korde Choudhari, et al. , World J. Surg. Oncol., 2013, 11, 118 CrossRef PubMed.
- A. P. Veith, et al. , Adv. Drug Delivery Rev., 2019, 146, 97–125 CrossRef.
- D. A. Popowich, et al. , Vascular, 2007, 15, 324–335 CrossRef.
- D. O. Schairer, et al. , Virulence, 2012, 3, 271–279 CrossRef PubMed.
- R. Y. Pelgrift, et al. , Adv. Drug Delivery Rev., 2013, 65, 1803–1815 CrossRef PubMed.
- X. Zhu, et al. , Nano Today, 2014, 9, 478–498 CrossRef.
- A. Simchi, et al. , Nanomedicine, 2011, 7, 22–39 CrossRef.
- Y. Yang, et al. , Biosurf. Biotribol., 2015, 1, 177–201 CrossRef.
- R. Ahmed, et al. , Biomed. Pharmacother., 2022, 149, 112707 CrossRef PubMed.
- J. L. Harding, et al. , J. Mater. Chem. B, 2014, 2, 2530–2536 RSC.
- L. Tan, et al. , Colloids Surf., B, 2021, 199, 111508 CrossRef PubMed.
- W. Fan, et al. , Angew. Chem., Int. Ed., 2018, 57, 8383–8394 CrossRef.
- G. Jin, et al. , Adv. Healthcare Mater., 2021, 10, 2001550 CrossRef.
- J. Cheng, et al. , Front. Chem., 2019, 7, 530 CrossRef PubMed.
- D. A. Riccio, et al. , Chem. Soc. Rev., 2012, 41, 3731–3741 RSC.
- J. H. Shin, et al. , J. Am. Chem. Soc., 2007, 129, 4612–4619 CrossRef PubMed.
- A. Farooq, et al. , J. Colloid Interface Sci., 2016, 478, 127–135 CrossRef PubMed.
- A. C. McKinlay, et al. , J. Am. Chem. Soc., 2008, 130, 10440–10444 CrossRef PubMed.
- M. A. Polizzi, et al. , Langmuir, 2007, 23, 4938–4943 CrossRef PubMed.
- R. V. Pinto, et al. , Angew. Chem., Int. Ed., 2020, 59, 5135–5143 CrossRef.
- A. C. McKinlay, et al. , Chem. Mater., 2013, 25, 1592–1599 CrossRef.
- A. R. Butler, et al. , Chem. Soc. Rev., 1987, 16, 361–380 RSC.
- P. Coppens, et al. , Chem. Rev., 2002, 102, 861–884 CrossRef PubMed.
- A. Gómez, et al. , Powder Diffr., 2007, 22, 27–34 CrossRef.
- J. Rodríguez-Hernández, et al. , Inorg. Chim. Acta, 2015, 428, 51–56 CrossRef.
- P. M. Crespo, et al. , J. Photochem. Photobiol., A, 2021, 412, 113244 CrossRef.
- G. A. F. Roberts, in Chitin Chemistry, ed. M. E., UK, London, 1992, pp. 54–84 Search PubMed.
- M. N. V. R. Kumar, et al. , Chem. Rev., 2004, 104, 6017–6084 CrossRef PubMed.
- R. Valentin, et al. , New J. Chem., 2003, 27, 1690–1692 RSC.
- F. Quignard, et al. , New J. Chem., 2008, 32, 1300–1310 RSC.
- Y. Guari, et al. , Chem. Commun., 2006, 2613–2615, 10.1039/B602460B,.
- J. Larionova, et al. , Angew. Chem., Int. Ed., 2008, 47, 8236–8240 CrossRef.
- Y. Guari, et al. , Dalton Trans., 2008, 3658–3660, 10.1039/B808221A,.
- B. Folch, et al. , Phys. Chem. Chem. Phys., 2010, 12, 12760–12770 RSC.
- E. Chelebaeva, et al. , Nanoscale, 2011, 3, 1200–1210 RSC.
- A. Tokarev, et al. , New J. Chem., 2013, 37, 3420–3432 RSC.
- D. F. Mullica, et al. , J. Crystallogr. Spectrosc. Res., 1991, 21, 81–85 CrossRef.
- D. Giustarini, et al., Methods in Enzymology, Academic Press, 2008, vol. 440, pp. 361–380 Search PubMed.
- J. Balmaseda, et al. , J. Phys. Chem. B, 2003, 107, 11360–11369 CrossRef.
- I. F. M. Rumengan, et al. , IOP Conf. Ser.: Earth Environ. Sci., 2017, 89, 012028 CrossRef.
- E. Reguera, et al. , Hyperfine Interact., 1993, 77, 1–10 CrossRef.
- P. M. Silva Filho, et al. , Mol. Pharmaceutics, 2019, 16, 2912–2921 CrossRef CAS.
- X. Zhou, et al. , Acta Biomater., 2017, 54, 128–137 CrossRef CAS.
- T. Feng, et al. , Biomaterials, 2019, 214, 119213 CrossRef CAS.
- M. J. R. Ahonen, et al. , Biomacromolecules, 2018, 19, 1189–1197 CrossRef CAS PubMed.
- A. W. Carpenter, et al. , Biomacromolecules, 2012, 13, 3334–3342 CrossRef CAS.
- F. G. Doro, et al. , J. Colloid Interface Sci., 2007, 307, 405–417 CrossRef PubMed.
- K. Razmjooee, et al. , Biomed. Mater., 2022, 17, 055013 CrossRef PubMed.
- J. Siepmann, et al. , Int. J. Pharm., 2011, 418, 6–12 CrossRef PubMed.
- S. N. Ghosh, J. Inorg. Nucl. Chem., 1974, 36, 2465–2466 CrossRef.
- R. Koncki, et al. , Anal. Chem., 1998, 70, 2544–2550 CrossRef PubMed.
- S. Sharma, et al. , Mater. Sci. Eng., C, 2020, 113, 110982 CrossRef CAS PubMed.
- S. Mukherjee, et al. , ACS Biomater. Sci. Eng., 2020, 6, 690–704 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj04711j |
| ‡ Current address: Departamento de Química Inorgánica, Facultad de Ciencias, Universidad de Granada, 18071 Granada, Spain. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |