
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChirality variation from self-assembly on Ullmann coupling for the DBCh adsorbate on Au(111) and Ag(111)†
Hongbing
Wang
ab,
Jinping
Hu
ab,
Zhaofeng
Liang
*c,
Huan
Zhang
ab,
Chaoqin
Huang
ab,
Lei
Xie
c,
Zheng
Jiang
 abc,
Han
Huang
d and
Fei
Song
*abc
abc,
Han
Huang
d and
Fei
Song
*abc
aShanghai Institute of Applied Physics, Zhangheng Road 239, Shanghai, 201000, China. E-mail: songfei@sinap.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100000, China
cShanghai Advanced Research Institute, Zhangheng Road 239, Shanghai, 201004, China. E-mail: liangzf@sari.ac.cn
dSchool of Physics and Electronics, Central South University, Changsha, 410073, China
First published on 26th January 2023
Abstract
On-surface Ullmann coupling has been considered an appealing approach for the precise fabrication of carbon-based covalent nanostructures under solution-free conditions. However, chirality has seldom been discussed in Ullmann reactions. In this report, self-assembled two-dimensional chiral networks are initially constructed in a large area on Au(111) and Ag(111) after adsorption of the prochiral precursor, 6,12-dibromochrysene (DBCh). Self-assembled phases are then transformed into organometallic (OM) oligomers after debromination, preserving the chirality; in particular, the formation of scarcely reported OM species on Au(111) is discovered herein. With the aryl–aryl bonding induced after intensive annealing, covalent chains are fabricated via the cyclodehydrogenation between chrysene blocks, resulting in the formation of 8-armchair graphene nanoribbons with staggered valleys on both sides. Before chiral polymer chains are constructed by chrysene blocks, the high structural flexibility of OM intermediates on Ag(111) is also revealed during reactions, which is derived from the twofold coordination of Ag atoms and conformationally flexible metal–carbon bonding. Our report not only provides solid evidence of atomically precise fabrication of covalent nanostructures with a feasible bottom-up approach but also sheds insights into the comprehensive investigation of chirality variation from monomers to artificial architectures via surface coupling reactions.
Introduction
Feasible fabrication of artificial low-dimensional architectures has been intensively investigated in the surface science field during the past decade considering their appealing applications in nanoelectronics, materials science, environmental science, etc.1–4 In this regard, on-surface synthesis emerges as a promising strategy towards the atomically precise construction of low-dimensional nanostructures in the framework of a bottom-up strategy.5–8 In particular, on-surface Ullmann coupling is revealed to be a controllable strategy to fabricate tailored covalent nanostructures on metal surfaces with well-defined width and edge structures based on specifically designed precursors in an ultrahigh vacuum (UHV) environment.9–16 As one of the key issues in molecular self-assembly, chirality has been elaborately investigated in the formation of versatile chiral nanostructures induced either by mirror-symmetry disruption upon adsorption of achiral molecules or preservation of molecular adsorbate chirality from prochiral monomers.17–22 However, the variation or preservation of adsorbate chirality during on-surface Ullmann coupling reactions is still less explored.With efforts devoted to the intrinsic prochirality of precursors, the structural transformation from prochiral reactants to advanced chiral polymers has been addressed through a sequence of single-molecule reactions,23–26 shedding new insights into on-surface chemistry.27,28 It is thus interesting to explore the crucial role of molecular prochirality and adsorbate chirality to on-surface Ullmann couplings with single-molecule-resolution imaging by monitoring variations of adsorbate chirality across polymerization reactions. On the other hand, it has been commonly recognized that metal surfaces possessing varying catalytic activities have significant influence on both the self-assembly and on-surface reactions.29–31 In this context, the chiral adsorption of precursors with rich enantiomers on different metal substrates is quite meaningful to unravel the variation of adsorption chirality across Ullmann couplings.
Within this context, 6,12-dibromochrysene (DBCh, Scheme 1) with the planar chirality, has been selected as the precursor to investigate the molecular chiral adsorption and chirality variation during surface Ullmann coupling on different metal substrates, namely Au(111) and Ag(111). Although a previous report has demonstrated the chiral adsorption of DBCh and the chiral arrangement of polymers on Au(111) and Cu(111),32 the process of chirality variation and preservation during reactions is still unclear due to the limited resolution of images. Recently, Cai et al. reported the formation of a chiral domain on Au(111) and one-dimensional metal–ligand chiral chains on Cu(111) with high-resolution scanning tunnelling microscopy (STM) and non-contact atomic force microscopy,33,34 and investigated the related electronic structures. However, the issue of chirality variation and structural variability of organometallic (OM) intermediates and polymers remains vague. As known, the activity of silver is between Au and Cu; Ag(111) is thus utilized, in addition to Au(111), to thoroughly explore the chiral molecular adsorbate and chirality variations during Ullmann coupling in this report. First, the chiral adsorption of DBCh with a hexagonal lattice is revealed on both Au(111) and Ag(111). Afterwards, transformation into chiral OM intermediates is revealed, including OM dimers on Au(111) and oligomers with flexible structures on Ag(111), which have not been properly addressed before and shall be attributed to the chiral adsorption of DBCh on the Au substrate and the flexible coordination of Ag, respectively. In the end, chiral nanoribbons (GNR) with alternating valleys on opposite sites are fabricated after overcoming the steric hindrance. Inspired by the high-resolution STM characterization and density functional theory (DFT) calculations, intermediate states with varied chirality during the reactions are clearly identified towards the fundamental understanding of chirality variation across Ullmann coupling.
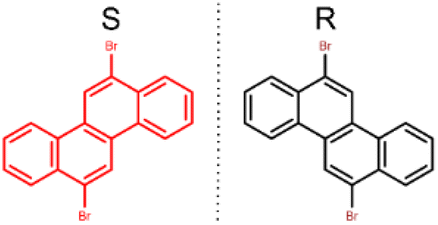 | ||
| Scheme 1 6,12-Dibromochrysene (DBCh) with mirror (S and R) enantiomers. | ||
Results and discussion
Self-assembly of DBCh on Au(111)
Generally, DBCh with Br substitution at the meta-position is prochiral and becomes chiral after adsorption on the solid surface. Following the deposition of 0.1 monolayer (ML) DBCh onto Au(111) kept at room temperature (RT), the hexagonal pattern is immediately formed with the nanopore embedded in the centre, as seen in Fig. 1a, which in principle is accommodated on the fcc sites of the Au(111) reconstruction and is analogous to the previous observation of DBCh on Au(111).33 Sequential deposition of 0.3 ML DBCh in total (Fig. 1b) results in the extension of the hexagonal domain along the high-symmetry direction of the substrate but still localized inside the fcc area. When the coverage is further increased to about 0.6 ML, as illustrated in Fig. 1c, the porous structure starts to cross over the herringbone line and grows into the hcp sites. Intriguingly, distinctive features are distinguished: the hexamer in the upper part of Fig. 1c is flexible and gets extended in accordance with the herringbone reconstruction of Au(111) (indicated by the dashed curve), however, the hexagonal domain in the lower part can hardly get bent around the elbow site, but instead, it steps over the reconstruction pattern as indicated by the straight dash line in Fig. 1c. Details of these two features are elaborately illustrated in Fig. 1d and e with molecule-resolved topography. While each DBCh molecule is imaged with two bright dots on both sides of the chrysene block due to the high density of states of Br atoms,35 the homochiral characteristic is embedded in the nanoporous structure of the self-assembled DBCh, namely, the clockwise arrangement (CW) in Fig. 1d and the anticlockwise (ACW) pattern in Fig. 1e. | ||
| Fig. 1 STM topography of self-assembled DBCh on Au(111) with the coverage of (a) 0.1 ML, (b) 0.3 ML, (c) 0.6 ML, respectively, revealing the tessellation process of the porous structure. (d) The typical hexagonal pattern of DBCh on Au(111) with the single-molecule resolution and (e) the mirror phase induced by the chiral adsorption. Molecular models are overlaid atop for a clear demonstration. (f) A tentative model for the CW chiral arrangement of trimeric DBCh on Au(111) stabilized by the halogen bonding motifs (highlighted with dashed lines). Scanning parameters: Ubias = −1.3 V, Itunneling (t) = 0.1 nA. | ||
As already mentioned, the ACW adsorption of DBCh enantiomers is rigid at the elbow site of reconstruction lines, as clearly confirmed in Fig. S1a in the ESI.† In contrast, the CW phase in Fig. 1d gets extended with flexibility at fcc sites of the herringbone reconstruction (see also in Fig. S1b in ESI†). Meanwhile, it can also be identified in Fig. 1d and e that the homochiral CW and ACW domains are constructed in order by intact DBCh building blocks with either the S or R adsorption chirality and are supposed to be stabilized mostly by the halogen bonding motifs in a triangular form as marked with dashed lines (the halogen bonding length is revealed to be about 0.33 ± 0.02 nm from statistics). It can thus be concluded that the as-formed nanostructure with the X3-synthon geometry36 is employed as the basic unit towards the fabrication of an extended porous network. Correspondingly, the tentative structural model is proposed in Fig. 1f for the CW domain (the model for the ACW phase is also shown in Fig. S1c in ESI†), highlighting the bonding motifs, which mediate the homochiral domains.
In fact, chains with limited length are also formed in connection to porous structures at the sub-monolayer coverage, as observed in Fig. 1b and c. Detailed information is shown in Fig. S1d and e, respectively, in ESI,† where precursors are closely packed with the intermolecular hydrogen and halogen bonding motifs. More intriguingly, R and S adsorbed enantiomers can also meet each other in the same chain, as presented in Fig. S1d.† Subsequently, once the coverage is increased to about 1 ML as presented in Fig. 2a, a two-dimensional (2D) porous network is ultimately established by expanding homochiral hexagonal domains, and regular nanopores are resolved within the network. Since the herringbone reconstruction can still be recognized at the full coverage, indicating the weak interaction between chiral adsorbates and substrate, the 2D hexagonal network is thus mediated by the delicate balance between the molecule–substrate (vdW force) and intermolecular (the halogen and hydrogen bonding) interactions. As inspired by the high-resolution STM in Fig. 2b, six DBCh molecules are tessellated into a hexagonal structure on the basis of the trimeric synthon, with a pore-to-pore distance of about 2.08 ± 0.02 nm (highlighted by the white rhomb). To verify the stability of homochiral domains, as an example, structural optimization of the CW arrangement has been performed with DFT calculations. As illustrated in Fig. 2c of the simulated STM hybridized with the model structure, the predicted adsorption configuration is, in general, consistent with the experimental observation, including topography (density of states) and lattice parameters. For instance, the periodicity of the CW domain is revealed to be 2.10 nm while the experimental value is 2.08 nm, and Br terminals are found to be interacting with neighbours in the form of X3 synthon geometry with the halogen bonding distance of 0.36 nm (0.33 nm from experiments). Nevertheless, the ACW homochiral phase is also discovered at the full monolayer coverage, which is presented in Fig. S1f in ESI.† In practice, the occurrence of CW and ACW phase is roughly equal from statistics, although they are locally separated from each other.
 | ||
| Fig. 2 (a) Increasing coverage to 1 ML leading to the linkage of hexagonal structure into 2D network in a large area, and (b) the high-resolution STM with the unit cell indicated. (c) The optimized configuration by DFT overlaid on top of the simulated STM image. Ubias = −1.3 V for (a) and 1.3 V for (b); It = 0.1 nA. | ||
Ullmann coupling of DBCh on the Au(111) surface
To initiate the Ullmann coupling of DBCh on Au(111), thermal annealing to 373 K is applied at beginning. Immediately, the pristine porous network on Au(111) collapses as observed in Fig. 3a. Meanwhile, it is interesting to discover the zig-zag structure with bright dots in the middle (highlighted by the dashed circle). | ||
| Fig. 3 Chirality variation in the dehalogenation of DBCh on Au(111). (a) Homochiral hexagonal phases are removed after annealing to 373 K with the formation of zig-zag species. A tentative model is also presented. Yellow circles: Au adatoms. (b) Zig-zag chains with considerable length are formed after further annealing to 423 K. (c) High-resolution STM of (b) suggests the configuration of covalent chains with the hindrance effect. DFT optimized structure with chirality is overlaid atop (both top and side views). (d) Sequential annealing to 573 K leads to the fabrication of planar chiral chains after cyclodehydrogenation. (e) and (f) Intensive annealing to 623 K and 673 K, respectively, indicating the robustness of ribbons with the armchair edge on Au(111). Ubias = −1.3 V for (a)–(d), Ubias = −1.5 V for (e) and (f); It = 0.1 nA. | ||
The new structure might be assigned to OM oligomers consisting of debrominated molecules with the same adsorption chirality (S) and Au adatoms in between,34 and a tentative model is overlaid on top of STM in Fig. 3a (yellow circle: Au atoms), which is thus expected to be chiral. Furthermore, the OM dimer is also discovered inside the self-assembled structure, as presented in Fig. S2a in ESI,† showing the structural transition from self-assembly to OM species. These observations suggest that OM oligomers with chiral adsorption can indeed be induced on Au(111), while OM intermediate states have been seldom reported in previous reports, suggesting the critical role of molecular chiral adsorbate towards the formation of OM species on Au(111) by capturing or confining Au adatoms in between.
Sequential annealing to 473 K induces the formation of linear oligomer chains with a large area, and alternative protrusions are identified on both sides of chains, as shown in Fig. 3b. The high-resolution STM in Fig. 3c undoubtedly shows that the protrusion actually consists of two parts with the opposite appearance: one part is imaged bright while the other one is relatively dim. Thus, one can conclude that while the covalent C–C bonding is formed between completely dehalogenated monomers, oligomer chains are not planar and apparently bear the chirality. As indicated by the structural model of covalent oligomers (both the side view and top view) overlaid atop in Fig. 3c, the steric hindrance between hydrogen atoms of neighbouring chrysene blocks results in the opposite tilting of consecutive units with respect to the substrate,34 which is consistent with the alternative bright and dim features in STM topography. Similar structures could also be found on the surface and are presented in Fig. S2b in ESI.† Subsequently, annealing to 573 K induces apparent changes to oligomer chains, as discovered in Fig. 3d. For instance, the previous feature with alternative brightness is missing, which might be assigned to the cyclodehydrogenation between neighbouring blocks by overcoming the steric hindrance and the corresponding formation of planar chains. The zoom-in view inset shows that the polymer chain is constructed by the alternating arrangement of S and R chrysene units with the armchair edge. Further annealing to 623 K makes the polymer chain clearly recognized with alternating valleys on the opposite site, namely, the N = 8 armchair graphene nanoribbon (8AGNR), as seen in Fig. 3e and S2c in ESI.† The DFT-optimized structure of the 1D chain is overlaid on top of STM in Fig. 3e, giving reasonable agreement with the experimental data (the distance between two neighbouring valleys on the same side is found to be 1.38 nm and 1.35 ± 0.02 nm from STM measurements). Further annealing to 673 K, no visible change is induced, as shown in Fig. S2d.† Interestingly, with intensive annealing to 723 K in Fig. 3f, the covalent chain is still robustly adsorbed on Au(111) with the inner structure clearly resolved. Consequently, it is reasonable to conclude that the self-assembly adsorbate homochirality on Au(111) is removed after Ullmann coupling, and OM chains with chirality can be induced as intermediate states.
Self-assembly of DBCh on Ag(111)
To explore the template effect towards the chirality variation, the deposition of DBCh onto Ag(111) has also been investigated. As shown in Fig. 4a, self-assembled DBCh molecules are closely packed on Ag(111) with the long-range order at RT. High-resolution STM in Fig. 4b reveals that the closely packed phase is actually constructed by the alternative adsorption of S and R enantiomers with a regular order on Ag(111), and enantiomers with the same prochirality are adsorbed within the same line, thus inducing the adsorption chirality. Again, DBCh precursors are imaged with two bright dots on both sides of the backbone due to the electron-rich characteristic of halogen atoms.35 Furthermore, the intactness of DBCh precursors on Ag(111) at RT is also confirmed by X-ray photoemission spectroscopy (XPS), where only the pristine carbon-bound Br component is resolved, as shown in Fig. S3 in ESI.† Moreover, dim dots are also resolved between DBCh molecules, which can only be assigned to surface Ag adatoms since no Br atoms are expected to fall off from precursors at this stage.37 Thus, the ordered structure is expected to be stabilized by both the metal-coordination bonding (Br⋯Ag⋯Br) and intermolecular attraction (CH⋯Br bonding, with length of about 0.3 nm) between DBCh molecules, as highlighted with white dashed lines in Fig. 4b. The simulated STM is represented in Fig. 4c and provides rational agreement with the experimental topography. Moreover, the unit cell parameter predicted by DFT calculations, for example, the lattice dimension, a = 2.32 nm and b = 1.16 nm, is also consistent with the experimental data (a = 2.30 ± 0.02 nm, b = 1.15 ± 0.02 nm). In addition, a different arrangement is also discovered occasionally inside the closely packed domain, as indicated by the white ellipse in Fig. 4a, and is presented in detail in Fig. S4a in ESI,† where both S and R enantiomers are found to be adsorbed in the same line. Besides, the unique domain consisting of only S or R chiral adsorbate is also discovered on the surface, as shown in Fig. S4b and c,† where precursors are exclusively packed in order without Ag adatoms involved.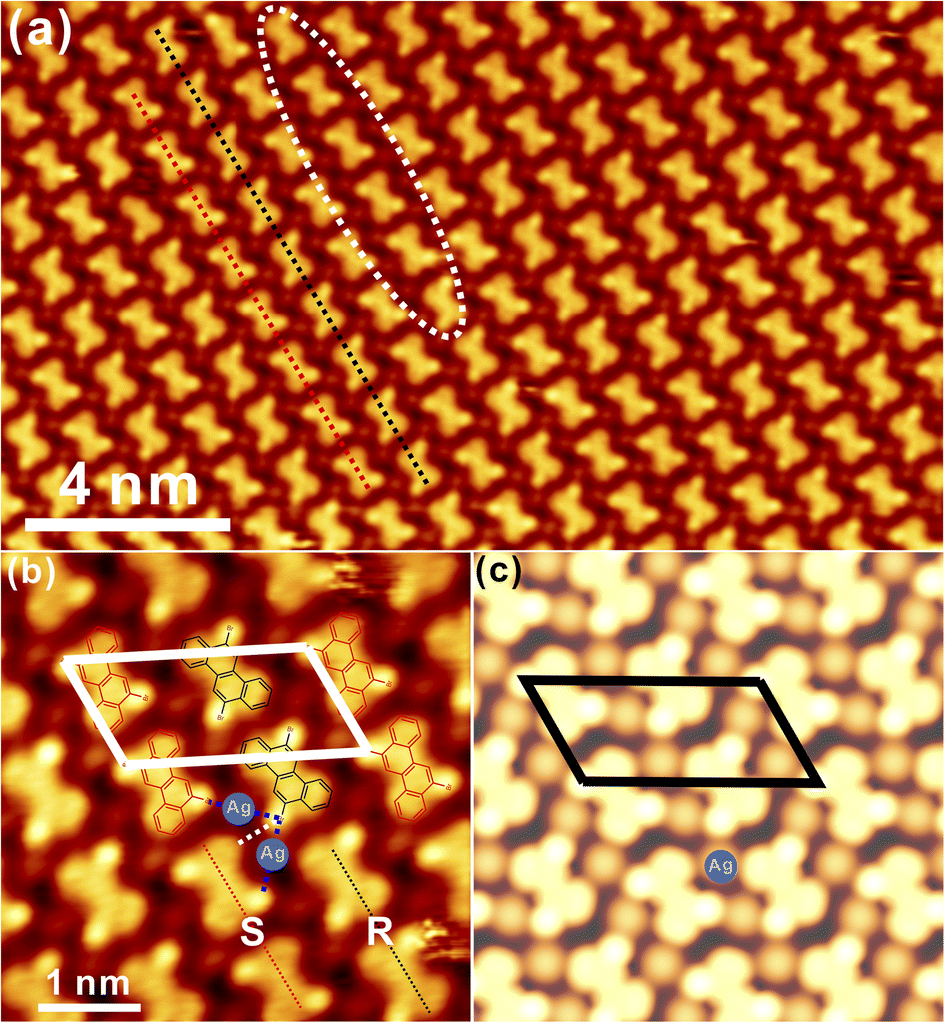 | ||
| Fig. 4 Self-assembly of DBCh on Ag(111) at RT. (a) STM topography of the fully DBCh covered surface and (b) the zoom-in STM image with the molecular model and unit cell labelled. Ag adatoms and intermolecular interaction are also highlighted as a guide to eyes. Ubias = −1.3 V, It = 0.1 nA. (c) The STM simulation in correspondence to panel (b). | ||
In addition to the closely packed phase, DBCh enantiomers can also be anchored on Ag(111) with other assembled patterns. As displayed in Fig. 5a, homochiral porous domains, including both CW and ACW arrangements, are also constructed on Ag(111), which is held slightly below RT (275 K) during deposition, and features are analogous to the self-assembly of DBCh on Au(111). More intriguingly, CW and ACW tessellations are found on the same terrace with the domain boundary clearly resolved. High-resolution STM images of CW and ACW phases are illustrated in Fig. 5b and c, respectively (additional STM images are also provided in Fig. S5a and b in ESI†). Once again, the halogen bonding (bonding length: 0.34 ± 0.02 nm) is proposed between bromine atoms of neighbouring DBCh enantiomers, which is responsible for the formation of trimeric X3-synthons, and homochiral phases are thus constructed from R and S chiral adsorbate enantiomers. As an example, DFT predicted configuration for the CW phase is illustrated in Fig. 5d, which in general provides a satisfactory agreement in both topography and unit cell parameters: the rhombohedral lattice is measured to be 2.09 ± 0.02 nm from STM images, and 2.12 nm from DFT calculations. In fact, these self-assembled structures are quite similar to those on Au(111), for example, the coexistence of both CW and ACW homochiral architectures with a roughly equal ratio.
 | ||
| Fig. 5 Homochiral domains from the self-assembly adsorption of DBCh on Ag(111) below RT. (a) Coexistence of CW and ACW hexagonal patterns as highlighted with black and red arrows. (b) and (c) Zoom-in views of CW and ACW hexamers with molecular models overlaid, indicating the bonding motifs stabilizing homochiral phases. (d) STM simulation of the CW hexagonal pattern as an example. Ubias = −1.3 V for (a), −0.8 V for (b), −1.3 V for (c), It = 0.1 nA. | ||
Intriguingly, the decorated porous structure is also observed connecting to normal pores, as shown in Fig. S4c† and elaborately illustrated in Fig. 6a. Apparently, extra trimeric X3-synthons (coloured dots) are constructed from the chiral adsorption of R or S enantiomers (the measured Br–Br distance: 0.37 ± 0.02 nm), embracing the original hexagonal patterns. STM images with the atomic resolution in Fig. 6b and c provide detailed information on the decorated hexagonal network, and the length of the rhombohedral unit cell is revealed to be 4.17 ± 0.02 nm. Obviously, homochiral hexamers are initially constructed by R and S chiral adsorbed enantiomers and surrounded by the homochiral X3-synthons with both the CW and ACW tessellations, as marked by the black or red circles in Fig. 6b and c. Surprisingly, regardless of its own homochirality, the inner hexamer is always surrounded by 6 homochiral X3-synthons with the regular alternation: 3 synthons are in the CW phase (marked with black dots) while another 3 are in the ACW phase (marked with blue dots). Nevertheless, quite a few exceptions are also found on the surface, where 4 and 5 synthons with the same adsorption chirality are positioned around the inner hexamer, as presented in Fig. S4d and e in ESI.† Notably, dim dots are resolved as well between DBCh precursors, which are assigned again to surface Ag adatoms, suggesting the existence of metal coordination bonding motifs. In contrast, the Ag coordination bonding is not evident in the regular porous phases in Fig. 5. Thus, one can conclude that the hexamers and trimeric clusters are stabilized together by the overall contribution from the halogen bonding, the intermolecular hydrogen bonding motifs as well as the metal coordinated bonds. To rationalize the multiple bonding motifs, DFT calculations have also been performed, and the optimized configuration corresponding to Fig. 6b is presented in Fig. 6d. The simulated STM image well corroborates the experimental findings where the dimension of the unit cell is calculated to be 4.22 nm, in good correspondence to the experimental value (4.17 ± 0.02 nm). It is also proposed by DFT calculations that CW and ACW trimeric synthons are adsorbed on different sites (top and hollow sites, respectively) on Ag(111). While the predominant self-assembled phase on Ag(111) at RT is the closely packed domain, the homochiral porous nanostructures are obtained with the substrate kept slightly below RT during deposition. Thus, it can be inferred that homochiral hexagonal phases with nanopores might be the preferred configuration on both Au(111) and Ag(111).
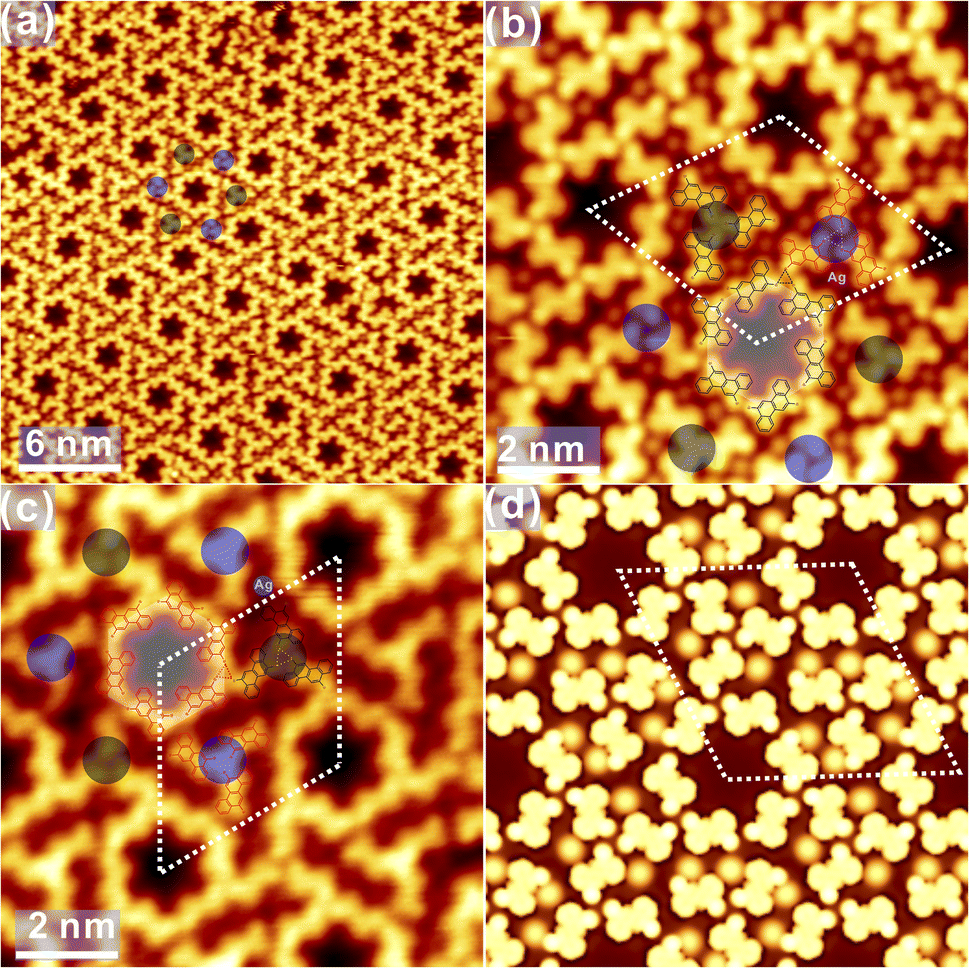 | ||
| Fig. 6 (a) Decorated hexamers in self-assembly on Ag(111). Decoration around hexamers (CW and ACW tessellation) is indicated with coloured dots. (b) The corresponding molecule-resolved image with the proposed adsorption configuration overlaid atop. (c) The homochiral phase to (b) with molecular model overlaid atop. (d) The simulated STM in correspondence to (b) as an example. Ubias = −1.4 V for (a), −0.7 V for (b) and (c), It = 0.1 nA. | ||
Ullmann coupling of DBCh on Ag(111)
After mild annealing to 323 K, immediately, disordered chains start to emerge, as discovered in Fig. 7a. At first glance, the handle-like species is identified, as marked by dashed circles in Fig. 7a. Detailed structural information is revealed from the high-resolution STM in Fig. 7b. The new species might be related to OM dimers, formed by the partially dehalogenated R and S enantiomers bound to surface Ag adatoms (imaged as bright protrusions in STM37), while similar structures have already been found on Au(111). The tentative model is overlaid atop STM for enhanced visualization, and the handle-shape structure is highlighted with the coloured cartoon. The distance between chrysene units in the handle-shape structure is measured to be 0.74 ± 0.02 nm, which agrees well with the formation of C–Ag–C bonding from the molecular model (length: 0.71 nm). Besides the formation of dimers, OM oligomers are also induced, most of which are constituted by debrominated molecules with the same adsorption chirality. Therefore, such OM oligomers can be considered as chiral locally. It is shown in Fig. 7c that subsequent annealing to 373 K leads to the considerable elongation of oligomers with the improved order as compared to the situation at 323 K. Meanwhile, these oligomer chains are still expected to be OM intermediate species and are flexibly packed on the surface. It is thus inferred that these chains can be tilted around the C–Ag–C bond within the surface plane and thus do not strictly follow high-symmetry directions of the substrate. Besides, regular dim dots confined in between OM oligomers are also recognized, which shall be assigned to detached Br atoms from precursors.37 The tentative model of OM oligomers is overlaid atop the STM image in Fig. 7c. In general, R and S chrysene blocks are randomly linked together to form OM oligomers at this stage; however, the chiral adsorption of oligomers can also be discovered at certain areas, as highlighted with the dashed circle in Fig. 7c.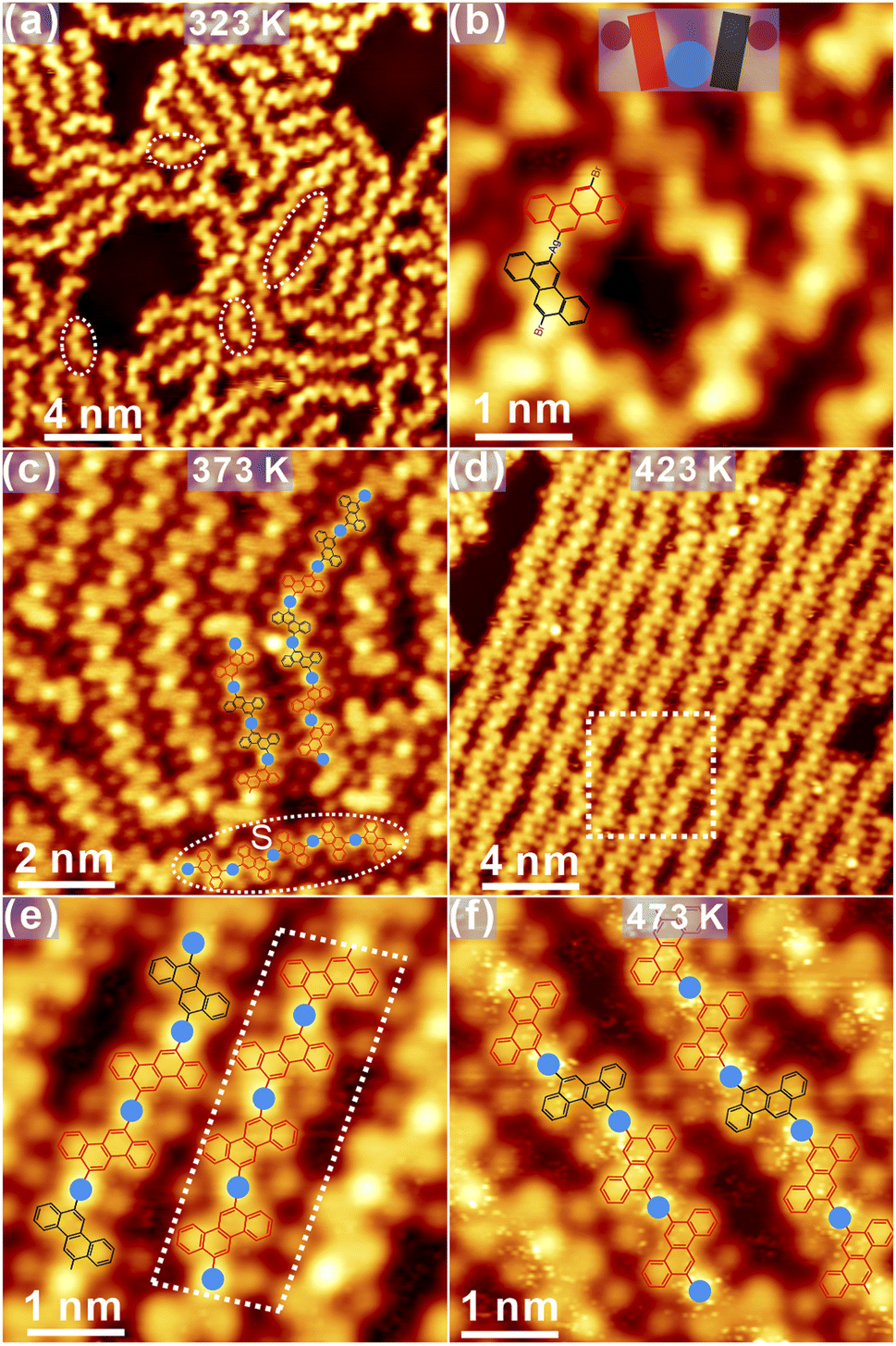 | ||
| Fig. 7 (a) OM dimers and oligomers are resolved after thermal annealing to 323 K as indicated with dashed circles. (b) The zoom-in view clearly shows the handle-shape structure of OM dimers with a cartoon model overlaid for easy illustration. (c) Further annealing to 373 K interlinks oligomers together into elongated chains. The coordination of Ag adatoms between building blocks is clearly imaged as bright protrusions. (d) OM oligomers with the long-range order are induced after annealing to 423 K. (e) The corresponding zoom-in view indicates that the coordination flexibility is still apparent in OM chains, and the local chirality in OM chains becomes visible. (f) Ordered OM chains are well preserved after additional annealing to 473 K, showing the random arrangement of R or S chrysene units. Ubias = −1.3 V, It = 0.1 nA. | ||
Notably, further annealing to 423 K results in the formation of substantially ordered OM chains on the surface, as shown in Fig. 7d. Apparently, detached Br atoms are still anchored in between oligomer chains. With the structural transition from tilted oligomers to linear chains, OM intermediates are still constituted by chrysene blocks bonded to Ag adatoms, as clearly visible in the zoom-in STM in Fig. 7e. While R or S chiral chrysene adsorbates are randomly arranged inside oligomers, a part with the only S chiral chrysene, for instance, is resolved as marked with the dashed quadrangle in Fig. 7e. Nevertheless, the local chirality can still be recognized regardless of the random connection of R or S chrysene blocks.
After further annealing to 473 K, OM chains are well preserved with an almost defect-free domain, as revealed in Fig. 7f. Notably, the characteristic of local chirality in OM chains becomes invisible since the majority of oligomer chains is now constructed by R or S chrysene without the specific order. Actually, OM species is revealed to be fairly stable up to 523 K annealing, and detached Br atoms are mediated by the metal coordination of Ag in combination with hydrogen bonding with the neighbouring chrysene blocks.37,38 Again, the C–Ag–C bond is still not linear at this stage with varied bonding angles. Thus, one can infer that the flexible coordination of Ag together with the rich conformations of oligomers, enable the high flexibility of OM chains on Ag(111). From the structural evolution illustrated in Fig. 7, one can also infer that the local chirality of OM oligomers declines with sequential annealing.
On the basis of flexible OM chains, more intensive annealing is applied to explore whether the structural transformation can occur from OM intermediate species to covalent polymers. Surprisingly, structure chaos is observed at the beginning after annealing to above 533 K (Fig. S6a in ESI†), and ordered nanostructures start to emerge only after annealing to 623 K, which prefer to accumulate along step edges as shown in Fig. 8a. Apparently, the total amount of species remaining on the surface gets significantly reduced due to desorption after intensive annealing. Moreover, a considerable number of bright dots are closely packed on both sides of polymers, which are attributed again to detached Br atoms as illustrated in detail in the zoom-in STM view in Fig. 8b. Meanwhile, such observation also agrees well with previous reports,31,37,38 where Br atoms falling off from precursors are stably adsorbed on the metal substrate.
 | ||
| Fig. 8 (a) Oligomers in a segment are observed on Ag(111) after annealing to 623 K. (b) The zoom-in view showing the formation of planar chains after the cyclodehydrogenation and aryl–aryl bonding between polymer dimers and (c) the closed ring structure. (d) The fabrication of polymer chain with staggered defects on both sides after annealing to 673 K. Ubias = −1.2 V for (a) and (b), −0.9 V for (c) and −1.3 V for (d); It = 0.1 nA. | ||
Furthermore, several unique nanostructures are induced after the high-temperature annealing, as presented in Fig. 8b and c. First, the cyclodehydrogenation is induced after kicking off the coordinated Ag atom, and the C–C covalent dimer is thus fabricated by covalently connecting chrysene units,32,33 as highlighted with the molecular model in Fig. 8b. Besides, covalent dimers can also be linked together into a double-dimer architecture induced by the aryl–aryl bonding after further dehydrogenation, and the tentative model is also proposed atop. One should also bear in mind that the rotation around the single C–C bond between chrysene units shall make dimers tilted with respect to the surface and thus the partial brightness in STM. Besides, the enclosed-ring architecture is discovered as well in Fig. 8c. Judged from its appearance, such enclosed architecture is probably induced by the aryl–aryl connection between a homochiral dimer and a chrysene unit after the selective dehydrogenation, and the possible structural model is proposed atop the STM image. In fact, both the double-dimer structure and the enclosed ring architecture are induced by the selective dehydrogenation of prochiral chrysene blocks at the meta position, followed by the aryl–aryl coupling. Other similar nanostructures can also be found and are summarized in Fig. S6b and c in ESI.† Further annealing to 673 K in Fig. 8d makes the Ag(111) surface tidied up, and only the covalent polymer chains with staggered defects are formed via the alternative linkage of R and S chrysene units, which have the feature of 8AGNR but bear valley defects on the opposite site.32 However, it is also notable that the total amount of 8AGNR is small with the limited length, which is probably caused by the substantial desorption after the intensive annealing.
In a word, evidenced by the high-resolution STM, chiral tessellations of DBCh enantiomers are induced on both Au(111) and Ag(111) into hexagonal pores with self-assembly, and OM oligomers with local chirality are discovered on Au(111) and Ag(111) (bearing flexible structures) during Ullmann coupling. While the unique homochiral arrangement of self-assembled DBCh is induced on Au(111), multiple homochiral structures are fabricated on Ag(111), implying the significant templating effect from the substrate. For example, the coordination motifs via surface adatoms are fairly apparent on Ag(111), but instead, it is occasionally observed on Au(111) due to the shortage of surface adatoms. Notably, no debromination of DBCh is observed on Ag(111) at RT, while partial debromination of other halogenated precursors has been frequently observed on Ag(111) at RT.14,37 Such difference might be assigned to the relatively high activation energy of the C–Br bonding in DBCh, which is higher than the activation barrier of the C–Br bonding in Br2Py37 by about 0.4 eV as predicted by DFT calculations. In addition, DFT calculations have also been employed in understanding the intermolecular bonding motifs by optimizing adsorption configurations and providing reasonable structures to the experimental data. Importantly, the key role of intermolecular halogen bonding and metal coordinated interactions is witnessed not only in the chiral adsorption of DBCh enantiomers, but also in mediating the stability of partially chiral oligomers constructed from chrysene blocks, especially flexible polymer chains on Ag(111). While OM chains with local chirality are identified on Au(111) and Ag(111) at certain areas, the ultimate formation of 8AGNR completely removes the pristine adsorption chirality by alternatively linking S and R chrysene units, as such a configuration is the most stable structure from the energy point of view.32 it is also obvious that the quality of graphene nanoribbons is substantially influenced by metal substrates. Based on these observations, the gradual fading of adsorption chirality from the self-assembly to covalent polymers is identified across on-surface Ullmann coupling.
Conclusions
In this work, chiral adsorption of DBCh enantiomers are witnessed on both Au(111) and Ag(111) by STM and DFT calculations, followed by the formation of locally chiral OM oligomers, especially with flexible arrangements on the silver substrate during Ullmann coupling. Generally, the shortage of Au adatoms on the surface makes it hard to form OM intermediates; however, chiral OM oligomers are resolved on Au(111), herein, probably due to the chiral adsorption of precursors. Meanwhile, OM oligomers consisting of R and S chrysene units are commonly observed on Ag(111) with varying configurations due to the flexible metal coordination of Ag. Even though structure chaos is encountered on both substrates during coupling reactions due to the steric hindrance between chrysene blocks, 8AGNR with staggered defects on both sides are eventually fabricated via cyclodehydrogenation after intensive annealing. Thus, the direct polymerization of chiral adsorbates of DBCh enantiomers into achiral carbon-based nanostructures is identified during surface Ullmann coupling, demonstrating the gradual fading of adsorbate chirality. It is also revealed that the self-assembly with coordinated bonding motifs on the surface presents a flexible way to manipulate intermediate states, and the chiral adsorbate is reserved locally within OM nanostructures in certain areas. Thus, this report provides encouraging opportunities for manipulating chiral/achiral nanostructures on a surface towards potential applications in related nanoelectronics.Methods
All STM experiments were performed in the FERMI system (Scienta-Omicron GmbH) under ultrahigh vacuum (UHV) conditions with a base pressure better than 5 × 10−10 mbar.35 STM imaging was performed in the constant-current mode at 77 K while the bias voltage was applied to the tip with the sample grounded. The STM tip was prepared by mechanically cutting the Pt/Ir (90% Pt, 10% Ir) wire. All STM images were processed afterwards with the WSxM software.39The Au(111) and Ag(111) (MaTecK, Germany) surfaces were cleaned by repeated Ar+ ion sputtering (alternating energy at 1.2 keV and 0.8 keV) and annealing cycles (623 K for Ag(111) and 673 K for Au(111), respectively). The DBCh precursor (Sigma-Aldrich, purity > 97%) was thoroughly degassed prior to deposition, and thermally evaporated onto the metal surface with a rate of 0.1 monolayer (ML) per min from the commercial evaporator (Omnivac, Germany). Herein, 1 ML is referred to the full coverage of DBCh molecules on the surface as judged from STM, and the evaporation rate was calibrated using a quartz crystal microbalance. Both the Au(111) and Ag(111) substrates were kept at RT (300 K) during deposition unless otherwise stated and were subsequently heated in sequence to initiate Ullmann coupling reactions.
DFT calculations were done by utilizing the Vienna ab initio simulation package (VASP).40,41 The PBE functional was adopted in combination with the third-generation van der Waals dispersion correction from Grimme (DFT-D3) and the projector-augmented wave (PAW) method for atomic cores.42–44 A plane-wave cut-off energy of 450 eV was employed, while the k-point sampling was chosen to be 3 × 3 × 1 to obtain a realistic and accurate picture of the energetics. Lattice parameters of Ag(111) and Au(111) after optimization were found to be 2.89 Å and 2.88 Å, respectively, in good agreement with previous reports.35,45 Optimized adsorption geometries for DBCh on the surface were obtained on the three-layer substrates based on the energy minimum, where the first layer and precursors are free to relax with the bottom two layers fixed until the forces on the active atoms dropped below 0.02 eV Å−1. The convergence criterion is 10−4 eV in total energy difference. STM simulations were performed in the framework of Tersoff–Hamann approximation with the gnuplot software by plotting partial charge densities, which were acquired by a static self-consistent calculation based on the DFT optimized configurations. In the end, the selection of a proper structure model is guided by the lattice parameters and STM features.
Author contributions
H. H. and F. S. proposed and designed the project. H. B. W. and J. P. H. performed LT-STM experiments and analysed experimental results. Z. F. L., L. X., H. Z., Z. J. and F. S. discussed the data. Z. F. and C. Q. H. performed the DFT calculations, H. B. W., H. H. and F. S. wrote the paper, and all authors discussed and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fruitful discussion with Prof. Dr Miao Yu is greatly appreciated. This project is financially supported by the National Natural Science Foundation of China (11874380, 22002183), National Key Research and Development Program of China (2021YFA1600800), the Photon Science Center for Carbon Neutrality of Chinese Academy of Sciences, and the Key Laboratory of Low-Carbon Conversion Science and Engineering of Chinese Academy of Sciences.Notes and references
- J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS PubMed.
- F. Song, J. W. Wells, K. Handrup, Z. S. Li, S. N. Bao, K. Schulte, M. Ahola-Tuomi, L. C. Mayor, J. C. Swarbrick, E. W. Perkins, L. Gammelgaard and P. Hofmann, Nat. Nanotechnol., 2009, 4, 373–376 CrossRef CAS PubMed.
- H. J. Gao and L. Gao, Prog. Surf. Sci., 2010, 85, 28–91 CrossRef CAS.
- W. F. Smith, Nat. Nanotechnol., 2007, 2, 77–78 CrossRef CAS PubMed.
- J. Björk and F. Hanke, Chem.–Eur. J., 2014, 20, 928–934 CrossRef PubMed.
- L. Dong, P. N. Liu and N. Lin, Acc. Chem. Res., 2015, 48, 2765–2774 CrossRef CAS PubMed.
- L. Talirz, P. Ruffieux and R. Fasel, Adv. Mater., 2016, 28, 6222–6231 CrossRef CAS PubMed.
- Q. Shen, H. Y. Gao and H. Fuchs, Nano Today, 2017, 13, 77–96 CrossRef CAS.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. L. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- M. Lackinger, Chem. Commun., 2017, 53, 7872–7885 RSC.
- A. Trandafir, G. Dan Pantoş and A. Ilie, Nanoscale, 2022, 14, 1929–1943 RSC.
- S. Clair and D. G. de Oteyza, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- Q. T. Fan, L. M. Liu, J. Y. Dai, T. Wang, H. X. Ju, J. Zhao, J. Kuttner, G. Hilt, J. M. Gottfried and J. F. Zhu, ACS Nano, 2018, 12, 2267–2274 CrossRef CAS PubMed.
- Q. Sun, B. V. Tran, L. Cai, H. Ma, X. Yu, C. Yuan, M. Stohr and W. Xu, Angew. Chem., Int. Ed., 2017, 56, 12165–12169 CrossRef CAS PubMed.
- C. Tao, L. Jiao, O. Yazyev, Y.-C. Chen, J. J. Feng, X. W. Zhang, R. B. Capaz, J. M. Tour, A. Zettl, S. G. Louie, H. J. Dai and M. F. Crommie, Nat. Phys., 2011, 7, 616–620 Search PubMed.
- K. E. Plass, A. L. Grzesiak and A. J. Matzger, Acc. Chem. Res., 2007, 40, 287–293 CrossRef CAS PubMed.
- C. Kulkarni, A. K. Mondal, T. K. Das, G. Grinbom, F. Tassinari, M. F. J. Mabesoone, E. W. Meijer and R. Naaman, Adv. Mater., 2020, 32, 1904965 CrossRef CAS PubMed.
- Y. Xu, J.-J. Duan, Z.-Y. Yi, K.-X. Zhang, T. Chen and D. Wang, Surf. Sci. Rep., 2021, 76, 100531 CrossRef CAS.
- H. H. Kong, Y. Y. Qian, X. B. Liu, X. L. Wan, S. Amirjalayer and H. Fuchs, Angew. Chem., Int. Ed., 2019, 59, 182–186 CrossRef PubMed.
- D. Han, T. Wang, J. M. Huang, X. Y. Li, Z. W. Zeng and J. F. Zhu, Nano Res., 2022, 15, 3753–3762 CrossRef CAS.
- B. Yang, N. Cao, H. X. Ju, H. P. Lin, Y. Y. Li, H. H. Ding, J. Q. Ding, J. J. Zhang, C. C. Peng, H. M. Zhang, J. F. Zhu, Q. Li and L. F. Chi, J. Am. Chem. Soc., 2019, 141, 168–174 CrossRef CAS PubMed.
- O. Stetsovych, M. Švec, J. Vacek, J. V. Chocholoušová, A. Jančařík, J. Rybáček, K. Kosmider, I. G. Stará, P. Jelínek and I. Starý, Nat. Chem., 2017, 9, 213–218 CrossRef CAS PubMed.
- X.-Y. Wang, J. I. Urgel, B. G. Barin, K. Eimre, M. Di Giovannantonio, A. Milani, M. Tommasini, C. A. Pignedoli, P. Ruffieux, X. L. Feng, R. Fasel, K. Müllen and A. Narita, J. Am. Chem. Soc., 2018, 140, 9104–9107 CrossRef CAS PubMed.
- N. Merino-Díez, M. S. G. Mohammed, J. Castro-Esteban, L. Colazzo, A. Berdonces-Layunta, J. Lawrence, J. I. Pascual, D. G. de Oteyza and D. Peña, Chem. Sci., 2020, 11, 5441–5446 RSC.
- M. Ammon, T. Sander and M. Sabine, J. Am. Chem. Soc., 2017, 139, 12976–12984 CrossRef CAS PubMed.
- C. Steiner, J. Gebhardt, M. Ammon, Z. C. Yang, A. Heidenreich, N. Hammer, A. Görling, M. Kivala and S. Maier, Nat. Commun., 2017, 8, 14765 CrossRef CAS PubMed.
- D. G. de Oteyza, A. Garcia-Lekue, M. Vilas-Varela, N. Merino-Diez, E. Carbonell-Sanroma, M. Corso, G. Vasseur, C. Rogero, E. Guitian, J. I. Pascual, J. E. Ortega, Y. Wakayama and D. Peña, ACS Nano, 2016, 10, 9000–9008 CrossRef CAS PubMed.
- M. Bieri, M.-T. Nguyen, O. Gröning, J. M. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669–16676 CrossRef CAS PubMed.
- B. Yang, B. Dong and L. F. Chi, ACS Nano, 2020, 14, 6376–6382 CrossRef CAS PubMed.
- M. Fritton, D. A. Duncan, P. S. Deimel, A. Rastgoo-Lahrood, F. Allegretti, J. V. Barth, W. M. Heckl, J. Björk and M. Lackinger, J. Am. Chem. Soc., 2019, 141, 4824–4832 CrossRef CAS PubMed.
- T. A. Pham, B. V. Tran, M.-T. Nguyen and M. Stöhr, Small, 2017, 13, 1603675 CrossRef PubMed.
- S. J. Sun, B. J. Li, B. Y. Fu, Z. L. Ruan, H. Zhang, W. Xiong, Y. Zhang, G. F. Niu, J. C. Lu, X. Q. Zuo, L. Gao and J. M. Cai, Chin. Chem. Lett., 2022, 33, 5142–5146 CrossRef CAS.
- S. J. Sun, Y. R. Guan, Z. L. Hao, Z. L. Ruan, H. Zhang, J. C. Lu, L. Gao, X. Q. Zuo and J. M. Cai, Nano Res., 2022, 15, 653–658 CrossRef CAS.
- J. B. Hu, J. P. Hu, Z. D. Zhang, K. C. Shen, Z. F. Liang, H. Zhang, Q. W. Tian, P. Wang, Z. Jiang, H. Huang, J. W. Wells and F. Song, Appl. Surf. Sci., 2020, 513, 145797 CrossRef CAS.
- D. Peyrot, M. G. Silly and F. Silly, Phys. Chem. Chem. Phys., 2018, 20, 3918–3924 RSC.
- J. P. Hu, Z. F. Liang, K. C. Shen, L. Xie, H. Zhang, C. Q. Huang, Y. B. Huang, H. Huang, J. X. Tang, Z. Jiang, M. Yu and F. Song, Nano Res., 2021, 14, 4704–4713 CrossRef CAS.
- Q. T. Fan, T. Wang, J. Y. Dai, J. L. Kuttner, G. Hilt, J. M. Gottfried and J. F. Zhu, ACS Nano, 2017, 11, 5070–5079 CrossRef CAS PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baró, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1998, 77, 3865–3868 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- Z. F. Liang, Y. Wang, C. Q. Hua, C. C. Xiao, M. G. Chen, Z. Jiang, R. Z. Tai, Y. H. Lu and F. Song, Nanoscale, 2019, 11, 14134–14140 RSC.
- F. De Marchi, G. Galeotti, M. Simenas, M. C. Gallagher, E. Hamzehpoor, O. MacLean, R. M. Rao, Y. Chen, D. Dettmann, G. Contini, E. E. Tornau, M. Ebrahimi, D. F. Perepichka and F. Rosei, Nanoscale, 2019, 11, 19468–19476 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional STM images of the self-assembly and the formation of various oligomers during Ullmann coupling. See DOI: https://doi.org/10.1039/d2na00789d |
| This journal is © The Royal Society of Chemistry 2023 |