 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the influence of substrate binding on photocatalytic dehalogenation with a heteroleptic supramolecular [M4La2Lb2] square containing PDI photosensitizers as ligands†
C. Jasslie
Nielsen
 a,
Petrus C. M.
Laan
a,
Raoul
Plessius
a,
Joost N. H.
Reek
a,
Jarl Ivar
van der Vlugt
ab and
Sonja
Pullen
*a
a,
Petrus C. M.
Laan
a,
Raoul
Plessius
a,
Joost N. H.
Reek
a,
Jarl Ivar
van der Vlugt
ab and
Sonja
Pullen
*a
aHomogeneous, Supramolecular and Bio-Inspired Catalysis, Van’t Hoff Institute for Molecular Sciences, Faculty of Natural Sciences, University of Amsterdam, P.O. Box 94720, 1090 GS Amsterdam, The Netherlands. E-mail: s.pullen@uva.nl
bBioinspired Coordination Chemistry & Catalysis, Institute of Chemistry, Carl von Ossietzky University Oldenburg, Carl-von-Ossietzky-Strasse 9-11, D-26129 Oldenburg, Germany
First published on 19th January 2023
Abstract
Photoredox catalysis is a valuable tool in a large variety of chemical reactions. Main challenges still to be overcome are photodegradation of photocatalysts and substrates, short lifetimes of reactive intermediates, and selectivity issues due to unwanted side reactions. A potential solution to these challenges is the pre-organization of the photosensitizer, substrate and (co)-catalyst in supramolecular self-assembled structures. In such architectures, (organic) dyes can be stabilized, and higher selectivity could potentially be achieved through pre-organizing desired reaction partners via non-covalent interactions. Perylene diimide (PDI) is an organic dye, which can be readily reduced to its mono- and dianion. Excitation of both anions leads to highly reducing excited states, which are able to reduce a variety of substrates via single electron transfer. The incorporation of PDI into a heteroleptic [M4La2Lb2] supramolecular square has been recently demonstrated. Herein we investigate its photophysical properties and demonstrate that incorporated PDI indeed features photocatalytic activity. Initial results suggest that the pre-organisation by binding positively affects the outcome.
Introduction
Supramolecular photocatalysis merges two exciting research fields and has led to the development of many fascinating complex systems that are utilizing non-covalent interactions to achieve improved selectivity and new reactivity.1–5 Light-driven catalysis has been proven very versatile in the development of novel reaction routes.6,7 Photons can be used to selectively activate one specific component in a complex mixture, thus initiating a reaction in a clean and selective fashion.8 With the use of photosensitizers (PS), visible light can be harvested to initiate a reaction. Through light absorption, a PS is brought into its excited state (PS*). From there, either energy transfer (EnT) to an energy acceptor (A),9,10 or photoinduced electron transfer (PET) can occur.11 In the latter case, depending on the quenching mechanism, either an oxidized (PS+) or reduced (PS−) species is generated that is subsequently returned to the original state (PS) via single electron transfer (SET). Through these one-electron processes, reactive radical species can be formed from activated substrates such as aryl iodides,12 or diazonium compounds.13 In the past decade, the development of organic dyes containing aryl amines such as perylene diimide (PDI, 1) (Scheme 1) has gained increasing attention,14 in order to find alternatives for noble metal transition metal photosensitizers.15 Such organic dyes are very promising for photocatalysis, as they enable the formation of significantly stronger reducing or oxidizing excited states, and thus the formation of radicals from less activated compounds.16,17 PDI 1 is a fluorescent organic dye that is commonly used as an organic photosensitizer due to its ability to generate highly reducing excited states that are able to transfer one electron to substrates such as aryl bromides that are otherwise difficult to reduce.18,19 In addition, the photophysical properties of PDI can be easily tuned and optimized i.e., by introducing substituents at the PDI core. As a single molecule photocatalyst, 1 has been used for reactions such as the dehalogenation of aryl bromides.19 In principle, the aryl radicals generated in this process may be used for arylation reactions, e.g. through C–C cross coupling reactions. However, due to competition of fast hydrogen atom transfer (HAT) from solvent or radical cations generated from oxidation of a sacrificial reductant (e.g. NEt3), such cross coupling reactions are limited to substrates such as pyrrole that, if present in high concentrations, feature a rapid reaction rate with the generated radicals.20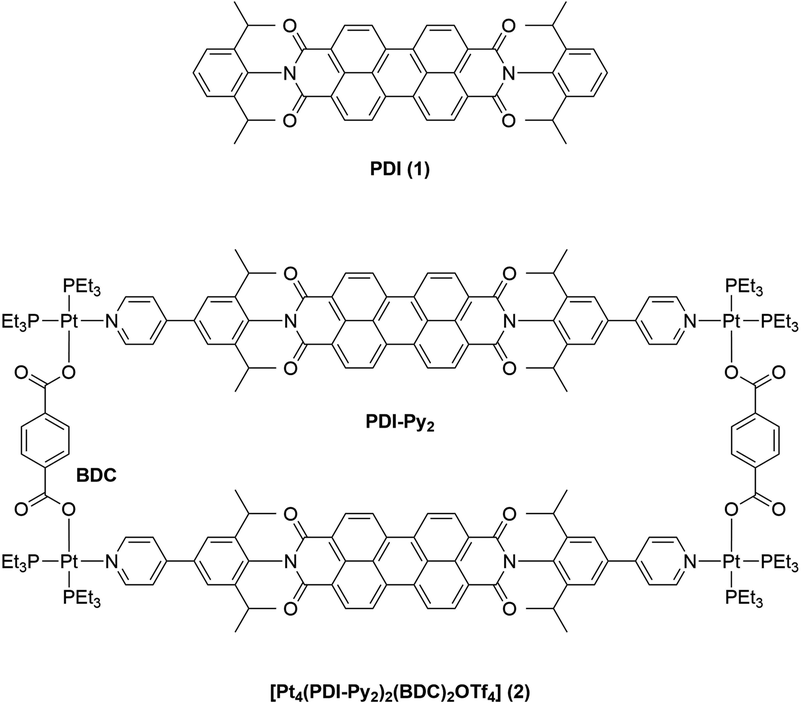 | ||
| Scheme 1 Molecular structures of PDI (1, top) and the heteroleptic square [Pt4(PDI-Py2)2(BDC)2OTf4] (2, bottom). | ||
Supramolecular self-assembly is a valuable tool to combine several functional ligands and metals in one architecture, creating a binding pocket that is available for substrate binding through non-covalent interactions.21–30 While most early supramolecular systems are homoleptic, e.g. consist of one type of ligand with two or more donor groups that are connected via metals, more recently heteroleptic systems with at least two different kinds of ligands have come into focus.31,32 Due to straightforward synthetic modification, photosensitizers such as 1 can be integrated as a ligand backbone into supramolecular constructs.18,33 In the past decade, several photoactive supramolecular constructs including various organic- and transition-metal-based dyes (i.e. 2D squares and 3D cages) have been developed and many of them show interesting applications in molecular sensing or light-driven catalysis.34–38 In a heteroleptic supramolecular construct, in addition to the photosensitizer, other functions can potentially be introduced, regulating the distance between photosensitizers39 or even acting as binding sites for substrates.
Würthner and co-workers incorporated a PDI moiety as linker into homoleptic supramolecular structures and showed that the dye maintains its photophysical and electrochemical properties.40–43 More recently, van der Vlugt and Reek demonstrated that in heteroleptic square [Pt4(PDI-Py2)2(BDC)2OTf4] (2) with two PDI-Py2 units separated by ligands of different length (Scheme 1), the distance between the two PDI units determines their rotational freedom.44 With smaller benzene-dicarboxylate (BDC) as ligand “B”, the distance between the two PDI units was estimated to be about 6.9 Å by molecular modelling. Flat aromatic molecule pyrene was shown to bind with a binding constant Ka of 964 M−1 (in CD3CN) in between the two PDI units, which is promoted by π–π-interactions. Increasing the distance to 11.3 Å by using larger ligand biphenyl-dicarboxylate (BPDC) has two effects: on the one hand binding of pyrene is significantly weaker (48 M−1, in CD3CN), and secondly, the PDI units are now far enough apart from each other so that rotation along the N–N axis is possible. It is known that PDI 1 undergoes two reversible reductions.45 Electrochemical characterization of heteroleptic square 2 in DCM revealed that the first reduction event for each PDI moiety occurs in two separate steps with a peak separation of 60 mV. With the larger spacer BPDC, this peak splitting was more significant, and the first reduction of the two PDI moieties occurs clearly in two individual, reversible events. The larger difference in peak separation was attributed to the increased rotational freedom of the two PDI units, rendering them chemically inequivalent. In contrast to the first reduction, the second reduction of both PDI moieties was shown to occur simultaneously. It was confirmed that overall a reversible four-electron reduction of 2 is possible. In the presence of pyrene guest, the peak separation between the two first redox events is less prominent, and it was concluded that the guest molecule clearly influences the redox properties of such supramolecular constructs.
Herein, we investigate the photocatalytic properties of supramolecular square 2, containing two PDI moieties and the shorter BDC ligand as spacer. Particularly, we were interested to see whether (and how) the substrate binding strength affects the overall catalytic performance. After presenting the results on photocatalytic dehalogenation of three different substrates, we will discuss how this system could be optimized and how our findings may contribute to future research on supramolecular photocatalysis.
Results and discussion
Synthesis
Heteroleptic supramolecular square [Pt4(PDI-Py2)2(BDC)2OTf4] (2) was synthesized according to literature procedures.44 Clean formation of the desired supramolecular construct was confirmed by 1H-NMR, 31P-NMR, DOSY-NMR and ESI-MS (see ESI Fig. S1–4†). As established by Stang and co-workers, such heteroleptic architectures form selectively due to a charge separation effect at Pt(II) nodes, rendering this species thermodynamically more stable than corresponding homoleptic assemblies.46Photophysical characterization
UV-vis absorption and fluorescence spectra were recorded in DMF (3.33 μM for 2 and 6.66 μM for PDI-Py2), the solvent which was also used for photocatalysis (Fig. 1).19 Characteristic absorption maxima at 528, 493 and 461 nm were found in accordance with literature.19,44 Only a minor bathochromic shift for the supramolecular square 2 in comparison with free ligand PDI-Py2 was observed, indicating that electronic properties of the organic dye do not change significantly upon integration in the supramolecular construct. This is in accordance with homoleptic and heteroleptic systems previously reported by Würthner,18 as well as data for 2 recorded in DCM by van der Vlugt and Reek.44 Furthermore, emission spectra showed that fluorescence of the PDI units is retained upon self-assembly, thus opening up the possibility to use these excited states for light-induced catalysis.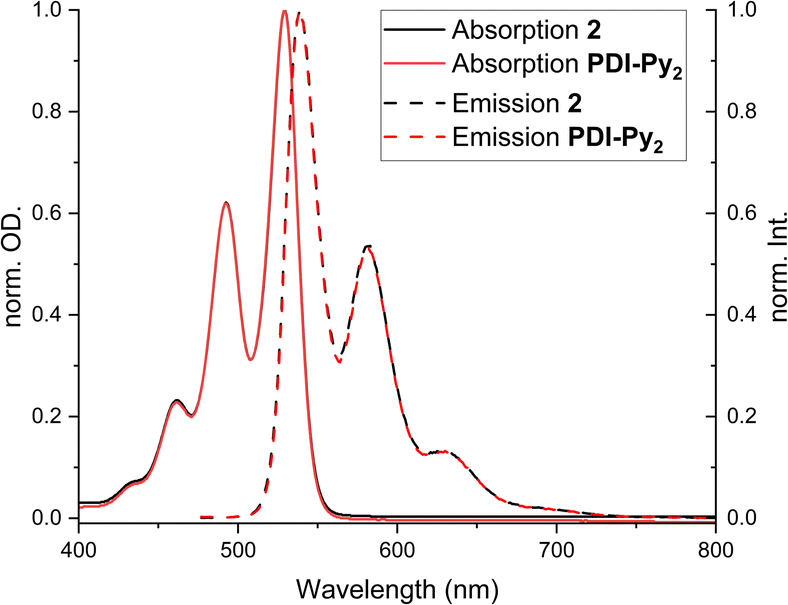 | ||
| Fig. 1 Normalized absorption and emission spectra of free PDI-Py2 ligand (6.66 μM in DMF) and the supramolecular square 2 (3.33 μM in DMF). | ||
Photocatalytic dehalogenation of 4-bromobenzaldehyde
Encouraged by the observation that the photophysical properties of PDI moieties incorporated in supramolecular square 2 are maintained, we set out to study the photocatalytic properties of this system. A reaction that has initially been reported by König and co-workers to work very well with single PDI 1 as photosensitizer is the dehalogenation of 4-bromobenzaldehyde 3 (Scheme 2).19 Catalysis was performed in DMF-d7 under irradiation with 465 nm light and in the presence of NEt3 (8 eq.) as sacrificial electron donor. All results are summarized in Table 1. With free ligand PDI-Py2, the reaction reaches almost full conversion to benzaldehyde 3b in nearly quantitative yield after 3 h. Using the parent PDI dye 1 gives slightly lower conversion but with a similarly high yield for the product. In contrast, using supramolecular square 2 as photocatalyst, only 51% of the starting material 3 is converted after 3 h with 45% yield of 3b. After 24 h of reaction time, full conversion is achieved with 71% yield of the desired product 3b. We wondered if the lower yield might be due to decomposition or further (side) reactions of product 3b. Indeed, irradiation of the reaction mixture containing free PDI 1 as photocatalyst for 24 h showed 100% conversion, but no benzaldehyde 3b was found in the 1H-NMR spectrum of the reaction mixture. In comparison, in the presence of 2, the product 3b seems more stable, as still 71% is found after 24 h irradiation. Furthermore, irradiation of the reaction mixture in the absence of any photocatalyst resulted in 25% conversion with 25% yield of 3b. Notably, performing the reaction in the dark did not show any significant conversion. These findings show on the one hand that several side reactions occur in the presence of light, and these occur less in the presence of 2. It is likely that the side reactions lead to dimerization products of benzaldehyde.47 On the other hand, the results clearly demonstrate that the supramolecular square 2 indeed acts as a photocatalyst in the dehalogenation of 4-bromobenzaldehyde 3, though featuring slower conversion and lower overall yield than the parent photocatalyst 1. Furthermore, we suspected that binding affinity between photocatalyst and substrate may have an influence on catalytic performance. | ||
| Scheme 2 Photocatalytic dehalogenation of 4-bromobenzaldehyde 3. | ||
| Entry | Substrate | Catalyst | Time | Conversion | Yield |
|---|---|---|---|---|---|
| a Experimental conditions: catalyst (0.83 mM for 1, 0.42 mM for 2), substrate (10 μmol), NEt3 (80 μmol), 456 nm Kessil lamp (34 W) stirred for 3 or 24 hours in 600 μL DMF-d7. Conversion and yield determined by 1H-NMR using 1,3,5-trimethoxybenzene as external standard. All experiments were performed in duplicate. Due to irradiation the reaction temperature was 38 °C; in the dark reaction an oil bath was used to mimic this temperature increase. | |||||
| 1 | 3 | PDI-Py2 | 3 h | 100 | 99 |
| 2 | 3 | 1 | 3 h | 92 | 92 |
| 3 | 3 | 2 | 3 h | 51 | 45 |
| 4 | 3 | 1 | 24 h | 100 | 0 |
| 5 | 3 | 2 | 24 h | 100 | 71 |
| 6 | 3 | None | 3 h | 25 | 25 |
| 7 | 3 | 1, dark | 3 h | 2 | 0 |
Substrate binding studies
In order to test our hypothesis that the reactivity may be influenced by binding strength of the substrate to the supramolecular square 2, we performed 1H-NMR titrations in DMF-d7 under the same conditions as used for catalysis, i.e. using a constant host concentration of 0.42 mM. Besides 4-bromobenzaldehyde 3, we also determined the binding constants of 9-bromphenanthrene 4 and 1-bromopyrene 5, which are both flat aromatic molecules that are potential substrates for dehalogenation and may feature a stronger binding affinity to the supramolecular square 2 (Scheme 3). | ||
| Scheme 3 Molecular structures of the three different substrates used for photocatalytic dehalogenation and host/guest binding studies. | ||
Addition of the different substrates (from 1 to 60 eq.) was performed in 10 steps and followed by 1H-NMR (see Fig. 2 and S5–8†). As expected, 4-bromobenzaldehyde 3 did not show any shifts, thus indicating no binding affinity to the host 2 under these conditions (Fig. 2a). In contrast, for both 9-bromphenanthrene 4 and 1-bromopyrene 5, up-field shifts for the PDI-hydrogens d and e of 9.26 and 8.82 ppm were observed (Fig. 2b). Fitting of the binding curves anticipating a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio for host/guest formation, resulted in binding constants Ka of 80 M−1 and 237 M−1 for substrates 4 and 5, respectively. It should be noted that the binding constants depend on the solvent used and might be higher in different solvents.
:1 ratio for host/guest formation, resulted in binding constants Ka of 80 M−1 and 237 M−1 for substrates 4 and 5, respectively. It should be noted that the binding constants depend on the solvent used and might be higher in different solvents.
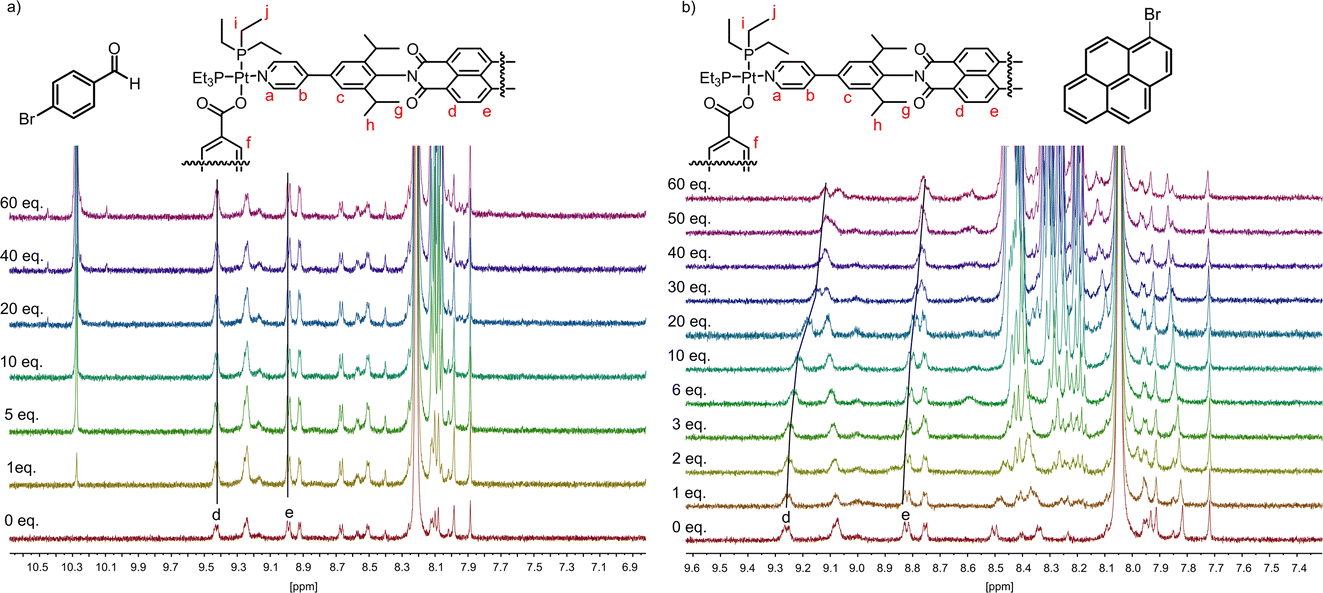 | ||
| Fig. 2 1H-NMR titration in DMF-d7 of (a) 0 to 60 eq. 4-bromobenzaldehyde 3 to host 2 (0.42 mM) and (b) 0 to 60 eq. 1-bromopyrene 5 to host 2 (0.42 mM). | ||
For instance, in the previous study by van der Vlugt and Reek a binding constant of 946 M−1 was obtained for pyrene in CD3CN.44 In addition to host/guest binding with supramolecular square 2, we were also interested in comparing the association constants of free PDI 1 and the two substrates 9-bromophenanthrene 4, and 1-bromopyrene 5, which both featured weak to moderate binding with the supramolecular square. In both cases, significantly smaller up-field shifts were observed for the PDI-hydrogens in the 1H-NMR spectrum (Fig. S9–12†), upon addition of a total of 60 equivalents of the respective guest molecules. Binding constants were determined to be 14.5 M−1 and 16 M−1, respectively, indicating significantly weaker association as compared to the supramolecular square 2.
Photocatalytic dehalogenation of 4 and 5
Having established that the two flat aromatic π-conjugated substrates 4 and 5 do bind significantly in between the two PDI moieties of supramolecular square 2, we were curious to see how binding would affect their reactivity in photocatalyzed dehalogenation using the supramolecular host as catalyst. The same experimental conditions as above were used for catalysis and the results are summarized in Table 2. For 9-bromophenanthrene 4, the conversion with photocatalyst PDI 1 after 3 h was 49% with a yield of 25%, which is slightly increased after 24 h. In comparison, conversion and yield using supramolecular square 2 as photocatalyst are only slightly lower. The observation that the difference is not as significant as for the previous substrate 1, clearly indicates a positive effect on catalysis induced by substrate binding into 2. Interestingly, even in the absence of any photosensitizer 33% conversion and 14% yield are observed after 24 h, indicating that the substrate is already directly activated by light. This is even more pronounced when using 1-bromopyrene 5 as substrate. Here, full conversion is achieved in the absence of photocatalyst, and the yield does not significantly change in the presence of either 1 or 2. In both cases, irradiation is crucial, as no conversion of the substrates in the dark is observed. Similarly to the case of benzaldehyde, side products are suspected to be dimerization products. The unsensitized dehalogenation of both substrates unfortunately complicates a fair comparison of free PDI 1 and supramolecular photocatalyst 2.| Entry | Substrate | Catalyst | Time | Conversion | Yield |
|---|---|---|---|---|---|
| a Experimental conditions same as in Table 1. | |||||
| 1 | 4 | 1 | 3 h | 49 | 25 |
| 2 | 4 | 1 | 24 h | 66 | 37 |
| 3 | 4 | 2 | 3 h | 32 | 16 |
| 4 | 4 | 2 | 24 h | 58 | 26 |
| 5 | 4 | None | 24 h | 33 | 14 |
| 6 | 4 | 1, dark | 3 h | 0 | 0 |
| 7 | 5 | 1 | 3 h | 100 | 61 |
| 8 | 5 | 1 | 24 h | 100 | 63 |
| 9 | 5 | 2 | 3 h | 100 | 75 |
| 10 | 5 | 2 | 24 h | 100 | 63 |
| 11 | 5 | None | 24 h | 100 | 65 |
| 12 | 5 | 1, dark | 3 h | 0 | 0 |
The next step will be to look into C–C cross coupling reactions with the generated aryl radicals. It is known that aryl radicals act as hydrogen atom acceptors and thus react very fast with solvents and radical cations generated from NEt3 to yield the dehalogenation products. For C–C-coupling reactions with more complex reaction partners, it may therefore be crucial to pre-organize the second substrate in proximity to the photosensitizer, so as to overcome diffusion limitations and promote coupling instead of unwanted HAT.
Mechanistic considerations
In the initial study by König and co-workers,19 a two-photon mechanism was anticipated. Initial excitation followed by reductive quenching of PDI 1 was hypothesized to generate monoanion PDI˙−, which would subsequently be excited by absorption of a second photon to yield excited PDI˙−*, which should have a reduction potential strong enough to reduce 4-bromobenzaldehyde to generate the aryl-radical. This initial mechanistic proposal has been further analyzed by Balzani and Ceroni,48 who found the PDI˙− monoanion to be photostable and concluded that PDI˙−* cannot be the active photocatalyst. Instead, they suggested that the reaction does not proceed via a two-photon process, but rather viaPDI˙−* decomposition products that remain to be identified. It has further been shown that PDI˙−* features a very short lifetime in the sub-ns timescale,49 which renders bimolecular diffusion of photocatalyst and substrate poorly compatible. Thus, pre-organized photocatalyst–substrate pairs may play an important role in optimizing aryl radical generation and stabilization of reactive intermediates. Wenger proposed alternatively to generate the PDI dianion by chemical reduction, which is a closed-shell species featuring significantly longer excited state lifetimes.17With respect to the different potential mechanisms that may occur, it will be interesting to look into binding of substrates to the singly or doubly reduced versions of the PDI units (i.e. overall doubly and four-fold reduced supramolecular square 2). Furthermore, mechanistic studies with 2 could be used to reveal if substrate binding does influence the lifetime of excited states and alters the photocatalytic mechanism taking place. In order to answer these questions, time-resolved fluorescence and absorption studies are required. We believe that such detailed mechanistic studies will bring important insights into the respective mechanism and thus on the influence of substrate–photocatalyst pre-organization through non-covalent interactions.
Conclusions
We have investigated the photocatalytic properties of a heteroleptic supramolecular construct containing organic dye PDI. The photophysical properties of PDI are retained after self-assembly, and as a result the supramolecular square 2 is still an active photocatalyst for the dehalogenation of substrates like 4-bromobenzaldehyde 3. However, if no significant binding between substrate and supramolecular photocatalyst is given, conversions are somewhat lower compared to the use of the parent PDI 1. 4-Bromobenzaldehyde 3 does not bind in the cavity and as such no pre-organization effect can be expected for this substrate. Interestingly, larger flat aromatic molecules such as 9-bromophenanthrene 4 and 1-bromopyrene 5 do bind with significant affinity in the pocket, and for these substrates the conversions are relatively improved, yet not as good as when using the free PDI as photocatalyst. Further strengthening of the binding constant may improve the pre-organization effect, which would likely be possible by simply changing the solvent as it does play an important role in non-covalent host/guest interactions. As demonstrated, the binding strength can already be improved by using substrates with extended π-systems, but the results with these substrates are blurred as they can be directly activated by light to undergo dehalogenation reactions. Overall, we established that supramolecular squares containing organic photosensitizer PDI as ligand backbone can be used as photocatalysts for organic transformations, and pre-organization of the substrate affects its efficiency. For further development the binding strength of the substrate should be improved that could be reached using various strategies that we are currently exploring.Experimental
Materials and methods
Synthetic procedures are described below. All other chemicals were purchased from commercial suppliers and used without further purification. Dry solvents were purified using an MBraun SPS-800. NMR spectra were recorded on a Bruker DRX 500, AMX 400 or DRX 300, ESI-MS data were obtained from an HR-ToF Bruker Daltonik GmbH Impact II. UV-vis spectra were recorded on a Shimadzu UV-2600 spectrophotometer and fluorescence measurements were performed on an Edinburgh Instruments Spectrofluorometer FS5.Synthesis
PDI-Py2 ligand50 and supramolecular heteroleptic square 2,44 were prepared according to literature procedures.Host–guest binding studies
:1 model and the Nelder–Mead method.
Catalysis
All experiments were performed in duplicate. For irradiation, a Kessil lamp (PR160L 456 nm 34 W) was used. The reaction mixture was irradiated in a 10 mL Biotage Microwave tube and the progress was monitored by 1H-NMR. Catalyst (0.83 mM for 1, 0.42 mM for 2), substrate (10 μmol), NEt3 (80 μmol), 465 nm Kessil lamp (34 W) stirred for 3 or 24 hours in 600 μL DMF-d7. Conversion and yield determined by 1H-NMR using 1,3,5-trimethoxybenzene as external standard. All experiments were performed in duplicate. Due to irradiation the reaction temperature was 38 °C; in the dark reaction an oil bath was used to mimic this temperature increase.Author contributions
C. J. N. performed all the experimental work, performed data analysis, and contributed to writing the paper. S. P. guided the experimental work, contributed to data analysis, and wrote the manuscript. P. C. M. L. and R. P. provided precursor compounds for the synthesis. J. I. v. d. V. and J. N. H. R. contributed to data analysis and to reviewing/editing the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Simon Matthew for support with fluorescence measurements and Ed Zuidinga for assistance with mass spectrometry.Notes and references
- A. Méndez-Ardoy and D. M. Bassani, Faraday Discuss., 2015, 185, 549–558 RSC.
- D. F. Cauble, V. Lynch and M. J. Krische, J. Org. Chem., 2003, 68, 15–21 CrossRef CAS PubMed.
- F. Burg and T. Bach, J. Org. Chem., 2019, 84, 8815–8836 CrossRef CAS PubMed.
- J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed.
- T. Keijer, T. Bouwens, J. Hessels and J. N. H. Reek, Chem. Sci., 2021, 12, 50–70 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- C. Prentice, J. Morrisson, A. D. Smith and E. Zysman-Colman, Beilstein J. Org. Chem., 2020, 16, 2363–2441 CrossRef PubMed.
- J. D. Nguyen, E. M. D’Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734–4743 CrossRef CAS PubMed.
- N. Noto and S. Saito, ACS Catal., 2022, 12, 15400–15415 CrossRef CAS.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- C. Rosso, G. Filippini and M. Prato, Eur. J. Org. Chem., 2021, 2021, 1193–1200 CrossRef CAS.
- H. Li and O. S. Wenger, Angew. Chem., Int. Ed., 2022, 61, e202110491 CAS.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- Z. Chami, M. Gareil, J. Pinson, J. M. Saveant and A. Thiebault, J. Org. Chem., 1991, 56, 586–595 CrossRef CAS.
- W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201–210 CrossRef CAS PubMed.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889–3919 RSC.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969–984 CrossRef CAS.
- P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- R. Plessius, N. Orth, I. Ivanović-Burmazović, M. A. Siegler, J. N. H. Reek and J. I. van der Vlugt, Chem. Commun., 2019, 55, 12619–12622 RSC.
- X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100–109 CrossRef CAS PubMed.
- Y. Jiao, Y. Zuo, H. Yang, X. Gao and C. Duan, Coord. Chem. Rev., 2021, 430, 213648 CrossRef CAS.
- D. Rota Martir and E. Zysman-Colman, Chem. Commun., 2019, 55, 139–158 RSC.
- D. Rota Martir and E. Zysman-Colman, Coord. Chem. Rev., 2018, 364, 86–117 CrossRef CAS.
- I. Regeni, B. Chen, M. Frank, A. Baksi, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2021, 60, 5673–5678 CrossRef CAS PubMed.
- N. P. E. Barry, J. Furrer, J. Freudenreich, G. Süss-Fink and B. Therrien, Eur. J. Inorg. Chem., 2010, 725–728 CrossRef CAS.
- F. Würthner, A. Sautter, D. Schmid and P. J. A. Weber, Chem.–Eur. J., 2001, 7, 894–902 CrossRef.
- K. Mahata, P. D. Frischmann and F. Würthner, J. Am. Chem. Soc., 2013, 135, 15656–15661 CrossRef CAS PubMed.
- P. Spenst and F. Würthner, J. Photochem. Photobiol., C, 2017, 31, 114–138 CrossRef CAS.
- P. Spenst, A. Sieblist and F. Würthner, Chem.–Eur. J., 2017, 23, 1667–1675 CrossRef CAS PubMed.
- R. Plessius, V. Deij, J. N. H. Reek and J. I. van der Vlugt, Chem.–Eur. J., 2020, 26, 13241–13248 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- Y.-R. Zheng, Z. Zhao, M. Wang, K. Ghosh, J. B. Pollock, T. R. Cook and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 16873–16882 CrossRef CAS PubMed.
- J. Anibal, A. Malkani and B. Xu, Catal. Sci. Technol., 2020, 10, 3181–3194 RSC.
- M. Marchini, A. Gualandi, L. Mengozzi, P. Franchi, M. Lucarini, P. G. Cozzi, V. Balzani and P. Ceroni, Phys. Chem. Chem. Phys., 2018, 20, 8071–8076 RSC.
- N. T. La Porte, J. F. Martinez, S. Chaudhuri, S. Hedström, V. S. Batista and M. R. Wasielewski, Coord. Chem. Rev., 2018, 361, 98–119 CrossRef CAS.
- P. D. Frischmann and F. Würthner, Org. Lett., 2013, 15, 4674–4677 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2fd00179a |
| This journal is © The Royal Society of Chemistry 2023 |