
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-efficiency electrosynthesis of urea over bacterial cellulose regulated Pd–Cu bimetallic catalyst†
Shengbo
Zhang‡
ab,
Jing
Geng‡
ab,
Zhong
Zhao
ab,
Meng
Jin
ab,
Wenyi
Li
ab,
Yixing
Ye
ab,
Ke
Li
*c,
Guozhong
Wang
 ab,
Yunxia
Zhang
ab,
Huajie
Yin
ab,
Haimin
Zhang
*ab and
Huijun
Zhao
d
ab,
Yunxia
Zhang
ab,
Huajie
Yin
ab,
Haimin
Zhang
*ab and
Huijun
Zhao
d
aKey Laboratory of Materials Physics, Centre for Environmental and Energy Nanomaterials, Anhui Key Laboratory of Nanomaterials and Nanotechnology, CAS Center for Excellence in Nanoscience, Institute of Solid State Physics, Chinese Academy of Sciences, Hefei 230031, China. E-mail: zhanghm@issp.ac.cn
bUniversity of Science and Technology of China, Hefei 230026, China
cKey Laboratory of Agricultural Sensors, Ministry of Agriculture, School of Information and Computer, Anhui Agricultural University, Hefei 230026, China. E-mail: kelee@ustc.edu.cn
dCenter for Catalysis and Clean Energy, Griffith University, Gold Coast Campus, QLD4222, Australia
First published on 28th October 2022
Abstract
Ambient synthesis of urea through electrocatalytic coupling reaction of carbon dioxide (CO2) with nitrate (NO3−) has been regarded as a promising means to substitute the industrial energy- and capital-concentrated Haber–Bosch and Bosch–Meiser processes. Here we report the fabrication of PdCu alloying nanoparticles (NPs) anchored on carbonized bacterial cellulose (CBC) (PdCu/CBC) for ambient electrosynthesis of urea. As the electrocatalyst, the PdCu/CBC exhibits superior electrocatalytic activity toward urea synthesis with CO2 and NO3−, affording a remarkable urea yield rate of 763.8 ± 42.8 μg h−1 mgcat.−1 at −0.50 V (vs. RHE) and an exceptional faradaic efficiency (FE) of 69.1 ± 3.8% at −0.40 V (vs. RHE) under ambient conditions. The theoretical calculations unveil that this alloying catalyst provides Pd and Cu dual active sites with favored internal electron transferability, enabling generation of key *NO2 and *CO2 intermediates to facilitate C–N coupling reaction for urea synthesis. The operando spectroscopy characterization studies support the theoretical calculation results. To ensure the accuracy of the analysis data in this work, we investigated in detail the influence of the by-products in urea synthesis on the diacetyl monoxime colorimetric method and 1H nuclear magnetic resonance method for urea quantitative determination.
Broader contextUrea is one of the most essential nitrogen-based fertilizers in agricultural production and other chemical synthesis with an output of ∼100 million tons per year. Currently, the industrial synthesis of urea is highly dependent on the traditional Haber–Bosch and Bosch–Meiser processes under harsh reaction conditions. Recent studies have demonstrated that the electrocatalytic coupling of N2 with CO2 provides the feasibility for direct urea production under ambient conditions. Despite some achievements, the urea yield rate and faradaic efficiency are still very low, due to the high N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond energy, very low N2 solubility in aqueous electrolyte, and competitive N2 hydrogenation and CO2 hydrogenation reactions and so on. The coupling of CO2 and NO3− (or NO2−) in electrocatalysis could be an effective means to solve the above issues and realize high-efficiency urea production with high current efficiency. Herein, we reported an adsorption-regulated synthetic approach utilizing bacterial cellulose as the adsorption regulator to fabricate a Pd–Cu bimetallic catalyst, which as an electrocatalyst exhibited superior electrocatalytic C–N coupling activity toward urea production with a very high faradaic efficiency of 69.1 ± 3.8% at −0.40 V (vs. RHE), surpassing almost all of the currently reported urea synthesis electrocatalysts. To ensure the accuracy of the analysis data in this work, we investigated in detail the influence of the by-products in urea synthesis on the diacetyl monoxime colorimetric method and 1H NMR method for urea determination. N bond energy, very low N2 solubility in aqueous electrolyte, and competitive N2 hydrogenation and CO2 hydrogenation reactions and so on. The coupling of CO2 and NO3− (or NO2−) in electrocatalysis could be an effective means to solve the above issues and realize high-efficiency urea production with high current efficiency. Herein, we reported an adsorption-regulated synthetic approach utilizing bacterial cellulose as the adsorption regulator to fabricate a Pd–Cu bimetallic catalyst, which as an electrocatalyst exhibited superior electrocatalytic C–N coupling activity toward urea production with a very high faradaic efficiency of 69.1 ± 3.8% at −0.40 V (vs. RHE), surpassing almost all of the currently reported urea synthesis electrocatalysts. To ensure the accuracy of the analysis data in this work, we investigated in detail the influence of the by-products in urea synthesis on the diacetyl monoxime colorimetric method and 1H NMR method for urea determination.
|
Introduction
Urea (NH2CONH2) is not only the most widely utilized nitrogen fertilizer in agriculture, but also serves as the critical feedstock to produce fine chemicals such as urea-formaldehyde and barbiturates in chemical synthesis.1,2 Currently, the industrial synthesis of urea is highly dependent on the energy- and capital-concentrated Haber–Bosch and Bosch–Meiser processes, namely, through N2 + 6H+ + 6e− → 2NH3 and 2NH3 + CO2 → NH2CONH2 + H2O reactions under extreme conditions.3,4 These two processes for urea production not only consume enormous fossil energy, but also produce huge emissions of CO2.5–7 Undoubtedly, the exploration of renewable-energy-driven new techniques for urea production is highly needed for the sustainable development of our society.Recently reported studies have demonstrated that the electrochemical synthesis of urea under ambient conditions through an electrocatalytic coupling reaction of carbon dioxide (CO2) with nitrogen-containing precursors (e.g., N2, NO, NO3−, NO2−) has been widely accepted as a promising approach to substitute the industrial urea production process.8–29 At the beginning, the electrocatalytic coupling of N2 with CO2 was first investigated on Pd1Cu1/TiO2-400 and Bi–BiVO4 catalysts for urea synthesis under ambient conditions.9,10 In their studies, N2 can be effectively adsorbed and activated to generate *N2 intermediates, which promote CO2 reduction to form *CO, and then the generated *CO further reacts with *N2 to produce the desirable *NCON* intermediates via a thermodynamically feasible electrochemical C–N coupling reaction.9,10 Despite some achievements, the electrocatalytic urea synthesis performance is still low (urea yield rate <6.0 mmol h−1 g−1, faradaic efficiency <13%) in a N2-incorporated reaction system, mainly owing to the high bond energy of NN without dipole, the low solubility of N2 in aqueous electrolyte, and the competitive hydrogen evolution reaction (HER).9,10,30 To solve these issues caused by the N2 precursor, nitrate (NO3−) and nitrite (NO2−) in aqueous electrolytes have been investigated as the nitrogen sources coupling with CO2 for electrocatalytic synthesis of urea under ambient conditions.16–28 In comparison with the N2 reactant, the superiorities of NO3− and NO2− as the reactants are their high solubility in aqueous solution and the lower dissociation energy of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (204 kJ mol−1), which enable more efficient C–N coupling reaction for urea production.16–28 Besides this, the development of high-efficiency electrocatalysts is also critically important for urea synthesis. To date, precious and non-precious metal based electrocatalysts have been fabricated and investigated for electrocatalytic coupling of NO3− and NO2− (or NO) and CO2 to efficiently synthesize urea under ambient conditions, exhibiting high electrocatalytic activities.16–29 In terms of catalytic mechanistic research, Zhang et al. reported that the formation of *CO and *NH2 intermediates through the adsorption and activation of CO2 and NO on a Zn nanobelt catalyst is critically important for subsequent C–N coupling synthesis of urea with a yield rate of 15.13 mmol h−1 g−1 and a faradaic efficiency of 11.26% at ∼40 mA cm−2.29 Meanwhile, Zhang et al. synthesized urea by coupling CO2 with NO2− on oxygen vacancy-rich ZnO porous nanosheets with a yield rate of 16.56 μmol h−1 and a faradaic efficiency of 23.26% at −0.79 V (vs. RHE).26 Yu et al. revealed that the key steps during electrocatalytic urea synthesis are the generation of *NO2 and *CO2 intermediates through the adsorption and activation of CO2 and NO3− on In(OH)3 with exposed {100} facets and InOOH with oxygen vacancies.17,18 Recently, Rose Amal and co-workers reported a single-atom electrocatalyst with Cu–N4 sites to couple CO2 and NO3− with a urea yield rate of 4.3 mmol s−1 cm−2 and a faradaic efficiency (FE) of 28% at −0.9 V (vs. RHE).22 On the basis of these reported works, it can be concluded that design and construction of dual active sites in an electrocatalyst are highly desirable for high-efficiency generation of CO2 and NO3− (or NO2−) derived active intermediates to synthesize urea under ambient conditions.
O bond (204 kJ mol−1), which enable more efficient C–N coupling reaction for urea production.16–28 Besides this, the development of high-efficiency electrocatalysts is also critically important for urea synthesis. To date, precious and non-precious metal based electrocatalysts have been fabricated and investigated for electrocatalytic coupling of NO3− and NO2− (or NO) and CO2 to efficiently synthesize urea under ambient conditions, exhibiting high electrocatalytic activities.16–29 In terms of catalytic mechanistic research, Zhang et al. reported that the formation of *CO and *NH2 intermediates through the adsorption and activation of CO2 and NO on a Zn nanobelt catalyst is critically important for subsequent C–N coupling synthesis of urea with a yield rate of 15.13 mmol h−1 g−1 and a faradaic efficiency of 11.26% at ∼40 mA cm−2.29 Meanwhile, Zhang et al. synthesized urea by coupling CO2 with NO2− on oxygen vacancy-rich ZnO porous nanosheets with a yield rate of 16.56 μmol h−1 and a faradaic efficiency of 23.26% at −0.79 V (vs. RHE).26 Yu et al. revealed that the key steps during electrocatalytic urea synthesis are the generation of *NO2 and *CO2 intermediates through the adsorption and activation of CO2 and NO3− on In(OH)3 with exposed {100} facets and InOOH with oxygen vacancies.17,18 Recently, Rose Amal and co-workers reported a single-atom electrocatalyst with Cu–N4 sites to couple CO2 and NO3− with a urea yield rate of 4.3 mmol s−1 cm−2 and a faradaic efficiency (FE) of 28% at −0.9 V (vs. RHE).22 On the basis of these reported works, it can be concluded that design and construction of dual active sites in an electrocatalyst are highly desirable for high-efficiency generation of CO2 and NO3− (or NO2−) derived active intermediates to synthesize urea under ambient conditions.
Herein, bacterial cellulose (BC) was used as the adsorption regulator to fabricate PdCu alloying nanoparticles anchored on the carbonized BC (PdCu/CBC). The as-synthesized PdCu/CBC shows a Pd to Cu molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and a surface area of 162.4 m2 g−1 with microporous and mesoporous structures. In the presence of CO2 and NO3−, the PdCu/CBC as the electrocatalyst exhibits superior electrocatalytic activity toward urea synthesis with a remarkable urea yield rate of 763.8 ± 42.8 μg h−1 mgcat.−1 and a faradaic efficiency (FE) of 59.7 ± 3.4% at −0.50 V (vs. RHE) under ambient conditions. The high urea synthesis performance can be ascribed to PdCu alloying catalysts with favored internal electron transfer properties, simultaneously providing Pd and Cu dual active sites for the adsorption and activation of CO2 and NO3− to generate their corresponding active intermediates for facilitating C–N coupling reaction, confirmed by the theoretical and experimental results. The findings in this work would be helpful for the design and development of high-efficiency electrocatalysts for electrochemical C–N coupling to synthesize urea.
:1 and a surface area of 162.4 m2 g−1 with microporous and mesoporous structures. In the presence of CO2 and NO3−, the PdCu/CBC as the electrocatalyst exhibits superior electrocatalytic activity toward urea synthesis with a remarkable urea yield rate of 763.8 ± 42.8 μg h−1 mgcat.−1 and a faradaic efficiency (FE) of 59.7 ± 3.4% at −0.50 V (vs. RHE) under ambient conditions. The high urea synthesis performance can be ascribed to PdCu alloying catalysts with favored internal electron transfer properties, simultaneously providing Pd and Cu dual active sites for the adsorption and activation of CO2 and NO3− to generate their corresponding active intermediates for facilitating C–N coupling reaction, confirmed by the theoretical and experimental results. The findings in this work would be helpful for the design and development of high-efficiency electrocatalysts for electrochemical C–N coupling to synthesize urea.
Results and discussion
In this work, bacterial cellulose (BC) was used as the adsorption regulator to synthesize PdCu alloying nanoparticles anchored on carbonized bacterial cellulose (CBC) (denoted PdCu/CBC) by a synthetic approach combining wet-chemistry impregnation with a carbonization fixation process (Fig. S1, ESI†).31,32 The pre-treated BC with rich oxygen groups (Fig. 1a) was used as the adsorption regulator to controllably impregnate Pd2+/Cu2+ ions. The scanning electron microscopy (SEM) image in Fig. 1b showed the maintenance of the fiber-like carbon structure of the CBC after anchoring the alloying nanoparticles. The transmission electron microscopy (TEM) image of the PdCu/CBC in Fig. 1c displays homogeneous PdCu nanoparticles loaded onto the carbon support. The high-resolution TEM (HRTEM) image indicates that the lattice distance of an individual nanoparticle is 0.22 nm (inset in Fig. 1c), attributed to the (111) interplanar distance of the face-centred cubic (FCC) PdCu alloy,33 and the HRTEM image of the nanoparticles located at different areas also shows the same results (Fig. S2, ESI†), confirming the formation of PdCu alloying nanoparticles on the CBC. For a meaningful comparison, Pd/CBC and Cu/CBC were also fabricated using a similar synthetic method to the PdCu/CBC except for only use of Pd2+ or Cu2+ as a precursor. Fig. S3 and S4 (ESI†) show the TEM images, elemental mapping images and X-ray photoelectron spectroscopy (XPS) analysis results of the Pd/CBC and Cu/CBC. To further confirm the formation of PdCu alloying nanoparticles on the CBC, the powder X-ray diffraction (XRD) measurements of the Pd/CBC, Cu/CBC and PdCu/CBC were performed, as shown in Fig. 1d. It can be clearly seen that the XRD patterns of the Pd/CBC, Cu/CBC and PdCu/CBC are consistent with the metallic Pd phase (JCPDS Card No. 46-1043), metallic Cu phase (JCPDS Card No. 04-0836) and PdCu alloying phase (JCPDS Card No. 48-1551), respectively.34–36 Moreover, the XRD patterns of the PdCu/CBC are obviously different from those of the Pd/CBC and Cu/CBC samples. In this work, the XPS technique was also employed to investigate the chemical composition and chemical state of the Pd/CBC, Cu/CBC and PdCu/CBC. The high-resolution C 1s (Fig. S3c, S4c and S5b, ESI†) and O 1s (Fig. S3d, S4d and S5c, ESI†) XPS spectra confirm the presence of rich O groups and the formation of Pd–O, Cu–O and Pd/Cu–O bonds in the Pd/CBC, Cu/CBC and PdCu/CBC, respectively.31,32 The high-resolution Pd 3d and Cu 2p XPS spectra were measured to analyze the electronic properties of PdCu alloy (Fig. 1e and f). Compared to the sole Pd/CBC and Cu/CBC, the binding energy of Pd 3d5/2 for the Pd/CBC is located at ∼335.7 eV, while the binding energy of the PdCu/CBC is shifted slightly toward the high binding energy direction to ∼336.0 eV.9,37 The binding energy of Cu 2p3/2 for the Cu/CBC is located at ∼932.9 eV, while a slight shift toward the low binding energy direction occurs for the PdCu/CBC (∼932.3 eV).9,37 The above XPS results indicate the formation of PdCu alloy with favored internal electron transfer between Pd and Cu,9,37 favourable for electrocatalysis. The elemental composition of the PdCu/CBC was further investigated by using energy-dispersive X-ray spectroscopy (EDX) (Fig. 1g) and inductively coupled plasma atomic emission spectrometry (ICP-AES) (Table S1, ESI†), and the obtained results suggest that the Pd to Cu molar ratio is about 1:1 for the PdCu/CBC. The corresponding elemental mapping images confirm that the Pd and Cu elements share similar distributions, implying the formation of PdCu alloying nanoparticles (Fig. 1g). The Brunauer–Emmett–Teller (BET) measurement indicates that the PdCu/CBC has a surface area of 162.4 m2 g−1 with microporous and mesoporous structures (Fig. S6, ESI†), which would be helpful for the exposure of catalytically active sites and mass transport during electrocatalysis.31,32
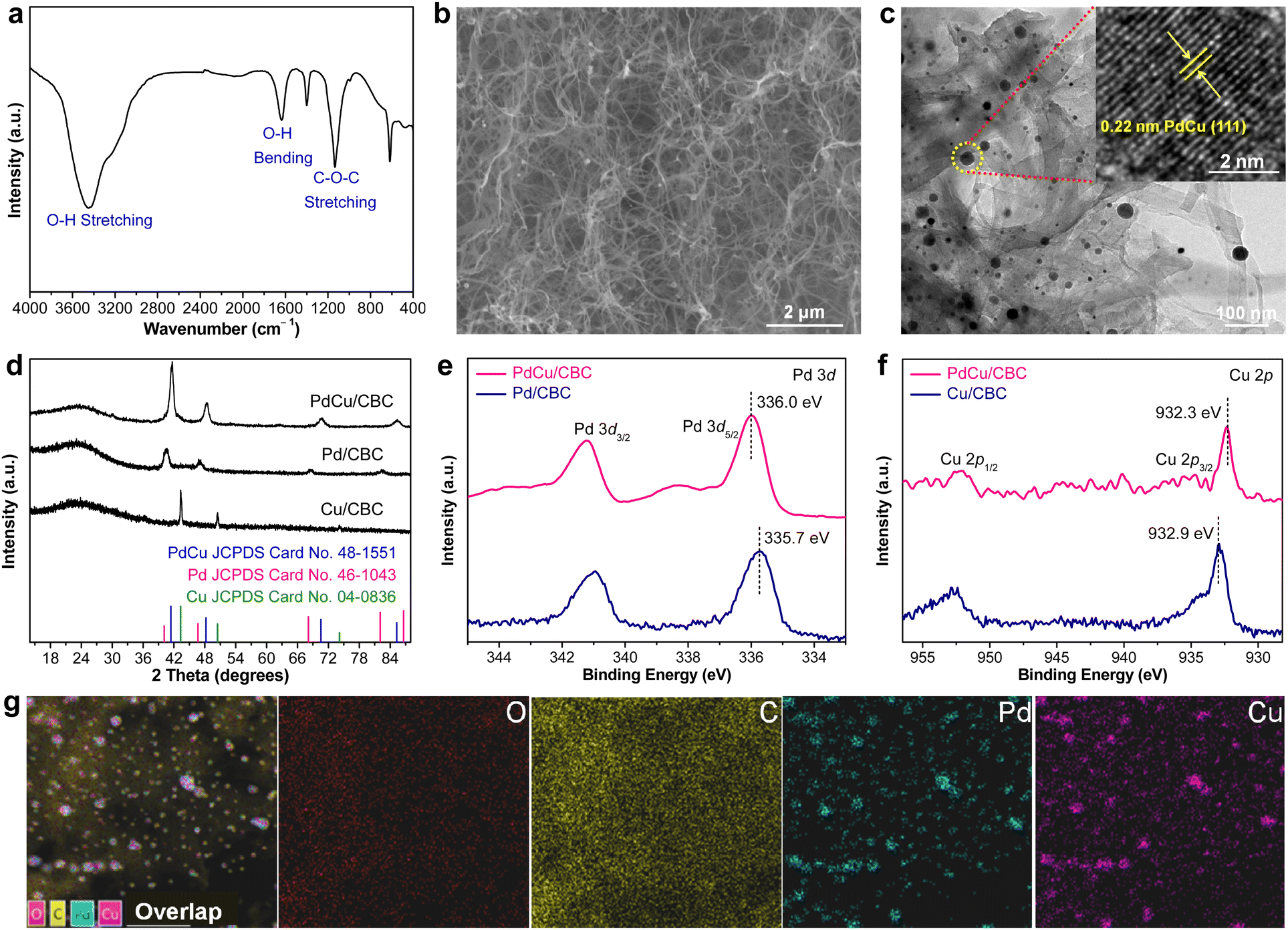 | ||
| Fig. 1 (a) FT-IR spectrum of BC. (b) SEM and (c) TEM images of the PdCu/CBC. (d) XRD patterns of the Pd/CBC, Cu/CBC and PdCu/CBC. (e) Pd 3d XPS spectra of the Pd/CBC and PdCu/CBC. (f) Cu 2p XPS spectra of the Cu/CBC and PdCu/CBC. (g) Corresponding elemental mapping images of the PdCu/CBC. | ||
The as-fabricated PdCu/CBC electrocatalyst was subsequently evaluated for the electrocatalytic urea synthesis by coupling NO3− and CO2 under ambient conditions. All electrochemical measurements were performed in Ar- or CO2-saturated 0.05 M KNO3 electrolyte using an H-type two-compartment electrochemical cell, which was separated with a Nafion 211 membrane and equipped with three-electrode configurations (see the Experimental section for details, ESI†). Fig. 2a presents the linear sweep voltammetry (LSV) curves of the PdCu/CBC in CO2- and Ar-saturated 0.05 M KNO3 electrolytes. Interestingly, it was found that the current density was lowered by CO2 feeding within the investigated potential range in comparison with that obtained in Ar-saturated electrolyte. In the Ar-saturated electrolyte, the obtained cathodic current densities at the given applied potentials could be ascribed to the NO3− reduction reaction (NO3−RR) and/or hydrogen evolution reaction (HER).17 With the introduction of CO2, the obviously decreased cathodic current densities at the given applied potentials could mean the effective inhibition of the NO3−RR and/or HER, thus facilitating the C–N coupling reaction for urea synthesis, consistent with recently reported works.17 To ensure the accuracy of the analysis data, we investigated in detail the reliability of the diacetyl monoxime colorimetric method and the 1H nuclear magnetic resonance (1H NMR) method for urea product determination in this work. The detailed experimental results and discussions are presented in Fig. S7–S12 (ESI†). The results demonstrate that during the electrocatalytic synthesis of urea, concurrently produced by-products such as NH3 and NO2− have important influences on the measurement results of urea using the diacetyl monoxime colorimetric method (Fig. S8–S10, ESI†), consistent with the recently reported work by Wang et al.21 In contrast, the influence of NH3 and NO2− by-products is ignorable for the urea quantitative determination using the 1H NMR method (Fig. S11 and S12, ESI†). Therefore, the 1H NMR method was employed to qualitatively and quantitatively measure the produced urea in this work. The above experimental results could be helpful for other researchers to avoid possible false positive results of the urea measurement. As shown in Fig. S12b (ESI†), the 1H NMR spectra of the products display three peaks identified as 14NH2CO14NH2 (∼5.6 ppm), d6-DMSO (as an internal standard) (∼2.5 ppm) and H2O (∼4.7 ppm), while no other liquid products can be detected, indicating the high selectivity of the PdCu/CBC toward urea synthesis through the C–N coupling reaction. Fig. 2b shows the dependence of the urea yield rate (Rurea) and faradaic efficiency (FE) on the applied potential in CO2-saturated 0.05 M KNO3 electrolyte over a 2 h period. The time-dependent current density curves at different potentials and the corresponding 1H NMR spectra of the samples are shown in Fig. S12 (ESI†). The as-synthesized PdCu/CBC exhibits high electrocatalytic activity toward urea synthesis, affording a large Rurea of 763.8 ± 42.8 μg h−1 mgcat.−1 at −0.50 V (vs. RHE) with a FE of 59.7 ± 3.4%. The highest FE that can be achieved was found to be 69.1 ± 3.8% at −0.40 V (vs. RHE). In order to ensure the accuracy of the 1H NMR results, the urease decomposition method was also adopted in this work (Fig. S13, ESI†). The quantitative analysis results based on the urease decomposition method demonstrate that the yielded urea is 785.2 ± 39.2 μg h−1 mgcat.−1, almost identical to that obtained from the NMR results. For a meaningful comparison, the Pd/CBC and Cu/CBC as electrocatalysts were also evaluated for urea synthesis. At −0.50 V (vs. RHE) in CO2-saturated 0.05 M KNO3 electrolyte over a 2 h period, the Rurea and FE are 215.1 ± 11.8 μg h−1 mgcat.−1 and 17.4 ± 1.0% for the Pd/CBC and 314.2 ± 19.5 μg h−1 mgcat.−1 and 27.0 ± 1.7% for the Cu/CBC, respectively (Fig. 2c and Fig. S14, ESI†). The electrocatalytic performance of urea synthesis with CO2 and NO3− exceeds those of most of the recently reported electrocatalysts of urea synthesis (Fig. 2d and Table S2, ESI†). Apparently, the PdCu/CBC displays far superior electrocatalytic urea synthesis performance, possibly owing to the existence of Pd and Cu dual sites with favored internal electron transferability. Besides the targeted urea product, the by-products NH3, NO2−, CO and H2 are inevitably produced during electrocatalysis, and also determined by the colorimetric methods and gas chromatography (GC) methods (Fig. S15–S18, ESI†). In contrast to the produced urea, the electrocatalytic activity toward the by-products NH3 and NO2− is obviously lower, meaning a higher electrocatalytic selectivity of the PdCu/CBC toward urea synthesis. Meanwhile, no HCOOH or N2H4 product was detected during electrocatalysis (Fig. S12 and S19, ESI†). On the basis of the detected products during electrocatalytic urea synthesis, the faradaic efficiency (FE) of each electrocatalytic product at different potentials was calculated and is shown in Fig. S20 (ESI†). Obviously, the produced urea is the primary product at potentials from −0.20 V to −0.60 V (vs. RHE), indicating a high selectivity of the PdCu/CBC toward urea synthesis. The stability test of the PdCu/CBC was conducted using CO2-saturated 0.05 M KNO3 electrolyte at −0.50 V (vs. RHE) over a 10 h period (Fig. 2e). As shown, the obtained chronoamperometric curve displays an almost insignificant change in current density over the entire 10 h testing period (Fig. 2e). Significantly, the urea yield shows a good linear relationship with reaction time (Fig. 2f and Fig. S21, ESI†); moreover, the change in the corresponding FE is slight (Fig. 2g), signifying high stability of the PdCu/CBC for electrocatalytic urea synthesis. The cycling stability of the PdCu/CBC was examined using CO2-saturated 0.05 M KNO3 electrolyte at −0.50 V (vs. RHE) with a 2 h testing cycle for 8 consecutive cycles (Fig. S22, ESI†). As shown in Fig. 2h, the obtained Rurea and FE for 8 consecutive cycles show slight changes, demonstrating good cycling stability. The high stability of the PdCu/CBC can be attributed to its structural stability as evidenced by the XRD patterns, and TEM and elemental mapping images of the PdCu/CBC after 8 consecutive cycles (Fig. S23, ESI†). To confirm the produced urea that originated from the PdCu/CBC catalyzed coupling reaction of CO2 with NO3−, the isotope labelling experiments were subsequently performed at −0.50 V (vs. RHE) for 2 h measurement using CO2-saturated 0.05 M K14NO3 and K15NO3 electrolytes. The 1H NMR spectra in Fig. 3a indicate that the produced sample that originated from 14NO3− shows a chemical shift at ∼5.59 ppm, consistent with the standard 14NH2CO14NH2, while the 15NO3− converted product displays chemical shifts at ∼5.48 and ∼5.70 ppm, completely consistent with the standard 15NH2CO15NH2. These isotope labelling experimental results verify that the urea produced in this work is indeed from the PdCu/CBC catalyzed coupling reaction of CO2 with NO3−. In addition, the quantitative analysis based on the 1H NMR spectra of the standard 14NH2CO14NH2 and 15NH2CO15NH2 (Fig. S11 and S24, ESI†) was also carried out in this work. The experimental results demonstrate that using CO2-saturated 0.05 M K14NO3 and K15NO3 electrolytes, the Rurea values of the yielded 14NH2CO14NH2 and 15NH2CO15NH2 are 763.8 ± 42.8 μg h−1 mgcat.−1 and 750.8 ± 34.3 μg h−1 mgcat.−1, respectively. As shown in Fig. 3b, the typical 14NH2CO14NH2/15NH2CO15NH2 signal at 162.6 ppm is detectable for the electrolyte sample collected after 2 h electrocatalysis in 13C NMR spectra. Furthermore, the typical 15NH2CO15NH2 signal at 76.8 ppm is detectable for the electrolyte sample collected after 2 h electrolysis in 15N NMR spectra (Fig. S25, ESI†). Thus, it can be concluded that the PdCu/CBC electrocatalyst exhibits high electrocatalytic activity toward ambient urea synthesis.
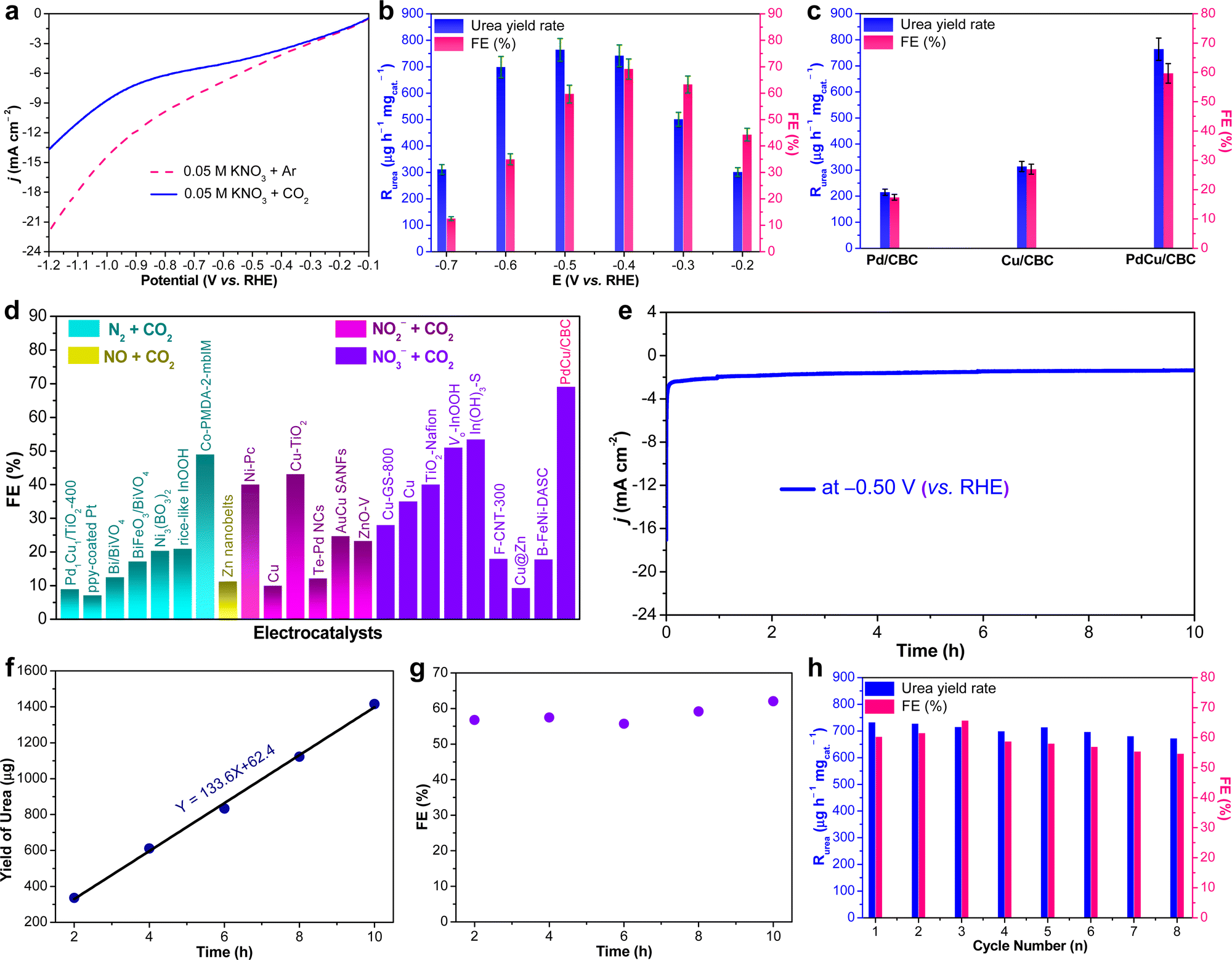 | ||
| Fig. 2 (a) LSV curves of the PdCu/CBC in 0.05 M KNO3 electrolyte with Ar or CO2 feeding gas. (b) Dependence of Rurea and FE on the applied potentials. (c) Rurea and FE of the PdCu/CBC, Cu/CBC and Pd/CBC at −0.50 V (vs. RHE) for 2 h reaction. (d) Comparison of the FEs of the reported electrocatalysts and the PdCu/CBC with different nitrogen sources. (e) Stability test of the PdCu/CBC at −0.50 V (vs. RHE). (f) The urea yield and (g) FE for urea production over the PdCu/CBC toward electrochemical coupling NO3− with CO2 with reaction time at −0.50 V (vs. RHE). (h) Recycling stability test of the PdCu/CBC. | ||
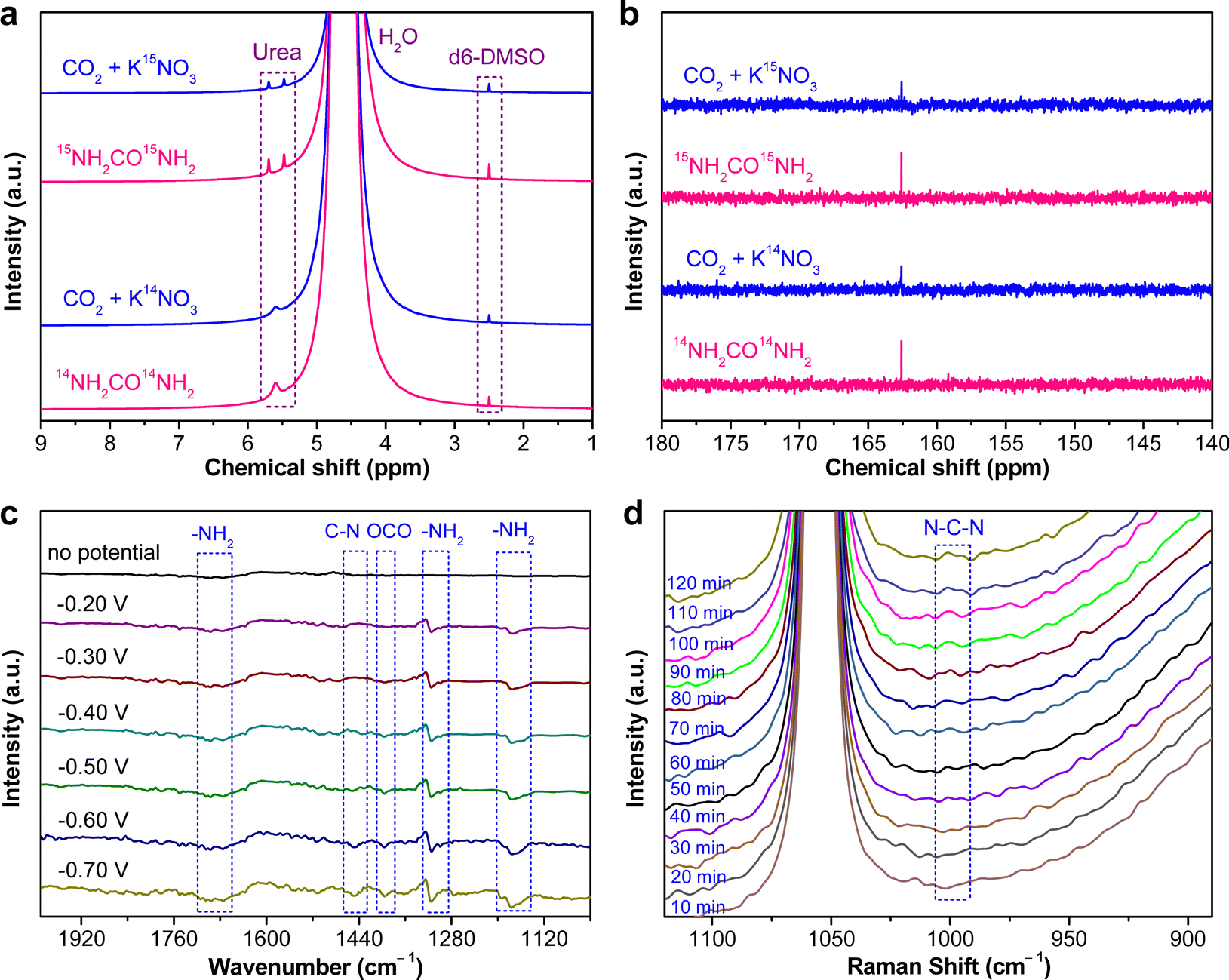 | ||
| Fig. 3 (a) 1H NMR spectra of 0.05 M 14KNO3/15KNO3 electrolytes saturated with CO2 after 2 h electrolysis and standard 14NH2CO14NH2/15NH2CO15NH2 solutions. (b) 13C NMR spectra of 0.05 M 14KNO3/15KNO3 electrolytes saturated with CO2 after 2 h electrolysis and standard 14NH2CO14NH2/15NH2CO15NH2 solutions. (c) Operando SR-FTIR spectroscopy measurements under various potentials for the PdCu/CBC in CO2-saturated 0.05 M KNO3 electrolyte. (d) Operando Raman spectroscopy measurements of the PdCu/CBC in CO2-saturated 0.05 M KNO3 electrolyte at −0.50 V (vs. RHE) for 2 h reaction. | ||
To eliminate the interferences of other factors on the produced urea, several controllable experiments were conducted as follows: (i) Ar-saturated 0.05 M KNO3; (ii) CO2-saturated 0.05 M KNO3 without any applied potential (open-circuit); (iii) CO2-saturated 0.05 M K2SO4 without any nitrogen-containing precursor; and (iv) carbon paper without the PdCu/CBC electrocatalyst. As shown in Fig. S26 (ESI†), only a negligible amount of urea product is detectable for all cases, further indicating that the produced urea is indeed from the electrocatalytic coupling reaction of CO2 with NO3− on the PdCu/CBC. To illustrate that the PdCu/CBC exhibited enhanced C–N coupling activity, the as-synthesized PdCu alloy nanoparticles anchored on the conventional carbon black (denoted PdCu/CB) and carbonized bacterial cellulose (CBC) (denoted CBC) were also used as the electrocatalysts for the C–N coupling reaction (Fig. S27, ESI†). At −0.50 V (vs. RHE) for 2 h, no urea product can be detected for the CBC, while the PdCu/CB shows a urea yield rate of 424.4 ± 24.6 μg h−1 mgcat.−1 and a FE of 33.3 ± 2.0% in CO2-saturated 0.05 M KNO3 electrolyte, apparently lower than those (763.8 ± 42.8 μg h−1 mgcat.−1 and 59.7 ± 3.4%, respectively) obtained from the PdCu/CBC (Fig. S27, ESI†). This is because the rich oxygen functional groups in the biomass-derived materials (i.e., bacterial cellulose) have a superior coordination ability with metal ions, which can be advantageously utilized to control the impregnation/loading of metal ions. More attractively, these chemically impregnated metal ions can be in situ converted to a nanoalloy anchored on the carbon support via M–O coordination configurations during the simple carbonization process. This unique oxygen coordination structure may be conducive to the adsorption and activation of CO2 and NO3− to generate their corresponding active intermediates for facilitating the C–N coupling reaction.
To unveil the electrocatalytic activity mechanism of C–N coupling to urea, operando spectroscopic studies such as the advanced operando synchrotron radiation FTIR (SR-FTIR) and operando Raman measurements were performed to study the electrocatalytic urea synthesis by coupling CO2 and NO3− on the PdCu/CBC over the potential range from −0.20 to −0.70 V (vs. RHE) in this work (Fig. S28 and S29, ESI†).17Fig. 3c shows the results of the SR-FTIR measurements. As shown, without the applied potential, there is no infrared absorption peak in the SR-FTIR spectra. In the investigated potential range, the infrared bands at ∼1685 and ∼1176 cm−1 are very obvious, which are attributed to the bending and rocking modes of –NH2.17,18,38 The wagging mode of –NH2 is also found at ∼1317 cm−1.17,18,39 With the applied potential increasing from −0.20 to −0.40 V (vs. RHE), a new weak vibration band at ∼1448 cm−1 appears, and its intensity is apparently increased with the potential further increasing from −0.40 to −0.70 V (vs. RHE), assignable to the stretching vibration of C–N.17,18,38,40 The operando SR-FTIR results show that the urea synthesis by the electrocatalytic C–N coupling reaction of CO2 with NO3− is successfully achievable, supportable for the electrocatalytic experimental results aforementioned. In addition, the infrared band belonging to *OCO* species adsorbed on the active sites appears at 1394 cm−1, corresponding to its symmetric stretching mode.17,18Fig. 3d shows the operando Raman measurements for the electrocatalytic urea synthesis at −0.50 V (vs. RHE) for 2 h. The Raman intensity of the peak at around 1000 cm−1, corresponding to the N–C–N stretching of urea, is increased obviously from 30 to 120 min, implying that the coupling reaction of CO2 with NO3− takes place gradually with reaction time under the given electrocatalytic conditions.41,42 The above operando SR-FTIR and Raman spectroscopy measurements confirm the successful realization of C–N coupling with CO2 and NO3− over the PdCu/CBC in this work.
To deeply understand the intrinsic reason for the high electrocatalytic urea synthesis activity of the PdCu/CBC, we conducted the CO2 temperature-programmed desorption (CO2-TPD) experiments for PdCu/CBC and CBC samples. As shown in Fig. 4a, the CBC substrate only shows a very weak CO2 chemisorption peak at ∼255 °C, while the PdCu/CBC exhibits substantial enhancements in the CO2 chemisorption at ∼360 and 436 °C, ascribed to the chemical adsorption of CO2 on PdCu sites. As such, the binding strength of CO2 chemisorption on the PdCu/CBC is remarkably stronger than that on the CBC, meaning the enhanced adsorption and activation of CO2 on the PdCu/CBC for high-efficiency urea synthesis. To deeply understand the C–N coupling mechanism of the PdCu/CBC, the density functional theory (DFT) calculations were performed in this work. Fig. 4b shows the optimized structural model of PdCu alloy with an exposed (111) plane, and the Pd and Cu sites in PdCu alloy are the catalytically active centers for the adsorption and activation of CO2 and NO3−, respectively. The Bader charge analysis shows that there is 1.65 e− charge transfer between Pd and Cu sites in the PdCu/CBC. The findings confirmed an apparent electron transfer between Cu and Pd for the formation of PdCu alloy, consistent with the XPS analysis. Such electron transfer changed the electronic states of metals and their d-band centers.43,44Fig. 4c shows the projected density of states (PDOS) of the Pd-4d or Cu-3d orbitals of the PdCu/CBC, pristine Pd and pristine Cu. It was noted that under the same strain conditions, the d-band centers of pristine-Pd/Cu and Pd/Cu in the PdCu/CBC differed from each other. The d-band center value of Pd atoms in the PdCu/CBC was calculated to be −1.406 eV, showing an upshift in comparison with that (−1.819 eV) for pristine Pd. Similarly, the d-band center value of Cu atoms upshifted from −2.313 eV to −1.528 eV after the formation of PdCu alloy. This is due to the fact that the position of the d-band center is modulated by the electron distribution due to the charge transfer.43,44 The increased d-band center value means that the PdCu nanoalloy can provide more d-bands to the adsorbate as compared with monometallic Pd or Cu, favoring the strong adsorption and activation of CO2 and NO3− to generate their corresponding active intermediates for facilitating the C–N coupling reaction. Fig. 4d shows the free energy profile and reaction pathway of the C–N coupling on the PdCu(111) plane, and the optimized intermediate structure corresponding to each reaction step is displayed in Fig. S30 and S31 (ESI†). The electrocatalytic urea synthesis on PdCu is triggered by the thermodynamically spontaneous reduction reaction from HNO3 to *HNO3 with an energy output of −3.38 eV. Then, *HNO3 is further reduced to *NO2 with an energy output of −0.87 eV. Moreover, we performed the calculations on *CO2 conversion to *COOH or *CO on the PdCu(111) plane (Fig. S32, ESI†).45 The results indicated that the corresponding kinetic energy barrier is 1.32 eV for the formation of *COOH on PdCu(111). Direct dissociation of CO2 into adsorbed *CO and *O is unlikely to occur on the PdCu(111) plane due to the very large kinetic energy barrier of 2.82 eV. The further conversion of *COOH to *CO by breaking the strong C–O bond is also slow in kinetics, and the decomposition of *COOH to *CO + *OH encounters a large kinetic energy barrier of 1.32 eV. The elementary steps of hydrogenation of *CO2 to *COOH and *COOH to *CO both have large energy barriers of ∼1.3 eV. Importantly, it was found that the process from *NO2 to *CO2NO2 with an energy output of –1.54 V is more thermodynamically favourable than the hydrogenation of *CO2 to *COOH or *CO with an energy output of 1.3 eV (Fig. S32, ESI†) and the protonation of *NO2 to *HNO2 with an energy output of −1.28 V (inset in Fig. 4d and Fig. S33, ESI†), meaning more favored C–N coupling reaction with high selectivity. For the subsequent steps from *CO2NO2 to *CONH2NH2, most of the protonation processes are thermodynamically spontaneous; only three steps are uphill in energy, namely, the protonation of *CO2NO2 to *CO2NOOH with an uphill energy of 0.87 V, *CO2NO to *CO2NHO with an uphill energy of 0.64 V, and *CO2NH2 to *COOHNH2 with an uphill energy of 0.20 V. Obviously, the protonation of *CO2NO2 to *CO2NOOH could be the determining step for the overall urea production process.
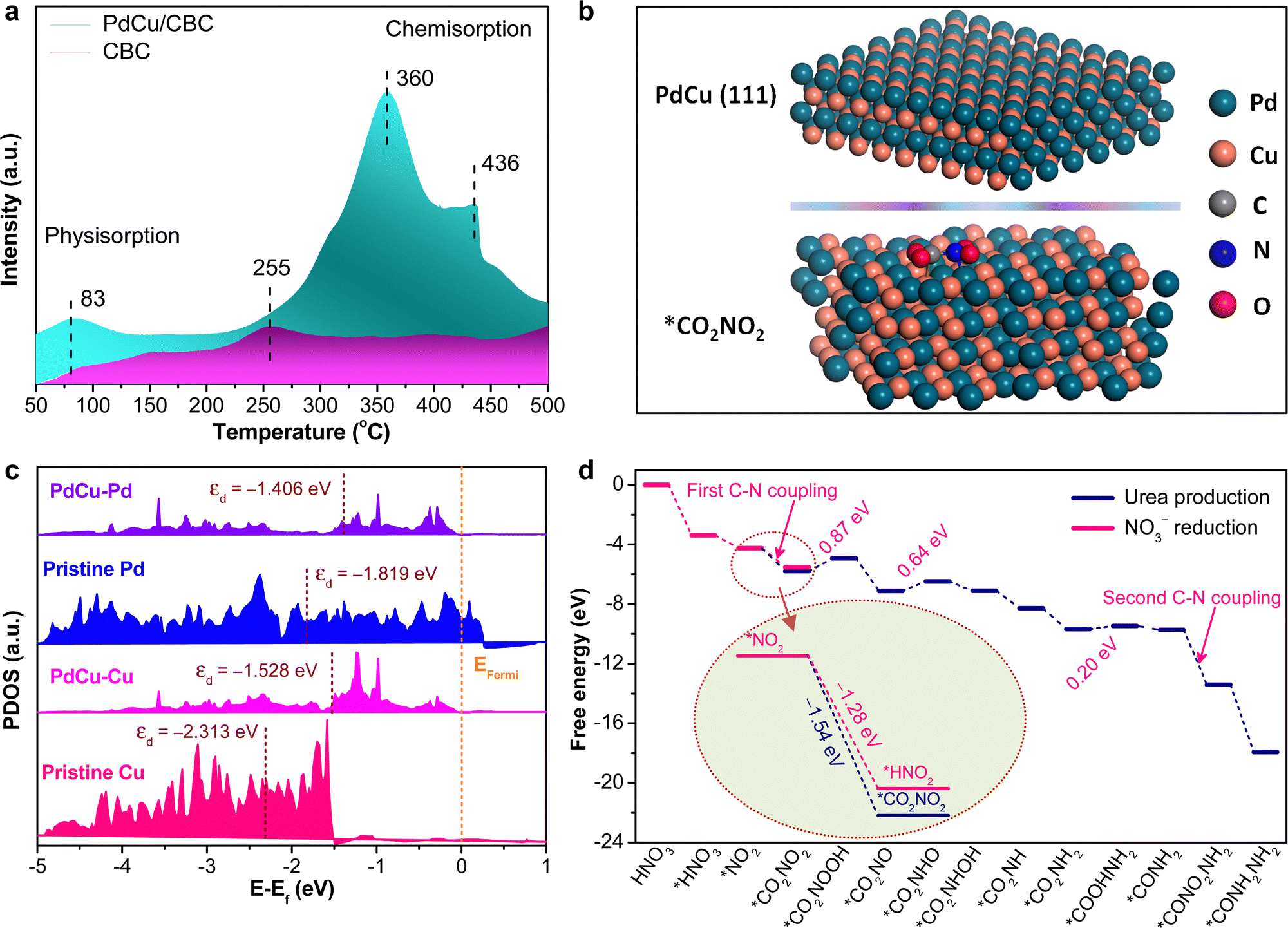 | ||
| Fig. 4 (a) CO2-TPD spectra of the PdCu/CBC and CBC. (b) DFT optimised configurations of the PdCu(111) plane and optimized geometric structure of the intermediate *CO2NO2 on PdCu(111). The green, orange, blue, red, white and grey balls represent Pd, Cu, N, O, H and C atoms, respectively. (c) The projected density of states (PDOS) onto the Pd-4d or Cu-3d orbitals of the PdCu/CBC, pristine Pd and pristine Cu. The dashed line represents the d-band center of Pd or Cu atoms. The Fermi level is set at 0 eV. (d) Free-energy diagram for urea production of the PdCu/CBC. | ||
Conclusions
In summary, we exemplified an adsorption-regulated synthetic strategy to fabricate PdCu alloying nanoparticles on carbonized bacterial cellulose (PdCu/CBC) for electrocatalytic synthesis of urea with CO2 and NO3− under ambient conditions. The results demonstrated that the as-synthesized PdCu/CBC provided Pd and Cu dual active sites with favored internal electron transferability, enabling adsorption and activation of CO2 and NO3− for high-efficiency electrocatalytic urea production under ambient conditions. Employing the PdCu/CBC electrocatalyst, the maximum urea yield rate achieved was found to be 763.8 ± 42.8 μg h−1 mgcat.−1 with a faradaic efficiency of 59.7 ± 3.4 at −0.50 V (vs. RHE) and high stability. The operando spectroscopy characterization studies combined with the theoretical calculations unveiled the catalytically active mechanisms of C–N coupling to generate urea with CO2 and NO3− through a coupling protonation process. The findings in this work would be helpful for the design and development of dual active site dominated electrocatalysts for high-efficiency C–N coupling to produce urea.Author contributions
H. Zhang and S. Z. conceived the concept and designed the experiments. S. Z. and J. G. fabricated the catalysts and performed the material characterization and electrochemical measurements. K. L. performed DFT calculations. Z. Z., M. J., W. L., Y. Y., G. W., Y. Z., H. Y. and H. Zhao contributed to the experimental design. H. Zhang and S. Z. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 52172106 and 51872292), Anhui Provincial Natural Science Foundation (Grant No. 2108085QB60 and 2108085QB61), CASHIPS Director's Fund (Grant No. YZJJ2021QN18 and YZJJ2021QN21), China Postdoctoral Science Foundation (Grant No. 2020M682057), and Special Research Assistant Program, Chinese Academy of Sciences.Notes and references
- X. Zhu, X. Zhou, Y. Jing and Y. Li, Nat. Commun., 2021, 12, 4080 CrossRef CAS PubMed.
- B. M. Comer, P. Fuentes, C. O. Dimkpa, Y.-H. Liu, C. A. Fernandez, P. Arora, M. Realff, U. Singh, M. C. Hatzell and A. J. Medford, Joule, 2019, 3, 1578–1605 CrossRef CAS.
- C. Zhu, M. Wang, C. Wen, M. Zhang, Y. Geng, G. Zhu and Z. Su, Adv. Sci., 2022, 9, 2105697 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- C. Chen, N. He and S. Wang, Small Sci., 2021, 1, 2100070 CrossRef CAS.
- C. Zhu, C. Wen, M. Wang, M. Zhang, Y. Geng and Z. Su, Chem. Sci., 2022, 13, 1342–1354 RSC.
- D. B. Kayan and F. Köleli, Appl. Catal., B, 2016, 181, 88–93 CrossRef CAS.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Shi, S. Li, X. Wang and G. Zhang, Chem. Sci., 2021, 12, 6048–6058 RSC.
- M. Yuan, J. Chen, Y. Xu, R. Liu, T. Zhao, J. Zhang, Z. Ren, Z. Liu, C. Streb, H. He, C. Yang, S. Zhang and G. Zhang, Energy Environ. Sci., 2021, 14, 6605–6615 RSC.
- M. Yuan, J. Chen, H. Zhang, Q. Li, L. Zhou, C. Yang, R. Liu, Z. Liu, S. Zhang and G. Zhang, Energy Environ. Sci., 2022, 15, 2084–2095 RSC.
- M. Yuan, H. Zhang, Y. Xu, R. Liu, R. Wang, T. Zhao, J. Zhang, Z. Liu, H. He, C. Yang, S. Zhang and G. Zhang, Chem. Catal., 2022, 2, 309–3207 CrossRef.
- D. B. Kayan and F. Köleli, Appl. Catal., B, 2016, 181, 88–93 CrossRef CAS.
- D. Saravanakumar, J. Song, S. Lee, N. H. Hur and W. Shin, ChemSusChem, 2017, 10, 3999–4003 CrossRef CAS PubMed.
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu and S. Li, Nat. Sustainability, 2021, 4, 868–876 CrossRef.
- C. Lv, C. Lee, L. Zhong, H. Liu, J. Liu, L. Yang, C. Yan, W. Yu, H. H. Hng, Z. Qi, L. Song, S. Li, K. P. Loh, Q. Yan and G. Yu, ACS Nano, 2022, 16, 8213–8222 CrossRef CAS.
- N. Meng, X. Ma, C. Wang, Y. Wang, R. Yang, J. Shao, Y. Huang, Y. Xu, B. Zhang and Y. Yu, ACS Nano, 2022, 16, 9095–9104 CrossRef CAS.
- X. Liu, P. V. Kumar, Q. Chen, L. Zhao, F. Ye, X. Ma, D. Liu, X. Chen, L. Dai and C. Hu, Appl. Catal., B, 2022, 316, 121618 CrossRef CAS.
- X. Wei, X. Wen, Y. Liu, C. Chen, C. Xie, D. Wang, M. Qiu, N. He, P. Zhou, W. Chen, J. Cheng, H. Lin, J. Jia, X.-Z. Fu and S. Wang, J. Am. Chem. Soc., 2022, 144, 11530–11535 CrossRef CAS PubMed.
- J. Leverett, T. Tran-Phu, J. A. Yuwono, P. Kumar, C. Kim, Q. Zhai, C. Han, J. Qu, J. Cairney, A. N. Simonov, R. K. Hocking, L. Dai, R. Daiyan and R. Amal, Adv. Energy Mater., 2022, 2201500, DOI:10.1002/aenm.202201500.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2016, 55, 3942 CrossRef CAS.
- X. Zhao, H. Yang, P. Jing, W. Shi, G. Yang and P. Cheng, Small, 2017, 13, 1603279 CrossRef PubMed.
- N. Cao, Y. Quan, A. Guan, C. Yang, Y. Ji, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2020, 577, 109–114 CrossRef CAS PubMed.
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS.
- S. Liu, S. Yin, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Cell Rep. Phys. Sci., 2022, 3, 100869 CrossRef CAS.
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen and J. Zheng, Nat. Commun., 2022, 13, 1 CrossRef PubMed.
- Y. Huang, R. Yang, C. Wang, N. Meng, Y. Shi, Y. Yu and B. Zhang, ACS Energy Lett., 2022, 7, 284–291 CrossRef CAS.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- S. Zhang, T. Shi, K. Li, Q. Sun, Y. Lin, L. R. Zheng, G. Wang, Y. Zhang, H. Yin and H. Zhang, J. Phys. Chem. C, 2022, 126, 965–973 CrossRef CAS.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS.
- Y. Qiu, L. Xin, Y. Li, I. T. McCrum, F. Guo, T. Ma, Y. Ren, Q. Liu, L. Zhou, S. Gu, M. J. Janik and W. Li, J. Am. Chem. Soc., 2018, 140, 16580–16588 CrossRef CAS PubMed.
- G.-T. Fu, X. Jiang, R. Wu, S.-H. Wei, D.-M. Sun, Y.-W. Tang, T.-H. Lu and Y. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 22790–22795 CrossRef CAS PubMed.
- Y. Zeng, P. X. Sun, Z. Pei, Q. Jin, X. Zhang, L. Yu and X. W. Lou, Adv. Mater., 2022, 34, 2200342 CrossRef CAS PubMed.
- H. Xu, H. Xu, Z. Chen, X. Ran, J. Fan, W. Luo, Z. Bian, W.-X. Zhang and J. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 3861–3868 CrossRef CAS PubMed.
- Q. Gao, H. S. Pillai, Y. Huang, S. Liu, Q. Mu, X. Han, Z. Yan, H. Zhou, Q. He, H. Xin and H. Zhu, Nat. Commun., 2022, 13, 2338 CrossRef CAS PubMed.
- R. Keuleers, H. O. Desseyn, B. Rousseau and C. Van Alsenoy, J. Phys. Chem. A, 1999, 103, 4621–4630 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS.
- M. Manivannan and S. Rajendran, Int. J. Eng. Sci. Technol., 2011, 3, 8048–8060 Search PubMed.
- R. L. Frost, J. Kristof, L. Rintoul and J. T. Kloprogge, Spectrochim. Acta, Part A, 2000, 56, 1681–1691 CrossRef.
- M. DelloStritto, M. L. Klein and E. Borguet, J. Phys. Chem. A, 2019, 123, 5378–5387 CrossRef CAS PubMed.
- Q. Zhao, L. Liu, R. Liu and L. Zhu, Chem. Eng. J., 2018, 353, 311–318 CrossRef CAS.
- A. A. B. Padama, A. P. S. Cristobal, J. D. Ocon, W. A. Diño and H. Kasai, J. Phys. Chem. C, 2017, 121, 17818 CrossRef CAS.
- X. Nie, X. Jiang, H. Wang, W. Luo, M. J. Janik, Y. Chen, X. Guo and C. Song, ACS Catal., 2018, 8, 4873 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the experimental process, XRD patterns, XPS analysis, calibration curves and electrochemical measurement results. See DOI: https://doi.org/10.1039/d2ey00038e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |