
Polymeric nanomaterial strategies to encapsulate and deliver biological drugs: points to consider between methods
Xiangxun
Chen
 ab,
Yuao
Wu
b,
Van Thanh
Dau
c,
Nam-Trung
Nguyen
b and
Hang Thu
Ta
*abd
ab,
Yuao
Wu
b,
Van Thanh
Dau
c,
Nam-Trung
Nguyen
b and
Hang Thu
Ta
*abd
aSchool of Environment and Science, Griffith University, Nathan Campus, Brisbane, Queensland 4111, Australia. E-mail: h.ta@griffith.edu.au; Web: https://hangta.group/ Web: https://experts.griffith.edu.au/27034-hang-ta
bQueensland Micro- and Nanotechnology Centre, Griffith University, Nathan Campus, Brisbane, Queensland 4111, Australia
cSchool of Engineering and Built Environment, Griffith University, Gold Coast, Queensland 4215, Australia
dAustralian Institute for Bioengineering and Nanotechnology, University of Queensland, St Lucia, QLD 4067, Australia
First published on 13th January 2023
Abstract
Biological drugs (BDs) play an increasingly irreplaceable role in treating various diseases such as cancer, and cardiovascular and neurodegenerative diseases. The market share of BDs is increasingly promising. However, the effectiveness of BDs is currently limited due to challenges in efficient administration and delivery, and issues with stability and degradation. Thus, the field is using nanotechnology to overcome these limitations. Specifically, polymeric nanomaterials are common BD carriers due to their biocompatibility and ease of synthesis. Different strategies are available for BD transportation, but the use of core–shell encapsulation is preferable for BDs. This review discusses recent articles on manufacturing methods for encapsulating BDs in polymeric materials, including emulsification, nanoprecipitation, self-encapsulation and coaxial electrospraying. The advantages and disadvantages of each method are analysed and discussed. We also explore the impact of critical synthesis parameters on BD activity, such as sonication in emulsifications. Lastly, we provide a vision of future challenges and perspectives for scale-up production and clinical translation.
 Xiangxun Chen | Mr Xiangxun Chen received his BSc in Pharmacology and MBiomedHlthSc (GPA: 4.0/4.0, awarded with distinction) from Monash University. He is a PhD candidate under the supervision of Assoc. Prof. Hang T. Ta, Prof. Nam-Trung Nguyen and Dr Shehzahdi S. Moonshi at Queensland Micro- and Nanotechnology Centre, Griffith University. He is a recipient of the ring-fenced scholarships issued from the Graduate Research School at Griffith University. His current research interest is the development of novel drug delivery strategies for thrombolysis. |
 Yuao Wu | Dr Yuao Wu is a research fellow in Assoc. Professor Hang T. Ta's group at Queensland Micro and Nanotechnology Centre, Griffith University, Queensland, Australia. He completed his PhD at the University of Queensland in 2021. Dr Yuao Wu is the author of 18 publications and has attended 16 conferences and symposiums until now. His current research direction is theranostic nanoparticles that contain treatment and diagnosis parts for inflammatory and Alzheimer's diseases. He has solid professional knowledge of nanoparticle synthesis, MRI, in vitro and in vivo studies, molecular biology, and drug design. Moreover, he has proficient lab skills in transmission electron microscope operation, in vivo and in vitro experiments, western blotting, qPCR, HPLC, and NMR. |
 Van Thanh Dau | Dr Van Thanh Dau received the BS degree in aerospace engineering from Hochiminh City University of Technology, Vietnam, in 2002, and the MS and PhD degrees in micro-mechatronics from Ritsumeikan University, Japan, in 2004 and 2007, respectively. From 2007 to 2009, he was a Postdoctoral Fellow with the Japan Society for the Promotion of Science (JSPS). From 2010 to 2018, he worked for a start-up in England and leading companies in Japan before joining Griffith University in 2019. He has published 150+ papers and 23 inventions (8 US patents) in the field of MEMS, electrohydrodynamics and nanoprinting. His inventions have led to 5+ commercial products. He is a senior member of IEEE and currently leads a team of 7 PhD students at the School of Engineering and Built Environment, Griffith University. His research focuses on MEMS sensors and actuators, electrohydrodynamics, flexible electronics, aerosols and respiratory medical devices. |
 Nam-Trung Nguyen | Nam-Trung Nguyen received his Dip-Ing, Dr Ing and Dr Ing Habil degrees from Chemnitz University of Technology, Germany, in 1993, 1997 and 2004, respectively. In 1998, he was a postdoctoral research engineer in the Berkeley Sensor and Actuator Center (University of California at Berkeley, USA). He is a Fellow of ASME and a Senior Member of IEEE. Nguyen's research is focused on microfluidics, nanofluidics, micro/nanomachining technologies, micro/nanoscale science, and instrumentation for biomedical applications. He published over 500 journal papers and filed 8 patents, of which 3 were granted. Among the books he has written, the first and second and third editions of the bestseller “Fundamentals and Applications of Microfluidics” were published in 2002, 2006 and 2019, respectively. |
 Hang Thu Ta | Hang Ta is an Associate Professor at the School of Environment and Science and Queensland Micro- and Nanotechnology Centre, Griffith University. She is a Heart Foundation Future Leader Fellow and currently leads a team of 14 students and postdocs working on (1) nanomaterials for the diagnosis and treatment of life-threatening diseases; (2) organ-on-the-chip, and (3) cell delivery and therapy. She has a unique skill set combining chemistry and biology skills. She got a PhD from University of Melbourne in 2009 and then worked at the Baker Heart and Diabetes Institute and University of Queensland before moving to Griffith University in 2020. A/Prof. Ta has been awarded a number of prizes, grants and prestigious fellowships such as a National Heart Foundation postdoctoral fellowship, NHMRC ECR fellowship and Heart Foundation Future Leader Fellowship. She has secured several competitive grants from national funding agencies for both discovery and infrastructure projects. |
1. Introduction
The development of new therapeutic agents expands the range of available biological drugs (BDs) for treating diseases or relieving relevant symptoms. BDs are a group of medicines originating from living organisms with complex structures.1,2 Generally, BDs involve protein or enzyme-based therapeutics.3 BDs offer advantages over low molecular weight drugs as they can target more precisely with high potency, resulting in reduced intrinsic toxicity.1,4 Thus, BDs are widely used for life-threatening diseases such as cancer, cardiovascular diseases, and other chronic diseases.5–7 Since peptide-based insulin as the first BD was approved by the U.S. Food and Drug Administration (FDA) in 1982 to treat diabetes, the FDA has approved 621 BDs up to 2021.8,9 In 2020, these BDs contributed to a US$253 billion pharmaceutical market and were forecast to double in 2025.10 However, due to their nature, BDs have disadvantages such as poor solubility, permeability, and bioavailability in oral delivery.11–13 Moreover, for intravenous administration, BD accumulation in the bloodstream may induce a secondary off-target effect that could be lethal. For instance, a high administration rate of tPA (the only approved treatment option for stroke) may lead to tPA accumulation in different organs and cause haemorrhages such as an intracranial haemorrhage in hyperglycaemia and diabetes patients.14,15 Therefore, there is a need to find a solution to overcome these challenges to make full use of BDs, which is beneficial for a broader population.16Nanoparticles (NPs) attract attention from the research community because of their wide range of applications, including diagnosis, analysis, imaging, and tissue engineering.17 Moreover, NPs can act as carriers to deliver therapeutic agents.18 The small size of NPs favours permeability through different membranes and the blood–brain barrier for a higher efficiency which has massively enhanced the therapeutic utility of the agents.19 Additionally, applying appropriate targeting and releasing strategies can further improve the precision and also the uptake rate of the original BD.20
In terms of availability for use, the FDA has approved over 20 NPs for clinical use, mainly inorganic NPs, liposomes, and polymeric NPs.19 Inorganic NPs such as iron, silica and gold can be precisely synthesised into nanospheres, nanobundles and nanoshells to carry BDs.21 Inorganic NPs such as 111In superparamagnetic iron oxide NPs can be employed as theranostic materials due to their unique radioactive and magnetic properties.22–38 However, the biodistribution and cytotoxicity of inorganic particles are not fully investigated.39–41 Moreover, the solubility of inorganic NPs is relatively low.42 Therefore, it is challenging to translate inorganic delivery systems into clinical use, especially when heavy metals are employed as a part of the carrier.
Liposome is made up of phospholipids, which mainly form a spherical structure. This spherical structure can be multilayered and carry hydrophilic, hydrophobic, or even lipophilic drugs.43 Since the FDA approved the first nanoliposomal drugs in 1995, many studies have been devoted to the use of liposomes in disease treatment.44 However, the drug encapsulation efficiency (EE) using liposomes as a delivery system is relatively low.45 Besides, liposome carriers are mainly absorbed by the spleen and kidneys and release the drug in these organs.46 This has undoubtedly limited BD nanodelivery to some extent. Since inorganic and liposome forms have certain disadvantages, other materials should be considered for the transportation and delivery of BDs.
Polymers are one of the most popular materials currently used for drug-loading nano-systems. Polymeric materials can be divided into non-biodegradable and biodegradable polymers.47 Non-biodegradable nanopolymers such as acrylic polymers are generally used for the local injection of antibodies or bone-implant components.47 But their low biocompatibility and toxicity can lead to irreversible health complications.47 Consequently, scientists focus more on developing biodegradable polymers such as polylactic acid (PLA) and poly lactic-co-glycolic acid (PLGA) to deliver drugs. Biodegradable polymeric materials do not produce harmful and non-biocompatible by-products upon degradation and therefore do not generally accumulate in major organs to produce cytotoxicity effects.48 They can also be combined with inorganic NPs for therapeutic and diagnostic purposes.49,50 Degradable polymers have been explored as candidates for the delivery of targeted BDs.
Methods to incorporate BDs into the nanoparticles include adsorption, conjugation and encapsulation.51 Adsorption is a method that does not involve the formation of chemical bonds or the alteration of polymers, relying on van der Waals forces, electrostatic forces and hydrophobic interactions to anchor the BD to the relevant areas containing the lipophilic components of the polymer.52 These interactions, such as van der Waals forces, may aggregate and impact the colloidal stability, thus not supporting the stable transportation of BD to the target.53,54 This may cause a reduction in the BD efficiency. The exposed BD structure may also be altered by temperature and acidity change, thus limiting the usefulness of this method.55
Conjugation is generally achieved by forming a covalent bond between the functional groups of the BD and the polymer.56 In this case, the BD is exposed at the surface of the nanoparticles to facilitate the targeting activity. Although conjugation may not impact chemical drugs, it may lead to BD denaturing and thus reduce BD efficacy.57–59 Specifically, the BD cannot be freely released to the target sites, and this binding may impact the conformational change of the BD to bind to the target.60 It would be helpful to retain BD activity while improving the targeting efficacy.
Encapsulation is considered as a more suitable method for BD transportation. BDs can be encapsulated into polymeric materials without the involvement of covalent bonds to retain their activity.61 Encapsulated BDs can also be protected from the body environment, pH, ions and enzymes. BD encapsulation combined with targeting strategies and stimulus-responsive strategies (e.g. pH or temperature) can enhance the use and efficacy of BDs.62
Each nanoparticle synthesis method offers its unique EE and results in a unique final product.63 Common methods are emulsification, nanoprecipitation, self-assembly, and electrospraying.64–66 Yet, the advantages and disadvantages of these methods have not been well documented. Therefore, this paper comprehensively reviews and compares methods to encapsulate BDs such as proteins, enzymes, plasmid deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), offering better insights and knowledge toward improved nanodelivery systems for BDs. The advantages and disadvantages of each method are analysed and discussed. We also explore the impact of critical synthesis parameters on BD activity, such as sonication in emulsification. Lastly, we provide a vision of future challenges and perspectives for scale-up production and clinical translation.
2. Emulsification
Emulsification is the process of dispersing two or more immiscible liquids to form a semi-stable mixture. The emulsification method allows one phase of the liquids to be dispersed in tiny droplets in another phase to create a non-homogeneous emulsion system. Nanoencapsulation using emulsions consists of a mixture of an organic or oil phase and an aqueous phase.67 Depending on the continuous phase and the dispersed phase, they are divided into (1) oil-in-water emulsions, where the continuous phase is oil, and the dispersed phase is an aqueous solution; and (2) water-in-oil emulsions, where the continuous phase is an aqueous solution and the dispersed phase is oil.67 Emulsification is generally classified into single emulsion and double emulsion for BD encapsulation.2.1. Single emulsion
Single emulsion is one of the most frequently utilised methods for encapsulating BDs.68Table 1 summarizes single emulsion studies for BD encapsulation in the last 5 years. Usually, this method utilises an aqueous solution of BDs followed by the addition of an amphiphilic polymer or a polymer with a stabiliser dispersed in the organic phase by homogenising techniques such as stirring or sonication. The help of an amphiphilic polymer or stabiliser allows the polymer to encapsulate protein. The particle solution from the previous step is processed by solvent evaporation to obtain the final NPs (Fig. 1).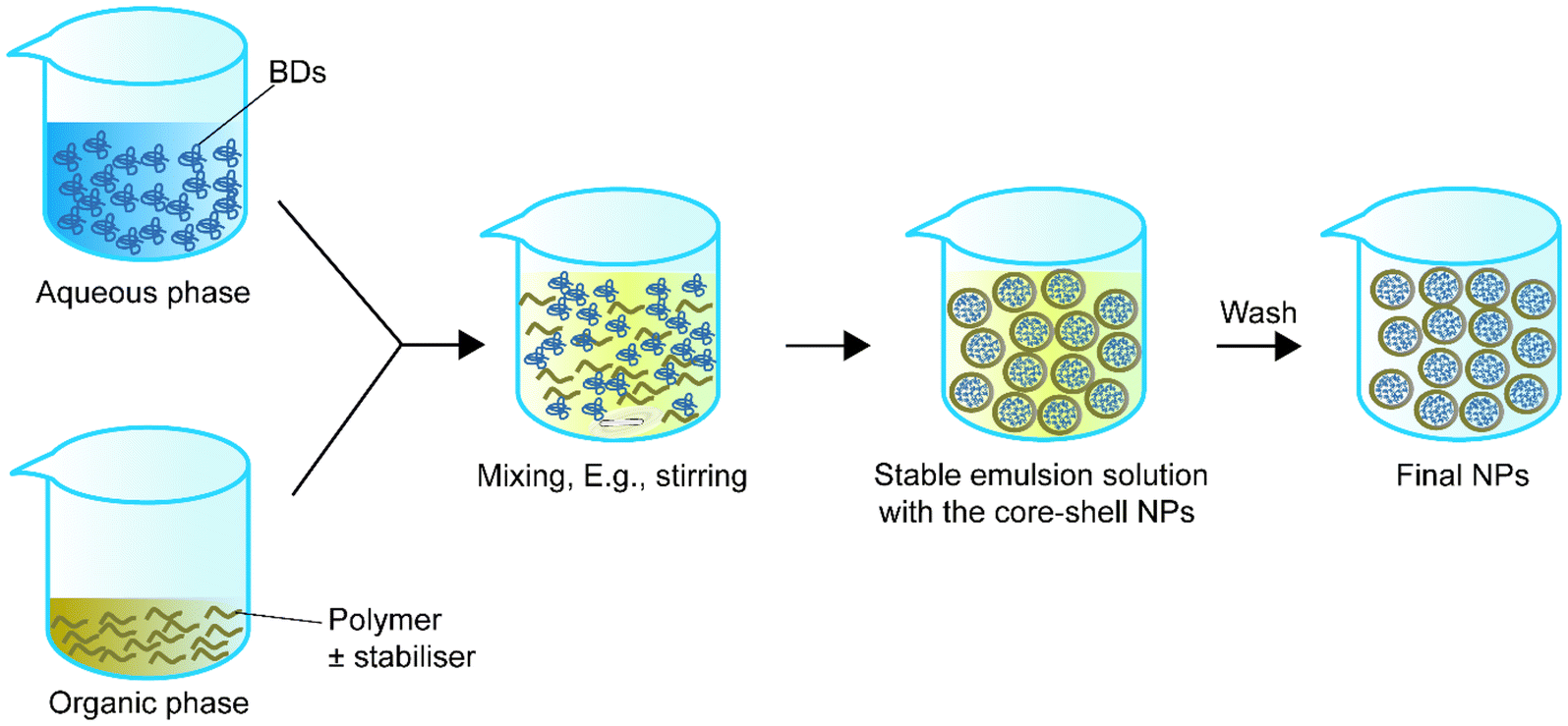 | ||
| Fig. 1 Schematic representation of single emulsion to synthesise core–shell nanoparticles. | ||
| Encapsulated compounds | Polymer | Surfactant or second polymer(s) | Applications | Size | Encapsulation efficiency, % | Loading capacity, % | Ref. |
|---|---|---|---|---|---|---|---|
| Proteins isolated from L. panamensis | PLGA | PVA | Vaccine | LpanUA.27.1860: 36.56 μm at 750 μg ml−1 | LpanUA.22.1260: 94.66 ± 4.86 | — | 74 |
| – Soluble LpanUA.27.1860 | LpanUA.22.1260: 44.97 μm at 750 μg ml−1 | LpanUA.27.1860: 89.03 ± 4.91 | |||||
| – Insoluble LpanUA.22.1260 | |||||||
| tPA | PEG–PLGA | — | Thrombolysis | 276.20 ± 27.58 nm | 94.78 ± 3.09 | 3.60 ± 0.01 | 69 |
| BVZ | mPEG–PAE for BVZ | — | Anti-cancer | BVZ loaded: 82.07 ± 0.19 nm, binary mixture: 132.60 ± 0.058 nm | BVZ loaded: 85.50 ± 0.33 | BVZ loaded: 0.54 ± 0.01 | 72 |
| NY-ESO-1 and IMM60 | PLGA | PVA | Anti-cancer | At PLGA 10 mg mL−1 and peptide content of NPs of 11.4, 13.4, and 11.5 μg mg−1: 227 ± 61 nm | — | — | 70 |
Zamanlu et al.69 demonstrated a single emulsion encapsulating protein with smaller particle sizes. PEGylated PLGA (PEG–PLGA) was employed to encapsulate tPA using a single emulsion solvent diffusion/evaporation method. The size of the NPs was in the range of 200–300 nm. The PEGylated PLGA-tPA NPs were 24.93 nm larger, and the EE was 4.83% higher than the PLGA-tPA group (89.85 ± 9.71%). Also, the loading capacity (LC) of the PEGylated PLGA-tPA NPs was higher than the PLGA-tPA group (3.60 ± 0.01% vs. 1.99 ± 0.01%). The cell viability and thrombolytic activity of the PEGlyated PLGA shell group were also higher than the PLGA only group. However, the study did not provide the in vitro release profiles for the PEGylated-PLGA-tPA group, PLGA-tPA group and tPA only group. The information about how NPs with different structures perform in a prolonged period is thus limited. Furthermore, Dölen et al.70 used a single emulsion with sonication for 2 minutes and 2.5% PVA as a surfactant. This study fabricated New York esophageal squamous cell carcinoma 1 (NY-ESO-1) peptide mix-loaded PLGA NPs of 227 ± 61 nm (PLGA 10 mg mL−1, α-GalCer analog IMM60 at 50 μg mL−1, and three immunogenic cancer germline antigens, NY-ESO-1 mix at 0.33 mg mL−1). The LC and EE were not reported. Similar levels of the immune response activated by the encapsulated IMM60, and NY-ESO-1 were found in vitro and in vivo, which was higher than the response from free IMM60 and peptide. The use of this NP has moved to a phase I clinical trial for safety and tolerability assessment in humans (NCT04751786).71 Notably, the organic solvent used in this study during the preparation was dichloromethane and dimethyl sulfoxide (DMSO), but the reactivity of the peptide mix was not impacted.
NPs synthesised by single emulsion can also be employed for the oral administration of BDs. Feng et al.72 used methoxy polyethylene glycol (mPEG)–poly(β-amino ester) [PAE] to encapsulate bevacizumab (BVZ) with a single emulsion solvent evaporation method. The NPs’ size was 132.60 ± 0.058 nm, with an EE of 85.50 ± 0.33% and an LC of 0.54 ± 0.01%. They also used a single emulsion to prepare a docetaxel-loaded carboxymethyl chitosan (CMC)–PLGA NP. The two NPs were physically mixed to form a binary mixture at 132.60 ± 0.058 nm for dual anti-cancer treatments. Compared with the free drug groups in mice, the NP mixture group had a higher absorption rate across the small intestine and a higher cytotoxic effect in non-small cell lung cancer cells. Moreover, the NP mixture group had a higher plasma concentration than the free drug group (7.85 μg ml−1 at 50 min vs. 148.24 ng ml−1 at 50 min).
The NPs in these studies had a high EE but the LC was lower than 10%. The low LC indicates that the BD cores formed in the core–shell NP structure are small and that the polymer shell makes up the bulk of the NPs. The low LC of hydrophilic BDs and the difficulty of scaling up have prevented this method from being widely used. Knowing these drawbacks, a modified emulsion method, double emulsion, was used to focus on core–shell encapsulation.73
2.2. Double emulsion
In the early 1980s, Matsumoto et al.75 introduced the concept of double emulsion. The double emulsion technique is an expanded version of a single emulsion with two common types, water–oil–water and oil–water–oil. For instance, in the water–oil–water type, the primary emulsion solution contains an aqueous phase with the BD and an organic phase with the polymer or stabiliser. Then the primary emulsion solution is added to a second aqueous phase. Sonication helps the secondary emulsion solution for stable core–shell structure NPs. Finally, the NPs in the secondary emulsion solution can be collected via solvent evaporation (Fig. 2). Table 2 summarizes double emulsion studies for BD encapsulation in the last 5 years.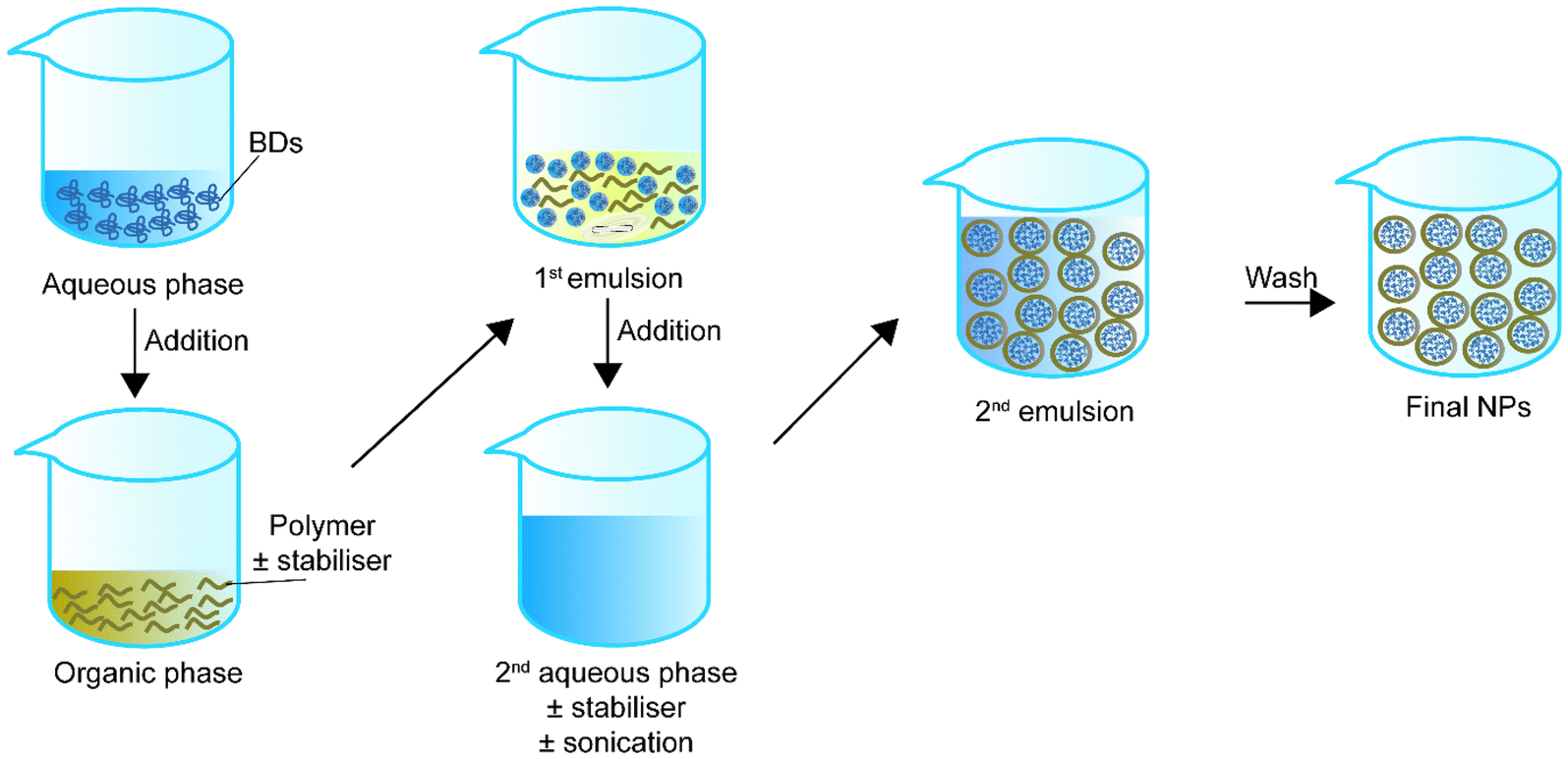 | ||
| Fig. 2 Schematic representation of double emulsion to synthesise core–shell nanoparticles. | ||
| Encapsulated compounds | Polymer | Surfactant or second polymer(s) | Applications | Size | Encapsulation efficiency, % | Loading capacity, % | Sonication settings | Ref. |
|---|---|---|---|---|---|---|---|---|
| Lysozyme | mPEG | — | Infection | ∼180–220 nm | 2.4 | 4.78 | 15 s followed by 30 s on ice, amplitude not included | 78 |
| siRNA, paclitaxel and G0-C14 | PEG–PLA | PVA as a stabiliser | Anti-cancer | 140.5 ± 1.67 nm | 91.03 ± 4.21 | — | 80 W, 30 s followed by 100 W, 2 min | 84 |
| Insulin/PEG6000 | PLGA | — | Diabetes | 240 nm | 50 | — | 50% amplitude, 50%, 1 min | 79 |
| 8-Mer Smac peptide and a 14-mer cell penetrating peptide | PLGA–mPEG2000 | — | Anti-cancer | 197–140 nm | 48 | 2.4 | 30% amplitude for 30 s followed by 10% amplitude for 1 minute, 3 min | 80 |
| KPV/PLGA | Chitosan-coated with HA | PVA as a stabiliser | Ulcerative colitis | 272.33 nm | 39.7 ± 3.6 | — | 6 × 10 s, amplitude not included, 30 s followed by 10% amplitude 1 minute | 81 |
| Oxidation-resistant griffithsin | PLGA | PVA as a stabiliser | HIV-1 infection | 98.5 ± 37.2 nm | 48.9 ± 12.7 | 48.9 ± 12.7 μg mg−1 | Did not specify | 83 |
| siRNA | PLGA–FA–PEG | — | Liver cancer | ∼90 nm | 64.3 ± 5.6 | — | 80 W for 2 min × 2 times, and 10 s × 6 times | 85 |
| Catalase | PLGA–PEG | PVA or CHA as stabilisers | Neurotherapeutic | For CHA: 58.4–70.8 nm | — | — | Sonicated for 15, 30, and 60 seconds for different groups, amplitude not included | 88 |
| For PVA: 109.8–113.3 nm | ||||||||
| cdGMP | PLGA | 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-PEG-maleimide | Middle East Respiratory Syndrome Coronavirus | 114.0 nm | ∼48% | — | 40% amplitude for 1 min, followed by 30% amplitude for 2 min | 86 |
| Vancomycin | PLA–PLGA | PVA as a stabiliser | Antibacterial | 312 nm | 53 ± 7 | 25 ± 3 | 40% amplitude for 15 s for the first emulsion, 40% amplitude for 30 s | 87 |
| Gliadin protein | PLGA | poly(ethyl methacrylate) or PVA | Anti-celiac disease | 529 ± 6.4 nm | — | 10.8 ± 1.5 | 100% amplitude for 30 s × 2 times | 89 |
Furthermore, sonication in a double emulsion does not influence the activity of small size BDs. For example, Priwitaningrum et al.80 prepared a second mitochondria-derived activator of caspase (Smac) loaded polymeric NPs with an average diameter of 197 nm for anti-tumour purposes. The first emulsion solution was formed by 30 seconds of sonication at 10% amplitude with the presence of Smac peptide saline solution, mPEG2000–PLGA and PLGA in an ice bath. PVA was added to the solution, followed by 1-minute sonication at 10% amplitude to stabilise the NPs. The Smac NPs achieved an EE of 48% and 2.4% LC. Sonication did not impact the in vitro and in vivo results. The in vitro results indicated that after 10% amplitude sonication, the NPs were still able to induce triple-negative 4T1 mouse breast tumour cells apoptosis rapidly. Besides, the NPs expressed a synergistic anticancer effect with doxorubicin in vivo.
Similarly, Xiao et al.81 utilised a PLGA–PVA–chitosan polymer with a hyaluronic acid (HA) functionalised coating to deliver lysine–proline–valine (KPV) using a double emulsion with sonication for ulcerative colitis (UC) treatment. KVP was dropwise added to the PLGA-containing organic phase to form the primary emulsion solution for synthesis. Sonication was applied to the primary emulsion solution six times for 10 seconds with PVA and depolymerised chitosan. With an average size of 272.3 nm, the NPs remained intact after oral administration across the stomach and aggregated towards the wound of colonic epithelial cells in a UC mouse model (Fig. 3). Even though the authors previously proved that the therapeutic efficacy of KPV in the non-functionalised NPs was 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold lower than the free KPV concentration,82 in this study, they did not directly compare the therapeutic efficacy of the non-functionalised NPs with the functionalised NPs. Xiao et al.81 only demonstrated that the tissue uptakes of the functionalised NPs were higher than the non-functionalised NPs at 4 hours and 18 hours.
000-fold lower than the free KPV concentration,82 in this study, they did not directly compare the therapeutic efficacy of the non-functionalised NPs with the functionalised NPs. Xiao et al.81 only demonstrated that the tissue uptakes of the functionalised NPs were higher than the non-functionalised NPs at 4 hours and 18 hours.
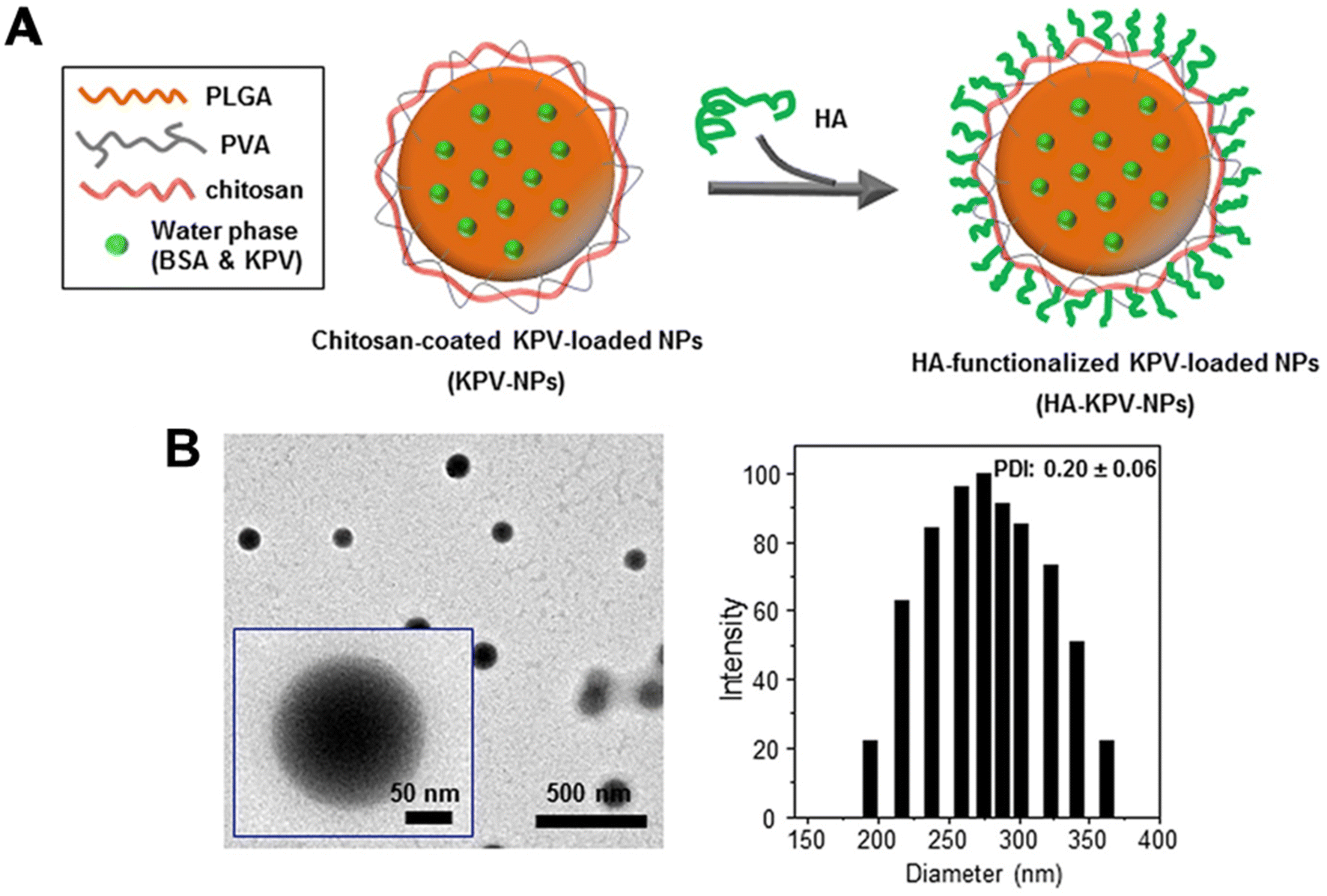 | ||
| Fig. 3 Preparation and characterization of KPV-loaded polymeric NPs. (A) Schematic illustration of the fabrication process of HA-KPV-NPs. (B) Representative TEM images and the corresponding size distribution of HA-KPV-NPs. Reprinted from ref. 81, copyright (2017), with permission from Cell Press. | ||
Minooei et al.83 demonstrated that two times of sonication (no amplitude shown) in double emulsion did not reduce the effect of an oxidation-resistant variant of griffithsin, an algae-derived antiviral BD for anti-human immunodeficiency virus-1 (HIV-1) infection in vitro. PVA was present in both the primary and secondary emulsion solutions as a stabiliser. The griffithsin variant was encapsulated in a PLGA core with a size of 98.5 ± 37.2 nm, accompanied by a 48.9 ± 12.7% EE and 48.9 ± 12.7 μg mg−1 LC. Also, the half-maximal inhibitory concentration (IC50) of the griffithsin variant NP was 4.09-fold lower than its free form (1.1 nM vs. 4.5 nM), which indicated that griffithsin variant NPs prevented HIV-1 infection better than the free griffithsin in vitro.
Double emulsion with sonication was able to load small interfering ribonucleic acid (siRNA).84 In the study they used a PEG–PLA with an amphiphilic dendrimer, G0-C14 and paclitaxel as the shell to load siRNA, for matrix metalloproteinase-rich tumour treatment. A 30 seconds 80 W sonication for the primary emulsion and 2 minutes 100 W sonication for the secondary emulsion did not impact the binding activity and stability of the siRNA. Later, Zhang et al.85 synthesised a 90 nm NP by double emulsion in an ice-bath with about 12% amplitude ultrasonication to encapsulate siRNA with folic acid (FA)-modified PLGA–thioketal (TK)–PEG. The NPs with the siRNA had a lower tumour volume from day 1 to day 16 than the free siRNA group and the non-specific siRNA group. The specific siRNA inhibits the translation of the corresponding protein by binding to the messenger RNA (mRNA) of the target gene.
There are a few studies successfully encapsulating small BDs using sonication in double emulsions. For example, Lin and his group loaded cyclic diguanylate monophosphate (cdGMP, 0.69 kDa) within hollow PLGA NPs (114 nm) for a Middle East Respiratory Syndrome Coronavirus vaccine.86 A 40% amplitude (1 minute) for the primary emulsion and then 30% amplitude (2 minutes) for the secondary emulsion did not impact the activity of cdGMP and it was able to achieve 48% EE (Fig. 4). Moreover, Ural et al.87 did a study using 40% amplitude sonication twice during the synthesis process to load vancomycin (1.45 kDa) into a PLA–PLGA shell. The first sonication was done for 15 s to the primary emulsion solution containing vancomycin and PLA–PLGA solution. Then the solution was added to sodium chloride solution with 0.5% w/v PVA to form the secondary emulsion. The secondary emulsion solution was sonicated at the same amplitude for 30 s. The optimised particles had a size of 319 nm, 50 ± 7% EE and 23 ± 3% LC. However, this study did not explore the in vitro and vivo therapeutic utility of the NPs. These results suggest that use of sonication can be translated to small BD encapsulation.
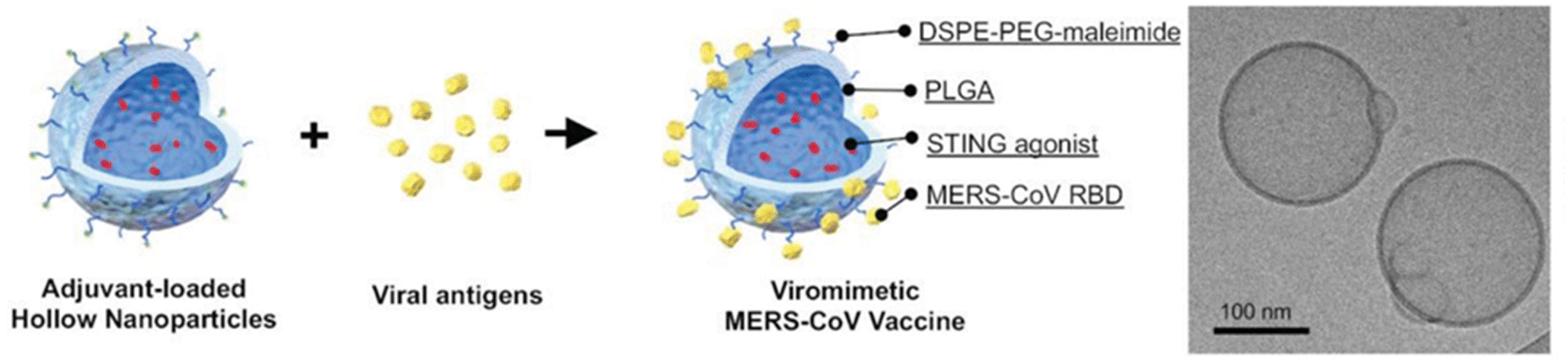 | ||
| Fig. 4 Characterization of adjuvant-loaded viromimetic nanoparticles. A schematic showing the preparation of a viromimetic nanoparticle vaccine. Hollow PLGA nanoparticles with encapsulated adjuvant and surface maleimide linkers were prepared using a double emulsion technique. Recombinant viral antigens were then conjugated to the surface of the nanoparticles via thiol-maleimide linkages. Cryo-electron microscopy of a cdGMP-loaded hollow nanoparticle. Reprinted from ref. 86, copyright (2019), with permission from Wiley. | ||
Liao et al.88 studied the size and activity of enzyme-loaded polymeric NPs prepared in either 1 wt% cholic/deoxycholic acid (CHA) sodium salt or 1 wt% PVA surfactants under different sonication conditions for the double emulsion. The PEG–PLGA-coated catalase NPs with CHA and sonicated for 15, 30, and 60 seconds were smaller than those with PVA prepared under the same settings. Their sizes at 15 s to 30 s and 60 s were similar for the CHA as well as PVA groups (∼80 nm for CHA and ∼120 nm for PVA at 15, 30 and 60 seconds of sonication). For the catalase activity, the group sonicated for 30 seconds had the highest catalase activity of about 45 units per ml and 80 units per ml for CHA and PVA, respectively. Further sonication, such as 60 seconds, reduced catalase activity by 30 units per ml compared with that of 30 seconds (for both the CHA and PVA groups). Sonication of less than 30 seconds also reduced the catalase activity. The 15 seconds sonication results for the CHA and PVA groups were about 10 units per ml and 5 units per ml, respectively, lower than the 60 seconds sonication results. Surprisingly, sonication for one second did not preserve the catalase activity for both groups but resulted in the least activity (∼5 units per ml and ∼40 units per ml for CHA and PVA, respectively).
Although the sonication time of some studies mentioned above was higher than the range reported by Liao et al.,88 the encapsulated BDs, regardless of their sizes, still provided beneficial effects in various applications. This point is greatly supported by a study encapsulating gliadin protein with PLGA using a double emulsion with two times of 100% amplitude sonication to produce an NP at a size of 529 ± 6.4 nm.89 The activity of the protein to reduce the inflammatory and enteropathy response was retained in three different celiac disease mice models.89 Notably, a phase I clinical trial and a double-blinded randomised phase II trial have been completed for this NP (NCT03486990, and NCT03738475, respectively).
However, we still suggest that low to moderate amplitude sonication (e.g., lower than 50% amplitude) with an ice bath for the double emulsion is beneficial for preserving BD activity. Future studies can assess the effect of sonication time on the dispersion of the droplets in the emulsion and the enzymatic activity after the sonication. The impact of prolonged sonication over an ice bath on BD activity and structure is also something to consider.
Admittedly, emulsions are one of the most established and widely used synthesis methods, with over 50% EE in most cases. However, parameters such as the extended exposure period of the BDs to the solvent in the emulsion also make it debatable whether the BDs can still perform their function. The current review suggests that a low temperature and amplitude and time of less than one minute can still keep the BDs functioning. But further exploration of the BDs’ secondary and tertiary structure after using sonication and an emulsifier/stabiliser is needed to support this suggestion. Moreover, we suggest that excessive exposure to the organic phase without rapid emulsion homogenisation during droplet formation may contribute more to BD denaturation than the effect of energetic sonication waves on BD denaturation.
3. Nanoprecipitation
Nanoprecipitation is one of the first drug encapsulation methods developed by Fessi et al.93 in 1989. At first, it was applied to synthesise polymeric encapsulated particles within 100 to 300 nm. The physicochemical principles of non-protein NP synthesis using nanoprecipitation, such as the mixing steps and the solvents, have been thoughtfully investigated.94 Moreover, the path to industrialisation has also been explored, such as modifications on an industrial scale and the establishment of controlled microfluidic systems.95–97 Therefore, this method is considered to have a high expectation for BD encapsulation due to its simplicity of operation and its ability to avoid exposure to different emulsifiers and surfactants under sonication conditions. Table 3 summarizes nanoprecipitation studies for BD encapsulation in the last 5 years.| Precipitation types | Encapsulated compounds | Polymer | Second polymer or stabiliser(s) | Applications | Size | Encapsulation efficiency, % | Loading capacity, % | Ref. |
|---|---|---|---|---|---|---|---|---|
| Simple nanoprecipitation | Lysozyme and α-chymotrypsin | PLGA | — | Improving protein delivery, specific disease application not specified | Lysozyme-loaded: 336 ± 40 nm | Lysozyme-loaded: 94 ± 5 | Theoretical loading: 5 | 98 |
| α-Chymotrypsin loaded: 440 ± 16 nm | α-Chymotrypsin loaded: 74 ± 4 | |||||||
| Simple nanoprecipitation | Amylase | PLGA | Pluronic F68 | Immunotherapeutic | 147.7 ± 4.3 nm | 24 ± 11.2 | — | 99 |
| Simple nanoprecipitation | Trypsin inhibitor isolated from tamarindo seeds | Whey protein conjugated chitosan | Tween 80 | Obesity-related inflammatory response | 109.40 ± 7.53 nm | 98.5 ± 1.95 | — | 100 |
| Simple nanoprecipitation | Catalase | PLGA–PEG | CHA or PVA | Neurotherapeutic | For CHA: 58.4–70.8 nm | — | — | 88 |
| For PVA: 109.8–113.3 nm | ||||||||
| Flash nanoprecipitation | Ovalbumin or HRP | PS-b-PEG | — | Improving protein delivery, specific disease not specified | 115 nm | — | 50 | 102 |
| Flash nanoprecipitation | Polymyxin B, HRP, lysosome, blue Dextran, vancomycin, RNA and β-galactosidase | PLA-b-PASP or PASP-b-PLA-PEG | PLA-b-PEG as stabilising coating when using PLA-b-PASP for encapsulation | General delivery | Polymyxin B loaded: 119 ± 7 nm | Polymyxin B loaded: 80 ± 2% | Polymyxin B loaded: 27% | 107 |
| HRP loaded: 76 ± 2 | HRP loaded: 82 ± 4% | HRP loaded: 18% | ||||||
| Lysosome loaded: 114 ± 14 nm | Lysosome loaded: 98 ± 0.4% | Lysosome loaded: 19% | ||||||
| Blue Dextran loaded: 167 ± 18 nm | Blue Dextran loaded: 50 ± 5 | BlueDextran loaded: 7% vancomycin loaded: 2% | ||||||
| Vancomycin loaded: 107 ± 16 nm | Vancomycin loaded: 12 ± 4% | RNA loaded: 11% | ||||||
| RNA loaded: 70 ± 6 nm | RNA loaded: 48 ± 7% | β-Galactosidase loaded: not reported | ||||||
| β-Galactosidase loaded: 130 ± 26 nm | β-Galactosidase loaded: not reported | |||||||
| Flash nanoprecipitation | Insulin | HA | DDAB | Diabetes | 45 nm | 94.2 ± 1.8 | 13.2 ± 0.6 | 109 |
| Flash nanoprecipitation | Insulin and soybean trypsin inhibitor (SBTI) | PS-b-PAA | HPMCAS, chitosan, PEG | Diabetes | Coated with HPMCAS: 322 ± 6 nm | >98 | — | 110 |
| Coated with chitosan: 321 ± 17 nm | ||||||||
| Coated with PS-b-PEG: 172 ± 8 nm | ||||||||
| iNCs/HPMCAS/Zn/C10: 486 ± 60 nm |
3.1. Simple nanoprecipitation
A homogeneous core–shell nanoparticle is usually made by the dropwise addition of the non-solvent solution of BD to the dissolved BD solution first, followed by the addition of the polymer in its solvent to the BD particle solution. While the mixture of the BD particles and polymer solution is stable, the non-solvent of the polymer can be added to the mixture solution under appropriate conditions such as stirring to form the BD–polymer core–shell NPs (Fig. 5).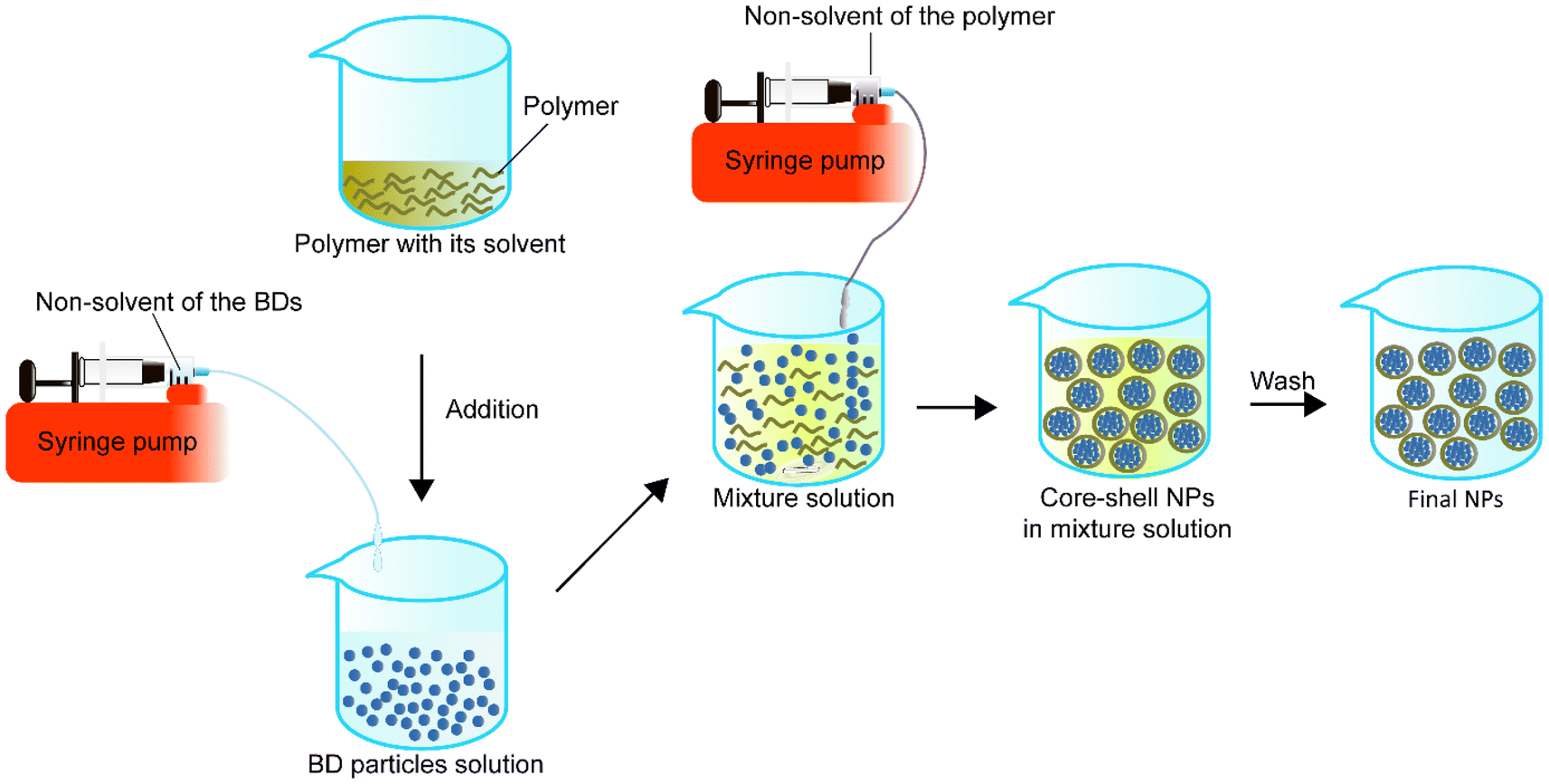 | ||
| Fig. 5 Schematic representation of simple nanoprecipitation to synthesise core–shell nanoparticles. | ||
In 2012, Morales-Cruz et al.98 employed a two-step precipitation method to encapsulate lysozyme and α-chymotrypsin. They demonstrated that acetonitrile could prevent irreversible denaturation of proteins during the first precipitation process, which might happen when using DMSO. Also, PLGA is soluble in acetonitrile, so in the mixture, both the protein precipitated particles and PLGA are miscible with each other. Later, when the mixture was added to water, PLGA automatically precipitated and wrapped around the precipitated proteins to form core–shell NPs. Their results indicated that this method had a theoretical protein LC of about 2% or 5% and achieved an EE of 94 ± 5% for lysozyme and 74 ± 4% for α-chymotrypsin. The size of the lysozyme-loaded PLGA NPs was 336 ± 40 nm, and the α-chymotrypsin-loaded PLGA NPs were 440 ± 16 nm. Two-step precipitation did not impact the residual activity of the lysosomes but influenced the α-chymotrypsin. The residual activity of the α-chymotrypsin decreased to 49%, while lysosome activity was 100%. They then encapsulated a horse heart cytochrome c (Cyt-c) with PLGA. After encapsulation, the encapsulated enzyme residual activity was 2% higher than bare precipitated enzyme particles (96%). The particle size was 242 nm with 72 ± 2% EE. Although they showed that α-chymotrypsin NPs were cytotoxic to human cervical cancer cell lines, there was no comparison of the effect of NPs with the free α-chymotrypsin and existing anticancer agent.
Nelemans et al.99 recently optimised a drug delivery system to encapsulate BSA and amylase with PLGA. This study first showed that only acetonitrile could precipitate BSA with a homogeneous particle distribution while acetone and ethanol caused a heterogeneous particle distribution. They found the particle size of BSA varied linearly when the concentration increased from 10 mg ml−1 to 25 mg ml−1 with 5 mg ml−1 increments (35.2 ± 0.2 nm, 46.2 ± 7.4, 54.3 ± 1.5 and 67.0 ± 4.1 mg ml−1 respectively). Only 2 mg ml−1 amylase was used for precipitating a 133.7 ± 4.2 nm particle. The authors eventually encapsulated amylase with PLGA and 1% Pluronic F68 (diameter: 147.7 ± 4.3 nm, including a 14 nm PLGA shell). However, Nelemans et al.99 only encapsulated about 24 ± 11.2% of the amylase into the PLGA NPs. They tested the initially precipitated proteins for activity by directly dissolving the proteins in PBS. The activity of the redispersed amylase was 1.1% higher than its stock solution. This result supported the view that using organic solvents such as acetonitrile to precipitate BDs does not hinder efficacy.
Queiroz et al.100 reported a different type of nanoprecipitation. Using a whey protein-conjugated chitosan as a shell, they loaded trypsin inhibitor isolated from tamarindo seeds (TTI). Instead of forming TTI particles first to keep it stable, they combined the TTI with the shell complex. Then Tween 80-containing ethanol solution was proportionally added to the solution. A core–shell structure of 109.40 ± 7.53 nm in diameter was formed by homogenising at 17500 rpm with an EE of 98.5 ± 1.95%, and no LC was reported. This work demonstrated that the use of a stabiliser and homogeniser in simple nanoprecipitation might not impact the performance of NPs. Also, the TTI-encapsulated NPs showed higher in vitro antitrypsin activity than the free TTIs at 40 °C and 60 °C. When the pH was increased from 2.0 to 6.0, the TTI-encapsulated NPs still had better antitrypsin activity than free TTI. However, their Fourier transform infrared spectroscopy results showed several more vibrational bands than free TTI, suggesting that an underlying chemical reaction had occurred. Overall, such an interaction may not be sufficient to change the activity profile of TTI, but whether the secondary and tertiary structure of TTI will be changed after encapsulation within the NP is worth exploring.
Furthermore, it is worth considering the effect of nanoprecipitation on BD activity compared with other methods. For example, Liao et al.88 synthesised catalase-loaded PLGA–PEG NPs using the double emulsion method with the sonication and nanoprecipitation method. Although there was no significant difference in particle size when comparing NPs prepared by nanoprecipitation and the double emulsion method with sonication, there was a significant reduction in catalase activity between the group using nanoprecipitation and the group using emulsion with 30 seconds’ sonication. Especially for the PVA as the surfactant groups, nanoprecipitation-prepared NPs were 35 units per ml lower in catalase activity than the NPs synthesised from a double emulsion with 30 seconds of sonication.88 Notably, the precipitation method that Liao et al.88 used to compare them was a one-step nanoprecipitation that did not precipitate catalase in NP form before mixing the catalase with the PLGA–PEG/acetone solution. Combined with the conclusion from Nelemans et al.,99 the acetone solution in PLGA may impact both the distribution and the activity of catalase.
Taken together, the selection of a non-solvent of BDs and polymers during the nanoprecipitation process contributes to the activity and characteristics of its final product. To gain a more comprehensive understanding of BD precipitation, small-sized BDs such as siRNA can be used for future studies. Following that, the secondary and tertiary structure of the precipitated large or small-size BDs can be examined to see whether a solvent such as acetonitrile causes any changes on their active sites. Future studies could also determine the impact of different organic solvents on BD structure. In addition to the future suggestions, the works reviewed above on simple nanoprecipitation reported fluctuating EE and did not report their LC. However, nanoprecipitation is a much simpler process than double emulsions, allowing an appropriate size control of the NPs.
3.2. Flash nanoprecipitation
Flash nanoprecipitation is used to reduce the effect of non-solvent on the BD. While regular flash nanoprecipitation is used to encapsulate hydrophobic cores, recent studies have utilised a similar principle to encapsulate hydrophilic cores, particularly BDs, in a process called inverse flash nanoprecipitation (iFNP).101–103 In particular, iFNP first dissolves the BD and polymer in solution form, meanwhile adding non-solvents of the polymer and ionic crosslinker to a mixing chamber such as confined impinging jets (CIJ) or a multi-inlet vortex mixer (MIVM) to synthesise NPs that are more homogenised than the conventional precipitation within seconds (Fig. 6).104,105 This method is considered to have a higher loading and EE than emulsification and conventional nanoprecipitation.101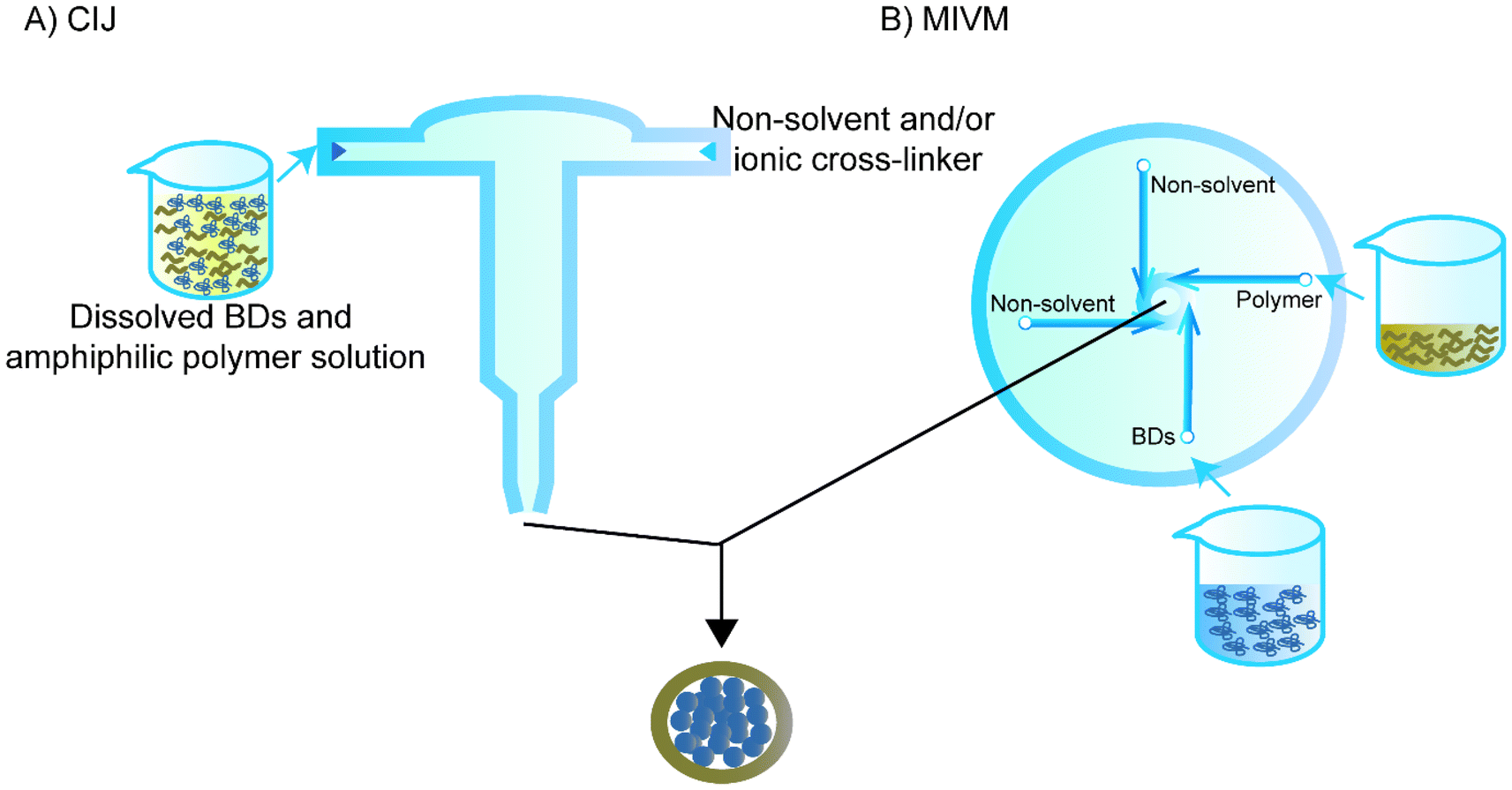 | ||
| Fig. 6 Schematic representation of inverse flash nanoprecipitation (iFNP) to synthesise simple core–shell nanoparticles. (A) The confined impinging jets (CIJ) and (B) a multi-inlet vortex mixer (MIVM). The NPs synthesised by iFNP are only a schematic sample, not the actual size. | ||
Since Pagels and Prud'homme103 introduced the use of iFNP for BD delivery in scaffolds, the group has intensively investigated the possibility of using iFNP to produce BD–polymer core–shells. They encapsulated tobramycin, peptide I, tryptophan, lysozyme, vancomycin, and glutathione separately using a CIJ.103 DMSO was used as a solvent for both the tobramycin and poly(n-butyl acrylate)-b-poly(acrylic acid), and 5% H2O was used for better dissolving peptide I and vancomycin, while 5% H2O and 5% acetic acid was added to the DMSO for tryptophan. The non-solvent was chloroform for all of the groups except for tobramycin, and acetone was quickly added to the other side of the CIJ. The diameters of the capsulated NPs were all within 200 nm. The group also used transmission electron microscopy to confirm the core–shell structure that was hypothesised for NPs prepared by iFNP containing hydrophilic cores and hydrophobic shells.
In a later study, they also used CIJ to encapsulate vaccine antigen ovalbumin (OVA) with PS-b-PEG and obtained a 147 ± 3 nm NP with 50% LC.102 They also used MIVM for OVA loading in PS-b-PEG and achieved a size of 115 nm. In addition, an NP using PS-b-PEG to encapsulate horseradish peroxidase (HRP) was synthesised with a size of 167 nm and 30% LC. However, the authors did not report the EEs for all the prepared NPs. Despite that, this design only required 0.2 mg (200 μl of 1 mg ml−1) of the BDs for each encapsulation run, while other studies such as Levit et al.106 needed to use at least 0.9 mg of BSA for preparation. Markwalter followed up with a PLGA–poly(aspartic acid) [PASP] shell to encapsulate polymyxin B, HRP, lysosome, blue Dextran, vancomycin, RNA and β-galactosidase.107 PASP has been employed in numerous biomedical applications such as drug delivery.108 For both CIJ and MIVM settings, they timely added solvent and anti-solvent with ion-crosslinker solution to different mixer entries. Later the primary BD/PLGA–PASP core–shell NPs in their non-solvent were incubated in an aqueous buffer. The aqueous phase was extracted for EE analysis. The primary NPs in their non-solvent were heat-treated to evaporate the previous solvent, and then a second copolymer, PLA-b-PEG in tetrahydrofuran, acetone or acetonitrile, was added to be the new solvent. The solution contained the primary NPs, and the second polymer was injected into a new mixer, and an aqueous non-solvent stream was added to the other inputs for a second iFNP. The PLA in the second polymer was self-assembled on the surface of the primary NPs, and the PEG tail provided a strong stability effect. They also simplified the process by directly using a PASP-b-PLA-b-PEG triblock polymer, and BDs dissolved in DMSO and then acetone as the non-solvent with an ion-crosslinker. When using a higher molecular weight PLA block, the NPs had sizes of 119 ± 7 nm, 76 ± 2, 114 ± 14 nm, 167 ± 18 nm, 107 ± 16 nm, 70 ± 6 nm, and 130 ± 26 nm for encapsulating polymyxin B, HRP, lysosome, blue Dextran, vancomycin, RNA and β-galactosidase, respectively. EE and theoretical LC values are presented in Table 3. This study offered the idea of a hydrogel as a core polymer followed by crosslink hardening of the intermediate polymer to sequester BDs in the polymer shell better. The study highlighted the importance of using a crosslinker during BD encapsulation. The absence of a crosslinker can sometimes result in a loose shell, which may increase the likelihood of package escape. For example, they have shown that polymyxin B without the NH3 as a crosslinker was 40 nm larger than that with the crosslinker. Without the crosslinker, they did not provide EE and LC results for polymyxin B. Nevertheless, we expect the result to be lower than those with the crosslinker. Zeng et al.109 also employed MIVM to load insulin with a natural linear polymer, HA, for oral delivery. Ionic surfactant dimethyl-dioctadecyl ammonium bromide (DDAB) was used to provide a hydrophobic ion pair with insulin for better encapsulation. The team designed three nanoparticles for 45 nm.109 The nanoparticles obtained an EE of 94.2 ± 1.8% and an LC of 13.2 ± 0.6%. The authors also tested the NPs in a diabetes rat model. The NPs provided a sustained release of insulin within a 9-hour timeframe, thus offering a better blood glucose concentration reduction than insulin solution and insulin powder capsules in a rat model.
McManus et al.110 used a CIJ mixer to perform iFNP to load insulin and soybean trypsin inhibitor with over 98% EE for oral delivery. The study used two flash precipitation steps. The first was done using a non-degradable polystyrene-b-polyacrylic acid (PS-b-PAA) to load insulin and soybean trypsin inhibitor. Then, an MIVM was used to form one more layer with hydroxypropyl methylcellulose acetate-succinate (HPMCAS) polymer and sodium caprate. In theory, double encapsulation protects against hydrolysis before it reaches the target sites, allowing for passing of the protein through the gastric barrier and stably releasing the encapsulated protein to reduce blood sugar, and the inhibitor to avoid degradation of the NPs in the mucus barrier. Further in vitro and in vivo studies are required to assess the effect of double-layer protection.
Overall, flash nanoprecipitation is better at encapsulating BDs, and the EE is higher than for simple nanoprecipitation. This is most likely due to the high speed of encapsulation compared with simple nanoprecipitation. Flash nanoprecipitation allows a more precise control of the process conditions than emulsification, which could be helpful for future large-scale production and faster translation to clinical use. However, there are similarities between flash nanoprecipitation, simple nanoprecipitation, and the emulsification method where organic solvents are used. The effects of organic solvents on BDs have been discussed in the previous section, and future studies should try to carefully examine the effects of organic solvents used on therapeutic outcomes for each BD.
4. Self-assembly methods
Self-assembly is the spontaneous generation of ordered core–shell structures through local interactions between a BD core and a polymeric shell.111 The interactions include non-polar linkages such as van der Waals forces, electrostatic forces, hydrogen bonding, π–π aromatic stacking, and hydrophobic forces. Past studies mainly used hydrophobic polymers with hydrophobic chemicals for self-assembly.112Fig. 7 shows one of the possible representative approaches for synthesising BD-loaded core–shell nanoparticles.113Table 4 summarises self-assembly studies for BD encapsulation in the last 5 years.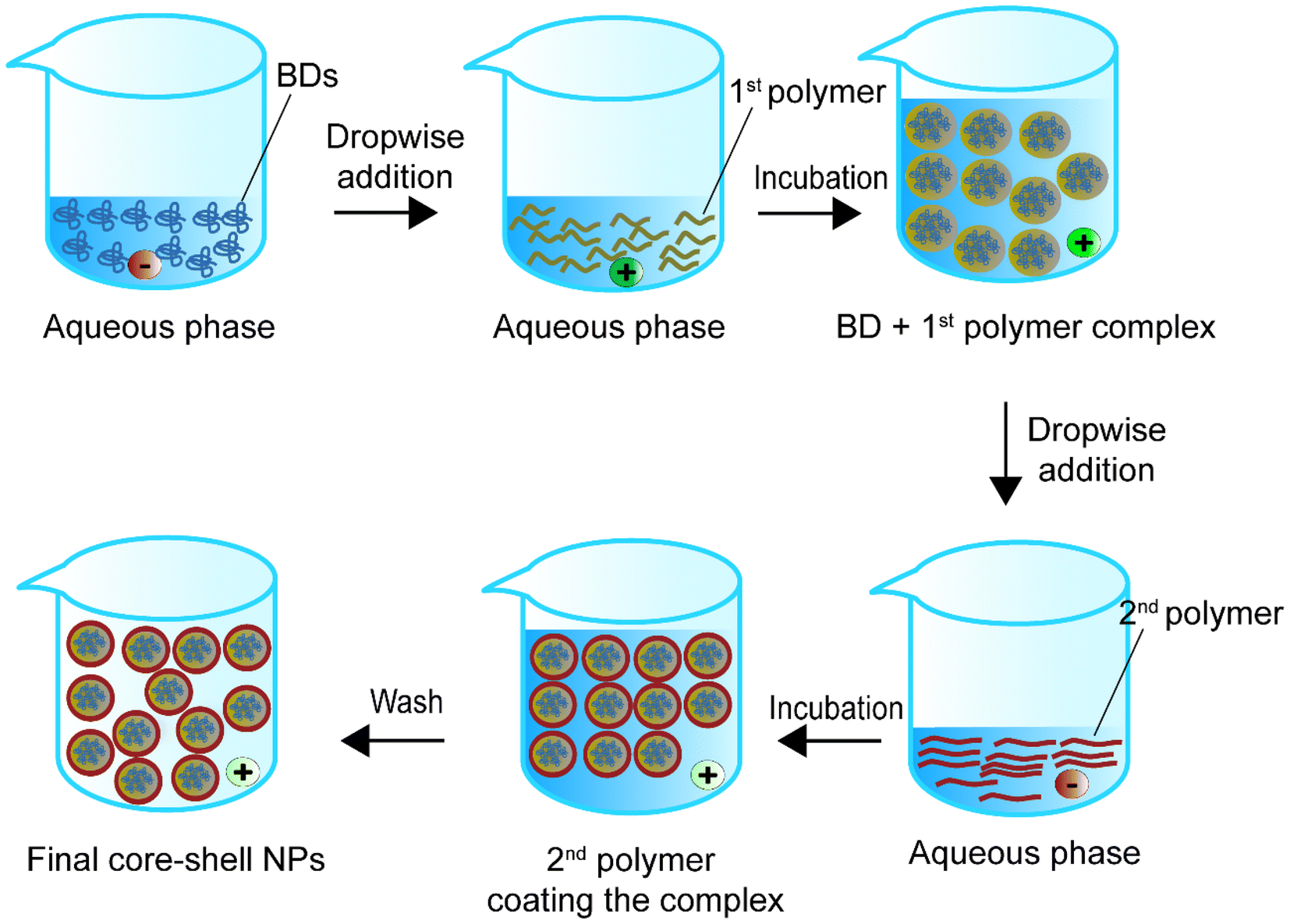 | ||
| Fig. 7 Schematic representation of the self-assembly method to synthesise core–shell nanoparticles. | ||
| Encapsulated compounds | Polymer | Second polymer or stabiliser(s) | Applications | Size | Encapsulation efficiency, % | Loading capacity, % | Ref. |
|---|---|---|---|---|---|---|---|
| Cyclic peptide CP(CPAETC)2 | pDPA | — | General BD delivery | 14 nm | — | — | 114 |
| Superoxide dismutase | Dopa-conjugated HA | — | Treating drug-induced hepatotoxicity and liver injury | 221.1 ± 13.1 nm | 73.4 | — | 115 |
| Plasmid DNA | HA | — | Anticancer | 193.1 ± 8.72 nm | — | — | 113 |
| PDCD4 | PCAH | PDA | Gene therapy | 110 nm | — | — | 140 |
| Enzyme-responsive polypeptide modified polyamide-amine (HPAA-peptide-Fc) | CD-HPG | HPAA | Anti-cancer | ∼300 nm | — | — | 138 |
| tPA | PAAc | PEG and PMNT | Thrombolysis | 48 ± 2 nm | — | — | 121 |
| Urokinase | PLGA | LSIPPKA peptide, beta-cyclodextrin and fucoidan | Thrombolysis | 575.6 nm | 62.61 | 22.89 | 122 |
| Insulin | p-Hydroxyphenylethyl anisate | 3-Acrylamidophenylboronic acid | Diabetes | 165.37 nm | ∼65 | ∼16 | 123 |
| Insulin | Poly(n-butylcyanoacrylate) | SDS and poloxamer 407 | Diabetes | ∼140 nm | 100% | ∼18 | 130 |
| BSA | TA | PS-b-PEG and PEI | Improving protein delivery, not specified | 10 kDa PEI: 153 ± 7 nm | 10 kDa PEI: 79 ± 7 | 10 kDa PEI: 13 ± 1 | 106 |
| Microtubule-associated sequences (MTAS) and nuclear localization signals and plasmid DNA | PBAE-447 | PGA | General gene therapy | 155 ± 40 nm | — | — | 142 |
| Synthetic mRNA | PBAE | PGA | General gene therapy | 109.6 ± 26.6 nm | — | — | 131 |
| Systemic siRNA | Modified PBAE | — | Brain glioblastoma | 67 nm | — | — | 133 |
| Luciferase mRNA | PASP | PEG | General intracerebroventicular delivery | 34 nm | — | — | 136 |
| In vitro transcribed mRNA and cotransfected single guide | PASP | — | Cerebral genome engineering | 100–130 nm | — | — | 134 |
| CRISPR-associated protein 9 mRNA and guide RNA | PASP | — | Cerebral genome engineering | 64.9 ± 5.1 nm | — | — | 135 |
| Double stranded RNA | Poly-L-lysine | — | Gene silencing | 46.2 ± 8.1 nm | — | — | 139 |
| SiRNA | Trimethyl and thiolated chitosan | HA | Anti-cancer | 110 nm | — | — | 143 |
| mRNA | Chitosan | PEG | Atherosclerotic lesions | ∼110 nm | 60% | — | 141 |
In terms of protein or enzyme encapsulation, Larnaudie et al.114 designed a drug delivery system with cyclic peptide CP(CPAETC)2 as the core and poly(2-(diisopropylamino)ethyl methacrylate) (pDPA) as the shell to form a 14 nm core–shell sphere at physiological pH. The paper only reported preliminary results and looked at the size and some physical properties of this new material. Later, Lee et al.115 employed a polysaccharides polymer, dopa-derivatised HA, to synthesise superoxide dismutase (SOD) loaded spherical NPs. Next, inorganic ultra-small size calcium phosphate (USCaP) nanocrystals were produced in situ on the spherical NP surface for a more efficient uptake in liver injury treatment. The final particles had an average diameter of 221.1 ± 13.1 nm and 73.4% EE.115 The authors suggested that the results gave an insight into the future use of different catechol-derivatised hydrophilic polymers (CDHPs) for surface charge management in different biomedical applications. The work reported both in vitro and in vivo experiments. In this study, the cytotoxicity of the NPs was evaluated in vitro only without examining whether the NPs would cause damage to organs in vivo. A limitation of these NPs is that SOD-encapsulated HA NPs should not be used in people with liver diseases. This is because serum HA is an important clinical non-invasive biomarker of liver fibrosis for chronic liver diseases such as chronic hepatitis infections and cirrhosis.116 For example, in cirrhosis or hepatitis C patients, the level of HA is higher than in healthy humans because the patients had increased liver fibrosis.117,118 Therefore, increasing the HA level at this time may obscure the true extent of liver damage of the patient and mislead clinical management.118–120 Mei et al.121 used mixtures of anionic poly(acrylic acid) (PAAc) and tPA in combination with cationic poly(ethylene glycol)-b-poly[4-(2,2,6,6-tetramethylpiperidine-1-oxyl)aminomethylstyrene] (PEG-b-PMNT) di-block amphiphilic copolymers to form nanoparticles in water using hydrophobic polycations. The copolymer had 4-amino-2,2,6,6-tetramethylpiperidine-1-oxyl (4-amino-TEMPO) on its side chains, which controls the reactive oxygen species, to aid the treatment. The team showed that their particles were effective compared with free tPA in reducing brain infarct volume and improving neurobehavioral results in a stroke mouse model. Furthermore, the prepared particles also reduced the area of tPA-induced subarachnoid haemorrhage by about 10% compared with free tPA, thus demonstrating that the particles could improve the brain haemorrhage previously caused by the use of tPA. However, the authors also did not report the LC and EE and whether this method is comparable to other encapsulation methods. Moreover, the representative images and the infarct volume results of the free tPA and tPA NPs without TEMPO group were very similar. The particles with TEMPO showed a lower infarct volume compared with the group with encapsulated tPA without TEMPO. The same happened for the haemorrhage area results between the two treatment groups. Also, the subarachnoid haemorrhage area of the tPA without TEMPO groups was not significantly different to the free tPA treatment. Combined together, TEMPO may have a higher therapeutical value than tPA and the redox NP shell.
Despite that, Zhang et al.122 also used a self-assembly method to load urokinase (UK) within an adamantane (ADA) conjugated fucoidan shell. The core contained tirofiban and a loaded small molecule peptide named LSIPPKA in PLGA beta-cyclodextrin (β-CD) NPs. Then UK was added to the β-CD, and LSIPPKA-modified PLGA aqueous solution. The final core–shell NPs with ADA–fucoidan shells were formed via the host–guest inclusion interaction of β-CD and ADA in an aqueous solution (Fig. 8). The size of the nanoparticles was larger (575.6 nm) compared with other studies due to the double layers. The LC and EE were reported as 22.89% and 62.61%, respectively.
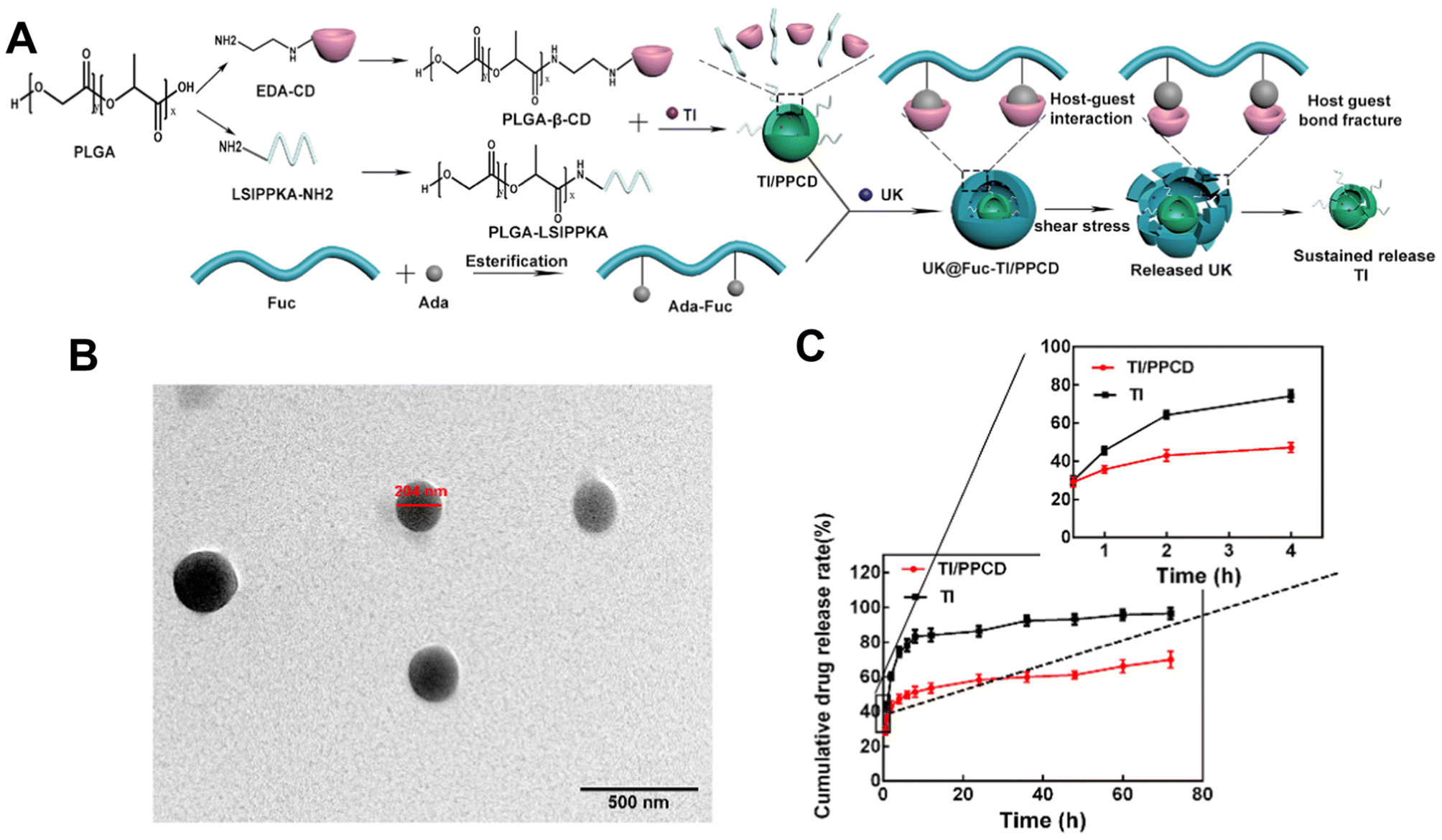 | ||
| Fig. 8 Synthesis route, thrombolytic mechanism and characteristics of UK@Fuc-TI/PPCD. (A) The synthesis route and drug release mechanism of urokinase UK@Fuc-TI/PPCD, (B) TEM image of UK@Fuc-TI/PPCD NPs after shearing (1000 dyne per cm2) for 20 min. (C) The sustained release profile of TI from TI/PPCD cores in PBS medium and free TI was used as the control. Reprinted from ref. 122, copyright (2021), with permission from Elsevier. | ||
Ma et al.123 employed a novel plant-derived p-hydroxyphenylethyl anisate and 3-acrylamidophenylboronic acid to form a shell to load insulin for diabetic treatment. The insulin core was self-assembly-encapsulated in the shell. As the NPs had a good EE and LC (65% 16%, respectively), this indicates that this novel polymer is promising for BD-encapsulated polymeric transportation. The NPs provided sustained insulin release for up to 28 hours in a diabetic mouse model. They had good biocompatibility that did not cause histological differences in major organs and did not show notable in vitro and in vivo toxicity after 14 days of injection.
Levit et al.106 combined iFNP with self-assembly to encapsulate BSA. They first formed a BSA–tannic acid (TA) bound complex in the presence of an amphiphilic block copolymer stabiliser, polystyrene-b-polyethylene glycol (PS-b-PEG) in a CIJ mixer. They then employed 10 kDa polyethylenimine (PEI) to coat the complex via electrostatic interaction to form a core–shell NP. They claimed that the TA did not affect the size or zeta potential but only helped increase the LC. In BSA–TA-loaded NPs with 10 kDa PEI, the addition of TA increased the EE from 8 ± 3% to 79 ± 7% and the LC from 1% to 13 ± 1%. Interestingly, when using a higher Mw PEI (750 kDa), an aggregate complex formed with a decreased EE by 29% and LC by 5% compared with PEI at 10 kDa. In this work, the TA–BSA complex was formed by hydrogen bonding. The authors did not assess whether the binding of TA affected the BSA. Moreover, it might be an issue if the model protein BSA is replaced by therapeutic BDs in the future, because hydrogen bonds might be critical for BD functionality. Namely, hydrogen bonds are actively involved in potential therapeutic targets or receptors of various diseases, such as severe acute respiratory syndrome coronavirus-2 (SARSr-CoV2) infection, Alzheimer's disease, and cardiovascular diseases.124–128 So binding TA or other agents with BDs via hydrogen bonds in the future may need a case-by-case analysis. We suggest that self-assembly may reduce the effect of BD structural changes that may happen in emulsion methods. However, the EE in this method may be lower than in those in other methods because the interaction forces between the self-assembled particles are weaker than those of particles synthesised by other methods. A major problem with the self-assembled polymeric drug delivery system is that the ionic interactions affect the NPs’ stability. The ion strength between the core–shell is heavily impacted by the concentration, and a diluted concentration may cause the structure to collapse.129 Thus, research has worked on applying other aids to overcome the loading problem in self-assembly. In particular, the NPs developed by Cheng et al.130 carried insulin with poly(n-butylcyanoacrylate) for oral delivery. The dispersibility of poly(n-butylcyanoacrylate) was enhanced by the addition of sodium dodecyl sulfate (SDS), whereas poloxamer 407 was used to raise the physical stability of the NPs for crossing the intestinal mucus layer. Following the addition of these stabilisers, 3 minutes (2 s on and 3 s off) at 200 W sonication was used for better loading of the insulin. The authors claimed that the stabilisers and sonication did not impact the insulin activity. The size of the NPs was about 140 nm. The study showed a sustainable insulin release and blood glucose level compared with the free insulin injection group. Future studies can follow up on the LC and EE of these particles to enrich their profiles and thus increase the credibility of using the self-assembly method to synthesise BD–polymer core–shell NPs.
Up-to-date, the potential of the self-assembly method to encapsulate BDs for gene therapy has been extensively explored for various diseases. For example, Smith et al.132 loaded a negatively charged plasmid with leukaemia-targeted chimeric antigen receptor genes (194-1BBz) and CD3ζ cytoplasmic signalling domains to an overall positively charged poly(beta-amino ester) [PBAE] polymer with microtubule-associated-nuclear localisation peptides. The primary positively charged complex was covered with a negatively charged polyglutamic acid shell with a targeting ligand to form NPs with a 155 ± 40 nm diameter. They showed that the lymphocyte antigen-recognised NPs could be translocated to T cell nuclei to prevent disease progression. This study also supports the ability of the self-assembly method to effectively load genetic information compared with the free gene group.
Their group also showed the potential of these carriers in mRNA delivery via self-assembly131 (Fig. 9). Synthetic mRNA was attached to the positively charged PBAE to form a complex, and then the complex was coated with an antibody-functionalised PGA to form NPs at a size of 109.6 ± 26.6 nm. They demonstrated that their self-assembled NPs had better viability, T-cell expression, and knock-out efficiency than the conventional electroporation for transfection. Moreover, the encapsulated mRNA can be encoded with other transcription factors for anti-tumour purposes and programmed for hematopoietic stem cell self-renewal.
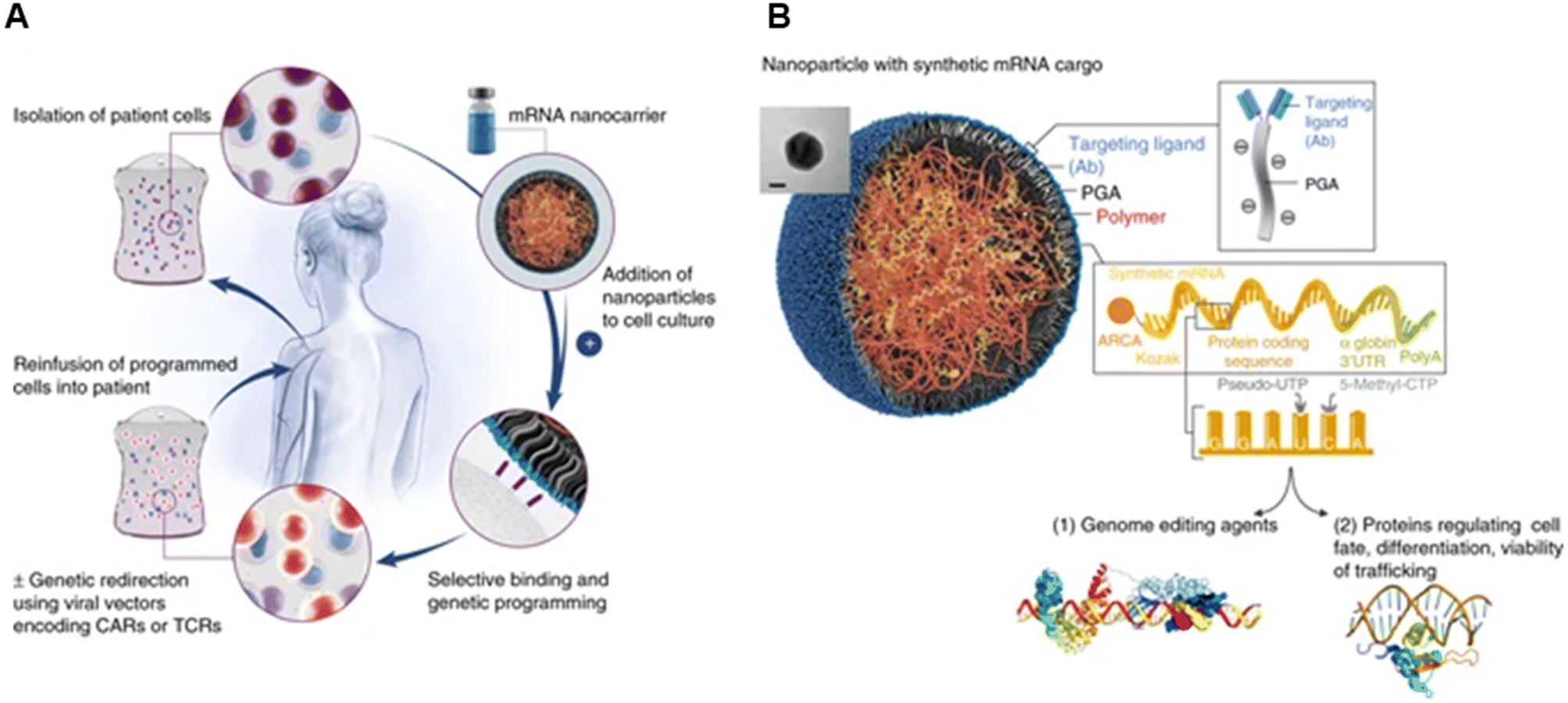 | ||
| Fig. 9 mRNA nanoparticles to program therapeutic T-cells. (A) Scheme explaining how cultured T-cells can be programmed to express therapeutically relevant transgenes carried by polymeric NPs. These particles are coated with ligands that target them to specific cell types, enabling them to introduce their mRNA cargoes and cause the targeted cells to express selected proteins (like transcription factors or genome-editing agents). (B) Design of targeted mRNA-carrying NPs. The inset shows a transmission electron micrograph of a representative NP; scale bar, 50 nm. Also depicted is the synthetic mRNA encapsulated in the NP, which is engineered to encode therapeutically relevant proteins. Reprinted from ref. 131, copyright (2017), with permission from Springer Nature. | ||
Karlsson et al.133 used the self-assembly method in an acidic environment to load systemic siRNA in modified PBAE with extra disulfide bonds to enhance the intracellular release of the siRNA for brain glioblastoma treatment. The study demonstrated that their self-assembled NPs (∼57 nm) could cross the barrier via a vesicular mechanism in an in vitro simulated blood–brain barrier model. In addition, after intravenously injecting the NPs, a significantly higher amount of the NPs was found in the brain of cancer-bearing mice compared with controls, without affecting the integrity of the blood–brain barrier. Other studies with the self-assembly method were also conducted after this one to deliver mRNA to the brain. Yet, they all used intracerebroventricular injection, a method to bypass the blood–brain barrier by injecting the NPs directly into the cerebrospinal fluid (see Table 4 for details of size and other parameters of the studies).134–136 We suspect this injection method was chosen to avoid the early release of self-assembled NPs due to ionisation or acid–base conditions. However, intracerebroventricular injections are prone to infection, bleeding, malposition and increased probability of nosocomial infections due to their catheter placement in clinical contexts, which should therefore be noted for clinical translation.137 Given that Karlsson et al.133 have demonstrated that self-assembled NPs of small size can cross the blood–brain barrier via transcytosis by a vesicular mechanism, future studies could try the subcutaneous injection of self-assembled NPs for brain diseases.
A recent paper reported NPs at a size of 300 nm using a cationic core consisting of a hyperbranched cationic polyamide-amine–peptide-amine (HPAA-peptide-Fc) bonded siRNA complex and coated by an anionic hyperbranched polyglycerol ether (HPG)-modified β-cyclodextrin derivative (CD-HPG) shell for cancer therapy.138 After the core polypeptide complex formed, the siRNA siPLK1 was dissolved and bound to the complex in a complex aqueous solution with the help of ultrasonication. The aqueous shell solution was added to the cationic siPLK1-containing complex for self-assembly NP formation. The particles were effectively down-regulated PLK1 protein expressed in breast cancer cells. However, these cancer-focused studies did not report the EE and LC of the methods. This raises a further concern that the method has relatively low loading and also concerns about the stability of the encapsulated BDs. Double-stranded RNA could also be encapsulated via the self-assembly method. The RNA was first mixed with an epigallocatechin gallate to form a complex, and then encapsulated by poly-L-lysine under 30 minutes’ vortexing and 5 minutes’ sonication in distilled water.139 With the aid of the polymer, the release period was extended compared with the free RNA group.
In terms of DNA delivery, Gu et al.113 synthesised retro-inverso D-peptide (RIF7)-modified HA/bioreducible hyperbranched poly(amidoamine) (RHB)/plasmid DNA (pDNA) ternary nanoparticles with a size of 193.1 ± 8.72 nm for cancer treatment. The authors claimed that the positively charged RHB coated the negatively charged pDNA via electrostatic interaction at a 5:1 weight ratio to form an RHB/pDNA complex. Due to the overall positive charge of the complex, a negatively charged RIF7-HA was employed to coat the complex to decrease the surface charge of the complex for a better circulation and targeting effect. Cui et al.140 utilised electrostatic interactions to synthesise BD-loaded polymeric core–shells for anti-tumour purposes. The team first mixed a gene called programmed cell death protein 4 (PDCD4) with a cationic poly(cystamine bisacrylamide)-urea-histamine (PCAH) carrier, which was then combined with the anionic polymer dextran-G-anhydride (PDA) to form particles with a diameter of approximately 110 nm. Compared with the free PDCD4 group, which had a minimum effect similar to PBS, the encapsulated PDCD4 group could reduce 4T1 tumour cell proliferation in vitro (100% vs. 60%). Consistently, the PDCD4-loaded PCAH NP group had a lower tumour volume than the free gene group. These results indicated the importance of the use of a carrier for anti-tumour gene therapy. While this work showed that the NPs effectively increased the transfection efficiency by 5 times as compared with the group without DNA (mean fluorescence intensity: 3 × 104) at pH 7, they did not include the transfection results of the free gene group as part of comparison.
Results from studies mentioned above support the idea that self-assembly can encapsulate different genetic materials and allow better targeting through polymeric NP carriers. Crosslinkers had been used to stabilise the nanoparticles. PEGylated-chitosan NPs (110 nm) were crosslinked by tripolyphosphate to deliver mRNA (miR-206 or miR-223, 60% EE).141 The expression of the ATP-binding cassette transporters A1 gene in macrophages was reduced both in vitro and in vivo. As a result, the cholesterol transport to the plasma, liver and feces in these crosslinked NP-treated mice was decreased. Furthermore, we suggest that self-assembly may reduce the effect of the BD structural changes that may happen in emulsion methods. However, the EE in this method may be lower than in those in other methods because the interaction forces between the self-assembled particles are weaker than those of particles synthesised by other methods. A major problem with the self-assembled polymeric drug delivery system is that the ionic interactions affect the NPs’ stability. The ion strength between the core–shell is heavily impacted by concentration, and a diluted concentration may cause the structure to collapse.129 Thus, research has worked on applying other aids to overcome the loading problem in self-assembly. In particular, the NPs developed by Cheng et al.130 carried insulin with poly(n-butylcyanoacrylate) for oral delivery. The dispersibility of poly(n-butylcyanoacrylate) was enhanced by the addition of sodium dodecyl sulfate (SDS), whereas poloxamer 407 was used to raise the physical stability of the NPs for crossing the intestinal mucus layer. Following the addition of these stabilisers, 3 minutes (2 s on and 3 s off) at 200 W sonication was used for better loading of the insulin. The authors claimed that stabilisers and sonication did not impact the insulin activity. The size of the NPs was about 140 nm. The study showed a sustainable insulin release and blood glucose level compared with the free insulin injection group. Future studies can follow up on the LC and EE of these particles to enrich their profiles and thus increase the credibility of using the self-assembly method to synthesise BD–polymer core–shell NPs.
4.1. Coaxial electrospraying
Coaxial electrospray is a novel method to encapsulate BDs, improving stability and producing small NPs Electrospraying technique is already applied in other areas, such as solar cells in large-scale production settings.144,145A coaxial electrospray includes a coaxial nozzle, a positive electrode applied at the tip of the nozzle, and a negative or ground electrode connected to the collector to create an electric field. The syringe pump pushes the core and shell material depending on the requirements. The potential difference pushes the liquids out to produce nanoscale particles (Fig. 10). This technique shares a similar electrohydrodynamic process to electrospinning.146 Electrospinning produces different morphologies of nanomaterial, such as nanofibers and nano-scaffolds to encapsulate chemical drugs and BDs.147–151 Due to the morphological interest, this review only focuses on the electrospraying method for synthesising BD-loaded polymeric NPs. Table 5 summarizes coaxial electrospraying studies for BD encapsulation in the last 5 years.
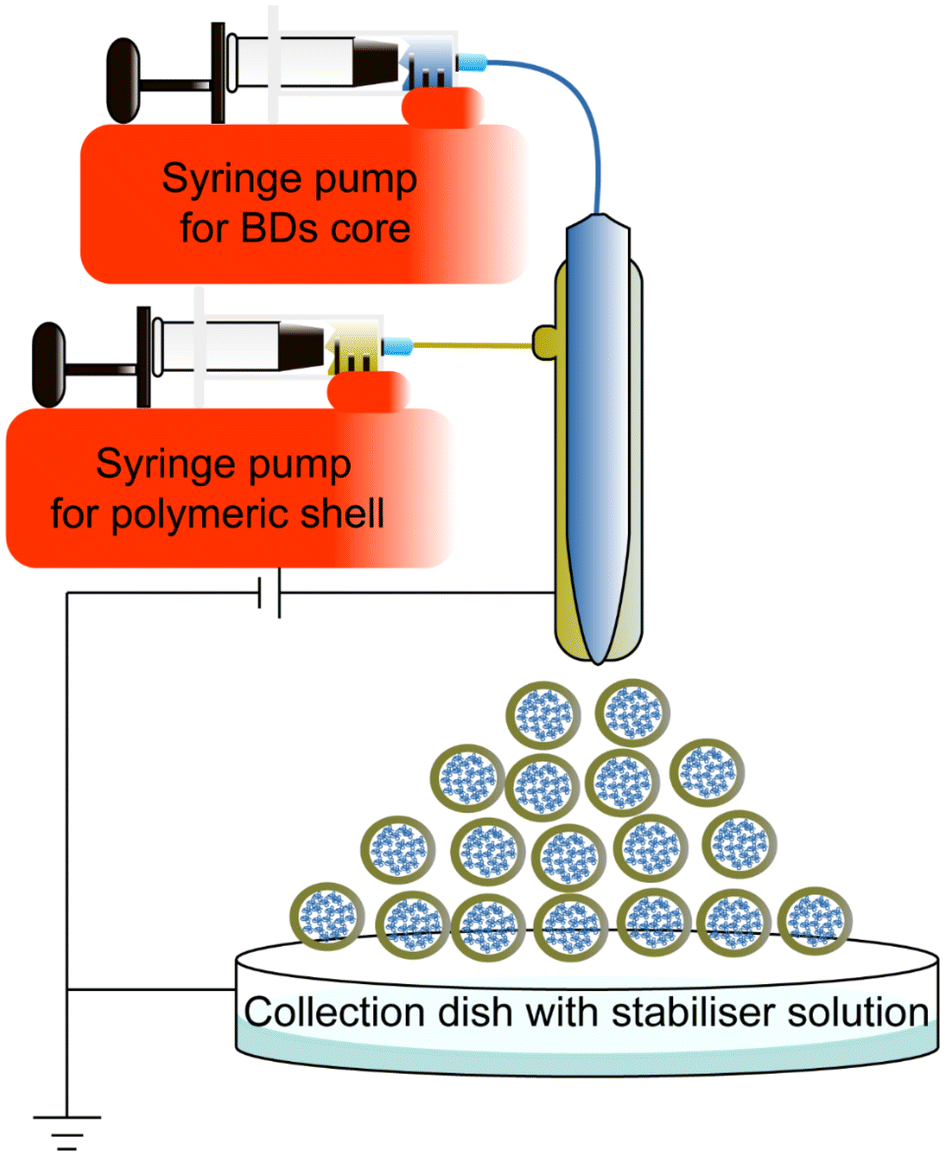 | ||
| Fig. 10 Schematic representation of the coaxial electrospraying method to synthesise core–shell nanoparticles. The NPs synthesised by coaxial electrospray in the scheme are only a schematic sample, not the actual size. | ||
Previous studies have focused on micron-sized BD core–shell encapsulation particles. However, only a limited number of nanoscale BD core–shell encapsulation studies were reported.152 Yaghoobi et al.153 reported the preparation of streptokinase (SK) loaded PLGA nanoparticles with a size of 37 ± 12 nm, 90% EE, and 8.2% LC. This is a pioneering study of BD–polymer core–shell nanoencapsulation. Subsequently, the team used mPEG–PLGA to encapsulate SK and obtained particles with a size of 194.3 ± 15.9 nm, EE of 83.3 ± 3.2% and LC of 8.2 ± 0.7%.154 This study demonstrated that NPs prepared by coaxial electrospray did not impact the bioactivity of BD and blood parameters such as red blood cells as well as coagulation factors, compared with free SK. The human umbilical vein endothelial cells and female rats treated with NPs group also had a better result in vitro and in vivo viability than those with commercial SK. However, the authors did not examine thrombolysis rates, which would provide a better evaluation of whether the synthetic nanoparticles were more effective than the free SK. Overall, the BD used in these two articles could be heavily loaded into hydrophobic polymers and form a dense shell. However the synthesised NPs might not be homogenous as reported by Hasanpour et al.;154 this coaxial electrospraying method yielded a polydispersity index of 0.73 ± 0.015. Future optimisation may be required to obtain more homogeneous nanoparticles.
Furthermore, there has been a disagreement on whether coaxial electrospraying can be used for high-throughput production in the industry.155 This method involves the intervention of electromagnetic fields and potential differences. For example, without good isolation of the magnetic field, some nanoparticles with positive charges interact with the surrounding air but do not drop to the collection plate, reducing the efficiency of coaxial electrospray to produce BD-loaded NPs.156 It was suggested that using ring electrodes for less distributed cone-jet sprayed NPs is one of the possible solutions.157 Further studies can also explore establishing a large-scale electronic static isolation shield to favour the mass production of the core–shell NPs.
5. Insights for future directions, perspectives, and translation of biomedical applications
Manufacturing methods for BD encapsulation summarised in the previous sections have successfully loaded various BDs such as proteins, enzymes, peptides, DNA, mRNA and siRNA. Typical methods to produce core–shell BD-loaded polymeric NPs include single emulsion, double emulsion, simple nanoprecipitation, iFNP, self-assembly and coaxial electrospray. Using polymers to encapsulate BDs with a single emulsion method exhibits a low EE and is challenging to control for scale-up production. The double emulsion improves the low EE issue of the single emulsion method and has been extensively investigated for various BDs. However, the effects of sonication and organic solvents on BDs have not been fully explored. The simple nanoprecipitation method shows fluctuations in EE for different BDs, and this method also involves organic solvents. iFNP produces core–shell NPs by rapidly introducing a non-solvent of the polymer into the cavity and mixing rapidly. The shortcomings of normal nanoprecipitation, such as the contact time of non-solvent to the BDs and polymer are improved by using iFNP. The self-assembly method also allows for the rapid formation of core–shell structures but can be affected by the concentration dilution, leading to the breakage of the nanomaterial, thus compromising its effectiveness. Coaxial electrospray allows precise control of the variables to achieve high LC and EE. However, coaxial electrospray is susceptible to the influence of surrounding static electromagnetic fields, leading to sample waste and poorly distributed sample results (as summarised in Table 6). Therefore, further optimisation should be carried out to address the shortcomings of these methods.| Synthesis methods | EE | LC | Advantages and disadvantages |
|---|---|---|---|
| Single emulsion | Low | Low | ✓ Commonly used methods that allow uniform shape and size distribution |
| Double emulsion | Moderate | Low | ✗ Use of sonication, stabiliser and organic solvents may impact BD stability |
| Simple nanoprecipitation | EE and LC are impacted by the size of the BDs | ✓ Fast and simple to prepare | |
| ✗ Use of organic solvents | |||
| iFNP | ✓ Reduce the contact/reaction time between the BDs and non-solvent improves NP characteristics | ||
| ✗ Unknown mixing mechanism | |||
| Self-assembly | High | Low | ✓ Variety of shape |
| ✗ Package leakage, and pre-release of BDs due to surrounding micro-environment change | |||
| Coaxial electro-spraying | High | Low | ✓ High encapsulation efficiency |
| ✗ Waste of sample due to collection condition | |||
The combination of microfluidic devices and conventional synthesis methods is a practical approach in advancing BD-loaded polymeric NP synthesis. Previously, microfluidics had mostly been studied at the micron level for BD encapsulation.158,159 However, the summarised iFNP method section supports the use of microfluidic devices in BD nano-encapsulation. Also, there are studies using microfluidic devices with self-assembly mechanisms to encapsulate siRNA in 7C1 and PEG2000 polymer NPs and successfully conduct endothelial gene silencing in mouse organs.160 These examples demonstrate the use of microfluidic devices in polymer-coated BD encapsulation.161 Further refinement of the devices may be achieved via modifying the structure of the device to reduce BD and non-solvent contact and also enhance the mixing ability. For example, the use of a Tesla structure in microfluidic devices has been shown to benefit insulin encapsulation in PLGA NPs; primary parameters such as flow rate and the concentration of PLGA and insulin do not interfere with the physical characteristics of the produced NPs.162 Whether these could be translated to general BD-loaded polymeric NP production with microfluidic devices still needs investigation. Overall, it is suggested that the combination of conventional synthesis methods with microfluidic devices can facilitate the use of polymers for BD encapsulation in the future. The use of microfluidics devices is also a potential solution for large-scale production and better control of the conditions under which the nanoparticles are formed. If future studies can overcome the disadvantage of slow production yield, the microfluidic-based method will benefit future mass production applications.163
Furthermore, one of the key challenges that BDs face in drug delivery is releasing, denaturing and clearing before reaching the target site.164,165 Future studies can explore other biodegradable materials to refine their properties and compare their effects with the currently available FDA-approved polymers for a more comprehensive understanding. Specifically, FDA-approved polymers, such as PLGA, may not be a suitable polymer shell for encapsulating BDs in acute disease treatment.166,167 PLGA has a slow hydrolysis profile in a physiological pH and temperature that releases encapsulated molecules after days to months,166–169 yet some studies targeting acute disease treatment are still using PLGA as a carrier. It is important to explore other novel polymers that can be used for acute and chronic diseases with minimum modification.
The polymer shell should be modified to tune the hydrolysis rate and drug release profile corresponding to a specific disease. For example, the polymeric shell used in cancer or diabetes should have a slow dissolution or hydrolysis rate to provide long-term sustained release of the therapeutic compounds. However, in acute diseases such as thrombosis and stroke, the shell should be hydrolysed or degraded quicker to offer sustained release over a shorter period.170
New types of polymer such as molecularly imprinted polymers (MIPs) can be employed for targeted drug delivery. MIPs are synthetic materials that contain recognition sites for specific binding to potential binding sites in the body.171 Based on the required disease's physiological conditions, functional and crosslinking monomers are selected based on their ability to bind to a template molecule via weak non-covalent or reversible covalent bonds. Cleavage of the bonds between the monomers and template can produce the required MIPS. MIPs can be degraded in physiological conditions with different degradation profiles that allow controlled release with the help of temperature and pH. Also, due to the specificity of the functional monomer, it has a high affinity to the targets.171,172 Several studies have demonstrated the potential use of MIPs for the delivery of chemical drugs or other therapeutic agents in biomedical applications.173–175 It is worth applying different modifications or inventions to the MIPs for different needs. Using MIPs as novel shells to encapsulate BDs or directly stitching BDs as part of the MIPs into a more stereospecific polymer cocktail will broaden the utility of synthesis methods and the application types.176
As can be seen from the articles reviewed in this paper, the size of the BDs may impact the efficiency of the encapsulation. For example, in Markwalter et al.,87,107 vancomycin is typically only 1.45 kDa in size, and thus can easily escape from the packaging envelope during the encapsulation process.177 So far, there are only a few studies encapsulating small molecular weight BDs with polymeric materials. Future explorations can adapt different production methods and materials, such as introducing MIPs for small molecular weight BD encapsulation to achieve better results for drug delivery. This review believes that the advance of polymeric materials can benefit the established synthesis methods for nanoparticle synthesis.
A Janus structure can be considered for polymeric nanoencapsulation for BDs. Janus-structured NPs are particles that contain multiple domains to simultaneously load a combination of drugs for various purposes, such as theranostics and delayed drug release.178–180 Combining the Janus structure with existing production methods is something that studies have already done. Synergistic analgesia by oral administration of Janus-structured NPs using a double emulsion of naja naja atra venom protein and resveratrol has been reported.181 Also, a study used cellulose acetate and polycaprolactone as carriers to transport sliver NPs and lavender oil.182 Currently, a limited number of studies are using a polymeric carrier to load BDs in a Janus structure. Whether such a structure can be combined with existing production methods and the use of microfluidic devices to benefit BD-loaded polymeric NP delivery is worth exploring in the future.
The BD-encapsulated NPs undergoing clinical trials are mainly lipid NPs such as SARSr-CoV2 vaccines, mRNA-1273, BNT162b2, and NVX-CoV2373 that were tested in phase III clinical trials.183–186 There are a limited number of polymeric BD-encapsulated NPs prepared by emulsion methods undergoing clinical trials as discussed in previous sections. Advanced microfluidic devices appear as a new potential and efficient approach to encapsulate BDs within polymeric nanoparticles.
6. Conclusion
This review first discussed the advantages and disadvantages of various available nanomaterials and then discussed the use of polymeric materials for BD encapsulation. Free BD is not efficiently absorbed in the body and is prone to degradation, clearance or aggregation, possibly leading to other complications. Using polymers to encapsulate BDs, rather than conjugate BDs, allows for better drug delivery by maximising the bioactivity of BDs while increasing efficacy. A core–shell structure is preferred to encapsulate BDs due to BD characteristics, high LC, EE, a controlled release manner and a higher stability of core–shell encapsulation. This review describes the various methods used to encapsulate BD with polymers in recent years.The information collected in this review is expected to benefit the development of future polymeric NPs for BD delivery by facilitating the selection of synthesis method and suitable polymeric materials. A combination of appropriate improvements to existing methods and development of novel materials could drive the advancement of drug delivery.
Abbreviations
| BD | Biological drug |
| BVZ | Bevacizumab |
| CHA | Cholic/deoxycholic acid |
| CIJ | Confined impinging jets |
| CMC | Carboxymethyl chitosan |
| CP(CPAETC)2 | Cyclic peptide |
| Cyt-c | Cytochrome c |
| DDAB | Dimethyl-dioctadecyl ammonium bromide |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| EE | Encapsulation efficiency |
| FA | Folic acid |
| FDA | Food and Drug Administration |
| HA | Hyaluronic acid |
| HIV-1 | Human immunodeficiency virus-1 |
| HPAA | Hyperbranched cationic polyamide-amine |
| HPG | Polyglycerol ether |
| HPMCAS | Hydroxypropyl methylcellulose acetate-succinate |
| HRP | Horseradish peroxidase |
| IC50 | Half-maximal inhibitory concentration |
| KPV | Lysine–proline–valine |
| LC | Loading capacity |
| M. luteus | Micrococcus luteus |
| MIPs | Molecularly imprinted polymers |
| MIVM | Multi-inlet vortex mixer |
| mPEG | Methoxy polyethylene glycol |
| mRNA | Messenger RNA |
| NP | Nanoparticles |
| NY-ESO-1 | New York esophageal squamous cell carcinoma 1 |
| OVA | Ovalbumin |
| PAAc | Poly(acrylic acid) |
| PAE | Poly(β-amino ester) |
| PASP | Poly(aspartic acid) |
| PBAE | Poly(beta-amino ester) |
| PCAH | Poly(cystamine bisacrylamide)-urea-histamine carrier |
| PDA | Polymer dextran-G-anhydride |
| pDNA | Plasmid DNA |
| pDPA | Poly(2-(diisopropylamino)ethyl methacrylate) |
| PEG | Polyethylene glycol |
| PEI | Polyethylenimine |
| PLA | Polylactic acid |
| PLGA | Poly lactic-co-glycolic acid |
| PMNT | Poly[4-(2,2,6,6-tetramethylpiperidine-1-oxyl)aminomethylstyrene] |
| PS-b-PAA | Polystyrene-b-polyacrylic acid |
| PS-b-PEG | Polystyrene-b-polyethylene glycol |
| PVA | Poly(vinyl alcohol) |
| RHB | Bioreducible hyperbranched poly(amido amine) |
| RIF7 | Retro-inverso D-peptide |
| RNA | Ribonucleic acid |
| SARSr-CoV2 | Severe acute respiratory syndrome coronavirus-2 |
| siRNA | Small interfering ribonucleic acid |
| SK | Streptokinase |
| Smac | Second mitochondria-derived activator of caspase |
| SOD | Superoxide dismutase |
| TA | Tannic acid |
| TEMPO | Tetramethylpiperidine-1-oxyl |
| TK | Thioketal |
| UC | Ulcerative colitis |
| UK | Urokinase |
| USCaP | Ultra-small size calcium phosphate |
| β-CD | β-Cyclodextrin |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Australian National Health and Medical Research Council (HTT: APP1037310, APP1182347, APP2002827) and National Heart Foundation of Australia (HTT: 102761). Xiangxun Chen is supported by a scholarship from Griffith University.References
- T. Morrow and L. H. Felcone, Biotechnol. Healthc., 2004, 1, 24–29 Search PubMed
.
- J. C. N. Chan and A. T. C. Chan, ESMO Open, 2017, 2, e000180 CrossRef
-
Therapeutic Antibody Engineering, ed. W. R. Strohl and L. M. Strohl, Woodhead Publishing, 2012, pp. 1–595. DOI:10.1533/9781908818096.1
- S. Marqus, E. Pirogova and T. J. Piva, J. Biomed. Sci., 2017, 24, 21 CrossRef PubMed
- A. Mahipal and A. Grothey, J. Oncol. Pract., 2016, 12, 1219–1228 CrossRef PubMed
- V. Schirrmacher, Int. J. Oncol., 2019, 54, 407–419 CAS
- J. S. Van Taunay, M. T. Albelda, J. C. Frias and M. J. Lipinski, J. Cardiovasc. Pharmacol., 2018, 72, 77–85 CrossRef CAS PubMed
- M. Bliss, Bull. Hist. Med., 1982, 56, 554–568 CAS
- U.S. Food and Drug Administration, 2021.
- PRNewswire, Global Biologics Market Analysis Report 2022-2025 & 2030 Featuring Merck & Co, AbbVie, F. Hoffmann-La Roche, Johnson & Johnson, & Pfizer, https://www.prnewswire.com/news-releases/global-biologics-market-analysis-report-2022-2025–2030-featuring-merck–co-abbvie-f-hoffmann-la-roche-johnson–johnson–pfizer-301530433.html, accessed 05.10, 2022.
- S. W. Chung, T. A. Hil-lal and Y. Byun, J. Drug Targeting, 2012, 20, 481–501 CrossRef CAS PubMed
- R. Bajracharya, J. G. Song, S. Y. Back and H.-K. Han, Comput. Struct. Biotechnol. J., 2019, 17, 1290–1308 CrossRef CAS PubMed
- B. Homayun, X. Lin and H.-J. Choi, Pharmaceutics, 2019, 11, 129 CrossRef CAS PubMed
- S. J. Won, X. N. Tang, S. W. Suh, M. A. Yenari and R. A. Swanson, Ann. Neurol., 2011, 70, 583–590 CrossRef CAS
- Y. Jiang, J. Han, P. Spencer, Y. Li, S. J. Vodovoz, M.-M. Ning, N. Liu, X. Wang and A. S. Dumont, Brain Hemorrh., 2021, 2, 116–123 CrossRef CAS
- H. Bardania, S. A. Shojaosadati, F. Kobarfard, F. Dorkoosh, M. E. Zadeh, M. Naraki and M. Faizi, J. Thromb. Thrombolysis, 2017, 43, 184–193 CrossRef CAS
-
S. Gouda, R. George Kerry, G. Das and J. Kumar Patra, in Nanomaterials in Plants, Algae and Microorganisms, ed. D. K. Tripathi, P. Ahmad, S. Sharma, D. K. Chauhan and N. K. Dubey, Academic Press, 2019, pp. 219–235. DOI:10.1016/B978-0-12-811488-9.00011-1
- S. Sim and N. K. Wong, Biomed. Rep., 2021, 14, 42–42 CrossRef CAS PubMed
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed
- J. K. Patra, Das, L. Fraceto, E. Campos, M. D. P. Rodríguez-Torres, L. Acosta-Torres, L. Diaz-Torres, R. Grillo, M. Swamy, S. Sharma, S. Habtemariam and H. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed
- H. C. Huang, S. Barua, G. Sharma, S. K. Dey and K. Rege, J. Controlled Release, 2011, 155, 344–357 CrossRef CAS PubMed
- R. Chakravarty, S. Goel, A. Dash and W. Cai, Q. J. Nucl. Med., 2017, 61, 181–204 Search PubMed
- H. Wang, R. Kumar, D. Nagesha, R. I. Duclos, S. Sridhar and S. J. Gatley, Nucl. Med. Biol., 2015, 42, 65–70 CrossRef CAS PubMed
- H. T. Ta, S. Prabhu, E. Leitner, F. Jia, D. von Elverfeldt, K. E. Jackson, T. Heidt, A. K. N. Nair, H. Pearce and C. Von Zur Muhlen, Circ. Res., 2011, 109, 365–373 CrossRef CAS PubMed
- H. T. Ta, Z. Li, C. E. Hagemeyer, G. Cowin, S. Zhang, J. Palasubramaniam, K. Alt, X. Wang, K. Peter and A. K. Whittaker, Biomaterials, 2017, 134, 31–42 CrossRef CAS PubMed
- H. T. Ta, N. Arndt, Y. Wu, H. J. Lim, S. Landeen, R. Zhang, D. Kamato, P. J. Little, A. K. Whittaker and Z. P. Xu, Nanoscale, 2018, 10, 15103–15115 RSC
- H. T. Ta, Z. Li, Y. Wu, G. Cowin, S. Zhang, A. Yago, A. K. Whittaker and Z. P. Xu, Mater. Res. Express, 2017, 4, 116105 CrossRef
- N. N. M. Yusof, A. McCann, P. J. Little and H. T. Ta, Thromb. Res., 2019, 177, 161–171 CrossRef CAS PubMed
- K. X. Vazquez-Prada, J. Lam, D. Kamato, Z. P. Xu, P. J. Little and H. T. Ta, Arterioscler., Thromb., Vasc. Biol., 2021, 41, 601–613 CrossRef CAS
- Y. Liu, Y. Wu, R. Zhang, J. Lam, J. C. Ng, Z. P. Xu, L. Li and H. T. Ta, ACS Appl. Bio Mater., 2019, 2, 5930–5940 CrossRef CAS
- N. Arndt, H. D. Tran, R. Zhang, Z. P. Xu and H. T. Ta, Adv. Sci., 2020, 7, 2001476 CrossRef CAS PubMed
- Y. Wu, K. X. Vazquez-Prada, Y. Liu, A. K. Whittaker, R. Zhang and H. T. Ta, Nanotheranostics, 2021, 5, 499 CrossRef PubMed
- H. Ta, S. Prabhu, E. Leitner, K. Putnam, F. Jia, N. Bassler, K. Peter and C. Hagemeyer, Heart, Lung Circ., 2010, 19, S10 CrossRef
- A. U. Rehman, Y. Wu, H. D. Tran, K. Vazquez-Prada, Y. Liu, H. Adelnia, N. D. Kurniawan, M. N. Anjum, S. S. Moonshi and H. T. Ta, ACS Appl. Nano Mater., 2021, 4, 10136–10147 CrossRef CAS
- S. S. Moonshi, Y. Wu and H. T. Ta, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, e1760 CAS
- Y. Wu, Y. Yang, W. Zhao, Z. P. Xu, P. J. Little, A. K. Whittaker, R. Zhang and H. T. Ta, J. Mater. Chem. B, 2018, 6, 4937–4951 RSC
- Y. Wu, R. Zhang, H. D. Tran, N. D. Kurniawan, S. S. Moonshi, A. K. Whittaker and H. T. Ta, ACS Appl. Nano Mater., 2021, 4, 3604–3618 CrossRef CAS
- Y. Wu, G. Cowin, S. S. Moonshi, H. D. Tran, N. A. Fithri, A. K. Whittaker, R. Zhang and H. T. Ta, Mater. Sci. Eng., C, 2021, 131, 112477 CrossRef CAS PubMed
- Z. Ferdous and A. Nemmar, Int. J. Mol. Sci., 2020, 21(7), 2375 CrossRef CAS PubMed
- M. Fisichella, F. Berenguer, G. Steinmetz, M. Auffan, J. Rose and O. Prat, BMC Genomics, 2014, 15, 1–15 CrossRef PubMed
- S. Aalapati, S. Ganapathy, S. Manapuram, G. Anumolu and B. M. Prakya, Nanotoxicology, 2014, 8, 786–798 CAS
- G. Yang, S. Z. F. Phua, A. K. Bindra and Y. Zhao, Adv. Mater., 2019, 31, 1805730 CrossRef PubMed
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102–102 CrossRef PubMed
- E. Beltrán-Gracia, A. López-Camacho, I. Higuera-Ciapara, J. B. Velázquez-Fernández and A. A. Vallejo-Cardona, Cancer Nanotechnol., 2019, 10, 11 CrossRef
-
M. Rahman, S. Beg, A. Verma, F. Anwar, A. Samad and V. Kumar, Nanotechnology-Based Approaches for Targeting and Delivery of Drugs and Genes, ed. V. Mishra, P. Kesharwani, M. C. I. Mohd Amin and A. Iyer, Academic Press, 2017, pp. 151–166. DOI:10.1016/B978-0-12-809717-5.00005-1
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Front. Pharmacol., 2015, 6, 286 Search PubMed
- H. Idrees, S. Z. J. Zaidi, A. Sabir, R. U. Khan, X. Zhang and S.-U. Hassan, Nanomaterials, 2020, 10, 1970 CrossRef CAS PubMed
- S. Agarwal, Macromol. Chem. Phys., 2020, 221, 2000017 CrossRef CAS
- S. Biswas, P. P. Deshpande, G. Navarro, N. S. Dodwadkar and V. P. Torchilin, Biomaterials, 2013, 34, 1289–1301 CrossRef CAS PubMed
- A. Gagliardi, E. Giuliano, E. Venkateswararao, M. Fresta, S. Bulotta, V. Awasthi and D. Cosco, Front. Pharmacol., 2021, 12, 601626–601626 CrossRef CAS PubMed
-
V. Fasiku, E. K. Amuhaya, K. M. Rajab and C. A. Omolo, Nano- and Microencapsulation – Techniques and Applications, 2021. DOI:10.5772/intechopen.93364
- Z. Zhou and M. Hartmann, Top. Catal., 2012, 55, 1081–1100 CrossRef CAS
- S. A. Edwards and D. R. M. Williams, Phys. Rev. Lett., 2004, 92, 248303 CrossRef CAS PubMed
- M. Boström, D. R. M. Williams and B. W. Ninham, Phys. Rev. Lett., 2001, 87, 168103 CrossRef PubMed
- H. H. Nguyen and M. Kim, Appl. Sci. Converg. Technol., 2017, 26, 157–163 CrossRef
- S. Smith, K. Goodge, M. Delaney, A. Struzyk, N. Tansey and M. Frey, Nanomaterials, 2020, 10(11), 2142 CrossRef CAS PubMed
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS PubMed
- F. Szoka and D. Papahadjopoulos, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4194–4198 CrossRef CAS PubMed
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed
- S. J. Park, Int. J. Nanomed., 2020, 15, 5783–5802 CrossRef CAS PubMed
- S. L. Perry and D. J. McClements, Molecules, 2020, 25, 1161 CrossRef CAS PubMed
- S. Wang, P. Huang and X. Chen, ACS Nano, 2016, 10, 2991–2994 CrossRef CAS PubMed
-
O. Martínez-Muñoz, L. Ospina-Giraldo and C.-E. Mora-Huertas, Nanoprecipitation: Applications for Entrapping Active Molecules of Interest in Pharmaceutics, IntechOpen, London, 2020. DOI:10.5772/intechopen.93338
- T. Jibowu, J. Nanomed. Nanotechnol., 2016, 7(3), 100379 Search PubMed
- D. Panigrahi, P. K. Sahu, S. Swain and R. K. Verma, SN Appl. Sci., 2021, 3, 638 CrossRef CAS
- C. Chen, W. Liu, P. Jiang and T. Hong, Micromachines, 2019, 10, 125 CrossRef PubMed
- B. K. Sabjan, M. S. Munawar, D. Rajendiran, K. S. Vinoji and K. Kasinathan, Curr. Drug Res. Rev., 2020, 12, 4–15 CrossRef PubMed
- D. Essa, P. P. D. Kondiah, Y. E. Choonara and V. Pillay, Front. Bioeng. Biotechnol., 2020, 8, 48 CrossRef PubMed
- M. Zamanlu, M. Eskandani, J. Barar, M. Jaymand, P. S. Pakchin and M. Farhoudi, J. Drug Delivery Sci. Technol., 2019, 53, 101165 CrossRef CAS
- Y. Dölen, U. Gileadi, J. L. Chen, M. Valente, J. H. A. Creemers, E. A. W. Van Dinther, N. K. van Riessen, E. Jäger, M. Hruby, V. Cerundolo, M. Diken, C. G. Figdor and I. J. M. de Vries, Front. Immunol., 2021, 12, 641703 CrossRef PubMed
- J. H. A. Creemers, I. Pawlitzky, K. Grosios, U. Gileadi, M. R. Middleton, W. R. Gerritsen, N. Mehra, L. Rivoltini, I. Walters, C. G. Figdor, P. B. Ottevanger and I. J. M. de Vries, BMJ Open, 2021, 11, e050725 CrossRef PubMed
- Q.-P. Feng, Y.-T. Zhu, Y.-Z. Yuan, W.-J. Li, H.-H. Yu, M.-Y. Hu, S.-Y. Xiang and S.-Q. Yu, Mater. Sci. Eng., C, 2021, 124, 112039 CrossRef CAS PubMed
- M. Iqbal, N. Zafar, H. Fessi and A. Elaissari, Int. J. Pharm., 2015, 496, 173–190 CrossRef CAS PubMed
- J. D. Ospina-Villa, C. Gómez-Hoyos, R. Zuluaga-Gallego and O. Triana-Chávez, J. Microbiol. Methods, 2019, 162, 1–7 CrossRef CAS PubMed
- S. Matsumoto, T. Inoue, M. Kohda and K. Ikura, J. Colloid Interface Sci., 1980, 77, 555–563 CrossRef CAS
- Y. F. Maa and C. C. Hsu, Pharm. Dev. Technol., 1999, 4, 233–240 CrossRef CAS PubMed
- J. Cai, X. Huai, S. Liang and X. Li, Front. Energy Power Eng. China, 2010, 4, 313–318 CrossRef
- E. Steiert, L. Radi, M. Fach and P. R. Wich, Macromol. Rapid Commun., 2018, 39, 1800186 CrossRef PubMed
- M. Boushra, S. Tous, G. Fetih, H.-Y. Xue and H.-L. Wong, J. Drug Delivery Sci. Technol., 2019, 49, 632–641 CrossRef CAS
- D. L. Priwitaningrum, J. Jentsch, R. Bansal, S. Rahimian, G. Storm, W. E. Hennink and J. Prakash, Int. J. Pharm., 2020, 585, 119535 CrossRef CAS PubMed
- B. Xiao, Z. Xu, E. Viennois, Y. Zhang, Z. Zhang, M. Zhang, M. K. Han, Y. Kang and D. Merlin, Mol. Ther., 2017, 25, 1628–1640 CrossRef CAS PubMed
- H. Laroui, G. Dalmasso, H. T. T. Nguyen, Y. Yan, S. V. Sitaraman and D. Merlin, Gastroenterology, 2010, 138, 843–853 CrossRef CAS PubMed
- F. Minooei, J. R. Fried, J. L. Fuqua, K. E. Palmer and J. M. Steinbach-Rankins, Int. J. Nanomed., 2021, 16, 1189–1206 CrossRef PubMed
- X. Li, A. n. Sun, Y.-j. Liu, W.-j. Zhang, N. Pang, S.-x. Cheng and X.-r. Qi, NPG Asia Mater., 2018, 10, 238–254 CrossRef CAS
- Z. Zhang, Y. Heng, W. Cheng, Y. Pan, S. Ni and H. Li, Mater. Des., 2021, 204, 109648 CrossRef CAS
- L. C. Lin, C. Y. Huang, B. Y. Yao, J. C. Lin, A. Agrawal, A. Algaissi, B. H. Peng, Y. H. Liu, P. H. Huang, R. H. Juang, Y. C. Chang, C. T. Tseng, H. W. Chen and C. J. Hu, Adv. Funct. Mater., 2019, 29, 1807616 CrossRef PubMed
- M. S. Ural, M. Menéndez-Miranda, G. Salzano, J. Mathurin, E. N. Aybeke, A. Deniset-Besseau, A. Dazzi, M. Porcino, C. Martineau-Corcos and R. Gref, Pharmaceutics, 2021, 13, 1992 CrossRef CAS PubMed
- R. Liao, J. Pon, M. Chungyoun and E. Nance, Biomaterials, 2020, 257, 120238 CrossRef CAS PubMed
- T. L. Freitag, J. R. Podojil, R. M. Pearson, F. J. Fokta, C. Sahl, M. Messing, L. C. Andersson, K. Leskinen, P. Saavalainen, L. I. Hoover, K. Huang, D. Phippard, S. Maleki, N. J. C. King, L. D. Shea, S. D. Miller, S. K. Meri and D. R. Getts, Gastroenterology, 2020, 158, 1667–1681 CrossRef CAS PubMed
- J. Su and A. Cavaco-Paulo, Ultrason. Sonochem., 2021, 76, 105653–105653 CrossRef CAS PubMed
- N. Leister and H. Karbstein, Colloids Interfaces, 2020, 4, 8 CrossRef CAS
- M. M. Rahman, B. Byanju, D. Grewell and B. P. Lamsal, Ultrason. Sonochem., 2020, 64, 105019 CrossRef CAS PubMed
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury and S. Benita, Int. J. Pharm., 1989, 55, R1–R4 CrossRef CAS
- A. Arizaga, G. Ibarz, R. Piñol and A. Urtizberea, J. Exp. Nanosci., 2014, 9, 561–569 CrossRef CAS
- J. Tao, S. F. Chow and Y. Zheng, Acta Pharm. Sin. B., 2018, 9, 4–18 CrossRef PubMed
- R. Rietscher, C. Thum, C.-M. Lehr and M. Schneider, Pharm. Res., 2015, 32, 1859–1863 CrossRef CAS PubMed
- J. Hiemer, A. Clausing, T. Schwarz and K. Stöwe, Chem. Eng. Technol., 2019, 42, 2018–2027 CrossRef CAS
- M. Morales-Cruz, G. M. Flores-Fernández, M. Morales-Cruz, E. A. Orellano, J. A. Rodriguez-Martinez, M. Ruiz and K. Griebenow, Results Pharma Sci., 2012, 2, 79–85 CrossRef PubMed
- L. C. Nelemans, M. Buzgo and A. Simaite, AIP Conf. Proc., 2021, 78(1), 29 Search PubMed
- J. L. C. De Queiroz, R. O. De Araújo Costa, L. L. Rodrigues Matias, A. F. De Medeiros, A. F. Teixeira Gomes, T. D. Santos Pais, T. S. Passos, B. L. L. Maciel, E. A. Dos Santos and A. H. De Araújo Morais, Food Hydrocolloids, 2018, 84, 247–256 CrossRef CAS
- Y. Liu, G. Yang, D. Zou, Y. Hui, K. Nigam, A. P. J. Middelberg and C.-X. Zhao, Ind. Eng. Chem. Res., 2020, 59, 4134–4149 CrossRef CAS
- C. E. Markwalter and R. K. Prud'homme, J. Pharm. Sci., 2018, 107, 2465–2471 CrossRef CAS PubMed
- R. F. Pagels and R. K. Prud'homme, J. Controlled Release, 2015, 219, 519–535 CrossRef CAS PubMed
- K. M. Pustulka, A. R. Wohl, H. S. Lee, A. R. Michel, J. Han, T. R. Hoye, A. V. McCormick, J. Panyam and C. W. Macosko, Mol. Pharm., 2013, 10, 4367–4377 CrossRef CAS PubMed
- Y. Liu, C. Cheng, R. K. Prud'homme and R. O. Fox, Chem. Eng. Sci., 2008, 63, 2829–2842 CrossRef CAS
- S. L. Levit, R. C. Walker and C. Tang, Polymers, 2019, 11(9), 1406 CrossRef CAS PubMed
- C. E. Markwalter, R. F. Pagels, A. N. Hejazi, A. G. R. Gordon, A. L. Thompson and R. K. Prud'homme, AAPS J., 2020, 22, 18 CrossRef CAS PubMed
- H. Adelnia, H. D. Tran, P. J. Little, I. Blakey and H. T. Ta, ACS Biomater. Sci. Eng., 2021, 7, 2083–2105 CrossRef CAS PubMed
- Z. Zeng, C. Dong, P. Zhao, Z. Liu, L. Liu, H.-Q. Mao, K. W. Leong, X. Gao and Y. Chen, Adv. Healthcare Mater., 2019, 8, 1801010 CrossRef PubMed
- S. McManus, Y. Zhang, B. Kim, B. Lee, M. ElSayed and R. Prud’homme, Precis. Nanomed., 2020, 3(4), 710–723, DOI:10.33218/001c.18519
- S. Liu, S. de Beer, K. M. Batenburg, H. Gojzewski, J. Duvigneau and G. J. Vancso, ACS Appl. Mater. Interfaces, 2021, 13, 17034–17045 CrossRef CAS PubMed
- A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 582–612 CAS
- J. Gu, X. Chen, X. Fang and X. Sha, Acta Biomater., 2017, 57, 156–169 CrossRef CAS PubMed
- S. C. Larnaudie, J. C. Brendel, K. A. Jolliffe and S. Perrier, ACS Macro Lett., 2017, 6, 1347–1351 CrossRef CAS PubMed
- M. S. Lee, N. W. Kim, J. E. Lee, M. G. Kim, Y. Yin, S. Y. Kim, B. S. Ko, A. Kim, J. H. Lee, S. Y. Lim, D. W. Lim, S. H. Kim, J. W. Park, Y. T. Lim and J. H. Jeong, Acta Biomater., 2018, 81, 231–241 CrossRef CAS PubMed
- O. H. Orasan, G. Ciulei, A. Cozma, M. Sava and D. L. Dumitrascu, Clujul Med., 2016, 89, 24–31 Search PubMed
- M. Gudowska, E. Gruszewska, A. Panasiuk, B. Cylwik, R. Flisiak, M. Świderska, M. Szmitkowski and L. Chrostek, Clin. Exp. Med., 2016, 16, 523–528 CrossRef CAS PubMed
- R. E. Avila, R. A. Carmo, P. Farah Kde, A. L. Teixeira, L. V. Coimbra, C. M. Antunes and J. R. Lambertucci, Braz. J. Infect. Dis., 2010, 14, 335–341 CrossRef PubMed
- D. C. Rockey and D. M. Bissell, Hepatology, 2006, 43, S113–S120 CrossRef CAS PubMed
- J. F. Hansen, K. M. Christiansen, B. Staugaard, B. K. Moessner, S. Lillevang, A. Krag and P. B. Christensen, PLoS One, 2019, 14, e0212036 CrossRef CAS PubMed
- T. Mei, A. Kim, L. B. Vong, A. Marushima, S. Puentes, Y. Matsumaru, A. Matsumura and Y. Nagasaki, Biomaterials, 2019, 215, 119209 CrossRef CAS PubMed
- H. Zhang, Y. Pei, L. Gao, Q. He, H. Zhang, L. Zhu, Z. Zhang and L. Hou, Nano Today, 2021, 38, 101186 CrossRef CAS
- Q. Ma, L. Bian, X. Zhao, X. Tian, H. Yin, Y. Wang, A. Shi and J. Wu, Mater. Today Bio, 2022, 13, 100181 CrossRef CAS PubMed
- B. J. Wall, M. F. Will, G. K. Yawson, P. J. Bothwell, D. C. Platt, C. F. Apuzzo, M. A. Jones, G. M. Ferrence and M. I. Webb, J. Med. Chem., 2021, 64, 10124–10138 CrossRef CAS PubMed
- S. B. Alvi, S. Ahmed, D. Sridharan, Z. Naseer, N. Pracha, H. Wang, K. D. Boudoulas, W. Zhu, N. Sayed and M. Khan, Front. Cardiovasc. Med., 2021, 8, 742315 CrossRef CAS PubMed
- Z. Jin, Y. Zhao, Y. Sun, B. Zhang, H. Wang, Y. Wu, Y. Zhu, C. Zhu, T. Hu and X. Du, Nat. Struct. Mol. Biol., 2020, 27, 529–532 CrossRef CAS PubMed
- W. Yan, Y. Zheng, X. Zeng, B. He and W. Cheng, Signal Transduction Targeted Ther., 2022, 7, 26 CrossRef CAS PubMed
- D. Giovinazzo, B. Bursac, I. Sbodio Juan, S. Nalluru, T. Vignane, M. Snowman Adele, M. Albacarys Lauren, W. Sedlak Thomas, R. Torregrossa, M. Whiteman, R. Filipovic Milos, H. Snyder Solomon and D. Paul Bindu, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017225118 CrossRef CAS PubMed
- M. S. Bahniuk, A. K. Alshememry, S. V. Elgersma and L. D. Unsworth, J. Nanobiotechnol., 2018, 16, 15 CrossRef PubMed
- H. Cheng, X. Zhang, L. Qin, Y. Huo, Z. Cui, C. Liu, Y. Sun, J. Guan and S. Mao, J. Controlled Release, 2020, 321, 641–653 CrossRef CAS PubMed
- H. F. Moffett, M. E. Coon, S. Radtke, S. B. Stephan, L. McKnight, A. Lambert, B. L. Stoddard, H. P. Kiem and M. T. Stephan, Nat. Commun., 2017, 8, 389 CrossRef CAS PubMed
- T. T. Smith, S. B. Stephan, H. F. Moffett, L. E. McKnight, W. Ji, D. Reiman, E. Bonagofski, M. E. Wohlfahrt, S. P. S. Pillai and M. T. Stephan, Nat. Nanotechnol., 2017, 12, 813–820 CrossRef CAS PubMed
- J. Karlsson, Y. Rui, K. L. Kozielski, A. L. Placone, O. Choi, S. Y. Tzeng, J. Kim, J. J. Keyes, M. I. Bogorad, K. Gabrielson, H. Guerrero-Cazares, A. Quiñones-Hinojosa, P. C. Searson and J. J. Green, Nanoscale, 2019, 11, 20045–20057 RSC
- H. J. Kim, S. Ogura, T. Otabe, R. Kamegawa, M. Sato, K. Kataoka and K. Miyata, ACS Cent. Sci., 2019, 5, 1866–1875 CrossRef CAS PubMed
- S. Abbasi, S. Uchida, K. Toh, T. A. Tockary, A. Dirisala, K. Hayashi, S. Fukushima and K. Kataoka, J. Controlled Release, 2021, 332, 260–268 CrossRef CAS PubMed
- L. Y. Chan, Y. L. Khung and C.-Y. Lin, Nanomaterials, 2019, 9(1), 67 CrossRef PubMed
- A. J. Atkinson, Jr., Transl. Clin. Pharmacol., 2017, 25, 117–124 CrossRef PubMed
- J. Liang, C. Wu, X. Zhou, Y. Shi, J. Xu, X. Cai, T. Fu, D. Ma and W. Xue, Colloids Surf., B, 2021, 205, 111918 CrossRef CAS PubMed
- R. K. Dhandapani, D. Gurusamy and S. R. Palli, ACS Appl. Bio Mater., 2021, 4, 4310–4318 CrossRef CAS PubMed
- P.-F. Cui, L.-Y. Qi, Y. Wang, R.-Y. Yu, Y.-J. He, L. Xing and H.-L. Jiang, J. Controlled Release, 2019, 303, 253–262 CrossRef CAS PubMed
- M.-A. Nguyen, H. Wyatt, L. Susser, M. Geoffrion, A. Rasheed, A.-C. Duchez, M. L. Cottee, E. Afolayan, E. Farah, Z. Kahiel, M. Côté, S. Gadde and K. J. Rayner, ACS Nano, 2019, 13, 6491–6505 CrossRef CAS PubMed
- T. T. Smith, S. B. Stephan, H. F. Moffett, L. E. McKnight, W. Ji, D. Reiman, E. Bonagofski, M. E. Wohlfahrt, S. P. S. Pillai and M. T. Stephan, Nat. Nanotechnol., 2017, 12, 813–820 CrossRef CAS PubMed
- S. Bastaki, S. Aravindhan, N. Ahmadpour Saheb, M. Afsari Kashani, A. Evgenievich Dorofeev, F. Karoon Kiani, H. Jahandideh, F. Beigi Dargani, M. Aksoun, A. Nikkhoo, A. Masjedi, A. Mahmoodpoor, M. Ahmadi, S. Dolati, S. Namvar Aghdash and F. Jadidi-Niaragh, Life Sci., 2021, 266, 118847 CrossRef CAS PubMed
- I. Bu, Y.-S. Fu, J.-F. Li and T.-F. Guo, RSC Adv., 2017, 7, 46651–46656 RSC
- L. Peltonen, H. Valo, R. Kolakovic, T. Laaksonen and J. Hirvonen, Expert Opin. Drug Delivery, 2010, 7, 705–719 CrossRef CAS PubMed
- J. A. Bhushani and C. Anandharamakrishnan, Trends Food Sci. Technol., 2014, 38, 21–33 CrossRef
- Y. Liu, X. Chen, Y. Gao, D.-G. Yu and P. Liu, J. Nanobiotechnol., 2022, 20, 244 CrossRef CAS PubMed
- W. Jiang, X. Zhang, P. Liu, Y. Zhang, W. Song, D.-G. Yu and X. Lu, Adv. Compos. Hybrid Mater., 2022, 5, 3045–3056 CrossRef CAS
- X. Liu, M. Zhang, W. Song, Y. Zhang, D.-G. Yu and Y. Liu, Gels, 2022, 8(6), 357 CrossRef CAS PubMed
- P. Mishra, P. Gupta and V. Pruthi, Mater. Sci. Eng., C, 2021, 119, 111450 CrossRef CAS PubMed
- D. Han, S. Sherman, S. Filocamo and A. J. Steckl, Acta Biomater., 2017, 53, 242–249 CrossRef CAS PubMed
- M. Zamani, M. P. Prabhakaran, E. S. Thian and S. Ramakrishna, Int. J. Pharm., 2014, 473, 134–143 CrossRef CAS PubMed
- N. Yaghoobi, R. Faridi Majidi, M. A. Faramarzi, H. Baharifar and A. Amani, Adv. Pharm. Bull., 2017, 7, 131–139 CrossRef CAS PubMed
- A. Hasanpour, F. Esmaeili, H. Hosseini and A. Amani, Mater. Sci. Eng., C, 2021, 118, 111427 CrossRef CAS PubMed
-
F. Bensebaa, in Interface Science and Technology, ed. F. Bensebaa, Elsevier, 2013, vol. 19, pp. 85–146 Search PubMed
-
G. Lüttgens and N. Wilson, in Electrostatic Hazards, ed. G. Lüttgens and N. Wilson, Butterworth-Heinemann, Oxford, 1997, pp. 14–38. DOI:10.1016/B978-075062782-5/50003-7
- T. Si, L. Zhang, G. Li, C. J. Roberts, X. Yin and R. Xu, J. Biomed. Opt., 2013, 18, 075003 CrossRef PubMed
- L. Yu, Q. Sun, Y. Hui, A. Seth, N. Petrovsky and C.-X. Zhao, J. Colloid Interface Sci., 2019, 539, 497–503 CrossRef CAS PubMed
- J. Pessi, H. A. Santos, I. Miroshnyk, J. Yliruusi, D. A. Weitz and S. Mirza, Int. J. Pharm., 2014, 472, 82–87 CrossRef CAS PubMed
- O. F. Khan, P. S. Kowalski, J. C. Doloff, J. K. Tsosie, V. Bakthavatchalu, C. B. Winn, J. Haupt, M. Jamiel, R. Langer and D. G. Anderson, Sci. Adv., 2018, 4, eaar8409 CrossRef PubMed
- S. Gimondi, C. F. Guimarães, S. F. Vieira, V. M. F. Gonçalves, M. E. Tiritan, R. L. Reis, H. Ferreira and N. M. Neves, Nanomedicine, 2022, 40, 102482 CrossRef CAS PubMed
- Y. Qiu, Y. Liu, Y. Xu, Z. Li and J. Chen, Electrophoresis, 2020, 41, 902–908 CrossRef CAS PubMed
- G.-Y. Ahn, I. Choi, M. Song, S. K. Han, K. Choi, Y.-H. Ryu, D.-H. Oh, H.-W. Kang and S.-W. Choi, ACS Macro Lett., 2022, 11, 127–134 CrossRef CAS PubMed
- X. Yang, Z. Pan, M. R. Choudhury, Z. Yuan, A. Anifowose, B. Yu, W. Wang and B. Wang, Med. Res. Rev., 2020, 40, 2682–2713 CrossRef PubMed
- V. Agrahari, V. Agrahari and A. K. Mitra, Ther. Delivery, 2016, 7, 257–278 CrossRef CAS PubMed
- B. S. Zolnik and D. J. Burgess, J. Controlled Release, 2007, 122, 338–344 CrossRef CAS PubMed
- M. A. Tracy, K. L. Ward, L. Firouzabadian, Y. Wang, N. Dong, R. Qian and Y. Zhang, Biomaterials, 1999, 20, 1057–1062 CrossRef CAS PubMed
- C. E. Holy, S. M. Dang, J. E. Davies and M. S. Shoichet, Biomaterials, 1999, 20, 1177–1185 CrossRef CAS PubMed
- M. Shameem, H. Lee and P. P. DeLuca, AAPS PharmSci, 1999, 1, 1–6 CrossRef PubMed
- S. A. Sufi, M. Hoda, S. Pajaniradje, V. Mukherjee, S. M. Coumar and R. Rajagopalan, Int. J. Pharm., 2020, 588, 119738 CrossRef CAS PubMed
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443–455 CrossRef CAS PubMed
- A. Motaharian, M. R. M. Hosseini and K. Naseri, Sens. Actuators, B, 2019, 288, 356–362 CrossRef CAS
- X. Wang, Y. Pei, Y. Hou and Z. Pei, Polymers, 2019, 11, 313 CrossRef PubMed
- X. Zhang, D. An, R. Zhang, Y. Huang and Z. Liu, Int. J. Pharm., 2022, 615, 121476 CrossRef CAS PubMed
- D. Yin, X. Li, Y. Ma and Z. Liu, Chem. Commun., 2017, 53(50), 6716–6719 RSC
- H. R. Culver and N. A. Peppas, Chem. Mater., 2017, 29, 5753–5761 CrossRef CAS PubMed
- R. Fekety, Med. Clin. North Am., 1982, 66, 175–181 CrossRef CAS PubMed
- H. Su, C. A. Hurd Price, L. Jing, Q. Tian, J. Liu and K. Qian, Mater. Today Bio, 2019, 4, 100033 CrossRef CAS PubMed
- K. Kim, J. Guo, Z. Liang and D. Fan, Adv. Funct. Mater., 2018, 28, 1705867 CrossRef
- H. Liu, H. Wang, X. Lu, V. Murugadoss, M. Huang, H. Yang, F. Wan, D.-G. Yu and Z. Guo, Adv. Compos. Hybrid Mater., 2022, 5, 1017–1029 CrossRef CAS
- L. Liu, W. Yao, X. Xie, J. Gao and X. Lu, J. Nanobiotechnol., 2021, 19, 235 CrossRef CAS PubMed
- M. Wang, D.-G. Yu, G. R. Williams and S. W. Bligh, Pharmaceutics, 2022, 14(6), 1208 CrossRef CAS PubMed
- L. R. Baden, H. M. El Sahly, B. Essink, K. Kotloff, S. Frey, R. Novak, D. Diemert, S. A. Spector, N. Rouphael, C. B. Creech, J. McGettigan, S. Khetan, N. Segall, J. Solis, A. Brosz, C. Fierro, H. Schwartz, K. Neuzil, L. Corey, P. Gilbert, H. Janes, D. Follmann, M. Marovich, J. Mascola, L. Polakowski, J. Ledgerwood, B. S. Graham, H. Bennett, R. Pajon, C. Knightly, B. Leav, W. Deng, H. Zhou, S. Han, M. Ivarsson, J. Miller and T. Zaks, N. Engl. J. Med., 2021, 384, 403–416 CrossRef CAS PubMed
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. Pérez Marc, E. D. Moreira, C. Zerbini, R. Bailey, K. A. Swanson, S. Roychoudhury, K. Koury, P. Li, W. V. Kalina, D. Cooper, R. W. Frenck, Jr., L. L. Hammitt, Ö. Türeci, H. Nell, A. Schaefer, S. Ünal, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Şahin, K. U. Jansen and W. C. Gruber, N. Engl. J. Med., 2020, 383, 2603–2615 CrossRef CAS PubMed
- S. J. Thomas, E. D. Moreira, Jr., N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. Pérez Marc, F. P. Polack, C. Zerbini, R. Bailey, K. A. Swanson, X. Xu, S. Roychoudhury, K. Koury, S. Bouguermouh, W. V. Kalina, D. Cooper, R. W. Frenck, Jr., L. L. Hammitt, Ö. Türeci, H. Nell, A. Schaefer, S. Ünal, Q. Yang, P. Liberator, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Şahin, W. C. Gruber and K. U. Jansen, N. Engl. J. Med., 2021, 385, 1761–1773 CrossRef CAS PubMed
- P. T. Heath, E. P. Galiza, D. N. Baxter, M. Boffito, D. Browne, F. Burns, D. R. Chadwick, R. Clark, C. Cosgrove and J. Galloway, N. Engl. J. Med., 2021, 385, 1172–1183 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |