
Modulating the triplet chromophore environment to prolong the emission lifetime of ultralong organic phosphorescence†
Guohui
Yang‡
,
Anqi
Lv‡
,
Zixuan
Xu
,
Zhicheng
Song
,
Kang
Shen
,
Chongyang
Lin
,
Guowei
Niu
,
Huili
Ma
 ,
Huifang
Shi
* and
Zhongfu
An
*
,
Huifang
Shi
* and
Zhongfu
An
*
Key Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing, China. E-mail: iamhfshi@njtech.edu.cn; iamzfan@njtech.edu.cn
First published on 5th May 2022
Abstract
The molecular environment plays a vital role in regulating the photophysical properties of optoelectronic functional materials. Here, in a series of phenoxazine derivatives with a triazine core and double-branched structures, we found that changes in the surroundings of the triplet chromophore are important to regulate the organic phosphorescence lifetime. The emission lifetime of the 10,10′-(6-methoxy-1,3,5-triazine-2,4-diyl)bis(10H-phenoxazine) (MT) molecule with the strongest interaction between the triplet chromophore reached 325.26 ms, which is 387 times longer than that of the 10,10′-(6-ethoxy-1,3,5-triazine-2,4-diyl)bis(10H-phenoxazine) (ET) molecule with the weakest interaction (0.84 ms). This work indicates that the modulation of the triplet chromophore environment is essential to the performance regulation of organic phosphorescence, which will provide guidance for the development of organic room temperature phosphorescent materials.
 Zhongfu An | Professor AN Zhongfu graduated from Nanjing University of Post and Telecommunication and received his PhD degree in 2014. After graduation, he went to Department of Chemistry, National University of Singapore for his post-doctoral research. He joined Institute of Advanced Materials, Nanjing Tech University and started his independent research at the end of 2015. Professor AN Zhongfu focuses on the research of purely organic phosphorescence materials and applications. He initially proposed and verified the design principle of the stabilization of the triplet excitons by H-aggregation for ultralong organic phosphorescence. Up to now, he has published over 100 journal papers including Nature Materials, Nature Photonics, Nature Communications, Angewandte Chemie-International Edition, Advanced Materials, Journal of American Chemical Society, and so forth with total citations over 5000 times. |
The molecular environment plays a crucial role in manipulating the optoelectronic performance of organic functional materials in various fields owing to the interactions between the functional units and surrounding molecules.1–4 For example, luminogens with D–A architectures show colourful emission along with fine spectral structure changes in different solvents, which is ascribed to diverse secondary bonds between the chromophores and the surrounding solvents, such as hydrogen bonds, dipole–dipole interactions, etc., that is the solvent effect.5–7 For a series of luminogens with aggregation-induced emission features, the photoluminescence (PL) quantum efficiency exhibits a significant improvement as the molecular environment changes from solution to solid state, since the molecular motions of the luminogens can be strictly confined by surrounding molecules in the solid state.8–10 Coincidently, there also exist similar luminescent behaviours in host–guest materials. By doping guest molecules into the different hosts, some luminescence properties can be manipulated greatly, such as the emission colours, quantum efficiency, even emission lifetimes and so on.11–13 In other words, the molecular environment has momentous impacts on the optoelectronic properties of functional materials, which is beneficial to enhance the performance of devices based on these functional materials.
Recently, room temperature phosphorescence (RTP) in purely organic materials has received widespread attention due to long-lived lifetimes, large Stokes shift, and rich excited states.14–16 To obtain efficient RTP, it needs two requirements. One is to generate abundant triplet excitons by promoting the intersystem crossing (ISC) process of excitons from the singlet excited states to the triplet excited states. The other is to make the triplet excitons as stable as possible by suppressing the non-radiative transition process. The inhibition of the non-radiative transition process is highly dependent on the molecular environment, especially in crystals.17–19 Therefore, great successes in understanding the relationship between intermolecular interactions and RTP performance have been achieved based on various molecular structures.20–23 Notably, each RTP molecule can be constructed with multiple units with different functions, such as chromophores responsible for emission, assembly groups in charge of intermolecular interactions, auxiliary units taking charge of the regulation of intermolecular stacking, etc., of which the environment is quite different.24,25 In the previous reports, some efforts were made to study the effect of the molecular environment on phosphorescent properties.26,27 In 2019, our group proposed that the effective stacking between triplet chromophores plays a crucial role in the generation of ultralong organic phosphorescence;28 however, the influence of the surrounding environment of the triplet chromophore on the properties of RTP has been rarely studied. Thus, it is critical to make the chromophore surroundings clear for improving RTP performance, especially for molecules containing more than one chromophore (Fig. 1a). However, there are barely any reports on understanding the influence of the chromophore environment on the RTP performance.
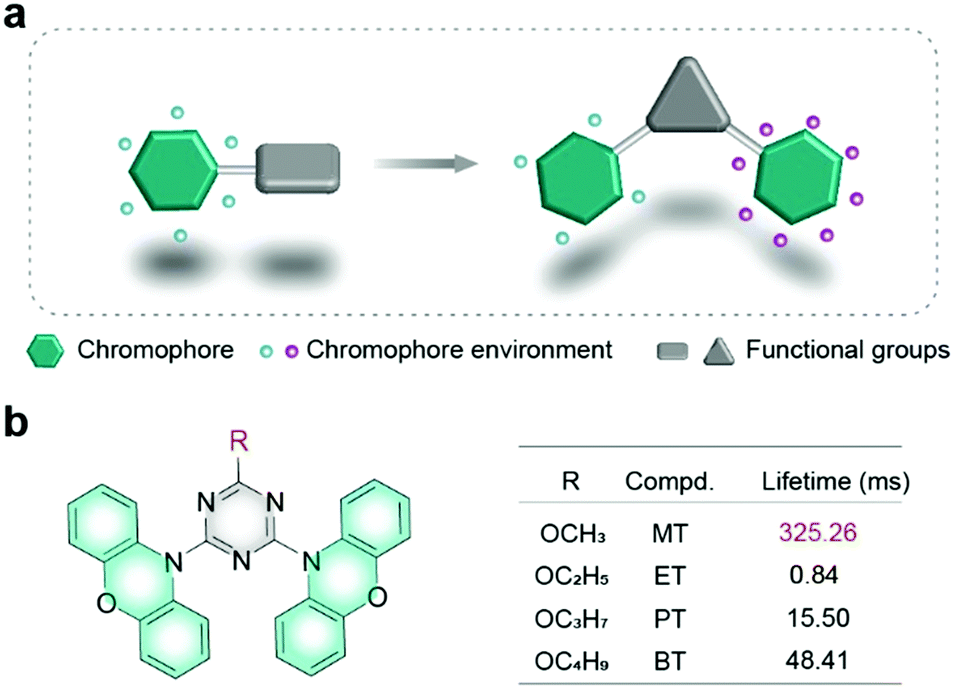 | ||
| Fig. 1 (a) Schematic illustration of the chromophore environment. (b) Chemical structures of organic phosphors with different phosphorescence lifetimes. | ||
Herein, we designed and synthesized a series of phenoxazine based molecules as shown in Fig. 1b, which were named as 10,10′-(6-methoxy-1,3,5-triazine-2,4-diyl)bis(10H-phenoxazine) (MT), 10,10′-(6-ethoxy-1,3,5-triazine-2,4-diyl)bis(10H-phenoxazine) (ET), 10,10′-(6-propoxy-1,3,5-triazine-2,4-diyl)bis(10H-phenoxazine) (PT) and 10,10′-(6-butoxy-1,3,5-triazine-2,4-diyl)bis(10H-phenoxazine) (BT). Phenoxazine was chosen as a chromophore, while triazine with multiple nitrogen atoms was selected as a functional unit to construct secondary interactions. Inspired by the research on alkyl chain engineering for regulating intermolecular stacking for high performance in organic electronics,29,30 alkyls were used as auxiliary units. What's more, alkyls have little effect on the molecular electronic configuration.31–33 Their molecular structures were confirmed by NMR (Fig. S1–S8, ESI†), elemental analysis, high-resolution mass spectrometry (HRMS) (Fig. S9–S12, ESI†), and X-ray single-crystal diffraction. By adjusting the length of the alkyl chain, the phosphorescence lifetime of MT is up to 325.26 ms, which is increased by 387 times compared with the ET molecule in the crystal. To the best of our knowledge, it is the longest lifetime for phenoxazine derivatives.34–36 Apart from the phosphorescence lifetime variation, phosphorescence colours were also tuned from yellow to green by manipulating the alkyl chains. Combining single-crystal analyses and theoretical calculations, we found that the precise environment changes of chromophore units played a vital role in manipulating the phosphorescence lifetime. This finding builds a bridge to understand the relationship between the chromophore environments and phosphorescence properties, which is beneficial to design novel UOP molecules with excellent performance.
Firstly, photophysical properties of the phenoxazine phosphors were investigated in a dilute solution and crystal. In a dilute dichloromethane solution, the absorption spectra of MT, ET, PT and BT molecules showed similar profiles with multiple peaks between 250 and 350 nm (Fig. S13, ESI†), which were attributed to π–π* → n−π*transitions. Meanwhile, the four compounds showed similar steady-state PL emission peaks at 300 and 400 nm in dichloromethane solution, which might be attributed to the local excited-state and charge-transfer state (Fig. S14, ESI†), due to their donor–acceptor (D–A) architectures, proved by the red-shift of the wide emission bands with an increase in the solvent polarity. Notably, the phosphorescence spectra of MT, ET, PT and BT in 2-methyltetrahydrofuran at 77 K were completely identical with peaks at 405 nm (Fig. S15, ESI†).
In the crystalline state, these four compounds showed sky blue fluorescent emission under UV-light excitation and afterglow emission after a delayed time of 8 ms. The phosphorescence nature of afterglow emission for these four phosphors in the crystal state was further confirmed by the evidence of the reduced intensity of emission with the environment changed from vacuum to oxygen (Fig. S17, ESI†). Moreover, the lifetimes of the four molecules in the short-wavelength range at around 450 nm are all in the nanosecond scale, indicating that the blue emission is assigned to the fluorescence (Fig. S18, ESI†). As shown in Fig. 2a, MT showed persistent luminescence after the UV lamp was switched off with an ultra-long lifetime as long as 325.26 ms (Fig. 2b, Table S2 and Fig. S19, ESI†), which is 387 times longer than that of the ET molecule (0.84 ms). And MT molecules, unlike the other three molecules, can be excited by visible light at around 420 nm, from the excitation-phosphorescence emission mappings (Fig. 2c and Fig. S16, ESI†), which is currently rare in RTP materials. Moreover, we found that the phosphorescence of MT in the crystal state showed a red-shift with bright yellow afterglow emission at 540 nm compared with the other three molecules. In addition, we checked the RTP performance in the amorphous state by doping the four phosphors into PMMA. No persistent luminescence was observed, indicating that the packing of the crystal has a key effect on the phosphorescence properties.
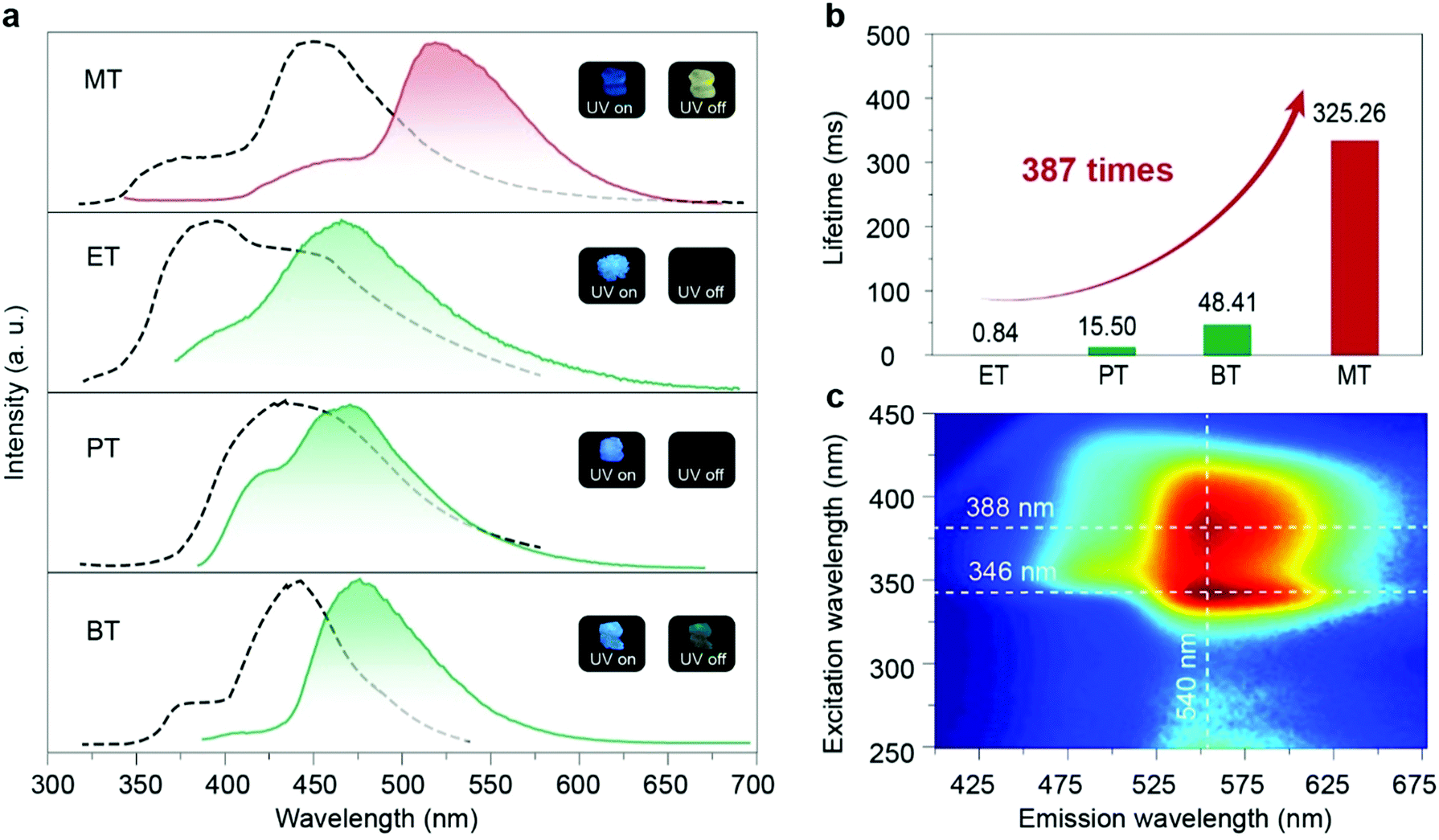 | ||
| Fig. 2 Photophysical properties of MT, ET, PT and BT in crystals under ambient conditions. (a) Normalized steady-state photoluminescence (dotted lines) and phosphorescence (solid lines) spectra of MT, ET, PT and BT crystals. Insets: Photographs of MT, ET, PT and BT crystals under 365 nm UV light on (left) and off (right). (b) Comparison of phosphorescence lifetimes for MT, ET, PT and BT in crystals. (c) Excitation-phosphorescence emission mapping of the MT crystal. | ||
To understand the difference of emission lifetimes within the similar molecular structures with different alkyl chain lengths, X-ray single crystal diffraction and theoretical calculations were carried out (Fig. S20–S25, ESI†). The crystal structures of the four compounds all showed a distorted asymmetric configuration due to the directive effect of the alkyl chain, and the angle between the triazine ring and the phenazine on the side of the alkyl chain benzene ring is inversely proportional to the lifetime (Fig. S20, ESI†). Moreover, we found that the triplet chromophore was distributed on the same side of phenazine directed by the alkyl chains via calculating the natural transition orbitals (NTOs) of the lowest triplet excited states (Fig. 3a and b and Fig. S25, ESI†). After analyzing the intermolecular interactions on individual molecules, we found that the intermolecular interactions on molecules modified with different alkyl chain lengths varied considerably. The central molecule of the longer-lived MT molecule is subjected to 20 types of interactions in total, including C–H⋯π, C–H⋯C–H, C–H⋯N, etc., with the distances ranging from 2.343 to 3.389 Å. Compared with the MT molecule, the shortest-lived ET molecule has less restrictive interactions around the molecule and relatively increased distances (2.585–3.310 Å) with relatively weaker strength (Fig. 3c and d). The strong confinement effect can suppress the nonradiative transition and thus prolong the lifetime of the material. By analyzing the distribution of triplet chromophores around a central triplet chromophore, it was found that the longest-lived methoxy molecule has seven triplet chromophores around a triplet excited state center, which was densely distributed. However, for the ET molecule, it has only three chromophores around a triplet chromophore center, which was a relatively loose distribution (Fig. S21, ESI†). The distance analysis between dimer and trimer triplet chromophores also proves that the interaction between triplet chromophores in MT molecules is more close (Fig. S22, ESI†). The triplet chromophores between the MT layers are connected head-to-head by the interaction of C–H⋯π, which was strong with the distance of 2.857 and 2.889 Å. However, for ET molecules there was no interaction between the triplet chromophores between the interlayers. And the triplet chromophore and the non-triplet chromophore in the other two molecules (PT and BT) interacted in the head-to-tail direction, separating the interaction between the triplet chromophores (Fig. S23, ESI†). Therefore, we speculated that the stable triplet excitons generated due to the enhanced interactions between the triplet chromophores, which constructed a relatively rigid environment to restrain the non-radiative transition of the triplet excitons, and thus to prolong the emission lifetime of the materials. Hence, we proposed that the rational modulation of the triplet chromophore environment is crucial for enhancing the ultralong organic phosphorescence lifetime.
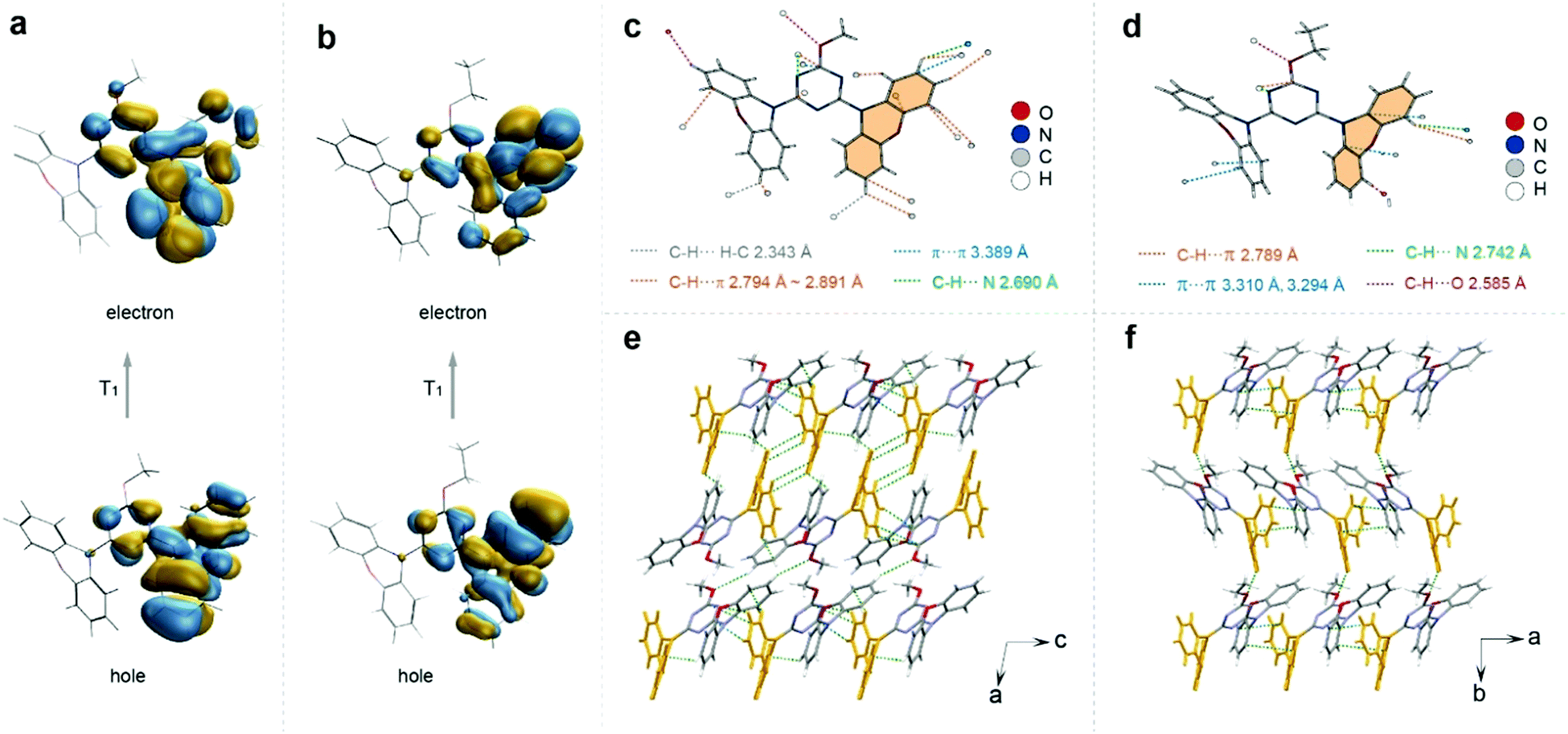 | ||
| Fig. 3 Theoretical calculation and crystal analysis of MT and ET molecules. Natural transition orbitals (NTOs) of T1 states for MT (a) and ET (b) molecules. Intermolecular interactions around one molecule of MT (c) and ET (d) in the crystal state. The packing modes of MT (e) and ET (f) crystals. The yellow labels in e and f represent the triplet chromophore. | ||
To further prove our hypothesis, we calculated the free volume distributions of the four molecules by using Materials Studio (MS) software (Fig. S24, ESI†), which provides more space for nonradiative decay by molecular motions. The results showed that the compactness of the entire molecular packing does have a great impact on the RTP performance. ET with the shortest lifetime has nearly twice the porosity volume compared to the other molecules, but the proportion of free volume in MT with the longest lifetime is slightly larger than that of the BT molecule, further proving the predominance of molecular interactions between triplet chromophores on the UOP enhancement.
To further explore the mechanism, we used a visual approach named reduced density gradient (RDG) to evaluate the weak intermolecular interactions in the dimer. It is regarded that the green flaky regions represent intermolecular interactions around the triplet chromophores. Obviously, only the MT molecule showed an extensive green area, suggesting the existence of weak intermolecular interactions in the dimer (Fig. S26, ESI†), which further stabilized the triplet excitons by restraining the non-radiative transition to prolong the emission lifetime (Fig. 4b). In contrast, there are no weak intermolecular interactions between the triplet chromophores in dimers of ET, PT, and BT, respectively (Fig. S26, ESI†). Furthermore, we calculated the excited-state electronic structures including the nature transitions orbitals (NTOs) and spin–orbit coupling (SOC) in the dimer at the B3LYP/def2-SVP level, indicating that the electron clouds of NTOs located on two MT molecules with a larger delocalization space, which stabilized the triplet excitons and prolongs their lifetime. The SOC value between T1 and S0 was decreased by two orders of magnitude from 1.687 cm−1 in ET to 0.01 cm−1 in MT (Fig. 4a), corresponding to less efficient nonradiative decay, which agrees well with the experimental results (Fig. 4a). These results show that the stacking between the triplet and the triplet chromophores causes NTOs in the dimers to be distributed more on phenoxazines. And the smaller SOC value causes the triplet electrons to have a larger delocalization space, which is beneficial to the UOP performance (Fig. 4b).
 | ||
| Fig. 4 Proposed mechanism for ultra-long phosphorescence (a). Natural transition orbitals (NTOs) of T1 states for the MT and ET dimer. (b) The mechanism of the different emission behaviours in the compound. | ||
Conclusions
In summary, we have developed an effective strategy to improve the properties of ultralong organic phosphorescence through the manipulation of the triplet chromophore environment. The MT molecule with the strongest triplet chromophore interactions showed an ultra-long lifetime of 325.26 ms, which is over 300 times more than its counterparts. Through the single crystal analysis and theoretical calculation, it was proved that the intense intermolecular interactions around the triplet chromophores stabilized the triplet excitons and prolonged the lifetime of the material. The modulation of the triplet chromophore environment will be beneficial for improving the ultralong organic phosphorescence This research will provide a design principle to realize UOP materials with high performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21975120, 62134007, 21875104, and 21973043), and the fund for Talented of Nanjing Tech University (No. 201983).Notes and references
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- Q. Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- D. W. Zhang, M. Li and C. F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- S. J. Liu, Y. Y. Li, R. T.-K. Kwok, J. W.-Y. Lam and B. Z. Tang, Chme. Sci., 2021, 12, 3427–3436 RSC.
- L. S. Ni, C. Shen and G. P. Yong, Mater. Chem. Phys., 2018, 205, 278–282 CrossRef CAS.
- B. Sk, E. Ravindran, U. Deori, N. Yadav, G. P. Nanda and P. Rajamalli, J. Mater. Chem., 2022, 10, 4886–4893 CAS.
- Y. Zhao, Q. Zhang, K. Chen, H. Gao, H. Qi, X. Shi, Y. Han, J. Wei and C. Zhang, J. Mater. Chem., 2017, 5, 4293–4301 CAS.
- D. Wang and B. Z. Tang, Acc. Chem. Res., 2019, 52, 2559–2570 CrossRef CAS PubMed.
- J. Li, J. X. Wang, H. X. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- Z. Zhao, H. K. Zhang, J. W.-Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- X. G. Yang, X. M. Lu, Z. M. Zhai, Y. Zhao, X. Y. Liu, L. F. Ma and S. Q. Zang, Chem. Commun., 2019, 55, 11099–11102 RSC.
- S. Guo, W. B. Dai, X. A. Chen, Y. X. Lei, J. B. Shi, B. Tong, Z. X. Cai and Y. P. Dong, ACS Mater. Lett., 2021, 3, 379–397 CrossRef CAS.
- D. Wang, Y. F. Xie, X. H. Wu, Y. X. Lei, Y. B. Zhou, Z. X. Cai, M. C. Liu, H. Y. Wu, X. B. Huang and Y. P. Dong, J. Phys. Chem. Lett., 2021, 12, 1814–1821 CrossRef CAS PubMed.
- S. Tian, H. Ma, X. Wang, A. Lv, H. Shi, Y. Geng, J. Li, F. Liang, Z.-M. Su, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 6645–6649 CrossRef CAS PubMed.
- Z. Lin, R. Kabe, K. Wang and C. Adachi, Nat. Commun., 2020, 11, 191 CrossRef CAS PubMed.
- P. Wei, X. Zhang, J. Liu, G.-G. Shan, H. Zhang, J. Qi, W. Zhao, H. H.-Y. Sung, I. D. Williams, J. W.-Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9293–9298 CrossRef CAS PubMed.
- S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W.-W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed.
- S. Cai, H. Shi, D. Tian, H. Ma, Z. Cheng, Q. Wu, M. Gu, L. Huang, Z. An, Q. Peng and W. Huang, Adv. Funct. Mater., 2018, 28, 1705045 CrossRef.
- K. Ling, H. Shi, H. Wang, L. Fu, A. Lv, K. Huang, W. Ye, M. Gu, C. Ma, X. Yao, W. Jia, J. Zhi, W. Yao, Z. An, H. Ma and W. Huang, Adv. Opt. Mater., 2019, 7, 1901076 CrossRef CAS.
- X. Yang and D. Yan, Adv. Opt. Mater., 2016, 4, 897–905 CrossRef CAS.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, J. Phys. Chem. Lett., 2017, 8, 1894–1898 CrossRef CAS PubMed.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- Z. Yang, C. Xu, W. L. Li, Z. Mao, X. Y. Ge, Q. Y. Huang, H. J. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. G. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed.
- W. Y. Jia, Q. Wang, H. F. Shi, Z. F. An and W. Huang, Chem. – Eur. J., 2020, 26, 4437–4448 CrossRef CAS PubMed.
- J. H. Zhi, Q. Zhou, H. F. Shi, Z. F. An and W. Huang, Chem. – Asian J., 2020, 15, 947–957 CrossRef CAS PubMed.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- J. Yang, X. Zhen, B. Wang, X. M. Gao, Z. C. Ren, J. Q. Wang, Y. J. Xie, J. R. Li, Q. Peng, K. Y. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- N. Gan, X. Wang, H. Ma, A. Lv, H. Wang, Q. Wang, M. Gu, S. Cai, Y. Zhang, L. Fu, M. Zhang, C. Dong, W. Yao, H. Shi, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 14140–14145 CrossRef CAS PubMed.
- L. Gu, H. F. Shi, M. X. Gu, K. Ling, H. L. Ma, S. Z. Cai, L. L. Song, C. Q. Ma, H. Li, G. C. Xing, X. C. Hang, J. W. Li, Y. R. Gao, W. Yao, Z. G. Shuai, Z. F. An, X. G. Liu and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 8425–8431 CrossRef CAS PubMed.
- X. Wang, H. L. Ma, M. X. Gu, C. Q. Lin, N. Gan, Z. L. Xie, H. Wang, L. F. Bian, L. S. Fu, S. Z. Cai, Z. G. Chi, W. Yao, Z. F. An, H. F. Shi and W. Huang, Chem. Mater., 2019, 31, 5584–5591 CrossRef CAS.
- Y. C. Zhang, K. Zhang, Y. Y. Ma, L. L. Lin, C. K. Wang and J. Z. Fan, PCCP, 2020, 22, 19746–19757 RSC.
- Q. Y. Liao, Q. Q. Li and Z. Li, ChemPhotoChem, 2021, 5, 694–701 CrossRef CAS.
- L. J. Tu, W. L. Che, S. H. Li, X. N. Li, Y. J. Xie and Z. Li, J. Mater. Chem., 2021, 9, 12124–12132 CAS.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC.
- Y. C. Duan, L. L. Wen, Y. Gao, Y. Wu, L. Zhao, Y. Geng, G. G. Shan, M. Zhang and Z. M. Su, J. Phys. Chem., 2018, 122, 23091–23101 CAS.
- F. Ni, Z. C. Zhu, X. Tong, M. J. Xie, Q. Zhao, C. Zhong, Y. Zou and C. L. Yang, Chem. Sci., 2018, 9, 6150–6155 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, additional spectra, and calculated data. CCDC 2155558–2155561. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc00836j |
| ‡ These authors are the co-first authors of this article. |
| This journal is © The Royal Society of Chemistry 2022 |