
A co-crystallization strategy toward high-performance n-type organic semiconductors through charge transport switching from p-type planar azaacene derivatives†
Zongrui
Wang
 *ab,
Renping
Li
c,
Kexiang
Zhao
d,
Fei
Yu
b,
Jianfeng
Zhao
*c,
Yonggang
Zhen
a and
Qichun
Zhang
*ef
*ab,
Renping
Li
c,
Kexiang
Zhao
d,
Fei
Yu
b,
Jianfeng
Zhao
*c,
Yonggang
Zhen
a and
Qichun
Zhang
*ef
aSchool of Materials Science and Engineering, Beijing University of Chemical Technology, Chaoyang District North Third Ring Road 15, Beijing 100029, P. R. China. E-mail: wangzr@mail.buct.edu.cn
bSchool of Materials Science and Engineering, Nanyang Technological University, 639798, Singapore
cInstitute of Advanced Materials (IAM), Nanjing Tech University, Nanjing 210000, P. R. China. E-mail: iamjfzhao@njtech.edu.cn
dCollege of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China
eDepartment of Materials Science and Engineering, City University of Hong Kong, Hong Kong SAR 999077, P. R. China. E-mail: qiczhang@cityu.edu.hk
fCenter of Super-Diamond and Advanced Films (COSDAF), City University of Hongkong, Hong Kong SAR 999077, P. R. China
First published on 20th October 2021
Abstract
In this work, we demonstrated that the co-crystallization strategy has offered an efficient and promising alternative route to achieve high-performance n-type semiconductors through charge-transport switching from pristine p-type systems. By using a simple “green synthesis” process through molecular “doping” with F4TCNQ into a p-type planar azaacene derivative TMIQ (0.27 cm2 V−1 s−1) host, charge transport characteristic switching occurs with a high electron mobility of 0.12 cm2 V−1 s−1 under atmospheric conditions obtained for the D–A complex TMF4TQ (cocrystal). The reasons for such switching lie in the ingenious energy level and molecular packing arrangement tailoring. Specifically, the insertion of F4TCNQ molecules has led to packing transformation from herringbone stacking (TMIQ) to a dense 2D brick arrangement and the low-lying LUMO levels (−4.55 eV) aligned to gold electrodes, thereby facilitating efficient electron injection and transport, and ensuring the air-stable nature, which is further confirmed using theoretical calculations. We believe that our work would provide new insights into high-performance air-stable n-type organic semiconductors exploration.
Introduction
Developing air-stable high-performance n-type organic semiconductors (OSCs) is of great significance due to their potential application in integrated circuits for large-area, flexible, low-cost and lightweight electronics1–3 including radio frequency tags,4,5 optical displays,6,7 electronic papers,8,9 wearable devices10 and artificial intelligent systems.11–14 However, compared to their p-type counterparts, the overall development of n-type organic semiconductors seriously lags behind in terms of mobility and ambient stability, on account of the high-lying LUMO (the lowest unoccupied molecular orbital) energy level and the large π-core barrier.15,16 Consequently, great efforts have been dedicated to overcoming these issues, aiming at realizing a good air-stable electron transport nature. The most favored path should be through molecular structure modification, i.e., introducing electron-withdrawing units like aromatic diimide,17–19 cyano,20,21 carbonyl,20,22 halogen species,23,24etc. into semiconductors as part of the π-system and/or the side attachments. Although it has been proven to be a practical way to achieve ideal n-type OSCs with high electron affinities (better for electron injection) and large π-conjugated planar backbones (for effective intermolecular orbital overlap), the accompanying complicated synthesis and purifying procedures make it a daunting prospect. More seriously, some efforts have been sacrificed due to the lack of accurate prediction of the concomitant properties of organic synthesis, resulting in a huge waste of resources for lots of unsatisfied products with a little bit of desired or even undesired/no performance. Therefore, developing an economic, simple, and highly effective strategy by directly decorating the known systems to fulfill the charge-transport switching and air-stable high electron transport is quite necessary.Doping suitable molecules into OSCs is recognized as a “green” and effective method to fine-control the transistor behavior via regulating the molecular orbitals and carrier density.25–27 In addition to the well-known effect of improving the material conductivity and device stability, molecular doping was also reported to be able to switch the charge transport nature from p-type to n-type due to the electron trap passivation resulting from the trap filling effect.28 But the resulting n-channel device was extremely unstable under ambient conditions.28 Despite the fact that air-stability could be improved by tailoring the LUMO level, the further increase in the doping ratio tends to produce the disordered molecular arrangement and impede the inherent intermolecular interactions, which instead induces ineffective transport channels. On this basis, recently, an orderly molecular “doping” strategy, i.e., co-crystallization, where the host and guest (donor and acceptor) co-assembled with a fixed stoichiometric ratio and long-range ordered packing structure, has been emerging as a promising alternative way to exploit the novel optoelectronic properties such as n-type/ambipolar transport,29 light emission,30,31 ferroelectricity,32,33 photoconductivity,34,35 and so on.36,37 Unlike the traditional synthetic procedures that require harsh and cumbersome synthesis conditions in virtue of covalent bond breaking and formation, the co-crystallization strategy is a “green synthesis” strategy where noncovalent interactions (e.g., π–π stacking, hydrogen/halogen bonding, electrostatic interactions, charge transfer interactions) act as the driving forces for cocrystal formation, which not only eliminates tedious chemical synthesis, but also enables a flexible control of molecular arrangement and energy levels. Specifically, in D–A complexes, the hybridization between the frontier orbitals of appropriate donors (D) and acceptors (A) could bring about new LUMO hybrid orbitals more aligned with the Fermi levels of gold electrodes, and when coupled with the super-exchange mechanism enabling the indirect hole/electron pathway along alternate packing columns, a charge-transport switching from hole to electron transport would be possible. In the previous work, Zhang et al. achieved charge conversion from a p-type conjugated semiconductor 2,7-di-tert-butyl-10,14-di(thiophen-2-yl)phenanthro[4,5-abc][1,2,5]thiadiazolo[3,4-i]phenazine (DTPTP) to n-type when co-assembled with tetracyanoquinodimethane (TCNQ), but unfortunately, the resulting cocrystal DTPTP2-TCNQ exhibited a relatively low electron mobility (∼3 × 10−3 cm2 V−1 s−1), which is far from meeting the requirements.38 Thus, up to now, it is still regarded as a big challenge to acquire high-performance air-stable n-type OSCs via this molecule-level “green synthesis” pathway.
Herein, the charge transport characteristics of a planar azaacene 8,8,18,18-tetramethyl-8,18-dihydroindolo[1,2,3-fg]indolo[3′,2′,1′:8,1]quinolino[2,3-b]acridine (TMIQ)34 were investigated and confirmed to be a p-type organic semiconductor with a good hole mobility of 0.27 cm2 V−1 s−1 (Fig. 1). It has been demonstrated that the large π-conjugated plane and N heteroatoms benefit from the effective intermolecular interaction, which is also conducive to guest molecule doping (co-crystallization). For this reason, electron-deficient 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) molecules were successfully assembled into the TMIQ host utilizing a simple and green solution-processed method (Fig. 1), where the noncovalent interactions including CT interactions, hydrogen bonding and π–π interactions act as the driving forces. An amazing charge-transport characteristic switching from hole to electron transport has been achieved through the ingenious energy level and molecular packing arrangement tailoring. The acceptor insertion on one hand has lowered the LUMO levels which favor the electron injection via the orbital hybridization of alternated arranged donors and acceptors due to the CT interactions, and also contributed to a dense two-dimensional (2D) brick stacking from the original herringbone structure. With the low-lying LUMO level (−4.55 eV) below the threshold for thermodynamically favorable electrochemical oxidation as well as the dense stacking by molecular rearrangement, a remarkable air-stable electron transport with mobility of up to 0.12 cm2 V−1 s−1 has been realized, which is among the best n-type performances in co-cocrystal systems. In-depth density functional theory (DFT) calculations confirm this charge transport transformation. This work informs that such a “green synthesis” method by molecular “doping” has offered an efficient and promising avenue for high-performance air-stable n-type OSC acquisition.
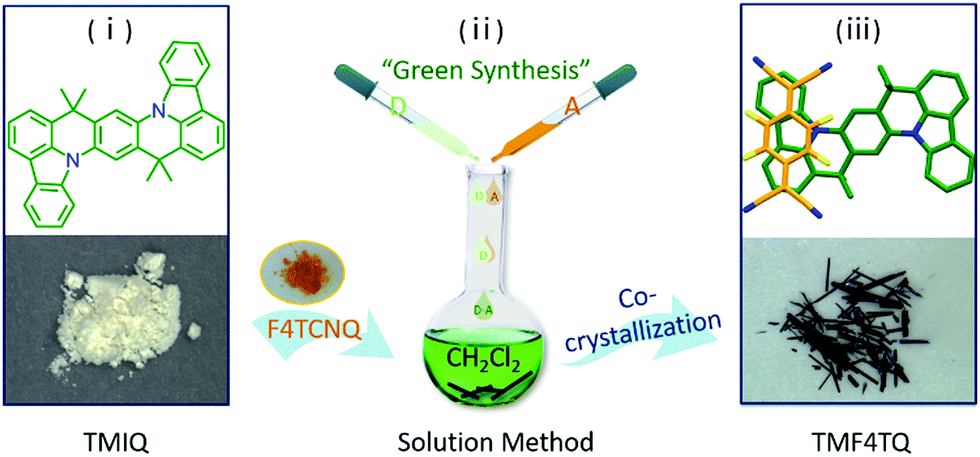 | ||
| Fig. 1 Schematic diagram of the synthetic procedure for cocrystal TMF4TQ and the corresponding chemical structures. | ||
Results and discussion
The quasi-2D planar azaacene molecule TMIQ was synthesized and characterized according to the reported procedure (Scheme S1, ESI†).34 UV-vis absorption spectroscopy and cyclic voltammetry (CV) measurements were performed in dichloromethane (CH2Cl2, DCM) solution to determine the energy band structure. The optical bandgap of TMIQ was estimated to be 3.22 eV from the onset of the absorption curve with the equation of Eg = 1240/λ (λ: 384 nm, Fig. S1, ESI†). The highest occupied molecular orbital (HOMO) was calculated to be −5.20 eV from the onset oxidation potential (0.8 V) of the CV curve (EHOMO = −(4.4 + Eonsetox) eV, Fig. S2, ESI†).39,40 The high-lying HOMO levels and relatively low bandgap indicate that TMIQ could be a good semiconductor material candidate for organic electronic devices favoring the hole transport. In order to lower the LUMO levels to open up the electron transport channel, the commonly used electron-deficient molecules F4TCNQ (A) were selected as p-dopants to be incorporated into the TMIQ host (D) via a simple and green synthetic route by merely mixing two components in a fixed molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by a solution processing method (Fig. 1). After natural standing for almost one day, black rod-like crystals (TMF4TQ) were precipitated from the mixed solution, which were distinctly different from the original reactants (Fig. 1 and Fig. S3, ESI†). The success of the co-assembly of the two components was preliminarily confirmed by X-ray photoelectron spectroscopy (XPS) and energy-dispersive X-ray spectroscopy (EDS) elemental mapping analysis for the coexistence of C, N, and F elements (Fig. 2c and Fig. S3, ESI†). X-ray powder diffraction (PXRD) analysis further proved this co-crystallization process that XRD profiles of TMF4TQ were in good agreement with its corresponding simulated crystallographic patterns but apparently different from that of the single components (Fig. 2d and Fig. S4, ESI†). UV-vis-NIR absorption and Fourier transform infrared spectroscopy (FTIR) were also characterized to give insight into the co-assembled behavior. From UV-vis-NIR spectroscopy analysis, although no obvious peak shifts or new peaks were observed in the mixed solution compared with that of the individual constitutes (Fig. S5, ESI†), in solid-state absorption, there appeared a significant new broad band at the infrared region (∼1200 nm), indicating the existence of charge transfer (CT) interactions (Fig. 2a). This CT characteristic switching was further investigated by FTIR, where the C
:1 by a solution processing method (Fig. 1). After natural standing for almost one day, black rod-like crystals (TMF4TQ) were precipitated from the mixed solution, which were distinctly different from the original reactants (Fig. 1 and Fig. S3, ESI†). The success of the co-assembly of the two components was preliminarily confirmed by X-ray photoelectron spectroscopy (XPS) and energy-dispersive X-ray spectroscopy (EDS) elemental mapping analysis for the coexistence of C, N, and F elements (Fig. 2c and Fig. S3, ESI†). X-ray powder diffraction (PXRD) analysis further proved this co-crystallization process that XRD profiles of TMF4TQ were in good agreement with its corresponding simulated crystallographic patterns but apparently different from that of the single components (Fig. 2d and Fig. S4, ESI†). UV-vis-NIR absorption and Fourier transform infrared spectroscopy (FTIR) were also characterized to give insight into the co-assembled behavior. From UV-vis-NIR spectroscopy analysis, although no obvious peak shifts or new peaks were observed in the mixed solution compared with that of the individual constitutes (Fig. S5, ESI†), in solid-state absorption, there appeared a significant new broad band at the infrared region (∼1200 nm), indicating the existence of charge transfer (CT) interactions (Fig. 2a). This CT characteristic switching was further investigated by FTIR, where the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibrations showed a distinct redshift of 6 cm−1 from the original F4TCNQ (2226 cm−1) to the cocrystal TMF4TQ (2220 cm−1), which can be ascribed to the CT transition from D to A (Fig. 2b). Moreover, the degree of charge transfer (DCT) was estimated from the geometry of F4TCNQ with the calculation of the bond length.41–43 By using the single-crystal X-ray diffraction (SCXRD) method, the DCT of TMF4TQ was calculated to be 0.128 (detailed in Fig. S6 and Table S1, ESI†), suggesting a partial charge transfer that is related to the frontier orbital hybridization between D and A components in a supramolecular complex.44 To more explicitly illustrate this CT nature on the energy levels, DFT calculations were conducted on the Gaussian 09 program at the B3LYP/6-31G** level.45 As depicted in Fig. S7 (ESI†), the frontier orbital hybridization occurring between the D (TMIQ) and A (F4TCNQ) molecules generates the novel hybrid HOMO (−5.21 eV) and LUMO (−4.55 eV) levels, as well as a narrowed bandgap (0.66 eV), which accounts for the sub-bandgap absorption in UV-vis-NIR spectra (Fig. 2a). To be mentioned, the hybrid HOMO is mainly localized on the TMIQ component, while the hybrid LUMO is partially distributed on the F4TCNQ, implying a better electronic coupling (Fig. S7, ESI†). Besides more criticality, the lowered LUMO level induced by the “doped” F4TCNQ makes it more aligned with the gold electrode, which has reduced the electron injection barrier and facilitated efficient electron transport, indicating that an electron transport would be achieved.
N stretching vibrations showed a distinct redshift of 6 cm−1 from the original F4TCNQ (2226 cm−1) to the cocrystal TMF4TQ (2220 cm−1), which can be ascribed to the CT transition from D to A (Fig. 2b). Moreover, the degree of charge transfer (DCT) was estimated from the geometry of F4TCNQ with the calculation of the bond length.41–43 By using the single-crystal X-ray diffraction (SCXRD) method, the DCT of TMF4TQ was calculated to be 0.128 (detailed in Fig. S6 and Table S1, ESI†), suggesting a partial charge transfer that is related to the frontier orbital hybridization between D and A components in a supramolecular complex.44 To more explicitly illustrate this CT nature on the energy levels, DFT calculations were conducted on the Gaussian 09 program at the B3LYP/6-31G** level.45 As depicted in Fig. S7 (ESI†), the frontier orbital hybridization occurring between the D (TMIQ) and A (F4TCNQ) molecules generates the novel hybrid HOMO (−5.21 eV) and LUMO (−4.55 eV) levels, as well as a narrowed bandgap (0.66 eV), which accounts for the sub-bandgap absorption in UV-vis-NIR spectra (Fig. 2a). To be mentioned, the hybrid HOMO is mainly localized on the TMIQ component, while the hybrid LUMO is partially distributed on the F4TCNQ, implying a better electronic coupling (Fig. S7, ESI†). Besides more criticality, the lowered LUMO level induced by the “doped” F4TCNQ makes it more aligned with the gold electrode, which has reduced the electron injection barrier and facilitated efficient electron transport, indicating that an electron transport would be achieved.
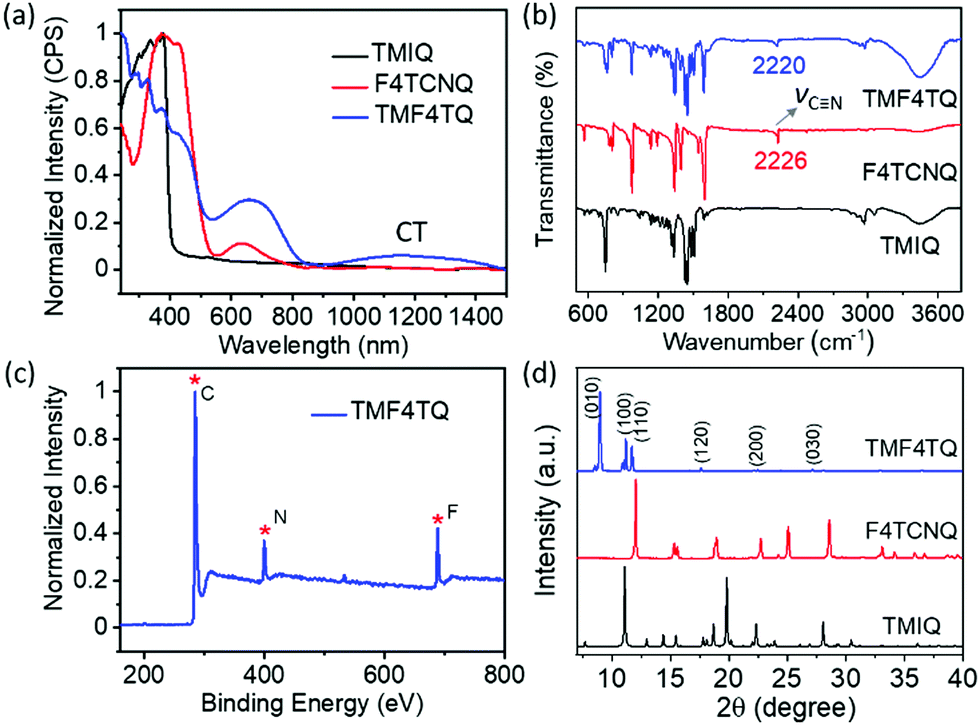 | ||
| Fig. 2 (a) UV-vis-NIR absorption, (b) FTIR spectra, and (d) PXRD patterns of powders for TMIQ, F4TCNQ, and TMF4TQ cocrystals. (c) Full scan of XPS for the cocrystal TMF4TQ. | ||
Changes in the crystal structure and molecular packing with the incorporation of acceptors were performed and investigated by single-crystal X-ray diffraction (SCXRD, Fig. 3 and CCDC† number: 2111795). The crystallographic data are summarized in Table S2 (ESI†). Compound TMIQ crystallizes into the monoclinic system in space group P21/c with unit-cell parameters of a = 6.4601(11) Å, b = 22.900(3) Å, c = 8.6053(14) Å, α = 90°, β = 98.884(8)°, and γ = 90°. As shown in Fig. 3a, the TMIQ molecules adopt a herringbone arrangement with the π–π stacking along the a-axis direction, resulting in an efficient electronic coupling and a priority electronic channel. Because there is a relatively large structure twist in the TMIQ molecule with a dihedral angle of up to 16°, the vertical distance between the adjacent molecules along the π–π stacking is not uniform, where the shortest distance is 3.44 Å. Besides, the neighboring π–π stacking columns are communicated by the C–H⋯π interactions, providing a stable crystal structure and additional charge transport channels. After co-assembled with F4TCNQ acceptors, a very different molecular packing is obtained with the crystal structure of the cocrystal TMF4TQ adopted in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/b_char_0031_0304.gif) (triclinic system) with unit-cell dimensions of a = 8.9947(4) Å, b = 10.5212(4) Å, c = 10.7845(4) Å, α = 79.6858(13)°, β = 67.3649(12)°, and γ = 68.3715(14)°. Herein, the TMIQ and F4TCNQ molecules are alternately arranged along the c-axis in a stoichiometry of 1:1 via the D–A interactions and hydrogen bonding, and in the a-axis direction, the TMIQ molecules form close packing through π–π interactions, both of which resulted in a 2D brick stacking configuration that is quite different from that for a single component (Fig. 3b). Apart from this, the insertion of acceptors also leads to a tighter crystal packing structure, in which the average π–π stacking distance between the adjacent TMIQ molecules is reduced to 3.40 Å, indicating a stronger electronic coupling. Meanwhile, the good overlap between the alternate D/A pairs as well as the short intermolecular D–A distance of 3.35 Å also denote the formation of an efficient charge transport route. On this basis, it can be inferred that this kind of change in the molecular arrangement would have a great impact on the final electrical properties.
(triclinic system) with unit-cell dimensions of a = 8.9947(4) Å, b = 10.5212(4) Å, c = 10.7845(4) Å, α = 79.6858(13)°, β = 67.3649(12)°, and γ = 68.3715(14)°. Herein, the TMIQ and F4TCNQ molecules are alternately arranged along the c-axis in a stoichiometry of 1:1 via the D–A interactions and hydrogen bonding, and in the a-axis direction, the TMIQ molecules form close packing through π–π interactions, both of which resulted in a 2D brick stacking configuration that is quite different from that for a single component (Fig. 3b). Apart from this, the insertion of acceptors also leads to a tighter crystal packing structure, in which the average π–π stacking distance between the adjacent TMIQ molecules is reduced to 3.40 Å, indicating a stronger electronic coupling. Meanwhile, the good overlap between the alternate D/A pairs as well as the short intermolecular D–A distance of 3.35 Å also denote the formation of an efficient charge transport route. On this basis, it can be inferred that this kind of change in the molecular arrangement would have a great impact on the final electrical properties.
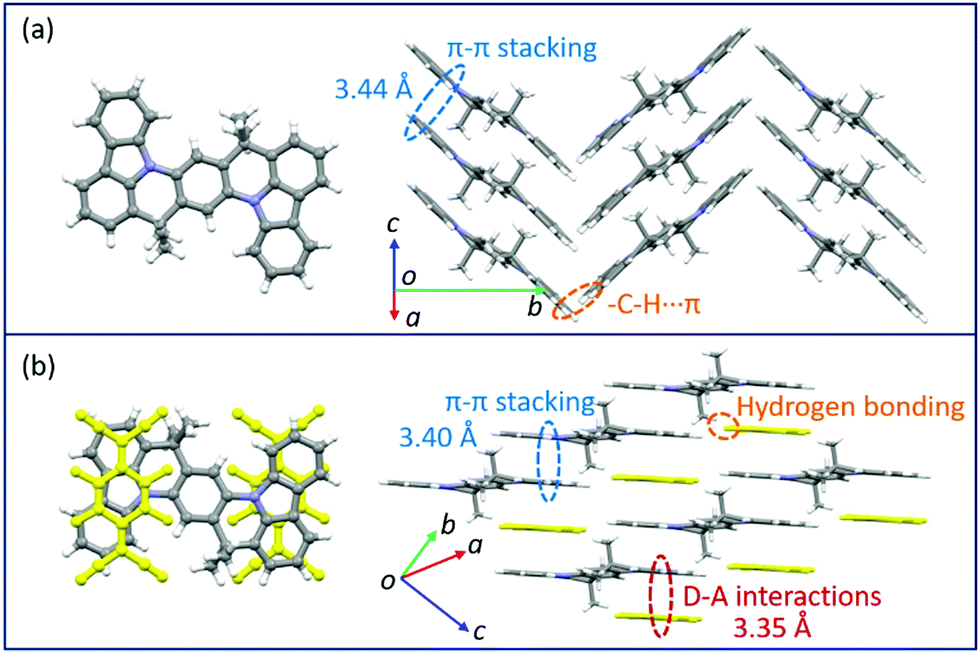 | ||
| Fig. 3 Crystal structures of TMIQ and TMF4TQ: (a) molecular structure and crystal packing for TMIQ (herringbone stacking). (b) Molecular structure and crystal packing for TMF4TQ (2D brick stacking). | ||
To explore the effect of energy levels and molecular arrangement regulation by F4TCNQ “doping” on the inherent charge transport properties, OFETs based on the micro/nanocrystals of the single component TMIQ and cocrystal TMF4TQ were fabricated and investigated (Fig. 4). Above all, the self-assembled behaviors of these two compounds on OTS-modified SiO2/Si substrates was discussed. Single-crystalline micro-ribbons with lengths of several tens to hundreds of micrometers were obtained by dropping their CH2Cl2 solution on the substrates (Fig. S8, ESI†). As depicted in Fig. 4a, the out-of-plane XRD patterns for TMIQ micro-ribbons exhibit intense peaks at (020), (011) and (040) within their crystallographic data, suggesting that the crystals roughly grow along the π–π stacking direction, which is also a preferred charge transport direction. Whereas, for TMF4TQ, a strong and sharp peak that appeared in the XRD profile could be indexed as (010), which indicates that the crystals grow with the ac plane parallel to the substrate, thereby inferring the charge transport likely adopting a 2D mode (Fig. 4b). Based on the as-prepared micro-ribbons, top-contact bottom-gate OFETs were constructed via evaporating gold source/drain electrodes onto the target microcrystals through “organic ribbon masks”.46 All device fabrication and characterization studies were carried out in air (relative humidity range 50%–75%) and ambient temperature. As shown by the typical transfer and output characteristics in Fig. 4, devices based on TMIQ micro-ribbons exhibited a hole-transporting feature with the maximum mobility of up to 0.27 cm2 V−1 s−1 and a high on/off ratio of 106 (Fig. 4c and e), which is well consistent with the aforementioned energy level analysis. While in contrast, as predicted, the introduction of equal proportional F4TCNQ molecules into the host has brought about an amazing conversion in the charge-transport nature (Fig. 4d and f). The devices based on the cocrystal TMF4TQ displayed the electron transport properties with μe extracted from the saturated region up to 0.12 cm2 V−1 s−1 (and 0.09 cm2 V−1 s−1 from the linear region at VSD of 30 V), which is among the best n-type performances in cocrystal systems.29,47 Besides, all the transistors showed good air stability with smooth curves under atmospheric conditions as well as the almost no-shifted threshold voltage in repeated measurements (Fig. S9, ESI†), due to the low-lying LUMO level (−4.55 eV) tailored by the dopant molecules.
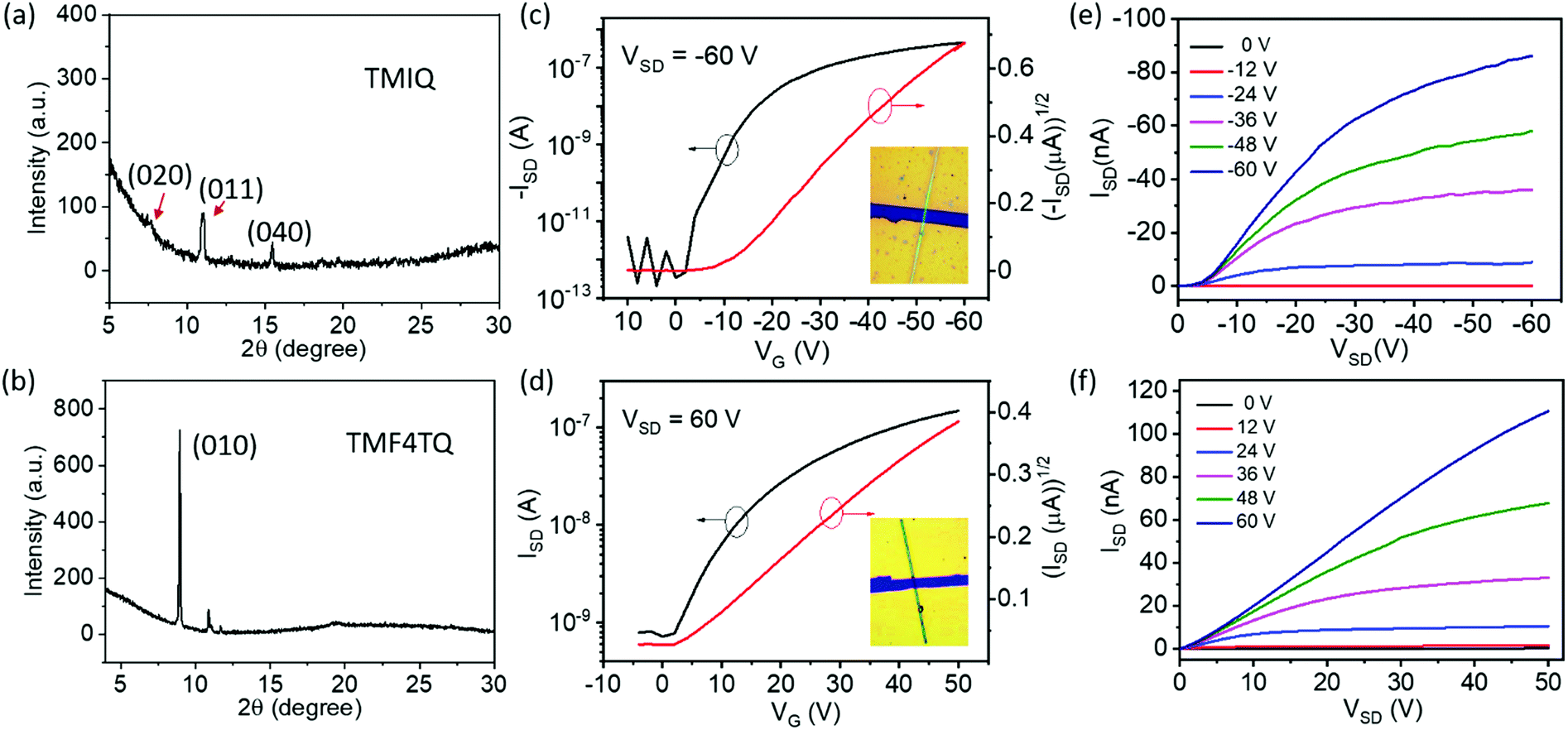 | ||
| Fig. 4 (a and b) XRD patterns of self-assembled microcrystals for (a) TMIQ and (b) TMF4TQ on OTS-modified SiO2/Si substrates. (c and d) Representative transfer and (e and f) output characteristics of OFET devices based on (c and e) TMIQ and (d and f) TMF4TQ micro-ribbons (inset: the corresponding device). | ||
To more explicitly understand this transition of charge transport characteristics and their corresponding structure–property relationships, the effective electronic couplings strongly related to the charge transport were estimated through the molecular energy-splitting method.48,49 The detailed calculation approach to compute the intermolecular transfer integrals (t) along the stacking directions is illustrated in Fig. S10–S12 (ESI†). Since it has been speculated that the π–π stacking directions are the prominent transport path as previously discussed in the pure TMIQ crystal (Fig. 5a), the transfer integrals (i) are calculated to be 12.25 meV for electrons and 28.57 meV for holes (Fig. S10, ESI†). The apparently larger thole than telectron, combined with the high-lying HOMO level (Fig. 5b), accounts for its hole-dominant transport. By comparison, the cocrystal TMF4TQ adopts a 2D transport mainly at the ac plane (Fig. 5c), involving direct electronic couplings along the π–π stacking (D–D, ii) and mixed-stacking directions (D–A–D/A–D–A, iii) via the super-exchange effect.50,51 Surprisingly, the insertion of F4TCNQ molecules, along with the evident structural rearrangement, is proven to have a significant impact on effective transfer integrals. Specifically, an obviously larger estimated telectron (35.24 meV) is obtained in the D–D directions (ii, Fig. 5c and Fig. S11, ESI†) than that of thole (17.28 meV). In addition, along the mixed-stacking direction (iii, Fig. 5c and Fig. S12, ESI†) also, there is a similar trend that a distinctly greater telectron (21.22 meV) has been observed (thole: 4.218 meV), both of which illustrate the formation of an effective electron transmission channel and thereby induce a charge transport conversion from holes to electrons. Beyond this, the downward LUMO level (−4.55 eV) aligned with the gold electrodes (Fig. 5d) has reduced the electron injection barrier, thereby collectively ensuring the high-performance air-stable electron transport.
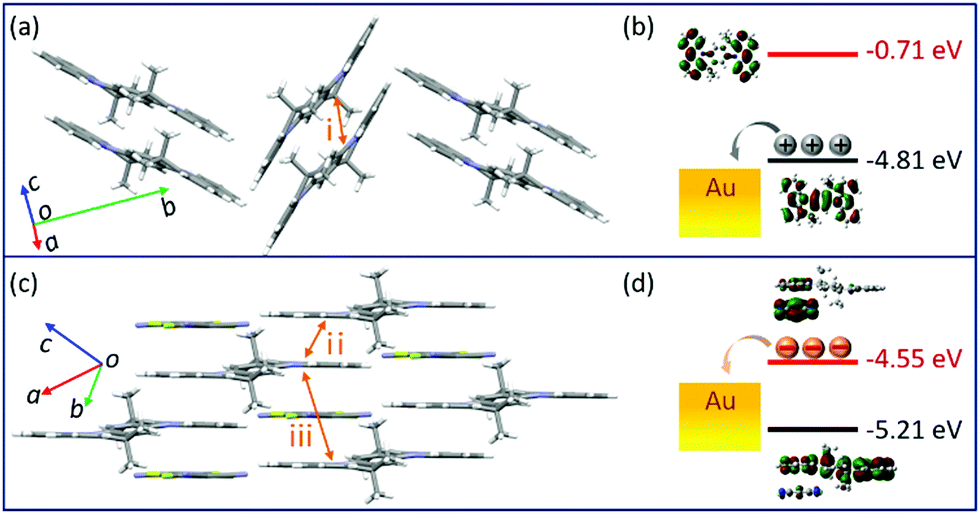 | ||
| Fig. 5 (a and c) The most favorable plane for charge transfer for (a) TMIQ and (c) TMF4TQ. (b and d) Calculated MO energy level diagram of (b) TMIQ and (d) TMF4TQ upon the Fermi level of gold electrodes. | ||
Conclusions
In summary, we have successfully realized the transition of charge transport from p-type to n-type through a simple and economic “green synthesis” method. That is, by “doping” electron-deficient molecules F4TCNQ into a p-type planar azaacene host TMIQ (μh = 0.27 cm2 V−1 s−1) in the molar ratio of 1:1 via a simple solution processing method, an ingenious energy level and molecular packing arrangement tailoring for charge-transport characteristic switching have been achieved. The intermolecular CT interactions, hydrogen bonding and π–π interactions induced by inserting F4TCNQ have led to a dense two-dimensional (2D) brick stacking, which is responsible for the effective electron transport channels. Meanwhile, the lowered LUMO levels (−4.55 eV) caused by the frontier orbital hybridization of alternated D and A molecules not only reduce the electron injection barrier favoring electron injection, but also block the electrochemical oxidation of water and oxygen under an ambient atmosphere, thereby resulting in a high-performance air-stable n-type cocrystal TMF4TQ with an electron mobility (μe) of up to 0.12 cm2 V−1 s−1 in the saturated region. DFT calculations further illustrate and confirm this charge transport characteristic switching. Our results have well proved the feasibility of developing novel high-performance air-stable n-type OSCs by this easy-to-process co-crystallization strategy, and are believed to retain great significance for low-cost devices and complementary metal oxide semiconductor (CMOS)-like circuits.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Fundamental Research Funds for the Central Universities (buctrc202128, buctrc202103), the National Natural Science Foundation of China (52103200, 21975126, 51822308, 21975263), and the starting-up funding from the City University of Hong Kong. We also acknowledge the State Key Laboratory of Organic–Inorganic Composites, and the Ministry of Education and Synergetic Innovation Center for Organic Electronics and Information Displays.Notes and references
- H. Sun, X. Guo and A. Facchetti, Chemistry, 2020, 6, 1310–1326 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2011, 112, 2208–2267 CrossRef PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS.
- B. Crone, A. Dodabalapur, Y. Y. Lin, R. W. Filas, Z. Bao, A. LaDuca, R. Sarpeshkar, H. E. Katz and W. Li, Nature, 2000, 403, 521–523 CrossRef CAS PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- R. Capelli, S. Toffanin, G. Generali, H. Usta, A. Facchetti and M. Muccini, Nat. Mater., 2010, 9, 496–503 CrossRef CAS PubMed.
- G. Gelinck, P. Heremans, K. Nomoto and T. D. Anthopoulos, Adv. Mater., 2010, 22, 3778–3798 CrossRef CAS.
- G. H. Gelinck, H. E. A. Huitema, E. van Veenendaal, E. Cantatore, L. Schrijnemakers, J. B. P. H. van der Putten, T. C. T. Geuns, M. Beenhakkers, J. B. Giesbers, B.-H. Huisman, E. J. Meijer, E. M. Benito, F. J. Touwslager, A. W. Marsman, B. J. E. van Rens and D. M. de Leeuw, Nat. Mater., 2004, 3, 106–110 CrossRef CAS.
- H. E. A. Huitema, G. H. Gelinck, J. B. P. H. van der Putten, K. E. Kuijk, C. M. Hart, E. Cantatore, P. T. Herwig, A. J. J. M. van Breemen and D. M. de Leeuw, Nature, 2001, 414, 599 CrossRef CAS.
- M. Y. Lee, H. R. Lee, C. H. Park, S. G. Han and J. H. Oh, Acc. Chem. Res., 2018, 51, 2829–2838 CrossRef CAS PubMed.
- J. Y. Oh, D. Son, T. Katsumata, Y. Lee, Y. Kim, J. Lopez, H.-C. Wu, J. Kang, J. Park, X. Gu, J. Mun, N. G.-J. Wang, Y. Yin, W. Cai, Y. Yun, J. B.-H. Tok and Z. Bao, Sci. Adv., 2019, 5, eaav3097 CrossRef CAS PubMed.
- W. Shi, Y. Guo and Y. Liu, Adv. Mater., 2020, 32, e1901493 CrossRef PubMed.
- S. Dai, Y. Zhao, Y. Wang, J. Zhang, L. Fang, S. Jin, Y. Shao and J. Huang, Adv. Funct. Mater., 2019, 29, 1903700 CrossRef CAS.
- Y. van de Burgt, A. Melianas, S. T. Keene, G. Malliaras and A. Salleo, Nat. Electron., 2018, 1, 386–397 CrossRef.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS.
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS.
- F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109–5115 RSC.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- Y. Qiao, Y. Guo, C. Yu, F. Zhang, W. Xu, Y. Liu and D. Zhu, J. Am. Chem. Soc., 2012, 134, 4084–4087 CrossRef CAS.
- A. Yassar, F. Demanze, A. Jaafari, M. El Idrissi and C. Coupry, Adv. Funct. Mater., 2002, 12, 699–708 CrossRef CAS.
- J. A. Letizia, A. Facchetti, C. L. Stern, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 13476–13477 CrossRef CAS.
- M. H. Yoon, A. Facchetti, C. E. Stern and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 5792–5801 CrossRef CAS PubMed.
- T. Lee, C. A. Landis, B. M. Dhar, B. J. Jung, J. Sun, A. Sarjeant, H.-J. Lee and H. E. Katz, J. Am. Chem. Soc., 2009, 131, 1692–1705 CrossRef CAS PubMed.
- B. Lüssem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef PubMed.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS.
- Z. Wang, Y. Zou, W. Chen, Y. Huang, C. Yao and Q. Zhang, Adv. Electron. Mater., 2019, 5, 1800547 CrossRef.
- B. D. Naab, S. Himmelberger, Y. Diao, K. Vandewal, P. Wei, B. Lussem, A. Salleo and Z. Bao, Adv. Mater., 2013, 25, 4663–4667 CrossRef CAS.
- J. Zhang, J. Jin, H. Xu, Q. Zhang and W. Huang, J. Mater. Chem. C, 2018, 6, 3485–3498 RSC.
- G. Bolla, Q. Liao, S. Amirjalayer, Z. Tu, S. Lv, J. Liu, S. Zhang, Y. Zhen, Y. Yi, X. Liu, H. Fu, H. Fuchs, H. Dong, Z. Wang and W. Hu, Angew. Chem., Int. Ed., 2021, 60, 281–289 CrossRef CAS PubMed.
- Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef.
- A. S. Tayi, A. Kaeser, M. Matsumoto, T. Aida and S. I. Stupp, Nat. Chem., 2015, 7, 281 CrossRef CAS.
- Z. Wang and Q. Zhang, Asian J. Org. Chem., 2020, 9, 1252–1261 CrossRef CAS.
- Z. Wang, F. Yu, J. Xie, J. Zhao, Y. Zou, Z. Wang and Q. Zhang, Chem. – Eur. J., 2020, 26, 3578–3585 CrossRef CAS PubMed.
- Q. Tang, G. Zhang, B. Jiang, D. Ji, H. Kong, K. Riehemann, Q. Ji and H. Fuchs, SmartMat, 2021, 2, 109–118 CrossRef.
- L. Sun, Y. Wang, F. Yang, X. Zhang and W. Hu, Adv. Mater., 2019, 31, 1902328 CrossRef PubMed.
- (a) Y. Huang, Z. Wang, Z. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2019, 58, 9696–9711 CrossRef CAS; (b) H. Ye, G. Liu, S. Liu, D. Casanova, X. Ye, X. Tao, Q. Zhang and Q. Xiong, Angew. Chem., Int. Ed., 2018, 57, 1928–1932 CrossRef CAS; (c) G. Liu, J. Liu, X. Ye, L. Nie, P. Gu, X. Tao and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 198–202 CrossRef CAS.
- J. Zhang, P. Y. Gu, G. K. Long, R. Ganguly, Y. X. Li, N. Aratani, H. Yamada and Q. C. Zhang, Chem. Sci., 2016, 7, 3851–3856 RSC.
- W. Zhang, X. Sun, P. Xia, J. Huang, G. Yu, M. S. Wong, Y. Liu and D. Zhu, Org. Lett., 2012, 14, 4382–4385 CrossRef CAS.
- G. Li, J. F. Zhao, S. F. Yang, Y. X. Li, G. Rakesh and Q. C. Zhang., Chem. – Asian J., 2018, 13, 250–254 CrossRef CAS.
- H. Jiang, P. Hu, J. Ye, K. K. Zhang, Y. Long, W. Hu and C. Kloc, J. Mater. Chem. C, 2018, 6, 1884–1902 RSC.
- P. Hu, K. Du, F. Wei, H. Jiang and C. Kloc, Cryst. Growth Des., 2016, 16, 3019–3027 CrossRef CAS.
- T. J. Kistenmacher, T. E. Phillips and D. O. Cowan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 763–768 CrossRef CAS.
- H. Mendez, G. Heimel, A. Opitz, K. Sauer, P. Barkowski, M. Oehzelt, J. Soeda, T. Okamoto, J. Takeya, J. B. Arlin, J. Y. Balandier, Y. Geerts, N. Koch and I. Salzmann, Angew. Chem., Int. Ed., 2013, 52, 7751–7755 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. P. Jr., F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- L. Jiang, J. Gao, E. Wang, H. Li, Z. Wang, W. Hu and L. Jiang, Adv. Mater., 2008, 20, 2735–2740 CrossRef CAS PubMed.
- J. Zhang, W. Xu, P. Sheng, G. Zhao and D. Zhu, Acc. Chem. Res., 2017, 50, 1654–1662 CrossRef CAS.
- M. Ottonelli, M. Piccardo, D. Duce, S. Thea and G. Dellepiane, Energy Procedia, 2012, 31, 31–37 CrossRef CAS.
- E. F. Valeev, V. Coropceanu, D. A. D. S. Filho, S. Salman and J.-L. Brédas, J. Am. Chem. Soc., 2006, 128, 9882–9886 CrossRef CAS PubMed.
- L. Zhu, Y. Yi, Y. Li, E.-G. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2012, 134, 2340–2347 CrossRef CAS PubMed.
- H. Geng, X. Zheng, Z. Shuai, L. Zhu and Y. Yi, Adv. Mater., 2015, 27, 1443–1449 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2111795. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc04610a |
| This journal is © The Royal Society of Chemistry 2022 |