
Molecular simulations of zwitterlation-induced conformation and dynamics variation of glucagon-like peptide-1 and insulin†
Qi
Qiao‡
 ,
Lirong
Cai‡
and
Qing
Shao
*
,
Lirong
Cai‡
and
Qing
Shao
*
Chemical and Materials Engineering Department, University of Kentucky, Lexington, Kentucky 40506, USA. E-mail: qshao@uky.edu
First published on 20th January 2022
Abstract
Zwitterionic materials have shown their ability to improve the circulation time and stability of proteins. Zwitterionic peptides present unique potential because genetic technology can fuse them to any wild-type protein. One critical question is the effect of the fusing zwitterionic peptides on the conformation and dynamics of the original protein domain. To shed light on this question, we investigate the conformation and dynamics of six artificial proteins composed of two small therapeutic polypeptide and protein (glucan-like peptide-1 and insulin) and a zwitterionic (glutamic acid-lysine)10 peptide in an explicit solvent using molecular dynamics simulations. The zwitterionic peptide is fused to the N- and C-terminal of the glucan-like peptide-1 and the chain A and B of the insulin. We analyze the conformation and dynamics variation of the polypeptide and protein domain using root mean square deviation, root mean square fluctuation, solvent accessible surface area, and secondary structure distributions. The simulation results show that the zwitterlation induces substantial changes in the conformation of the glucan-like peptide-1 and a moderate change in the conformation of the insulin, while the two polypeptide and protein remain folded. The glucan-like peptide-1 presents a full α-helix conformation when zwitterlated at the C-terminal. The zwitterionic location also plays a role in the conformational change. These zwitterlation-induced conformation variations indicate a comprehensive relationship between zwitterlation and protein stability and activity.
1. Introduction
Zwitterionic (ZW) materials emerge for their ability to stabilize proteins.1–7 Zwitterionic materials consist of motifs possessing both cationic and anionic groups.1,8–11 This particular structure provides strong hydration that can be leveraged to enhance the stability of a protein.12–17 Among various zwitterionic materials, peptides stand out as a promising candidate to protect protein because they can connect to a specific location on the protein using genetic technology and the final products have the potential to be manufactured at a large scale.18–22 Thus, zwitterionic peptides open a gate to design and fabricate novel artificial proteins by fusing the peptide with a targeted protein. However, the design of such artificial proteins needs a fundamental understanding of the effect of ZW peptides on the structural and dynamic properties.One open question is the effect of zwitterionic peptides on small proteins’ structural and dynamic properties. Here we refer small proteins to those with <50 amino acid residues. Small proteins have their unique role in therapeutics because they are generally easier to transport across the cell membranes than big proteins. Small proteins are also a great ingredient when designing artificial proteins. The small proteins may be more vulnerable than big ones because they have less nonbonded interactions to hold the structure and a fast pace to unfold. Even though the small proteins may remain folded,2,23 the fusing zwitterionic peptide may still affect their conformation and dynamics due to a lower thermodynamic barrier for the conformation variation. Thus, we must investigate the effect of zwitterionic peptides on the structural and dynamic properties of small proteins. The harvested knowledge can be used to develop principles for designing artificial proteins based on zwitterionic peptides and small proteins.
The studies of zwitterionic peptides are much less compared to those for synthetic zwitterionic polymers. These efforts illustrate the potential of zwitterionic peptides in protecting protein stability. For instance, Liu et al.20 investigated the effect of poly(glutamic acid-lysine) (ploy(EK)) peptides on the stability of a native β-lactamase and its destabilized TEM-19 mutant by fusing the peptides on the C-terminal of the proteins. They found that the attached poly(EK) peptides could maintain the biological activity of the proteins and significantly increase their stability at harsh conditions, including high temperature and high-salt solutions. Banskot et al.24 designed a zwitterionic peptide with (VPX1X2G)n motif, where X1 and X2 represent different pairs of cationic and anionic amino acids. They investigated the performance of their design by fusing the peptide with different pairs of X1 and X2 on with glucan-like peptide-1 (GLP-1). They found that (VPKEG)n-modified GLP-1 shows the best performance in a mouse model. Some experimental work focuses on the anti-biofouling performance of zwitterionic peptides.1,7,8,25–29 They showed that zwitterionic peptide surface coatings could resist the adsorption of proteins.30–32 Many anti-biofouling materials have shown their ability to protect proteins, ranging from polyethylene glycol33–41 to synthetic zwitterionic polymers.3–8 Thus, these studies also imply the potential of zwitterionic peptides in protecting protein stability.
The simulations of zwitterionic peptides are even less compared to experimental studies. Our previous molecular simulation2 shows that the (EK)10 peptides could maintain the stability of the ubiquitin protein and resist the denaturing effect of high temperature. Teng et al.23 investigated the interaction of GLP-1 with unconnected zwitterionic peptides (VPX1X2G)n using MD simulation. They found that the (VPX1X2G)n could stabilize GLP-1 by forming a packing shell around it.
This paper aims to investigate the impact of a zwitterionic peptide on small therapeutic polypeptide and protein's structural and dynamic properties. This work selects GLP-1 (30 amino acid residues) and insulin (51 amino acid residues) as the small polypeptide and protein models. These two are important protein therapeutics for diabetes. We will investigate the structural and dynamic properties of the two wild-type GLP-1, insulin and the six GLP-1-Zwitterion and insulin-Zwitterion fusions. The six fusions include the two for GLP-1 and four for insulin. The two GLP-1-Zwitterion fusions are prepared by fusing a zwitterionic (EK)10 peptide to the N- and C-terminal. The four insulin-Zwitterion fusions are prepared by fusing a zwitterionic peptide on the N- and C-terminal of Chain A and B. It is worth noting that the tertiary structure obtained after cleaved C peptide will assist the insulin fold into the original native structure. Therefore, it is impossible to get the fusion at C terminal of Chain A or N terminal at Chain B in experiments. We included these two models in our simulations in order to study the insulin–peptide fusions systematically. The rest of this paper is organized as follows. Section 2 is the molecular model and simulation detail. Section 3 is the results and discussion, and Section 4 is the conclusion.
2. Molecular model and simulation detail
2.1. Molecular model
The all-atom models are used to describe the wild-type GLP-1, insulin, GLP-1-peptide and insulin–peptide fusions, water molecules and ions. The initial configuration of wild-type insulin is obtained from the Protein Databank (PDB ID: 3I40 Human insulin). The initial configuration of the wild-type GLP-1 is predicted by Rosetta online server42 using its sequences43 (Table S1, ESI†). Fig. 1 shows the initial configurations of wild-type GLP-1 and insulin. The GLP-1-Zwitterion and insulin-Zwitterion fusions are created by adding (EK)10 to the C-terminal or N-terminal of the wild-type GLP-1 and insulin. The initial configurations of the GLP-1-Zwitterion and insulin-Zwitterion fusions are predicted by Rosetta using their sequences. Fig. S1 (ESI†) shows the initial configurations of the six GLP-1-Zwitterion and insulin-Zwitterion fusions. Table 1 lists the label of the eight GLP-1, insulin, GLP-1-Zwitterion and insulin-Zwitterion fusions.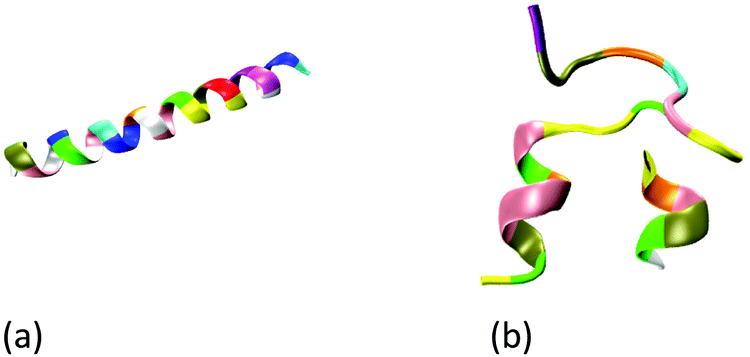 | ||
| Fig. 1 Initial configuration of wild-type (a) GLP-1, and (b) insulin. The GLP-1 and insulin: new-cartoon model, colored based on the residue name. | ||
| System | Description |
|---|---|
| GLP-1-wt | Wild-type GLP-1 |
| (EK)10-N-GLP-1 | (EK)10 peptide attached to N-terminal of the GLP-1 |
| GLP-1-C-(EK)10 | (EK)10 peptide attached to N-terminal of the GLP-1 |
| Insulin-wt | Wild-type insulin |
| (EK)10-N-A | (EK)10 peptide attached to N-terminal of the insulin A-chain |
| A-C-(EK)10 | (EK)10 peptide attached to C-terminal of the insulin A-chain |
| (EK)10-N-B | (EK)10 peptide attached to N-terminal of the insulin B-chain |
| B-C-(EK)10 | (EK)10 peptide attached to C-terminal of the insulin B-chain |
The simulation systems are created by placing the GLP-1-wt, insulin-wt or their zwitterlated fusions in a cubic box and filling the box with 0.15 M NaCl solution and extra counter-ions to neutralize the system. The 0.15 M NaCl solution simulates the real human body environment. The initial size of the simulation box is 5.0 × 8.2 × 5.8 nm3 for wild-type GLP-1 and 6.5 × 6.5 × 6.5 nm3 for wild-type insulin. These sizes of the boxes prevent the GLP-1, insulin, or their zwitterlated fusions from interacting with their mirrors and allow enough bulk water molecules. Fig. 2 shows the snapshot of an insulin B-C-(EK)10 fusion in a 0.15 M NaCl solution box. The systems in this work were simulated using the AMBER 14 force field.44 The nonbonded interactions are a sum of short-range Lennard–Jones 12-6 potential and long-range coulombic potential, as shown in eqn (1). The bonded interactions are a sum of the bond, angle, and dihedral potentials, as described in the force field. The classical TIP3P water model was used in this work.
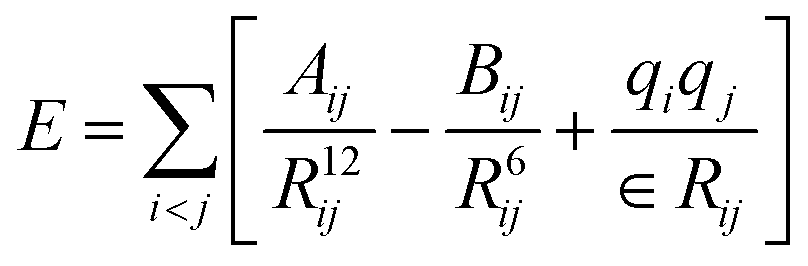 | (1) |
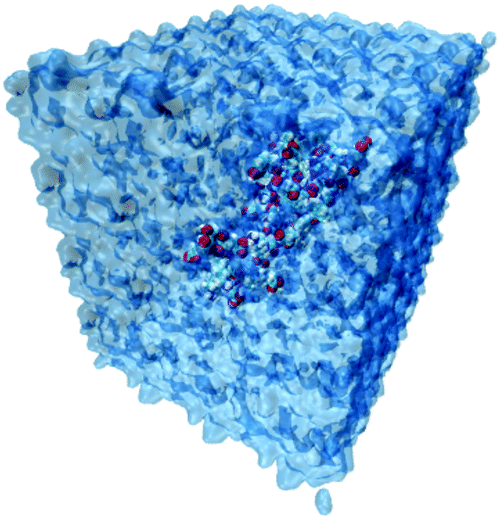 | ||
| Fig. 2 The snapshot of a simulation system containing insulin B-C-(EK)10 fusion. The water box is shown in blue. The insulin: VDW model, colored based on the atom name. The water molecules: quickSurf model, colored based on the atom name. | ||
2.2. Simulation detail
The molecular dynamics (MD) simulation includes three steps. First, the energy minimization was conducted to remove any too-close contacts between atoms. The minimization was carried out using the steepest descent algorithm (force tolerance = 10.0 kJ (mol−1·nm−1)) with a maximal step of 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Second, a 300 ns isobaric-isothermal (NPT, T = 310 K, P = 1 bar) ensembled MD simulation with a 2 fs integral step was run to reach a thermodynamic equilibrium of the system. The Berendsen method45 is used to control the temperature and pressure in this step. Third, a 200 ns canonical ensembled MD simulation (NPT, T = 310 K, P = 1 bar, and 2 fs integral step) was conducted for data collection at a frequency of 100 ps. The velocity-rescaling method46 is used to control the temperature. The Parrinello–Rahman method47 is used for pressure coupling in this step. The short-range van der Waals interactions use a 1.2 nm cut-off and the long-range electrostatic interactions were calculated using particle mesh Ewald (PME) sum.48 All bonds involving H atoms were constrained during the simulations.
000. Second, a 300 ns isobaric-isothermal (NPT, T = 310 K, P = 1 bar) ensembled MD simulation with a 2 fs integral step was run to reach a thermodynamic equilibrium of the system. The Berendsen method45 is used to control the temperature and pressure in this step. Third, a 200 ns canonical ensembled MD simulation (NPT, T = 310 K, P = 1 bar, and 2 fs integral step) was conducted for data collection at a frequency of 100 ps. The velocity-rescaling method46 is used to control the temperature. The Parrinello–Rahman method47 is used for pressure coupling in this step. The short-range van der Waals interactions use a 1.2 nm cut-off and the long-range electrostatic interactions were calculated using particle mesh Ewald (PME) sum.48 All bonds involving H atoms were constrained during the simulations.
3. Results and discussion
3.1. Root mean square deviation
We first analyzed the effect of the fusing ZW peptide on the conformation of the GLP-1 using root mean square deviation (RMSD) of Cα atoms. Fig. 3a shows the Rosetta-predicted structure (pink) and the MD simulation derived structure (orange) for a wild-type GLP-1. The Rosetta predicted structure is similar to those reported in receptor-protein complexes, while the MD derived structure is consistent with that in the literature.23 Such a difference of the GLP-1 structures could be due to the factor that Rosetta and MD simulations deploy distinct prediction approaches. Rosetta uses the similar PDB templates from the Protein Databank as the initial configuration. The Protein Databank contains many GLP-1 structures binding to the other proteins. Thus, Rosetta may likely predict the conformation of a GLP-1 chain when binding to the other proteins. On the other hand, MD simulations predict the GLP-1 conformation based on the balance between the bonded and non-bonded molecular interactions. The MD simulations tend to predict the conformation of GLP-1 when just presenting by itself. Thus, we use the structure in orange in Fig. 3a as the reference in the RMSD analysis.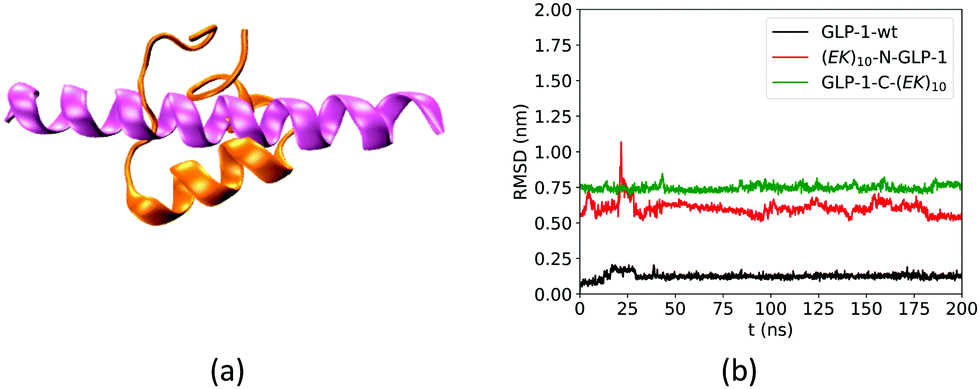 | ||
| Fig. 3 (a) The configuration of wild-type GLP-1 that predicted by Rosetta (pink) and a 300 ns NPT MD simulation (orange) and (b) RMSD of Cα atoms of amino acid residues on wild-type GLP-1 and its zwitterlated fusions. | ||
The fusing (EK)10 peptide changes the conformation of GLP-1. Fig. 3b shows the RMSD of Cα atoms of amino acid residues on the wild-type GLP-1 and the GLP-1 domain on the two zwitterlated fusions. The RMSD for the wild-type GLP-1 ranges from 0.04 to 0.2 nm, mostly around 0.1 nm. This RMSD range indicates that the wild-type GLP-1 fluctuates around the reference configuration. The two zwitterlated GLP-1 fusions present an RMSD larger than 0.5 nm. Such a large RMSD indicates that the GLP-1 possesses a distinct conformation from the reference.
The zwitterlation also affects the conformation of insulin in a moderate manner. Fig. 4 shows the RMSD curves for Chain A and B on the wild-type insulin and the insulin domain in the four artificial proteins. The structure of PDB ID: 3I40 is used as the reference. As shown in Fig. 4a, the RMSD for Chain A in the four artificial proteins ranges from 0.1 to 0.3 nm, similar to the wild-type one. This similarity in the RMSD scope indicates that Chain A in the four zwitterlated insulin fusions may present conformations close to those for the wild-type one. These conformations fluctuate around the reference. The Chain B in the four zwitterlated insulins could also possess conformations similar to the wild-type one. As shown in Fig. 4b, the scopes of the RMSD curves for the zwitterlated fusions overlap with that for the wild-type one. The one exception could be the Chain B in the (EK)10-N-B fusion. Its RMSD curve shows a value around 0.5 nm during 100 to 200 ns. This high RMSD value indicates that the insulin domain may present conformations that deviate from the reference.
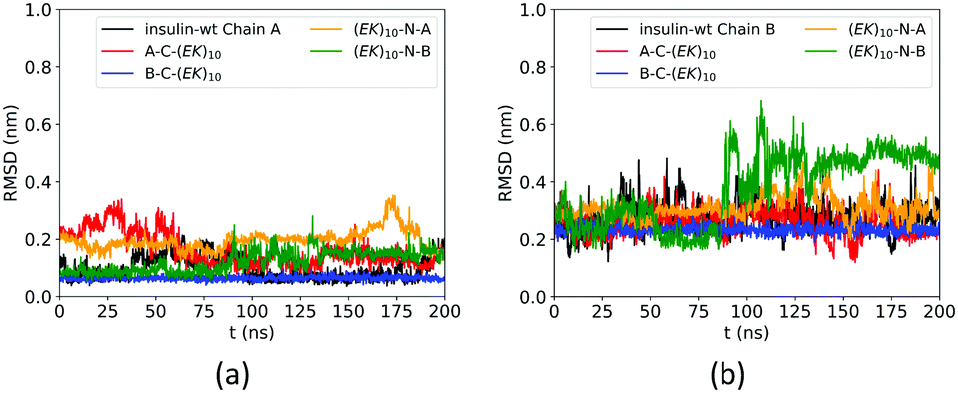 | ||
| Fig. 4 RMSD of Cα atoms of amino acid residues on (a) Chain A and (b) Chain B of wild-type insulin and its zwitterlated fusions. | ||
3.2. Root mean square fluctuation
We investigate the zwitterlation-induced variation in polypeptide and protein dynamics based on the root mean square fluctuation (RMSF) of Cα atoms on the wild-type GLP-1 and the corresponding polypeptide and protein domain on the zwitterlated GLP-1 fusions. The value of ΔRMSFi is calculated as RMSFi (fusion) – RMSFi (wild-type), where i refers to the Cα atom on amino acid residue i. A positive ΔRMSF value means that the zwitterlation increases polypeptide and protein dynamics, while a negative ΔRMSF indicates that the zwitterlation decreases polypeptide and protein dynamics.The C- and N-terminal zwitterlation induces distinct effects on the dynamics of the GLP-1. As shown in Fig. 5, many residues of GLP-1-C-(EK)10 present ΔRMSF close to zero. The near-zero ΔRMSF indicates that the major of the GLP-1 domain remains the dynamics of the wild-type one after being zwitterlated on the C-terminal. On the other hand, many residues of (EK)10-N-GLP-1 present positive ΔRMSF ranging from 0.1 to 0.3 nm. The majority of the GLP-1 domain becomes more dynamic after being zwitterlated on the N-terminal.
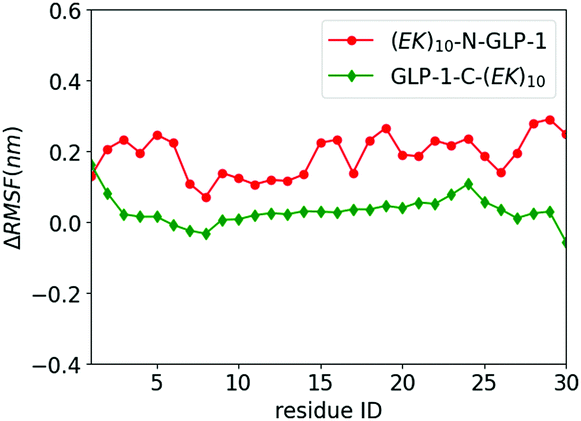 | ||
| Fig. 5 ΔRMSF of Cα atoms on GLP-1 and its zwitterlated fusions. Fig. S2 (ESI†) shows the original RMSF of GLP-1 and its fusions. | ||
The zwitterlation also shows the distinct effects on the dynamics of the insulin protein when fusing the (EK)10 peptide on the different locations. Fig. 6 shows the values of ΔRMSF for the residues on chains A and B of the insulin protein. As shown in Fig. 6a, the value of ΔRMSF keeps close to zero for all four insulin–peptide fusions. Chain A remains its dynamics regardless of the zwitterlation location. The majority of Chain B also remain the dynamics of a wild-type one when the zwitterlation occurs on the two terminals of Chain A and the C-terminal of chain B. However, the whole chain B becomes more dynamic when the zwitterlation occurs on the N-terminal of Chain B.
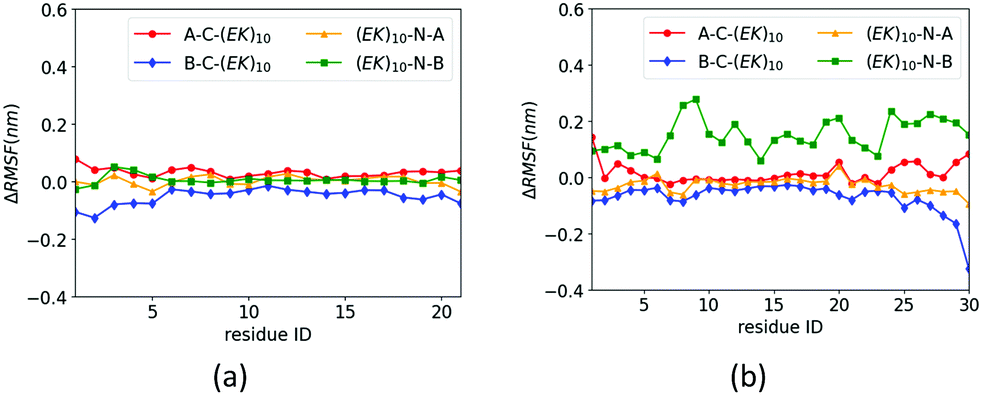 | ||
| Fig. 6 ΔRMSF of Cα atoms on insulin (a) chain A and (b) chain B and its zwitterlated fusions. Fig. S3 (ESI†) shows the original RMSF of insulin and its fusions. | ||
The distinct zwitterlation-induced dynamics variations for the GLP-1 and insulin indicate a comprehensive effect of the (EK)10 peptide on molecular interactions and conformation of the proteins. The dynamics of individual residues should be a synergistic consequence of the geometric constrain and non-bond molecular interactions. Thus, a change in the protein dynamics may indicate a variation of the conformation or molecular interactions. The distinct ΔRMSF curves in Fig. 6 suggest that zwitterlation could be used as a method to regulate the properties of a protein.
3.3. Solvent accessible surface area
Fusing peptides could cover the specific polypeptide and protein domain. We analyzed the impact of zwitterlation on the exposure area of the polypeptide and protein surface using the solvent accessible surface area (SASA) on the polypeptide and protein domain. The whole polypeptide and protein domain is divided into the hydrophobic and hydrophilic domains. The hydrophobic domain consists of ALA, ILE, LEU, MET, PHE, VAL, PRO, and GLY residues, and the hydrophilic domain consists of the rest amino acid residues. Fig. 7 shows SASA of the hydrophobic and hydrophilic domains and the whole polypeptide and protein for the six zwitterlated fusions and the two wild-type GLP-1 and insulin.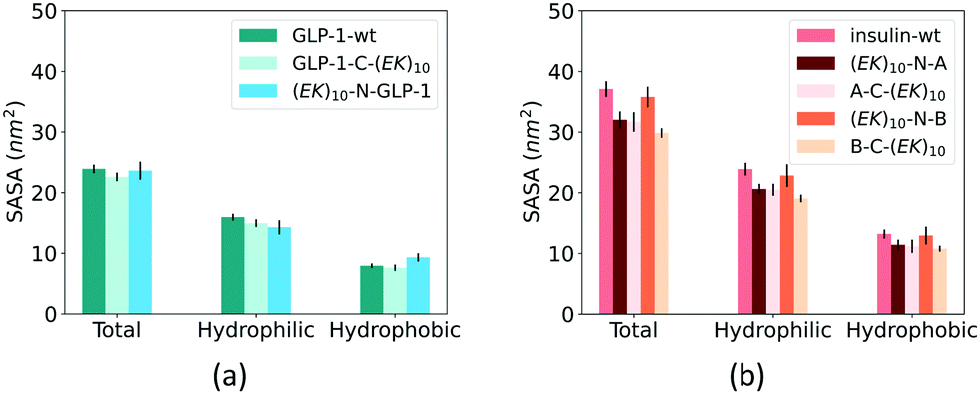 | ||
| Fig. 7 SASA of (a) GLP-1 and (b) insulin and their zwitterlated fusions. | ||
Zwitterlation only induces a small change in the surface area of the GLP-1 and insulin. Fig. 7a shows the SASA of the hydrophobic and hydrophilic domains and their sum for the GLP-1 with and without zwitterlation. The little difference between the SASAs illustrates that the hydrophobic and hydrophilic domains of the GLP-1 remain their surface area upon zwitterlation. Fig. 7b shows the SASA of the hydrophobic and hydrophilic domains, and their sum for the insulin protein with and without zwitterlation. Three zwitterlation cases show a decrease of SASA compared to that of the wild-type insulin: (EK)10-N-A, A-C-(EK)10 and B-C-(EK)10. The decrease is moderate and occurs in both hydrophobic and hydrophilic domains. The (EK)10-N-B case presents SASAs similar to those of the wild-type protein. The analysis of SASA indicates that zwitterionic peptide would not cover the surface of the wild-type GLP-1 and insulin. The zwitterlation may not induce any conformational changes leading to a significant variation in surface area. This consistency in SASA may help the polypeptide and protein keep its activity upon zwitterlation.
3.4. Secondary structure
At last, we analyzed the secondary structure of the GLP-1 and insulin with and without zwitterlation. Fig. 8 shows the number of amino acid residues in the five secondary structure categories for the GLP-1 and insulin domains in the six polypeptide and protein fusions and the two wild-type GLP-1 and insulin. The secondary structure is determined based on the hydrogen bond topology using the DSSP algorithm.49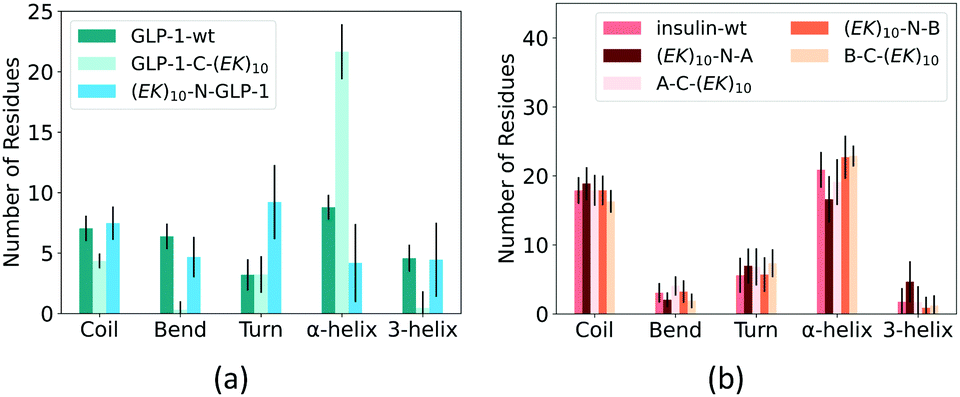 | ||
| Fig. 8 Percentage of secondary structure on (a) GLP-1 and (b) insulin and their zwitterlated fusions. | ||
Zwitterlation induces a significant change in the secondary structure of the GLP-1. Fig. 8a shows that the number of α-helix increases to around 20 when fusing an (EK)10 peptide to the C-terminal of the GLP-1, about twice as compared with the wild-type GLP-1. Meanwhile, the numbers of 3-helix and Bend decrease to almost zero and Coil slightly decreases to 4. The number of the Turn structure remains unchanged. The N-terminal fusing (EK)10 peptide shows an opposite effect on the number of amino acid residues taking the α-helix structure. As shown in Fig. 8a, the number of α-helix residues decreases to 4 for (EK)10-GLP-1. Meanwhile, the number of the Turn structure increase from 4 to 9 and the other secondary structures remain at the same level as the wild-type GLP-1 for the (EK)10-GLP-1 fusion.
Zwitterlation induces a moderate change in the secondary structure of the insulin. Fig. 8b shows that fusing an (EK)10 peptide to the N terminal of the insulin protein induces the number of the α-helix structure to decrease from 21 to 17 and the 3-helix increases from 2 to 5. The other three fusions present a small change in secondary structure distribution.
We then investigated the detailed secondary structure changes of individual residues during the final 200 ns. Fig. 9 shows the secondary structure of individual amino acid residues as a function of time for the GLP-1-wt and the two zwitterlated fusions. Indeed, the (EK)10 changes most of the secondary structure on the GLP-1 domain to the α-helix structure when attached to the C-terminal of the GLP-1. For instance, the GLP-1-C-(EK)10 fusion presents two major α-helix domains: RES5-19 and RES24-29, while the wild-type GLP-1 only presents one major α-helix domain: RES10-19. The C-terminal zwitterlation expands the original α-helix domain and induces the formation of a new α-helix domain.
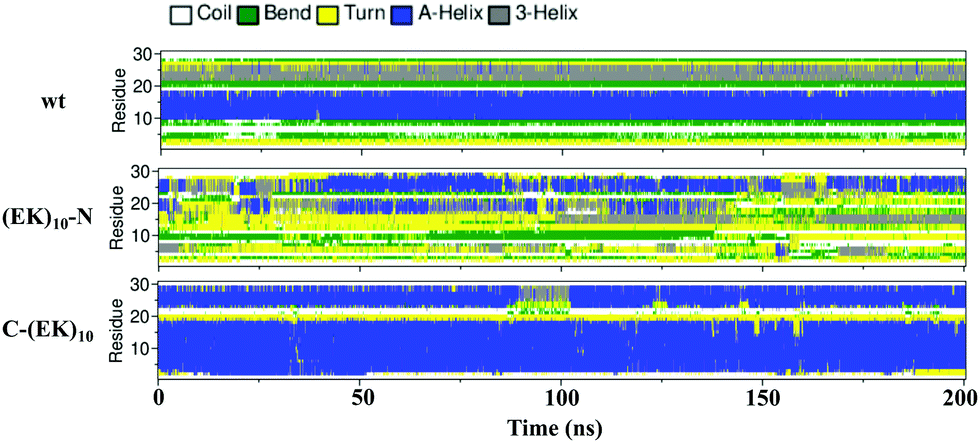 | ||
| Fig. 9 The secondary structure of individual residues as a function of time for the wild-type GLP-1 and its zwitterlated fusions. | ||
The N-terminal zwitterlation decreases the number of residues with the α-helix structures and alters the position of the α-helix domain. As shown in Fig. 9, the (EK)10-N-GLP-1 possess an α-helix domain range in RES19-26, different from the location of the α-helix domain on the wild-type GLP-1 (RES10-19). The RES10-19 domain mostly shows the Turn structure. Only a few α-helix structures are observed during the simulation.
Zwitterlation shows a moderate effect on the secondary structure of individual residues on the insulin protein. Fig. 10 shows the secondary structure of individual amino acid residues as a function of time for the insulin-wt and the two (EK)10-N-A and B-C-(EK)10 zwitterlated fusions in the final 200 ns. The data for the other two fusions are shown in Fig. S4 (ESI†). As shown in Fig. 10, the N-terminal zwitterlation on Chain A shifts RES1-9 from α-helix dominating to Turn dominating. The rest of the residues of (EK)10-N-A retain their main secondary structures during the simulation. Such a conformational change may be due to the direct effect of the zwitterionic peptide because the zwitterlation location is close to these residues. However, some zwitterlation induced conformational change may be distant from its location. For instance, the B-C-(EK)10 case shows more Bend/Turn around RES9-10 compared with the wild-type protein. The other amino acids maintain similar secondary structures compared with the wild-type insulin.
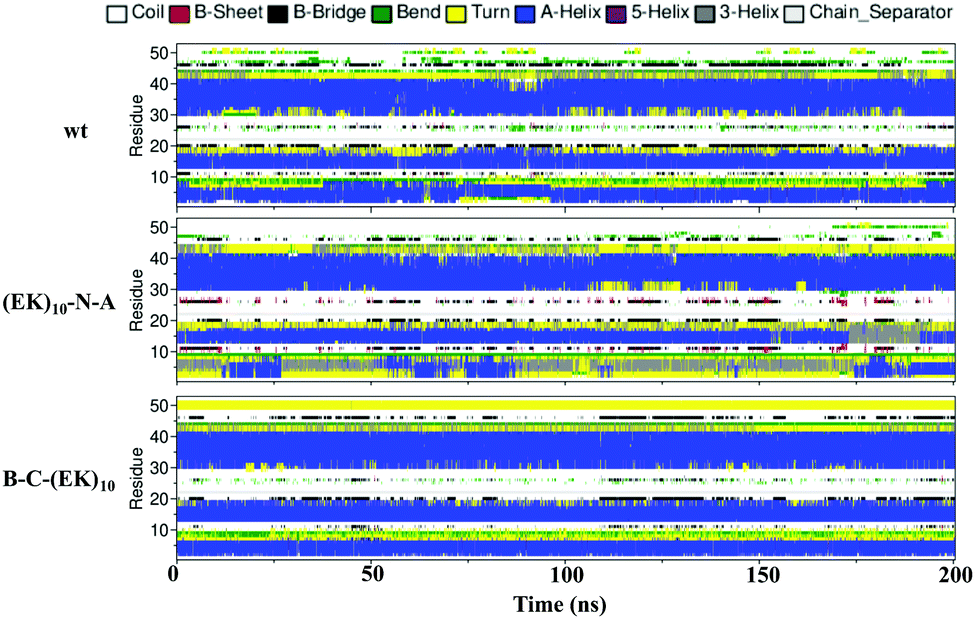 | ||
| Fig. 10 The secondary structure of individual residues as a function of time for the wild-type insulin and the (EK)10-N-A and B-C-(EK)10 protein-peptide fusions. | ||
The distinct zwitterlation-induced secondary structure variations imply a comprehensive relationship between zwitterlation, polypeptide and protein stability and functions. The above simulation data have shown that the GLP-1 and insulin remain their folded structure upon zwitterlation. The folded structures are characterized by the number of amino acid residues in the ordered secondary structure and the surface area. However, zwitterlation could shift the preference of the secondary structure for individual residues. The most significant example is the C-terminal zwitterlation induce a full α-helix structure for the GLP-1. Such shift in secondary structure could affect the biological fate of the polypeptide and proteins in multiple aspects: such as stability, binding affinity and reactivity. Thus, the ability of zwitterlation to induce the variation in secondary structure suggests that it can be used as a more profound technology than just improving the protein stability.
4. Conclusion
This work investigates the effect of fusing a zwitterionic peptide on the structural and dynamic properties of two small polypeptide and protein, GLP-1 and insulin using MD simulations. We analyzed the root mean square deviation and root mean square fluctuation of Cα atoms, solvent accessible surface area of the polypeptide and protein surface and secondary structure of individual residues of the wild-type GLP-1 and insulin and their six zwitterlated fusions. The simulation results show that zwitterlation induces a significant change in the conformation of GLP-1. Fusing an (EK)10 peptide to the C-terminal causes the GLP-1 to take a full α-helix structure. The insulin also presents some moderate changes in conformation when fused with the zwitterionic peptide. These simulation results imply that zwitterlation may enhance the stability of a protein but alter its preferred conformation. Such a comprehensive zwitterlation-conformation relationship would challenge the design of zwitterionic peptides suitable to protect specific proteins but also open a gate for designing zwitterionic peptides that can regulate the activity of proteins in a complex environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the Startup Funds of the University of Kentucky. We would also like to thank the University of Kentucky Center for Computational Sciences and Information Technology Services Research Computing for their support and use of the Lipscomb Compute Cluster and associated research computing resources.Notes and references
- J. B. Schlenoff, Zwitteration: coating Surfaces with Zwitterionic Functionality to Reduce Nonspecific Adsorption, Langmuir, 2014, 30(32), 9625–9636 CrossRef CAS PubMed.
- Q. Shao, Effect of conjugated (EK) 10 peptide on structural and dynamic properties of ubiquitin protein: a molecular dynamics simulation study, J. Mater. Chem. B, 2020, 8(31), 6934–6943 RSC.
- B. Kaupbayeva and A. J. Russell, Polymer-enhanced biomacromolecules, Prog. Polym. Sci., 2020, 101, 101194 CrossRef CAS.
- A. Munasinghe, S. L. Baker, P. Lin, A. J. Russell and C. M. Colina, Structure-function-dynamics of α-chymotrypsin based conjugates as a function of polymer charge, Soft Matter, 2020, 16(2), 456–465 RSC.
- Y. Han, Z. Yuan, P. Zhang and S. Jiang, Zwitterlation mitigates protein bioactivity loss in vitro over PEGylation, Chem. Sci., 2018, 9(45), 8561–8566 RSC.
- A. J. Keefe and S. Jiang, Poly (zwitterionic) protein conjugates offer increased stability without sacrificing binding affinity or bioactivity, Nat. Chem., 2012, 4(1), 59–63 CrossRef CAS PubMed.
- P. Zhang, P. Jain and C. Tsao, et al., Polypeptides with high zwitterion density for safe and effective therapeutics, Angew. Chem., 2018, 130(26), 7869–7873 CrossRef.
- Q. Shao and S. Jiang, Molecular understanding and design of zwitterionic materials, Adv. Mater., 2015, 27(1), 15–26 CrossRef CAS PubMed.
- S. Jiang and Z. Cao, Ultralow-fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications, Adv. Mater., 2010, 22(9), 920–932 CrossRef CAS PubMed.
- L. D. Blackman, P. A. Gunatillake, P. Cass and K. E. Locock, An introduction to zwitterionic polymer behavior and applications in solution and at surfaces, Chem. Soc. Rev., 2019, 48(3), 757–770 RSC.
- A. Wu, Y. Gao and L. Zheng, Zwitterionic amphiphiles: their aggregation behavior and applications, Green Chem., 2019, 21(16), 4290–4312 RSC.
- S. Chen, L. Li, C. Zhao and J. Zheng, Surface hydration: principles and applications toward low-fouling/nonfouling biomaterials, Polymer, 2010, 51(23), 5283–5293 CrossRef CAS.
- C. Leng, H.-C. Hung and S. Sun, et al., Probing the surface hydration of nonfouling zwitterionic and PEG materials in contact with proteins, ACS Appl. Mater. Interfaces, 2015, 7(30), 16881–16888 CrossRef CAS PubMed.
- C. Leng, S. Sun, K. Zhang, S. Jiang and Z. Chen, Molecular level studies on interfacial hydration of zwitterionic and other antifouling polymers in situ, Acta Biomater., 2016, 40, 6–15 CrossRef CAS PubMed.
- H. Huang, C. Zhang and R. Crisci, et al., Strong Surface Hydration and Salt Resistant Mechanism of a New Nonfouling Zwitterionic Polymer Based on Protein Stabilizer TMAO, J. Am. Chem. Soc., 2021, 143(40), 16786–16795 CrossRef CAS PubMed.
- C. Leng, X. Han and Q. Shao, et al., In situ probing of the surface hydration of zwitterionic polymer brushes: structural and environmental effects, J. Mater. Chem. C, 2014, 118(29), 15840–15845 CAS.
- C. A. Del Grosso, C. Leng and K. Zhang, et al., Surface hydration for antifouling and bio-adhesion, Chem. Sci., 2020, 11(38), 10367–10377 RSC.
- D. W. Romanini and M. B. Francis, Attachment of peptide building blocks to proteins through tyrosine bioconjugation, Bioconjugate Chem., 2008, 19(1), 153–157 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Advances in Chemical Protein Modification, Chem. Rev., 2015, 115(5), 2174–2195 CrossRef CAS PubMed.
- E. J. Liu, A. Sinclair and A. J. Keefe, et al., EKylation: addition of an Alternating-Charge Peptide Stabilizes Proteins, Biomacromolecules, 2015, 16(10), 3357–3361 CrossRef CAS PubMed.
- B. Li, Y. Wu and W. Zhang, et al., Efficient synthesis of amino acid polymers for protein stabilization, Biomater. Sci., 2019, 7(9), 3675–3682 RSC.
- Y. Hou and H. Lu, Protein pepylation: a new paradigm of protein–polymer conjugation, Bioconjugate Chem., 2019, 30(6), 1604–1616 CrossRef CAS PubMed.
- J. Teng, Y. Liu, Z. Shen, W. Lv and Y. Chen, Molecular simulation of zwitterionic polypeptides on protecting glucagon-like peptide-1 (GLP-1), Int. J. Biol. Macromol., 2021, 174, 519–526 CrossRef CAS PubMed.
- S. Banskota, P. Yousefpour, N. Kirmani, X. Li and A. Chilkoti, Long circulating genetically encoded intrinsically disordered zwitterionic polypeptides for drug delivery, Biomaterials, 2019, 192, 475–485 CrossRef CAS PubMed.
- M. He, K. Gao and L. Zhou, et al., Zwitterionic materials for antifouling membrane surface construction, Acta Biomater., 2016, 40, 142–152 CrossRef CAS PubMed.
- Y. Zhang, Y. Liu and B. Ren, et al., Fundamentals and applications of zwitterionic antifouling polymers, J. Phys. D: Appl. Phys., 2019, 52(40), 403001 CrossRef CAS.
- S. K. Lau and W. F. Yong, Recent Progress of Zwitterionic Materials as Antifouling Membranes for Ultrafiltration, Nanofiltration, and Reverse Osmosis, ACS Appl. Polym. Mater., 2021, 3(9), 4390–4412 CrossRef CAS.
- S. Jiang and Z. Cao, Ultralow-Fouling, Functionalizable, and Hydrolyzable Zwitterionic Materials and Their Derivatives for Biological Applications, Adv. Mater., 2010, 22(9), 920–932 CrossRef CAS PubMed.
- L. Mi and S. Jiang, Integrated Antimicrobial and Nonfouling Zwitterionic Polymers, Angew. Chem., Int. Ed., 2014, 53(7), 1746–1754 CrossRef CAS PubMed.
- H. Ye, L. Wang and R. Huang, et al., Superior antifouling performance of a zwitterionic peptide compared to an amphiphilic, non-ionic peptide, ACS Appl. Mater. Interfaces, 2015, 7(40), 22448–22457 CrossRef CAS PubMed.
- C. Li, C. Liu and M. Li, et al., Structures and Antifouling Properties of Self-Assembled Zwitterionic Peptide Monolayers: effects of Peptide Charge Distributions and Divalent Cations, Biomacromolecules, 2020, 21(6), 2087–2095 CrossRef CAS PubMed.
- J. A. Walker, K. J. Robinson and C. Munro, et al., Antibody-Binding, Antifouling Surface Coatings Based on Recombinant Expression of Zwitterionic EK Peptides, Langmuir, 2019, 35(5), 1266–1272 CrossRef CAS PubMed.
- F. M. Veronese, Peptide and protein PEGylation: a review of problems and solutions, Biomaterials, 2001, 22(5), 405–417 CrossRef CAS PubMed.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, PEGylation of biopharmaceuticals: a review of chemistry and nonclinical safety information of approved drugs, J. Pharm. Sci., 2016, 105(2), 460–475 CrossRef CAS PubMed.
- S. Jevševar, M. Kunstelj and V. G. Porekar, PEGylation of therapeutic proteins, Biotechnol. J., 2010, 5(1), 113–128 CrossRef PubMed.
- N. Nischan and C. P. Hackenberger, Site-specific PEGylation of proteins: recent developments, J. Org. Chem., 2014, 79(22), 10727–10733 CrossRef CAS PubMed.
- D. Xu, N. Smolin and R. K. Shaw, et al., Molecular insights into the improved clinical performance of PEGylated interferon therapeutics: a molecular dynamics perspective, RSC Adv., 2018, 8(5), 2315–2322 RSC.
- A. Zaghmi, E. Mendez-Villuendas, A. Greschner, J. Liu, H. de Haan and M. Gauthier, Mechanisms of activity loss for a multi-PEGylated protein by experiment and simulation, Mater. Today Chem., 2019, 12, 121–131 CrossRef CAS.
- E. M. Pelegri-O’Day, E.-W. Lin and H. D. Maynard, Therapeutic protein–polymer conjugates: advancing beyond PEGylation, J. Am. Chem. Soc., 2014, 136(41), 14323–14332 CrossRef PubMed.
- Y. Qi and A. Chilkoti, Protein–polymer conjugation—moving beyond PEGylation, Curr. Opin. Chem. Biol., 2015, 28, 181–193 CrossRef CAS PubMed.
- X. Liu, J. Sun and W. Gao, Site-selective protein modification with polymers for advanced biomedical applications, Biomaterials, 2018, 178, 413–434 CrossRef CAS PubMed.
- R. Moretti, S. Lyskov, R. Das, J. Meiler and J. J. Gray, Web-accessible molecular modeling with Rosetta: the Rosetta Online Server that Includes Everyone (ROSIE), Protein Sci., 2018, 27(1), 259–268 CrossRef CAS PubMed.
- B. Manandhar and J.-M. Ahn, Glucagon-like peptide-1 (GLP-1) analogs: recent advances, new possibilities, and therapeutic implications, J. Med. Chem., 2015, 58(3), 1020–1037 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81(8), 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126(1), 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: a new molecular dynamics method, J. Appl. Phys., 1981, 52(12), 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: AnN·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- W. Kabsch and C. Sander, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features, Biopolymers, 1983, 22(12), 2577–2637 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02561a |
| ‡ The two authors contribute equally to this research. |
| This journal is © The Royal Society of Chemistry 2022 |