
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeting extracellular lectins of Pseudomonas aeruginosa with glycomimetic liposomes†
Olga
Metelkina
abc,
Benedikt
Huck
de,
Jonathan S.
O'Connor
fg,
Marcus
Koch
h,
Andreas
Manz
 fg,
Claus-Michael
Lehr
de and
Alexander
Titz
*abc
fg,
Claus-Michael
Lehr
de and
Alexander
Titz
*abc
aChemical Biology of Carbohydrates, Helmholtz Institute for Pharmaceutical Research Saarland, Helmholtz Centre for Infection Research, 66123 Saarbrücken, Germany. E-mail: alexander.titz@helmholtz-hzi.de
bDeutsches Zentrum für Infektionsforschung (DZIF), Standort Hannover-Braunschweig, 38124 Braunschweig, Germany
cDepartment of Chemistry, Saarland University, 66123 Saarbrücken, Germany
dDrug Delivery, Helmholtz Institute for Pharmaceutical Research Saarland, Helmholtz Centre for Infection Research, 66123 Saarbrücken, Germany
eDepartment of Pharmacy, Saarland University, 66123 Saarbrücken, Germany
fKIST Europe, 66123 Saarbrücken, Germany
gDepartment of Systems Engineering, Saarland University, 66123 Saarbrücken, Germany
hINM – Leibniz Institute for New Materials, 66123 Saarbrücken, Germany
First published on 20th December 2021
Abstract
The antimicrobial resistance crisis requires novel approaches for the therapy of infections especially with Gram-negative pathogens. Pseudomonas aeruginosa is defined as priority 1 pathogen by the WHO and thus of particular interest. Its drug resistance is primarily associated with biofilm formation and essential constituents of its extracellular biofilm matrix are the two lectins, LecA and LecB. Here, we report microbial lectin-specific targeted nanovehicles based on liposomes. LecA- and LecB-targeted phospholipids were synthesized and used for the preparation of liposomes. These liposomes with varying surface ligand density were then analyzed for their competitive and direct lectin binding activity. We have further developed a microfluidic device that allowed the optical detection of the targeting process to the bacterial lectins. Our data showed that the targeted liposomes are specifically binding to their respective lectin and remain firmly attached to surfaces containing these lectins. This synthetic and biophysical study provides the basis for future application in targeted antibiotic delivery to overcome antimicrobial resistance.
Introduction
Resistance to antimicrobials is rising and expected to lead to more deaths from infections than from cancer in 2050 as stated by the WHO.1Pseudomonas aeruginosa are particularly problematic Gram-negative bacteria belonging to the ESKAPE pathogens, the major etiological agents of drug-resistant nosocomial infections. The main difficulty for treating P. aeruginosa infections is associated with its ability to form biofilms, a social lifestyle where the bacteria are embedded in a self-produced, resistance-conferring matrix.2 This biofilm increases antimicrobial resistance against antibiotics by a factor of 10–1000 and thereby renders these drugs ineffective.3 Therefore, the identification and development of new drugs and delivery strategies is a highly active research field.2The formulation of antibiotics as nanomedicines4–7 has been widely studied to overcome the different limitations of the free drugs, such as toxicity, solubility and bioavailability associated with sustained drug release,8 efficient mucus penetration9 and the ability of some formulations, e.g. liposomes, to fuse with the bacterial cell envelopes.10,11 Especially the liposomes have evolved as potent nanovehicles for antimicrobials.6 Arikayce (Insmed, Inc.), the liposomal formulation of amikacin, has been recently approved by the FDA for the treatment of Mycobacterium avium complex (MAC) lung infections.
Liposomes are hollow microscopic spherical lipid-based nanoparticles.12 They can be produced using various types of phospholipids to form lipid bilayers and supplementation with cholesterol changes fluidity and rigidity of the liposomal membrane. The advantage of liposomes responsible for their high popularity is the fact that diverse drugs covering a broad physicochemical property range can be encapsulated within liposomes. Relatively large amounts can be loaded into the inner sphere to overcome low drug solubility and provide stability under physiological conditions.
Moreover, the precise routing of a drug to the site of the disease can further improve the drug's availability at the diseased tissue and thereby increase pharmacodynamic efficacy. Thus, targeted drug delivery plays an important role in modern medicine for numerous diseases, such as tumors, infections and others. The increased local concentration of a targeted drug enables a decrease of total dosage and consequently a reduction of side effects.13,14
Targeting of therapeutic molecules generally follows two approaches: covalent conjugation of drug and targeting ligand15 or non-covalent drug encapsulation in surface-modified nanovehicles. The presence of a covalent bond between drug and targeting ligand has the advantage that fast dissociation and release of the untargeted drug is avoided. Careful conjugate design is important since the covalent modification of the drug can, on the other hand, also result in a loss of therapeutic activity. Prominent examples are antibody–drug conjugates used primarily in cancer and the recently approved cefiderocol, a cephalosporin antibiotic conjugated to a substrate for the bacterial cell uptake machinery. A comprehensive overview on conjugates of antimicrobials has recently been provided by Brönstrup and Klahn.16
Non-covalent drug encapsulation overcomes this hurdle since the targeting ligand is attached to the vehicle and the cargo remains unchanged. The widely used liposomes have another unique advantage due to their accessibility for chemical functionalization at the molecular level. Production of liposomes using varying ratios of targeted and untargeted lipids then allows the defined modification of the liposomal surface with targeting moieties.17 It was demonstrated that functionalization of the liposomal surface with carbohydrate moieties is a powerful approach allowing to target certain types of human cells, such as hepatocytes18,19 and various immune cells.20,21 Rademacher et al. reported the delivery of vaccines to dermal Langerhans cells using a targeted liposome decorated with a glycomimetic ligand for binding to their surface receptor Langerin.22 Additionally, the opportunity to use carbohydrate-decorated liposomes as vaccines against infections and cancer have been extensively studied in the last decade.23–26 The high potency and efficacy of the liposomes functionalized with carbohydrate moieties in inhibiting corresponding lectins is provided by their multivalent interaction with the proteins, similar to those observed for other multivalent systems.27 Rademacher et al. have further reported on heteromultivalent liposomes that enable efficient co-targeting of the same protein, i.e. DC-SIGN, through synergistic allosteric activation resulting in efficient binding to its carbohydrate recognition site.28
The targeting of antibiotics to biofilm-associated infections of P. aeruginosa is an active field of research. Extracellular and biofilm-associated proteins are the first potential targets encountered by an antimicrobial drug. Among many other biomolecules, P. aeruginosa expresses two extracellular lectins, LecA (PA-IL) and LecB (PA-IIL), that are crucial for establishing a mature biofilm structure.29,30 These proteins are carbohydrate-binding agents with LecA recognizing galactosides and LecB binding firmly to mannosides and fucosides. Both lectins are secreted homotetrameric proteins, a property which allows to efficiently bind and cross-link numerous glycoconjugates, e.g. host cell surface glycans, bacterial surface polysaccharides and the various exopolysaccharides secreted by P. aeruginosa. In addition to the biofilm-deficient phenotypes of the individual genetic LecA or LecB mutants, the localization of LecB towards the surface of the biofilm has been experimentally demonstrated.29–31 The resulting crosslinks between lectins and glycoconjugates are believed to be responsible for the stabilization of the biofilm matrix.32 We and others have therefore previously used these lectins as targets for antibiofilm agents.2,33–37 Our group has developed diverse glycomimetic inhibitors for LecA ranging from various galactosides to catechols38–40 and for LecB originating from modified mannosides35,41,42 and evolving into C-glycosidic sulfonamides.35,36,43 Furthermore, we have shown that a LecA-directed fluorescein-conjugate of a covalently binding epoxygalactoheptose moiety can be used to image bacterial biofilms in vitro.44
These results and the current lack of new antibiotics encouraged us to further develop these glycomimetics into targeting moieties for antibiotic delivery. In a first work reported in 2020, we have covalently attached either galactosides targeting LecA or C-fucosides targeting LecB to the widely used antibiotic ciprofloxacin.33 While we showed that these conjugates bind to the respective lectins, retain activity on their target bacterial gyrase, and are enriched at a biofilm compared to the parent antibiotic, their antibacterial activity has been reduced. This reduction is a consequence of the covalent modification of the drug that results in a decreased cellular uptake, and thus a reduced availability of the drug.
Here, we circumvented the covalent modification of the drug to be routed to the site of infection by developing new shuttles based on LecA- and LecB-targeted liposomes. These liposomes possess several copies of the targeting ligands on their surfaces which results in a multivalent presentation of these lectin ligands, leading to an increased affinity to their respective lectins.
Conventional methods to investigate the interactions between lectins and carbohydrates usually include fluorescence polarization assay (FP), enzyme-linked lectin assay (ELLA) and isothermal titration calorimetry (ITC).27 Although all these methods demonstrated their reliability for quantifying the affinity of monovalent ligands, the possible aggregation of multivalent lectins in presence of multivalent ligands may affect the assay readout and therefore such results should be carefully interpreted. On the other hand, surface plasmon resonance (SPR) avoids aggregation and precipitation, but data evaluation and quantification can be cumbersome as a result of the various interaction models.45
Microfluidics has been an important tool for the development of in vitro systems surrounding biofilm formation, analysis and drug treatments, especially for those concerning the circulatory system.46–48 The majority of the human circulatory system presents a laminar flow at a relaxed heart rate, in accordance to the low Reynolds number resulting from the small diameters of blood vessels.49 Medium sized arteries range from 0.8 mm to 1.8 mm in diameter and experience a blood flow rate of 3 mL min−1 at a relaxed heart rate, whilst corresponding veins experience 1.2 mL min−1 at a relaxed heart rate.50 Such diameters and flow rates compute to Reynolds numbers that should only reach around 2000 during mild exercise, which represents the transitional phase in a perfect undeformed channel. As such, microfluidic systems are ideal for mimicking the laminar flow within blood vessels at flow rates similar to a relaxed heart rate.51,52
We therefore developed a microfluidic system as a reductionistic model for a biofilm infection and intravenous liposome administration. We demonstrated that the targeted liposomes are specifically retained on a lectin-coated surface from a passing liquid stream. The loading of unmodified antibiotics into these targeted liposomes will further avoid the previously observed reduction of their pharmacodynamic effects, which was induced by the previous covalent conjugation.
Results and discussion
Synthesis of phospholipid–lectin ligand conjugates
Phospholipids are the main building blocks of liposomes. To obtain lectin-targeted phospholipids, we developed a synthetic route for the covalent conjugation of phospholipids with lectin ligands. Here, the two designed lectin-targeting groups were based on our previous reports on lectin inhibitors:35,36,39 the LecA-targeted ligand 8 (Scheme 1) consists of a β-linked galactoside carrying an aromatic aglycon and a thioglycosidic linkage for increased stability, while the LecB targeting ligand 15 (Scheme 2) is based on a hybrid-molecule between the two LecB ligands, mannose and fucose, and is further modified with a phenylsulfonamide substituent. For both cases, the aryl moiety is substituted in its para-position where the phospholipid can later be attached via amide bond formation (Scheme 3).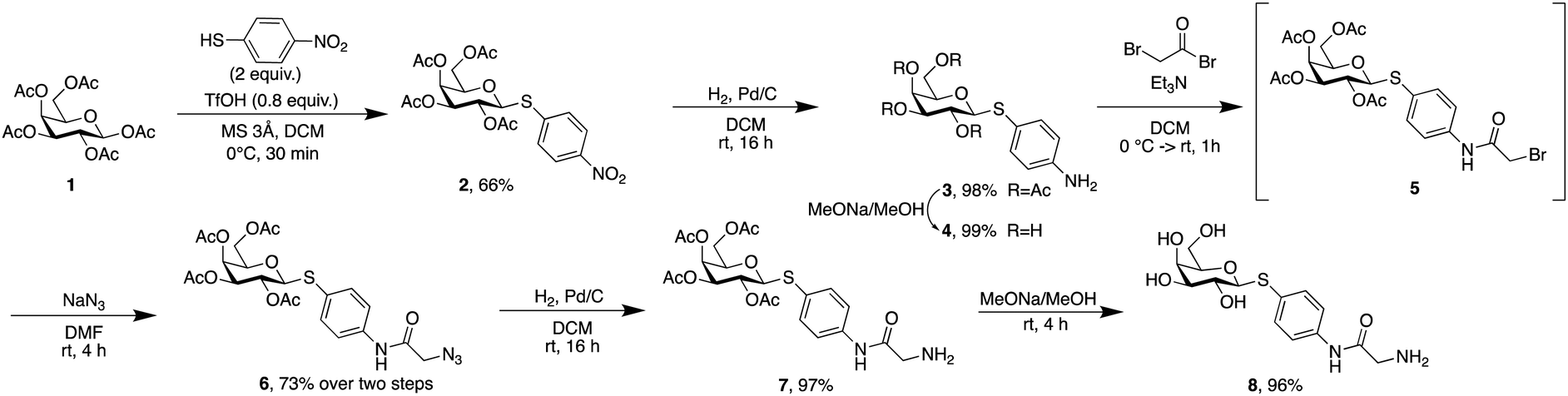 | ||
| Scheme 1 Synthesis of the LecA-targeting ligand thiogalactoside 8. | ||
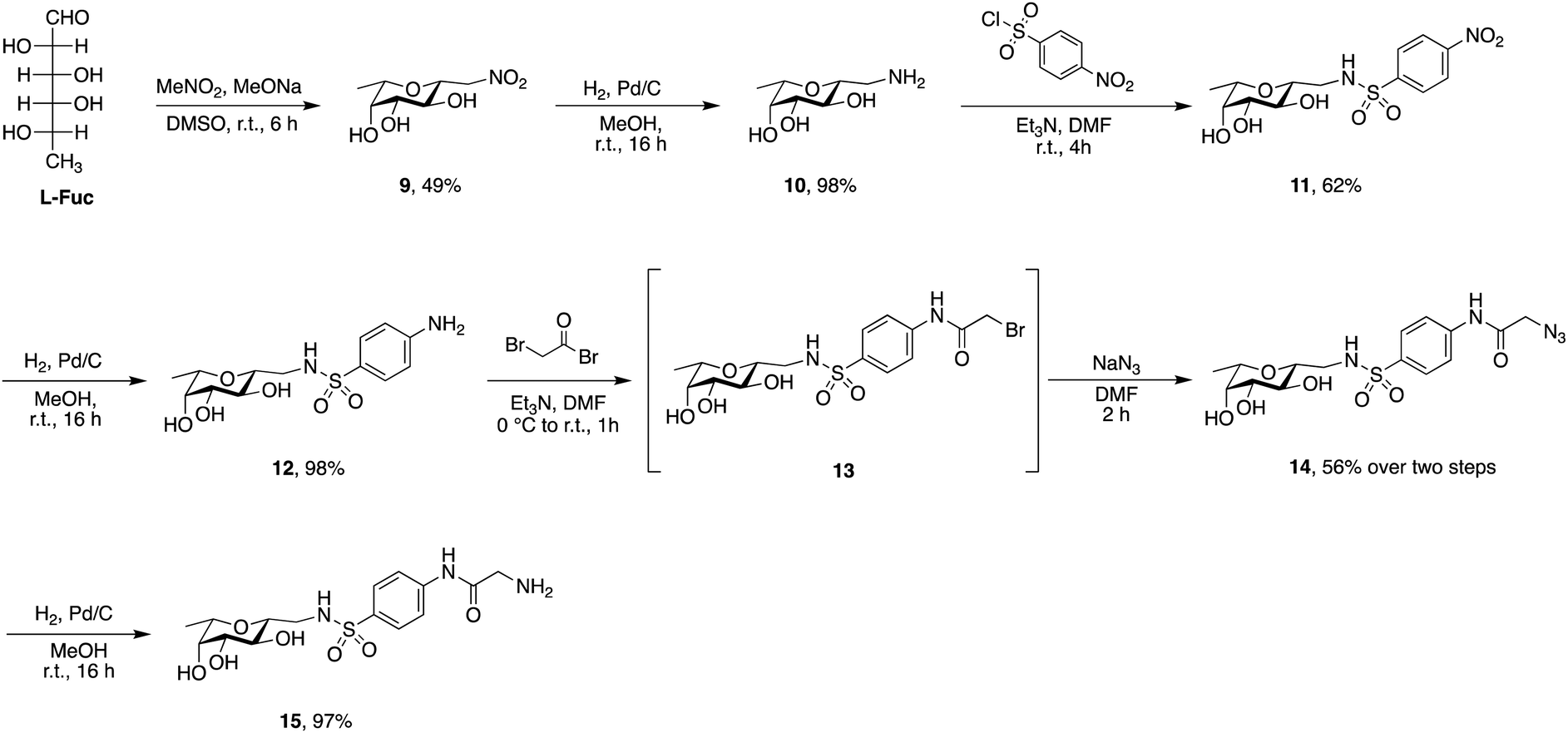 | ||
| Scheme 2 Synthesis of the LecB-targeting ligand C-fucoside 15. | ||
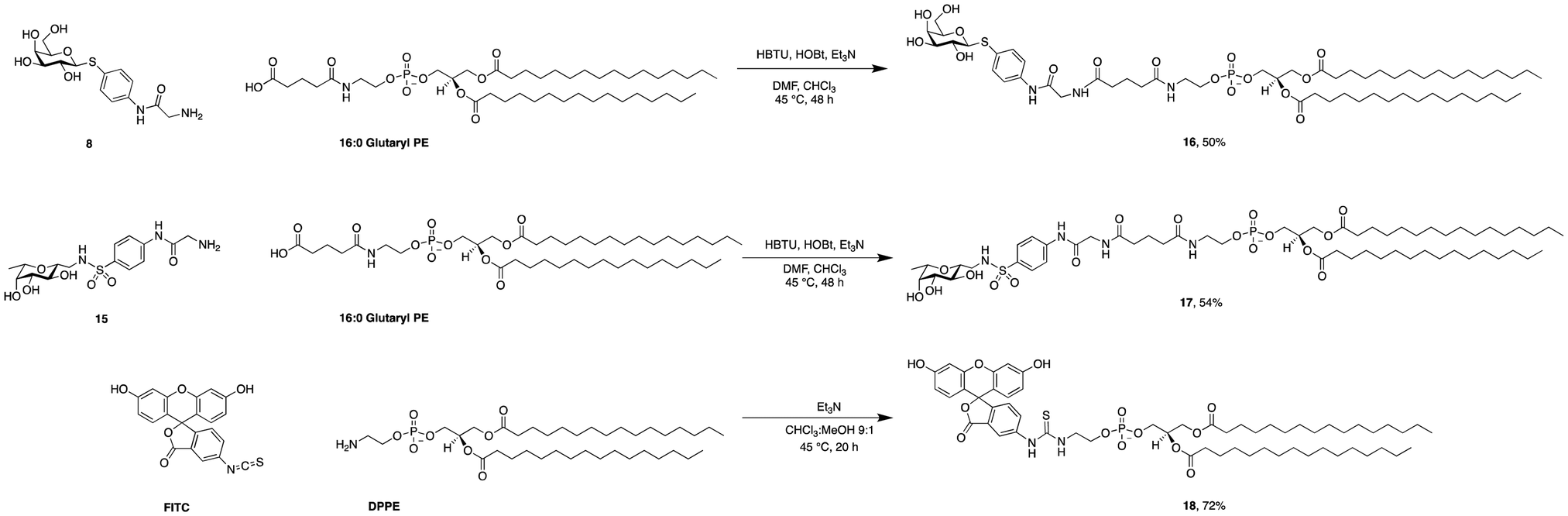 | ||
| Scheme 3 Synthesis of phospholipid conjugates: lectin-targeted lipids 16 and 17 and fluorescein-labelled lipid 18. | ||
For the synthesis of the LecA–ligand 8 (Scheme 1) β-D-galactose pentaacetate was reacted with 4-nitrothiophenol in a triflic acid-mediated glycosylation in 66% yield. The nitro group in 2 was then reduced using Pd/C and hydrogen to give aniline 3 quantitatively. Bromoacetylation of the per-O-acetylated aniline, followed by nucleophilic substitution of the bromide with sodium azide gave 6 in 73% yield over two steps. The latter compound was then hydrogenolytically reduced to amine 7 which was finally deacetylated under Zemplén conditions to provide LecA–ligand 8 in near quantitative yields.
Sulfonamide derivatives of C-fucosides demonstrate high affinity to LecB35,36 and they were therefore chosen as suitable ligands for targeting liposomes. Starting from L-fucose, the C-glycoside was installed via a Henry43,53 reaction with nitromethane to give 9 in 49% yield (Scheme 2). Catalytic hydrogenation of the nitro group gave amine 10 which was then transformed into sulfonamide 11 with 4-nitrophenylsulfonyl chloride and triethylamine. A second hydrogenation step yielded aniline 12 which was directly bromoacetylated. Crude 13 was subjected to a nucleophilic substitution with sodium azide to give C-fucosylated azide 14 in 56% yield over two steps. A third catalytic hydrogenation yielded amine 15 quantitatively.
Then, both carbohydrate-based targeting ligands, 8 and 15, were coupled to phospholipids in order to graft the targeting ligands onto the surfaces of the liposomes. The chosen lipid for conjugation was 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 PE) which was modified as an amide with glutaric acid. This 16:0 glutaryl phosphoethanolamine (16:0 glutaryl PE) offers one carboxylic acid available for conjugation with the targeting ligands. The amide bond between this carboxylic acid and the amine of the corresponding glycomimetics 8 and 15 was established using HBTU/HOBt to give conjugates 16 and 17. Optimization of the reaction conditions revealed a mixture of chloroform and dimethylformamide (ratio 2:1) as an optimal solvent for this reaction. The use of HOBt is crucial for the success of the reaction since the intermediate active ester of the phospholipid ensures complete solubility. Purification of 16 and 17 was successful when performed under reverse phase chromatography using a C18-column and a gradient of the solvents isopropanol/water/MeOH (ratio 5:4:1) and isopropanol, both supplemented with 0.2% formic acid. Furthermore, fluorescein isothiocyanate was conjugated with 16:0 PE in compound 18 as a tool to produce fluorescently labelled liposomes.
:0 PE) which was modified as an amide with glutaric acid. This 16:0 glutaryl phosphoethanolamine (16:0 glutaryl PE) offers one carboxylic acid available for conjugation with the targeting ligands. The amide bond between this carboxylic acid and the amine of the corresponding glycomimetics 8 and 15 was established using HBTU/HOBt to give conjugates 16 and 17. Optimization of the reaction conditions revealed a mixture of chloroform and dimethylformamide (ratio 2:1) as an optimal solvent for this reaction. The use of HOBt is crucial for the success of the reaction since the intermediate active ester of the phospholipid ensures complete solubility. Purification of 16 and 17 was successful when performed under reverse phase chromatography using a C18-column and a gradient of the solvents isopropanol/water/MeOH (ratio 5:4:1) and isopropanol, both supplemented with 0.2% formic acid. Furthermore, fluorescein isothiocyanate was conjugated with 16:0 PE in compound 18 as a tool to produce fluorescently labelled liposomes.
Preparation and characterization of lectin-targeted liposomes
With the various lipids in hand, an array of liposomes was produced using DSPC/cholesterol and various ratios of targeting ligands 16 and 17 and incorporation of fluorescein-containing lipid 18 (Table 1). All liposomes were prepared by mixing the corresponding lipids at the indicated ratios followed by their self-assembly via thin-film hydration and extrusion. In general, the sizes of all liposomes were comparable at approx. 200 nm (range 176–208 nm) (Fig. S1, ESI†) with a rather low polydispersity index ranging from 0.077 to 0.158. All liposomes showed a negative zeta potential in the range of −20 to −52 mV.| Type | Formulation | Mass ratio | Size (nm) | PdI | Zeta (mV) | Particle concentration (particles per mL) |
|---|---|---|---|---|---|---|
| Plain | DSPC:Chol |
2:1 |
207 ± 4 | 0.133 ± 0.024 | −20 ± 0.5 | |
| Plain, fluorescent | DSPC:18:Chol |
2:0.02:1 |
208 ± 2 | 0.077 ± 0.018 | −43 ± 0.2 | 1.55 × 108 ± 7.90 × 106 |
| LecA-targeted, 1% 16 | DSPC:16:Chol |
1.98:0.02:1 |
203 ± 3 | 0.158 ± 0.008 | −29 ± 1 | |
| LecA-targeted, 15% 16 | DSPC:16:Chol |
1.7:0.3:1 |
194 ± 6 | 0.149 ± 0.011 | −51 ± 2 | |
| LecA-targeted, 1% 16, fluorescent | DSPC:16:18:Chol |
1.96:0.02:0.02:1 |
182 ± 2 | 0.149 ± 0.015 | −32 ± 0.2 | |
| LecA-targeted, 15% 16, fluorescent | DSPC:16:18:Chol |
1.68:0.3:0.02:1 |
186 ± 2 | 0.097 ± 0.044 | −46 ± 2 | 7.35 × 108 ± 4.38 × 107 |
| LecB-targeted, 1% 17 | DSPC:17:Chol |
1.98:0.02:1 |
202 ± 2 | 0.131 ± 0.022 | −31 ± 2 | |
| LecB-targeted, 15% 17 | DSPC:17:Chol |
1.7:0.3:1 |
208 ± 1 | 0.102 ± 0.004 | −49 ± 3 | |
| LecB-targeted, 1% 17, fluorescent | DSPC:17:18:Chol |
1.96:0.02:0.02:1 |
176 ± 1 | 0.111 ± 0.02 | −40 ± 2 | |
| LecB-targeted, 15% 17, fluorescent | DSPC:17:18:Chol |
1.68:0.3:0.02:1 |
204 ± 1 | 0.099 ± 0.035 | −52 ± 1 | 5.65 × 108 ± 7.60 × 107 |
With the liposomes in hand, we first studied their binding to the lectins LecA or LecB in established competitive binding assays39–41 (Fig. 1). The specificity of the LecA-targeted liposomes carrying glycolipid 16 for LecA and the LecB-targeted liposomes carrying glycolipid 17 for LecB was established, untargeted plain liposomes were added at comparable DSPC/Chol concentrations and showed no sign of inhibition. Monovalent non-lipidated controls 4 and 12 have further been included for referencing the increase in affinity due to their multivalent presentation on the liposomal surface. Furthermore, the impact of the targeting ligand's surface density on target binding was also studied using liposomes with 1% or 15% (w/w) of the phospholipids carrying the lectin-targeted ligand. In all cases, a concentration-dependent inhibition curve was obtained for the targeting liposomes or the controls 4 and 12 with their corresponding lectins (Fig. 1).
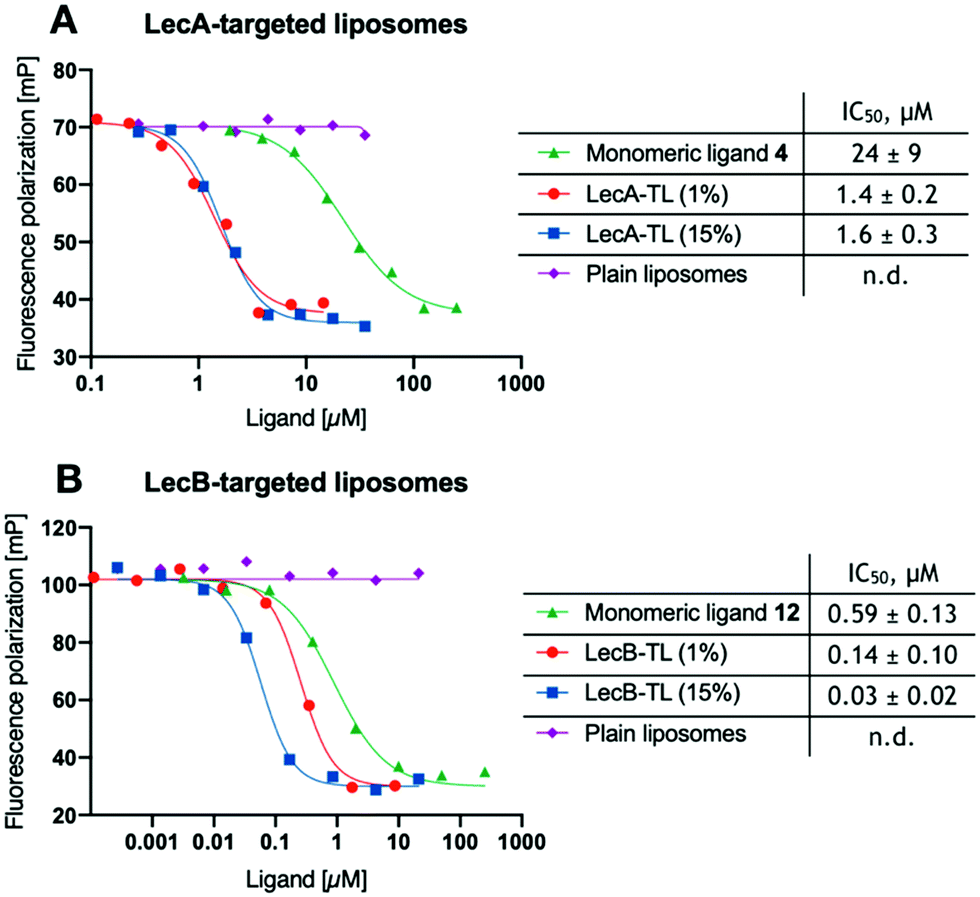 | ||
| Fig. 1 Competitive binding of lectin-targeted liposomes to LecA (A) or LecB (B). Plain liposomes do not contain targeting ligand and have therefore been used at total lipid concentrations comparable to the targeted liposomes (TL). IC50s for liposomes are normalized and depicted per targeting group, i.e. the affinity of a liposome carrying several copies of the targeting ligand is much higher. Averages and standard deviations from at least 3 independent experiments of technical triplicates each. | ||
To obtain comparable IC50 values after curve fitting, those values of the targeted liposomes were normalized to the molar concentration of lectin-targeted phospholipids 16 and 17. The concentration of the targeting ligands was quantified by LC-MS using a calibration curve for 16 and 17. The inhibition of both lectins with the targeted lipids displayed multivalently in the liposomes showed a significant boost compared to the free monomeric ligands 4 or 12. While the increase in relative potency was over 15-fold for LecA, it reached a nearly 20-fold increase for LecB. Surprisingly, varying the ligand density from 1% to 15% LecA-targeted lipid 16 did not change the molar inhibition, which could be an intrinsic trait of these liposomes or a result of having possibly reached the assay wall of these very potent multivalent systems. In contrast the increase of LecB-targeting lipid 17 from 1% to 15% in the resulting liposomes led to a further increase in normalized affinity.
The interaction of multivalent ligands with multivalent receptors often leads to oligomerization induced aggregation. In the present case, the targeted liposomes constitute a highly multivalent ligand system interacting with tetravalent lectins. To analyze the effect of aggregation the liposomal solutions were studied in presence of the corresponding lectins using dynamic light scattering (DLS) and with fluorescently labelled liposomes by nanoparticle tracking analysis (NTA) or fluorescence microscopy.
Liposomes containing 0, 1 or 15% LecA- or LecB-targeted phospholipids at a total lipid concentration of 800 μg mL−1 in aqueous buffer were incubated with LecA or LecB at varying concentrations (Fig. 2). Subsequently, these mixtures were diluted 1:10 with buffer and analyzed by DLS to determine Z-average size and polydispersity index (PdI) of the particle solution. The average size of plain liposomes did not change with increasing lectin concentration, while the observed average sizes of lectin-targeted liposomes increased from 200 nm to larger than 1 μm with increasing LecA concentrations as a consequence of multivalency-induced aggregation. Finally, aggregates became colloidally unstable and precipitated.
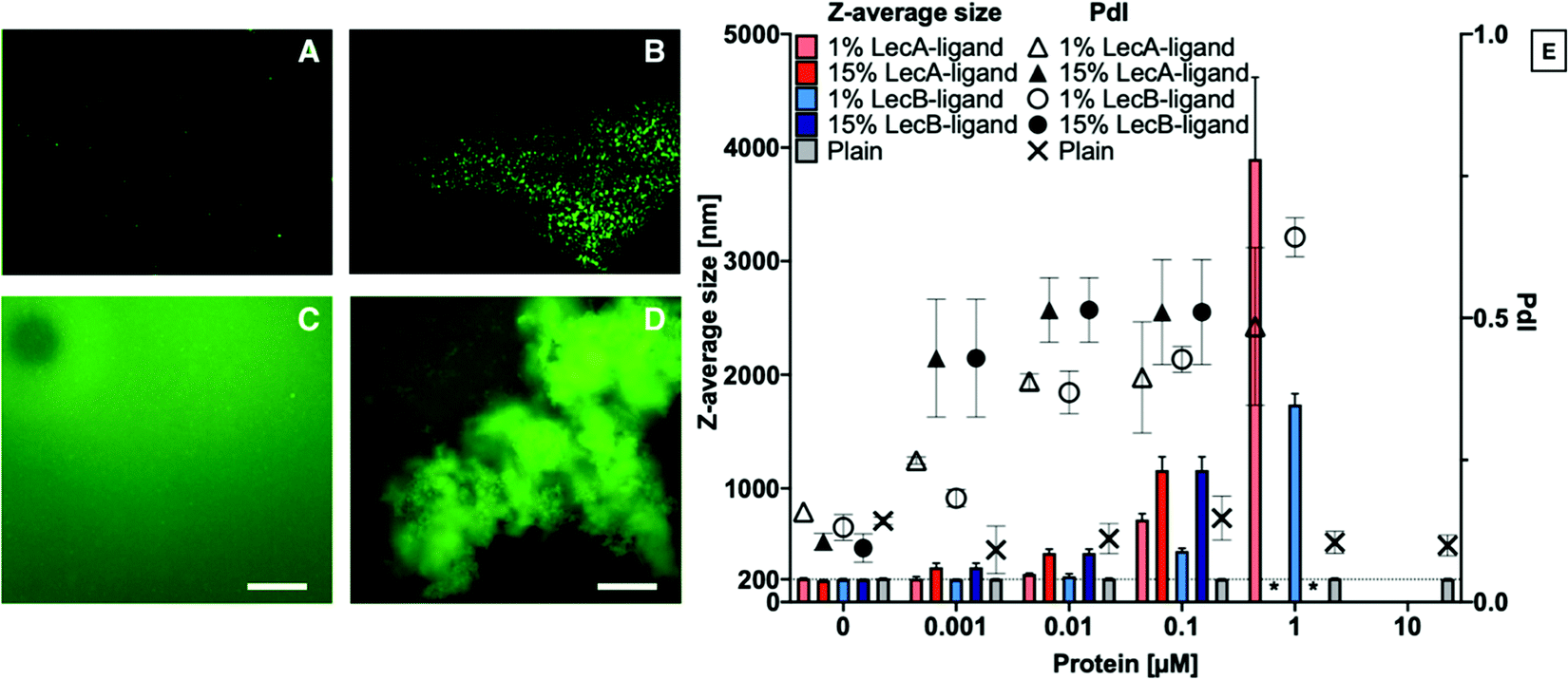 | ||
| Fig. 2 Lectin-targeted liposomes are specifically aggregated with lectins in solution. (A) Plain liposomes and (B) LecB-targeted liposomes preincubated with LecB for 30 min, protein to targeting ligand molar ratio of 2:1 – images taken with a NTA camera; (C) plain liposomes and (D) LecA targeted-liposomes incubated with LecA for 15 min – images were taken by fluorescence microscope; (E) correlation of Z-average size and polydispersity index (PdI) of LecA- or LecB-targeted and plain liposomes and the concentration of the corresponding lectin. Increase of the protein concentration led to liposomal aggregation. * – sample precipitated, in case of 10 μM lectin concentration all lectin-targeted samples precipitated. Averaged values from 2 independent experiments with 6–12 technical replicates, error bars correspond to standard deviation. For C and D: scale bar corresponds to 200 μm. | ||
For further analysis of the liposome/lectin interaction, we produced LecA-, LecB-, and untargeted liposomes spiked with 1% of the fluorescent lipid 18. The aggregation of targeted liposomes with their lectins in solution was studied using fluorescence detection methods, such as NTA and fluorescence microscopy. The clearly observable aggregation for the system LecA/LecA-targeted liposomes analyzed by DLS (Fig. 2E), could be confirmed using NTA analysis where no aggregation of the fluorescently spiked plain liposomes was observed whereas the LecA-targeted analogs showed aggregation (Fig. 2A and B). Due to the related tetrameric structure of LecB, a similar tendency of LecB-targeted liposomes to aggregate in presence of LecB was observed using fluorescence microscopy while plain liposomes remained stably dispersed and were not aggregated by either of the lectins (Fig. 2A and C–E).
The lectin-targeted liposomes are designed to attach to the biofilm of P. aeruginosa in the infected patient and release their antibiotic cargo at the site of the infection. Clearance mechanisms are enhanced by the dynamic flow of body fluids, e.g. in the vascular system. To analyze the successful targeting of liposomes under flow, we developed a model system where the lectins LecA or LecB have been immobilized onto an abiotic surface and the specific retention of the liposomes onto this surface was analyzed under flow conditions in vitro.
For this purpose, we engineered a polydimethylsiloxane (PDMS)–glass microfluidic device system that consisted of four flow cells upon the same hydrogel-coated support as shown in Fig. 3. 3D printing was implemented in the production of the devices to allow for rapid prototyping of different architectures of devices at a vastly reduced cost compared to other methods. Only the PDMS was plasma treated to avoid damaging the hydrogel prior to experimentation. This treatment resulted in weaker bonding between the glass and PDMS, which was mitigated by using epoxy sealant around the flow cell. We chose multiple cells on the same support to allow for simultaneous experiments and comparison between the cells without needing to compensate for hydrogel variation between different devices. Separation of the four cells also eliminated any opportunity of cross contamination between experiments and interfering illumination from neighbouring cells. PDMS was chosen due to its optical transparency (Fig. S3, ESI†) and chemical inertness to the liposomes and other capillary treatments within this work, thus removing the possibility of residual liposome adsorption hence eliminating background noise. Flow cells channel dimensions were designed to be approximately 5.75 × 1 × 0.5 mm (length × width × depth), with a corresponding volume of 2.82 μL (Fig. S2, ESI†). These dimensions were chosen to mimic medium to small arteries/veins found in vivo. Flow cells ruptured with flow rates above 2.4 mL min−1. Laminar flow was observed throughout all experimentation, with flow rates up to 500 μL min−1. Therefore; the fabricated devices were suitable for modeling flow through similar sized veins at low flow rates.
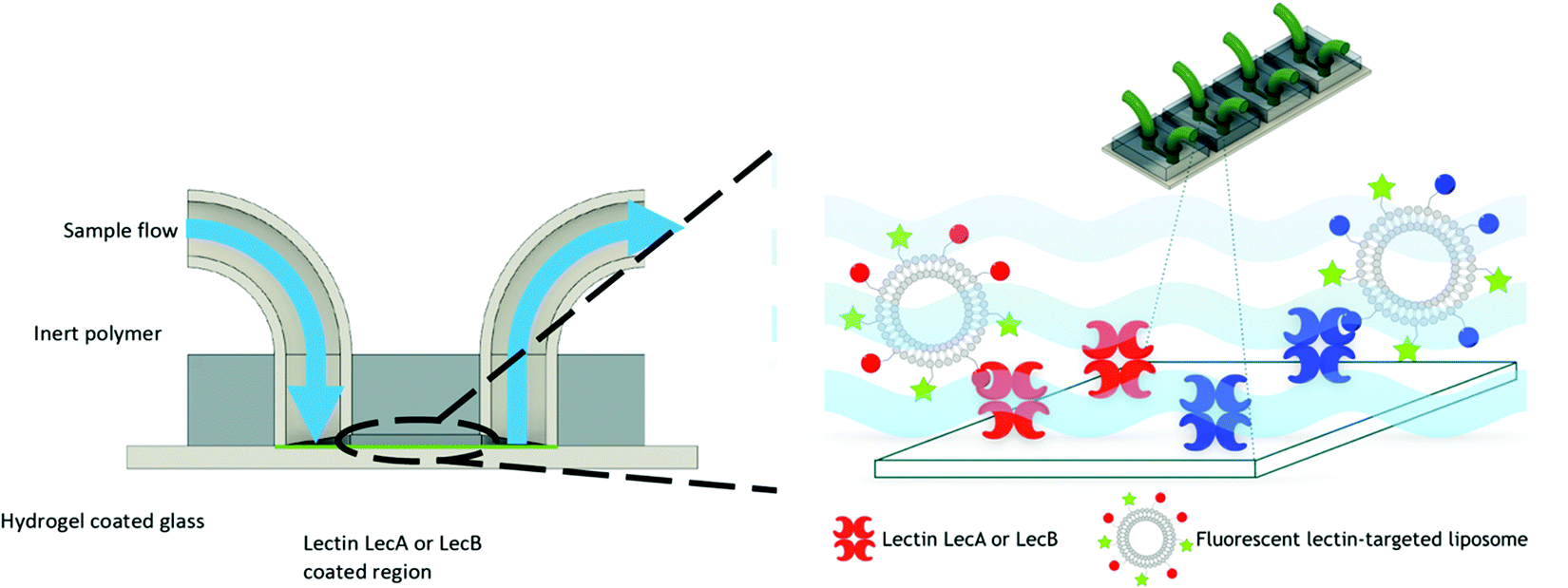 | ||
| Fig. 3 Concept of the in vitro microfluidic device. Fluorescent lectin-targeted liposomes interact with the proteins LecA or LecB that have been covalently immobilized on the glass surface. The liposomes are retained on the surface due to their specific ligand–lectin interaction and targeting was quantified by microscopy. | ||
The obtained flow channels were subsequently coated with the lectins LecA or LecB via covalent amide coupling to the N-hydroxysuccinimide ester activated glass slide surface. After thorough equilibration of the lectin-coated surfaces with buffer to remove blocking agent and unbound proteins, fluorescent LecA-, LecB- or untargeted liposomes were injected and the flow cell was imaged by confocal fluorescence microscopy (Fig. 4 and 5).
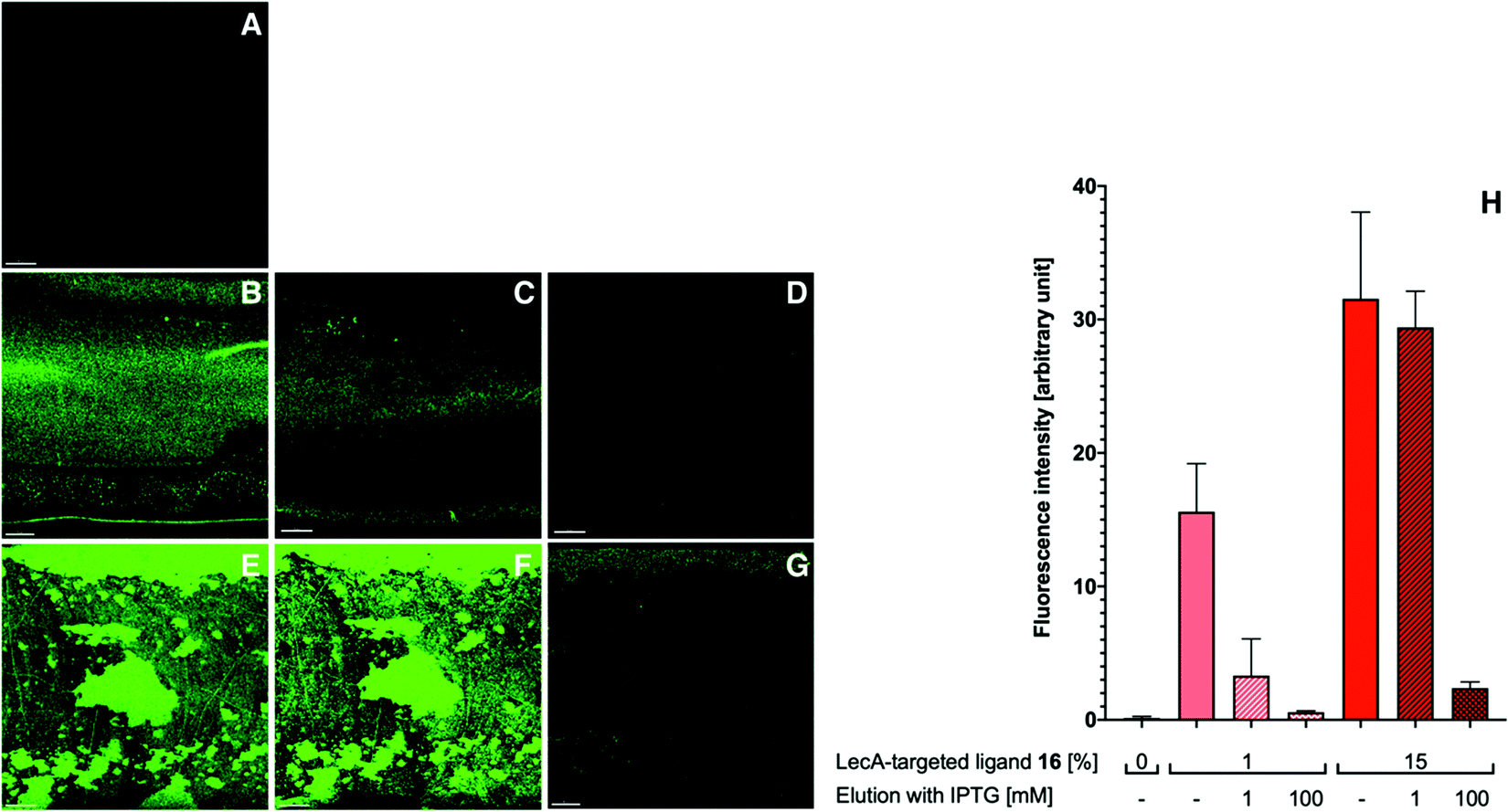 | ||
| Fig. 4 Targeting liposomes to a LecA-coated surface under flow. Injection of liposomes (5 min) and analysis by confocal microscopy after washing out unbound liposomes with buffer (5 min): Fluorescent liposomes without targeting ligand (A); fluorescent liposomes with 1% LecA-targeted phospholipid 16 (B); fluorescent liposomes with 1% LecA-targeted phospholipid 16 eluted with 1 mM IPTG (C) and 100 mM IPTG (D); fluorescent liposomes with 15% LecA-targeted phospholipid 16 (E); fluorescent liposomes with 15% LecA-targeted phospholipid 16 after elution with 1 mM IPTG (F) and 100 mM IPTG (G); quantification of the interaction between LecA-targeted liposomes with LecA-modified surface under flow conditions. Averages from 3 independent experiments with 9–12 technical replicates, error bars correspond to standard deviation. For A–G: scale bar = 50 μm. | ||
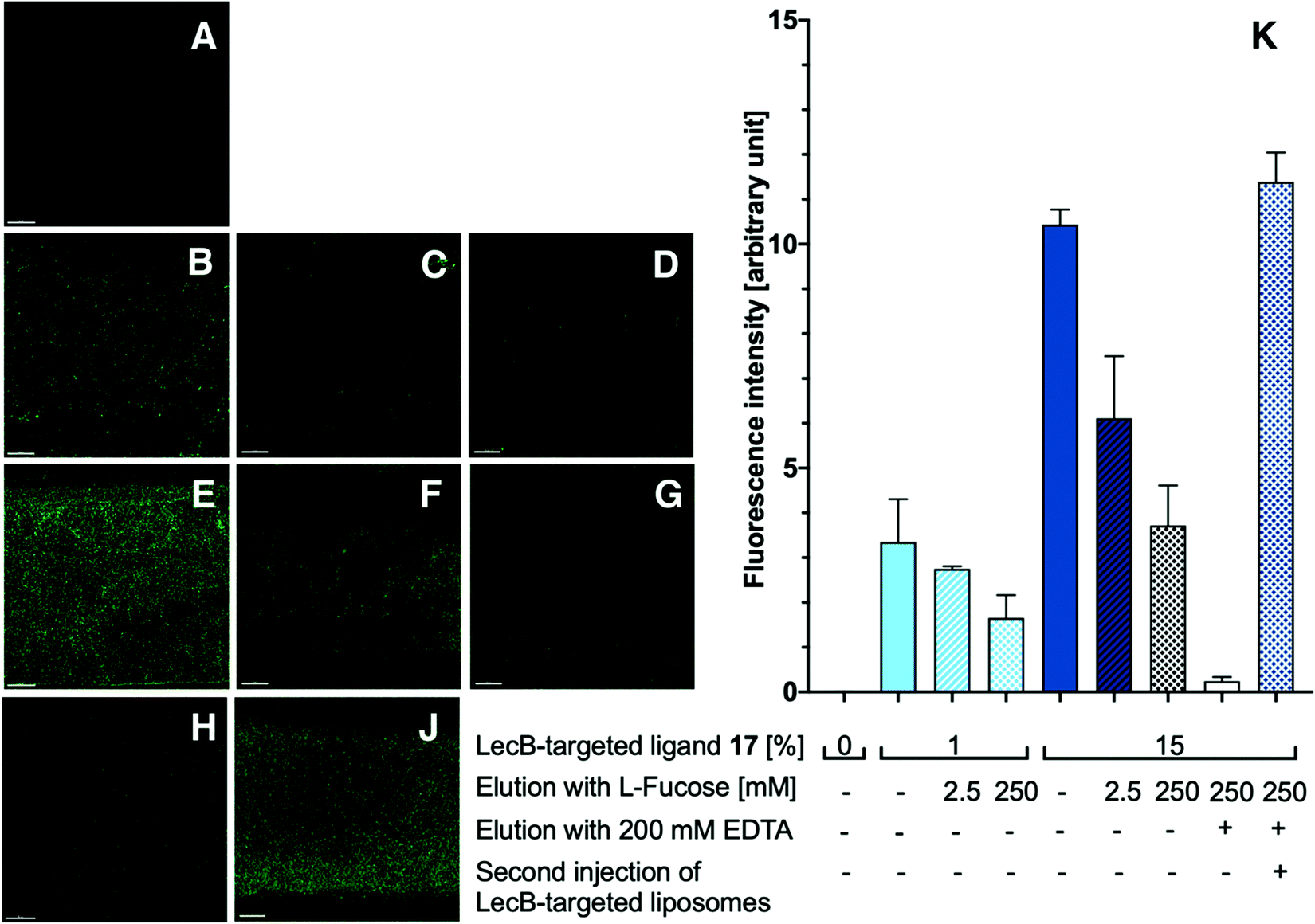 | ||
| Fig. 5 Targeting liposomes to a LecB-coated surface under flow. Injection of liposomes (5 min) and analysis by confocal microscopy after washing out unbound liposomes with buffer (5 min): Fluorescent liposomes without targeting ligand (A); fluorescent liposomes with 1% LecB-targeted phospholipid 17 (B); fluorescent liposomes with 1% LecB-targeted phospholipid 17 eluted with 2.5 mM L-fucose (C) and 250 mM L-fucose (D); fluorescent liposomes with 15% LecB-targeted phospholipid 17 (E); fluorescent liposomes with 15% LecB-targeted phospholipid 17 after elution with 250 mM L-fucose for 5 min (F) and 10 min (G); fluorescent liposomes with 15% LecB-targeted phospholipid 17 eluted with 200 mM EDTA to regenerate the glass surface (H); regenerated chamber after equilibration with PBS/Ca for 10 min and repeated injection of the liposomes with 15% LecB-targeted phospholipid 17 (J); quantification of the fluorescence image analyses to assess the interaction between LecB-targeted liposomes with LecB-coated surface using the microfluidic device (K). Averages from 3 independent experiments with 9–12 technical replicates, error bars correspond to standard deviation. Scale bars correspond to 50 μm. | ||
Surface targeting-ligand density can be an important factor for targeted nanomaterials and their interaction with their receptors. Since the targeted lectins are multivalent, varying ligand density may affect binding and thus targeting efficiency. For the above described competitive binding assay (Fig. 1), aggregation induced by the multivalent ligand/receptor system is also likely to occur as demonstrated by DLS and fluorescence detection in solution (Fig. 2). Because the lectins are covalently bound to the surface in the microfluidics system, aggregation induced precipitation from the solution is reduced and the observed bound fluorescence intensity directly correlates to the strength of the targeting efficiency which allows to study the influence of the targeting ligand density.
With the appropriately coated microfluidics device in hand, we studied the targeting effects of the various fluorescent liposomes with different targeting ligand densities, i.e. 1% or 15% of the targeted phospholipids 16 and 17 (Fig. 4 and 5). To assess the carbohydrate-dependency of the targeting process, the competitive inhibition of their interaction with the lectin-coated surfaces was studied after addition of isopropyl β-D-thiogalactoside (IPTG) for LecA or L-fucose for LecB. Fluorescence intensities of all recorded images were further measured and the effects could be quantified.
Drastic differences in fluorescence intensities were measured between targeted and plain liposomes for the respective lectins. In case of the glycomimetic-functionalized liposomes, the targeting to lectin-coated surfaces was highly stable under flow conditions as detected by the remaining strong fluorescence signal even after continuous washing with buffer for 1 h. In contrast, plain liposomes were fully removed indicating that the interaction between liposomes and the coated surfaces is carbohydrate-dependent.
In both cases, an increase in targeting ligand density from 1% to 15% significantly increased the detected fluorescence signal. This observation suggests that liposomes containing 15% of the targeting ligand are more efficient in both cases. Furthermore, a tendency to also form aggregates on the lectin-coated surface was observed for the more densely decorated LecA-targeted liposomes in particular. The latter corresponded to the increased efficacy of the multivalent interaction for LecA when compared to the monovalent ligand in the competitive binding assay. In case of LecB, differences in fluorescence intensity between 1% and 15% targeted liposomes could be observed, but the aggregation was significantly lower than for LecA and a more homogenous distribution of the liposomes along the entire channel was observed. Apart from the unknown quantity of immobilized lectin, one explanation for this phenomenon is the different spatial orientation of binding sites in the LecB tetramer which disfavors simultaneous binding compared to LecA where the sites are adjacent and optimally oriented for the multivalent binding to a surface localized pair of ligands.
The carbohydrate specificity of the interaction between lectin-targeted liposomes and surface-attached lectins was confirmed by the efficient removal of LecA-targeted liposomes after injection of IPTG or LecB-targeted liposomes after injection of L-fucose (Fig. 4G and 5G). The displacement of LecB-targeted liposomes was difficult and required increased amounts of competitive inhibitor L-fucose (>250 mM) compared to an efficient displacement of the LecA-targeted system and its competitor IPTG (100 mM), which is in agreement with the increased binding affinity of LecB towards its ligands compared to LecA. Regeneration of the lectin-coated microfluidics channels was achieved washing with 200 mM aqueous EDTA and after re-equilibration with calcium(II)-containing buffer, the system could be reused for analysis (Fig. 5J and K).
With the data from the competitive binding assay and the microfluidic flow assay with optical detection, we clearly observed a specific binding of the targeted liposomes to their lectin targets and a good retention under flow conditions. Both of these observations indicate our targeted liposome's potential for applications.
However, in both of these assays, we could not quantify the binding affinity of the targeted liposomes for their lectins, since it is likely that the lower assay limit was reached for the competitive binding assay (Fig. 1). Therefore, we established a surface-plasmon resonance experiment with LecA-targeted liposomes and surface-immobilized LecA. Carboxylic acids present on hydrophilic SPR chips were activated and LecA was immobilized at high density through amide coupling. To validate the system, the monovalent LecA ligand para-nitrophenyl β-D-galactoside (pNP-Gal) was injected in a multicycle experiment at concentrations ranging from 1.56 to 400 μM (Fig. 6A and B). Fast association (kon = 4.32 × 103 M−1 s−1) and dissociation kinetics (koff = 0.0831 s−1) were determined and equilibrium analysis revealed a Kd of 17.9 μM which is in agreement with literature reports (ITC: Kd = 14.1 μM, competitive binding assay IC50 = 19.0 μM).39,54,55
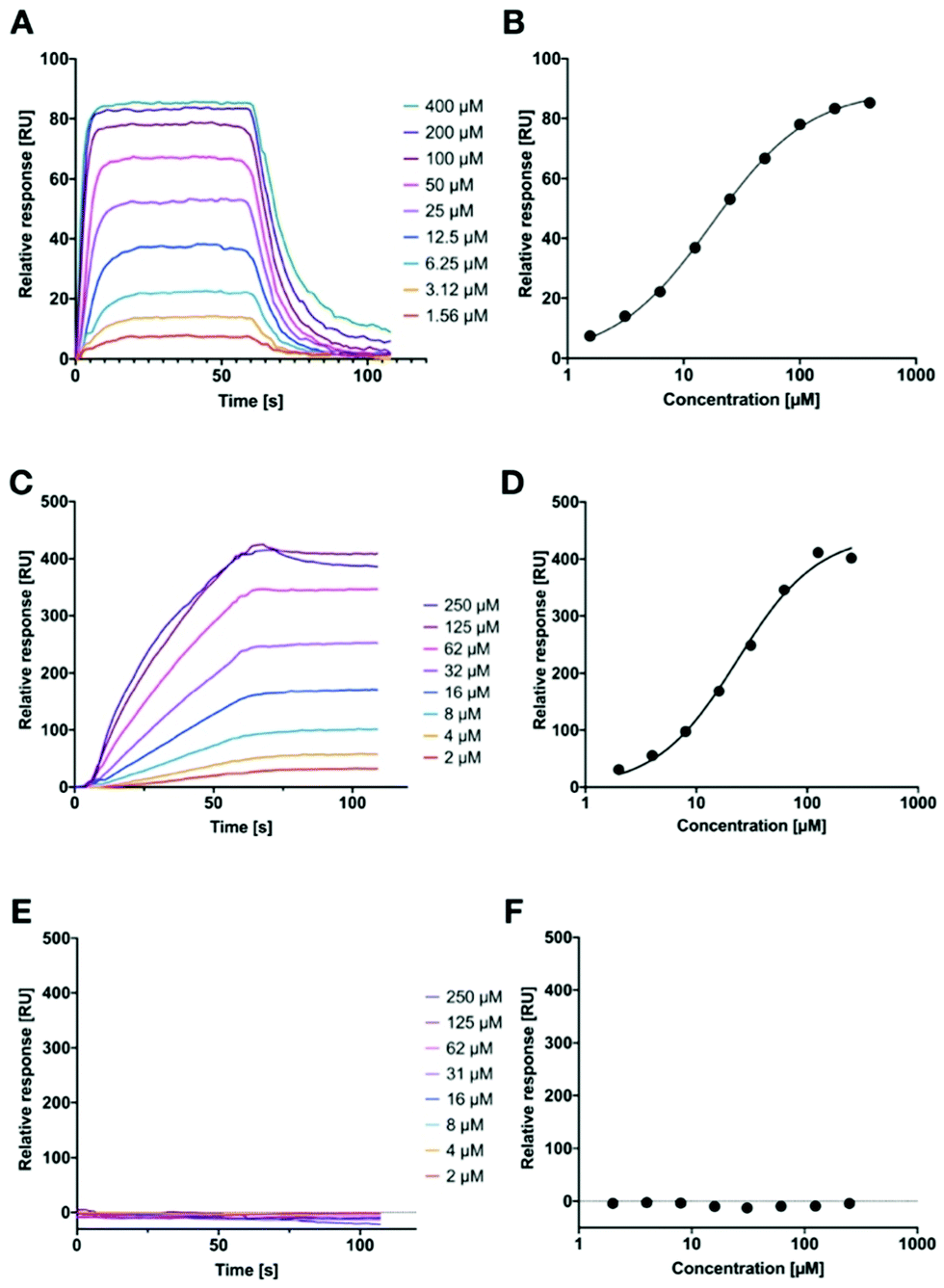 | ||
| Fig. 6 Surface plasmon resonance analysis of the interaction of surface-immobilized LecA with pNP-Gal (A and B) or 15% LecA-targeted liposomes alone (C and D) and in presence of 50 mM IPTG in the running buffer as competitor (E and F). SPR sensorgrams are shown in (A), (C) and (E); indicated concentrations correspond to the monovalent pNP-Gal in (A and B), and the targeted phospholipid in (C–F); maximal obtained response for the different concentrations is plotted for affinity analysis in (B), (D), and (F). | ||
Subsequently, we tested LecA-targeted liposomes decorated with 15% of targeting ligand 16 on the same SPR chip (Fig. 6C and D). The LecA-targeted liposomes were injected at a calculated concentration of 16 ranging from 2 to 250 μM in a multicycle experiment. Strongly different sensorgrams were obtained indicating a slow association and a virtually absent dissociation from immobilized LecA. In fact, regeneration of the chip surface required the use of EDTA and the presence of a detergent for efficient liposome removal. The association of the liposomes with the surface was so slow that it did not reach a saturation within 60 seconds of injection time, and further extension of the association phase to 120 seconds did also not lead to saturation (120 s data not shown). We also studied binding kinetics at a higher flow rate of 100 μL min−1 and did not observe mass transport effects impacting the binding of the liposomes with immobilized LecA (Fig. S5A, ESI†).45 Thus, the affinity analysis was performed on the highest response obtained and indicated a comparable affinity per molecule of targeting ligand to pNP-Gal. However, these fitting data must be considered with care as no saturation was achieved and they also contradict the approx. 20-fold higher potency of inhibition in the competitive binding assay. It can nevertheless be generalized, that these LecA-targeted liposomes are very potent binders, which once trapped at their surface-exposed target LecA will not detach again for very long time spans (Fig. S5B, ESI†). This fact can be assigned to their highly multivalent binding and provides an excellent basis for future use of the liposomes for targeted antibiotic delivery and furthermore for infection imaging experiments.
Since we could not reach saturation with the targeted liposomes, we tested the specificity of their binding to the surface in presence of a small galactoside as inhibitor of the LecA binding sites. The chip was now equilibrated with running buffer containing 50 mM IPTG and the injection of the liposomes was repeated as before (Fig. 6E and F). In presence of IPTG, the LecA-targeted liposomes did not bind to the LecA-surface in the SPR experiment. In addition, the LecB-targeted liposomes were also tested on the LecA-chip and no binding was detected (Fig. S4, ESI†). Both observations are further corroborating the high carbohydrate specificity of binding.
Conclusions
In conclusion, we have developed highly specific and highly potent nanovehicles for the targeting of biofilm associated lectins of P. aeruginosa, LecA and LecB. After coupling synthetic glycomimetics to phospholipids, a set of LecA- and LecB-targeted liposomes with varying lipid composition have been produced and characterized. These targeted liposomes showed high specificity to their respective targets and exhibited very strong carbohydrate-dependent binding to surfaces coated with these lectins. Microscopic analysis of targeted fluorescent liposomes in a microfluidic device demonstrated their potential for imaging applications. Future work will address the use of these targeted delivery systems for antibiotic delivery to break antimicrobial resistance and reduce toxicity of untargeted drugs.Materials and methods
General methods, chemical synthesis and NMR spectra can be found in the ESI.†Preparation and characterization of liposomes
For liposome preparation 20 mg phospholipids and 10 mg cholesterol were dissolved in a 5 mL mixture of chloroform/methanol 2:1. Targeting lipids 16 or 17 and/or fluorescein labelled lipid 18 were added at the indicated ratios. After complete removal of organic solvents using a rotary evaporator under reduced pressure at 70 °C, the obtained thin lipid film was hydrated with 5 mL PBS/Ca buffer (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.4 supplemented with 1 mM CaCl2), under rotation for 1 h at 65 °C followed by sonication for 30 sec at 65 °C. The resulting colloidal solution was extruded 10 times through a 0.2 μm pore size polycarbonate membrane (Polycarbonate track-etched membrane, Sartorius, Germany) at 70 °C via a Lipofast L-50 extruder (Avestin, Germany). For DLS analysis, the obtained liposomes were diluted 100-fold in MilliQ water to determine the Z-average size and polydispersity index (PdI) by dynamic light scattering (DLS) and measure the ζ-potential by electrophoretic light scattering using a Zetasizer Nano ZS (Malvern Panalytical Ltd, UK). Data are presented in Table 1. To measure the exact concentration of the LecA- or LecB-targeting ligands in the obtained solutions, liposomes were diluted 100-fold in MeOH and analyzed by LC-MS using the solvent system A: iPrOH/MeOH/H2O (5:4:1) + 0.05% HCOOH, B: iPrOH + 0.05% HCOOH and a gradient of 5–40% B.
Imaging of liposomes using cryo transmission electron microscopy
Cryo-TEM imaging of the obtained liposomal solutions was conducted by placing a 3 μL droplet of the liposomal solution onto a S147-4 holey carbon film (Plano, Germany) before blotting the sample to a thin liquid film for 2 s and plunging into liquid ethane at T = 108 K using a Gatan (Pleasonton, USA) CP3 cryo plunge system. The vitrified sample was transferred under liquid nitrogen to a Gatan model 914 cryo-TEM holder and visualized at T = 100 K using a JEOL (Akishima, Japan) JEM-2100 LaB6 TEM operating at an accelerating voltage of 200 kV at low-dose conditions. Images are shown in Fig. S1 (ESI†).Quantification of nanoparticle concentration and stability
Sizes and concentrations of the fluorescent liposomes were determined by nanoparticle tracking analysis (NTA, LM-10, Malvern, UK). Samples at a total phospholipid concentration of 4 mg mL−1 were diluted 1:10.000 in PBS/Ca, 200 μL aliquots were injected into a chamber illuminated by a laser beam at 532 nm and three 30 sec long videos were recorded. The data were processed and analyzed using NanoSight 3.1 software.
Additionally, LecB-targeted fluorescent liposomes (15% phospholipid 17) in PBS/Ca were incubated with LecB (molar ratio ligand/protein 1:1, corresponds to 50 μM), for 1 h at 37 °C and the obtained mixture was diluted 1:10.000 in PBS/Ca and studied by NTA as described above (Fig. 2A and B).
Competitive binding assay
The procedure for fluorescence polarisation-based competitive binding assays for both lectins, LecA and LecB, was adopted form Joachim et al. for LecA and Sommer et al. for LecB.39,56 The assay was performed in PBS/Ca2+ buffer (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.4 supplemented with 1 mM CaCl2). The final protein concentrations in the assays were 1 μM for LecA or 20 nM for LecB. Fluorescence intensities were recorded on a PheraStar FS plate reader (BMG Labtech GmbH, Germany) and polarization was calculated and plotted. IC50 values were obtained from a four-parameter curve fitting procedure. Averages and standard deviations were calculated from at least three independent experiments of technical triplicates each. pNP-Gal was used as a positive control and the obtained IC50 was 21 ± 6 μM, which agrees with literature data.39Microfluidics device preparation
The design and dimensions of the microfluidic device is shown in Fig S2 (ESI†). Instrumentation: 3D computer aided drawings were drawn on Autodesk Fusion 360 (Autodesk, California, USA), exported as .STL files and sliced into GCode by Ultimaker Cura (Ultimaker, Utrecht, Netherlands) and printed using a fused filament 3D printer (Ultimaker2 Extended+, Ultimaker, Utrecht, Netherlands). All molds were printed with a 0.25 mm nozzle. The print speed was 20 mm s−1, layer height was 0.1 mm, print temperature was 210 °C, bed temperature was set to 60 °C. A PICO low pressure plasma system (Diener Electronic GmbH & Co. KG, Ebhausen, Germany) was used for the plasma treatment of PDMS. Device fabrication: Molds were fabricated by the direct 3D printing of PLA onto cleaned microscope slides. The print height of the first layer on the glass slide was adjusted by the height of the microscope slide used, typically 100 μm plus 10 μm for nozzle clearance. The total print time of the mold was two hours and 29 minutes. PDMS was prepared in a 10:1 ratio (polymer:crosslinker) by weight. The PLA-glass mold was then sealed with hot glue and then filled with PDMS. The PDMS was left to cure overnight at 40 °C. The PDMS was then removed from the mold and was cleaned with isopropanol and deionized water. The PDMS was then plasma treated with oxygen, 350 W for 1 minute and immediately brought into contact with the NHS-activated glass slide (SL HCX, XanTec bioanalytics GmbH, Düsseldorf, Germany). The PDMS–glass device was then placed under a 2 kg mass and baked for 2 hours. Epoxy glue was applied to the PDMS–glass edge to help secure and avoid the peeling off of the PDMS. Silicone tubing was then inserted into the inlets/outlets and sealed in place with black PDMS and left to cure at 40 °C for 1 hour or longer if not cured. Devices were sterilized with ethanol and dried before use.
In vitro flow assay preparation
The microfluidics device was assembled as described above. For rehydration of the hydrogel surface, each channel was rinsed with MilliQ H2O (500 μL min−1, 10 min). Then, the channel was loaded with LecA or LecB (140 μM) in 10 mM sodium acetate buffer pH 4.5 and incubated for 90 min at room temperature. The channels were consequently treated with 1 M ethanolamine hydrochloride at pH 8.5 in MilliQ water (500 μL min−1, 15 min) followed by reequilibration of the channels with PBS/Ca buffer (500 μL min−1, 45 min).For the experiments with fluorescently labelled liposomes, the corresponding channel was flushed with a solution of targeted or plain fluorescein-labelled liposomes (total phospholipid concentration = 70 μg mL−1) in PBS/Ca buffer (500 μL min−1, 5 min) followed by elution of unbound material with PBS/Ca buffer (500 μL min−1, 10 min). To test for carbohydrate-dependent elution, a channel with bound targeted liposomes was additionally rinsed with a solution of the respective competitive inhibitor (IPTG for LecA or L-fucose for LecB) dissolved at the indicated concentrations in PBS/Ca buffer (500 μL min−1, 10 min).
Confocal fluorescence microscopy
Fluorescence images were acquired immediately after eluting unbound material from the channel with PBS–Ca2+-buffer with a Leica DMi8 Confocal Fluorescence Microscope (Leica Microsystems, Germany) equipped with a 25× water immersion objective. Fluorescein was excited with a 488 nm laser. Images were acquired at the bottom of the channels. Images were background corrected based on the blank control and the mean fluorescence intensity was measured using ImageJ (2.1.0/1.53c) software.57 The obtained mean values were plotted using GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA).Inverted fluorescence microscopy
To study the stability of liposomes in presence of LecA, plain (1% of lipid 18) and LecA-targeted (15% of lipid 16, 1% of lipid 18) fluorescent liposomes in PBS/Ca (40 μg mL−1 total phospholipid concentration) were preincubated with 14 μM LecA for 15 min in PDMS channels at static conditions. The channels were subsequently investigated with an inverted fluorescence microscope (Nikon Eclipse Ti-S) equipped with a Nikon Intensilight 130 W mercury lamp and 10× plan Nikon objective with a numerical aperture of 0.25. Images were acquired with the attached Orca R2 monochrome 1.3 MP CCD camera (Hamamatsu, Japan) at a resolution of 1024 × 1024 with 0.547 μm per pixel. Images were processed using ImageJ for background correction (Fig. 2C and D).Surface plasmon resonance
SPR experiments were performed on a Reichert 2-channel SPR SR7500DC (Reichert Technologies Life Sciences, Buffalo, NY, USA) at 25 °C. For LecA immobilization, the surface of a HC1000M sensor chip (XanTec bioanalytics GmbH, Düsseldorf, Germany) was equilibrated with borate elution buffer (1 M NaCl, 0.1 M sodium borate pH 9.0), followed by activation of the carboxylic acids with 0.2 M N-hydroxysuccinimide (NHS) and 0.4 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC*HCl) in MilliQ water channel 1 and 2 (contact time of 480 s, flow rate 10 μL min−1) until the binding response reached 800 RU. LecA (100 μg mL−1) in 10 mM sodium acetate pH 3.6 was then injected to the activated chip surface on channel 2 (contact time 660 s, flow rate 10 μL min−1) and 5500 RU of LecA were immobilized. Remaining NHS esters in both channels were quenched with 1 M ethanolamine hydrochloride in MilliQ water pH 8.5 (contact time 360 s, flow rate 10 μL min−1). Liposome stock solutions (at 500 μM of targeted phospholipid in PBS/Ca) were subsequently diluted in running buffer (10 mM phosphate buffer pH 7.4, 2.7 mM KCl, 137 mM NaCl, 100 μM CaCl2). Liposomes were injected (contact time of 60 s and dissociation time of 60 s, flow rate 50 μL min−1) at a concentration of the targeted phospholipid of 2, 4, 8, 16, 31, 62, 125 and 250 μM. The chip surface was regenerated after each liposome injection by 3 injections of 1% CHAPS and 100 mM EDTA in MilliQ water followed by 4 injections of the running buffer (contact time of 120 s, flow rate 50 μL min−1). 100 μM pNP-Gal in running buffer was injected before and after each analyte to monitor chip regeneration and protein activity.Affinity/equilibrium analysis was performed for pNP-Gal on the same chip. A pNP-Gal stock solution (10 mM in PBS/Ca) was prepared, then diluted to the required concentrations in running buffer and injected at 1.56, 3.125, 6.25, 12.5, 25, 50, 100, 200 and 400 μM (injection time of 60 s, flow rate 50 μL min−1) without regeneration steps. Data analysis was performed using Scrubber 2.0 software (Biologic Software Pty Ltd) using a 1:1 binding model to fit the experimental data.
Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
A. T. acknowledges funding from the European Research Council for an ERC Starting Grant (Sweetbullets, 716311). We further thank Dr Jelena Konstantinovic and Dr Andreas Kany (HIPS) for HRMS measurements.References
- J. O’Neill, Tackling drug-resistant infections globally: final report and recommendations, Government of the United Kingdom, 2016 Search PubMed.
- S. Wagner, R. Sommer, S. Hinsberger, C. Lu, R. W. Hartmann, M. Empting and A. Titz, J. Med. Chem., 2016, 59, 5929–5969 CrossRef CAS PubMed.
- D. Monroe, PLoS Biol., 2007, 5, e307 CrossRef PubMed.
- A. Gonzalez Gomez and Z. Hosseinidoust, ACS Infect. Dis., 2020, 6, 896–908 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- K. Forier, K. Raemdonck, S. C. De Smedt, J. Demeester, T. Coenye and K. Braeckmans, J. Controlled Release, 2014, 190, 607–623 CrossRef CAS PubMed.
- W. Gao, S. Thamphiwatana, P. Angsantikul and L. Zhang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 532–547 CAS.
- S. B. Lim, A. Banerjee and H. Önyüksel, J. Controlled Release, 2012, 163, 34–45 CrossRef CAS PubMed.
- T. Yu, K. W. Y. Chan, A. Anonuevo, X. Song, B. S. Schuster, S. Chattopadhyay, Q. Xu, N. Oskolkov, H. Patel, L. M. Ensign, P. C. M. van Zjil, M. T. McMahon and J. Hanes, Nanomedicine, 2015, 11, 401–405 CrossRef CAS PubMed.
- R. E. W. Hancock and F. S. L. Brinkman, Annu. Rev. Microbiol., 2002, 56, 17–38 CrossRef CAS PubMed.
- Z. Drulis-Kawa, A. Dorotkiewicz-Jach, J. Gubernator, G. Gula, T. Bocer and W. Doroszkiewicz, Int. J. Pharm., 2009, 367, 211–219 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- Y.-C. Yeh, T.-H. Huang, S.-C. Yang, C.-C. Chen and J.-Y. Fang, Front. Chem., 2020, 8, 286 CrossRef CAS PubMed.
- W. Gao, Y. Chen, Y. Zhang, Q. Zhang and L. Zhang, Adv. Drug Delivery Rev., 2018, 127, 46–57 CrossRef CAS PubMed.
- M. Srinivasarao and P. S. Low, Chem. Rev., 2017, 117, 12133–12164 CrossRef CAS PubMed.
- P. Klahn and M. Brönstrup, Nat. Prod. Rep., 2017, 34, 832–885 RSC.
- A. A. Khan, K. S. Allemailem, S. A. Almatroodi, A. Almatroudi and A. H. Rahmani, 3 Biotech., 2020, 10, 163 CrossRef PubMed.
- G. J. L. Bernardes, R. Kikkeri, M. Maglinao, P. Laurino, M. Collot, S. Y. Hong, B. Lepenies and P. H. Seeberger, Org. Biomol. Chem., 2010, 8, 4987–4996 RSC.
- S. Kawakami and M. Hashida, J. Controlled Release, 2014, 190, 542–555 CrossRef CAS PubMed.
- P.-L. Jiang, H.-J. Lin, H.-W. Wang, W.-Y. Tsai, S.-F. Lin, M.-Y. Chien, P.-H. Liang, Y.-Y. Huang and D.-Z. Liu, Acta Biomater., 2015, 11, 356–367 CrossRef CAS PubMed.
- C. M. Nycholat, S. Duan, E. Knuplez, C. Worth, M. Elich, A. Yao, J. O’Sullivan, R. McBride, Y. Wei, S. M. Fernandes, Z. Zhu, R. L. Schnaar, B. S. Bochner and J. C. Paulson, J. Am. Chem. Soc., 2019, 141, 14032–14037 CrossRef CAS PubMed.
- E.-C. Wamhoff, J. Schulze, L. Bellmann, M. Rentzsch, G. Bachem, F. F. Fuchsberger, J. Rademacher, M. Hermann, B. Del Frari, R. van Dalen, D. Hartmann, N. M. van Sorge, O. Seitz, P. Stoitzner and C. Rademacher, ACS Cent. Sci., 2019, 5, 808–820 CrossRef CAS PubMed.
- S. Deng, L. Bai, R. Reboulet, R. Matthew, D. A. Engler, L. Teyton, A. Bendelac and P. B. Savage, Chem. Sci., 2014, 5, 1437–1441 RSC.
- F. Broecker, S. Götze, J. Hudon, D. C. K. Rathwell, C. L. Pereira, P. Stallforth, C. Anish and P. H. Seeberger, J. Med. Chem., 2018, 61, 4918–4927 CrossRef CAS PubMed.
- G. Liao, Z. Zhou, S. Suryawanshi, M. A. Mondal and Z. Guo, ACS Cent. Sci., 2016, 2, 210–218 CrossRef CAS PubMed.
- D. Fayolle, N. Berthet, B. Doumeche, O. Renaudet, P. Strazewski and M. Fiore, Beilstein J. Org. Chem., 2019, 15, 937–946 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- R. Wawrzinek, E.-C. Wamhoff, J. Lefebre, M. Rentzsch, G. Bachem, G. Domeniconi, J. Schulze, F. F. Fuchsberger, H. Zhang, C. Modenutti, L. Schnirch, M. A. Marti, O. Schwardt, M. Bräutigam, M. Guberman, D. Hauck, P. H. Seeberger, O. Seitz, A. Titz, B. Ernst and C. Rademacher, J. Am. Chem. Soc., 2021, 143, 18977–18988 CrossRef CAS PubMed.
- S. P. Diggle, R. E. Stacey, C. Dodd, M. Cámara, P. Williams and K. Winzer, Environ. Microbiol., 2006, 8, 1095–1104 CrossRef CAS PubMed.
- D. Tielker, S. Hacker, R. Loris, M. Strathmann, J. Wingender, S. Wilhelm, F. Rosenau and K.-E. Jaeger, Microbiology, 2005, 151, 1313–1323 CrossRef CAS PubMed.
- D. Passos da Silva, M. L. Matwichuk, D. O. Townsend, C. Reichhardt, D. Lamba, D. J. Wozniak and M. R. Parsek, Nat. Commun., 2019, 10, 2183 CrossRef PubMed.
- J. Meiers, E. Siebs, E. Zahorska and A. Titz, Curr. Opin. Chem. Biol., 2019, 53, 51–67 CrossRef CAS PubMed.
- J. Meiers, E. Zahorska, T. Röhrig, D. Hauck, S. Wagner and A. Titz, J. Med. Chem., 2020, 63, 11707–11724 CrossRef CAS PubMed.
- M. B. Calvert, V. R. Jumde and A. Titz, Beilstein J. Org. Chem., 2018, 14, 2607–2617 CrossRef CAS PubMed.
- R. Sommer, K. Rox, S. Wagner, D. Hauck, S. S. Henrikus, S. Newsad, T. Arnold, T. Ryckmans, M. Brönstrup, A. Imberty, A. Varrot, R. W. Hartmann and A. Titz, J. Med. Chem., 2019, 62, 9201–9216 CrossRef CAS PubMed.
- R. Sommer, S. Wagner, K. Rox, A. Varrot, D. Hauck, E.-C. Wamhoff, J. Schreiber, T. Ryckmans, T. Brunner, C. Rademacher, R. W. Hartmann, M. Brönstrup, A. Imberty and A. Titz, J. Am. Chem. Soc., 2018, 140, 2537–2545 CrossRef CAS PubMed.
- V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492–4503 RSC.
- E. Zahorska, S. Kuhaudomlarp, S. Minervini, S. Yousaf, M. Lepsik, T. Kinsinger, A. K. H. Hirsch, A. Imberty and A. Titz, Chem. Commun., 2020, 56, 8822–8825 RSC.
- I. Joachim, S. Rikker, D. Hauck, D. Ponader, S. Boden, R. Sommer, L. Hartmann and A. Titz, Org. Biomol. Chem., 2016, 14, 7933–7948 RSC.
- S. Kuhaudomlarp, E. Siebs, E. Shanina, J. Topin, I. Joachim, P. da Silva Figueiredo Celestino Gomes, A. Varrot, D. Rognan, C. Rademacher, A. Imberty and A. Titz, Angew. Chem., Int. Ed., 2021, 60, 8104–8114 CrossRef CAS PubMed.
- D. Hauck, I. Joachim, B. Frommeyer, A. Varrot, B. Philipp, H. M. Möller, A. Imberty, T. E. Exner and A. Titz, ACS Chem. Biol., 2013, 8, 1775–1784 CrossRef CAS PubMed.
- R. Sommer, D. Hauck, A. Varrot, S. Wagner, A. Audfray, A. Prestel, H. M. Möller, A. Imberty and A. Titz, ChemistryOpen, 2015, 4, 756–767 CrossRef CAS PubMed.
- R. Sommer, T. E. Exner and A. Titz, PLoS One, 2014, 9, e112822 CrossRef PubMed.
- S. Wagner, D. Hauck, M. Hoffmann, R. Sommer, I. Joachim, R. Müller, A. Imberty, A. Varrot and A. Titz, Angew. Chem., Int. Ed., 2017, 56, 16559–16564 CrossRef CAS PubMed.
- E. M. Munoz, J. Correa, R. Riguera and E. Fernandez-Megia, J. Am. Chem. Soc., 2013, 135, 5966–5969 CrossRef CAS PubMed.
- M. T. Meyer, V. Roy, W. E. Bentley and R. Ghodssi, J. Micromech. Microeng., 2011, 21, 10 CrossRef.
- A. Thomas, S. Wang, S. Sohrabi, C. Orr, R. He, W. Shi and Y. Liu, Biomicrofluidics, 2017, 11, 024102 CrossRef PubMed.
- M. Ejiugwo, Y. Rochev, G. Gethin and G. O’Connor, Tissue Eng., Part C, 2021, 27, 77–88 CrossRef PubMed.
- E. P. Helps and D. A. McDonald, J. Physiol., 1954, 124, 631–639 CrossRef CAS PubMed.
- M. Klarhöfer, B. Csapo, C. Balassy, J. C. Szeles and E. Moser, Magn. Reson. Med., 2001, 45, 716–719 CrossRef PubMed.
- B. Majhy, P. Priyadarshini and A. K. Sen, RSC Adv., 2021, 11, 15467–15476 RSC.
- V. M. Dominical, D. M. Vital, F. O’Dowd, S. T. O. Saad, F. F. Costa and N. Conran, Exp. Hematol., 2015, 43, 223–228 CrossRef PubMed.
- D. Hauck, V. R. Jumde, C. J. Crawford and A. Titz, in Carbohydrate Chemistry: Proven Synthetic Methods, ed. P. Kosma, T. Wrodnigg and A. Stütz, CRC Press, Boca Raton, FL, 1st edn, 2021, vol. 5, pp. 17–22 Search PubMed.
- R. U. Kadam, D. Garg, J. Schwartz, R. Visini, M. Sattler, A. Stocker, T. Darbre and J.-L. Reymond, ACS Chem. Biol., 2013, 8, 1925–1930 CrossRef CAS PubMed.
- S. Kuhaudomlarp, E. Gillon, A. Varrot and A. Imberty, in Lectin Purification and Analysis: Methods and Protocols, ed. J. Hirabayashi, Springer US, New York, NY, 2020, pp. 257–266 Search PubMed.
- R. Sommer, S. Wagner, A. Varrot, C. M. Nycholat, A. Khaledi, S. Häussler, J. C. Paulson, A. Imberty and A. Titz, Chem. Sci., 2016, 7, 4990–5001 RSC.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena and K. W. Eliceiri, BMC Bioinf., 2017, 18, 529 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02086b |
| This journal is © The Royal Society of Chemistry 2022 |