
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of Ir modification on the durability of FeNC catalysts under start-up and shutdown cycle conditions†
Carolin
Prössl
a,
Markus
Kübler
a,
Stephen
Paul
a,
Lingmei
Ni
ab,
Simon-Johannes
Kinkelin
c,
Nils
Heppe
a,
Klaus
Eberhardt
d,
Christopher
Geppert
d,
Wolfram
Jaegermann
b,
Robert W.
Stark
b,
Michael
Bron
 c and
Ulrike I.
Kramm
*ab
c and
Ulrike I.
Kramm
*ab
aDepartment of Chemistry, Technical University of Darmstadt, Catalysts and Electrocatalysts group, Alarich-Weiss-Str. 4, 64287 Darmstadt, Germany. E-mail: ulrike.kramm@tu-darmstadt.de
bDepartment of Materials- and Earth Sciences, Technical University of Darmstadt, Alarich-Weiss-Str. 2, 64287 Darmstadt, Germany
cInstitute of Chemistry, Martin Luther University Halle-Wittenberg, von-Danckelmann-Platz 4, D-06120 Halle (Saale), Germany
dTRIGA Research Reactor, Johannes Gutenberg University of Mainz, Fritz-Straßmann-Weg 2, 55128 Mainz, Germany
First published on 29th October 2021
Abstract
A common problem associated with FeNC catalysts is their poor stability dominated by the carbon oxidation reaction (COR). In this work, the feasibility of stabilizing FeNC catalysts with small quantities of Ir was explored. With iridium being present, instead of COR the oxygen evolution reaction should be favored. The impact on structure and morphology was investigated by 57Fe Mössbauer spectroscopy, X-ray photoelectron spectroscopy, Raman spectroscopy and transmission electron microscopy. The catalytic activity and durability for the oxygen reduction reaction was evaluated by rotating ring disc electrode experiments and accelerated stress tests mimicking the start-up and shutdown cycle (SSC) conditions, respectively. For selected samples the stability was analysed for SSCs in proton exchange membrane fuel cells. Moreover, the faradaic efficiency towards oxygen evolution reaction vs. COR was determined and the resistance towards COR analysed by in situ Raman spectroscopy. The results indicate indeed a suppression of the COR; however, specifically for fuel cell applications, further optimization is necessary.
Introduction
The world faces an increasing demand of energy in the sectors transportation, building and industry. At the same time, greenhouse gas emissions, specifically the CO2 release must be reduced to huge extent.1 Regarding the transportation sector, the proton exchange membrane fuel cell (PEM-FC) may find application in passenger cars, bus transport, light- and heavy-duty vehicles. In PEM-FCs, platinum-based catalysts accelerate the electrochemical reactions. However, the cost of the platinum-based technology so far impedes broader fuel cell electric vehicle (FCEV) commercialization.2 Besides, platinum (Pt) is characterized by volatile production volumes, scarcity and geopolitically criticality. Pt catalyses the hydrogen oxidation reaction (HOR) in an excellent way. However, with respect to commercialization, the high quantities that are especially important for the cathodic oxygen reduction reaction (ORR) strongly limit the integration of FCEV in the market. They are usually fabricated in form of nanoparticles which are dispersed on a high surface area carbon support (Pt/C).3As an alternative to Pt, non-precious metal catalysts (NPMCs) have been researched extensively because using non-critical elements helps to cut materials costs and to avoid precious metal price fluctuations. Iron-nitrogen-carbon (FeNC) catalysts belong to the class of carbon-based NPMCs and show a promising activity for the ORR in acidic media.4–6 They are also of interest for the ORR in alkaline environment as e.g. of relevance for alkaline fuel cells or metal air batteries,7–9 the hydrogen evolution reaction10,11 or CO2 reduction reaction.12,13 This work focusses on their utilization as ORR catalyst for acidic conditions, as in PEM-FC. Besides the activity, the stability (potential holds) and durability (potential cycling) are of great importance. Four main degradation mechanisms have been identified for FeNC: (1) micropore flooding; (2) protonation followed by anion adsorption of co-catalytic moieties; (3) demetallation and (4) the carbon oxidation reaction (COR).3,5,14–17
The reverse-current decay mechanism is a process which is relevant during the start-up and shutdown of a PEM-FC, for example between automotive drive cycles. Why is it called reverse current mode? Under usual condition the electron transport is from anode (HOR) to cathode (ORR). Between automotive drive cycles, air penetrates the anodic compartment and gets trapped in the porous catalyst layer. As Pt/C catalysts are capable to catalyse both HOR and ORR, in such regions the ORR can proceed. While maintaining the potential difference between anode and cathode, the system searches for a counter reaction (oxidation) that can occur on the cathodic site. Thus, during this degradation the current flow is reversed.18,19 Under these conditions, potentials of 1.5 V can easily occur at the cathode. In consequence, to address related catalyst degradation phenomena, stress tests mimicking these conditions between automotive drive cycles are summarized as start-up and shutdown cycle (SSC) conditions. Severe performance losses have been observed especially after accelerated stress tests (ASTs) mimicking the SSC.20,21 The main activity loss is explicitly attributed to COR.17,22 Thermodynamically the COR has a standard potential of 0.2 VRHE for direct oxidation, but the kinetic hindrance shifts the potential up to 0.9–1.0 VRHE.22 Using more stable carbon allotropes to implement FeN4 centres is one possible strategy to shift the onset for COR, while the implementation in PEM-FC is difficult.23 By applying differential electrochemical mass spectrometry (DEMS) studies, Choi et al.17 observed CO2 evolution from MNC catalysts starting at 0.9 VRHE. The electrochemical burn-off of the carbon support affects the performance by changing the electrical conductivity and hydrophilicity of the catalyst layer.14 Besides, for FeNC catalysts the oxidation of the carbon support leads to the destruction of the hosted FeN4 sites which is detrimental to the ORR activity.14,24 The loss of iron content upon high voltage polarization is a descriptor for the decay of mass activity in PEM-FC.14 Based on this insight, strategies need to be developed to overcome the degradation.
Another important factor is to minimize Fe leaching from spectator species e.g. by an advanced acid treatment.17,25,26
Improving the carbon structure in terms of graphitization enhances its stability towards COR but goes hand in hand with a trade-off in lower yield of active FeN4 sites and BET surface area.3
Thus, a stabilization strategy is desired that enables to keep the benefits of high surface area and site density in the preparation of today's most active FeNC catalysts. Therefore, in this work an approach has been adapted which is known to work for Pt/C ORR catalysts. Crowtz et al.27 showed that the addition of Ir or Ru OER catalysts led to improved stability of a Pt/C catalyst during 1400 SSC/load/idle pulses in form of subsequent constant current (0.942 mA cm−2) and potential (0.65 and 0.95 VRHE) holds in rotating disc electrode (RDE) experiments. The stabilization during 1.6 V potential holds for 30 min in PEM-FC was observed by Oh et al.28 for Pt/C catalysts with 1–5 wt% IrO2 added. Similar observations were made by Atanasoski et al.29 and Cullen et al.30 for Ir and Ru additions (note: under SSC conditions both will transform to Ru and Ir oxides).
Motivated by these findings, the aim of this study was to evaluate the suitability of small amount iridium (Ir) modified cathode catalysts in order to enhance the durability at high electrode potentials. While the modification of MNC with small amounts of platinum is described in different works,31,32 to the best of our knowledge there are so far no reports on Ir/FeNC catalysts.
Furthermore, different methods for combining iridium and the FeNC material were tested. Structural characterization was done by 57Fe Mössbauer spectroscopy, X-ray photoelectron spectroscopy (XPS), nitrogen sorption measurements, transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy (EDS) as well as neutron activation analysis (NAA) to follow the impact on morphology and iron/iridium speciation.
In order to mimic the SSC on a laboratory scale, rotating ring disc electrode (RRDE) experiments employing an accelerated stress test (AST) protocol were conducted and evaluated for a reference FeNC catalyst and the modifications. The degradation of the catalyst in acidic media was investigated by in situ Raman spectroscopy as a technique which provides information about the changes of defective carbon structures due to COR. In addition, PEM-FC measurements adopting the SSC protocol were performed for selected samples and enable conclusions on the impact of application conditions RRDE vs. PEM-FC.
Results and discussion
Influence of different iridium modifications on morphology and surface area
In order to obtain iridium modified FeNC catalysts three different approaches as schematically illustrated in Fig. 1a were investigated: (1) direct addition of iridium in the precursor mixture (Pyro), (2) galvanic displacement of (inactive) iron by iridium during the first acid leaching (GD) and (3) post-preparation addition of independently prepared iridium nanoparticles (Ref-x% Ir). In this case, a variation of the iridium loading on the FeNC (Ref) was performed (intended loadings of 1 wt%, 2 wt% and 4 wt%). The iridium nanoparticles were prepared by a polyol approach, as described previously.33 The adsorption and desorption isotherms can be found in the ESI, Fig. S1.† As indicated by the BET surface area values in Fig. 1b all catalysts had surface areas between 520 and 666 m2 g−1. Based on this, an Ir/C reference catalyst was prepared using KetjenBlack 300 as carbon support (BET ≈ 800 m2 g−1).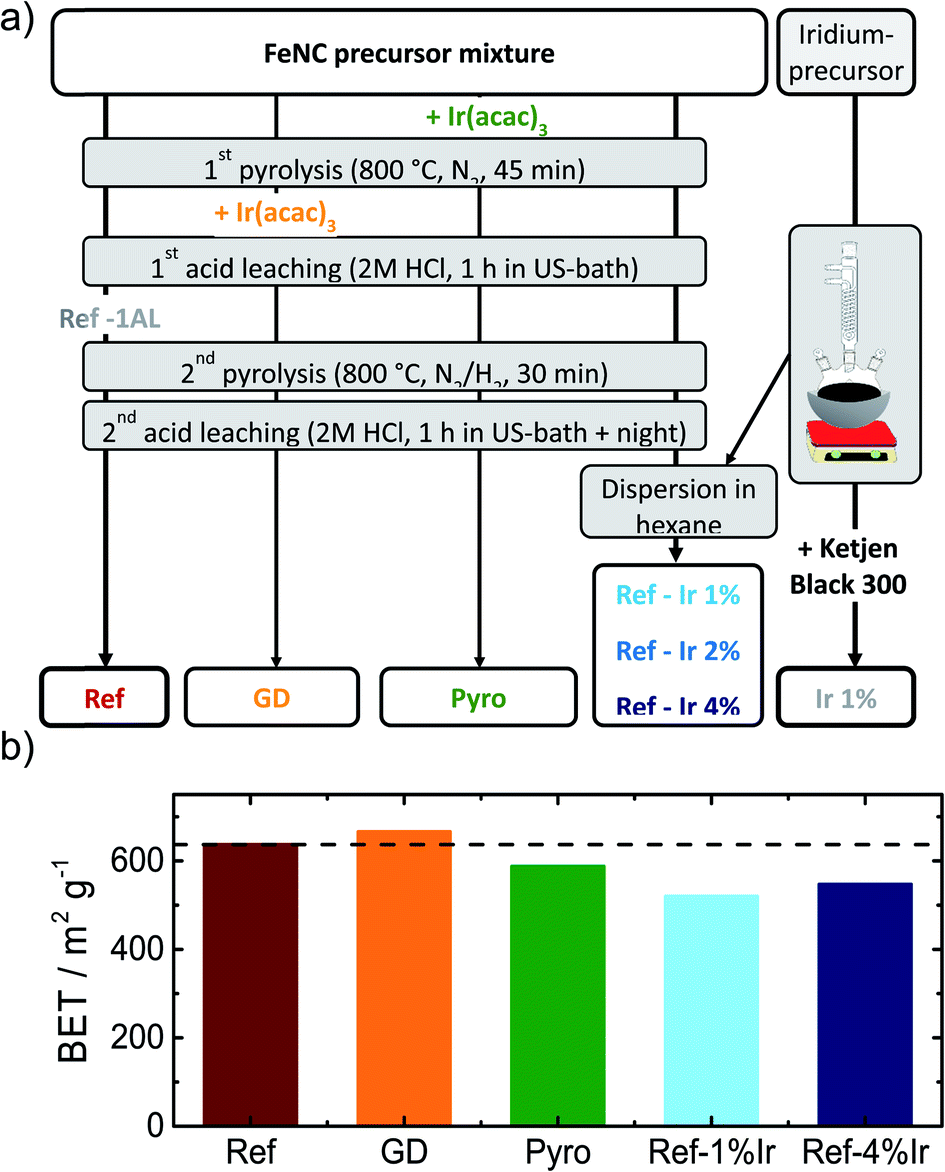 | ||
| Fig. 1 (a) Scheme of the different preparation routes to obtain the FeNC reference catalyst (Ref) as well as the iridium modified catalysts and an Ir/C catalyst, (b) influence of the different modification procedures on the BET surface area of the catalysts. | ||
In order to compare homogeneity of the iridium distribution on the FeNC catalyst, transmission electron microscopy (TEM) images were recorded (Fig. 2), additional overview images can be found in the ESI, Fig. S2 and S3.† The FeNC reference catalyst labelled with ‘Ref’ exhibits a carbon pore structure qualitatively corresponding to a mesoporous material. This observation is in line with the BET results. As intended by the applied purification treatment,25 no inorganic particles can be detected.
 | ||
| Fig. 2 TEM images of the FeNC catalyst Ref as well as the iridium modified FeNC catalysts and Ir/C. | ||
Moreover, no graphitized regions could be observed but loosely jointed graphene layers. The FeNC reference catalyst seems therefore relative pure in composition, by means of absent inorganic particles, and exhibits an amorphous, porous carbon structure. The related EDS analysis is summarized in ESI, Table S1† confirmed the existence of all precursor elements, carbon, nitrogen, oxygen, sulfur and chlorine from the acid leaching. The calculated elemental ratio N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe was 4.4. This would enable for all iron ions to have a nominal fourfold coordination to nitrogen (Fe–N4).
:Fe was 4.4. This would enable for all iron ions to have a nominal fourfold coordination to nitrogen (Fe–N4).
For the GD catalyst no inorganic particles, neither from iron-based species nor iridium were found. The porous carbon structure is comparable to the reference catalyst with a slightly more developed pore network. For our sample, it is generally assumed that during galvanic displacement metallic iron that is formed during the pyrolysis34 gets oxidized by IrCl3, with the consequence of Ir3+ reduction and Ir0 deposition on the catalyst. Although during acid leaching so much iridium was present that in total 2 wt% of iridium could have been deposited, no distinct Ir peaks could be detected in the EDS spectra. No particles were visible in TEM. Nevertheless, it will be shown later that indeed a small quantity of iridium can be found in the catalyst. Also the Pyro sample exhibits amorphous carbon with high degree of porosity similar to the GD sample. In contrast, this sample clearly displays the formation of about 5 nm-large nanoparticles which are randomly distributed over the catalyst together with some agglomerates of 20 nm.
The images of the Ref-1% Ir catalyst confirm the precipitation of Ir NPs on the FeNC catalyst. This sample is composed of regions with aggregated NPs and parts were almost no particles were deposited. The situation is similar for Ref-4% Ir. The dispersion of particles is still inhomogeneous. The manner of inhomogeneous deposition raises the question of preferential locating on special sites, e.g. defects or heteroatoms, or mechanical dispersion as origin of the irregularities. In case of the former the NPs located at defects could also take effect on the very same during accelerated stress tests (AST). The deposition of 2 nm Pt NPs on carbon blacks demonstrated a preferred deposition at surface defects and heteroatoms.35 For the 1% Ir/C reference system the dispersion of nanoparticles is improved compared to the post preparation modified catalysts, this might be an effect of larger surface area or more homogeneously distributed anchor points (e.g. defects).
The amounts of iridium and iron (only for the reference sample) were determined by NAA. Ir is one of the most chemically stable metals. It does not dissolve in aqua regia and elaborate digestion procedures are necessary.36 Based on this, indirect methods that pre-require dissolution of the catalyst, tend to underestimate the content. NAA enables a non-destructive determination of Ir and Fe,37 and is thus favored. The iron content of Ref was 2.59 wt% and was within the error margin compared to previous values.25 The iridium reference sample Ir/C contained 0.38 wt% Ir, that was much less compared to the intended 1 wt%. Similarly, also for Ref-2% Ir and Ref-4% Ir the measured values were much smaller with 0.61 wt% and 1.27 wt% Ir, respectively. The comparison of these three samples illustrates that the yield of precipitate during polyol synthesis of the NP was much smaller than intended (compare Experimental section).
The Ir value of 0.74 wt% obtained for the Pyro sample was slightly larger compared to the nominal value, considering typical yields and the initial Ir amount in the precursor.25 The smallest Ir value was found for GD with only 0.11 wt%. If all iridium would have been deposited on the catalyst during galvanic displacement, a maximum of 2 wt% would have been possible. This illustrates, that only a partial fraction was integrated. Independent of the low amounts of iridium it is clear that the achieved Ir loadings were typically much less than intended with GD < Ir/C < Ref-2% Ir < Pyro, Ref-4% Ir. As shown later, the trend is almost similar in XPS.
Raman spectra were recorded to identify the carbon morphology and to estimate the ratio of ordered and disordered carbon. For each catalyst, spectra were recorded at different spots and averaged spectra are shown in ESI Fig. S4.† Fig. S5† provides the fits of these spectra for all catalysts. The spectra were deconvoluted using five Voigt profiles with varying Lorentzian and Gaussian contributions. The main features of the obtained Raman spectra in Fig. S5† are broad D- and G-bands at around 1340 cm−1 and 1593 cm−1, respectively, as expected for amorphous sp2-bound carbons.38 They are matching the characteristics of FeNC catalysts.20,22,39 The Raman bands are related to scattering at graphene layers doped with heteroatoms and layers including defective structures. Although metallic Fe is known to catalyze graphitization during pyrolysis,40 the spectra suggest a highly amorphous and defective carbonaceous material with graphene as the structural unit. For the present synthesis route the addition of sulfur in combination with a pyrolysis temperature of 800 °C yielded highly amorphous carbon.39 The bands in Fig. S5† are originating from different vibrations and defective modes: the G-band arises from in-plane C–C bond stretching E2g modes, that can be visualized as vibration of the sub-lattices of graphene against each other.38,41,42 The D-band is attributed to the A1g breathing mode near the K point allowed at crystallite boundaries.38,42 The D2-band is assigned to a modulated E2g mode near the Γ point of boundary graphite layers.43,44 Thus, while the G-band is associated with interior graphite layers, the D2-band is associated with surface layers of graphene. The D3-band is assigned to organic functionalities and fragments in amorphous carbons but also to interstitial defects and interstitial heteroatoms between the layers or structural units.20,44 The D4-band as shoulder of the D-band is related to mixed sp2–sp3 bonds, polyene structures and general organic compounds.45,46 It may arise from hydrocarbons or aliphatic moieties linked to the carbon layers.47
For all catalysts the relative areas associated with the bands (normalized to the G-band area) are similar within the error margin confirming that the iridium modification had no significant impact on the carbon morphology.
Impact of iridium modification on the catalytic activity
In the following it will be discussed to what extent the addition of iridium can affect the electrochemical characteristics. Fig. 3a provides cyclic voltammograms (CVs) in N2 saturated electrolyte after reaching steady state conditions. The FeNC reference catalyst shows the highest double layer capacitance, and a well-pronounced redox peak at ∼0.62 V. In a previous work, for a similar preparation, this redox peak was assigned to quinone/hydro-quinone (Q/HQ) on the basis of pH-dependent CV measurements.48 Therefore, we assume a similar origin, here. The redox peak is also present in the CVs of all other samples, although it is less pronounced. While the capacitance of Ref-2% Ir was almost similar to Ref, all other samples reached significantly lower capacitance. The Pyro catalyst was the only sample that exhibited clearly pronounced hydrogen adsorption and desorption peaks within the CV. This is surprising considering the fact, that Ref-4% Ir contained almost the double amount of iridium. The last named sample was prepared by a polyol synthesis, based on this, a first hypothesis could be that the capping agents still block the surface of the catalyst. However, after deposition an intensive cleaning was performed. Together with the initial potential cycling prior to these CVs a clean surface can be assumed. As also a similar capacitance compared to the other samples was reached, we do not assume that the capping agents account for the missing Hads/Hdes regions. Further origins could be found in the different sizes of nanoparticles or a much better electronic interaction of Ir with carbon in case of Pyro. From these two options, we assume that the improved interaction with the carbon support in case of Pyro is more likely at the origin of the observed Hads/Hdes in this sample.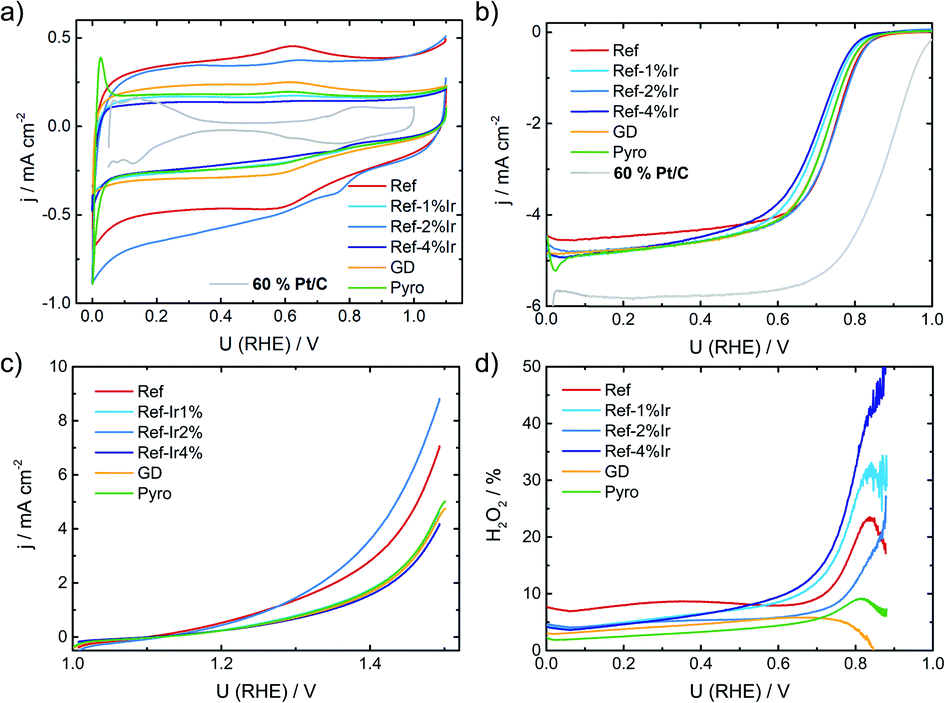 | ||
| Fig. 3 (a) Cyclic voltammetry of the different catalysts in N2 saturated 0.1 M H2SO4, (b) RDE measurements in O2 saturated electrolyte at 1500 rpm, (c) test for oxidative current density in the SSC potential range and (d) relative formation of hydrogen peroxide, determined from the RRDE measurement at 1500 rpm. All measurements performed with a catalyst loading of 0.13 mg cm−2 and a sweep rate of 10 mV s−1. For reasons of comparison CV and RDE of a commercial Pt/C catalyst are added to (a) and (b), concrete measurement conditions are given in Kübler et al.23 | ||
In Fig. 3b the RDE measurements in O2 saturated electrolyte are shown. Induced by iridium addition the half-wave potential E1/2 shifted to less positive values, indicating lower catalytic activity towards the ORR. In the ESI, Fig. S6† the electrochemical measurements on Ir/C are given for comparison. In the CV, the hydrogen adsorption and desorption currents are visible again. Related to the ORR, the onset of Ir/C was far below 0.5 V. Nonetheless, as it contributes only marginally to the overall composition of the catalyst (in terms of weight percentage) no pronounced effect on the activity has been expected for the modified catalysts. In order to elucidate possible origins of the lowering in ORR activity, structural characterization, as discussed below, was performed. For reasons of comparison in Fig. 3a and b the CV and RDE measurement of a 60% Pt/C is added, as taken from Kübler et al.23
Fig. 3c provides information on the oxidation capability (oxygen evolution reaction (OER) and carbon oxidation reaction (COR)) of the catalysts in the start-up and shutdown cycle (SSC) condition applied during accelerated stress tests (ASTs), as discussed below. In Fig. 3d the selectivity towards hydrogen peroxide formation during ORR is given. Please note, that these measurements were made at a comparatively small loading in relation to most published data on FeNC catalysts. Due to the possibility of a 2 × 2 electron reduction with peroxide as the intermediate, measurements at high loading underestimate the true peroxide formation.49–53 As peroxide formation can lead to a further oxidation of the carbon support or might attack the active sites,54 peroxide formation was tracked during activity measurements and between the ASTs (see below). The peroxide formation was largest for the FeNC reference catalyst Ref with approx. 8% H2O2 and decreased for the post preparation modified samples, while it was lowest for the Pyro sample at a level <5% H2O2. For reasons of comparison, the often as benchmarking catalyst considered “Fe0.5” by Jaouen's group produces 20% H2O2 at a similar loading.21
Insights into iron signature and electronic state of iridium
In order to characterize iridium on the surface of the catalyst, X-ray photoelectron spectroscopy (XPS) was performed of the iridium modified catalysts. In Fig. 4 the Ir 4f, C 1s and N 1s finescan regions are given (see ESI Fig. S7† for survey scans and N 1s and C 1s fine scan regions of Ref as well as Fig. S8† for all Fe 2p fine scan regions). Table 1 summarizes the near-surface elemental composition in atomic percentage, Table S2† summarizes iridium and iron contents in wt% as well as a rough estimate of the ratios of Ir4+ to Ir0. The iron content was similar for all samples.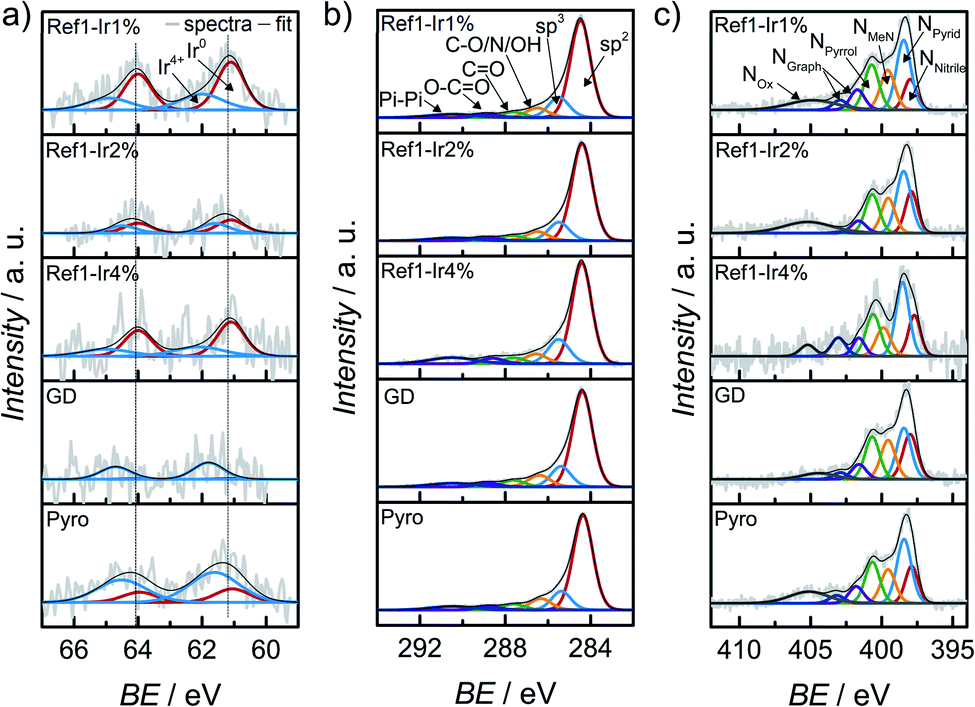 | ||
| Fig. 4 X-ray photoelectron spectroscopy (a) Ir 4f, (b) C 1s and (c) N 1s finescan regions for all catalysts modified with iridium. The dashed lines in (a) represent the position for Ir0. | ||
| In at% | Fe | Ir | N | O | C | S | Cl |
|---|---|---|---|---|---|---|---|
| Ref | 0.27 | — | 4.05 | 5.41 | 88.90 | 0.53 | n.d. |
| Ref-Ir 1% | 0.25 | 0.03 | 4.58 | 3.89 | 90.02 | 0.60 | 0.62 |
| Ref-Ir 2% | 0.31 | 0.04 | 4.37 | 4.28 | 89.72 | 0.50 | 0.78 |
| Ref-Ir 4% | 0.31 | 0.12 | 4.14 | 4.01 | 90.23 | 0.80 | 0.39 |
| GD | 0.25 | 0.01 | 3.87 | 5.90 | 88.58 | 0.35 | 1.02 |
| Pyro | 0.37 | 0.06 | 4.69 | 4.66 | 88.32 | 1.10 | 0.79 |
Considering the Ir 4f XP spectra in Fig. 4a mixed metallic and oxidized Ir phases are detected for the Ir NP modifications Ref-1% Ir, and Ref-4% Ir with 33–43% of oxidic Ir. Similar to NAA, the near-surface iridium loading obtained from XPS was lower than the nominal intended amounts, however, the trends are the same for both techniques.
The Ir 4f narrow scan in Fig. 4a of the GD catalyst shows a high signal-to-noise ratio. The higher binding energy position might indicate a higher degree of oxidation as compared to Ref-x% Ir (see the dashed lines as indicator of the main contribution of Ref-x% Ir). Even though not as pronounced, also the Pyro sample exhibits a shift to higher binding energies. Thus, while the metallic state seems more favorable for the Ir NP synthesized by the polyol method, GD and Pyro seem to favor higher oxidic Ir contributions.
Fig. 4b shows the C 1s XPS narrow spectra with speciation assignment. The main component is sp2 bonded carbon corresponding to graphene layers. However, besides sp2 (61–69%) significant amounts of sp3 (13–16%) within the carbon are present as well. The nitrogen bound carbon C–N is not distinguishable from C–O and C–OH with conventional XPS. The combined portion of C–O/N/OH accounts for 7–9% within the carbon C 1s signal. For all samples the N/Fe ratio from XPS was ≫ 4.
It is interesting to note, that the GD sample has the lowest nitrogen content. While the iridium chloride addition during the acid leaching was supposed to enhance the removal of iron with simultaneous deposition of Ir, it was not assumed that the nitrogen content will be affected on the basis of previous results in which we compared non-leached and acid leached samples.55 This could indicate that the processes during acid-leaching were possibly changed by the presence of iridium chloride.
XPS narrow scans are discussed in the following to elucidate the nitrogen chemical states in detail. The main species in all spectra is NPyrid with area ratios of 24–31% within the N 1s peak. Furthermore, the NMeN with 13–18% and NPyrrol with 17–20% can also contribute to the ORR activity. Jaouen et al.48 reported comparable shares of nitrogen species for another catalyst obtained from the oxalate supported pyrolysis of porphyrins with 17% for NMeN, 34% for NPyrid and 15% for NPyrrol within the N 1s peak. Kramm et al.55 observed the development of NPyrid and NPyrrol after pyrolysis at 800 °C and a subsequent acid leaching and assigned them to distorted FeNx-centers besides the NMeN. Recently, Marschall-Roth et al. observed a shift of the metal nitrogen binding peak to more positive values for the pyridinic in comparison to the pyrrolic nitrogen coordination, for their Fe(phen2N2) complex.56 In consequence, the overall region up to ∼402 eV could be associated with different forms of FeN4 moieties.
The iron speciation of the catalysts was evaluated by applying 57Fe Mössbauer spectroscopy (Fig. 5). All spectra are of equal appearance and were fitted assuming the same three doublet sites, which can be assigned to different FeN4 environments.49 Nevertheless an overlay of inorganic clusters or nanoparticles cannot be ruled out as contributions to the doublets by RT measurements.57 The Mössbauer parameters together with the assignment to iron species are provided in the ESI, Table S3.†Fig. 5f compares the relative absorption areas of the catalysts. While a similar composition was expected for the reference catalyst and the two post pyrolysis modified catalysts, it was unclear to what extent GD and Pyro might be affected by the presence of iridium during the acid leaching, respectively, pyrolysis step. Neither during pyrolysis nor during acid leaching, the addition of iridium had a significant impact on the relative absorption areas of the doublets.
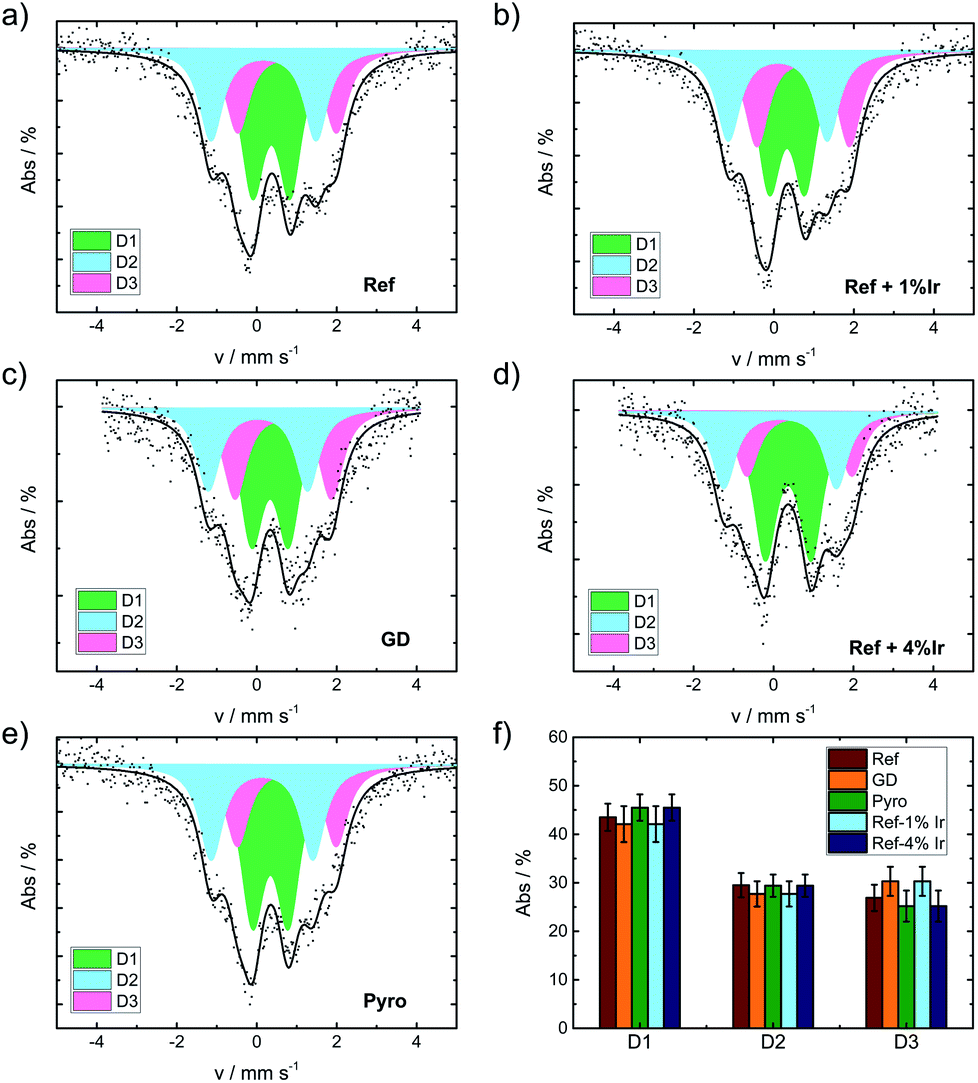 | ||
| Fig. 5 Mössbauer spectra of the catalysts as indicated (a) Ref, (b) Ref+1% Ir, (c) GD, (d) Ref+4% Ir, (e) Pyro and (f) comparison of the relative absorption areas for the different catalysts. | ||
In order to track possible changes of the Mössbauer parameters, Fig. 6a compares isomer shift and quadrupole splitting of all the doublets of the five catalysts. The quadrupole splitting ΔEQ is a measure of the electric field gradient at the iron nucleus. In case of D1 only marginal changes were observable except for Ref-4% Ir which had a larger quadrupole splitting. For D2 there was a slight decrease of ΔEQ what might indicate a brief variation of stacking of the FeN4 moieties in the graphene sheets, in similarity to variations observed for α- and β-iron phthalocyanine.58
 | ||
| Fig. 6 (a) Comparison of the Mössbauer parameters of the three doublets for the different catalysts in quadrupole splitting vs. isomer shift plotting. With dashed lines the isomer shift values of the FeNC reference catalyst Ref are highlighted, in (b) the relation between the balance point of the Mössbauer spectra (as average measure of electron density on all iron species) and the amount of iridium is given. | ||
The isomer shift δiso is related to the electron density on the iron center and therefore of particular interest in the view on catalysis.59 For D2 and D3 the isomer shift is highest for Ref and decreases for all iridium modified catalysts. The presence of a new iron species, e.g. in form of an iridium – iron system can lead to a change of the isomer shift.60 Mößbauer et al.60 observed an increase in isomer shift by increasing Ir content that was attributed to an increase in electron density for metallic Fe–Ir samples. Based on the preparation conditions, however, we do not expect the formation of any iron iridium bimetallic components. Such a bimetallic component would then also be expected to contribute with a new iron signature in the Mössbauer spectra.
As a consequence, it can be assumed that the interaction of iridium with FeN4 moieties with the carbon matrix either directly or indirectly causes slight electronic variations on the iron signatures. (Please note: as direct interaction we contemplate the presence of iridium in close vicinity to the FeN4 centers and as indirect one can consider an interaction with the carbon black which in turn affects the properties of FeN4 sites). Indeed, support effects are well known in catalysis specifically also for molecular sites.61,62 Thus, the question arises how bulk-like the character of nanoparticles must be to inducing similar kind of interaction as a support?
Being aware of the limitations of RT Mössbauer spectroscopy specifically for this group of catalysts; however, the parameters of the individual doublets should be considered carefully as iron and iron oxide clusters or nanoparticles could possibly overlay (particularly with D1).57 Based on this, in Fig. 6b the balance point as measure of the average electronic state of iron is plotted as a function of the iridium content from XPS. For doublet sites the balance points are identical to their respective isomer shifts. In order to determine the balance point of the overall spectra, the integrated absorption area was determined and the balance point equals the velocity where half of the area is obtained.
Moreover, possible correlation attempts between the Mössbauer balance point or the balance point in N 1s and Ir0 as well as correlation graphs for individual sites can be found in the ESI, Fig. S9.†
There was a trend of decreasing isomer shift with increasing iridium content. As the trend was opposite to the observations made by Mößbauer et al.60 two aspects should be noted: the type of interaction is different in nature. Induced by Ir modification, herein a decrease of the isomer shift and increase in electron density was found, while at the same time with respect to Fig. 3b a decrease of E1/2 was observed.
The GD sample is falling out of trend. A possible origin could be a much smaller size of iridium particles in this catalyst. The impact of iridium modification on the iron signatures in FeNC catalysts is certainly an aspect of great interest for future work as it seems connected to the ORR activity. But identifying the origin of it requires so much additional characterization that it goes beyond the scope of this work.
Influence of iridium addition on the degradation of FeNC catalysts in accelerated stress tests mimicking the start-up and shutdown conditions
For the purpose of investigating the degradation and durability of FeNC catalysts, ASTs in a RRDE setup were performed. The experimental conditions were selected such that they mimic the SSC conditions that can appear in a proton exchange membrane fuel cell (PEM-FC) during automotive applications (see experimental part for details). After a specific number of cycles, CV and RRDE were performed and at the end of the test, again CV, RRDE and the oxidative current in the 1.0–1.5 V potential range were recorded. Fig. 7 compares the Beginning of Test (BoT) and End of Test (EoT) electrochemical data. In the ESI† the changes over the number of SSC cycles are shown for all samples with respect to CVs in N2, RDE, and H2O2 (Fig. S10–S12†).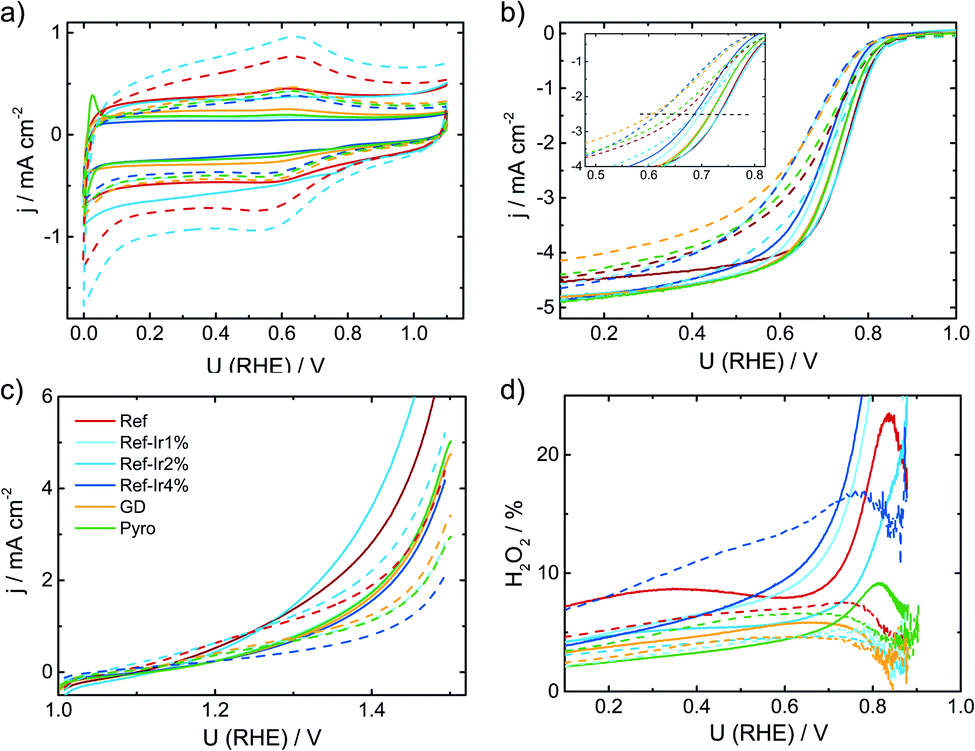 | ||
| Fig. 7 Comparison of beginning of test and end of test (+10k cycles in SSC range, dashed line) electrochemical data of all investigated catalysts. (a) Cyclic voltammetry in N2 saturated 0.1 M H2SO4, (b) RDE measurements in O2 saturated electrolyte at 1500 rpm, (c) test for oxidative current density in the SSC potential range and (d) relative formation of hydrogen peroxide, determined from the RRDE measurement at 1500 rpm. All measurements performed with a catalyst loading of 0.13 mg cm−2 and a sweep rate of 10 mV s−1. | ||
During the SSC cycles, new pores can be formed if the carbon oxidation reaction (COR) is the dominating current contributor. An increase in capacitance is caused by an increased accessible surface area in course of the formation of new pores or utilization of existing surface area (e.g. by enhanced wetting).22 Pseudo capacitive redox groups (Q/HQ) are created by partial oxidation of surface carbon.
Kumar et al.22 observed for SSCs at 80 °C of metal-NC catalysts an initial increase in capacitance but after excessive cycling the capacitance was declining as newly formed pores could not compensate for the loss of carbon as CO2/CO.22 For a more systematic discussion of the changes, in Fig. 8a the capacitance is plotted as a function of cycle number, Fig. 8b gives the relative change in capacitance.
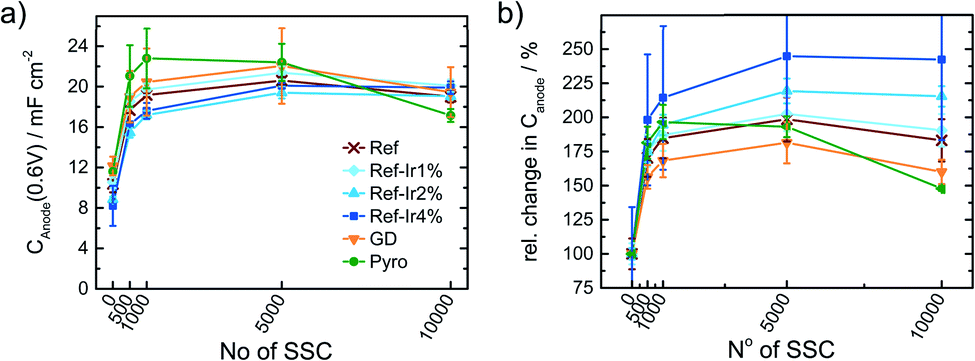 | ||
| Fig. 8 (a) Change in capacitance and relative change in capacitance as a function of the number of SSC cycles applied to the catalysts. Values extracted from CV measurements in N2 saturated 0.1 M H2SO4. | ||
Similar to Kumar et al.,22Fig. 8a depicts an increase in capacitance as visible for the first 0.5–1k cycles with a decline after completing 10k cycles.
Exploring the relative changes in capacitance in Fig. 8b, the subsequent addition of Ir NP to the FeNC causes a large and systematic increase in capacitive current. The increase in double layer capacitance under N2 is an indication for carbon oxidation at the surface. However, a relation from the capacitance change to possible destruction/oxidation of the active site is not straightforward.63 The performance loss is not directly correlating with the growth of capacitance. Considering the absolute development of the capacitance for Ref-x% Ir, the lower initial values could also result from residual capping agents from the polyol method. In course of the high potential cycling, those are oxidatively removed and more carbon surface is exposed to the electrolyte.64 Typical platinum group metal (PGM) features that are normally present in the hydrogen adsorption/desorption region are partly observed for the Ir modifications.
In the Ref-x% Ir series only for the Ref-4% Ir at the beginning of life (BoT) before AST a slight hydrogen underpotential deposition (HUPD) region is noticeable in Fig. 7a. The absence of features can be explained by the very low loading of Ir determined by XPS and NAA and the small particle size. For the Pyro sample more pronounced features are existing which also diminish after 1k of SSC cycles. From the electrochemical response this sample contained significantly larger Ir particles than the polyol synthesized ones. Interestingly, this sample also exhibited a typical capacitance increase but the greatest fall off at the EoT. This could indicate a catalytic role of Ir during SSC cycling.
For the more detailed analysis of the changes in catalytic activity as a function of SSCs, Fig. 9 provides the absolute and relative changes in kinetic current density, half wave potential and hydrogen peroxide formation. During the AST the main losses in ORR activity are visible for the first 0.5–1k cycles for all investigated catalysts. The development of the kinetic current density jkin at 0.75 V follows an exponential decay, as it has been observed for other FeNC catalysts.5,22,65 Ref exhibits a decrease of 60% from BoT to EoT (after 10k SSCs) kinetic current density. In previous studies losses of ORR activity scaled from 15% (5k SSCs, 25 °C), 84% (150 square wave 0.9–1.4 V, 80 °C) to 85% (500 SSCs, 80 °C), while the temperature is a main contributor for the deactivation.20,21 The main activity loss during SSC is explicitly attributed to the destruction of Fe–Nx sites by COR of the matrix.14,22,24,26
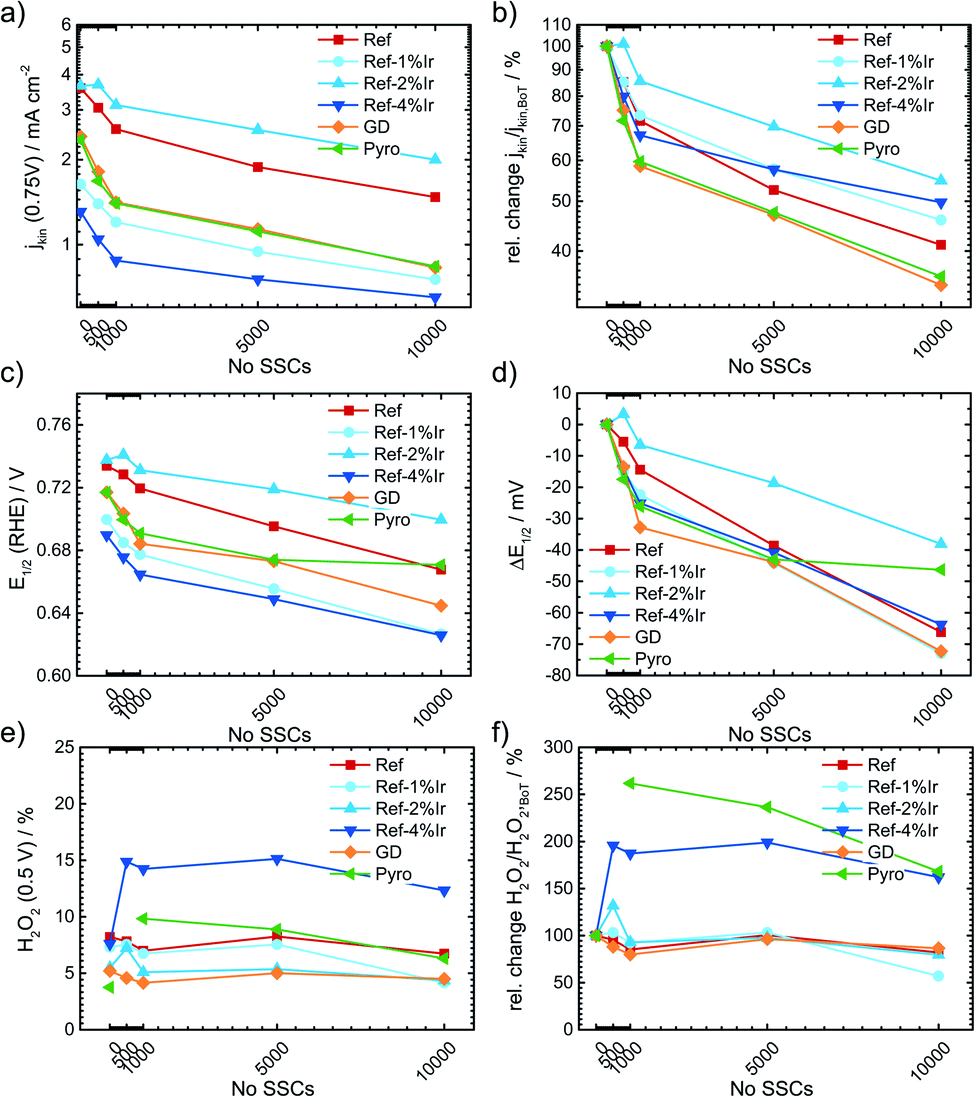 | ||
| Fig. 9 Change in kinetic current density (a) absolute scale, (b) relative scale, half wave potential (c) absolute value, (d) ΔE1/2 and hydrogen peroxide formation (e) absolute and (f) relative as a function of the number of SSCs. | ||
For the iridium modified catalysts only Ref-2% Ir provided the same starting activity compared to Ref, while at EoT even a larger fraction of activity is retained (55% vs. 40%). Indeed, with respect to the overall loss EoT/BoT all samples with post preparation addition of iridium revealed a larger remaining relative activity than Ref. In case of GD and Pyro samples the retentions at EoT were worse compared to Ref. The half-wave potential E1/2 was determined as the potential where half of the diffusion limiting current density was reached. An inspection of E1/2 underlines the conclusions as obtained on basis of the kinetic current density. The half wave potential was also prone to SSC and was decreasing by 66 mV from 0.734 to 0.668 V. The smallest change in half wave potential was again found for Ref-2% Ir with <40 mV. It is interesting to note, that the selectivity did not change for most of the catalysts. Only, in the case of Ref-4% Ir and Pyro it increases by a factor of 2–3 depending on the number of SSCs. Notably, these two samples exhibited the largest quantity of iridium.
Raman spectra were also recorded after SSCs and analyzed at EoT condition. The spectra are already integrated in ESI, Fig. S5.† For all samples a decrease in the regions associated with the D-band, D3-band and D4-band occurred. These changes were smallest for Pyro and Ref-2% Ir that also exhibit the smallest change in half-wave potential. This indicates that the iridium modification can indeed prevent the oxidation of carbon while the diverse behavior of the samples does not allow a concrete conclusion which structural attribute of Ir is most important for the effect.
For the Ref catalyst, Ref-2% Ir, and Ref-4% Ir the selectivity towards oxygen evolution reaction (OER) in the oxidative current density region was further evaluated by RRDE technique. As under the given experimental conditions only COR and OER are expected but no other reaction products are known to contribute to the current density, it can be assumed that the remaining percentages to 100% are associated with any form of COR. Even though it is known that this approach typically underestimates the true faradaic efficiency towards OER,66 it will provide the possibility to deduce trends, thus a qualitative rather than a quantitative comparison between the samples.
In Fig. 10a and b the disc together with the ring currents as a function of the potential and the faradaic efficiency towards OER are plotted, respectively. For each experiment two cycles were measured after activation of the electrodes, while the efficiencies are only provided for the 2nd scan. The contribution of OER to the overall current was with <5% still rather low. However, an enhancement through Ir addition can be clearly seen in the plot.
 | ||
| Fig. 10 (a) RRDE measurements to determine the faradaic efficiency for the OER in the overall oxidative current at iDisc, iRing was kept at 0.4 V and 0.2 V during the first and second cycle; respectively, and relates to the ORR. In (b) the faradaic efficiencies for 1 mA cm−2, 3 mA cm−2 and 5 mA cm−2 are provided for the 2nd scan. All measurements in N2 saturated 0.1 M H2SO4 at a loading of 0.13 mg cm−2. | ||
In situ and locally resolved Raman spectroscopy
To get a better idea, whether the iridium modification of the reference catalysts indeed affected carbon oxidation during SSC conditions, locally resolved in situ Raman spectra were taken for Ref and Ref-4% Ir.The cyclic voltammetry of both samples and a picture of the setup are shown in ESI, Fig. S13.† While the absolute current density values were higher than in the measurements in the standard setup, the trends remained the same. The higher current densities can be explained by the five times larger sweep rate. In total 5000 SSCs were performed and spectral changes were followed on 49 spots at BoT, after 1000 SSCs and EoT. In Fig. 11, the averaged Raman spectra are provided together with the fits. Considering the standard deviation, it can be noted, that over the 49 measurement sites the Raman spectra vary to a distinct extend. Nonetheless, it becomes clear, that changes related to the D3-band and D4-band are more pronounced for Ref compared to Ref-4% Ir. For the D-band the changes look less intensive, but close to each other. This is also confirmed when the areas of the individual bands are compared (Fig. 11c and d).
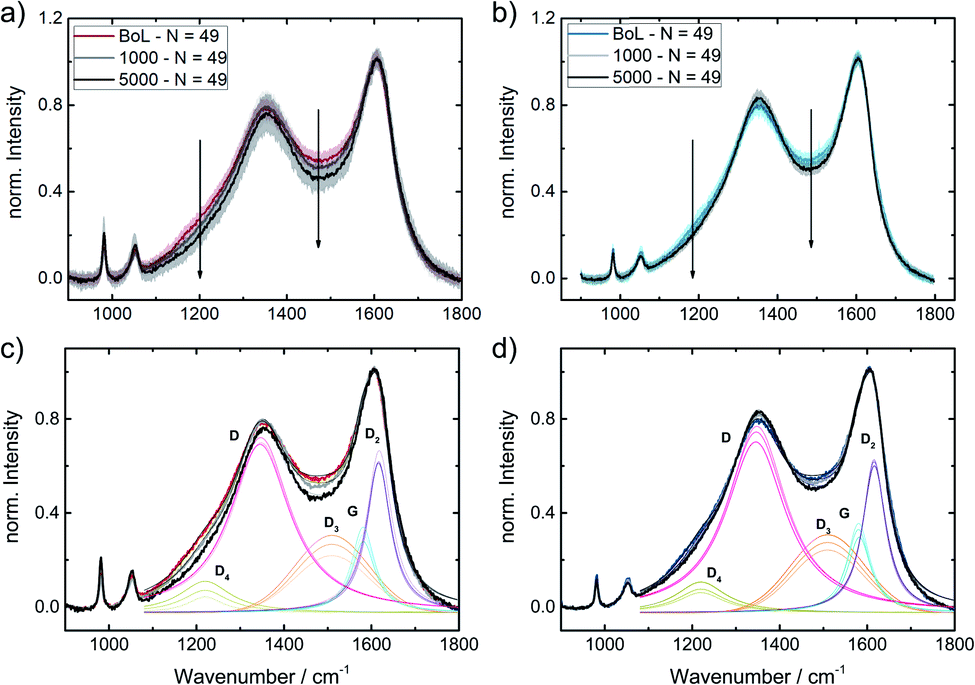 | ||
| Fig. 11 Averaged spectra including standard deviation for (a) Ref and (b) Ref-4% Ir catalysts and fitted spectra of (c) Ref and (d) Ref-4% Ir catalysts. Spectra were obtained BoT, after 1000 and 5000 SSCs at RT in the in situ Raman setup described in the experimental section. | ||
Based on these considerations, we can conclude that iridium modification lowers the oxidation of amorphous carbon connected with heteroatoms (D3) and hydrocarbons (D4). It seems likely that to some extent this diminution of carbon oxidation is linked to the larger faradaic efficiency for OER observed for the iridium modified catalysts. Nonetheless, it is likely that larger εFaraday would be required for a complete suppression of degradation related to carbon oxidation in this range.
Influence of iridium addition on SSCs in fuel cell application
Finally, it is discussed to what extent the positive effect of Ir addition on the stability of FeNC during SSCs can also be transferred to fuel cell condition. Therefore, Ref, Ref-2% Ir and Ref-4% Ir were investigated with respect to degradation in PEM-FC. Fig. 12 compares the polarization curves at BoT, after 50, 100 and 150 SSCs.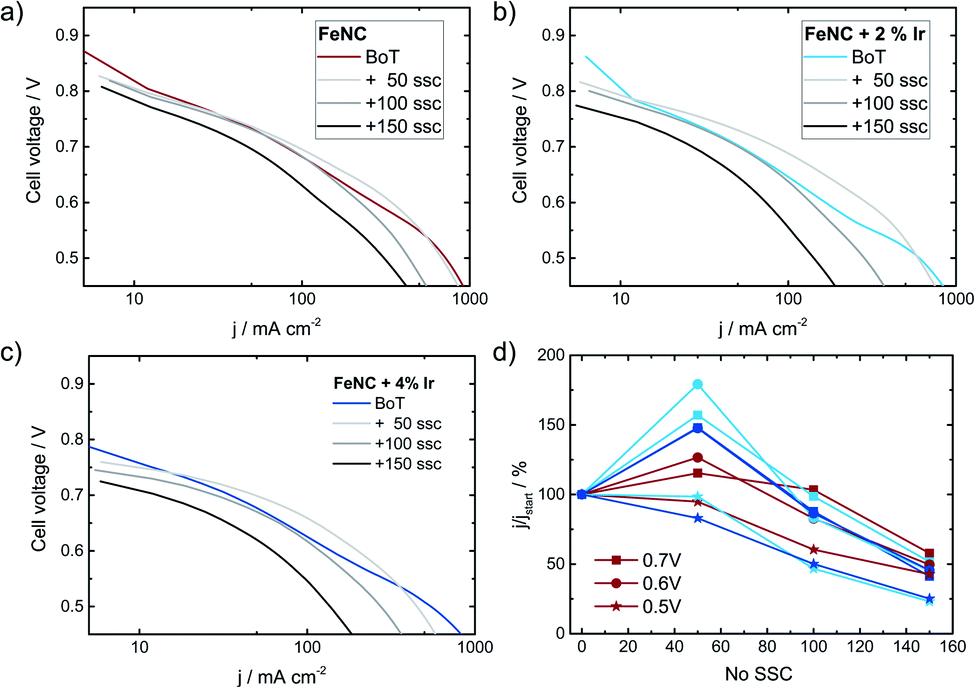 | ||
| Fig. 12 Fuel cell polarization curves in H2/O2 at 80 °C for (a) Ref, (b) Ref-2% Ir and (c) Ref-4% Ir at a cathode catalyst loading of 3.5 mg cm−2 (both Ir added catalysts), 4.1 mg cm−2 for Ref. Measurements were made with 1 bar backpressure and a Nafion 212 membrane. In (d) the change in current density at cell voltages of 0.7, 0.6 and 0.5 V is given. The provided curves are averaged values out of two individual measurements. SSCs were performed with H2 gas flow at the anode and N2 gas flow at the cathode. | ||
The highest open circuit voltage was recorded for Ref followed by Ref-2% Ir and Ref-4% Ir. All catalysts exhibited a pronounced degradation over increasing number of SSCs. In Fig. 12d the relative change in current density is compared for three cell voltages. For all catalysts, induced during the first 50 SSCs an increase in current density is observed at U > 0.5 V. This indicates an improved performance during these first cycles. Under further cycling, however, all catalysts lose performance so that after 150 SSCs only 25–50% of the initial current density remained (depending on cell voltage). How the benefit of iridium modification can be kept for >50 SSCs and why the performance goes down need to be addressed in future work. As shown by Choi et al.17 for another FeNC catalyst the oxidation current associated with the COR appears already at much smaller potentials when the temperature is raised from 20 °C to 70 °C. In consequence, while the oxidative current is neglectable at 1.5 V and 20 °C it is approximately 10 fold at 70 °C.
This might explain why the transfer of the results obtained at RT to FC condition was not successful: the reaction rate for COR seems to increase faster with increasing temperature as compared to the OER. In consequence, while the improvement is visible at RT in RDE, further optimization is required for FC application.
Conclusions
An iridium-based approach to potentially stabilize FeNC against carbon oxidation was investigated. Iridium was either added to the precursor mixture (Pyro), during the acid leaching (GD) or post preparation (Ref-x% Ir). The iridium loadings were intended to reach approx. 1–4 wt% for all samples. NAA indicated final quantities between 0.11 wt% (GD) and 1.27 wt% (Ref-4% Ir).The size and the distribution of Ir particles depends on the procedure how Ir was added while the morphology of the carbon matrix remained unchanged. Independent of the procedure, the iridium addition led to a lowering of the initial activity in terms of half-wave potential. A possible origin might be found in a decrease of average electron density on iron as expressed by the balance point of the Mössbauer spectra. This could indicate some electronic interaction which needs to be investigated in more detail in future work as such electronic interaction might also apply to other metals as e.g. Pt in Pt/FeNC or Pt/CoNC hybrid catalysts.
The durability of the catalysts under SSC conditions was evaluated for 10k cycles in RRDE experiments and accompanied by OER selectivity measurements and in situ Raman spectroscopy of selected samples. Under these wet-chemical conditions, Ir led indeed to a slight improvement of the durability. A larger faradaic efficiency towards the OER and less carbon oxidation were found. However, transferring the approach to FC condition provided an improvement only within the first 50 SSCs whereas at EoT (150 SSC) the relative change in current compared to BoT remained the same. The unfavorable improvement of the kinetics of the COR over OER might be at the origin of this. The results illustrate that further optimization and possibly a better dispersion of iridium on FeNC is required for stabilization during SSC under FC conditions. Nonetheless, the trends during RDE are promising and could provide an alternative way of FeNC stabilization, while the carbon morphology can remain the same.
Experimental
Preparation of the catalysts
In order to prepare the catalysts, all chemicals were used as received. Iron(II) oxalate dihydrate (99%, Alfa Aesar), 5,10,15,20-tetrakis-(4-methoxyphenyl)-21,23H-porphyrin (H2TMPP, Porphyrin Laboratories) and sulfur (99.5%, Carl Roth) were used as Fe/C/N/S precursors for the FeNC catalyst. The pyrolysis took place in nitrogen atmosphere followed by an acid leaching. Then the catalyst was treated with forming gas consisting of 10 ± 1% H2 in N2 (ARCAL F10, Air Liquide). The acid leaching (AL) was performed in aqueous HCl which was diluted from concentrated HCl (37%). For the galvanic displacement a 1 g L−1 iridium ICP standard solution (99.999%, Carl Roth) from IrCl3 in 10% HCl was used.For the preparation of Ir NPs, the metal salt iridium acetylacetonate (Ir(acac)3, 98%, Strem Chemicals) was used as precursor in diethylene glycol dibutyl ether (DEDB) (99%, Sigma Aldrich) with the reducing substance 1,2-tetradecanediol (90%, Sigma-Aldrich) and the capping agents oleylamine (70%, Sigma-Aldrich) and oleic acid (Ph. Eur., Sigma-Aldrich). Additional chemicals are acetone (Fisher Scientific) and n-hexane (95%, Carl Roth).
000 rpm and the supernatant discarded. To remove residual organics, the material was washed twice with 5 mL n-hexane, dispersed for 2 min in the ultrasonic bath, precipitated with 5 mL acetone and collected via centrifugation for 20 min at 10000 rpm. The dark brown to clear supernatant was discarded. The as prepared NPs were dried in air and mortared for homogenization. The yield was 74% and 31% for the first and second batch, respectively. From previous projects, Ir NPs yielded 100% in relation to the metal mass in the precursor but are known to be coated with residual capping agents (15–40%). For further modification an iridium content of 80% was estimated, i.e. 20% of the weight in NPs were supposedly side products from the synthesis.
After cooling down below 100 °C the material was transferred into 2 M HCl which was accompanied by a strong H2S evolution. The acid leaching was performed for 1 h in an ultrasonic bath (model: Emmi H 120, EMAG AG) and leached overnight.
The catalyst was filtered with a 0.1 μm PVDF membrane (Durapore®, Millipore), dried in air at 60 °C and mortared for homogenization. The yield after this step in relation to the overall precursor mass was 8%, in agreement with previous reports.34,67
A forming gas treatment was performed in quartz glass boats in the same furnace with a ramp of 600 °C h−1 up to 800 °C and was held for 30 min. The heating and cooling was performed under nitrogen atmosphere. A subsequent acid leaching in 2 M HCl was carried out, again in an ultrasonic bath for 1 h with leaching overnight.
The FeNC catalyst was again separated by filtration, dried in air at 60 °C and mortared for homogenization. At this stage the catalyst is termed “Ref”. The yield was 7% in relation to the total precursor mass and in agreement with previous work.25
000 rpm. The catalysts are labeled “Ref-1% Ir”, “Ref-2% Ir” and “Ref-4% Ir” according to the intended loading of iridium. Yields of 94% for Ref-1% Ir, 97–101% for Ref-2% Ir (2 batches), 96–97% for Ref-4% Ir (2 batches) were obtained.
Electrochemical characterization
The 0.1 M H2SO4 electrolyte was prepared from concentrated sulfuric acid (98%) and deionized water (house line or 18.2 MΩ cm−1 Smart2Pure UV, Thermo Scientific Barnstead). For the ink in half cell measurements water (house line or 18.2 MΩ cm−1 Smart2Pure UV, Thermo Scientific Barnstead), Nafion® solution (5 wt%, Sigma Aldrich or QuinTech) and isopropanol (99.5%, Fisher Scientific or Carl Roth, 99.95%) were used.For the ink in PEM-FC measurements water (18.2 MΩ cm−1 Smart2Pure UV, Thermo Scientific Barnstead), Nafion® solution (5 wt%, QuinTech) and isopropanol (Carl Roth, 99.95%) were used.
The gases for the fuel cell measurements were Alphagaz™ 1 N2 (99.999%, Air Liquide), Alphagaz™ 1 O2 (99.998%, Air Liquide) and Alphagaz™ 1 H2 (99.998%, Air Liquide). For the MEA Nafion® NM-212 (QuinTech) was used in combination with a 250 μm thick GDL (H23 C9, Freudenberg). The membrane was pretreated by boiling in 3 wt% H2O2 and subsequently activated in 0.05 M H2SO4 (diluted sulfuric acid 98%) and deionized water (18.2 MΩ cm−1 Smart2Pure UV, Thermo Scientific Barnstead) to remove residual organic species and impurities. As anode catalyst layer Pt/C (HisPEC 3000, 20 wt% Pt/C) was sprayed on a GDL. Gaskets were fabricated from PTFE foil (BOLA).
In order to obtain a catalyst loading of 0.13 mg cm−2 a suspension was prepared and coated on a GC disc. Therefore, 5 mg of catalyst powder was mixed with 615 μL water, 25 μL Nafion® solution and 360 μL isopropanol. The ink was dispersed twice for 45 min in the ultrasonic bath with intermittent 1–2 min mixing in the vortex shaker, while cooling the former with ice to keep the temperature below 30 °C. An aliquot of 6 μL ink was drop-casted on the GC disc of the RRDE electrode and dried at room temperature.
The potential drop Δφ between the working and reference electrode is dependent on the distance, the specific conductivity of the electrolyte and the current. In the present RRDE setup the positions of the electrodes to each other are fixed and an aqueous 0.1 M H2SO4 electrolyte is used. The averaged ohmic resistance is RΩ = 21.6 ± 1.5 Ω (15 iR-interrupt measurements) and assumed to be valid for all obtained RRDE experiments. The iR-free potential EiR-corr is calculated by subtracting the product of the current i and the resistance RΩ.
The used bipotentiostat has limited processing capabilities for continuous iR-corrected data acquisition and consequently the iR-correction was performed as post treatment on OER measurements only. A post-treatment becomes necessary for high currents, non-ideal cell geometry and low conductivity.
| C = djdν−1 | (1) |
Quality descriptors from the ORR measurements discussed in this work are the half wave potential E1/2, the kinetic current density jkin at 0.75 V, and the selectivity towards H2O2 formation.
In the probed potential window not only OER is taking place but also the undesired COR. By rotating the working electrode, formed oxygen is transported from the disc to the ring by convection. The ring potential is set to an ORR active potential (0.4 or 0.2 V) at which attained oxygen is reduced and a negative current recorded. To quantify the oxygen formation during the sweep, the faradaic efficiencies for j equal 1 mA cm−2, 3 mA cm−2 and 5 mA cm−2 are calculated with the ring current iR, the disc current iD and the collection efficiency Neff by eqn (2).
| εFaraday = (iRNeff−1)iD−1 | (2) |
As evaluation parameters the faradaic efficiency εFaraday, and related potential for the given current densities were considered.
The MEA was assembled from a FeNC cathode layer on the respective GDL, a Pt/C (0.12–0.16 mgPt cm−2) anode layer on the respective GDL and an activated Nafion® N-212 membrane. It was pressed at 125 °C with 5 kN for 2 min and cooled down under sustained pressure. A 4.82 cm2 cell was assembled with the MEA and PTFE gaskets aiming for a compression of 17–28% with a torque of 8 Nm.
A fuel cell test station (model: 885V, Scribner Associates) equipped with a potentiostat (model: 850e, Scribner Associates) was used with the software FuelCell® (version: 4.3e, Scribner Associates). The cell was operated at 80 °C with 100% humidified H2/O2. The flows of the gases were fixed to 0.2 L min−1 with 1 bar backpressure. Ohmic (iR) voltage losses were continuously measured with the current interrupt method.
The fuel cell testing protocol was initiated by determination of the OCV and a polarization curve was acquired. An AST protocol consisting of 50 SSC cycles between 1.0–1.5 V with 0.5 V s−1 was applied and the cell performance was tested after cycling. The SSC protocol was successively repeated three times yielding performance evaluations after 50, 100 and 150 cycles.
The performance of the PEM-FC is characterized by voltage vs. current density plots, with current density at 0.5 V, 0.6 V and 0.7 V.
Structural characterization
The spectra were eliminated of cosmic rays and corrected by a linear baseline taking the intensity between 900–927 cm−1 and 1773–1800 cm−1 into account. For further processing, a normalization around the maximum intensity (Imax = 1575–1625 cm−1) was performed. For this purpose, the previously determined maximum was averaged over ±7 cm−1 to account for fluctuations in the intensity. Averaged spectra from 48–49 points were deconvoluted using four Lorentzian profile line shapes for D1 at ∼1345 cm−1, G at 1582 cm−1, D2 at ∼1615 cm−1, D4 at 1150 cm−1 and a Gaussian peak for D3 at ∼1530 cm−1.
The following fitting constraints in Table 2 were applied with the respective fitting parameters.
| Band | Parameter | G | D1 | D2 | D3 | D4 |
|---|---|---|---|---|---|---|
| x c/cm−1 | Constraint | 1582* | 1337–1354 | 1606–1623 | 1450–1545 | 1140–1220 |
| FWHM/cm−1 | Constraint | 24–94 | 6–235 | 6–94 | 6–235 | 6–318 |
A cooled ultrasonic bath (21 °C) (model: SONOCOOL 255, Bandelin) was used for dispersing the ink. To obtain a loading of 0.13 mg cm−2, an aliquot of 5 μL was placed on top of a GC electrode with a disc diameter of ddisc = 5 mm (Adisc = 0.1967 cm2) inside a PTFE shroud. This electrode was used as working electrodes in an in situ setup. The electrode surface was prepared by polishing first with 0.3 μm alumina polish on a polish pad followed by polishing without any polish solution and finished by rinsing any residues with deionized water. A piece of carbon cloth and a Ag|AgCl|sat. KCl (model: SE11, Meinsberger) served as counter and reference electrodes. To reference the Ag|AgCl|sat. KCl electrode in respect to the RHE potential a hydrogen reference electrode (model: HydroFlex, Gaskatel) was used. All given potentials refer to the RHE. The measurements were conducted in 0.1 M H2SO4 electrolyte at room temperature using a potentiostat (Autolab PGSTAT128N, Metrohm Autolab B.V) with the software NOVA (version: 1.10.5, Metrohm Autolab B.V.) in a three-electrode setup. To protect the objective lens from acid electrolyte splashes, a quartz glass slide was placed between the lens and the electrolyte. Analogous to the RRDE experiments the electrochemical procedure before the subsequent in situ Raman measurements was carried out. The activity protocol was adopted for the CVs in argon saturated electrolyte. ORR activity measurements were not possible, instead the in situ Raman mapping of the BoT catalyst was performed at this stage. Then 1000 and further 5000 SSC cycles were performed and finished with subsequent CVs in argon according to the short activity protocol followed by Raman mapping of 49 individual spots.
Quantification of the spectra was done with CasaXPS (version: 2.3.16, Casa Software Ltd) offering a typical accuracy of ±10%. For the high-resolution scans a five-point Shirley background was adapted to account for inelastic electron scattering and the signals deconvoluted with mixed Gaussian/Lorentzian (70/30) line shapes. The fitted peak areas were corrected by atomic sensitivity factors implemented in the software. The oxygen 1s peak was corrected for the In–O contribution species from the indium foil on the sample holder.
Internal calibration to the peak of adventitious carbon at 284.6 eV C 1s spectra was not performed. The present samples contain significant amounts of oxygenated, graphitic or clustered carbon. These are influencing the global maximum peak of the C 1s spectra. A charge correction based on the maximum C 1s peak could therefore lead to false assignment or distribution of nitrogen components like pyrrolic and pyridinic N species.68 However, the position of the In peak from the In-foil was in agreement with the expected value.
The Fe 2p3/2 and Fe 2p1/2 signals were evaluated taking nitrogen bound iron (Fe–Nx) and iron oxides (Fe–Ox) with the corresponding satellites into account as shown in ESI, Table S4.†
The Ir 4f exhibits a low orbital splitting of 2.9 eV and to fit the spectra accurately both peaks of the superimposed doublet were fitted with additional peak area ratio constraint of 3:4, a peak separation of 2.9 eV and equal fwhm of the doublet. For the quantification the 4f7/2 contribution was evaluated. Two species, metallic iridium and iridium oxide, were considered in binding energy ranges of 60.6–61.1 and 61.6–62.1 eV, respectively, both with same constrains for fwhm (1.0–1.8 eV).
Table S5† lists the assignment of the N 1s species and fitting constraints. The N 1s region was evaluated according to Artyshkova et al.68 and Serov et al.69 in pyrolyzed CoTMPP and Fe–Ac/bipyridine-based ORR catalysts, Me–Nx was situated at 399.8 eV corresponding to a BE shift of 0.9–1.1 eV in relation to pyridinic nitrogen.68
This shift in BE was consolidated by density-functional theory (DFT) calculations for Me–N2 and Me–N4 as vacancy-and-substitution defects in graphene sheets.68 Ambient pressure XPS showed the MeN4 moiety is preferential over the MeN2 under oxidizing conditions.68 In spectra acquired with the resolution of a standard system MeN2 and MeN4 cannot be distinguished, but the overlapping peak position could influence the simulated peaks and relative amount of pyrrolic and pyridinic nitrogen making correlation with ORR activity from electrochemical experiments inaccurate.
The oxygen 1s peak was assigned to metal species (Me–O, Me–OH) with iridium or indium in the lower binding energy region. Organic oxygen was identified as hydroxyl (OH), ether bound (O–R), carbonyl and carboxyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and adsorbed water. The possible binding partners besides carbon are sulfur and nitrogen. However, they cannot be distinguished. The model constraints and species are depicted in Table S6.†
O) and adsorbed water. The possible binding partners besides carbon are sulfur and nitrogen. However, they cannot be distinguished. The model constraints and species are depicted in Table S6.†
Carbon is the main component of Fe–N–C catalysts and is a composite of the graphene layers (CC, sp2), amorphous fractions (C–H, sp3) and a variation of heteroatom compounds. Fe–N–C active FeNx-centers require C–N bounds that cannot be discriminated from oxygen or sulfur components in the present spectra. The identified components are listed in Table S7.†
Sulfur that is added to the precursor mixture can have various poor resolved species. The quantification is performed assuming the species in Table S8.† The low orbital splitting of 1.2 eV for the S 2p core level causes a strong overlap of the components and the mean energy level for 2p3/2 and 2p1/2 is evaluated.
The local chemical composition of the samples was obtained by EDS (for 60 s) and quantified using the software INCA (version: 4.15, Oxford Instruments) with a typical precision of ±5%.
Author contributions
The work was designed and supervised by UIK, CP performed all preparation and most of the characterization, so far not noted differently. MK was responsible for TEM and PEM-FC experiments, XPS was performed by SP and NH under supervision of WJ. SP performed BET measurements. LN performed the Mössbauer measurements and analysis. Standard Raman spectroscopy was performed under supervision of RWS, in situ Raman spectroscopy was performed by SJK and CP under supervision of MB. KE and CG performed NAA. The manuscript was written by CP and UIK. All authors contributed to the reviewing of the manuscript and have given their approval of its final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the BMBF (03XP0092), DFG (PAK981, KR3980/8-1) and the Stiftung Industrieforschung is acknowledged. The authors thank Charlotte Gallenkamp for providing the structure image used in the TOC graphic and Humera Khatoon Siddiqui and Nicole Segura Salas for weighting in the samples for NAA.Notes and references
- BP Energy Outlook: 2020 edition, https://www.bp.com/content/dam/bp/business-sites/en/global/corporate/pdfs/energy-economics/energy-outlook/bp-energy-outlook-2020.pdf Search PubMed.
- W. Bernhart, S. Riederle and M. Yoon, Fuel cells - a relatistic alternative for zero emission?, 2013, https://www.rolandberger.com/publications/publication_pdf/roland_berger_fuel_cells_20140113.pdf.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Recent Advances in Electrocatalysts for Oxygen Reduction Reaction, Chem. Rev., 2016, 116(6), 3594–3657 CrossRef CAS PubMed.
- P. Zelenay and D. Myers, ElectroCat (Electrocatalysis Consortium), US Department of Energy, Arington, 2019 Search PubMed.
- R. Chenitz, U. I. Kramm, M. Lefevre, V. Glibin, G. Zhang, S. Sun and J.-P. Dodelet, Energy Environ. Sci., 2018, 11, 365–382 RSC.
- E. Proietti, F. Jaouen, M. Lefévre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- Q. Yu, S. Lian, J. Li, R. Yu, S. Xi, J. Wu, D. Zhao, L. Mai and L. Zhou, J. Mater. Chem. A, 2020, 8, 6076–6082 RSC.
- S. Haller, V. Gridin, K. Hofmann, R. W. Stark, B. Albert and U. I. Kramm, Energy Technol., 2021, 9, 2001106 CrossRef CAS.
- A. Shahraei, M. Kübler, I. Martinaiou, K. A. Creutz, W. D. Z. Wallace, M. A. Nowroozi, S. Paul, N. Weidler, R. W. Stark, O. Clemens and U. I. Kramm, J. Mater. Chem. A, 2018, 6, 22310–22319 RSC.
- A. Shahraei, A. Moradabadi, I. Martinaiou, S. Lauterbach, S. Klemenz, S. J. Dolique, H.-J. Kleebe, P. Kaghazchi and U. I. Kramm, ACS Appl. Mater. Interfaces, 2017, 9, 25184–25193 CrossRef CAS PubMed.
- A. Morozan, V. Goellner, Y. Nedellec, J. Hannauer and F. Jaouen, J. Electrochem. Soc., 2015, 162, H719–H726 CrossRef CAS.
- S. Paul, Y.-L. Kao, L. Ni, R. Ehnert, I. Herrmann-Geppert, R. van de Krol, R. W. Stark, W. Jaegermann, U. I. Kramm and P. Bogdanoff, ACS Catal., 2021, 11, 5850–5864 CrossRef CAS.
- L. Delafontaine, T. Asset and P. Atanassov, ChemSusChem, 2020, 13, 1688–1698 CrossRef CAS PubMed.
- U. I. Kramm, M. Lefèvre, P. Bogdanoff, D. Schmeißer and J.-P. Dodelet, J. Phys. Chem. Lett., 2014, 3750–3756, DOI:10.1021/jz501955g.
- J. Herranz, F. Jaouen, M. Lefevre, U. I. Kramm, E. Proietti, J.-P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, P. Bertrand, T. Arruda and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 16087–16097 CrossRef CAS PubMed.
- G. Zhang, X. Yang, M. Dubois, M. Herraiz, R. Chenitz, M. Lefèvre, M. Cherif, F. Vidal, V. P. Glibin, S. Sun and J.-P. Dodelet, Energy Environ. Sci., 2019, 12, 3015–3037 RSC.
- C. H. Choi, C. Baldizzone, J.-P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed.
- C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, J. D. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273–A276 CrossRef CAS.
- A. Ohma, K. Shinohara, A. Iiyama, T. Yoshida and A. Daimaru, ECS Trans., 2011, 41, 775–784 CrossRef CAS.
- I. Martinaiou, A. Shahraei, F. Grimm, H. Zhang, C. Wittich, S. Klemenz, S. J. Dolique, H.-J. Kleebe, R. W. Stark and U. I. Kramm, Electrochim. Acta, 2017, 243, 183–196 CrossRef CAS.
- K. Kumar, L. Dubau, M. Mermoux, J. Li, A. Zitolo, J. Nelayah, F. Jaouen and F. Maillard, Angew. Chem., Int. Ed., 2020, 59, 3235–3243 CrossRef CAS PubMed.
- K. Kumar, P. Gairola, M. Lions, N. Ranjbar-Sahraie, M. Mermoux, L. Dubau, A. Zitolo, F. Jaouen and F. Maillard, ACS Catal., 2018, 8, 11264–11276 CrossRef CAS.
- M. Kübler, S. Wagner, T. Jurzinsky, S. Paul, N. Weidler, E. D. Gomez Villa, C. Cremers and U. I. Kramm, Energy Technol., 2020, 8, 2000433 CrossRef.
- V. Goellner, C. Baldizzone, A. Schuppert, M. T. Sougrati, K. Mayrhofer and F. Jaouen, Phys. Chem. Chem. Phys., 2014, 16, 18454–18462 RSC.
- U. I. Kramm, I. Herrmann-Geppert, J. Behrends, K. Lips, S. Fiechter and P. Bogdanoff, J. Am. Chem. Soc., 2016, 138, 635–640 CrossRef CAS PubMed.
- C. H. Choi, C. Baldizzone, G. Polymeros, E. Pizzutilo, O. Kasian, A. K. Schuppert, N. Ranjbar Sahraie, M.-T. Sougrati, K. J. J. Mayrhofer and F. Jaouen, ACS Catal., 2016, 6, 3136–3146 CrossRef CAS.
- T. C. Crowtz, D. A. Stevens, R. J. Sanderson, J. E. Harlow, G. D. Vernstrom, L. L. Atanasoska, G. M. Haugen, R. T. Atanasoski and J. R. Dahn, J. Electrochem. Soc., 2014, 161, F961–F968 CrossRef CAS.
- J.-G. Oh, W. H. Lee and H. Kim, Int. J. Hydrogen Energy, 2012, 37, 2455–2461 CrossRef CAS.
- R. T. Atanasoski, L. L. Atanasoska, D. A. Cullen, G. M. Haugen, K. L. More and G. D. Vernstrom, Electrocatalysis, 2012, 3, 284–297 CrossRef CAS.
- D. A. Cullen, K. L. More, L. L. Atanasoska and R. T. Atanasoski, J. Power Sources, 2014, 269, 671–681 CrossRef CAS.
- S.-M. Hwang, Y. Choi, M. G. Kim, Y.-J. Sohn, J. Y. Cheon, S. H. Joo, S.-D. Yim, K. A. Kuttiyiel, K. Sasaki, R. R. Adzic and G.-G. Park, J. Mater. Chem. A, 2016, 4, 5869–5876 RSC.
- A. K. Mechler, N. R. Sahraie, V. Armel, A. Zitolo, M. T. Sougrati, J. N. Schwämmlein, D. J. Jones and F. Jaouen, J. Electrochem. Soc., 2018, 165, F1084–F1091 CrossRef CAS.
- C. Prössl, M. Kübler, M. A. Nowroozi, S. Paul, O. Clemens and U. I. Kramm, Phys. Chem. Chem. Phys., 2021, 23, 563–573 RSC.
- U. I. Kramm, I. Herrmann-Geppert, S. Fiechter, G. Zehl, I. Zizak, I. Dorbandt, D. Schmeißer and P. Bogdanoff, J. Mater. Chem. A, 2014, 2, 2663–2670 RSC.
- J. Speder, A. Zana, I. Spanos, J. J. K. Kirkensgaard, K. Mortensen, M. Hanzlik and M. Arenz, J. Power Sources, 2014, 261, 14–22 CrossRef CAS.
- G. Bauer, W. Fresenius, W. Geibel, G. Jander and H. Oberländer, Elemente der achten Nebengruppe - III Platinmetalle Platin, Palladium, Rhodium, Iridium, Ruthenium, Osmium, Springer, Heidelberg, 1953 Search PubMed.
- K. Eberhardt and C. Geppert, Radiochim. Acta, 2019, 107, 535–546 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- U. I. Kramm, A. Zana, T. Vosch, S. Fiechter, M. Arenz and D. Schmeißer, J. Solid State Electrochem., 2016, 20, 969–981 CrossRef CAS.
- K. Kinoshita, Carbon - Electrochemical and Physicochemical Properties, 1st edn, Wiley-Interscience, 1987 Search PubMed.
- N. Larouche and B. L. Stansfield, Carbon, 2010, 48, 620–629 CrossRef CAS.
- F. Tuinstra and J. L. König, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- J. Robertson, Mater. Sci. Eng. R Rep., 2002, 37, 129–281 CrossRef.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- B. Dippel, H. Jander and J. Heintzenberg, Phys. Chem. Chem. Phys., 1999, 1, 4707–4712 RSC.
- M. N. Ess, D. Ferry, E. D. Kireeva, R. Niessner, F. X. Ouf and N. P. Ivleva, Carbon, 2016, 105, 572–585 CrossRef CAS.
- M. Pawlyta, J.-N. Rouzaud and S. Duber, Carbon, 2015, 84, 479–490 CrossRef CAS.
- F. Jaouen, J. Herranz, M. Lefèvre, J.-P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka, A. Garsuch, J. R. Dahn, T. S. Olson, S. Pylypenko, P. Atanassov and E. A. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623–1639 CrossRef CAS PubMed.
- L. Ni, C. Gallenkamp, S. Paul, M. Kuebler, P. Theis, S. Chabbra, K. Hofmann, E. Bill, A. Schnegg, B. Albert, V. Krewald and I. Kramm Ulrike, Advanced Energy and Sustainability Research, 2021, 2, 2000064 CrossRef.
- U. I. Kramm, R. Marschall and M. Rose, ChemCatChem, 2019, 11, 2563–2574 CrossRef CAS.
- A. Bonakdarpour, M. Lefevre, R. Yang, F. Jaouen, T. Dahn, J.-P. Dodelet and J. R. Dahn, Electrochem. Solid-State Lett., 2008, 11, B105 CrossRef CAS.
- N. D. Leonard and S. C. Barton, J. Electrochem. Soc., 2014, 161, H3100–H3105 CrossRef.
- M. Bron, S. Fiechter, P. Bogdanoff and H. Tributsch, Fuel Cells, 2002, 2, 137–142 CrossRef CAS.
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- U. I. Kramm, I. Abs-Wurmbach, I. Herrmann-Geppert, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2011, 158, B69–B78 CrossRef CAS.
- T. Marshall-Roth, N. J. Libretto, A. T. Wrobel, K. J. Anderton, M. L. Pegis, N. D. Ricke, T. V. Voorhis, J. T. Miller and Y. Surendranath, Nat. Commun., 2020, 11, 5283 CrossRef CAS PubMed.
- S. Wagner, H. Auerbach, C. E. Tait, I. Martinaiou, S. C. N. Kumar, C. Kübel, I. Sergeev, H.-C. Wille, J. Behrends, J. A. Wolny, V. Schünemann and U. I. Kramm, Angew. Chem., Int. Ed., 2019, 58, 10486–10492 CrossRef CAS PubMed.
- E. Kuzmann, A. Nath, V. Chechersky, S. Li, Y. Wei, X. Chen, J. Li, Z. Homonnay, M. Gál, V. K. Garg, Z. Klencsár and A. Vértes, Hyperfine Interact., 2002, 139/140, 631–639 CrossRef CAS.
- U. I. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, 1805623 CrossRef PubMed.
- R. L. Mößbauer, M. Lengsfield, W. van Lieres, W. Potzel, P. Teschner, F. E. Wagner and G. Kaindle, Z. Naturforsch., A: Phys. Sci., 1971, 26, 343–352 Search PubMed.
- A. B. Suryamas, G. M. Anilkumar, S. Sago, T. Ogi and K. Okuyama, Catal. Commun., 2013, 33, 11–14 CrossRef CAS.
- N. Cheng, L. Zhang, K. Doyle-Davis and X. Sun, Electrochem. Energy Rev., 2019, 2, 539–573 CrossRef.
- J.-Y. Choi, L. Yang, T. Kishimoto, X. Fu, S. Ye, Z. Chen and D. Banham, Energy Environ. Sci., 2017, 10, 296–305 RSC.
- I. A. Safo and M. Oezaslan, Electrochim. Acta, 2017, 241, 544–552 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef CAS.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CrossRef CAS.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlogl and P. Atanassov, Chem. Commun., 2013, 49, 2539–2541 RSC.
- A. Serov, K. Artyushkova and P. Atanassov, Adv. Energy Mater., 2014, 4, 1301735 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta04668c |
| This journal is © The Royal Society of Chemistry 2022 |