
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Capture of activated dioxygen intermediates at the copper-active site of a lytic polysaccharide monooxygenase†
Gabriela C.
Schröder
 ab,
William B.
O'Dell‡
ab,
Simon P.
Webb
c,
Pratul K.
Agarwal
*d and
Flora
Meilleur
*ab
ab,
William B.
O'Dell‡
ab,
Simon P.
Webb
c,
Pratul K.
Agarwal
*d and
Flora
Meilleur
*ab
aDepartment of Molecular and Structural Biochemistry, North Carolina State University, Raleigh, NC 27695, USA. E-mail: fmeille@ncsu.edu
bNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
cVeraChem LLC, 12850 Middlebrook Rd. Ste 205, Germantown, MD 20874-5244, USA
dDepartment of Physiological Sciences and High-Performance Computing Center, Oklahoma State University, Stillwater, OK 74078, USA. E-mail: pratul.agarwal@okstate.edu
First published on 2nd November 2022
Abstract
Metalloproteins perform a diverse array of redox-related reactions facilitated by the increased chemical functionality afforded by their metallocofactors. Lytic polysaccharide monooxygenases (LPMOs) are a class of copper-dependent enzymes that are responsible for the breakdown of recalcitrant polysaccharides via oxidative cleavage at the glycosidic bond. The activated copper-oxygen intermediates and their mechanism of formation remains to be established. Neutron protein crystallography which permits direct visualization of protonation states was used to investigate the initial steps of oxygen activation directly following active site copper reduction in Neurospora crassa LPMO9D. Herein, we cryo-trap an activated dioxygen intermediate in a mixture of superoxo and hydroperoxo states, and we identify the conserved second coordination shell residue His157 as the proton donor. Density functional theory calculations indicate that both superoxo and hydroperoxo active site states are stable. The hydroperoxo formed is potentially an early LPMO catalytic reaction intermediate or the first step in the mechanism of hydrogen peroxide formation in the absence of substrate. We observe that the N-terminal amino group of the copper coordinating His1 remains doubly protonated directly following molecular oxygen reduction by copper. Aided by molecular dynamics and mining minima free energy calculations we establish that the conserved second-shell His161 in MtPMO3* displays conformational flexibility in solution and that this flexibility is also observed, though to a lesser extent, in His157 of NcLPMO9D. The imidazolate form of His157 observed in our structure following oxygen intermediate protonation can be attributed to abolished His157 flexibility due steric hindrance in the crystal as well as the solvent-occluded active site environment due to crystal packing. A neutron crystal structure of NcLPMO9D at low pH further supports occlusion of the active site since His157 remains singly protonated even at acidic conditions.
Introduction
Lytic polysaccharide monooxygenases (LPMOs; also designated PMOs) are copper metalloenzymes that perform unique redox chemistry resulting in disruption of polysaccharide chains by oxygen atom insertion at the glycosidic bond.1 LPMOs originate from a range of organisms including bacteria, fungi, algae as well as animals and are classified according to Auxiliary Activity (AA),2 namely the AA9–AA11 and AA13–16 families. Herein we study activity of an AA9 LPMO, a family of LPMOs of fungal origin that are active on cellulose. The LPMO active site is highly conserved and contains a mononuclear Cu2+ center within a “histidine brace” motif in which the copper is coordinated by the N-terminal histidine amino group, the Nδ atom of His1 and the Nε atom of a second conserved histidine in the equatorial plane.3 The LPMO axial position is occupied by a Tyr residue 2.5–3.0 Å from the copper center, with the exception of some AA10 LPMOs where the Tyr is replaced by a Phe.4 In the Cu2+ resting state, the remaining equatorial and axial positions are each occupied by a water molecule.5,6Of particular commercial and biotechnological interest is the ability of fungal LPMOs to catalyze the oxidative cleavage of recalcitrant crystalline cellulose at the C1 and/or C4 position, thereby enhancing polysaccharide depolymerization by increasing glucan access to hydrolytic enzymes and ensuring efficient bioconversion.7,8 The overall reaction of LPMOs for oxidative cleavage of the glycosidic bond proceeds as shown in Scheme 1 in which oxygen is inserted at the C1 or C4 position which destabilizes the glycosidic bond resulting in bond breakage via an elimination reaction.9
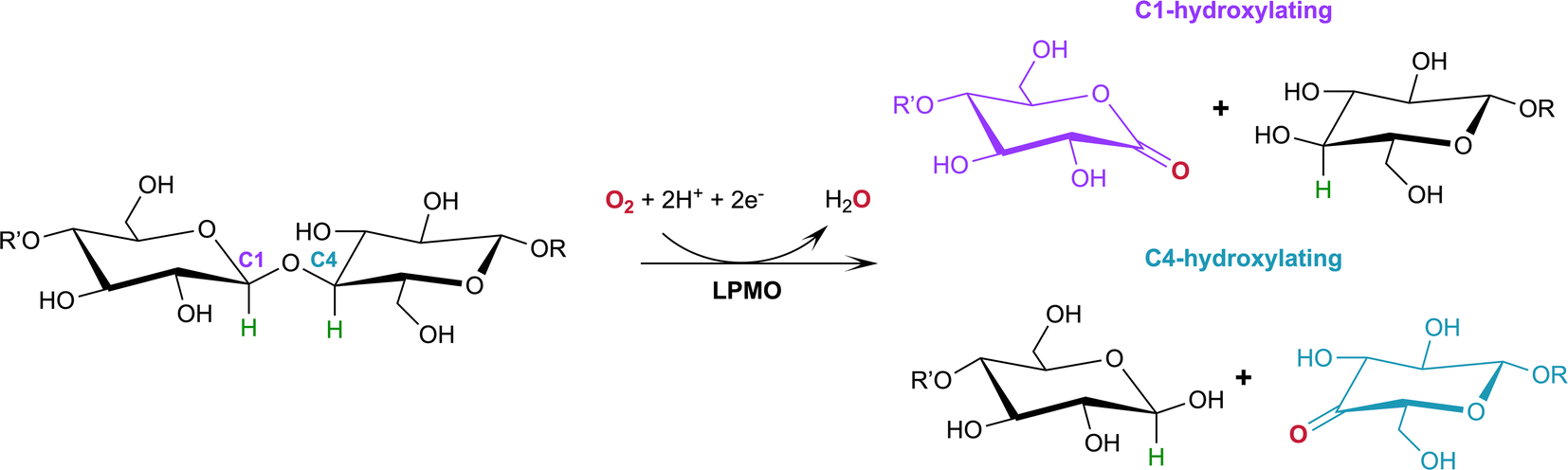 | ||
| Scheme 1 Oxidation of the C1/C4 carbon in the glycosidic bond catalyzed by LPMOs with oxygen as the co-substrate. H atoms to be abstracted by the reactive oxygen species shown in green. | ||
While H2O2 has also been shown to be a LPMO co-substrate,10–12 the work presented here focuses on the activation of O2, the first characterized co-substrate in polysaccharide oxidation and the substrate in the synthesis of H2O2 in the absence of polysaccharide substrate.9,13,14 One-electron reduction of the copper active site to Cu1+ and binding of the molecular oxygen co-substrate initiates the reaction cycle with the generally accepted formation of a Cu2+ superoxide, [Cu–O2]+.9,13,15,16 In the current consensus for the O2-based mechanism, following the rapid reduction of molecular dioxygen to the superoxide, the LPMO reaction mechanism requires further delivery of electrons and protons to form a reactive oxygen intermediate which oxidizes the polysaccharide substrate.17 The identity of the activated oxygen species responsible for hydrogen atom abstraction (HAA) from the polysaccharide substrate leading to subsequent substrate oxidation, is however still a matter of ongoing research.18 Intensive studies encompassing X-ray crystallographic, computational and spectroscopic examinations of mononuclear copper monooxygenases as well as biomimetic copper complexes have proposed dioxygen intermediates such as the superoxide [Cu–O2]+ and hydroperoxo [CuOOH]+ species where the O–O bond of molecular oxygen remains intact as the reactive intermediate, however the superoxide species is unlikely to act as the HAA since this has been shown to be energetically unfavorable.16,19–23 Furthermore, formation of the hydroxide [CuOH]2+ and oxyl [CuO]+ intermediates after O–O bond scission, with subsequent involvement in HAA, have been proposed.23–31 Formation of a potential hydroperoxo, hydroxy or oxyl HAA species would require further protonation and reduction of the superoxide; however, the identity of the proton donor is still debated. Several second-shell residues including histidine (His147 in LsLPMO9A/His161 in MtPMO3*) as well as glutamine (Gln167 in MtPMO3*) and glutamate (Glu201 in JdLPMO10A) have been proposed to be involved in intermediate protonation.31–33 QM/MM calculations by Hedegård et al. propose that a positively charged second-shell His147 in LsLPMO9A is involved in protonation of the superoxo intermediate.34 Mutagenesis and electron paramagnetic resonance (EPR) studies of His161 in MtPMO3* by Span et al. found that His161 plays a role as a proton donor during oxygen activation. QM/MM studies of JdLPMO10A have also proposed Glu201, a residue in the vicinity of the copper active site as a potential proton donor.33 In addition to protonation of the initially formed superoxide species via a second shell residue, recent quantum mechanics/molecular mechanics (QM/MM) calculations have also shown that the superoxide species can abstract a hydrogen atom from an exogenous reducing agent such as ascorbic acid to form a hydroperoxo species.35
Given their role in boosting polysaccharide degradation for downstream industrial applications, elucidation of intermediates in the LPMO reaction pathway will pave the way for improved utilization of LPMOs. While there have been extensive studies on the LPMO mechanism, direct characterization of catalytic intermediates remains elusive given the insoluble nature of the polysaccharide substrate and the risk of radiation damage-induced artefacts at the copper active site. Neutron protein crystallography is particularly well suited to the determination of protonation states within a protein due to the technique's unique sensitivity to hydrogen atoms and protons often invisible to X-ray diffraction.36,37 Neutron protein crystallography is also a non-destructive technique ideally suited for the study of radiation-sensitive metalloproteins for characterization of catalytic intermediates.38 The application of cryo-neutron protein crystallography to study the protonation states of activated oxygen catalytic intermediates has been demonstrated by Raven, Moody and coworkers for the characterization of compound I and compound II.39,40 In particular, a neutron crystallography study of cytochrome c peroxidase (CcP) resulted in cryo-capture and characterization of a deprotonated Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O compound I species in a 2.5 Å resolution structure.41 In further study, a potential protonated Fe(IV)–OH compound II species was observed in an dioxygen activated and cryocooled structure of ascorbate peroxidase (APX) at a 2.2 Å resolution.42
O compound I species in a 2.5 Å resolution structure.41 In further study, a potential protonated Fe(IV)–OH compound II species was observed in an dioxygen activated and cryocooled structure of ascorbate peroxidase (APX) at a 2.2 Å resolution.42
We report here two neutron models of a carbohydrate-free C4-hydroxylating Neurospora crassa LPMO9D: a structure of the activated, ascorbate-reduced form and a structure of the resting state at pH 4.4 (pD 4.8) to further address open questions regarding initial molecular oxygen activation. The neutron crystallographic structure of the reduced NcLPMO9D shows cryo-capture of activated dioxygen at the copper active site modelled as a superoxo and protonated hydroperoxo species with a population ratio of 0.30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.70. The neutron scattering length density maps of the conserved secondary coordination shell His157 is best represented as 70% imidazolate and a 30% singly protonated form, indicating the His157 acts as a proton donor during early dioxygen intermediate formation. This imidazolate intermediate state was found to be stable following DFT calculations. The copper coordinating N-terminal amino group His1 remains in a doubly protonated ND2 state following copper reduction and molecular oxygen activation. We probed the flexibility of His157 in our structure and the corresponding His161 in MtPMO3* using molecular dynamics (MD) simulations to further explore their function during initial oxygen activation. His161 in MtPMO3* displayed mobility between an inward and outward conformation, with His157 in our structure displaying similar mobility but to a lesser extent. Mining minima free energy calculations further indicate His157 flexibility in solution which is absent in the crystal structure due to binding-face to binding-face crystal packing. Furthermore, the neutron and X-ray crystal structures of the resting state NcLPMO9D at low pH show that His157 remains singly protonated when exposed to acidic conditions. The additive effect of limited mobility and active site occlusion due to crystal packing have therefore permitted trapping of a hydroperoxo intermediate and designation of the second-shell His157 as the proton donor.
:0.70. The neutron scattering length density maps of the conserved secondary coordination shell His157 is best represented as 70% imidazolate and a 30% singly protonated form, indicating the His157 acts as a proton donor during early dioxygen intermediate formation. This imidazolate intermediate state was found to be stable following DFT calculations. The copper coordinating N-terminal amino group His1 remains in a doubly protonated ND2 state following copper reduction and molecular oxygen activation. We probed the flexibility of His157 in our structure and the corresponding His161 in MtPMO3* using molecular dynamics (MD) simulations to further explore their function during initial oxygen activation. His161 in MtPMO3* displayed mobility between an inward and outward conformation, with His157 in our structure displaying similar mobility but to a lesser extent. Mining minima free energy calculations further indicate His157 flexibility in solution which is absent in the crystal structure due to binding-face to binding-face crystal packing. Furthermore, the neutron and X-ray crystal structures of the resting state NcLPMO9D at low pH show that His157 remains singly protonated when exposed to acidic conditions. The additive effect of limited mobility and active site occlusion due to crystal packing have therefore permitted trapping of a hydroperoxo intermediate and designation of the second-shell His157 as the proton donor.
Results and discussion
Crystallographic features of the NcLPMO9D neutron structures
Both the ascorbate-reduced and low pH neutron crystals of NcLPMO9D belonged to the monoclinic P21 space group. The crystal structures include two protein molecules per asymmetric unit (molecule A and molecule B) related by non-crystallographic symmetry (NCS). Analysis of the crystallographic contacts shows that the two NCS related NcLPMO9D molecules pack with their planar polysaccharide binding interfaces against each other in a binding-face to binding-face configuration also seen in PDB structures 4EIR, 5TKG, 5TKH and 5TKI (Fig. 1A).15,43,44 While present in a dimeric conformation in the crystal structure, LPMOs including NcLPMO9D function as single-domain monomeric proteins in solution, where they associate with planar crystalline cellulose as confirmed by early LPMO structural studies,8,15,45 and single-molecule observations of LPMO using atomic force microscopy (AFM).46 Within this binding-face to binding-face packing architecture, the two coordinated coppers are ∼12 Å apart, precluding direct interaction. Binding interface residues Tyr25, Tyr206 and Trp207 adopt distinct conformations in molecule A and B, contributing to different microenvironments near the Cu2+ active site (Fig. 1B). Structural alignment of our NcLPMO9D molecule A or molecule B with the oligosaccharide-bound form of LsLPMO9A (PDB 5ACI),47 indicates that the substrate pyranose rings of the LsLPMO9A structure superimpose with the proline rings Pro43 and Pro163 for both molecule A or molecule B of NcLPMO9D (Fig. 1C). The alignment further indicates that Tyr206 of molecule A is positioned to interact with substrate via its phenol ring. This suggests that the crystallographic environment of the active site in molecule A more closely mimics surface residue interaction with the pyranose ring of the glucose units and creates an occluded active site, possibly analogous to the bulk solvent-protected environment created when the planar active site of LPMO binds to its crystalline linear polysaccharide substrate. Such crystal packing in which active site residues mimic carbohydrate substrate pyranose rings has also been observed in the crystal packing of NcLPMO-3 (PDB 4EIS).15 Representation of the protein surface additionally illustrates the active site occlusion due to crystal packing (Fig. 2).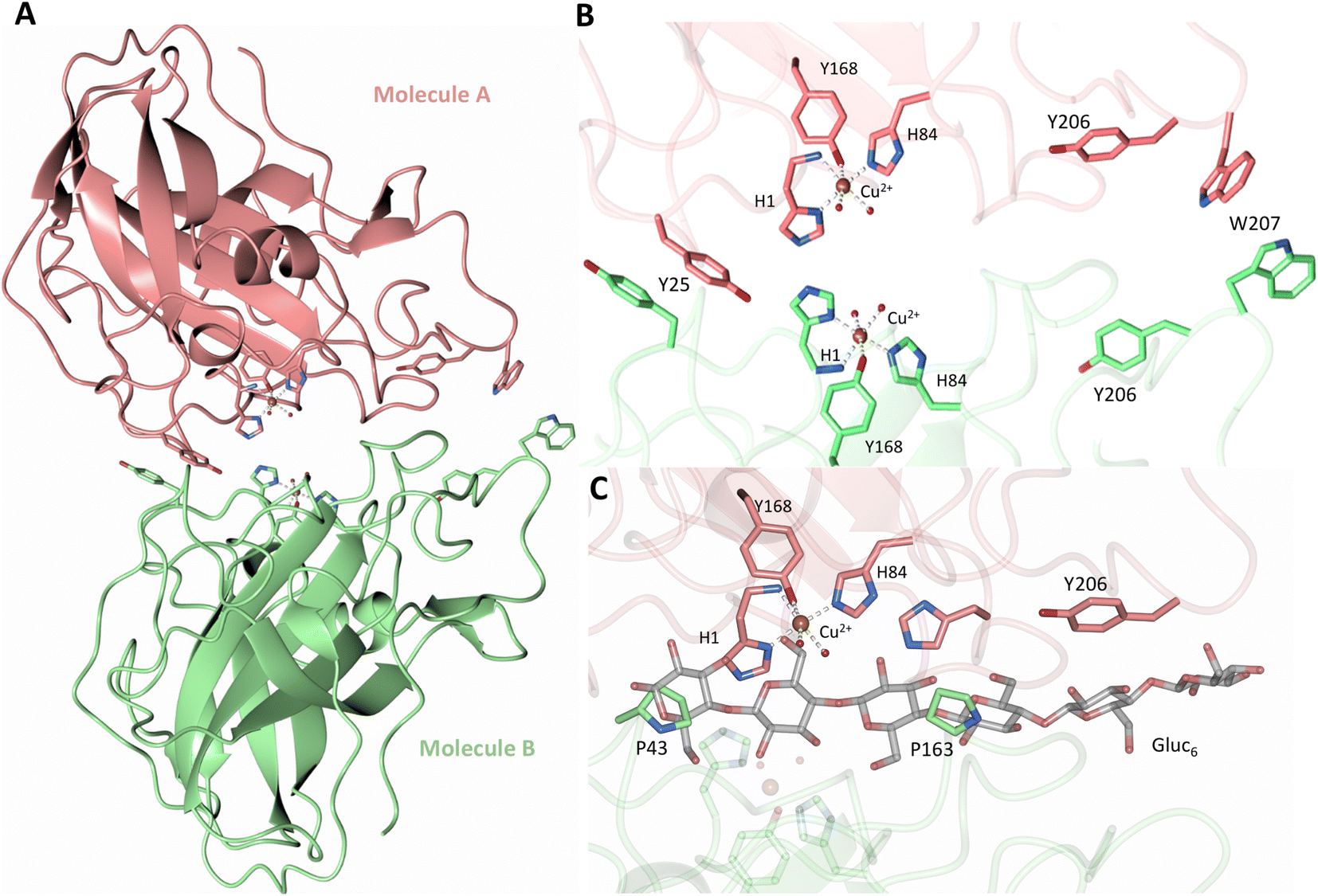 | ||
| Fig. 1 Crystal packing of molecule A and molecule B in the NcLPMO9D crystals. (A) Cartoon of molecule A (coral) and molecule B (light green) of the planar active site surfaces packed facing each other. (B) Crystal symmetry in molecule A and B is broken by alternate conformations of Tyr 25, Tyr206 and Trp207. (C) Alignment of molecule B of NcLPMO9D with the Gluc6 bound LsLPMO9A (PDB 5ACI) in grey to illustrate superposition of proline residues with glucose pyranose rings. | ||
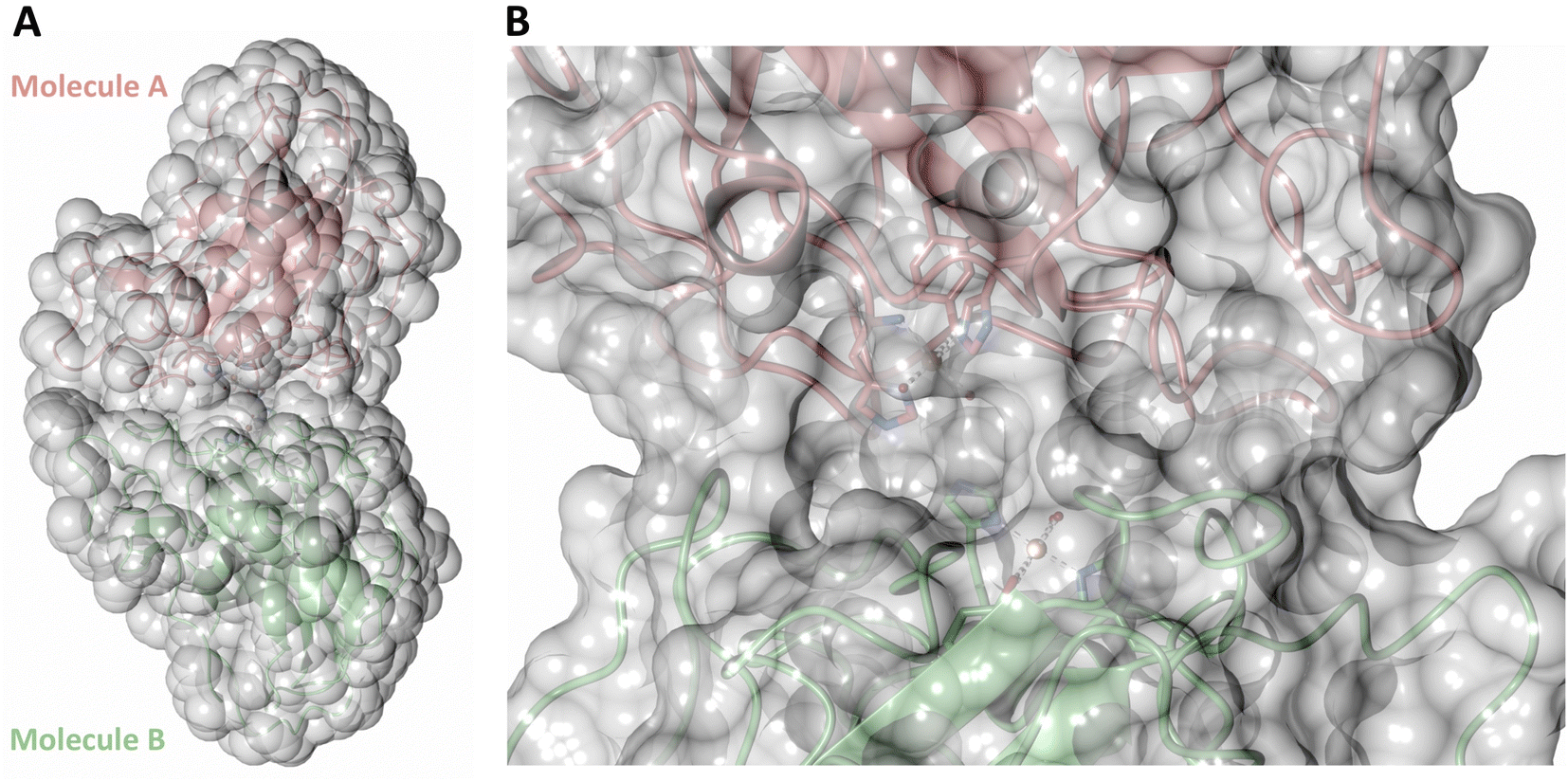 | ||
| Fig. 2 Solvent accessible surface in the crystal packing of molecule A and molecule B of the asymmetric unit of NcLPMO9D. Surface displayed with a probe radius of 1.5 Å. | ||
Ascorbate-reduced copper active site
To probe the protonation states of key catalytic residues and intermediates in the NcLPMO9D active site, a neutron diffraction dataset to 2.4 Å resolution was collected at 100 K on a crystal of NcLPMO9D reduced with ascorbate at pD 6.0 (pH 5.6) in the presence of atmospheric oxygen. Structure refinement was performed solely against neutron data to circumvent potential artefacts induced by X-ray radiation damage. The axial position is vacant, supporting displacement of both the equatorial and axial water molecules upon Cu2+ reduction and dioxygen activation as demonstrated by Kjaergaard et al. using extended X-ray absorption fine structure (EXAFS), X-ray Absorption Near Edge Spectroscopy (XANES) and density functional theory (DFT) calculations.16 The neutron scattering length density maps at the active site of molecule A indicate the presence of a fully occupied equatorially coordinated dioxygen species in a mixed state of a two-atom superoxo species and a three-atom hydroperoxo species with occupancies of 0.30 and 0.70, respectively (Fig. 3A and B). The intermediates have η1 end-on geometry at a Cu–O1 distance of ∼1.96 Å and ∼1.98 Å for the superoxo and hydroperoxo species, respectively. The superoxo species is modelled with an O–O bond length of 1.28 Å while the hydroperoxo species is modelled with a bond length of 1.46 Å. The Cu–O1–O2 angles are ∼147° and 151° for the superoxo and hydroperoxo species respectively. A Glu30 residue from molecule B is modeled 2.06 Å and 2.08 Å away from the O2 of the superoxo and hydroperoxo species, respectively, however it displays a disordered conformation with limited neutron scattering length density, as has been observed for NcLPMO9D structures with the same crystal packing.15,43,44 The modeling of an activated dioxygen species is in good accordance with our previous high resolution X-ray structures which indicated the presence of a peroxo species with a 1.44 ± 0.06 Å bond length with a Cu–O1–O2 angle of 140.52° and 1.90 Å from the Cu following ascorbate reduction (Fig. 3D).43 Activated dioxygen species have been modelled in further LPMO structures including NcLPMO9D and NcPMO-3 from Li et al., JdLPMO10A from Bacik et al. as well as in an artificial copper protein from Mann et al. (Table 1).15,20,33,43,48 Most recently, an EPR/electron–nuclear double resonance (ENDOR) spectroscopy study by Davydov et al. of copper coordination complexes captured a Cu(I)-superoxo species that undergoes internal electron transfer to form a Cu(II)-peroxo species which is protonated to form the Cu(II)-hydroperoxo species.49 These species were captured in end-on copper coordination corresponding to the equatorial end-on coordination observed for the hydroperoxo species in the structure presented here. In our previous high-resolution X-ray structure, discrimination between a putative superoxo and peroxo species was based on bond length determination due to the lack of visibility of hydrogen atoms. In addition, the occupancy of the activated dioxygen species only refined to 0.59 and an axial water remained present with an occupancy of 0.48. We reason that photoreduction due to X-ray beam exposure resulted in an intermediate with reduced occupancy. The NcLPMO9D active site presented here is absent of radiation damage since neutron protein crystallography is a non-destructive technique permitting capture of a mixed occupancy of a superoxo and hydroperoxo species.37 The observed hydroperoxo species may represent an early dioxygen intermediate in the LPMO catalytic mechanism, or it may be an intermediate in LPMO-catalyzed H2O2 formation, since LPMOs are known to produce H2O2 in the absence of substrate.50 Caldararu et al. have further shown it is more energetically favorable for H2O2 formation to proceed by dissociation of H2O2 from the copper active site than HO2.33 This higher dissociation energy of HO2 is supported by the observed binding stability of the hydroperoxo species in our structure.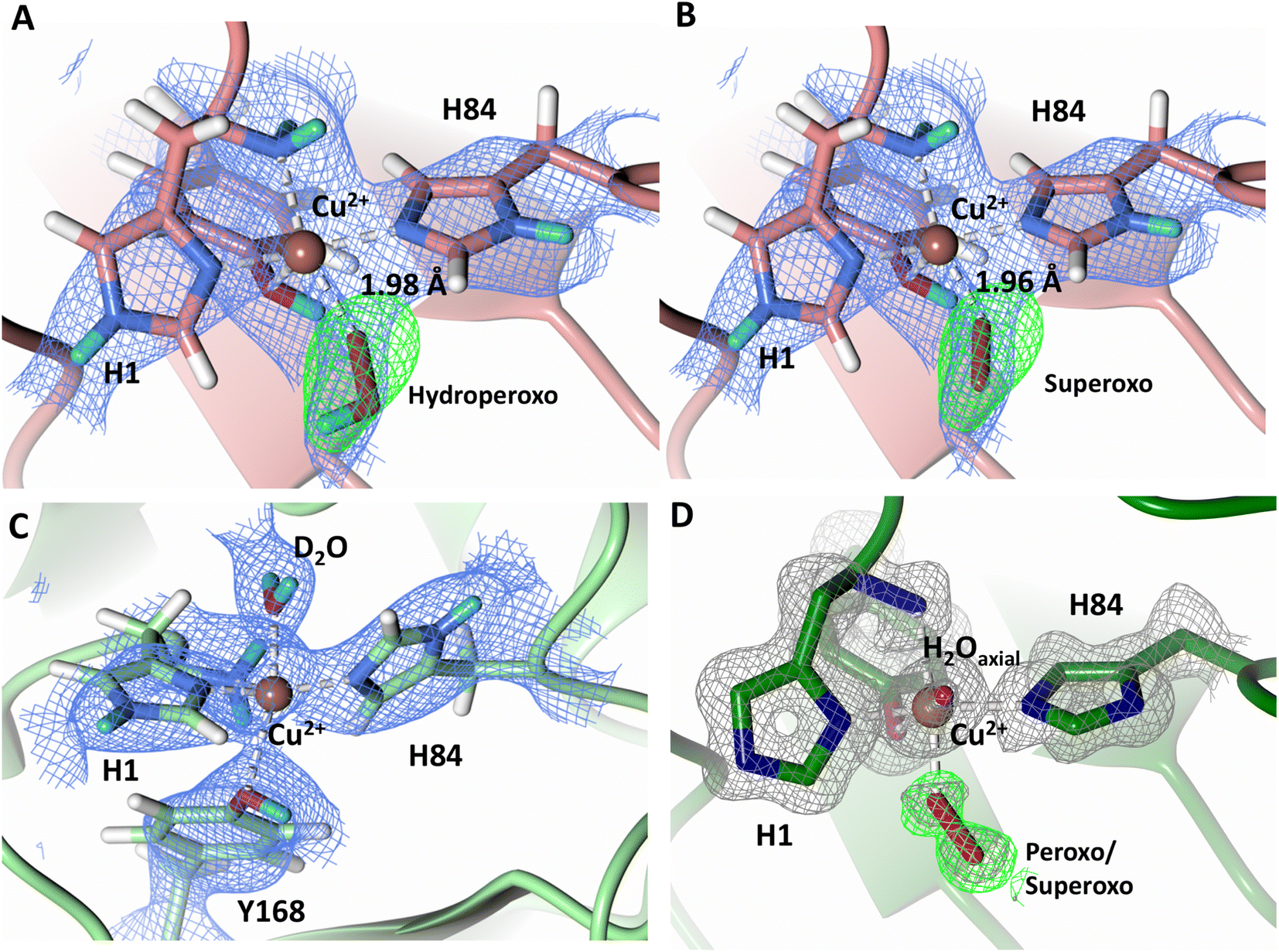 | ||
| Fig. 3 Neutron diffraction structure of the ascorbate reduced NcLPMO9D crystal (PDB 7T5D). (A) Active site of molecule A (coral) with a hydroperoxo species bound at the equatorial position. (B) Active site of molecule A (coral) with a superoxo species bound at the equatorial position. (C) Active site of molecule B (light green) with a water bound at the axial position. (D) Equatorially coordinated activated dioxygen superoxo/peroxo in X-ray diffraction structure of ascorbate reduced NcLPMO9D (green) by O'Dell et al.43 (PDB 5TKH). (A–C) Neutron scattering length density 2Fo–Fc maps displayed in blue at a 1.0 σ cut-off. Neutron scattering length density Fo–Fc maps displayed in green at a 2.5 σ cut-off. H atoms displayed in white and D atoms displayed in turquoise. (D) Electron density 2Fo–Fc maps displayed in grey at a 1.0 σ cut-off. Electron density Fo–Fc maps displayed in green at a 3.0 σ cut-off. | ||
| LPMO | PDB | Dioxygen species | Position | Cu–O (Å) | O1–O2 (Å) | Cu–O–O (°) |
|---|---|---|---|---|---|---|
| NcLPMO9D | 4EIR – chain A | Superoxide – end-on | Axial | 2.96 | 1.16 | 147.75 |
| 4EIR – chain B | Superoxide – end-on | Axial | 2.92 | 1.15 | 142.11 | |
| NcPMO-3 | 4EIS | Peroxo – end-on | Axial | 3.44 | 1.49 | 117.16 |
| JdLPMO10A | 5VG0 – Chain A | Peroxo – side-on | Equatorial | O1 – 2.14, O2 – 1.84 | 1.48 | O1 – 57.67, O2 – 79.39 |
| JdLPMO10A | 5VG0 – Chain A | Peroxo – end-on | Equatorial | 1.83 | 1.46 | 109.52 |
| JdLPMO10A | 5VG1 – Chain B | Peroxo – end-on | Equatorial | 2.49 | 1.45 | 115.32 |
| 5VG0/5VG1 joint quantum refinement (ComQum-X) – Chain A | Superoxide – end-on | Equatorial | 2.31 | 1.25 | 108.6 | |
| 5VG0/5VG1 joint quantum refinement (ComQum-X) – Chain B | Superoxide – end-on | Equatorial | 2.27 | 1.25 | 113.6 | |
| NcLPMO9D | 5TKH – Chain A | Peroxo – end-on | Equatorial | 1.90 | 1.44 | 140.52 |
| Artificial copper protein | 6ANX | Hydroperoxo | Intermediate | 1.94 | 1.52 | 137.60 |
| NcLPMO9D (current neutron structure) | 7T5D – Chain A | Superoxo | Equatorial | 1.96 | 1.28 | 147 |
| 7T5D – Chain A | Hydroperoxo | Equatorial | 1.98 | 1.46 | 151 |
The molecule B copper active site contains a water molecule coordinated in the axial position with a partial occupancy of 0.65 and a Cu–O distance of ∼2.2 Å while the equatorial position is vacant (Fig. 3C). This geometry, while not chemically relevant, has been observed in partially photo-reduced LPMO structures,6 suggesting that the active site of molecule B underwent only partial reduction during the ascorbate soak. We attribute the differences between molecule A and molecule B to the conformations of the Tyr residues at the binding interfaces.
Protonation state of His157
Testing of different protonation states and visual examination of the 2Fo–Fc and Fo–Fc omit neutron scattering length density maps of the molecule A secondary coordination shell His157 suggest that this residue is best modelled by the neutral Nε-protonated form and the imidazolate form with occupancies of 0.30 and 0.70, respectively (Fig. 4A and B). Analysis of the molecule B neutron scattering length density maps indicate that His157 is singly Nε-protonated at full occupancy (Fig. 4C). The Nε nitrogen of His157 in molecule A is observed at a distance of ∼3.0 Å from the distal oxygen O2 of the equatorially bound superoxo species and hydroperoxo species. These findings indicate that His157 plays a role in protonation of the activated dioxygen species observed in the barricaded active site presented here. While an imidazolate species may be an unusual catalytic intermediate, it has been determined that the pKa of a deprotonated His is 14, while the peroxo group is very basic with a pKa of up to 24 (as found in hydroperoxide dicopper complex), supporting the donor role of a neutral His proposed herein.51,52 Activity and structural studies of MtPMO3* as well as QM/MM calculations of LsLPMO9A have implicated this conserved histidine residue in proton transfer to the copper-bound superoxide during LPMO catalysis (His161 and His147 in MtPMO3* and LsLPMO9A, respectively).31,32 QM/MM studies have also indicated that the Glu201 residue near the copper active site in JdLPMO10A is capable of donating a proton to a bound superoxo to form a hydroperoxo species.33 Computational studies have suggested that ascorbate may function as a proton donor to the superoxo species;35 however, the active site occlusion due to crystal packing in our structure does not permit ascorbate to access the copper active site, and there was no evidence in the neutron scattering length density maps for an ascorbate molecule near the active site. Following formation of the superoxide in NcLPMO9D, the species we observe may result from protonation by His157 to form the Cu(II)-hydroperoxyl species [CuOOH]2+, the Cu(III)-hydroperoxo [CuOOH]2+, or upon further reduction a Cu(II)-hydroperoxo [CuOOH]+, i.e.| Superoxo: [Cu]+ + O2 → [Cu–O2]+ with Cu(II) center |
| Hydroperoxyl radical/Hydroperoxo: [Cu–O2]+ + H+ → [Cu–OOH]2+ with Cu(II)/Cu(III) center |
| Hydroperoxo: [Cu–O2]+ + H+ + e− → [Cu–OOH]+ with Cu(II) center |
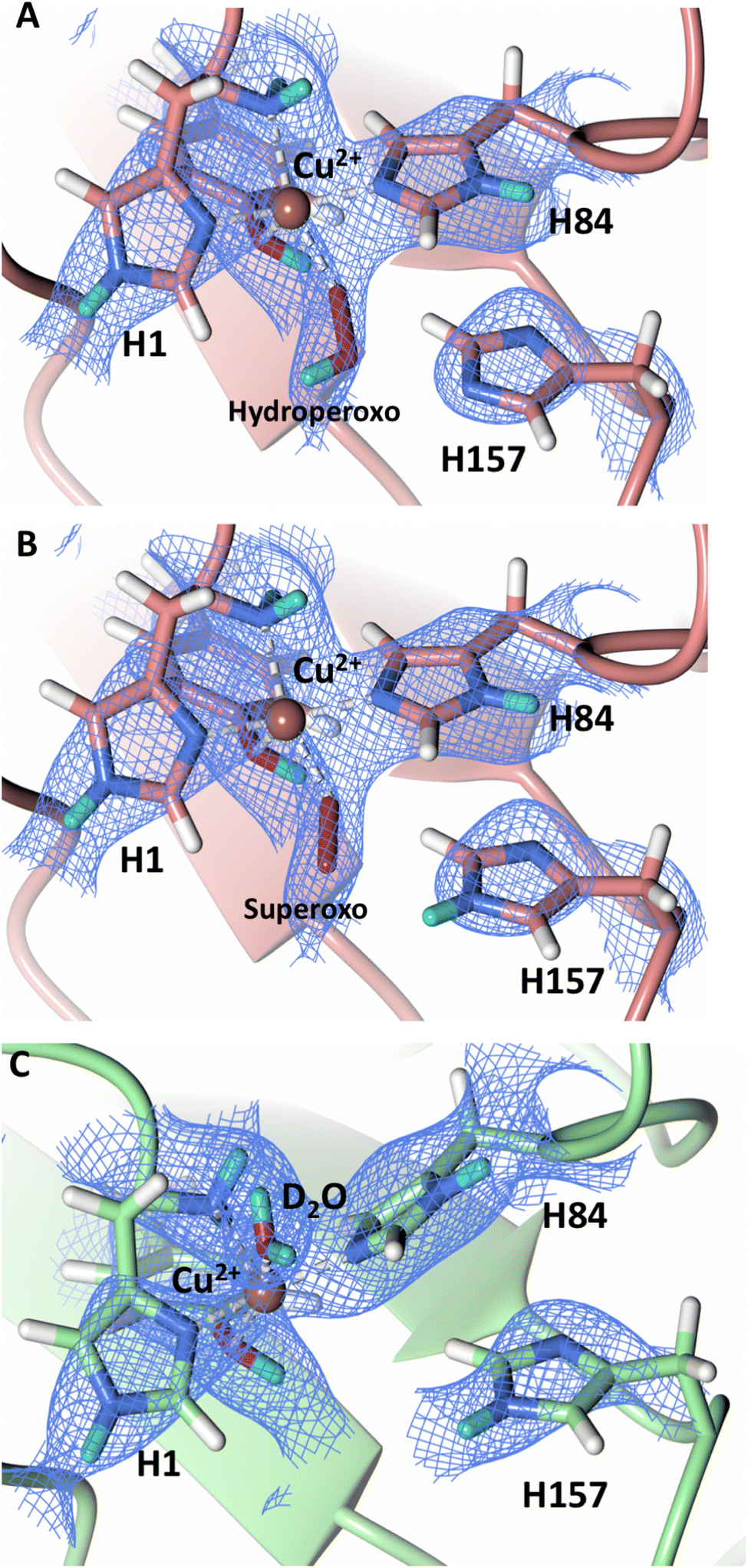 | ||
| Fig. 4 Mixed protonation state His157 in ascorbate reduced NcLPMO9D (PDB 7T5D). (A) The molecule A (coral) second shell His157 is deprotonated with an equatorial hydroperoxo species coordinated to Cu2+. (B) The molecule A second shell His157 is singly Nε-protonated with an equatorial superoxo species coordinated to Cu2+. (C) The molecule B (light green) second shell His157 is singly Nε-protonated with an axial water coordinated to Cu2+. Neutron scattering length density 2Fo–Fc maps displayed in blue at a 1.0 σ cut-off. H atoms displayed in white and D atoms displayed in turquoise. | ||
All these species have been posited as viable reaction intermediates in computational and small molecule studies.20,23,49,53–55
| Model | DFT active site residues | Starting proton position | System net charge | Multiplicity | Final His157 state |
|---|---|---|---|---|---|
| Superoxo, His157 NE2-protonated | His1, His84, His157, Gln166, Tyr168 | His157 NE2 | +1 | Triplet | His157 singly NE2-protonated |
| Hydroperoxyl radical, His157 doubly deprotonated | His1, His84, His157, Gln166, Tyr168 | O–O–H | +1 | Triplet | Double deprotonated His157 |
| Hydroperoxo, His157 doubly deprotonated | His1, His84, His157, Gln166, Tyr168 | O–O–H | 0 | Doublet | Double deprotonated His157 |
| Hydroperoxo, His157 doubly deprotonated | His1, His84, His157, Gln166, Tyr168 | O–O–H | −1 | Singlet | Double deprotonated His157 |
The DFT calculation results are summarized in Table 2. All the active-site models considered showed energy and energy-gradient convergence to a stationary point on the potential energy surface with the proton still located at its initial position and so are considered stable and none are ruled out. More details of the energy convergence profiles are provided in the ESI.† We note that the current DFT calculations/‘cutout’ models do not provide a means of directly comparing the relative energies of the proposed intermediates due to the different combinations of total charge and spin states across the four models. Nor can the possible Cu(III)-hydroperoxo with imidazolate His157 intermediate be adequately examined, due to probable multi-determinant character.58 Accurate calculated energy differences between all the intermediates will require a multi-determinant QM treatment due to propensity of copper centers to exhibit multiple near degenerate electronic states,58,59 as well as a more complex structural model e.g. inclusion of an electron donor molecule.23 This will be addressed in planned future calculations using QM/MM with a multi-determinant wavefunction QM method.
Active site protonation following initial dioxygen activation
The N-terminal amino group of the histidine brace has been postulated to play a role in intermediate formation and stabilization during LPMO catalysis.60 A previous neutron protein crystallography study of unreduced JdLPMO10A, a bacterial LPMO, suggested that the amino terminal is present as a mixture of the -ND− and -ND2 protonation states.48 The JdLPMO10A neutron structure was, however, later revisited and a joint X-ray–neutron quantum refinement concluded that the amino terminal is present solely in the -ND2 state.33 We therefore sought to determine the protonation state of the N-terminal amino group upon NcLPMO9D reduction by ascorbate in the presence of atmospheric O2. The neutron scattering length density map indicates that both molecule A and molecule B contain the amino terminal in the -ND2 protonation state. Fo–Fc omit difference maps calculated confirm the ND2 state in both molecule A and molecule B (Fig. 5A–D). The role of possible deprotonation of the N-terminal histidine amino group came to the forefront when it was proposed to promote reactive intermediate formation and stabilization based on findings from a small molecule copper complex.61 Structural and spectroscopic studies of a substrate-bound LsLPMO9A suggested that the two protons of the N-terminus are exposed to different chemical environments, with one being involved in a hydrogen bond network, which potentially promotes N-terminal deprotonation during LPMO catalysis.47 Our neutron protein crystallography structures show that the N-terminal amine is not involved in protonation upon initial Cu2+ reduction and molecular dioxygen activation. However, a mechanistic role for the N-terminal amine in other contexts cannot be conclusively ruled out by our experiments.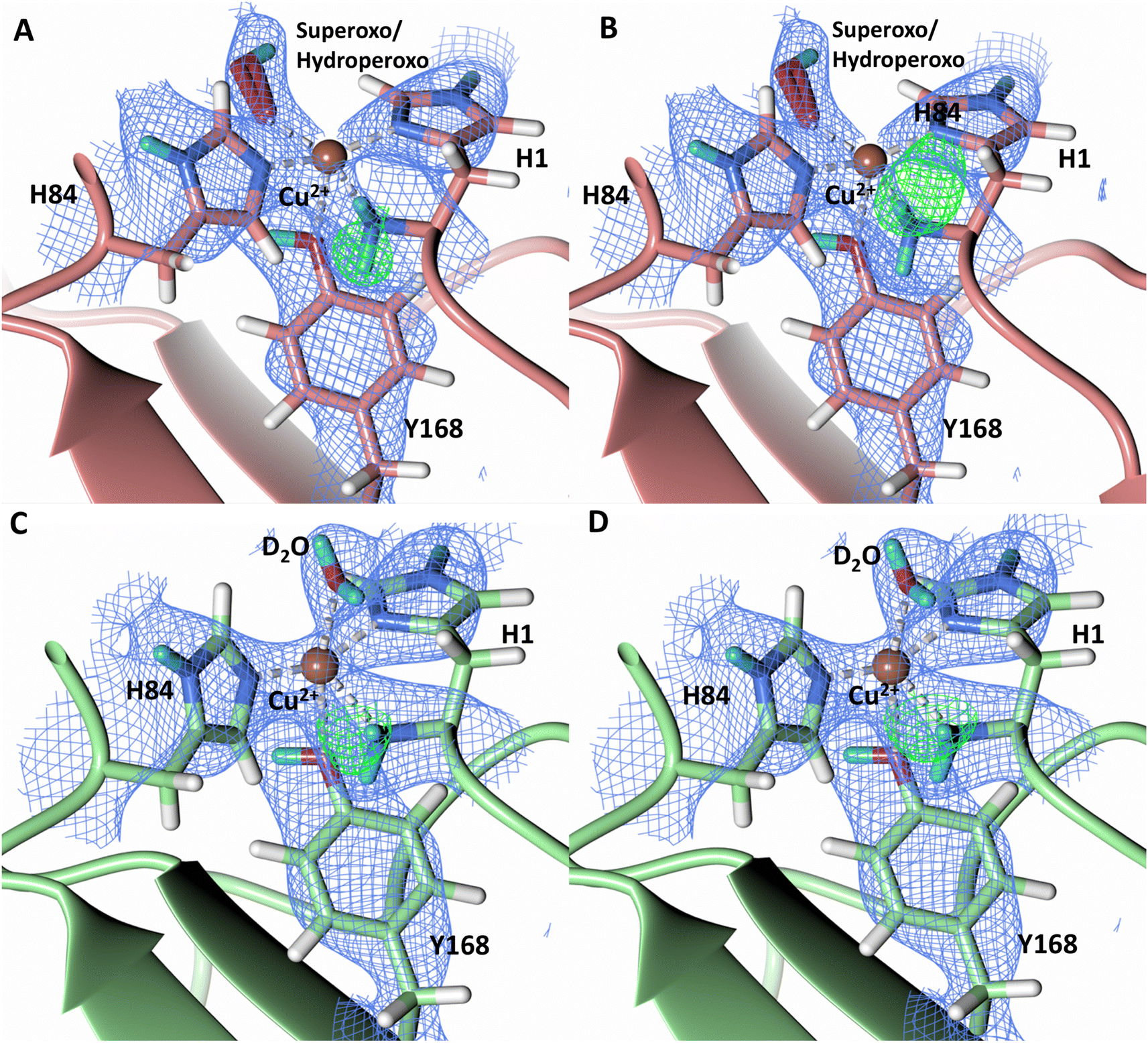 | ||
| Fig. 5 Protonation state of the N-terminal His157 amino group in the ascorbate reduced NcLPMO9D crystal (PDB 7T5D). (A) Molecule A (coral) omit map displaying D2 proton and (B) D3 proton of the N-terminal nitrogen. (C) Molecule B (light green) omit map displaying D2 proton and (D) D3 proton of the N-terminal nitrogen. Neutron scattering length density 2Fo–Fc maps displayed in blue at a 1.0 σ cut-off. Neutron scattering length density Fo–Fc maps displayed in green at a 3.0 σ cut-off. H atoms displayed in white and D atoms displayed in turquoise. | ||
A study by Paradisi et al. on LsAA9 spectroscopically detected a radical Cu2+-tyrosyl complex in which the axial Tyr164 was present as a deprotonated radical following reaction with H2O2 following ascorbate reduction.62 This tyrosyl radical intermediate formed irreversibly and was inactive toward substrate, indicating that it may play a role in active site protection during uncoupled turnover. A further spectroscopic study on TaLPMO9A by Singh et al. isolated a Cu2+-tyrosyl radical intermediate following ascorbate reduction and addition of H2O2.63 The long-lived Tyr175 radical intermediate was spectroscopically distinct from the tyrosyl radical observed in LsAA9 and was proposed to play a role in active site protection from oxidation or be involved in substrate oxidation. To address the observation of the two radical species, McEvoy et al. performed QM/MM to model the feasibility of both deprotonated tyrosyl radical species.64 The intermediate observed by Singh et al. was proposed to be to a cis-[Tyr-CuOH]+ formed by hydrogen transfer from the tyrosine hydroxy to an oxyl species in the equatorial plane. The intermediate observed by Paradisi et al. was characterized as a trans-[Tyr-CuOH]+ which can be formed through internal hydrogen transfer from a water molecule axially coordinated to the copper. Calculations of reactivity of these tyrosyl radical species toward the substrate however indicated limited reactivity, further supporting a protective role for tyrosine to prevent oxidative damage. Given these observations of a tyrosine acting as a proton donor to form a deprotonated phenoxyl species, we further examined the protonation state of Ty168 in the copper axial position of NcLPMO9D. Fo–Fc omit difference maps in both molecule A and B indicate Tyr168 remains in the protonated form (Fig. 6), excluding it as a proton donor at the early stages of dioxygen activation.
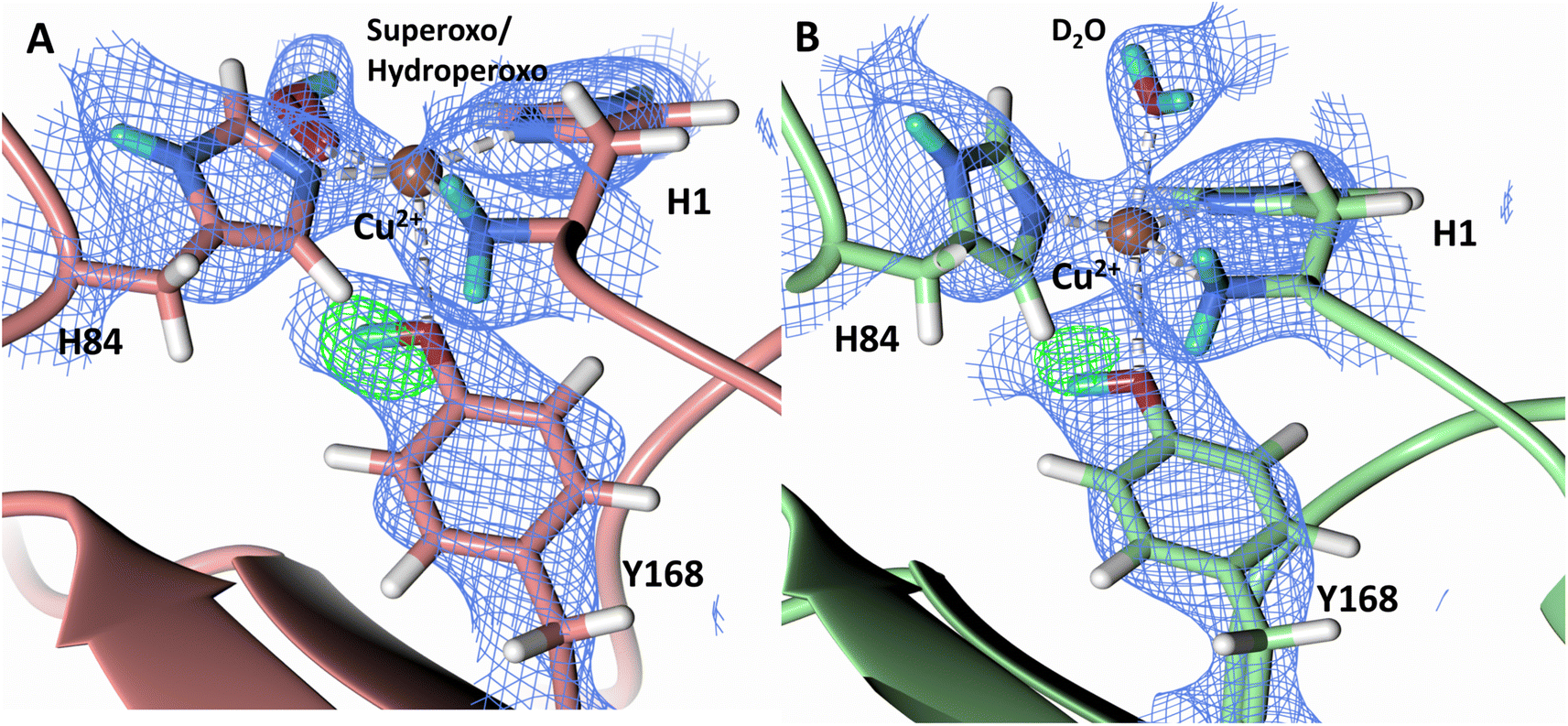 | ||
| Fig. 6 Protonation state of the Tyr168 in the ascorbate reduced NcLPMO9D crystal (PDB 7T5D). (A) Molecule A (coral) omit map displaying Oη protonation and (B) Molecule B (light green) omit map displaying Oη protonation. Neutron scattering length density Fo–Fc maps displayed in green at a 3.0 σ cut-off. H atoms displayed in white and D atoms displayed in turquoise. | ||
Histidine protonation at acidic conditions
During LPMO catalysis, a second shell histidine has been proposed to be involved in protonation of active site intermediates with QM/MM studies by Hedegård and Ryde on LsAA9A suggesting that His147 in a doubly protonated state can act as a proton donor.31 Additionally, Span et al. have proposed that His161 in MtPMO3 plays a role as a proton donor based on structural, mutagenesis and spectroscopic studies as well as activity assays.32 LPMOs are expected to catalyze oxygen insertion under acidic conditions since they function synergistically alongside cellulases which show optimum activity at pH 3.5–5.5.65–67 Given the surprising role of a neutral His157 as a proton donor to the activated dioxygen species in our structure, we sought to probe the protonation state of His157 when exposed to acidic buffer conditions. We collected a 2.14 Å room-temperature neutron diffraction dataset under low pH conditions (pH 4.4/pD 4.8) achieved by vapor exchange of the crystal with acidified buffer. Analysis of the neutron scattering length density maps indicate that the His157 residue remains singly protonated at the Nε-position at this pH, which is supported by Fo–Fc omit difference maps in both molecule A and B (Fig. 7). To further support our acidic pH structure obtained by vapor exchange, a 1.5 Å X-ray dataset was collected on a crystal directly soaked in acidic buffer. The position of His157 at pH 4.4 superimposes to its position at pH 6.0. In this position, Nδ-protonation would sterically clash with the backbone amide proton, further supporting the single Nε-protonation state observed in our neutron structure (Fig. S6 and S7†) which coincides with unrestrained refinement studies of the protonation state of second-shell His residues.68 The barricaded active site observed in crystallo and electropositive environment of the copper potentially play a role in maintaining the observed single histidine protonation state and prevent formation of protonation pathways to the active site. In both the neutron and X-ray low pH structures, the copper remains coordinated in the histidine brace, contrary to the disordered His78 observed in LsLPMO9A at pH 3.5 (PDB 5N04).69 The low pH structure of LsLPMO9A was obtained using high NaCl concentrations, however, resulting in His78 displacement by a chloride anion. The absence of NaCl in our crystallization conditions precludes coordinating histidine residue displacement by a chloride anion.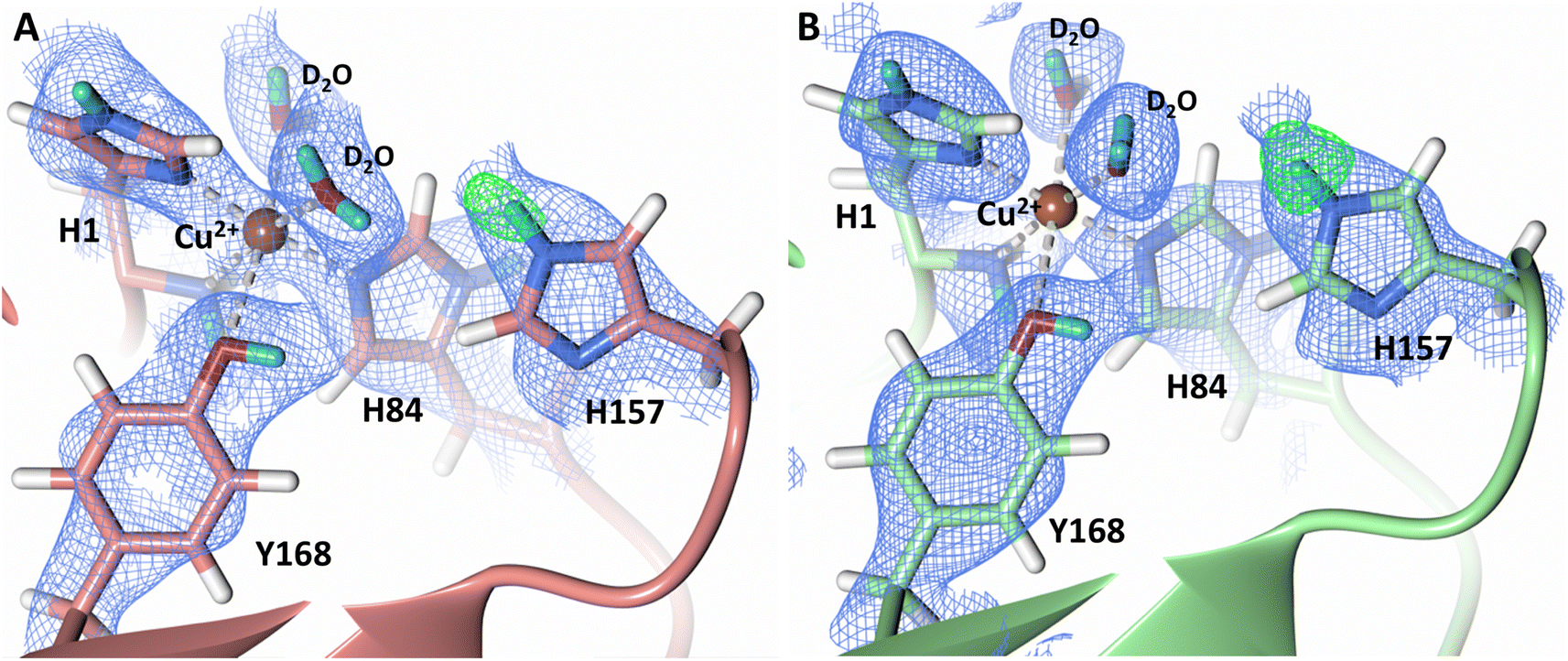 | ||
| Fig. 7 Protonation state of second-shell His157 at acidic conditions (PDB 7T5E). (A) Molecule A (coral) His157 Nε-protonation. (B) Molecule B (light green) His157 Nε-protonation. Neutron scattering length density 2Fo–Fc maps displayed in blue at a 1.0 σ cut-off. Neutron scattering length density Fo–Fc maps displayed in green at a 3.0 σ cut-off. H atoms displayed in white and D atoms displayed in turquoise. | ||
Conformational flexibility of His157
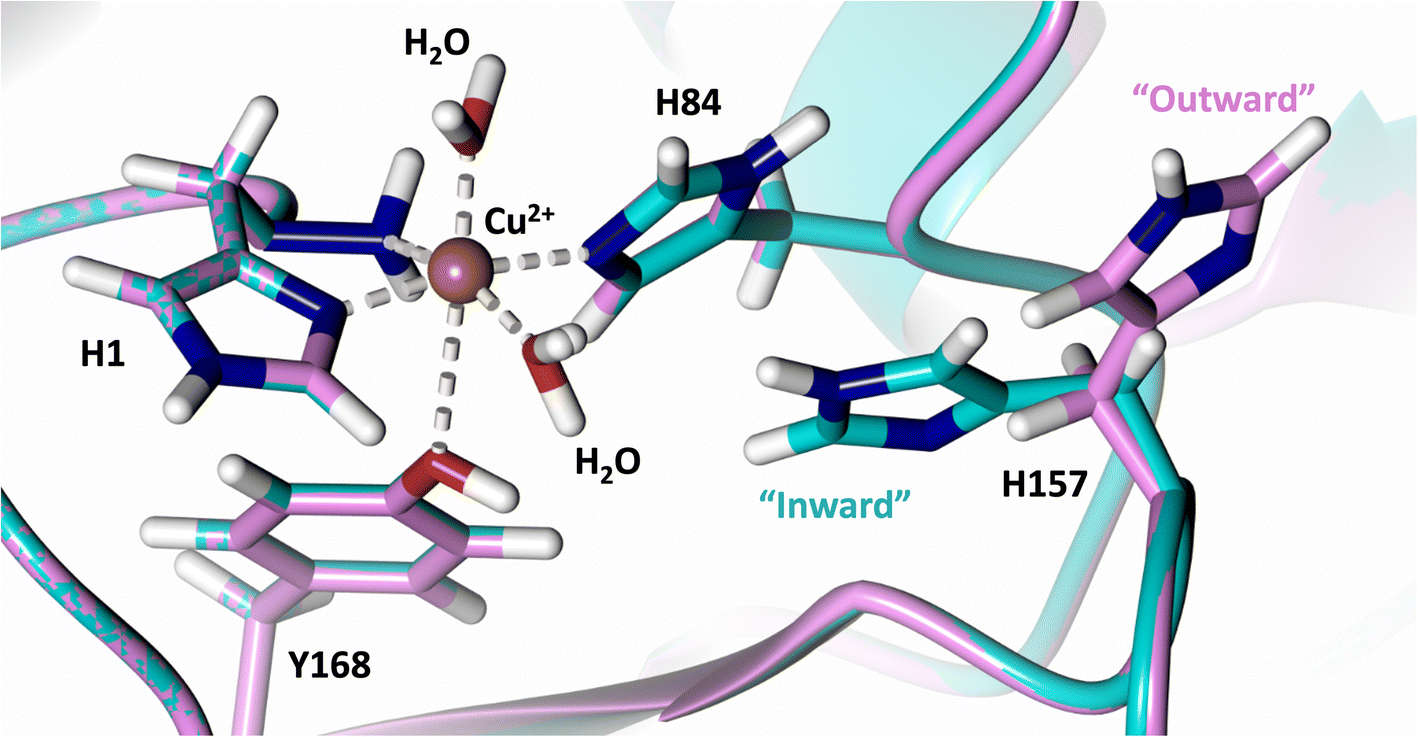
Analysis of the crystal structure of MtPMO3* deposited by Span et al. indicates that the second shell His161, corresponding to NcLPMO9D His157, is present in two conformations: an “inward” copper-facing conformation with a with a Cu–Nε distance of ∼5 Å, and an “outward” copper-distant conformation with a Cu–Nε distance of ∼10 Å (Fig. 8).32 The experimentally observed existence of both “inward” and “outward” conformations indicates mobility of the conserved second shell histidine, at least when the copper active site is solvent exposed. It may also suggest a mechanism whereby the “outward” second shell histidine can “collect” a proton from solvent (becoming doubly protonated) and, in turn, the “inward” conformation donates a proton in the active site.31,32 In this work we have established that in the dimeric packing of NcLPMO9D in the crystal, unusually, neutral His157 donates a proton to active site intermediates, and, furthermore, that it cannot become doubly protonated even under acidic conditions. We therefore conjectured (see previous section) that in the NcLPMO9D dimer, His157 is blocked (or barricaded) from “collecting” a proton from solvent. It does, however, seem possible, that in the unhindered monomer form of NcLPMO9D, a flexible His157 conformation dependent proton transport mechanism would be available.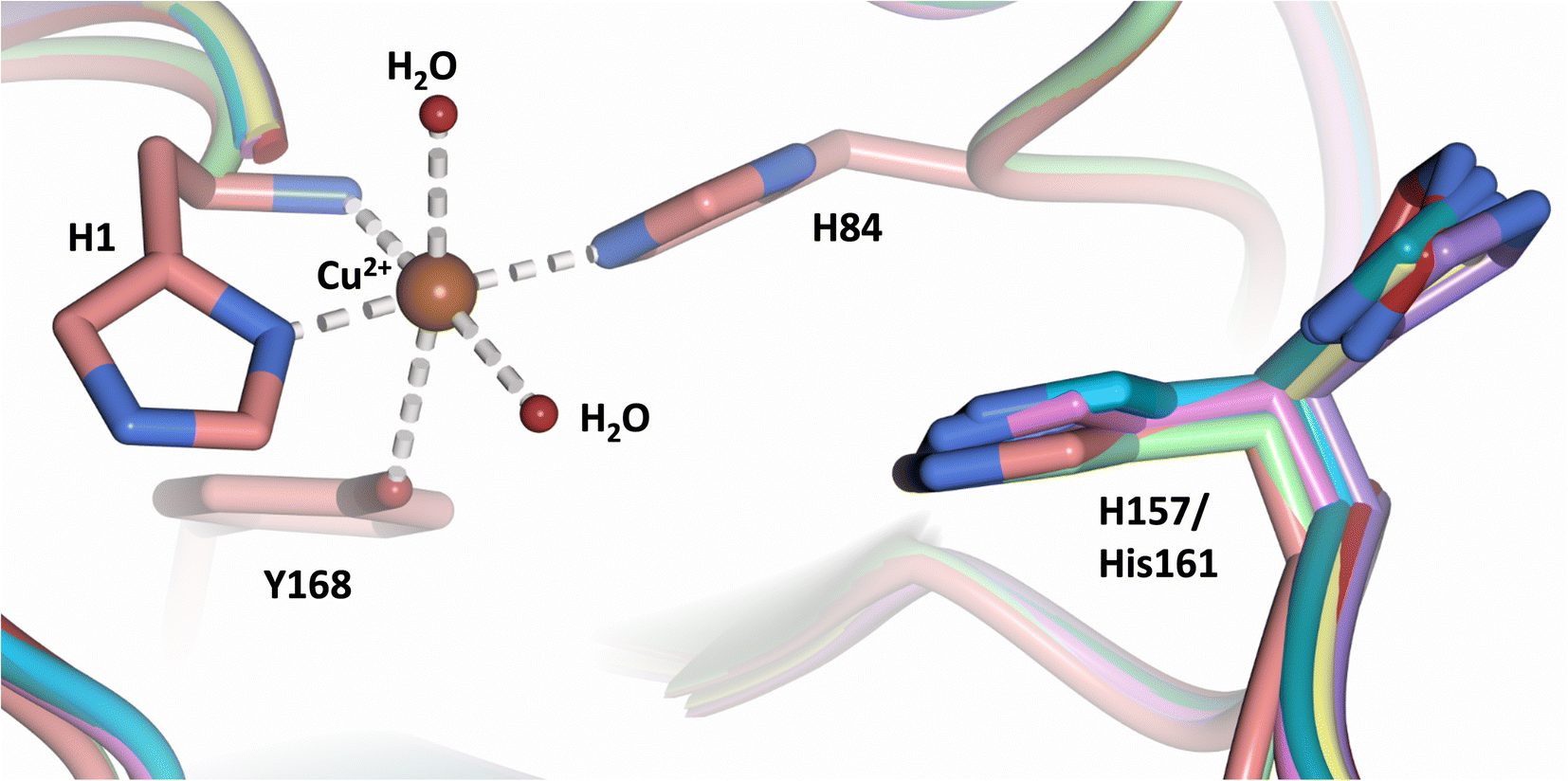 | ||
| Fig. 8 Alignment of a second shell residue His157 in the “inward” and “outward” conformation. Alignment of molecule A MtPMO3* His161 (PDB 5UFV) molecule A (teal), molecule B (yellow), molecule C (pink), molecule D (purple), molecule E (turquoise) and molecule F (red) with NcLPMO9D His157 (PDB 7T5C) from molecule A (coral) and molecule B (green). | ||
To examine computationally the possibility that NcLPMO9D His157 can exhibit “inward” and “outward” conformations when in monomeric form, and whether it is likely that dimer crystal packing occludes such flexibility, we used two different approaches: MD simulations and VM2 mining minima free energy calculations – see Methods section. The goal of the MD calculations was to establish whether inward/outward conversion is possible on the microsecond timescale. The mining minima calculations, unlike MD, can easily traverse high energy barriers during conformational searches. Therefore, it was employed to establish, regardless of kinetic barrier/timescale, whether inward/outward conformations exist or not in the monomer and dimer forms, and if so, what their relative energies are. It was also used to estimate an energy barrier to interconversion in the monomer.
The first 1.0 μs MD simulation, starting from the neutron structure reported here, showed a stable “inward” His157 conformation, staying in this original “inward” conformation during the entire MD trajectory. We note that although restraints were applied to keep Cu in the active site, the interacting residue conformations seen in the simulation were not adversely affected and are consistent with their positions in the neutron structure. The most flexible regions of the protein are 41–45, 117–120 and 197–214 (Fig. 9A). Representative conformations of the active site are shown in (Fig. 9B) and indicate little movement in that region.
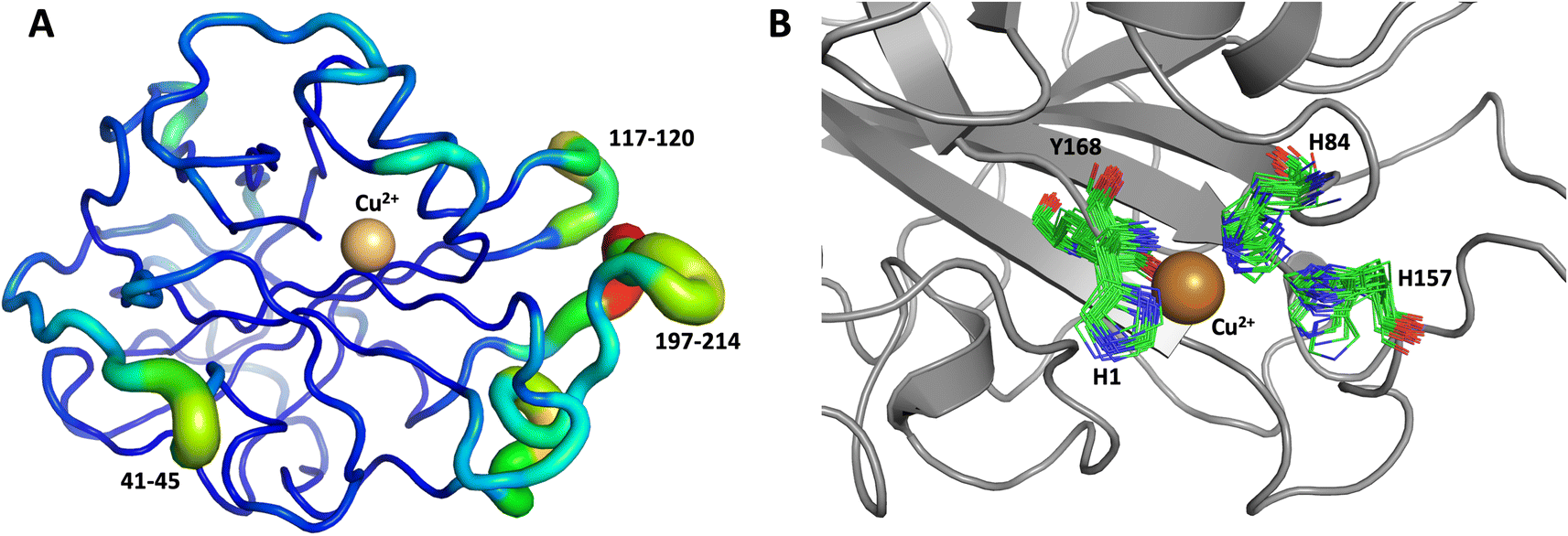 | ||
| Fig. 9 Conformational flexibility of LPMO and active-site residues. Results from 1 μs MD simulation, starting from the reported neutron structure. (A) Conformational flexibility of the entire protein calculated based on root mean square fluctuations of slowest 10 modes (RMSF10). Regions shown as thin blue tubes are rigid while colored regions with thicker green/yellow/red tubes show highest flexibility (regions labeled show largest flexibility). (B) Active site residues (only 20 frames spaced at regular interval of 50 ns are shown). | ||
To obtain further insight, another 1.0 μs MD simulation, based on the related LPMO MtPMO3* (PDB ID = 5UFV, monomer A), was performed. This MD simulation indicated that His161 (at the equivalent position to His157 in our neutron structure) is able to freely interconvert between the “inward” and “outward” conformations at the microsecond timescale (Fig. 10). Notably, the active site shows significantly more movement, and more than 10 transitions between inward and outward conformations were observed over the course of the whole simulation (Fig. 10B). Therefore, we hypothesized that His157 in our structure is possibly trapped in a conformation that makes it difficult to explore outward conformations at the microsecond timescale, i.e., there is a larger kinetic barrier to transition than for 5UFV. Importantly, for the MD simulation based on 5UFV, restraints between Cu and the surrounding residues were equivalent to those used in the first simulation, which excludes the possibility that the lack of interconversion between the inward and outward conformation in MD based on our structure is due to the use of distance restraints. Overall, we observed that MtPMO3* (5UFV) is, according to the simulation, a lot more flexible than our neutron diffraction structure, with the regions 18–25, 32–37, 110–122, 159–168 and 205–216 showing high flexibility (Fig. 10A). Given that the forcefield and water model are identical, and the only difference is the protein sequence, we are confident that the MtPMO3* second coordination shell residues display more overall flexibility than NcLPMO9D at the microsecond timescale. This difference in flexibility is consistent with the interconversion behavior of His157/161 as well. (See ESI† for representative conformations of the active site).
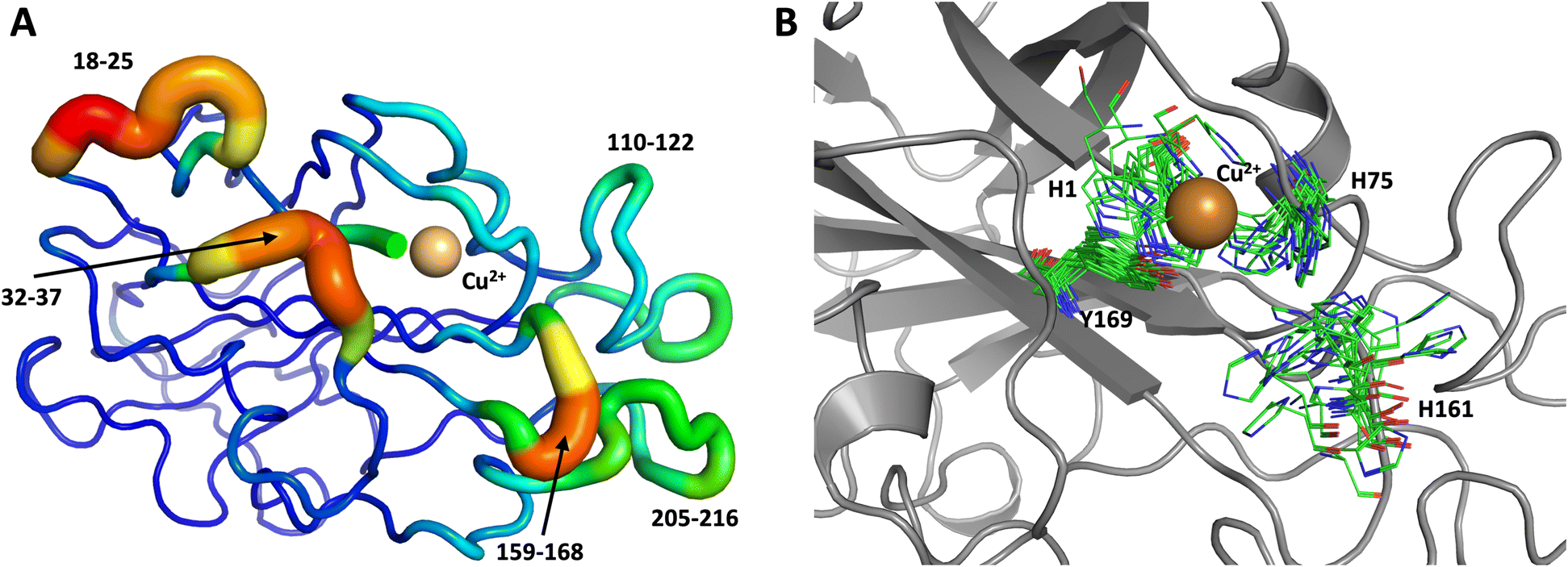 | ||
| Fig. 10 Conformational flexibility of a related LPMO and active-site residues (MtPMO3* PDB 5UFV) results from 1 μs MD simulation. (A) Conformational flexibility of based on RMSF10. (B) Active site residues. His161 (equivalent to His157) is highly flexible and transition back and forth between the inward and outward conformations during the 1 μs MD simulation (see Fig. 9 legend for more details). | ||
We performed another 1.0 μs MD simulation based on our neutron scattering LPMO structure but with an outward His157 conformation, in order to characterize the flexibility of His157 when starting from the outward conformation. The starting outward conformation was generated using the VM2 mining minima software, as described in detail below. This system is identical to the first MD simulation discussed above (same topology file with the same number of atoms) except for the starting conformation. In this simulation, His157 was able to interconvert to the inward conformation at the microsecond timescale only twice (Fig. 11), confirming our hypothesis of some dynamic interconversion of His157 (Fig. 11B) albeit at a lower rate than that observed for MtPMO3*. Once fully in the inward conformation, His157 stayed locked in that position, suggesting the presence of an energy barrier to interconversion that limits occurrence on the 1.0 μs timescale at least. It is interesting that the two related LPMOs show a difference in the rates of His157 (and equivalent His161) conformation interconversion. This interconversion rate could have a functional role, as it may affect the sampling of certain conformational events required in the catalytic cycle, as has been observed in several other enzyme systems.70
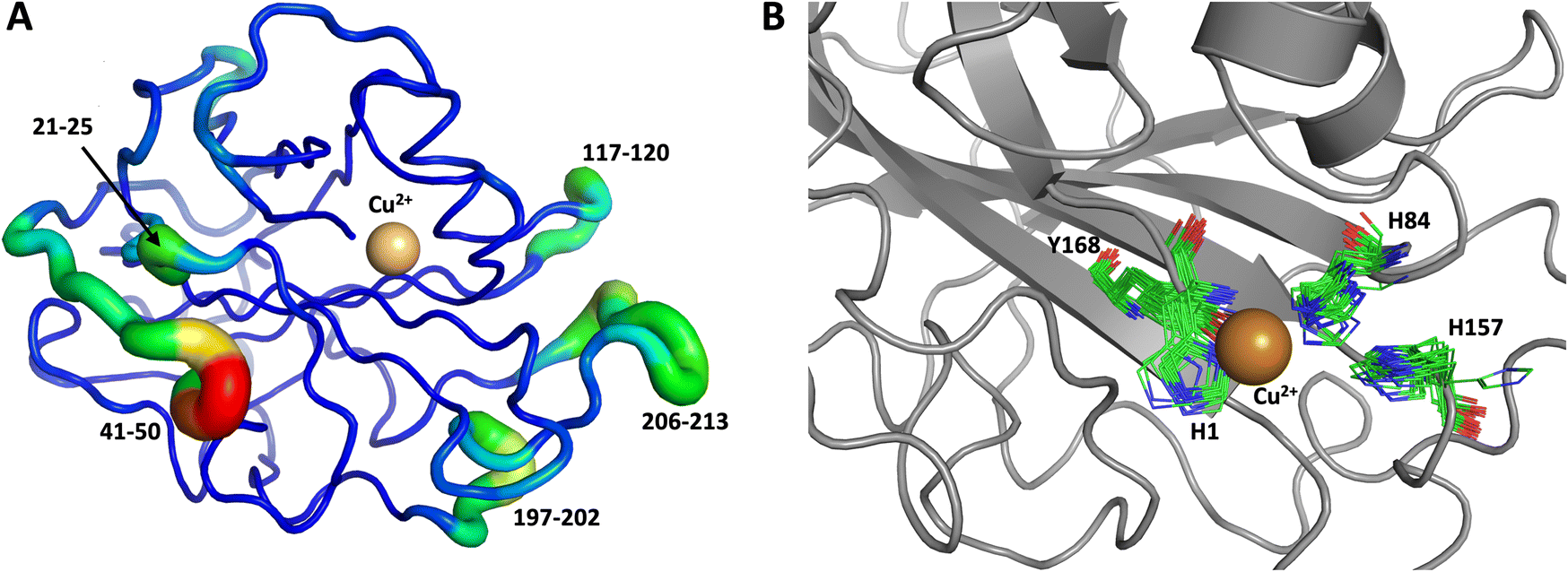 | ||
| Fig. 11 Conformational flexibility of LPMO and active-site residues (starting from His157 in outward conformation). Results from 1 μs MD simulation. (A) Conformational flexibility of based on RMSF10. (B) Active site residues. His157 is able to explore different conformations and transition into the inward conformation during the 1 μs MD simulation. See Fig. 9 legend for more details. | ||
When applied to NcLPMO9D with its active site exposed to solvent (i.e. the monomeric structure), the VM2 method, which performs extensive molecular conformational searching and provides a total free energy via a Boltzmann weighted ensemble of the resultant low energy conformers, predicts that both “inward” and “outward” His157 confirmations are populated (a representative structure from these calculations with His157 in the outward conformation was used as the starting model of the MD simulations previously described). Focusing only on the two lowest energy conformers found by VM2 (Fig. 12), the first conformer is “inward” and the second conformer is “outward” with a free energy difference of only 0.11 kcal mol−1 between them. Furthermore, summing the probabilities of each type of conformation populated, so that 99.0% of the total population is included, His157 has a predicted 3:1 “inward” to “outward” probability distribution. In contrast, when applied to the NcLPMO9D dimer, as per the crystal structure (Fig. 1A), VM2 finds only conformations where His157 remains fully “inward”. This is despite VM2's ability to drive molecular conformations over large energy barriers, as well as a free energy window set at 10 kcal mol−1 for retaining minima found by the conformational search. This result strongly supports preclusion of movement of His157 to an “outward” conformation due to the barricaded active site present in the crystal structure. An estimate of the inward/outward conversion barrier in the monomer was obtained by generation of an interpolated reaction path using the Morph Conformations, with corkscrew interpolation, capability in UCSF Chimera59,76 between the lowest free energy “inward” and “outward” conformations found by VM2. For each reaction path structure, a constrained conformational search was carried out, providing an energy path for the conversion process. These calculations suggest that the barrier is ∼11.0 kcal mol−1 from the inward to outward conformation, and ∼12.1 kcal mol−1 from the outward to the inward conformation. More details are available in the ESI – see Table S6 and Fig. S1.†
 | ||
| Fig. 12 Second shell His157 conformations from VM2 mining minima free energy calculations. For NcLPMO9D with its active site exposed to solvent His157 adopts an “inward” (cyan) and “outward” (pink) conformation. The inward and outward conformations are separated by a 0.11 kcal mol−1. H atoms displayed in white. | ||
We conclude, then, that the steric obstruction resulting from crystal packing of the dimer has significantly limited the conformational flexibility of His157 and suggest that the likely mobility of this residue in the monomer, which has a solvent exposed active site, may facilitate its protonation and serve a functional role. Taken together with the crystal structure active site occlusion, these factors may explain the single Nε-protonation of His157 observed even at acidic conditions. However, the single protonation of His157 solely at the Nε position in our crystal structures does not rule out formation of the doubly protonated second shell histidine residue under similar acidic operando conditions where the active site would not be barricaded by crystal packing.
Conclusion
The neutron structure presented here provides a full-atom snapshot of the early catalytic intermediates in the LPMO reaction pathway. Our results provide the first direct observation of a cryo-trapped protonated copper–dioxygen species at the LPMO active site. The unique occluded active site environment found in the crystal structure presented here has resulted in stabilization of an activated dioxygen species at the NcLPMO9D active site with joint occupancy as a superoxo intermediate and a hydroperoxo intermediate. A second-shell histidine was further observed in a single Nε-protonation state and in the imidazolate form, leading us to postulate that it plays a role in the early stages of dioxygen intermediate protonation to form an end-on coordinated hydroperoxo species, as observed in small molecule copper complexes.49 The unique barricaded environment provided by the binding-face to binding-face crystal packing in our structure facilitated trapping of the superoxo/hydroperoxo active site intermediate and identification of His157 as the proton donor. While it is not generally established which reactive intermediate is involved in HAA, the observed hydroperoxo species may perform substrate hydrogen abstraction or undergo reduction and protonation to form a potential Cu oxyl species responsible for substrate hydrogen abstraction in the LPMO reaction cycle. Given the absence of substrate in the current structure the peroxo species may, however, constitute an intermediate in the formation of H2O2 in a non-productive LPMO catalytic pathway. Although unusual, a neutral histidine functioning as a general acid has been mechanistically proposed for the His95 residue in triose phosphate isomerase and the His98 residue in methylglyoxal synthase.77–79 It remains to be determined whether the role of the second shell His157 in protonation of the activated dioxygen species prevails operando when NcLPMO9D is solvent-exposed and intermittently binds substrate. The computational studies presented here further describe novel flexibility of LPMO second shell His residues. The flexibility of MtPMO3* His161, as well as that observed for NcLPMO9D His157 to a lesser extent, may serve a functional role in catalysis that must be further explored.Experimental and computational methods section
A detailed account of the experimental procedure, data collection and refinement strategy has been described.80 Provided here are the experimental details in brief as well as descriptions of the computational methods employed.Protein expression and crystallization
Neurospora crassa LPMO9D (NcPMO-2) was heterologously expressed in the Pichia pastoris SuperMan5 strain and purified and crystallized as described by O'Dell et al. previously43,44 Crystal quality was assessed on beamline CG4-D, the IMAGINE instrument at the High Flux Isotope Reactor at Oak Ridge National Laboratory.81Crystal ascorbate soak and data collection
To reduce the copper active site, a large crystal was harvested and placed in a deuterated buffer containing 100 mM HEPES pD 6.0 (pH 5.6), 25% PEG 3350 and 100 mM ascorbic acid pD 6.0 (pH 5.6) at room temperature. The crystal was incubated for two hours after which it was flash-frozen in liquid nitrogen using the deuterated buffer supplemented with 25% glycerol as cryoprotectant. Time-of-flight neutron diffraction data were collected on beamline 11B, the Macromolecular Neutron Diffractometer (MaNDi) at the Spallation Neutron Source (SNS) at Oak Ridge National Laboratory.82Crystal low pH vapor exchange data collection
To obtain low pH conditions in the LPMO crystal, a large crystal was harvested and mounted in a quartz capillary to which a deuterated low pH buffer was added to facilitate vapor exchange in three stages.83,84 The first vapor exchange was performed in 22% PEG 3350 and 100 mM sodium acetate pD 5.6 (pH 5.2) for three days, followed by 22% PEG 3350 and 100 mM sodium acetate pD 5.2 (pH 4.8) for six days and 22% PEG 3350 and 100 mM sodium acetate pD 4.8 (pH 4.4) for 16 days. Time-of-flight neutron diffraction data were collected on beamline 11B, the Macromolecular Neutron Diffractometer (MaNDi) at the Spallation Neutron Source (SNS) at Oak Ridge National Laboratory.82 Following this, an X-ray dataset was collected on the same crystal at 298 K using a copper rotating-anode home source.Crystal low pH direct soak and data collection
LPMO crystals were directly exposed to acidic conditions by soaking crystals in low pH buffer. A crystal was transferred to a buffer composed of 22% PEG 3350, 100 mM and 100 mM sodium acetate with sequential incubation at pH 5.6, pH 5.2 and finally pH 4.8 for ten minutes each. Crystals were subsequently flash-frozen in liquid nitrogen with the pH 4.8 acidic buffer supplemented with 25% glycerol used as a cryoprotectant. Data were collected at 100 K using a copper rotating-anode home source.Model refinement
X-ray data were indexed, integrated and scaled with CrysAlisPRO and the CCP4 suite.85,86 Neutron diffraction data were reduced using the Mantid data analysis and visualization package.87 The time-of-flight diffraction data were integrated using three-dimensional profile fitting of the Bragg peaks.88 Structure solution and refinement of the X-ray diffraction data of the low pH structures as well the neutron diffraction data of the ascorbate-soaked structure were performed by utilizing the PHENIX software suite with manual model building to fit density maps performed in Coot.89–91 Dioxygen species were modelled using restrained bond length refinements.Electronic structure (density functional theory) calculations
The active site models used for DFT calculation were derived by extracting coordinates from the resting state X-ray structure (PDB 5TKG, O'Dell et al.43) including residues His1, His84, His157, Gln166 and Tyr168 along with the active site copper(II) ion, pre-bound molecular O2 and the axial and equatorial water molecules. To reduce the total number of atoms in these models, the main chain of residues His84, His157, Gln166 and Tyr168 were truncated at Cβ which was modeled as a methyl group. DFT calculations were performed with Gaussian 09 using the B3LYP functional with the 6-31G** basis set applied to all atoms. The model was implicitly solvated using the polarizable continuum model as implemented in Gaussian 09 with a dielectric constant of 4.24 (scrf keyword solvent = DiethylEther). For geometry optimization, the coordinates of one heavy atom per residue (as performed previously in DFT calculation of NcLPMO9D in O'Dell et al.43) were constrained to their starting values to maintain the relative conformation imposed by the protein backbone.MD simulations
Molecular dynamics (MD) simulations were performed for LPMO in explicit water solvent, using a protocol developed in our lab.92 Model preparation and simulations were performed using the AMBER v16 suite of programs for biomolecular simulations.93 AMBER's ff14SB force-fields were used for all simulations.94 MD simulations were performed using NVIDIA graphical processing units (GPUs) and AMBER's pmemd.cuda simulation engine using our lab protocols published previously.95,96A total of 4 separate simulations were performed for LPMO in the monomeric form. The monomer form was used to avoid crystal packing artifacts. The first MD simulation was based on the structure determined in this study with His157 in the inward conformation, a second MD simulation was started with His157 in outward conformation obtained from VM2 calculations – see below. The third MD simulation was based on a related LPMO structure with PDB code 5UFW (first protomer only). To rule out any difference in results between the monomeric and dimeric forms, a fourth MD simulation was performed with LPMO in dimeric form based on the structure reported here. (The results of the dimeric complexes were similar to the monomeric form and were excluded from further analysis).
The missing hydrogen atoms were added by AMBER's tleap program. After processing the coordinates of the protein and substrate, all systems were neutralized by addition of counter-ions and the resulting system were solvated in a rectangular box of SPC/E water, with a 10 Å minimum distance between the protein and the edge of the periodic box. The prepared systems were equilibrated using a protocol described previously.96 To keep Cu in the active-site for each protomer, 6 distance restraints (5.0 kcal mol−1 A−2) were applied between the following atoms: Cu-His1N, Cu-His1ND1, Cu-His84NE1, Cu-168TyrOH, and Cu with the two crystallographic water oxygen atoms. For 5UFW based system: Cu-His1N, Cu-His1ND1, Cu-His75NE1, Cu-169TyrOH, and Cu with the two crystallographic water oxygen atoms.
The equilibrated systems were then used to run 1.0 μs of production MD under constant energy conditions (NVE ensemble) and the 6 distance restraints per protomer. The use of the NVE ensemble is preferred as it offers better computational stability and performance.97 The production simulations were performed at a temperature of 300 K. As the NVE ensemble was used for production runs, these values correspond to the initial temperature at start of simulations. A temperature adjusting thermostat was not used in the simulations; over the course of 1.0 μs simulations the temperature fluctuated close to 300 K with RMS fluctuations between 2 and 4 K, which is typical for well equilibrated systems. A total of 1000 conformational snapshots (stored every 1000 ps) were collected for each system and were used for analysis. Structures were visualized using PyMOL.
VM2 calculations
VM2 is an implementation of the second-generation Mining Minima (M2) method originally developed by Gilson et al.100–105 It is a free energy calculation method, which combines extensive molecular conformational searching71,106 with a rigorous statistical mechanics approach.100 Molecular configuration integrals over all space are closely approximated in a tractable manner by summation of local configuration integrals associated with the low energy minima of the system.107,108 VM2's search algorithm can drive molecular conformations over high-energy barriers, via a mode-distort-minimize procedure, to find relevant minima even when they structurally diverse. As such, VM2 is well suited for exploration of thermally accessible conformations of His157 in monomeric and dimeric NcLPMO9D, respectively, as carried out in this study.We applied molecular mechanics (MM) based VM2, taking the AMBER ff14SB parameter files (prmtop etc.) generated by the setup for MD simulations (see above) and converting them to formats readable by the VM2 software. It should be noted that for computational efficiency, VM2 includes solvation effects through continuum models (see below for details), so only the water molecules coordinating directly with active site copper centers were explicitly included. To further manage computational expense, and to focus on the region of interest, not all protein atoms were included in the VM2 calculation. Rather, a ‘cutout’ approach was taken, where all residues containing an atom within 14 Å of the His157 alpha carbon (CA) were present in the energy model, and of these all atoms within 12 Å of His157 CA were set as mobile. For the monomer, this resulted in 904 atoms present in the calculation, with 476 of these mobile; for the dimer, this resulted in 1256 atoms present, with 600 of these mobile. To maintain the basic arrangement of the active site copper first-shell coordination residues (histidine brace, tyrosine, and two water molecules) during aggressive conformational searching, a flat-bottomed energy well restraint potential was applied to their coordinating atoms, as well as the central copper atom itself.
VM2 is an iterative method,71 with each iteration comprising a conformational search, with removal of any repeat conformers produced, calculation of the configuration integrals at 300 K for the resulting set of new conformers,107,108 addition of this set of conformers to the pool already established, and then determination of the total free energy by summation of the local configuration integrals of all conformers (minima) currently in the pool. Once the conformational search no longer finds new low energy conformers the total free energy stops changing between iterations. The calculations in this study were deemed converged when the absolute value of the free energy change between iterations was less than 0.01 kcal mol−1. The conformational search carried out during each VM2 iteration included 1200 mode-distort-minimize procedures. Molecular distortions were from atom displacements along single-modes produced by diagonalization of the Hessian matrix in torsional space and, in addition, atom displacements based on random combinations of pairs of these modes.71,106 For energy-derivative involved steps, e.g. the mode-distort-minimize procedure and the Hessian (energy 2nd-derivative) calculation, required for configurational entropy terms, solvation energy was included using the generalized Born (GB) continuum model.109,110 The Poisson–Boltzmann Surface Area (PBSA) method111 was applied to provide a more accurate single-point solvation energy correction at GB determined geometries. On completion, VM2 calculations output the total free energy, and in addition the relative free energy of each individual conformer along with its Boltzmann distribution probability. Also outputted are PDB and mol2 formatted files containing all protein conformers produced within 10 kcal mol−1 of the lowest energy conformer, up to a maximum of 1000, allowing for structural visualization and analysis.
Data availability
Crystallographic data for NcLPMO9D at low pH (X-ray structure), ascorbate soaked (neutron structure), low pH vapor exchange (joint X-ray/neutron structure) have been deposited at the Protein Data Bank under PDB IDs 7T5C, 7T5D and 7T5E, respectively, and can be obtained from https://doi.org/10.2210/pdb7t5c/pdb, https://doi.org/10.2210/pdb7t5d/pdb and https://doi.org/10.2210/pdb7t5e/pdb. Molecular dynamics (MD) trajectory data for this paper are available at the Data Archiving and Networked Services (DANS) at https://doi.org/10.17026/dans-zkr-pyrc. Further datasets supporting this article have been uploaded as part of the ESI.†Author contributions
GCS, WBO and FM conceived the project. WBO produced and crystallized the protein. GS and FM collected the neutron and X-ray diffraction data. GCS refined the structures. SPW and PKA performed the DFT calculations, free energy calculations and MD simulations. GCS wrote the original draft, and all authors edited and reviewed the paper.Conflicts of interest
S. P. W. declares an equity interest in VeraChem LLC, which develops the VM2 free energy software package. The remaining authors declare no conflict of interest.Acknowledgements
Protein expression, purification and crystallization experiments were conducted at the Center for Structural Molecular Biology (CSMB), a U.S. Department of Energy Biological and Environmental Research User Facility at Oak Ridge National Laboratory. Neutron diffraction data was collected at BL-11B MaNDi at the Spallation Neutron Source (SNS) at ORNL which is sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. The authors thank Brendan Sullivan for assistance with data reduction. X-ray diffraction data was collected at the Molecular Education, Technology, and Research Innovation Center (METRIC) facilities at North Carolina State University, which is supported by the State of North Carolina. GCS acknowledges support in part from the National Research Foundation (NRF), South Africa and the Oak Ridge National Laboratory Graduate Opportunities (GO!) program. F. M. acknowledges support from USDA NIFA Hatch 211001. This research was supported by the National Institute of General Medical Science R01-GM105978 to P. K. A.References
- K. K. Meier, S. M. Jones, T. Kaper, H. Hansson, M. J. Koetsier, S. Karkehabadi, E. I. Solomon, M. Sandgren and B. Kelemen, Chem. Rev., 2018, 118, 2593–2635 CrossRef CAS PubMed.
- A. Levasseur, E. Drula, V. Lombard, P. M. Coutinho and B. Henrissat, Biotechnol. Biofuels, 2013, 6, 1–14 CrossRef PubMed.
- R. J. Quinlan, M. D. Sweeney, L. Lo Leggio, H. Otten, J. C. N. Poulsen, K. S. Johansen, K. B. R. M. Krogh, C. I. Jørgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15079–15084 CrossRef CAS PubMed.
- R. C. Gregory, G. R. Hemsworth, J. P. Turkenburg, S. J. Hart, P. H. Walton and G. J. Davies, Dalton Trans., 2016, 45, 16904–16912 RSC.
- E. A. Span and M. A. Marletta, Curr. Opin. Struct. Biol., 2015, 35, 93–99 CrossRef CAS PubMed.
- M. Gudmundsson, S. Kim, M. Wu, T. Ishida, M. H. Momeni, G. Vaaje-Kolstad, D. Lundberg, A. Royant, J. Ståhlberg, V. G. H. Eijsink, G. T. Beckham and M. Sandgren, J. Biol. Chem., 2014, 289, 18782–18792 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, Science, 2010, 330, 219–222 CrossRef CAS PubMed.
- P. V. Harris, D. Welner, K. C. McFarland, E. Re, J. C. Navarro Poulsen, K. Brown, R. Salbo, H. Ding, E. Vlasenko, S. Merino, F. Xu, J. Cherry, S. Larsen and L. Lo Leggio, Biochemistry, 2010, 49, 3305–3316 CrossRef CAS PubMed.
- C. M. Phillips, W. T. Beeson, J. H. Cate and M. A. Marletta, ACS Chem. Biol., 2011, 6, 1399–1406 CrossRef CAS PubMed.
- B. Bissaro, Å. K. Røhr, G. Müller, P. Chylenski, M. Skaugen, Z. Forsberg, S. J. Horn, G. Vaaje-Kolstad and V. G. H. Eijsink, Nat. Chem. Biol., 2017, 13, 1123–1128 CrossRef CAS PubMed.
- F. Filandr, P. Man, P. Halada, H. Chang, R. Ludwig and D. Kracher, Biotechnol. Biofuels, 2020, 13, 1–13 CrossRef PubMed.
- A. A. Stepnov, Z. Forsberg, M. Sørlie, G. S. Nguyen, A. Wentzel, Å. K. Røhr and V. G. H. Eijsink, Biotechnol. Biofuels, 2021, 14, 1–14 CrossRef PubMed.
- W. T. Beeson, C. M. Phillips, J. H. D. Cate and M. A. Marletta, J. Am. Chem. Soc., 2012, 134, 890–892 CrossRef CAS PubMed.
- W. T. Beeson, V. V. Vu, E. A. Span, C. M. Phillips and M. A. Marletta, Annu. Rev. Biochem., 2015, 84, 923–946 CrossRef CAS PubMed.
- X. Li, W. T. Beeson, C. M. Phillips, M. A. Marletta and J. H. D. Cate, Structure, 2012, 20, 1051–1061 CrossRef CAS PubMed.
- C. H. Kjaergaard, M. F. Qayyum, S. D. Wong, F. Xu, G. R. Hemsworth, D. J. Walton, N. A. Young, G. J. Davies, P. H. Walton, K. S. Johansen, K. O. Hodgson, B. Hedman and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8797–8802 CrossRef CAS PubMed.
- J. A. Hangasky, T. C. Detomasi and M. A. Marletta, Trends Chem., 2019, 1, 198–209 CrossRef CAS.
- P. Chylenski, B. Bissaro, M. Sørlie, Å. K. Røhr, A. Várnai, S. J. Horn and V. G. H. Eijsink, ACS Catal., 2019, 9, 4970–4991 CrossRef CAS.
- R. L. Peterson, R. A. Himes, H. Kotani, T. Suenobu, L. Tian, M. A. Siegler, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2011, 133, 1702–1705 CrossRef CAS PubMed.
- S. I. Mann, T. Heinisch, T. R. Ward and A. S. Borovik, J. Am. Chem. Soc., 2017, 139, 17289–17292 CrossRef CAS PubMed.
- R. Trammell, Y. Y. See, A. T. Herrmann, N. Xie, D. E. Díaz, M. A. Siegler, P. S. Baran and I. Garcia-Bosch, J. Org. Chem., 2017, 82, 7887–7904 CrossRef CAS PubMed.
- I. Garcia-Bosch and M. A. Siegler, Angew. Chem., Int. Ed., 2016, 55, 12873–12876 CrossRef CAS PubMed.
- L. Bertini, R. Breglia, M. Lambrughi, P. Fantucci, L. De Gioia, M. Borsari, M. Sola, C. A. Bortolotti and M. Bruschi, Inorg. Chem., 2018, 57, 86–97 CrossRef CAS PubMed.
- P. J. Donoghue, J. Tehranchi, C. J. Cramer, R. Sarangi, E. I. Solomon and W. B. Tolman, J. Am. Chem. Soc., 2011, 133, 17602–17605 CrossRef CAS PubMed.
- D. Dhar, G. M. Yee, A. D. Spaeth, D. W. Boyce, H. Zhang, B. Dereli, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2016, 138, 356–368 CrossRef CAS PubMed.
- D. Schröder, M. C. Holthausen and H. Schwarz, J. Phys. Chem. B, 2004, 108, 14407–14416 CrossRef.
- K. Yoshizawa, N. Kihara, T. Kamachi and Y. Shiota, Inorg. Chem., 2006, 45, 3034–3041 CrossRef CAS PubMed.
- N. Dietl, C. Van Der Linde, M. Schlangen, M. K. Beyer and H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 4966–4969 CrossRef CAS PubMed.
- S. Kim, J. Ståhlberg, M. Sandgren, R. S. Paton and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 149–154 CrossRef CAS PubMed.
- E. D. Hedegård and U. Ryde, J. Biol. Inorg Chem., 2017, 22, 1029–1037 CrossRef PubMed.
- E. D. Hedegård and U. Ryde, Chem. Sci., 2018, 9, 3866–3880 RSC.
- E. A. Span, D. L. M. Suess, M. C. Deller, R. D. Britt and M. A. Marletta, ACS Chem. Biol., 2017, 12, 1095–1103 CrossRef CAS PubMed.
- O. Caldararu, E. Oksanen, U. Ryde and E. D. Hedegård, Chem. Sci., 2019, 10, 576–586 RSC.
- E. D. Hedegård and U. Ryde, Chem. Sci., 2018, 9, 3866–3880 RSC.
- B. Wang, P. H. Walton and C. Rovira, ACS Catal., 2019, 9, 4958–4969 CrossRef CAS PubMed.
- G. C. Schröder, W. B. O'Dell, D. A. A. Myles, A. Kovalevsky and F. Meilleur, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 778–786 CrossRef PubMed.
- W. B. O'Dell, A. M. Bodenheimer and F. Meilleur, Arch. Biochem. Biophys., 2016, 602, 48–60 CrossRef PubMed.
- G. C. Schröder and F. Meilleur, Acta Crystallogr., Sect. D: Struct. Biol., 2021, 77, 1251–1269 CrossRef PubMed.
- P. C. E. Moody and E. L. Raven, Acc. Chem. Res., 2018, 51, 427–435 CrossRef CAS PubMed.
- H. Kwon, T. E. Schrader, A. Ostermann, M. P. Blakeley, E. L. Raven and P. C. E. Moody, in Neutron Crystallography in Structural Biology, Elsevier Inc., 1st edn, 2020, vol. 634, pp. 379–389 Search PubMed.
- C. M. Casadei, A. Gumiero, C. L. Metcalfe, E. J. Murphy, J. Basran, M. G. Concilio, S. C. M. Teixeira, T. E. Schrader, A. J. Fielding, A. Ostermann, M. P. Blakeley, E. L. Raven and P. C. E. Moody, Science, 2014, 345, 193–197 CrossRef CAS PubMed.
- H. Kwon, J. Basran, C. M. Casadei, A. J. Fielding, T. E. Schrader, A. Ostermann, J. M. Devos, P. Aller, M. P. Blakeley, P. C. E. Moody and E. L. Raven, Nat. Commun., 2016, 7, 13445 CrossRef CAS PubMed.
- W. B. O'Dell, P. K. Agarwal and F. Meilleur, Angew. Chem., Int. Ed., 2017, 56, 767–770 CrossRef PubMed.
- W. B. O'Dell, P. D. Swartz, K. L. Weiss and F. Meilleur, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2017, 73, 70–78 CrossRef PubMed.
- S. Karkehabadi, H. Hansson, S. Kim, K. Piens, C. Mitchinson and M. Sandgren, J. Mol. Biol., 2008, 383, 144–154 CrossRef CAS PubMed.
- M. Eibinger, J. Sattelkow, T. Ganner, H. Plank and B. Nidetzky, Nat. Commun., 2017, 8, 894 CrossRef PubMed.
- K. E. H. Frandsen, T. J. Simmons, P. Dupree, J. C. N. Poulsen, G. R. Hemsworth, L. Ciano, E. M. Johnston, M. Tovborg, K. S. Johansen, P. Von Freiesleben, L. Marmuse, S. Fort, S. Cottaz, H. Driguez, B. Henrissat, N. Lenfant, F. Tuna, A. Baldansuren, G. J. Davies, L. Lo Leggio and P. H. Walton, Nat. Chem. Biol., 2016, 12, 298–303 CrossRef CAS PubMed.
- J. P. Bacik, S. Mekasha, Z. Forsberg, A. Y. Kovalevsky, G. Vaaje-Kolstad, V. G. H. Eijsink, J. C. Nix, L. Coates, M. J. Cuneo, C. J. Unkefer and J. C. H. Chen, Biochemistry, 2017, 56, 2529–2532 CrossRef CAS PubMed.
- R. Davydov, A. E. Herzog, R. J. Jodts, K. D. Karlin and B. M. Hoffman, J. Am. Chem. Soc., 2022, 144, 377–389 CrossRef CAS PubMed.
- T. Isaksen, B. Westereng, F. L. Aachmann, J. W. Agger, D. Kracher, R. Kittl, R. Ludwig, D. Haltrich, V. G. H. Eijsink and S. J. Horn, J. Biol. Chem., 2014, 289, 2632–2642 CrossRef CAS PubMed.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS.
- D. A. Quist, M. A. Ehudin, A. W. Schaefer, G. L. Schneider, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2019, 141, 12682–12696 CrossRef CAS.
- B. D. Neisen, N. L. Gagnon, D. Dhar, A. D. Spaeth and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 10220–10223 CrossRef CAS PubMed.
- B. Kim, D. Jeong, T. Ohta and J. Cho, Commun. Chem., 2019, 2, 1–6 CrossRef.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed.
- B. Westereng, D. Cannella, J. Wittrup Agger, H. Jørgensen, M. Larsen Andersen, V. G. H. Eijsink and C. Felby, Sci. Rep., 2015, 5, 1–9 Search PubMed.
- G. Courtade, R. Wimmer, Å. K. Røhr, M. Preims, A. K. G. Felice, M. Dimarogona, G. Vaaje-Kolstad, M. Sørlie, M. Sandgren, R. Ludwig, V. G. H. Eijsink and F. L. Aachmann, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5922–5927 CrossRef CAS PubMed.
- E. D. Larsson, G. Dong, V. Veryazov, U. Ryde and E. D. Hedegård, Dalton Trans., 2020, 49, 1501–1512 RSC.
- B. O. Roos, P. R. Taylor and P. E. M. Sigbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- L. Ciano, G. J. Davies, W. B. Tolman and P. H. Walton, Nat. Catal., 2018, 1, 571–577 CrossRef CAS.
- D. Dhar and W. B. Tolman, J. Am. Chem. Soc., 2015, 137, 1322–1329 CrossRef CAS PubMed.
- A. Paradisi, E. M. Johnston, M. Tovborg, C. R. Nicoll, L. Ciano, A. Dowle, J. Mcmaster, Y. Hancock, G. J. Davies and P. H. Walton, J. Am. Chem. Soc., 2019, 141, 18585–18599 CrossRef CAS.
- R. K. Singh, B. M. Blossom, D. A. Russo, R. Singh, H. Weihe, N. H. Andersen, M. K. Tiwari, P. E. Jensen, C. Felby and M. J. Bjerrum, Chem.–Eur. J., 2020, 26, 454–463 CrossRef CAS PubMed.
- A. McEvoy, J. Creutzberg, R. K. Singh, M. J. Bjerrum and E. D. Hedegård, Chem. Sci., 2021, 12, 352–362 RSC.
- M. Schülein, J. Biotechnol., 1997, 57, 71–81 CrossRef.
- H. Boer and A. Koivula, Eur. J. Biochem., 2003, 270, 841–848 CrossRef CAS PubMed.
- S. M. Cragg, G. T. Beckham, N. C. Bruce, T. D. H. Bugg, D. L. Distel, P. Dupree, A. G. Etxabe, B. S. Goodell, J. Jellison, J. E. McGeehan, S. J. McQueen-Mason, K. Schnorr, P. H. Walton, J. E. M. Watts and M. Zimmer, Curr. Opin. Chem. Biol., 2015, 29, 108–119 CrossRef CAS PubMed.
- S. Banerjee, S. J. Muderspach, T. Tandrup, K. E. H. Frandsen, R. K. Singh, J. Ø. Ipsen, C. Hernández-Rollán, M. H. H. Nørholm, M. J. Bjerrum, K. S. Johansen and L. Lo Leggio, Biomolecules , 2022, 12, 194 CrossRef CAS PubMed.
- K. E. H. Frandsen, J. C. N. Poulsen, T. Tandrup and L. Lo Leggio, Carbohydr. Res., 2017, 448, 187–190 CrossRef CAS PubMed.
- A. Ramanathan, A. Savol, V. Burger, C. S. Chennubhotla and P. K. Agarwal, Acc. Chem. Res., 2014, 47, 149–156 CrossRef CAS PubMed.
- W. Chen, M. K. Gilson, S. P. Webb and M. J. Potter, J. Chem. Theory Comput., 2010, 6, 3540–3557 CrossRef CAS PubMed.
- Y. M. M. Huang, W. Chen, M. J. Potter and C. E. A. Chang, Biophys. J., 2012, 103, 342–351 CrossRef CAS PubMed.
- W. You, Y. m. M. Huang, S. Kizhake, A. Natarajan and C. en A. Chang, PLoS Comput. Biol., 2016, 12, 1–25 CrossRef PubMed.
- P. Xu, T. Sattasathuchana, E. Guidez, S. P. Webb, K. Montgomery, H. Yasini, I. F. M. Pedreira and M. S. Gordon, J. Chem. Phys., 2021, 154, 104122 CrossRef CAS PubMed.
- W. Chen, X. Ren and C. en A. Chang, ChemMedChem, 2019, 14, 107–118 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- P. J. Lodi and J. R. Knowles, Biochemistry, 1991, 30, 6948–6956 CrossRef CAS PubMed.
- C. Lennartz, A. Schäfer, F. Terstegen and W. Thiel, J. Phys. Chem. B, 2002, 106, 1758–1767 CrossRef CAS.
- G. T. Marks, M. Susler and D. H. T. Harrison, Biochemistry, 2004, 43, 3802–3813 CrossRef CAS PubMed.
- G. C. Schröder, W. B. O'Dell, P. D. Swartz and F. Meilleur, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2021, 77, 128–133 CrossRef PubMed.
- F. Meilleur, A. Kovalevsky and D. A. A. Myles, in Methods in enzymology, Elsevier Inc., 1st edn, 2020, vol. 634, pp. 69–85 Search PubMed.
- L. Coates, A. D. Stoica, C. Hoffmann, J. Richards and R. Cooper, J. Appl. Crystallogr., 2010, 43, 570–577 CrossRef CAS.
- Q. Wan, J. M. Parks, B. L. Hanson, S. Z. Fisher, A. Ostermann, T. E. Schrader, D. E. Graham, L. Coates, P. Langan and A. Kovalevsky, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12384–12389 CrossRef CAS.
- O. Gerlits, T. Wymore, A. Das, C. H. Shen, J. M. Parks, J. C. Smith, K. L. Weiss, D. A. Keen, M. P. Blakeley, J. M. Louis, P. Langan, I. T. Weber and A. Kovalevsky, Angew. Chem., Int. Ed., 2016, 55, 4924–4927 CrossRef CAS PubMed.
- C. P. Kubicek, Fungi and Lignocellulosic Biomass, Wiley-Blackwell, Oxford, UK, 2012 Search PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- B. Sullivan, R. Archibald, P. S. Langan, H. Dobbek, M. Bommer, R. L. McFeeters, L. Coates, X. Wang, F. Gallmeier, J. M. Carpenter, V. Lynch and P. Langan, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 1085–1095 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367 CrossRef CAS PubMed.
- C. Narayanan, D. N. Bernard, K. Bafna, D. Gagné, C. S. Chennubhotla, N. Doucet and P. K. Agarwal, Structure, 2018, 26, 426–436 CrossRef CAS.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER 15, University of California, San Francisco, 2015 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- A. Ramanathan, P. K. Agarwal, M. Kurnikova and C. J. Langmead, J. Comput. Biol., 2010, 17, 309–324 CrossRef CAS PubMed.
- M. R. Duff, J. M. Borreguero, M. J. Cuneo, A. Ramanathan, J. He, G. Kamath, S. C. Chennubhotla, F. Meilleur, E. E. Howell, K. W. Herwig, D. A. A. Myles and P. K. Agarwal, Biochemistry, 2018, 57, 4263–4275 CrossRef CAS PubMed.
- S. Shukla, K. Bafna, C. Gullett, D. A. A. Myles, P. K. Agarwal and M. J. Cuneo, Biochemistry, 2018, 57, 5864–5876 CrossRef CAS PubMed.
- A. Ramanathan and P. K. Agarwal, J. Phys. Chem. B, 2009, 113, 16669–16680 CrossRef CAS PubMed.
- K. Bafna, C. Narayanan, S. Chakra Chennubhotla, N. Doucet and P. K. Agarwal, PLoS One, 2019, 14, 1–25 CrossRef PubMed.
- M. K. Gilson, J. A. Given, B. L. Bush and J. A. McCammon, Biophys. J., 1997, 72, 1047–1069 CrossRef CAS PubMed.
- M. S. Head, J. A. Given and M. K. Gilson, J. Phys. Chem. A, 1997, 101, 1609–1618 CrossRef CAS.
- L. David, R. Luo and M. K. Gilson, J. Comput.-Aided Mol. Des., 2001, 15, 157–171 CrossRef CAS PubMed.
- V. Kairys and M. K. Gilson, J. Comput. Chem., 2002, 23, 1656–1670 CrossRef CAS PubMed.
- C. E. Chang and M. K. Gilson, J. Am. Chem. Soc., 2004, 126, 13156–13164 CrossRef CAS PubMed.
- W. Chen, C. E. Chang and M. K. Gilson, Biophys. J., 2004, 87, 3035–3049 CrossRef CAS PubMed.
- C. E. Chang and M. K. Gilson, J. Comput. Chem., 2003, 24, 1987–1998 CrossRef CAS PubMed.
- C. E. Chang, M. J. Potter and M. K. Gilson, J. Phys. Chem. B, 2003, 107, 1048–1055 CrossRef CAS.
- M. J. Potter and M. K. Gilson, J. Phys. Chem. A, 2002, 106, 563–566 CrossRef CAS.
- D. Qiu, P. S. Shenkin, F. P. Hollinger and W. C. Still, J. Phys. Chem. A, 1997, 101, 3005–3014 CrossRef CAS.
- W. C. Still, A. Tempczyk, R. C. Hawley and T. Hendrickson, J. Am. Chem. Soc., 1990, 112, 6127–6129 CrossRef CAS.
- R. Luo, L. David and M. K. Gilson, J. Comput. Chem., 2002, 23, 1244–1253 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05031e |
| ‡ Current address: Biomolecular Measurement Division, National Institute of Standards and Technology, Gaithersburg, MD 20899; Biomolecular Labeling Laboratory, Institute for Bioscience and Biotechnology Research, Rockville, MD 20850. |
| This journal is © The Royal Society of Chemistry 2022 |