
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled monodefluorination and alkylation of C(sp3)–F bonds by lanthanide photocatalysts: importance of metal–ligand cooperativity†‡
Amy E.
Kynman
 ab,
Luca K.
Elghanayan
a,
Addison N.
Desnoyer
a,
Yan
Yang
c,
Laurent
Sévery
a,
Andrea
Di Giuseppe
a,
T. Don
Tilley
a,
Laurent
Maron
*c and
Polly L.
Arnold
*ab
ab,
Luca K.
Elghanayan
a,
Addison N.
Desnoyer
a,
Yan
Yang
c,
Laurent
Sévery
a,
Andrea
Di Giuseppe
a,
T. Don
Tilley
a,
Laurent
Maron
*c and
Polly L.
Arnold
*ab
aDepartment of Chemistry, University of California, Berkeley, Berkeley, CA 94720-1460, USA. E-mail: pla@berkeley.edu
bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cLPCNO, Université de Toulouse, 135 Avenue de Rangueil, 31077 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 8th November 2022
Abstract
The controlled functionalization of a single fluorine in a CF3 group is difficult and rare. Photochemical C–F bond functionalization of the sp3-C–H bond in trifluorotoluene, PhCF3, is achieved using catalysts made from earth-abundant lanthanides, (CpMe4)2Ln(2-O-3,5-tBu2-C6H2)(1-C{N(CH)2N(iPr)}) (Ln = La, Ce, Nd and Sm, CpMe4 = C5Me4H). The Ce complex is the most effective at mediating hydrodefluorination and defluoroalkylative coupling of PhCF3 with alkenes; addition of magnesium dialkyls enables catalytic C–F bond cleavage and C–C bond formation by all the complexes. Mechanistic experiments confirm the essential role of the Lewis acidic metal and support an inner-sphere mechanism of C–F activation. Computational studies agree that coordination of the C–F substrate is essential for C–F bond cleavage. The unexpected catalytic activity for all members is made possible by the light-absorbing ability of the redox non-innocent ligands. The results described herein underscore the importance of metal–ligand cooperativity, specifically the synergy between the metal and ligand in both light absorption and redox reactivity, in organometallic photocatalysis.
Photoredox catalysis is a powerful synthetic method for the functionalization of inert molecules using single electron transfer (SET) reactivity1–3 under irradiation with visible light.4 This has enabled challenging transformations under mild conditions including C–H activation,5–7 radical cross-coupling,8–11 and the valorization of lignin.12,13 However, detailed mechanistic studies of photoredox systems are difficult due to their inherent complexity and the short lifetimes of photoexcited intermediates.
Many lanthanides are more abundant in the environment than copper and their salts are less toxic than those of iron, so their potential for applications in catalysis merits exploration.14–17 In 1990, divalent Sm, Eu, and Yb complexes Ln(Cp*)2 (Cp* = C5Me5), were shown to more efficiently cleave vinylic C–F bonds when photolyzed, stoichiometrically forming Ln(III) halide complexes, and suggesting the value of increasing the reducing power of the LnII excited state.18 Subsequently, analogous reactions to cleave the weaker C–Cl and C–Br bonds could be made catalytic in Ln(II) halide (Ln = Sm, Eu, Yb), under near UV-photolysis conditions, by the addition of sacrificial reductant such as Zn or Al.19,20 The addition of simple donor ligands enabled benzylic C–Cl cleavage by EuII under blue light irradiation.21 The addition of an organic photocatalyst or a photo-absorbing substrate to Lewis acidic LnX3 salts (X = halide, triflate) has also been used to enhance the catalysis.22 Ln centers (Ln = Nd, Dy, Lu) with light-absorbing ligands such as porphyrins or phthalocyanins have been used to stoichiometrically dechlorinate phenols.23
Few reports of lanthanide photoredox catalysis exist with CeIII complexes receiving the most attention. Ce possesses both an accessible III/IV redox couple and an allowed excitation from the 4f1 ground state to the 5d1 excited state, which can give rise to luminescent behaviour. It is also the cheapest and most readily isolated of the rare earths, offering a promising alternative to current precious metal photocatalysts.
Building on the pioneering work on stoichiometric photoluminescent Ce chemistry,24,25 in 2015 Schelter and co-workers demonstrated the utility of CeIII in photocatalysis.26,27 Their CeIII amido complexes were catalysts for chlorine atom abstraction from benzyl chloride (Fig. 1, top), with both NaN(SiMe3)2 and additional Ce0 required for turnover.28
 | ||
| Fig. 1 Previous examples of photocatalytic C–X (X = halide) bond cleavage, and this work. | ||
They proposed an inner-sphere mechanism involving Ce⋯ClCR3 adduct formation that provides an additional thermodynamic driving force to a bond cleavage that was otherwise out of range of the reducing power of the Ce excited state. A more sterically congested CeIII tris(guanidinate) operates via an outer-sphere single electron transfer (SET) mechanism to cleave aryl iodides,28 highlighting the mechanistic diversity that is possible in these systems.29
To date, ligands that support lanthanide-centered photocatalysts have been limited to halides, pseudohalides, and simple N-donors.30,31 No organometallic lanthanide photocatalyst has yet been reported that combines the photoexcitable Ce cation with multidentate, tunable ligands. We have developed organometallic lanthanide complexes as sustainable catalysts,16,17,32,33 and considered that those capable of forming an inner-sphere adduct, and absorbing light, could achieve the unusual and difficult, selective catalytic conversion of strong sp3 C–F bonds.
Fluorine forms the strongest single bond to carbon and the C–F bond is ca. 25 kcal mol−1 stronger than the C–Cl bond in monohaloalkanes, and the C–H bond in alkanes.34 The selective activation and functionalization of C–F bonds is important, both due to the high bioaccumulation and toxicity of many perfluorinated compounds,35 and the utility of fluorinated pharmaceuticals.36 However, stoichiometric C(sp3)–F bond activation reactions are rare.37–40 In particular, it is difficult to facilitate the controlled cleavage of a single C–F bond as the C(sp3)–F bond strength decreases as each F is removed and the remaining C–F bonds lengthen.41,42
This obstacle makes a radical methodology more attractive.43–49 Jui and co-workers have demonstrated that some common photocatalysts can selectively activate a single C–F bond to form the putative ArCF2˙ radical, which can either be quenched directly via H atom transfer (HAT), or coupled with an alkene followed by HAT to generate difluoroalkanes (Fig. 1, middle).50,51 Gschwind and König have shown the photochemical functionalization of electron-poor trifluoromethylarenes.52 Nishimoto and Yasuda have described related C–F coupling protocols of perfluoroalkylarenes using tin reagents and an iridium photocatalyst.53
Here we show how selective, catalytic C–F bond functionalization can be achieved using a new family of LnIII compounds supported by a light-absorbing aryloxide-tethered N-heterocyclic carbene, CpMe4, and pseudohalide ligands (Fig. 1, lower). We show that visible light-irradiated Ce complexes can selectively abstract a single fluoride from PhCF3 and catalyze its alkylation by MgR2 to afford PhCF2R. The PhCF2˙ can also be quenched to selectively form PhCF2H or further alkylated via coupling with an alkene or other metal alkyls. We use combined experiment and density functional theory (DFT) computations to show the importance of coordination of the fluorinated substrate to the Lewis acidic metal in C–F activation, and the utility of the ligand in enabling photoredox catalysis for other lanthanide congeners.
Synthesis and light absorption studies of the complexes
We first made the Ce complex (CpMe4)2Ce(L) (1-Ce, CpMe4 = C5Me4H), where L is the bidentate aryloxide-N-heterocyclic carbene (NHC) ligand [2-O-3,5-tBu2-C6H2(1-C{N(CH)2N(iPr)})], Scheme 1, top.16,17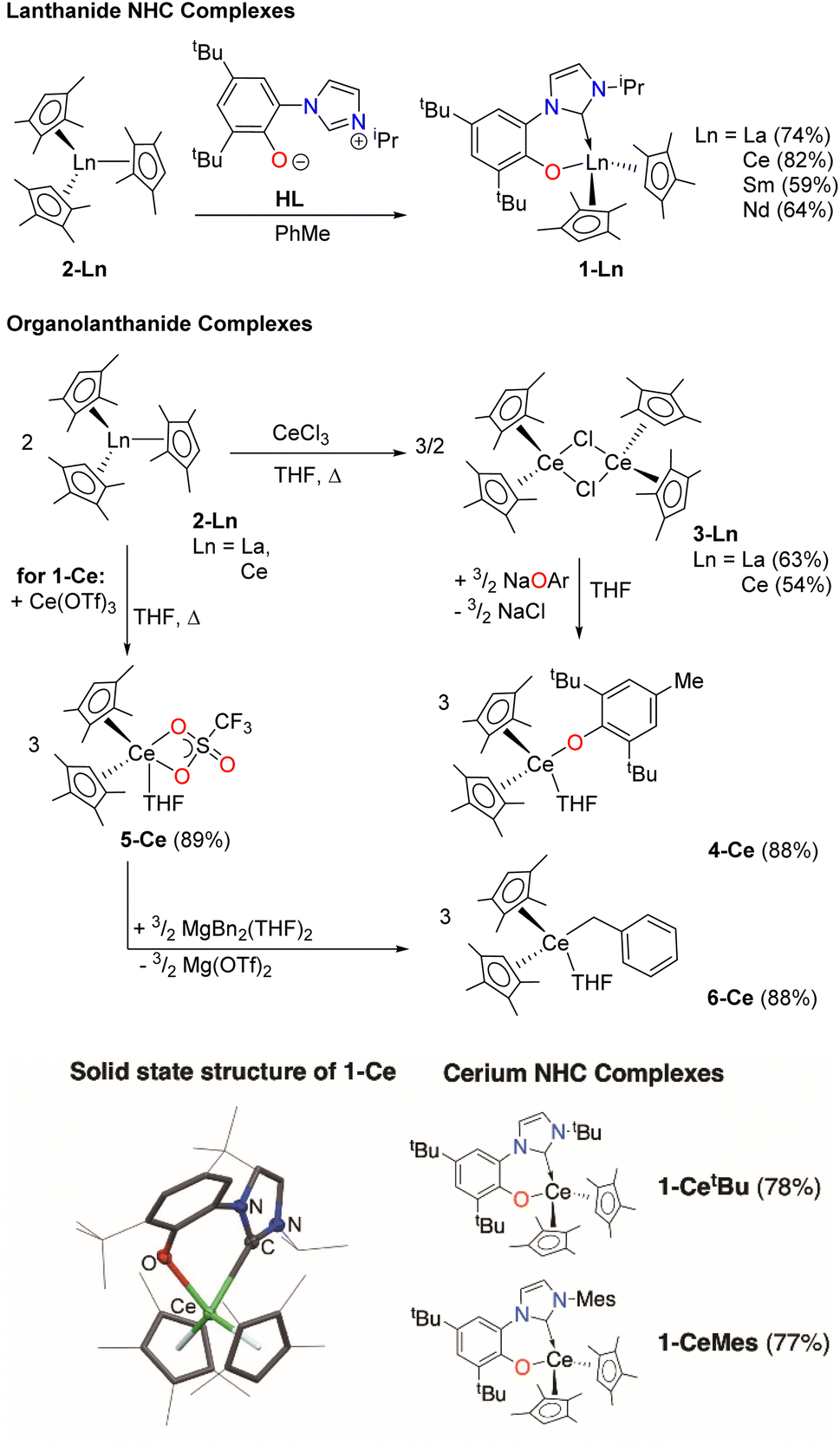 | ||
| Scheme 1 Syntheses of the complexes examined for C–F bond functionalization in this work (upper) and the solid-state structure of 1-Ce, H omitted for clarity, alongside the ligand variations studied for 1-Ce (lower). | ||
This ligand has two important characteristics that could open up photoredox catalysis to typically redox-innocent lanthanides with other electronic configurations than Ce. Like some planar aromatic heterocycles,54–56 the aryloxy-NHC L absorbs visible light when bound as a rigid bidentate ligand. The aryloxide group also has the potential to engage in one-electron redox chemistry by forming a phenoxy ligand radical.57–59 To probe the roles of both metal and ligand, we prepared differently substituted aryloxy-NHC ligand adducts with a variety of electronic and steric profiles16 (CpMe4)2Ce(LMes) (1-CeMes) LMes = 2-O-3,5-tBu2-C6H2(1-C{N(CH)2N(Mes)}) and (CpMe4)2Ce(LtBu) (1-CetBu) LMes = 2-O-3,5-tBu2-C6H2(1-C{N(CH)2N(tBu)}), Scheme 1, bottom, and a series of LnIII complexes – f0 La, f2 Nd and f5 Sm analogues (CpMe4)2Ln(L) (1-La, 1-Nd and 1-Sm), Ln(CpMe4)3 (2-La and 2-Ce) [(CpMe4)2Ln(μ-Cl)]2,24 (3-La and 3-Ce), (CpMe4)2Ce(OAr) (4-Ce, where OAr = O-2,6-tBu2-4-Me-C6H2),60 (CpMe4)2Ce(OTf)(THF) (5-Ce),61 and (CpMe4)2Ce(Bn)(THF) (6-Ce, Bn = CH2Ph),62,63Scheme 1, middle. All new complexes were fully characterized, see ESI.‡
The UV-Vis spectrum of the f1 molecule 1-Ce is shown in Fig. 2, overlaid with that of the f0 congener 1-La, proligand HL, and 1-CeMes. All 1-Ln luminesce under visible light excitation, with absorptions in the range 350–510 nm, see Fig. S23–S26‡ for the other 1-Ln spectra, and the excitation and emission spectra of 1-Ce. The two absorption maxima displayed by 1-CeMes are shifted closer in energy to each other, while the magnitude of the ligand-centered absorption of 1-CetBu (Fig. S24‡) is larger than that of 1-Ce and 1-CeMes. This suggests the possibility for further catalyst optimization by generating a broader envelope of light absorption or improving quantum yield.
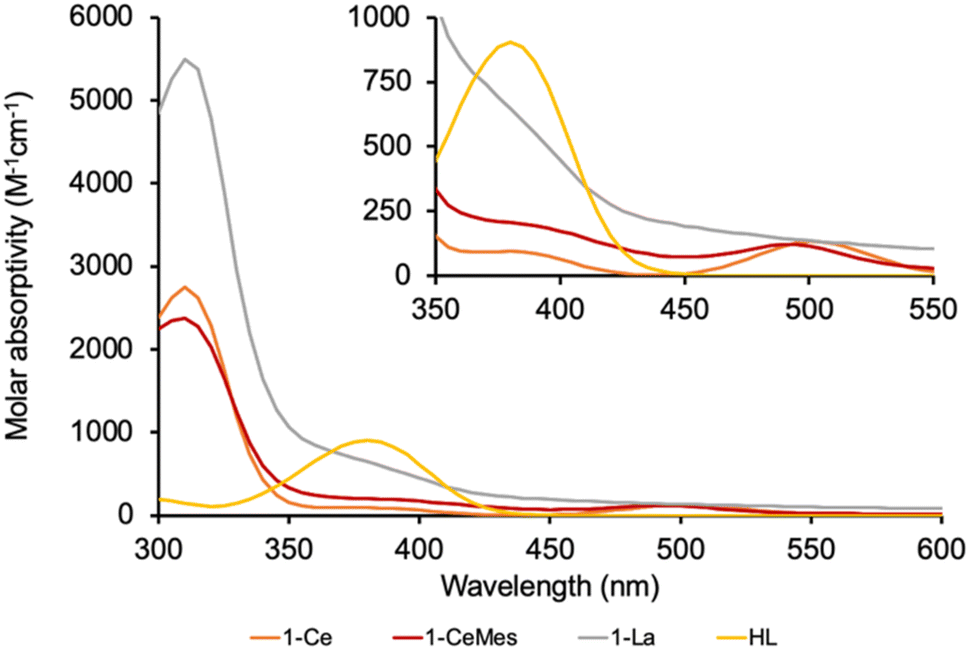 | ||
| Fig. 2 The absorption spectra of 1-Ce, 1-CeMes, 1-La and proligand HL. The absorption spectrum for the tert-Bu substituted analogue 1-CetBu, is also shown overlayed in the ESI.‡ The computed spectrum of 1-Ce using TD-DFT is included in the ESI.‡ | ||
TD-DFT analysis of the frontier orbitals involved in the light absorption
Time-dependent DFT (TD-DFT; B3PW91) calculations were performed on both 1-Ce and 1-La to explore the orbitals involved in their photoexcitation. The calculated and experimental absorption spectra agree well for 1-Ce (see Fig. S134‡). As expected, the SOMO of 1-Ce is comprised predominately of 4f character with minor orbital contributions from the aryloxide ligand (Fig. 3).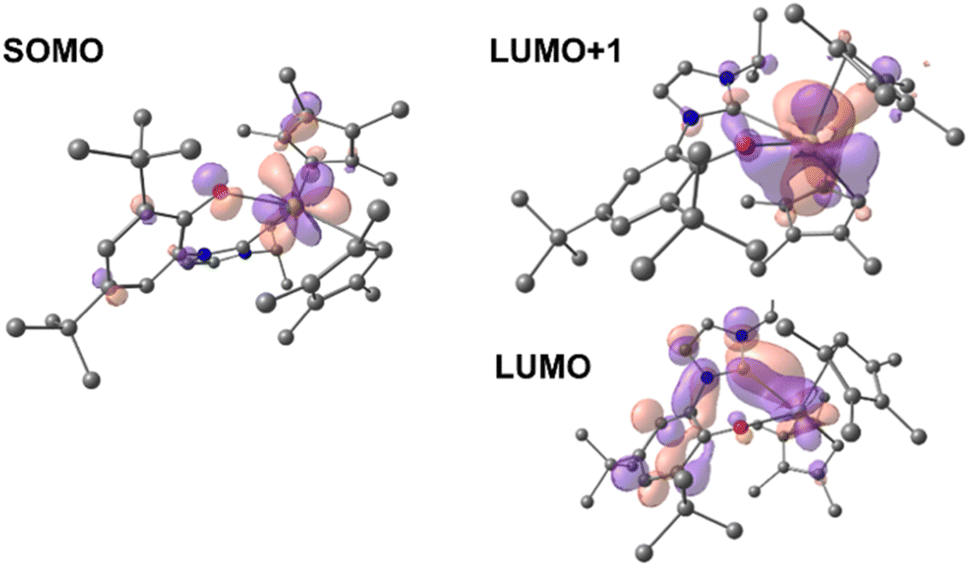 | ||
| Fig. 3 Depictions of the TD-DFT-calculated SOMO (left) and LUMO+1 (right, upper) and LUMO (right, lower) orbitals of 1-Ce. | ||
The absorption at 504 nm corresponds mainly to the SOMO–LUMO and SOMO–LUMO+1 transitions (see ESI‡ for the details of the states and the other frontier MOs). This is an excitation from an f-orbital into either the Ce–NHC or Ce–CpMe4 orbitals. The LUMO+1 is calculated to be a Ce-based (89%) hybrid of f- and d-orbitals (54% 5d and 46% 4f mixture) with no significant contributions from the ligands (11%). Notably, the LUMO, which also contributes to the calculated absorbance bands at 504 and 384 nm, is primarily of p-character and delocalized over both the aromatic ring and NHC moiety of the ligand L.
In the case of 1-La, the HOMO was calculated to be composed of significant π symmetry orbital contributions from both the CpMe4 and L ligands, Fig. 4. As expected for a formally f0 complex, there is no appreciable contribution from any La-based orbital to the HOMO. However, the LUMO+1 is predominately d-character (63% 5d and 23% 4f). TD-DFT predicts that the transition from HOMO to LUMO+1 results in a band at 325 nm. For both 1-La and 1-Ce, transitions are calculated in the higher energy region (between 320 and 390 nm) that involve π-type orbitals (Ln–CpMe4, Ln–NHC).
 | ||
| Fig. 4 Depictions of the TD-DFT-calculated HOMO (left) and LUMO+1 (right) for 1-La. | ||
Cyclic voltammetry and the excited state reduction potential of 1-Ce
The Rehm–Weller formalism is often used to estimate the excited state reduction potential (E1/2*) of photocatalysts.2,28 Excitation of a THF solution of 1-Ce with a 390 nm laser produces an emission band at 600 nm. Cyclic voltammetry (CV) experiments on 1-Ce (see Section S4.1‡) at fast scan rates (ν = 0.5 V s−1) display a quasi-reversible CeIII/IV redox couple at −0.15 V vs. Fc0/+, Fig. 5 purple trace. The Rehm–Weller formalism (Section S4.2‡) predicts the E1/2* of 1-Ce* as −2.20 V. This value is similar to those calculated for CeIII N-ligand complexes,28 and more positive than the literature ground state E1/2 of PhCF3 (−3.23 V vs. Fc0/+ in THF). This implies that the pre-coordination of the PhCF3 to the Lewis acid LnIII center is essential to provide the additional driving force.28,64 See also the DFT calculations below.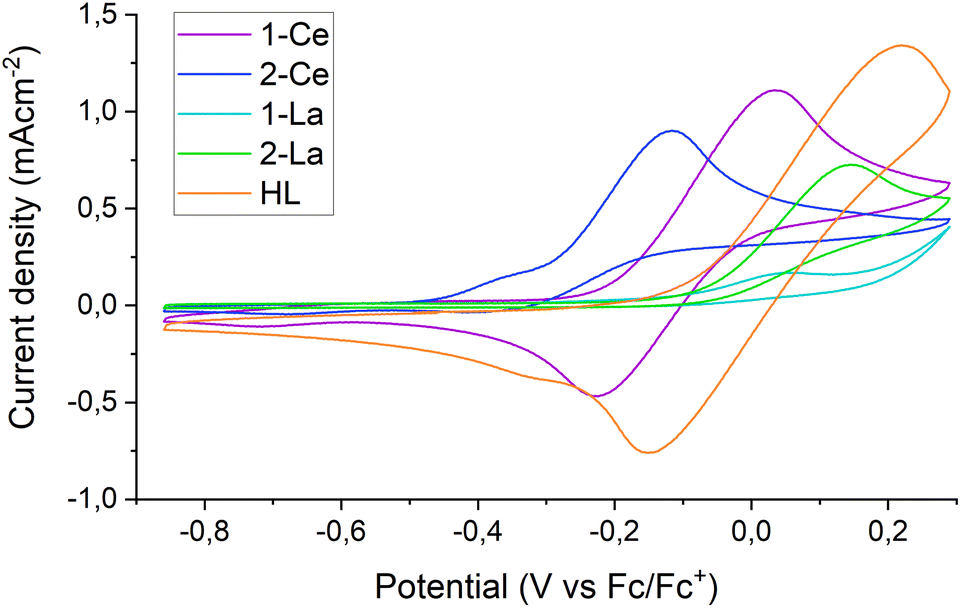 | ||
| Fig. 5 Cyclic voltammograms of 1-Ce (purple), 2-Ce (dark blue), 1-La (light blue), 2-La (green) and HL (orange) in THF with 0.085 M [nBu4N][BPh4] supporting electrolyte. [Analyte] = ca. 5 mM; ν = 0.5 V s−1. | ||
Further CV measurements were performed to analyze the potentials and reversibility of the oxidations of complexes 2-Ce, 1-La and 2-La as well as of the ligand HL (S4‡) in tetrabutylammonium tetraphenylborate [NBu4][BPh4] THF solutions.
In the case of HL, a redox peak around 0 V (vs. Fc/Fc+) is observed upon scanning oxidatively, displaying partial reversibility upon increasing the scan speed. This can be explained by a slow, irreversible reaction of the oxidized ligand. A similar redox feature around −0.15 V is observed for 1-Ce; the position of the oxidation at a more negative potential than that of free ligand implies a contribution from both the ligand and the metal center to this oxidation. This supports the hypothesis that electrons can be photoexcited from both metal and ligand.
In comparison, 1-La shows an onset of oxidation of similar amplitude but around 0.15 V, close to the limit of the electrolyte stability window. There is also a smaller feature at −0.15 V, attributed to small amounts of free ligand that are released after the first oxidative scan of the bulk solution. 2-Ce and 2-La both display fully irreversible oxidations with onsets around −0.3 V and −0.05 V, respectively. The presence of additional redox features may indicate the formation of electrochemically active degradation products of the oxidation. The voltammograms of 2-La and 2-Ce have recently been reported and show that the oxidation of lanthanide tris-CpMe4 complexes leads to decomposition at most standard scan rates.65 Here in contrast, the oxidation of 1-Ce is partially reversible at a scan rate of 500 mV s−1 (Fig. 5).
Stoichiometric C–F activation of PhCF3 to afford PhCF2H
First, complexes 1-Ce to 6-Ce were tested in a simple reaction with one equivalent of the substrate PhCF3, Scheme 2. It is expected to proceed by homolytic C–F bond scission, with the PhCF2˙ subsequently abstracting a H atom from the solvent THF, a good H atom donor. Little conversion was seen in these reactions, hypothesized to be due to binding competition between PhCF3 and THF solvent. Optimized reactions with five equivalents of PhCF3 in THF solution, irradiated with a 40 W Kessil A160WE Tuna Blue lamp in a Young's tap-valved NMR tube, were then monitored by 19F NMR spectroscopy. | ||
| Scheme 2 General reaction scheme for C–F activation and hydrodefluorination by Ln complexes 1-Ln to 3-Ln and cleavage by the Ce photocatalysts 1-Ce–6-Ce with PhCF3 (X = the monoanionic ligand CpMe4, Cl, OAr, OTf, Bn) n = 1–3. | ||
Complex 1-Ce performs best, forming the product PhCF2H in approximately 75% yield after 120 hours of irradiation at room temperature, Table S2.‡ The reaction does not proceed in the absence of light. Complex 2-Ce which does not contain L makes PhCF2H in only 5% yield under the same conditions, underlining the utility of the aryloxy-NHC ligand. The La congener 1-La is less effective than 1-Ce, giving 10% conversion.
Prior to irradiation, the interaction between PhCF3 and 1-Ln found in the computational study is suggested by a small change in chemical shift in the 19F NMR spectrum of PhCF3 added to a solution of 1-La or 1-Ce compared to an isolated solution of PhCF3 in a capillary, but chemical shifts in 19F NMR spectroscopy can be very sensitive to concentration effects (Scheme 2 and S8.1, Fig. S97‡). This Lewis acid activation of the C–F bond should facilitate its cleavage when the complex is photo-excited.
We expect the photoexcited adduct [1-Ce–F3CPh] to fragment into the CeIV–F intermediate in Scheme 2, releasing the PhCF2˙ radical. Alternatively, the f0 complex 1-La can show hydrodefluorination reactivity by homolysis of one La–ligand bond under irradiation of [1-La–F3CPh], forming a radical La fragment that can formally abstract F˙. While few prior examples exist, the photoinduced homolysis of Ln–C bonds has been reported.18,66
Catalytic defluoroalkylation of C(sp3)–F bonds: conversion of PhCF3 to PhCF2CH2CH2R
Photocatalysts 1–6-Ce can also facilitate defluoroalkylation, using commonly available Mg dialkyls as both the source of alkyl group and fluoride acceptor to turn over the catalyst, Scheme 3. MgBn2(THF)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46 gives the most effective alkylation of the ArCF2˙ and is therefore used in the reactions described here. Products were identified as PhCF2R (R = H, Bn) by GC/MS and 19F NMR spectroscopy, with minor amounts of the by-product PhCF2H and bibenzyl observed, presumably formed via homocoupling of benzyl radical.
46 gives the most effective alkylation of the ArCF2˙ and is therefore used in the reactions described here. Products were identified as PhCF2R (R = H, Bn) by GC/MS and 19F NMR spectroscopy, with minor amounts of the by-product PhCF2H and bibenzyl observed, presumably formed via homocoupling of benzyl radical.
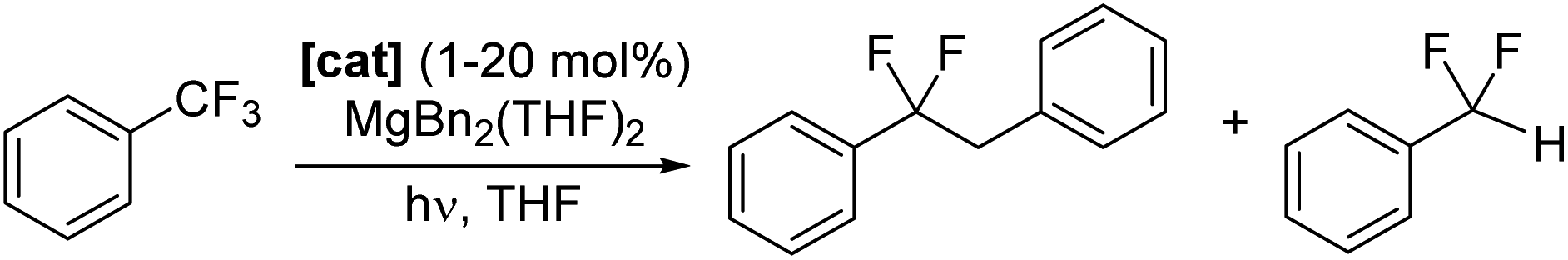 | ||
| Scheme 3 Photocatalytic defluoroalkylation of trifluorobenzene with 1–6-Ce and Mg(Bn)2(THF)2. | ||
Catalytic alkylation/arylation reactions are also possible using coupling partners Mg(allyl)2, MgPh2(THF)2, or nBu3Sn(allyl), (S6.2‡). Control reactions are described in Section S8.‡
1-Ce is the best of the Ce photocatalysts, giving 91% conversion to product after a total of 80 hours irradiation (Table 1). The series 1-Ln Ln = La, Ce, Nd, Sm was also tested using the same reaction conditions. After 48 hours irradiation all formed products, with yields that decrease from 67% for La to 13% for Sm, in alignment with decreasing atomic radius of the lanthanide. This is consistent with a mechanism that relies on substrate binding to the photoactivated complex and light absorption by the ligand L as well as (or instead of) the metal.
| Entry | Catalyst | Time (h) | Yieldb (%) |
|---|---|---|---|
| a 20 mol% catalyst loading, 1 equiv. MgBn2(THF)2 and 1 equiv. PhCF3, RT, THF-H8, irradiation with a 40 W Kessil A160WE Tuna Blue lamp. b Combined yield of PhCF2CH2Ph and PhCF2H. | |||
| 1 | 1-Ce | 48 | 51 |
| 2 | 1-Ce | 80 | 91 |
| 3 | 2-Ce | 48 | 6 |
| 4 | 4-Ce | 48 | 41 |
| 5 | 4-Ce | 80 | 65 |
| 6 | 1-La | 48 | 67 |
| 7 | 1-La | 60 | 72 |
| 8 | 2-La | 48 | 19 |
| 9 | 1-Nd | 48 | 32 |
| 10 | 1-Nd | 140 | 87 |
| 11 | 1-Sm | 48 | 12 |
| 12 | 7-Mg | 48 | 24 |
| 13 | 7-Mg | 80 | 56 |
| 14 | None | 260 | 22 |
There is a very small amount of transfer of L from Ce to Mg during the catalysis with 1-Ce observed by 1H NMR spectroscopy, forming [MgBn(L)]2 (7-Mg),67 which we have independently synthesized. We note that 7-Mg can also generate ArCF2˙ by F abstraction, in a similar manner to 1-La. In catalysis, 7-Mg forms a mixture of products in 56% yield after 80 hours irradiation. The bis(ligand) complex [Mg(L)2]2 (8-Mg) (see ESI‡) is much less reactive, presumably due to the greater steric crowding relative to 7-Mg.
Some transfer of benzyl anion from the reagent MgBn2(THF)2 to 2–5-Ce is also observed over time, with a gradual conversion to 6-Ce as the reactions progress, see Fig. S113.‡ This ligand exchange is more significant for 2–5-Ce than 1-Ce, the most robust catalyst.
We also targeted the group 2 analogue [CaBn(L)] since CaII has a similar ionic radius to LnIII (rcov 6 coordinate Ce = 1.15; Ca = 1.14; Nd = 1.123 Å)68 and would provide a catalyst with similar Lewis acidity but much less accessible d-orbitals at the metal. However, Schlenk equilibria prevented the isolation of a pure complex that could inform the mechanistic studies.
The conversion of PhCF3 to PhCF2CH2Ph can be catalyzed at catalyst loadings as low as 1 mol%. Table 2, and Fig. S69‡ show the data for the catalyzed conversion of PhCF3 to PhCF2CH2Ph in THF-H8, mediated by 1 mol% loading of 1-Ce, 1-La, 1-Nd, 1-Sm or 7-Mg. Following 63 hours irradiation, 1-Ce produces a combined yield of 28% with a PhCF2CH2Ph:PhCF2H ratio of 5:1. 1-La yields 42% conversion to PhCF2CH2Ph and PhCF2H after 63 hours irradiation, which increases to 62% following a further 21 hours, albeit with a product ratio of 2:1.
| Entry | Catalyst | Time (h) | Turnover | Ratiob |
|---|---|---|---|---|
| a 1 mol% catalyst loading, 1 equiv. MgBn2(THF)2 and 1 equiv. PhCF3, RT, THF-H8, irradiation with a 40 W Kessil A160WE Tuna Blue lamp; data at 84 hours unable to be collected for 1-Ce, 1-Nd and 1-Sm due to polymerization of THF solvent. b Ratio of PhCF2CH2Ph and PhCF2H products. | ||||
| 1 | 1-Ce | 63 | 28 | 5:1 |
| 2 | 1-La | 63 | 42 | |
| 3 | 1-La | 84 | 62 | 2:1 |
| 4 | 1-Nd | 63 | 20 | 3:1 |
| 5 | 1-Sm | 63 | 21 | 3:1 |
| 6 | 7-Mg | 63 | 23 | |
| 7 | 7-Mg | 84 | 27 | 2:1 |
Significantly, although 1-La lacks the accessible metal-based 4f1/5d1 excitation or III/IV redox couple, it can defluoroalkylate PhCF3 faster than 1-Ce. However, 1-Ce is more selective in this transformation, capable of giving higher ratio of PhCF2CH2Ph to PhCF2H than 1-La (see Table 2).
There appears to be a large kinetic isotope effect in these reactions as the formation of PhCF2D as a side-product is suppressed in THF-D8. For example, the ratio of PhCF2CH2Ph:PhCF2H formed by 1-Ce improves from 5:1 in THF-H8 to 11:1 in THF-D8.
Additional experiments in which mixtures of 1-Ce and PhCF3 are irradiated with lower energy light (525 nm) give significantly lower yields of PhCF2H in agreement with our expectation that the higher energy absorption of 1 gives most efficient access to the photoexcited state. However, further reactions of 1-La illuminated with the higher-energy 390 nm light result only in rapid decomposition of the lanthanum complex, Fig. S106.‡
Computational analysis of PhCF3 binding and C–F bond homolysis
Computational approaches (DFT, B3PW91 functional) were again used to gain insight into the PhCF3 coordination and C–F bond activation. The reaction, shown in Scheme 2, is calculated to begin by the photoexcitation of the 1-Ce ground state to 1-Ce*, a transition which is calculated to be 81.3 kcal mol−1, since the PhCF3 coordination is weak. This corresponds to absorption of a 350 nm photon. From this intermediate, a transition state (TS1) was located on the Potential Energy Surface (PES) that corresponds to C–F activation of the PhCF3 substrate, Fig. 6. The associated barrier from 1-Ce is high (125.7 kcal mol−1); that is, 44.4 kcal mol−1 from the photoexcited CeIII state, 1-Ce*. The TS1 was located on the doublet spin state surface and Natural Population Analysis (NPA) shows TS1 is clearly a transfer of fluoride rather than fluorine radical, Scheme 4. The charge of the transferred F is −0.94, consistent with a fluoride. Concurrently, the charge at the Ce center is +3.85, in line with the formation of Ce(IV) center. This is similar to that demonstrated by 1,2,4-(tBu)3-(C5H2)2CeH in which Ce abstracts an F from Ce-bound CHnF3−n to form Ce–F + CHnF2−n radical.63 | ||
| Fig. 6 Computed C–F activation pathway for the reaction of 1-Ce with PhCF3, including TS1. Those for 1-La and 7-Mg are in the ESI.‡ | ||
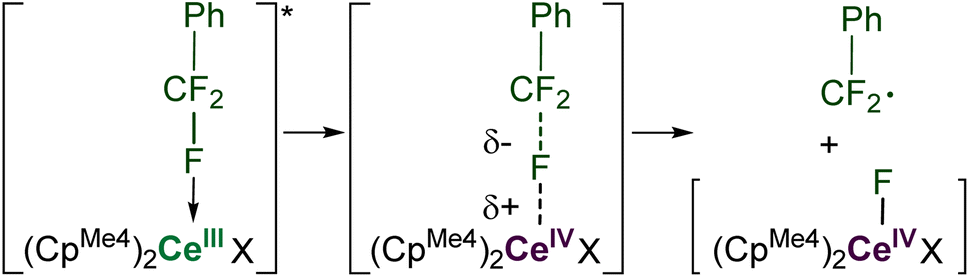 | ||
| Scheme 4 Pathway for the abstraction of fluoride (rather than F˙) to generate Ce(IV)–F and ArCF2˙. | ||
Following the intrinsic reaction coordinate (IRC) leads to the formation of a CeIV–F complex (INT1) and free PhCF2˙ which is endothermic by 28.2 kcal mol−1 from the photoexcited 1-Ce* state. The formation of the product PhCF2H via hydrogen atom abstraction from the solvent THF is computed to be athermic. This mechanism is distinct from the associative interchange process previously calculated for the reaction of (Cp′)2CeCH2Ph (Cp′ = 1,2,4-tri-t-butylcyclopentadienyl) with methyl halides.62
The triplet transition state TS1 computed for the d0f01-La analogue, shown in the ESI,‡ is only 7.9 kcal above the 1-La* excited state energy. The barrier for the excited state of the MgII complex 7-Mg*, also in the ESI,‡ is around 35 kcal mol−1 so that the reaction can occur but should be relatively slow, especially since the overall reaction is endothermic by 11.4 kcal mol−1. The reaction energy could also be rendered exothermic by trapping the TFT radical by another molecule of 7-Mg* or MgBn2(THF)2. The low barrier calculated for the 1-La reaction corresponds less well to the observed reactivity, so it may be that the light absorption probability for 1-La is lower, since it involves a form of ligand-to-ligand charge transfer that may well be dipole forbidden.
Experimental evidence for the formation of an ArCF2˙ radical intermediate
Seeking experimental support for the formation of a discrete PhCF2˙ intermediate, we prepared ortho-allylbenzotrifluoride 1-CF3-2-(C3H5)C6H4 (9),69 hypothesizing that the pendant alkene group would serve as an intramolecular radical trap for the proposed ArCF2˙ aryl radical. Irradiating a THF solution of 1-Ce and 9 results in the slow formation of one major new product, assigned based on its 19F NMR spectroscopic data, and comparison with an independently synthesized sample, Section S8.2,‡ as the ring-closed 10 (Scheme 5, S8.2‡). The distinctive 19F NMR spectra show large 2JF,F coupling constants of 250 Hz. The reaction of 1-Ce with 9 is significantly slower than with PhCF3, which we attribute to the greater steric hindrance about the activated CF3 group in 9.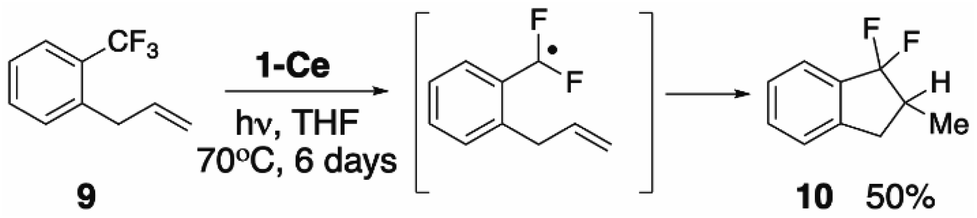 | ||
| Scheme 5 The C–F bond activation of 1-CF3-2-(C3H5)C6H4 mediated by 1-Ce. | ||
Oxidation of the metal and/or ligand
The formation of the ArCF2˙ catalyzed by 1-Ln should lead to an oxidized F-containing intermediate, [1-Ln–F]. The redox active and innocent metals can achieve this in different ways, shown in Scheme 6. For 1-Ce we expect the CeIII/IV redox couple to be involved, forming the fluoride intermediate CeIV [Ce(F)(L)CpMe42] 1-Ce-F, although we recognize that oxidation on the L or CpMe4 ligand is also possible. For 1-La and the other lanthanides which must remain LnIII we expect either [Ln(F)(L˙)(CpMe4)2] or [Ln(F)(L)(CpMe4˙)(CpMe4)]. We assume 7-Mg will form [Mg(F)(L˙)(Bn)] or [Mg(F)(L)] and ˙Bn.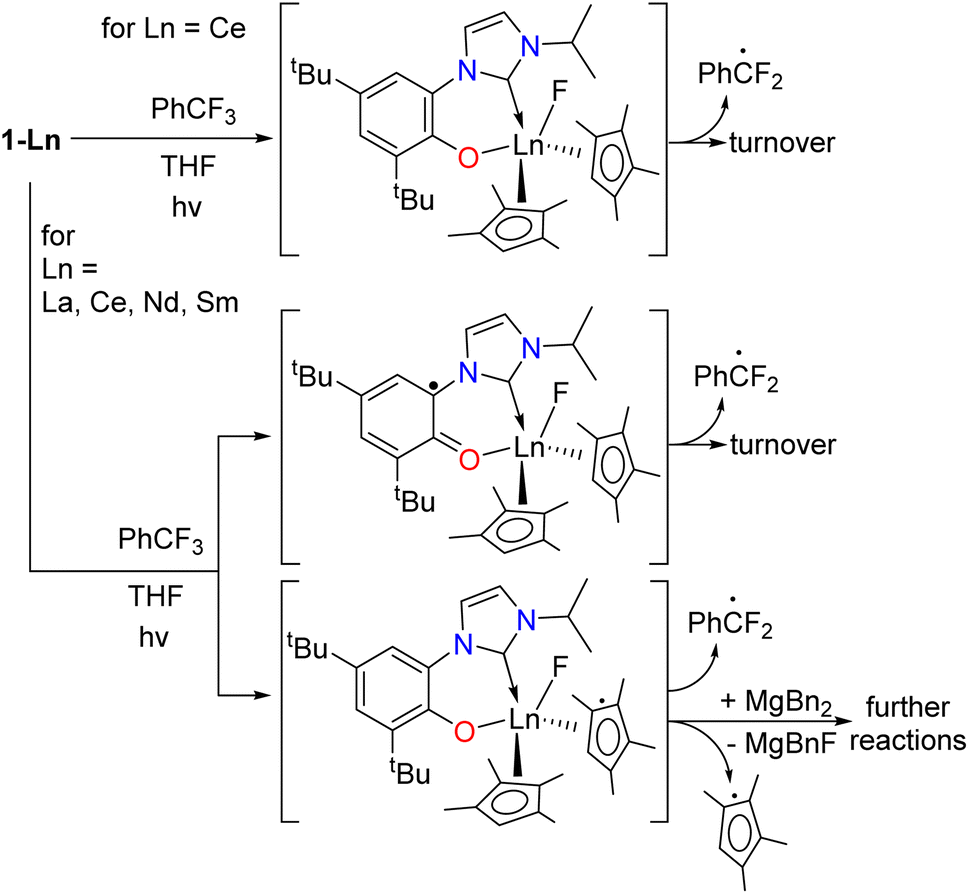 | ||
| Scheme 6 The proposed pathways of oxidation of 1-Ln that lead to turnover or catalyst degradation for Ln that have no accessible +IV oxidation state, i.e. Ln = La, Nd, and Sm. | ||
Cerium(IV) fluorides are uncommon and difficult to work with, and if a fluoride was formed in the catalytic cycle, we did not expect it would be isolable.73 The chemical oxidation of 1-Ce and 1-La was targeted using XeF2 or the hypervalent iodine reagent PhICl2 which has previously been used with success in organometallic CeIV chemistry.70–72 Reactions between 1-Ce and PhICl2 resulted in an immediate color change to the characteristic purple of molecular CeIV but the mixture soon bleaches to pale pink. The other reactions gave mixtures of products that could not be identified by NMR or IR spectroscopy, or separated by fractional crystallization. A reaction of 6-Ce with BF3 to target a CeIII–F complex which we planned to oxidize, yielded only the BF4 complex [(CpMe4)2Ce(BF4)(THF)]2 whose X-ray structure is included in the ESI.‡
Calculations show that CpMe4 is spontaneously released as a radical from the La center upon oxidation (S10.3‡) and providing a possible reason for the instability of 1-La in catalysis particularly when using higher energy lamps; there is also precedent for the homolysis of Ln–Cp bonds.74 Section S10.4‡ shows how for photoexcited 1-La* the unpaired spin density is mainly located at the O–NHC ligand, but localizes on the Cp ring and develops on the coordinated PhCF3 approaching the C–F activation step, finishing with unpaired spin density on both the PhCF2˙ radical and the Cp ligand.
The C–C bond forming step
Conventional organometallic CeIV alkyl complexes are not stable due to the reducing nature of the hydrocarbyl ligand.71 In line with this, DFT calculations indicate that the formation of a discrete [CeIV]–Bn is energetically unfavorable, so we hypothesized that the C–C bond is formed by coupling of ArCF2˙ directly with the reagent MgBn2(THF)2. Intrinsic reaction coordinate (IRC) calculations on this coupling (Fig. 7) identify an achievable transition state at 16.3 kcal mol−1 above the ground state. We also calculated the alternative possibility in which L transfer to MgBn2(THF)2 generates 7-Mg which we have observed as a by-product. Using the monomeric, THF-solvated form 7-Mg* in the calculation, the barrier for this all-Mg mediated reaction, Fig. 8, is 3.5 kcal mol−1 higher. The transition state is 19.8 kcal mol−1 above the energy of 7-Mg* and PhCF2˙.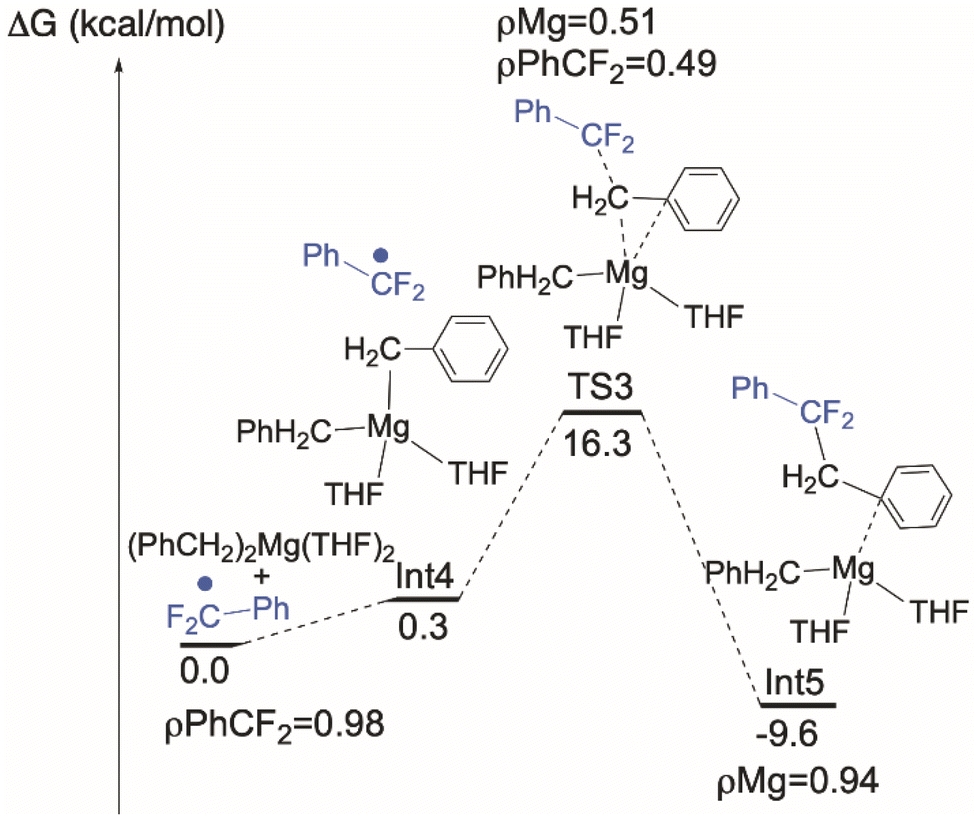 | ||
| Fig. 7 Computed C–C coupling pathway at MgBn2(THF)2. | ||
 | ||
| Fig. 8 Computed C–C coupling pathway at 7-Mg. | ||
The unpaired spin density rho (ρ) is reported on Fig. 7. As can be seen and as expected, the unpaired electron is first located on the PhCF2˙. At TS3, the simultaneous Mg–C bond breaking and the C–C bond formation suggests that the unpaired electron is delocalized between the PhCF2˙ and the Mg center. This clearly indicates that there is an homolytic cleavage of the Mg–C bond. This is further corroborated by the calculation showing that the unpaired spin density is localized at the Mg center in the final product. These results indicate that C–C bond formation is likely occurring at MgBn2(THF)2, which is also at significantly higher concentrations than 7-Mg in solution during catalysis.
Mechanistic discussion
The reactivity described here is notable in that it is not limited to Ce, which is the only rare earth with an accessible (III/IV) redox couple and f–d orbital transition. It is these properties that have led to the recent and widespread interest in photoredox catalysis with Ce complexes. Of all the complexes that are capable catalysts described here, the Ce congener shows the longest catalyst life, and cleanest product formations. We suggest that this is because for the photoactivated CeIII/IV system there is a simple, metal-based process for redox shuttling that enables the fluoride to be captured and removed, as well as the opportunity for both ligand and metal orbitals to contribute to the light absorption and bonding of the complex. For La and other metals, the redox part of the cycle relies on the redox reactivity of an ancillary ligand. The involvement of both phenoxide and cyclopentadienyl ligands in redox reactions has been previously documented,57–59,66,74 and cyclic voltammetry (CV) experiments show that pure HL possesses a pseudo-reversible redox event.Our proposed mechanism is outlined in Scheme 7. First, a weak but observable interaction between a fluoride of PhCF3 and the Ln metal, as observed in 19F NMR experiments for both Ce and La, serves to bring the substrate into the active site. C–F → LnIII (Ln = La, Sm, Yb) interactions have previously been hypothesized in the functionalization of C–F bonds mediated by lanthanide complexes.37–39
 | ||
| Scheme 7 Proposed mechanism of photocatalytic C–C coupling. | ||
Photoexcitation to a more reducing excited state3,75 follows, which leads to C–F cleavage and the generation of an ArCF2 radical, as supported by experiments with a radical trapping substrate, Scheme 4. The nature of the resulting putative Ln–F species depends on the identity of the rare earth element. For Ce, a discrete CeIV species is the most likely intermediate. For La, the ligands (either L or CpMe4) serve as the electron source through ligand-based redox non-innocence.
Calculations suggest that the ease of M–CpMe4 bond homolysis is greater for La versus Ce, and CV experiments imply that loss of a CpMe4 ligand from 1-La may lead to a cascading degradation process where the release of radical fragments accelerate decomposition (see Section S4‡). This supports the observation that complex 1-Ce is more effective for C–F activation while complex 1-La is faster for the coupling of the radical to form benzylated product, acting as a source of radicals that can facilitate C–F bond cleavage at the coordinated substrate. Moreover, 1-Ce is the most thermally robust of 1-Ln, able to operate at elevated temperatures (70 °C).
Reduction likely occurs when the generated PhCF2˙ couples with MgBn2 to form the C–C bond, with the resulting transient Mg(I) complex serving to reduce either the metal (in the case of Ce) or the ligand (in the case of La) back to the initial catalytic species.
Calculations agree with the proposed mechanism and with the need for inner-sphere binding of the fluorinated substrate to Lewis acidic metal to provide sufficient weakening of the C–X bond to enable photochemical cleavage. They also show how the ligands can contribute to light absorption to generate the reactive excited state, and how oxidation of the complex can be localized on the metal (for Ce) or ligand (for the other metals) to enable turnover.
The NHC ligand L is not labile in this system;16,76,77 variable temperature 1H NMR spectra, see S8.9,‡ do not show any interactions between the NHC of 1-Ce and MgBn2(THF)2 in THF solutions and all computed barriers increase in energy where the NHC is unbound or ligates to Mg or another reagent. Unsurprisingly, the calculations do not support the presence of a [CeIV–Bn] complex at any point.
Conclusions
Cleavage and functionalization of the strong and inert sp3 C–F bond of PhCF3 can be readily achieved using single component, earth-abundant organometallic photocatalysts, requiring both the Lewis acidity of the metal to bind the substrate, and the visible light-absorbing capabilities of the rigid aromatic ligands. The reactivity described here is notable in that it is not limited to Ce, which has been the focus of a recent surge of interest in earth-abundant metal photocatalysts.The photochemical hydrodefluorination and defluoroalkylation of PhCF3 is feasible for typically photoinactive metals including La and Mg because the rigid aryloxy-NHC ligand facilitates visible light absorption. Experiments with a radical-trapping substrate support the formation of ArCF2 radicals after C–F bond activation, and catalytic turnover in C–C bond forming reactions is most readily achieved using reagents such as MgBn2(THF)2 which can provide both the alkyl group and reduction of the catalyst intermediate. Computational experiments highlight the synergy between ligand-based and metal-based orbitals in the photoexcitation process. The complexes tested are also capable for homogeneous C(sp3)–Cl bond activation and functionalization. Studies to increase the rates and scope of the reactivity of these systems, and further tuning of ligand- and metal-based absorptions in photocatalysts are currently underway in our laboratory.
Data availability
Additional experimental, computational, and crystallographic data (PDF) are in the ESI.‡ The crystallographic cif data are deposited with the CCDC, codes 2121204–2121215 and 2217133. Raw datasets are in the open data archive DOI: 10.17632/8bx8zxbdgm.1.Author contributions
A. E. K.: investigation (experimental), visualization, analysis, writing – original draft, review & editing. L. K. E.: investigation (experimental). A. N. D.: investigation (experimental), visualization, analysis, writing – original draft, review & editing. Y. Y.: investigation (computation). L. S.: investigation (cyclic voltammetry), analysis, writing – original draft. A. D. G.: investigation (experimental). T. D. T.: funding aquisition, supervision. L. M.: funding acquisition, supervision, analysis, writing – review & editing. P. L. A.: funding acquisition, supervision, visualization, analysis, writing – original draft, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The synthetic, catalytic, and crystallographic parts of this research were supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division at the Lawrence Berkeley National Laboratory under Contract DE-AC02-05CH11231. The Catalysis Laboratory in the DOE Catalysis Program also provided resource for instrumentation used in this work. We also thank the University of Edinburgh. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 740311, P. L. A.) and the EPSRC are acknowledged for research funding through the UK Catalysis Hub (EP/K014714/1, P. L. A.). We acknowledge the National Institutes of Health (NIH) for funding the UC Berkeley College of Chemistry NMR facility under grant no. SRR023679A, S10OD024998 and 1S10RR016634-01, and Beamline 12.2.1 of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. L. M. is a senior member of the Institut Universitaire de France. The Chinese Academy of Science and the Chinese Scholarship Council are acknowledged for financial support and CALMIP is thanked for a generous grant of computing time. The authors thank Rex C. Handford, Francis Y. T. Lam, Nicholas J. Katzer, Gabe Herrera, Dr Amy N. Price and Dr Anthony R. Wong for crystallographic and/or experimental help, as well as Prof. Rebecca J. Abergel, Dr Leticia Arnedo Sanchez and Dr Jennifer Wacker for help with photophysical measurements and Mr Gabriel Herrera for the LCMS and HRMS data.References
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 8–11 CrossRef PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- K. P. Shing Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef CAS PubMed.
- D. Mazzarella, G. E. M. Crisenza and P. Melchiorre, J. Am. Chem. Soc., 2018, 140, 8439–8443 CrossRef CAS PubMed.
- D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002–7006 RSC.
- A. Hu, J. J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. Macmillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed.
- G. Goti, B. Bieszczad, A. Vega-Peñaloza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 1213–1217 CrossRef CAS PubMed.
- C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed.
- S. T. Nguyen, P. R. D. Murray and R. R. Knowles, ACS Catal., 2020, 10, 800–805 CrossRef CAS.
- Y. Zhou, D. Hu, D. Li and X. Jiang, JACS Au, 2021, 1, 1141–1146 CrossRef CAS PubMed.
- J. J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319–15322 CrossRef CAS PubMed.
- T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- P. L. Arnold, R. W. F. Kerr, C. Weetman, S. R. Docherty, J. Rieb, F. L. Cruickshank, K. Wang, C. Jandl, M. W. McMullon, A. Pöthig, F. E. Kühn and A. D. Smith, Chem. Sci., 2018, 9, 8035–8045 RSC.
- R. W. F. Kerr, P. M. D. A. Ewing, S. K. Raman, A. D. Smith, C. K. Williams and P. L. Arnold, ACS Catal., 2021, 11, 1563–1569 CrossRef CAS.
- P. L. Watson, T. H. Tulip and I. Williams, Organometallics, 1990, 9, 1999–2009 CrossRef CAS.
- T. Kondo, M. Akazome and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1991, 757 RSC.
- A. Ogawa, S. Ohya, Y. Sumino, N. Sonoda and T. Hirao, Tetrahedron Lett., 1997, 38, 9017–9018 CrossRef CAS.
- T. C. Jenks, M. D. Bailey, J. L. Hovey, S. Fernando, G. Basnayake, M. E. Cross, W. Li and M. J. Allen, Chem. Sci., 2018, 9, 1273–1278 RSC.
- J. Ma, F. Schäfers, C. Daniliuc, K. Bergander, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 9639–9645 CrossRef CAS PubMed.
- N. Nensala and T. Nyokong, Polyhedron, 1997, 16, 2971–2978 CrossRef CAS.
- M. D. Rausch, K. J. Moriarty, J. L. Atwood, J. A. Weeks, W. E. Hunter and H. G. Brittain, Organometallics, 1986, 5, 1281–1283 CrossRef CAS.
- P. N. Hazin, J. W. Bruno and H. G. Brittain, Organometallics, 1987, 6, 913–918 CrossRef CAS.
- H. Yin, P. J. Carroll, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2015, 137, 9234–9237 CrossRef PubMed.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef PubMed.
- H. Yin, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 5984–5993 CrossRef PubMed.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef PubMed.
- A. Prieto and F. Jaroschik, Curr. Org. Chem., 2022, 26, 6–41 CrossRef CAS.
- Y. Qiao, Q. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999–11003 CrossRef CAS PubMed.
- S. J. Gray, K. Brown, F. Y. T. Lam, J. A. Garden and P. L. Arnold, Organometallics, 2021, 40, 948–958 CrossRef CAS.
- F. Sinclair, J. A. Hlina, J. A. L. Wells, M. P. Shaver and P. L. Arnold, Dalton Trans., 2017, 46, 10786–10790 RSC.
- B. E. Smart, in Organofluorine Chemistry: Principles and Commercial Applications, ed. R. E. Banks, B. E. Smart and J. C. Tatlow, Springer US, Boston, MA, 1994, pp. 57–88 Search PubMed.
- Y. Wang and A. Liu, Chem. Soc. Rev., 2020, 49, 4906–4925 RSC.
- T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS.
- M. Janjetovic, A. M. Träff, T. Ankner, J. Wettergrena and G. Hilmersson, Chem. Commun., 2013, 49, 1826–1828 RSC.
- A. M. Träff, M. Janjetovic, L. Ta and G. Hilmersson, Angew. Chem., Int. Ed., 2013, 52, 12073–12076 CrossRef.
- M. Janjetovic, A. M. Träff and G. Hilmersson, Chem.–Eur. J., 2015, 21, 3772–3777 CrossRef.
- F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486–13505 RSC.
- H. Iwamoto, H. Imiya, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2020, 142, 19360–19367 CrossRef PubMed.
- X. Gong, Q. Zhou and G. Yan, Org. Biomol. Chem., 2022, 20, 5365–5376 RSC.
- Y. J. Yu, F. L. Zhang, T. Y. Peng, C. L. Wang, J. Cheng, C. Chen, K. N. Houk and Y. F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS PubMed.
- J. B. I. Sap, N. J. W. Straathof, T. Knauber, C. F. Meyer, C. F. Meyer, M. Médebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noël, C. W. Am Ende and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 9181–9187 CrossRef CAS.
- Y. C. Luo, F. F. Tong, Y. Zhang, C. Y. He and X. Zhang, J. Am. Chem. Soc., 2021, 143, 13971–13979 CrossRef CAS PubMed.
- C. Luo and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 14120–14125 CrossRef CAS.
- D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572–2578 CrossRef CAS PubMed.
- G. Yan, Chem.–Eur. J., 2022, 28, e202200231 CAS.
- G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC.
- D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef.
- H. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163–166 CrossRef PubMed.
- K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef PubMed.
- N. Sugihara, K. Suzuki, Y. Nishimoto and M. Yasuda, J. Am. Chem. Soc., 2021, 143, 9308–9313 CrossRef PubMed.
- P. Pinter, C. M. Schüßlbauer, F. A. Watt, N. Dickmann, R. Herbst-Irmer, B. Morgenstern, A. Grünwald, T. Ullrich, M. Zimmer, S. Hohloch, D. M. Guldi and D. Munz, Chem. Sci., 2021, 12, 7401–7410 RSC.
- H. Yamashita, T. Ikezawa, Y. Kobayashi and J. Abe, J. Am. Chem. Soc., 2015, 137, 4952–4955 CrossRef CAS.
- J. Li, C. Huang and C. Li, Chem, 2022, 1–13 Search PubMed.
- S. Shirase, K. Shinohara, H. Tsurugi and K. Mashima, ACS Catal., 2018, 8, 6939–6947 CrossRef CAS.
- M. R. Ringenberg, S. L. Kokatam, Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 788–789 CrossRef CAS PubMed.
- L. Benisvy, A. J. Blake, D. Collison, E. S. Davies, C. D. Garner, E. J. L. McInnes, J. McMaster, G. Whittaker and C. Wilson, Dalton Trans., 2003, 1975–1985 RSC.
- V. F. Quiroga Norambuena, A. Heeres, H. J. Heeres, A. Meetsma, J. H. Teuben and B. Hessen, Organometallics, 2008, 27, 5672–5683 CrossRef CAS.
- E. L. Werkema, L. Castro, L. Maron, O. Eisenstein and R. A. Andersen, New J. Chem., 2013, 37, 132–142 RSC.
- E. L. Werkema, R. A. Andersen, L. Maron and O. Eisenstein, Dalton Trans., 2010, 39, 6648 RSC.
- L. Maron, E. L. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279–292 CrossRef PubMed.
- H. Yin, A. V. Zabula and E. J. Schelter, Dalton Trans., 2016, 45, 6313–6323 RSC.
- M. T. Trinh, J. C. Wedal and W. J. Evans, Dalton Trans., 2021, 50, 14384–14389 RSC.
- M. E. Fieser, C. W. Johnson, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, Organometallics, 2015, 34, 4387–4393 CrossRef CAS.
- J. B. Waters and J. M. Goicoechea, Dalton Trans., 2014, 43, 14239–14248 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P. Clavel, G. Lessene, C. Biran, M. Bordeau, N. Roques, S. Trévin and D. de Montauzon, J. Fluorine Chem., 2001, 107, 301–310 CrossRef CAS.
- P. Dröse, A. R. Crozier, S. Lashkari, J. Gottfriedsen, S. Blaurock, C. G. Hrib, C. Maichle-Mössmer, C. Schädle, R. Anwander and F. T. Edelmann, J. Am. Chem. Soc., 2010, 132, 14046–14047 CrossRef.
- R. Anwander, M. Dolg and F. T. Edelmann, Chem. Soc. Rev., 2017, 46, 6697–6709 RSC.
- D. Schneider, N. Harmgarth, F. T. Edelmann and R. Anwander, Chem.–Eur. J., 2017, 23, 12243–12252 CrossRef CAS.
- U. J. Williams, J. R. Robinson, A. J. Lewis, P. J. Carroll, P. J. Walsh and E. J. Schelter, Inorg. Chem., 2014, 53, 27–29 CrossRef CAS.
- T. J. Mueller, M. E. Fieser, J. W. Ziller and W. J. Evans, Chem. Sci., 2011, 2, 1992–1996 RSC.
- Y. H. Wang, Q. Yang, P. J. Walsh and E. J. Schelter, Org. Chem. Front., 2022, 2612–2620 RSC.
- Z. R. Turner, R. Bellabarba, R. P. Tooze and P. L. Arnold, J. Am. Chem. Soc., 2010, 132, 4050–4051 CrossRef CAS PubMed.
- I. J. Casely, S. T. Liddle, A. J. Blake, C. Wilson and P. L. Arnold, Chem. Commun., 2007, 5037–5039 RSC.
Footnotes |
| † This paper is dedicated to the memory of Luca Elghanayan (2002–2022), who made important contributions to this research. |
| ‡ Electronic supplementary information (ESI) available: Additional experimental, computational, and crystallographic data (PDF). Raw datasets are in the open data archive doi: https://10.17632/8bx8zxbdgm.1. CCDC 2121204–2121215 and 2217133. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04192h. |
| This journal is © The Royal Society of Chemistry 2022 |