
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly stereoselective and enantiodivergent synthesis of cyclopropylphosphonates with engineered carbene transferases†
Xinkun
Ren‡
 a,
Ajay L.
Chandgude
a,
Daniela M.
Carminati
a,
Zhuofan
Shen
b,
Sagar D.
Khare
b and
Rudi
Fasan
*a
a,
Ajay L.
Chandgude
a,
Daniela M.
Carminati
a,
Zhuofan
Shen
b,
Sagar D.
Khare
b and
Rudi
Fasan
*a
aDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA. E-mail: rfasan@ur.rochester.edu
bDepartment of Chemistry and Chemical Biology, Rutgers University, New Brunswick, New Jersey 08854, USA
First published on 6th June 2022
Abstract
Organophosphonate compounds have represented a rich source of biologically active compounds, including enzyme inhibitors, antibiotics, and antimalarial agents. Here, we report the development of a highly stereoselective strategy for olefin cyclopropanation in the presence of a phosphonyl diazo reagent as carbene precursor. In combination with a ‘substrate walking’ protein engineering strategy, two sets of efficient and enantiodivergent myoglobin-based biocatalysts were developed for the synthesis of both (1R,2S) and (1S,2R) enantiomeric forms of the desired cyclopropylphosphonate ester products. This methodology enables the efficient transformation of a broad range of vinylarene substrates at a preparative scale (i.e. gram scale) with up to 99% de and ee. Mechanistic studies provide insights into factors that contribute to make this reaction inherently more challenging than hemoprotein-catalyzed olefin cyclopropanation with ethyl diazoacetate investigated previously. This work expands the range of synthetically useful, enzyme-catalyzed transformations and paves the way to the development of metalloprotein catalysts for abiological carbene transfer reactions involving non-canonical carbene donor reagents.
Introduction
Phosphorus-containing compounds are essential to all known forms of life1 and organophosphorus compounds have represented an important source of biologically active compounds, including enzyme inhibitors, antibiotics, and plant regulators (Fig. 1).2–6 For example, the phosphonyl/cyclopropane-containing adenosine monophosphate MRS2339 was identified as a potent drug for the treatment of heart failure.2 Other phosphonyl-functionalized molecules such as tenofovir3 and fosmidomycin analogs4 (Fig. 1) were developed as therapeutic agents for HIV and malaria, respectively, with the corresponding phosphonate ester derivatives being used as prodrugs with enhanced bioavailability. In nature, a variety of bioactive natural products incorporate phosphonate groups,6 including the clinically used antibiotic fosfomycin,7 the antifungal tripeptide rhizocticin,8 and the herbicide phosphinothricin.9 In view of the biological and pharmaceutical importance of organophosphorus compounds, the development of biocatalytic methods for the synthesis of optically active phosphonate-containing molecules is highly desirable.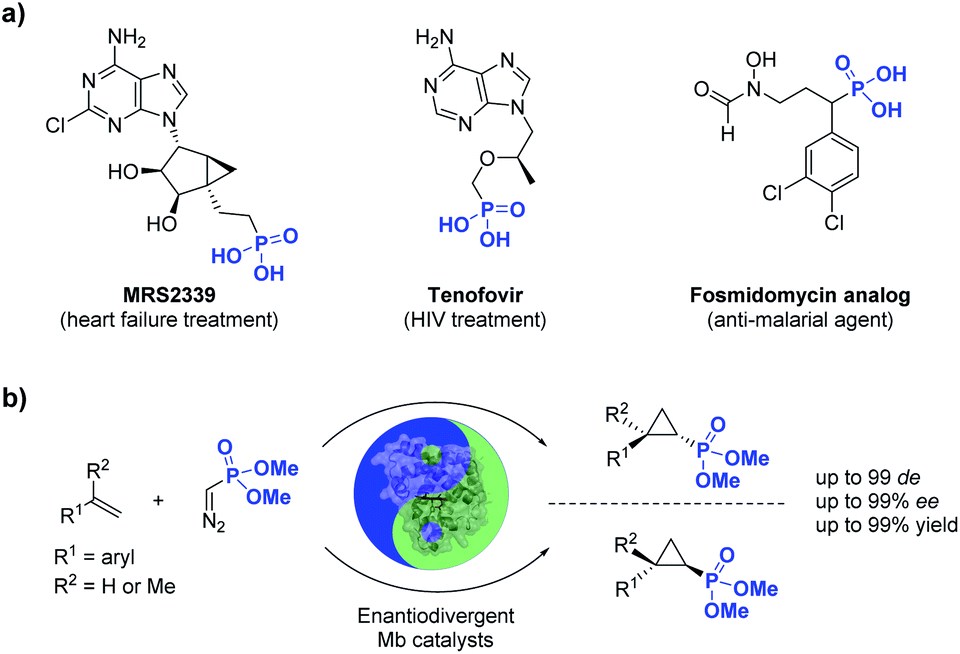 | ||
| Fig. 1 (a) Biologically active organophosphonates and (b) biocatalytic method for the stereoselective synthesis of cyclopropylphosphonates (this work). | ||
Among the various methodologies reported for the synthesis of organophosphonates, including methods for direct C–P bond formation,10 the asymmetric cyclopropanation of olefins with phosphonyl-containing diazo compounds represents an attractive approach for the synthesis of cyclopropane rings decorated with a phosphonyl group.11 While these reactions have been traditionally addressed using organo-transition metal catalysts, asymmetric cyclopropanation with acceptor-only phosphonyl diazo compounds has proven challenging.11 In this context, enzyme-mediated carbene transfer catalysis has recently emerged as a promising and sustainable alternative for the stereoselective synthesis of cyclopropane molecules.12–23 In particular, our group and others have demonstrated that engineered hemoproteins (e.g. myoglobin, cytochrome P450s)12–23 and artificial metalloenzymes24–31 can promote stereoselective cyclopropanations in both intermolecular and intramolecular settings. While various acceptor-only diazo compounds have been successfully employed for these biocatalytic transformations,12–31 the scope of these reactions has been thus far limited to diazo reagents that incorporate carbon-based electron withdrawing groups (i.e., RCHN2, where R = CO2R, CN, CF3, COR), which restricts the diversity of cyclopropane products obtainable through biocatalysis.
Here, we report the successful development of a biocatalytic strategy for promoting highly diastereo- and enantioselective olefin cyclopropanation in the presence of a phosphonyl diazo compound, which remains unprecedented for metalloenzymes (Fig. 1b). This methodology enables the efficient transformation of a broad range of olefin substrates to yield enantioenriched phosphonyl-functionalized cyclopropanes in both trans-(1S,2R) and trans-(1R,2S) configurations with high stereoselectivity and at a synthetically useful scale.
Results and discussion
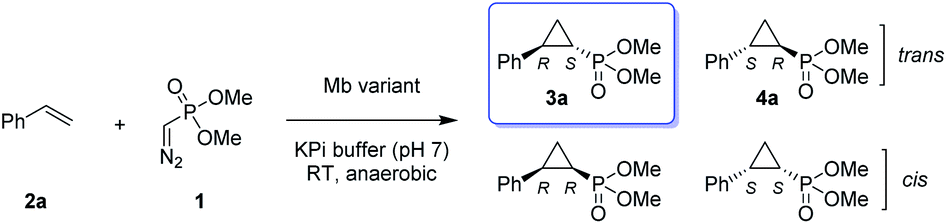
Given our prior success in developing hemoprotein-based catalysts for cyclopropanation reactions with acceptor-only diazo compounds, we envisioned the employment of Seyferth–Gilbert reagent dimethyl (diazomethyl)phosphonate32 (1) as carbene donor for the biocatalytic construction of phosphorus-containing cyclopropanes. Accordingly, we tested a model reaction with styrene (2a) and 1 in the presence of seven different hemoproteins, including myoglobin (Mb), Mb(H64V,V68A),13 P450BM3, catalase, cytochrome c from equine heart and from Hydrogenobacter thermophilus, P450 XplA, and P450 BezE (Table S1†). A control reaction containing hemin as the catalyst did not produce any product (Table 1, entry 1). In contrast, four of the tested hemoproteins showed detectable to moderate activity in this reaction (0.4% to 23% conversion), highlighting the importance of the protein environments for orchestrating this challenging transformation. XplA, a cytochrome P450 from Rhodococcus sp.33 previously identified as an efficient nitrene transferase for C–H amination,34 showed the highest level of activity, giving 23% yield (GC) but only moderate level of enantioselectivity (33% ee, entry 8, Table S1†) for the formation of (1R, 2S)-configured cyclopropanation product 4a. BezE, another P450-based nitrene transferase,34,35 exhibited excellent diastereoselectivity (93% de) but poor enantioselectivity (15% ee, entry 9, Table S1†) toward the formation of (1S, 2R)-configured stereoisomer 3a. While wild-type Mb shows minimal activity in the reaction (<1%; Table 1, entry 2), Mb(H64V,V68A), a Mb variant previously optimized for intermolecular styrene cyclopropanation with ethyl diazoacetate,13 was found to exhibit excellent stereoselectivity (>99% de and 98% ee for 3a; Table 1, entry 3), albeit still with very low activity (2%).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yieldb | TON | dec [%] | eed [%] |
| a Reaction conditions: 2.5 mM dimethyl (diazomethyl)phosphonate (1), 5 mM styrene (2a), 20 μM Mb variant in KPi buffer (50 mM, pH 7), 10 mM Na2S2O4, r.t., 16 h in sealed anaerobic crimp vials. See also Tables S1 and S2. b GC yield based on the calibration curves prepared using authentic standards. Yields of isolated products are reported in brackets. c % de Values: (trans − cis)/(trans + cis). d Trans% ee values: [(1S,2R) − (1R,2S)]/[(1R,2S) + (1S,2R)]. e Using 40 μM Mb, 20 mM 1, and 10 mM styrene. f Using 10 μM Mb, 20 mM 1, and 10 mM styrene. g Using whole cells at OD600 = 80, 20 mM 1, and 10 mM styrene. | |||||
| 1 | Hemin | 0% | 0 | n.d. | n.d. |
| 2 | Mb | 0.4% | 1 | 90 | n.d. |
| 3 | Mb(H64V,V68A) | 1.6% | 2 | >99 | 98 |
| 4 | Mb(H64G,V68A) | 67% | 83 | >99 | >99 |
| 5 | Mb(H64A,V68G) | 18% | 23 | >99 | 79 |
| 6 | Mb(H64V,V68G) | 13% | 16 | >99 | 97 |
| 7 | Mb(H64A,V68G,I107L) | 35% | 44 | >99 | 97 |
| 8e | Mb(H64G,V68A) | 99% (94%) | 250 | >99 | >99 |
| 9f | Mb(H64G,V68A) | 47% | 470 | >99 | >99 |
| 10g | Mb(H64G,V68A) | 99% | 187 | >99 | >99 |
Based on these results, we decided to further explore the Mb scaffold toward developing improved biocatalysts for this transformation. Accordingly, we screened a diverse panel of Mb variants (∼40) featuring one to four mutations within the protein active site (i.e., at positions Leu29, Phe43, His64, Val68, Ile107; Fig. S1 and Tables S2 and S3†). From this screen, Mb(H64G,V68A), a variant identified previously for olefin cyclopropanation with bulky diazoketone reagents,23 was found to offer superior performance over Mb(H64V,V68A) and the other Mb variants, producing 3a with excellent diastereo- and enantioselectivity in 67% yield (>99% de and ee; Table 1, entry 4; Fig. 2a). Interestingly, the single-site variants Mb(H64G) and Mb(V68A) displayed minimal to no activity under identical reaction conditions (0–4% yield; Table S2†, entries 3 & 6), indicating that these mutations exert a synergistic effect in enhancing the reactivity of the biocatalyst. To further examine the role of these two ‘hot spots’, the activity and selectivity of Mb(V68A)-containing variants featuring residues of varying size at the level of the distal His64 position were compared (Table S3†).
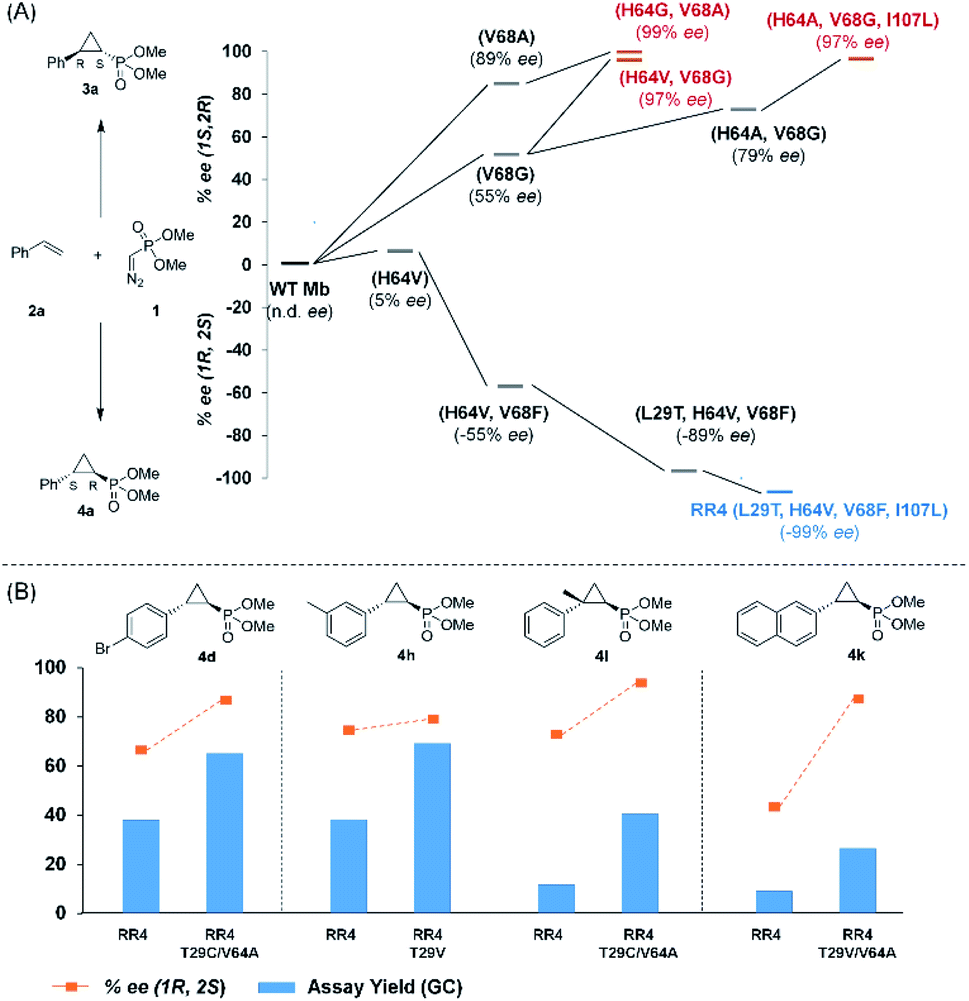 | ||
| Fig. 2 (A) Structure–activity analysis of the (1S,2R)- and (1R,2S)-selective cyclopropanation biocatalysts. See Fig. S4† for complete activity and selectivity data. (B) Improved (1R,2S)-selective biocatalysts obtained by directed evolution of Mb variant RR4 via a substrate walking approach. | ||
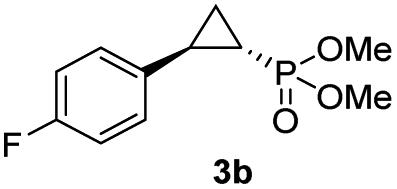
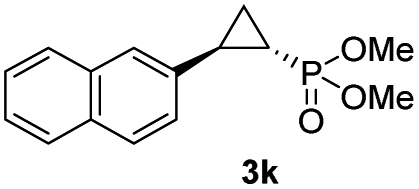
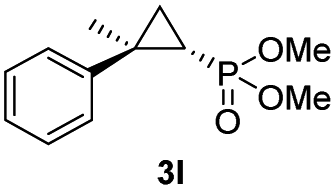
While all the double variants showed comparably high enantioselectivity (98 and >99% ee), a strong correlation was apparent between catalytic activity and a decreasing size (and thus steric hindrance) at the 64 position (1.6% (Val) → 30% (Ala) → 67% (Gly); Table S3†, entries 2–4). We hypothesize that a larger cavity at the distal histidine (64) position could better facilitate access of the bulky diazo reagent to the heme pocket and/or better accommodate the bulky phosphonyl group in the heme-bound carbene intermediate expected to mediate these reactions.36 Another interesting structure–activity trend emerging from the catalyst screening is the favorable effect of a Gly mutation at position 68 (i.e., V68G), as suggested by the functionality in the cyclopropanation reaction of various Mb variants incorporating this mutation (13–30% yield; Table S2†, entries 21–27). Using Mb(V68G) as background, a similar trend was observed with respect to an increase in catalytic activity upon mutation of His64 with smaller residues (Table S3†, entries 5–7). The most active catalyst in this group, Mb(H64A,V68G), was then subjected to a round of directed evolution via parallel site-saturation mutagenesis at positions 29, 43 and 107 which surround the heme active site (Fig. S1†). Screening of these libraries in whole cells led to the identification of Mb(H64A,V68G,I107L) variant with improved activity (18 → 35% yield) and enantioselectivity (79 → 97% ee(1S,2R)) compared to the parent enzyme (Table 1, entry 5 vs. 7; Fig. 2a). While this variant remained an inferior catalyst compared to Mb(H64G,V68A) in the reaction with styrene and 1, it later proved more effective for the transformation of challenging α, α-disubstituted styrenes. Next, the Mb(H64G,V68A)-catalyzed cyclopropanation reaction with the phosphonyl diazo reagent 1 was further optimized by varying the substrate and catalyst loading and the diazo:styrene ratio (Table S4†). While the initial conditions utilized an excess of styrene over the diazo reagent, these experiments showed that high to quantitative yields (86–99%) could be obtained in the presence of the olefin as the limiting reagent (i.e., styrene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1:2), while maintaining excellent diastereo- and enantioselectivity (>99% de and ee, Table 1, entry 8). In addition, under similar conditions, the reaction could be carried out with equally high efficiency and stereoselectivity using whole cells (E. coli) expressing the Mb(H64G,V68A) variant (Table 1, entry 10). The (1S,2R)-configuration of 3a was assigned based on the crystallographic analysis of the related product 3da (Fig. S5; Table S8†). Time-course experiments showed that this Mb(H64G,V68A)-catalyzed reaction reaches ∼80% conversion in ∼2 min and quantitative conversion of 2a to 3a in less than 5 min (Fig. S2†), indicating fast reaction kinetics. Using this variant, up to 470 catalytic turnovers were measured under catalyst-limited conditions (Table 1, entry 9). The substrate scope of the Mb(H64G,V68A) catalyst was then probed against a diverse panel of styrene derivatives and vinylarenes under the optimized conditions described above and at a preparative scale (0.5 mmol) (Table 2). These experiments showed that styrene derivatives carrying para, meta and ortho substitutions (2b–2k) are efficiently processed to give the corresponding cyclopropanation products 3b–3k in good to excellent yields (44–99%) and with excellent levels of diastereo (>99% de) and enantioselectivity (98–99% ee) (Table 2). Both electron-withdrawing and electron-donating groups on benzene ring were well tolerated (2b–2f) as well as large substrates such as 2-vinylnaphthalene (2k). Using Mb(H64A,V68G,I107L) as the catalyst, conversion of α-methyl-styrene into the trisubstituted phosphonyl-functionalized cyclopropane 3l could be also accomplished with high enantioselectivity (99% ee), albeit with moderate yield and diastereoselectivity (Table 2, entry 11).
:1 ratio of 1:2), while maintaining excellent diastereo- and enantioselectivity (>99% de and ee, Table 1, entry 8). In addition, under similar conditions, the reaction could be carried out with equally high efficiency and stereoselectivity using whole cells (E. coli) expressing the Mb(H64G,V68A) variant (Table 1, entry 10). The (1S,2R)-configuration of 3a was assigned based on the crystallographic analysis of the related product 3da (Fig. S5; Table S8†). Time-course experiments showed that this Mb(H64G,V68A)-catalyzed reaction reaches ∼80% conversion in ∼2 min and quantitative conversion of 2a to 3a in less than 5 min (Fig. S2†), indicating fast reaction kinetics. Using this variant, up to 470 catalytic turnovers were measured under catalyst-limited conditions (Table 1, entry 9). The substrate scope of the Mb(H64G,V68A) catalyst was then probed against a diverse panel of styrene derivatives and vinylarenes under the optimized conditions described above and at a preparative scale (0.5 mmol) (Table 2). These experiments showed that styrene derivatives carrying para, meta and ortho substitutions (2b–2k) are efficiently processed to give the corresponding cyclopropanation products 3b–3k in good to excellent yields (44–99%) and with excellent levels of diastereo (>99% de) and enantioselectivity (98–99% ee) (Table 2). Both electron-withdrawing and electron-donating groups on benzene ring were well tolerated (2b–2f) as well as large substrates such as 2-vinylnaphthalene (2k). Using Mb(H64A,V68G,I107L) as the catalyst, conversion of α-methyl-styrene into the trisubstituted phosphonyl-functionalized cyclopropane 3l could be also accomplished with high enantioselectivity (99% ee), albeit with moderate yield and diastereoselectivity (Table 2, entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | Product | Yieldb | % de | % ee |
| a Reaction conditions: 10 mM olefin, 20 mM dimethyl (diazomethyl)phosphonate (1), 40 μM Mb(H64G,V68A) purified protein in KPi buffer (50 mM, pH 7), 50 mL-scale, RT, 16 h. b Product conversion as determined by GC. Yields of isolated products are reported in brackets. Errors are within 10%. c Reaction volume: 600 mL. d Using Mb(H64V,V68G) as catalyst. e Using 5 mM olefin and 10 mM dimethyl (diazomethyl)phosphonate. f Using Mb(H64A,V68G,I107L). | ||||
| 1 |
|
99% (90%) | >99 | >99 |
| 2 |
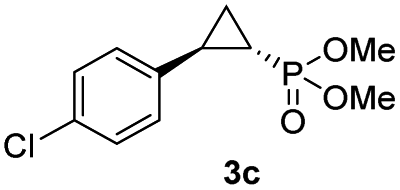
|
65% (59%) | >99 | >99 |
| 3c |
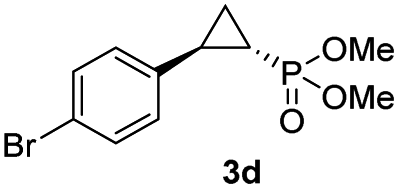
|
73% (61%) | >99 | >99 |
| 4d |
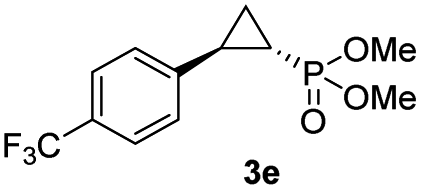
|
63% (52%) | >99 | 98 |
| 5 |
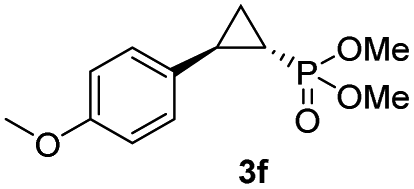
|
93% (87%) | >99 | >99 |
| 6 |
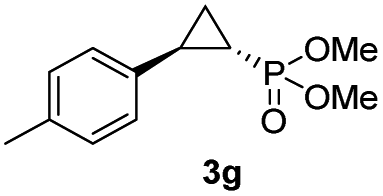
|
48% (41%) | >99 | 99 |
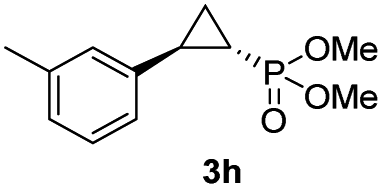
| 7 |
|
95% (85%) | >99 | >99 |
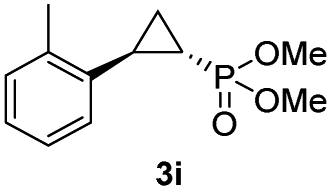
| 8 |
|
56% (51%) | >99 | >99 |
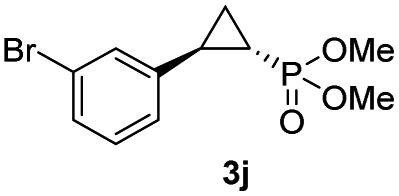
| 9 |
|
44% (36%) | >99 | >99 |
| 10d,e |
|
46% (41%) | >99 | 99 |
| 11f |
|
20% (15%) | 24 | >99 |
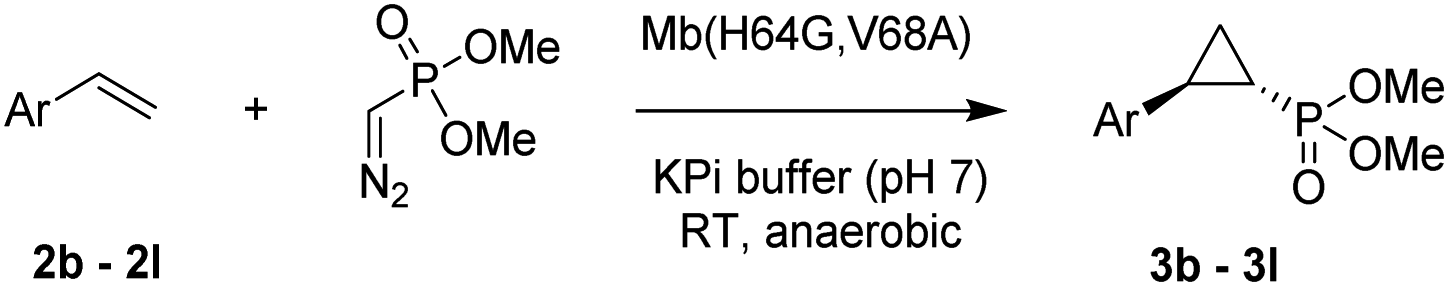
The Mb(H64G,V68A)-catalyzed biotransformation could be further scaled up to isolate 1.1 g of enantiopure 3d (61% isolated yield), which further demonstrated the robustness and scalability of this biocatalytic method (Scheme 1). This product was further derivatized via Suzuki–Miyaura cross-coupling (Scheme 1) to produce 3da (66% isolated yield), which could be crystallized for assignment of the stereochemical configuration of the cyclopropane ring. Of note, the Mb(H64G,V68A) variant exhibited a consistent (1S,2R)-stereoselectivity across the diverse panel of olefin substrates, as derived by the similar behavior of the products on chiral GC and SFC compared to the reference compound 3da.
 | ||
| Scheme 1 Gram-scale biocatalytic synthesis of 3d and further functionalization to 3da for crystallographic analysis. See Fig. S5 and Table S8† for crystallographic data. | ||
Having developed a general biocatalyst for the synthesis of (1S,2R)-configured cyclopropylphosphonate esters, we sought to identify variants that can provide access to the (1R,2S)-configured product, since enantiodivergent biocatalysts are highly desirable for medicinal chemistry and other synthetic applications. To that end, we expanded the panel of catalysts for initial screening to include a series of so-called ‘RR’ variants (i.e., RR1 to RR5), which we previously engineered for the enantiodivergent cyclopropanation of styrene and EDA.14 To our delight, all these five catalysts were found to favor the formation of the (1R,2S)-configured product 4a (73–99% ee(1R,2S)), albeit mostly with low activity (Table S2†, entries 38–42). Among them, Mb variant RR4 (= Mb(L29T,H64V,V68F,I107L)) displayed the most promising activity (17% yield), along with excellent stereoselectivity (99% de and ee; Table S2†, entry 41). Mutational ‘deconstruction’ of this variant indicated that the mutations at position 29, 68, and 107 are primarily responsible for the inversion of enantioselectivity (Fig. 2a). Upon optimization of the reaction conditions, nearly quantitative yield of 4a (96%), along with excellent de and ee, was achieved using whole cells expressing Mb variant RR4 in the presence of a slight (2-fold) excess of diazo reagent over the olefin (Table 3, entry 1). This Mb variant was then challenged with the same set of olefin substrates described in Table 2. All the substrates were successfully converted to the desired (1R,2S)-configured cyclopropane products (Table 3). However, only four of the 12 products were obtained in high yields (72–99%) and suboptimal levels of diastereo- and/or enantioselectivity were observed for various substrates, such as 4-bromo-styrene (2d), 3-methyl-styrene (2h), α-methyl-styrene (2l), and 2-vinylnaphthalene (2k).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Mutations (vs. RR4) | Product | Yieldb | % de | % ee |
| a Reaction conditions: 5 mM olefin, 10 mM dimethyl (diazomethyl)phosphonate (1), Mb-expressing E. coli (OD600 = 100) in KPi buffer (50 mM, pH 7), 1 mL-scale, RT, 16 h. b Product conversion as determined by GC. Errors are within 10%. | |||||
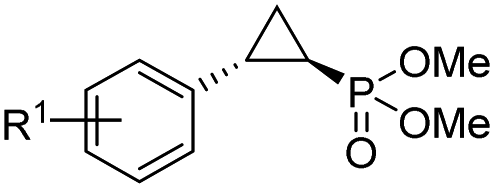
|
|||||
| 1 | RR4 | R1 = H (4a) | 96% | 99% | >99% |
| 2 | RR4 | R1 = 4-F (4b) | 72% | 99% | 91% |
| 3 | RR4 | R1 = 4-Cl (4c) | 38% | 99% | 82% |
| T29C,V64A | 64% | 99% | 92% | ||
| 4 | RR4 | R1 = 4-Br (4d) | 38% | 99% | 65% |
| T29C,V64A | 63% | 99% | 84% | ||
| 5 | RR4 | R1 = 4-CF3 (4e) | 9% | 99% | 33% |
| T29C,V64A | 24% | 99% | 56% | ||
| 6 | RR4 | R1 = 4-OMe (4f) | 99% | 99% | 59% |
| 7 | RR4 | R1 = 4-Me (4g) | 22% | 99% | 57% |
| T29C,V64A | 41% | 99% | 76% | ||
| 8 | RR4 | R1 = 3-Me (4h) | 40% | 27% | 72% |
| T29V | 71% | 91% | 76% | ||
| 9 | RR4 | R1 = 2-Me (4i) | 99% | 99% | >99% |
| 10 | RR4 | R1 = 3-Br (4j) | 40% | 46% | 88% |
| T29V | 99% | 94% | 89% | ||
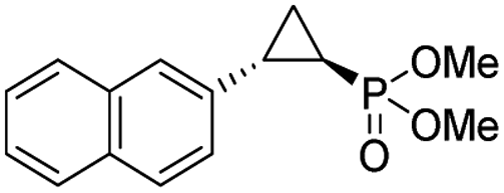
| 11 | RR4 |
 (4k) (4k) |
12% | 99% | 45% |
| T29V,V64A | 24% | 99% | 81% | ||
| 12 | RR4 |
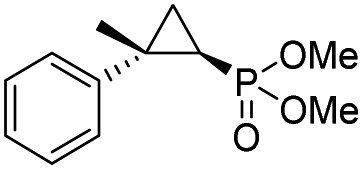 (4l) (4l) |
14% | 99% | 71% |
| T29C,V64A | 45% | 86% | 94% | ||
To overcome this limitation, we decided to further evolve RR4 using a “substrate walking” approach,37–39 whereby increased activity against the substrates found to be challenging for this biocatalyst, namely, compound 2d, 2h, 2l, and 2k (Fig. 2b), was used as the screening criterion for further evolution. Accordingly, RR4 was subjected to sequential rounds of site saturation mutagenesis at the active site residues Leu29 and His64, followed by library screening against the four aforementioned olefin substrates. This process led to identification of four new Mb variants, namely RR4/T29C, RR4/T29V, RR4/T29C/V64A, RR4/T29V/V64A, which feature improved catalytic activity as well as higher stereoselectivity toward the synthesis of the desired (1R,2S)-cyclopropylphosphonate products (Fig. 2b). The performance of these variants was then assessed against the entire set of eight ‘difficult’ substrates in the presence of RR4 as the catalyst (Table S6†). Gratifyingly, important enhancements in yield and/or stereoselectivity were generally obtained for all of these substrates. For example, using RR4/T29V, the synthesis of the meta-substituted products 4h and 4j could be achieved with significantly higher diastereoselectivity compared to RR4 (27–46% → 91–94% de; Table 3, entries 8 & 10). Using RR4(T29C,V64A), on the other hand, both improved yields and higher enantioselectivity could be obtained for the synthesis of the para-substituted cyclopropylphosphonates 4-e and 4g, and 4l derived from α-methyl-styrene (Table 3, entries 3–5, 7 and 12). From a structure–activity standpoint, these results also revealed a key role of the 29 position alone and of the combined 29/64 positions in controlling trans-selectivity and (1R,2S)-enantioselectivity in the presence of meta-substituted styrenes. Finally, a notable improvement in enantioselectivity (45 → 85% ee) for the (1R,2S)-selective cyclopropanation of 2-vinyl-naphthalene with phosphonyl diazo 1 was obtained using the evolved variant RR4(T29V,V64A).
Interestingly, the present studies showed that the Mb-catalyzed cyclopropanation of styrene with the phosphonyl diazo 1 represents a more challenging reaction than the same reaction in the presence of EDA, as investigated previously.13 This is evident from the lack of reactivity of wild-type Mb in the former reaction, compared to ∼200 TON with EDA, and the much lower TON supported by the optimized biocatalyst Mb(H64G, V68A) for the present reaction compared to the related Mb variant Mb(H64V,V68A) developed for styrene cyclopropanation with EDA (∼500 (Table 1) vs. > 10000 TON13). To better understand the basis for this differential reactivity, we investigated the reaction mechanism experimentally and computationally. Previous studies on the Mb-catalyzed styrene cyclopropanation with EDA indicated that this reaction involves a concerted carbene insertion mechanism,40 as opposed to a stepwise radical one.31 Similar to prior results with EDA, the addition of the radical spin trapping reagent 5,5-dimethyl-1-pyrroline N-oxide (DMPO) showed no detectable inhibition of the Mb(H64G,V68A)-catalyzed styrene cyclopropanation with 1 (Fig. 3a). In addition, no cis → trans isomerization was observed in the Mb(H64G,V68A)-catalyzed cyclopropanation of cis-β-deuterostyrene with the diazophosphonate reagent (Fig. 3b and S6†). Altogether, these results indicated that the two reactions shared a similar cyclopropanation mechanism involving a concerted carbene insertion step. To gain further insights into differences between the cyclopropanation reactions with diazophosphonate vs. EDA, these reactions were analyzed via Density Function Theory (DFT) using a model of histidine-ligated iron protoporphyrin system, as done previously.40 As shown in Fig. 3c, the energy profile for the heme-catalyzed cyclopropanation with EDA is consistent with that determined in previous studies,40 pointing at diazo compound activation to form the heme-bound carbene as the rate-determining step along the reaction coordinate. When compared with the reaction in the presence of 1, the two reactions displayed an overall similar energy profile but with some notable differences. For the initial step involving binding and activation of the diazo compound by the iron center, the transition state in the reaction with 1 (TS1diazophosphonate) was calculated to be 1.0 kcal mol−1 higher in energy than that of EDA (20.7 kcal mol−1vs. 19.7 kcal mol−1). Upon release of molecular dinitrogen, the iron-carbenoid intermediate formed in the presence of the diazo phosphonate reagent showed a 2.4 kcal mol−1 lower relative energy compared to that derived from EDA, indicating a higher stability and therefore lower reactivity than the latter. For the cyclopropanation step, a concerted mechanism was considered based on the experimental data. For this step, the energy barrier for cyclopropanation with 1 (TS2diazophosphonate) was found to be 2.1 kcal mol−1 higher in energy than that with EDA (TS2EDA) (ΔG = 12.0 kcal mol−1vs. 9.9 kcal mol−1). Overall, these studies suggest that the cyclopropanation reaction with the phosphoryl diazo compound 1 as catalyzed by the heme cofactor is less facile than that in the presence of EDA, which is in line with the general trend observed experimentally using hemin alone (0 vs. 14513 TON; Table 1), wild type Mb (1 vs. 18013 TON; Table 1), and the engineered Mb variants as noted above.
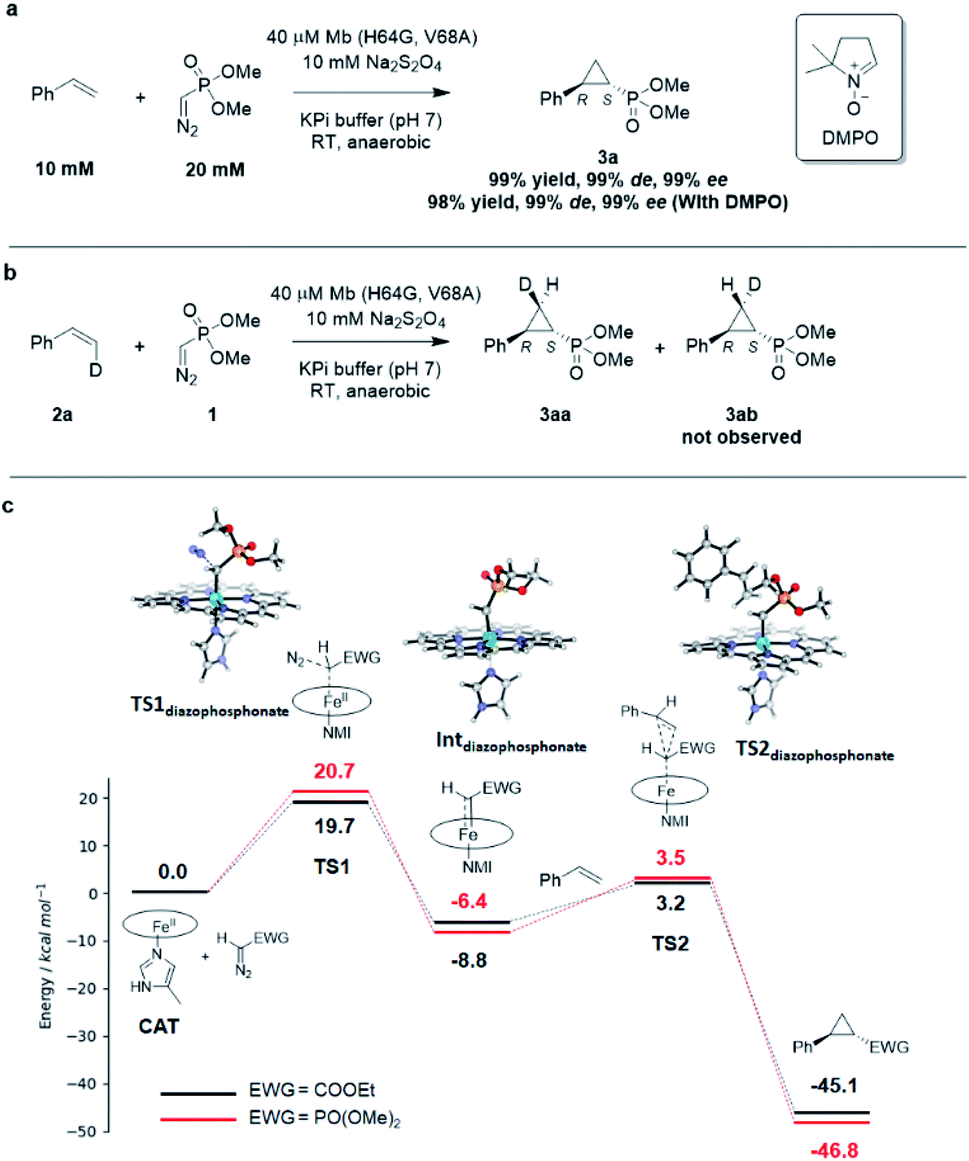 | ||
| Fig. 3 (A) Radical trapping experiment with styrene and dimethyl (diazomethyl)phosphonate. (B) Enzymatic cyclopropanation reaction with cis-β-deutero-styrene. (C) Gibbs free energy diagram for the heme-catalyzed styrene cyclopropanation reaction with dimethyl (diazomethyl)phosphonate (1) and EDA. Molecular models of key TS and intermediates in the reaction pathway with 1 are shown. ΔG values calculated based on the UB3LYP/6-311G** + SDD//UB3LYP-D3BJ/def2-TZVP + SDD method. See Table S8† for further details. NMI = 5-methyl-imidazole. | ||
To further investigate the difference in reactivity of the Mb catalysts in the cyclopropanation reaction with EDA vs.1, the transition states for the rate-determining step in these reactions as determined via DFT (i.e., TS1EDA and TS1diazophosphonate; Fig. 3c) were docked into the crystal structure of Mb(H64V,V68A)36 and a model of Mb(H64G,V68A), respectively, using the Rosetta software suite.41 After structure and energy optimization, the protein complex with the heme-bound dimethyl (diazomethyl)phosphonate substrate was found to be higher in energy than the protein complex with heme-bound EDA (Fig. S8†). Examination of these complexes and corresponding energy terms indicated that the dimethyl phosphonate group in reagent 1 causes greater steric repulsion with the protein environment compared to less bulky ethyl acetate moiety in the EDA reagent (Fig. S8†). These protein-dependent steric effects along with the higher energy barriers associated with the reaction with 1 (Fig. 3c) likely contribute to make the Mb-catalyzed cyclopropanation with the diazophoshonate reagent inherently more challenging that the same reaction in the presence of EDA as the carbene donor.
Conclusion
In summary, we have developed an efficient biocatalytic platform to perform the first example of intermolecular cyclopropanation using phosphorus-containing diazo compounds as carbene precursors. This biocatalytic strategy offers efficient, enantioselective, and scalable access to enantioenriched cyclopropylphosphonate esters, which can serve as key chiral building blocks for medicinal chemistry and other applications. Furthermore, enantiodivergent biocatalysts could be developed for this transformation, which enhances the synthetic value of this approach. For the (1R,2S)-selective biocatalyst, a substrate walking strategy proved effective to enable the stereoselective transformation of a broad range of vinyl arene substrates. Finally, our mechanistic and computational studies have yielded insights into similarities and differences between hemoprotein-catalyzed olefin cyclopropanation in the presence of the phosphonyl diazo reagent vs. diazoacetate, indicating how both kinetic and steric factors contribute to make the former reaction inherently more challenging than the latter. This work expands the catalytic repertoire of enzyme-catalyzed abiological transformations and opens the way toward the development of carbene transfer biocatalysts that utilize non-canonical carbene precursors.Data availability
The ESI† contains detailed descriptions of the experimental procedures, product characterization data, NMR spectra, screening data, crystallographic data and data related to the computational studies.Author contributions
X. R. and R. F. conceived the project; X. R., A. L. C, and R. F. designed the experiments; X. R., A. L. C., and D. M. C. performed the experiments; Z. S. and S. D. K designed and performed the computational studies; X. R. and R. F. wrote the manuscript with input from all the other co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. National Institute of Health grant GM098628 (R. F.) and U.S. National Science Foundation grants CBET-1929256 (R. F.) and CBET 1929237 (S. D. K.). The authors are grateful to Dr William Brennessel for assistance with crystallographic analyses. MS and X-ray instrumentation are supported by U.S. National Science Foundation grants CHE-0946653 and CHE-1725028 and the U.S. National Institute of Health grant S10OD030302.References
- C. Fernandez-Garcia, A. J. Coggins and M. W. Powner, Life, 2017, 7(3), 31 CrossRef PubMed.
- T. S. Kumar, S. Y. Zhou, B. V. Joshi, R. Balasubramanian, T. H. Yang, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2010, 53, 2562–2576 CrossRef CAS PubMed.
- A. Ustianowski and J. E. Arends, Infect. Dis. Ther., 2015, 4, 145–157 CrossRef PubMed.
- T. Haemers, J. Wiesner, S. Van Poecke, J. Goeman, D. Henschker, E. Beck, H. Jomaa and S. Van Calenbergh, Bioorg. Med. Chem. Lett., 2006, 16, 1888–1891 CrossRef CAS PubMed.
- F. Orsini, G. Sello and M. Sisti, Curr. Med. Chem., 2010, 17, 264–289 CrossRef CAS PubMed.
- H. Seto and T. Kuzuyama, Nat. Prod. Rep., 1999, 16, 589–596 RSC.
- R. D. Woodyer, Z. Y. Shao, P. M. Thomas, N. L. Kelleher, J. A. V. Blodgett, W. W. Metcalf, W. A. Van der Donk and H. M. Zhao, Chem. Biol., 2006, 13, 1171–1182 CrossRef CAS PubMed.
- S. A. Borisova, B. T. Circello, J. K. Zhang, W. A. van der Donk and W. W. Metcalf, Chem. Biol., 2010, 17, 28–37 CrossRef CAS PubMed.
- J. A. V. Blodgett, P. M. Thomas, G. Y. Li, J. E. Velasquez, W. A. van der Donk, N. L. Kelleher and W. W. Metcalf, Nat. Chem. Biol., 2007, 3, 480–485 CrossRef CAS PubMed.
- C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed.
- M. Marinozzi, F. Pertusati and M. Serpi, Chem. Rev., 2016, 116, 13991–14055 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- M. Bordeaux, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2015, 54, 1744–1748 CrossRef CAS PubMed.
- P. Bajaj, G. Sreenilayam, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2016, 55, 16110–16114 CrossRef CAS PubMed.
- D. Vargas, R. Khade, Y. Zhang and R. Fasan, Angew. Chem., Int. Ed., 2019, 58, 10148–10152 CrossRef CAS PubMed.
- A. M. Knight, S. B. J. Kan, R. D. Lewis, O. F. Brandenberg, K. Chen and F. H. Arnold, ACS Cent. Sci., 2018, 4, 372–377 CrossRef CAS PubMed.
- O. F. Brandenberg, C. K. Prier, K. Chen, A. M. Knight, Z. Wu and F. H. Arnold, ACS Catal., 2018, 8, 2629–2634 CrossRef CAS.
- K. Chen, S. Q. Zhang, O. F. Brandenberg, X. Hong and F. H. Arnold, J. Am. Chem. Soc., 2018, 140, 16402–16407 CrossRef CAS PubMed.
- A. L. Chandgude, X. Ren and R. Fasan, J. Am. Chem. Soc., 2019, 141, 9145–9150 CrossRef PubMed.
- X. Ren, A. L. Chandgude and R. Fasan, ACS Catal., 2020, 10, 2308–2313 CrossRef CAS PubMed.
- X. K. Ren, N. Y. Liu, A. L. Chandgude and R. Fasan, Angew. Chem., Int. Ed., 2020, 59, 21634–21639 CrossRef CAS PubMed.
- D. M. Carminati, J. Decaens, S. Couve-Bonnaire, P. Jubault and R. Fasan, Angew. Chem., Int. Ed., 2021, 60, 7072–7076 CrossRef CAS PubMed.
- D. Nam, V. Steck, R. J. Potenzino and R. Fasan, J. Am. Chem. Soc., 2021, 143, 2221–2231 CrossRef CAS PubMed.
- P. Srivastava, H. Yang, K. Ellis-Guardiola and J. C. Lewis, Nat. Commun., 2015, 6, 7789 CrossRef CAS PubMed.
- G. Sreenilayam, E. J. Moore, V. Steck and R. Fasan, Adv. Synth. Catal., 2017, 359, 2076–2089 CrossRef CAS PubMed.
- G. Sreenilayam, E. J. Moore, V. Steck and R. Fasan, ACS Catal., 2017, 7, 7629–7633 CrossRef CAS PubMed.
- P. Dydio, H. M. Key, A. Nazarenko, J. Y. Rha, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, Science, 2016, 354, 102–106 CrossRef CAS PubMed.
- K. Ohora, H. Meichin, L. M. Zhao, M. W. Wolf, A. Nakayama, J. Hasegawa, N. Lehnert and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 17265–17268 CrossRef PubMed.
- L. Villarino, K. E. Splan, E. Reddem, L. Alonso-Cotchico, C. G. de Souza, A. Lledos, J. D. Marechal, A. M. W. H. Thunnissen and G. Roelfes, Angew. Chem., Int. Ed., 2018, 57, 7785–7789 CrossRef PubMed.
- J. M. Zhao, D. G. Bachmann, M. Lenz, D. G. Gillingham and T. R. Ward, Catal. Sci. Technol., 2018, 8, 2294–2298 RSC.
- D. M. Carminati and R. Fasan, ACS Catal., 2019, 9, 9683–9697 CrossRef CAS PubMed.
- D. Seyferth and R. S. Marmor, Tetrahedron Lett., 1970, 2493–2496 CrossRef CAS.
- E. L. Rylott, R. G. Jackson, J. Edwards, G. L. Womack, H. M. B. Seth-Smith, D. A. Rathbone, S. E. Strand and N. C. Bruce, Nat. Biotechnol., 2006, 24, 216–219 CrossRef CAS PubMed.
- V. Steck, J. N. Kolev, X. Ren and R. Fasan, J. Am. Chem. Soc., 2020, 142, 10343–10357 CrossRef CAS PubMed.
- H. Tsutsumi, Y. Katsuyama, M. Izumikawa, M. Takagi, M. Fujie, N. Satoh, K. Shin-Ya and Y. Ohnishi, J. Am. Chem. Soc., 2018, 140, 6631–6639 CrossRef CAS PubMed.
- A. Tinoco, Y. Wei, J.-P. Bacik, D. M. Carminati, E. J. Moore, N. Ando, Y. Zhang and R. Fasan, ACS Catal., 2019, 9, 1514–1524 CrossRef CAS PubMed.
- R. Fasan, Y. T. Meharenna, C. D. Snow, T. L. Poulos and F. H. Arnold, J. Mol. Biol., 2008, 383, 1069–1080 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- J. T. Payne, C. B. Poor and J. C. Lewis, Angew. Chem., Int. Ed., 2015, 54, 4226–4230 CrossRef CAS PubMed.
- Y. Wei, A. Tinoco, V. Steck, R. Fasan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 1649–1662 CrossRef CAS PubMed.
- R. F. Alford, A. Leaver-Fay, J. R. Jeliazkov, M. J. O'Meara, F. P. DiMaio, H. Park, M. V. Shapovalov, P. D. Renfrew, V. K. Mulligan, K. Kappel, J. W. Labonte, M. S. Pacella, R. Bonneau, P. Bradley, R. L. Dunbrack, R. Das, D. Baker, B. Kuhlman, T. Kortemme and J. J. Gray, J. Chem. Theory Comput., 2017, 13, 3031–3048 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, synthetic procedures, additional MS spectra and LC-MS chromatograms, NMR spectra. CCDC [2105528]. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01965e |
| ‡ Current address: College of Engineering and Applied Sciences, Nanjing University, Nanjing, Jiangsu Province 210023, China. |
| This journal is © The Royal Society of Chemistry 2022 |