
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimulus-responsive self-assembled prodrugs in cancer therapy
Xiao
Dong
*a,
Rajeev K.
Brahma
b,
Chao
Fang
c and
Shao Q.
Yao
 *b
*b
aDepartment of Pharmacy, School of Medicine, Shanghai University, Shanghai 200444, China. E-mail: dong-xiao@shu.edu.cn
bDepartment of Chemistry, National University of Singapore, Singapore 117543, Singapore. E-mail: chmyaosq@nus.edu.sg
cState Key Laboratory of Oncogenes and Related Genes, Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China
First published on 18th March 2022
Abstract
Small-molecule prodrugs have become the main toolbox to improve the unfavorable physicochemical properties of potential therapeutic compounds in contemporary anti-cancer drug development. Many approved small-molecule prodrugs, however, still face key challenges in their pharmacokinetic (PK) and pharmacodynamic (PD) properties, thus severely restricting their further clinical applications. Self-assembled prodrugs thus emerged as they could take advantage of key benefits in both prodrug design and nanomedicine, so as to maximize drug loading, reduce premature leakage, and improve PK/PD parameters and targeting ability. Notably, temporally and spatially controlled release of drugs at cancerous sites could be achieved by encoding various activable linkers that are sensitive to chemical or biological stimuli in the tumor microenvironment (TME). In this review, we have comprehensively summarized the recent progress made in the development of single/multiple-stimulus-responsive self-assembled prodrugs for mono- and combinatorial therapy. A special focus was placed on various prodrug conjugation strategies (polymer–drug conjugates, drug–drug conjugates, etc.) that facilitated the engineering of self-assembled prodrugs, and various linker chemistries that enabled selective controlled release of active drugs at tumor sites. Furthermore, some polymeric nano-prodrugs that entered clinical trials have also been elaborated here. Finally, we have discussed the bottlenecks in the field of prodrug nanoassembly and offered potential solutions to overcome them. We believe that this review will provide a comprehensive reference for the rational design of effective prodrug nanoassemblies that have clinic translation potential.
 Xiao Dong | Xiao Dong received his PhD (2019) from the Shanghai Jiao Tong University School of Medicine, China, under the direction of Prof. Chao Fang. He worked as a Postdoctoral Research Fellow (2019–2021) in Prof. Shao Q. Yao's group at the National University of Singapore (NUS). He joined Shanghai University in 2022 and is currently an associate professor in the School of Medicine. His current research focuses on the development of supramolecular self-assembled nanomaterials for combinatorial cancer therapy. |
 Shao Q. Yao | Shao Q. Yao received his PhD from Purdue University (1998) and post-doctoral training (1998–2001) from the University of California, Berkeley and The Scripps Research Institute. He joined the National University of Singapore (NUS) in 2001 and is currently a Full Professor at the Department of Chemistry, NUS. His current research interest is in catalomics – the proteome-wide interrogation of enzymes and their associated molecules by using techniques in chemistry, biology and materials science. He is currently on the scientific advisory board of numerous international journals. |
1. Introduction
Cancer is one of the most lethal diseases that continues to affect hundreds of millions of patients in the world on a daily basis.1,2 The common cancer treatment paradigm includes chemotherapy, surgery, radiation therapy, and biological therapy.3–5 Amongst the various therapeutic options, cytoreductive chemotherapy that uses highly cytotoxic small-molecule drugs remains the dominant method for treatment of different cancers in the clinic, especially those in advanced or metastatic stages.6–8 Although much progress has been made in recent years in the fields of chemotherapy, small-molecule chemotherapeutics still have key drawbacks that need to be circumvented, including a narrow therapeutic index due to severe off-target effects and low drug bioavailability due to significant hurdles in absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties.9–11 Therefore, highly efficient tumor-localized delivery of chemotherapeutic agents with a temporal/spatial controlled-release ability (thus causing minimal exposure to non-diseased cells/tissues) has always been an important strategy in the field of drug delivery that needs to be transformative.Various strategies, including the use of prodrugs and physical encapsulation of drugs within nanostructures, have been exploited to address the aforementioned challenges associated with conventional small-molecule drugs (Fig. 1A and B).12–15 Following its introduction in 1958 by Albert, the concept of prodrugs remained the dominant approach to ameliorate the limitations in the ADMET properties of a parent drug without compromising its pharmacological activities (Fig. 1A).16–18 Between 2008 and 2017, prodrugs accounted for approximately 12% of all approved small-molecule drugs on the market.19 Despite the great progress that has been made in small-molecule prodrugs, they still suffer from challenges mainly associated with inherent small-molecule characteristics, including hepatic metabolism, short blood circulation time, fast renal clearance, premature activation, etc. Thus, the development of more innovative strategies is urgently needed.
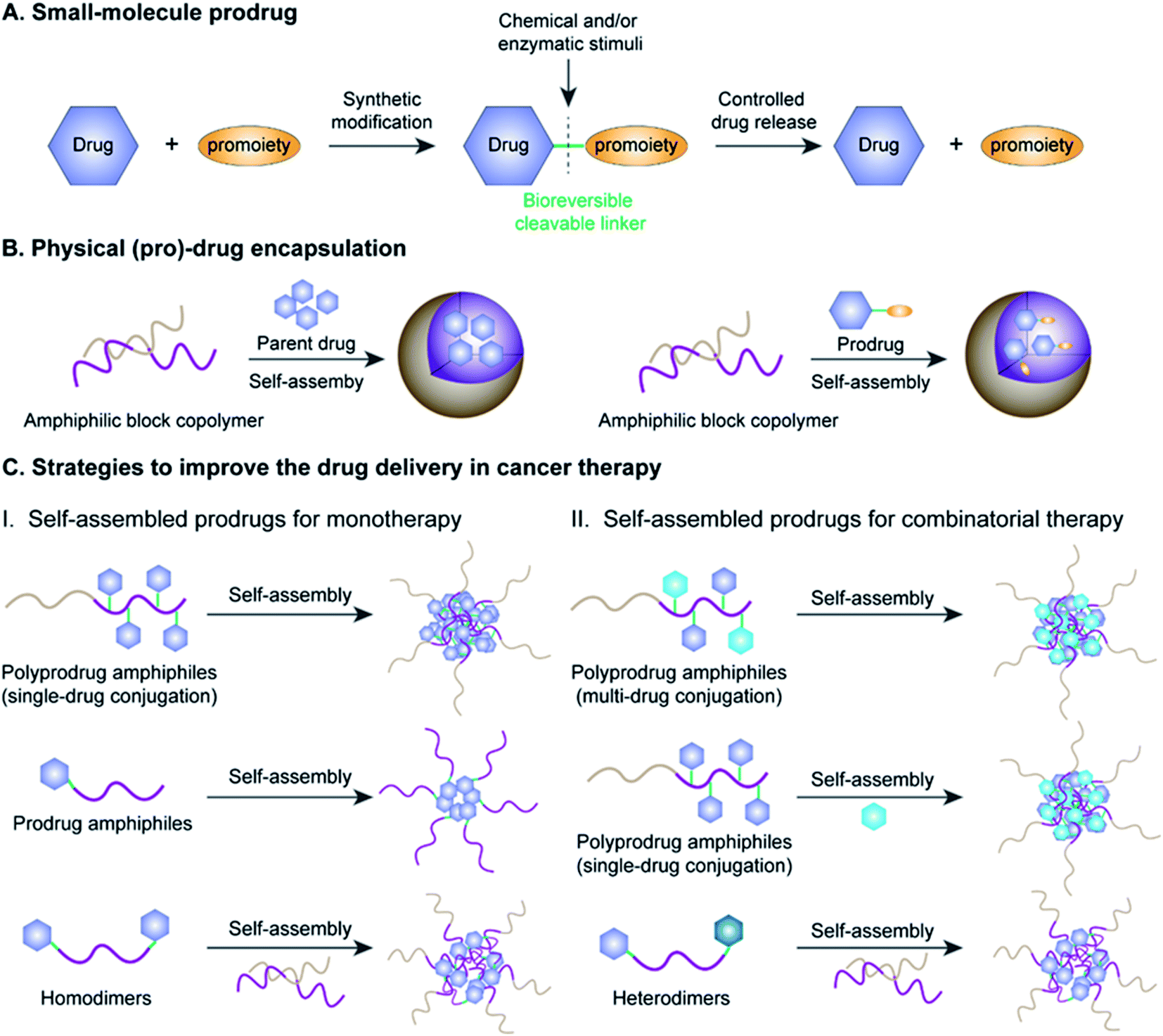 | ||
| Fig. 1 Schematic illustration of (A) the overall concept of small-molecule prodrug design; (B) physical loading of (pro)-drugs onto a nanoparticle (NP); (C) strategies to improve drug delivery efficiency for tumor (I) monotherapy or (II) combinatorial therapy by using different self-assembled NPs. | ||
Physical encapsulation of small-molecule (pro)-drugs within various organic nanocarriers (e.g. liposomes, micelles, polymers, nanogels, and dendrimers) or inorganic nanocarriers (e.g. silica nanocapsules) has recently emerged as a highly viable strategy to improve the ADMET properties of small-molecule (pro)-drugs in tumor therapy (Fig. 1B).20–24 Such nanocarriers effectively shield the small-molecule (pro)-drugs, resulting in an improvement in their stability, solubility, bioavailability and biodistribution.25 Notably, nanometer-sized vehicles can cause small-molecule (pro)-drugs to selectively accumulate near tumor tissues through the well-known effect of enhanced permeability and retention (EPR), which works by passively targeting the leaky tumor vascular system.26–28 These nanocarriers can additionally achieve active tumor targeting by surface modifications of the nanoparticle (NP) with suitable cancer-targeting ligands.29–31 Despite the many obvious benefits of these drug-loaded nanosystems, there still exist significant obstacles hindering their widespread clinical application, including low drug-loading capacity (<10% in most cases), premature drug release during blood circulation, bursting or poor drug release at target sites and carrier material-related biosafety issues.32–34
Fortunately with new advances in materials science and conjugation chemistry, self-assembled nanostructures of (poly)-prodrug amphiphiles or amphiphilized homo/heterodimers have emerged as a new paradigm for tumor-specific drug delivery.35–37 In recent years, various versatile stimulus-responsive prodrug nanoassemblies have been successfully developed for chemotherapy, phototherapy, immunotherapy, theranostics, and combinatorial therapy (Table 1). This review starts by introducing the concept of prodrugs, the design of prodrug nanoassemblies and commonly used methods in nano-prodrug preparation. Next, we summarize the rational design of self-assembled prodrugs on the basis of different conjugation approaches (i.e. polymer–drug conjugates, drug–drug conjugates, etc.) for tumor treatments. We emphasize the various approaches for controlled release of drugs based on innovative linker designs that exploit various tumor-associated stimuli (pH, ROS, GSH, enzyme, hypoxia, etc.; see Table 2). Finally, we discuss the various emerging challenges and future prospects of self-assembled prodrugs in relation to cancer therapy. Metal-catalysed nano-prodrug and prodrug-based hydrogels in tumor therapy, which have received increasing attention in recent years, are beyond the scope of this review. We expect that the inimitable and clear logical framework in this review will provide a comprehensive reference for readers, especially novice readers.
| Drug | Linkage | Therapy | Ref. |
|---|---|---|---|
| DOX | Hydrazone | Chemotherapy | 66, 68, 137, 138 and 148 |
| Chemotherapy & immunotherapy | 67 | ||
| Imine bond | Chemotherapy | 70 and 71 | |
| Enamine | Chemotherapy | 72 | |
| Dihydrazide | Chemotherapy | 82 | |
| Carbamate | Chemotherapy | 83 | |
| Anhydride | Chemotherapy | 84 | |
| Ketal | Chemotherapy & photodynamic therapy | 95 | |
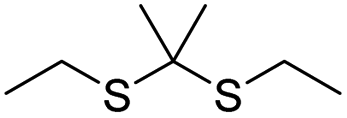
| Disulfide | Chemotherapy | 109 | |
| Thioether/disulfide/trisulfide | Chemotherapy | 115 | |
| Peptide | Chemotherapy, photodynamic therapy & immunotherapy | 126 | |
| Epacadostat | Peptide | Photothermo-immunotherapy | 125 |
| Epothilone B | Ketal | Chemotherapy | 93 |
| Paclitaxel | Ketal | Chemotherapy | 74 |
| Thioether | Chemotherapy & photodynamic therapy | 91 and 99 | |
| Chemotherapy & oxidation therapy | 92 and 100 | ||
| Disulfide | Chemotherapy & photodynamic therapy | 113 and 114 | |
| Azobenzene | Chemotherapy & photodynamic therapy | 132 | |
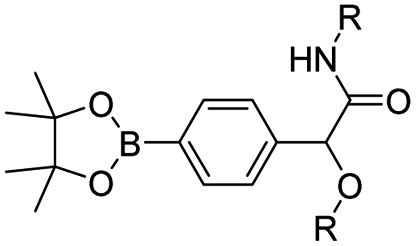
| Boronate ester | Chemotherapy & photodynamic therapy | 97 | |
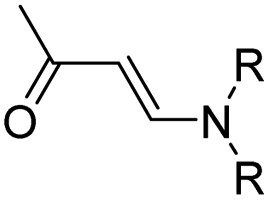
| Aminoacrylate | Chemotherapy & photodynamic therapy | 99 | |
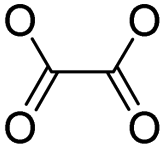
| Peroxalate ester | Chemotherapy, photodynamic therapy & photothermal therapy | 100 | |
| Peptide | Chemotherapy & photodynamic therapy | 124 | |
| Dithiomaleimide (DTM) | Chemotherapy & gene therapy | 139 | |
| Thioether/Disulfide/Selenoether/Diselenide | Chemotherapy | 113 | |
| Selenoether/Te/Thioether | Chemotherapy | 140 | |
| Gemcitabine | Ketal | Chemotherapy | 75 |
| Acetal | Chemotherapy | 76 | |
| Indomethacin | Ortho-ester | Chemotherapy | 85 |
| Capecitabine | Boronate esters | Theranostics | 93 |
| Bortezomib | Boronate esters | Chemotherapy | 79 and 81 |
| Boronic acid–salicylhydroxamate | Chemotherapy | 80 | |
| Camptothecin | Thioketal | Chemotherapy & photodynamic therapy | 90 and 103 |
| Chemotherapy & oxidation therapy | 94 | ||
| Disulfide | Chemotherapy | 110 and 135 | |
| Chemotherapy & photothermal therapy | 111 | ||
| Aspirin | p-Boronabenzyl ester | Immunotherapy | 96 |
| Gefitinib | Disulfide | Chemotherapy | 116 |
| Pt | Polyphenol–metal coordination | Chemotherapy | 136 |
| Cabazitaxel | Thioether | Chemotherapy & photodynamic therapy | 101 |
| Imidazoquinoline | Ester bond | Immunotherapy | 122 |
| IPM-Br | 2-Nitroimidazole | Chemotherapy & photodynamic therapy | 131 |
| Stimulus | Linkage | Chemical structure | Ref. |
|---|---|---|---|
| pH | Hydrazone |
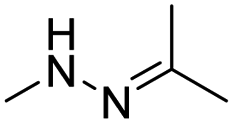
|
66–68, 137, 138 and 148 |
| Imine |
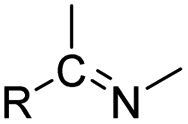
|
70 and 71 | |
| Enamine |
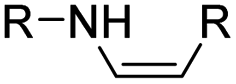
|
72 | |
| Ketal |
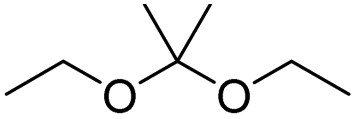
|
74 and 75 | |
| Acetal |
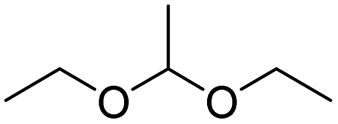
|
76 | |
| Boronate esters |
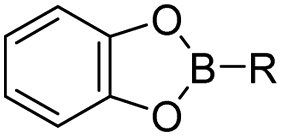
|
79 and 81 | |
| Boronic acid–salicylhydroxamate |
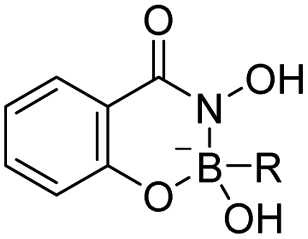
|
80 | |
| Dihydrazide |
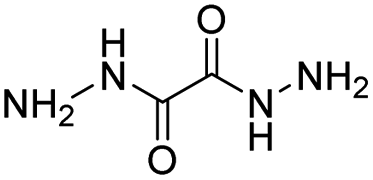
|
82 | |
| Carbamate |
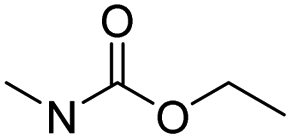
|
83 | |
| Anhydride |
|
84 | |
| Ortho-ester |
|
85 | |
| ROS | Thioketal |
|
90, 93–95 and 103 |
| Boronate ester |
|
96–98 | |
| Aminoacrylate |
|
99 | |
| Peroxalate ester |
|
100 | |
| GSH | Disulfide |
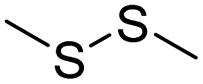
|
109–111, 113–116, 134 and 135 |
| Diselenide |
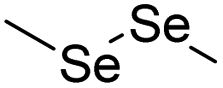
|
113 | |
| Selenoether |

|
113 and 140 | |
| Trisulfide |
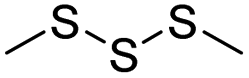
|
115 | |
| GSH/ROS | Thioether |
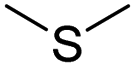
|
91, 92, 101, 102, 113, 115 and 140 |
| Te |

|
140 | |
| Enzyme | Ester bond |
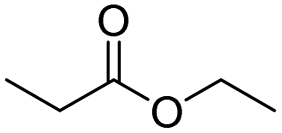
|
122 |
| Peptide | GL2/GFLG/PVGLLG/FRRG | 123–126 | |
| L-γ-glutamylglycine |
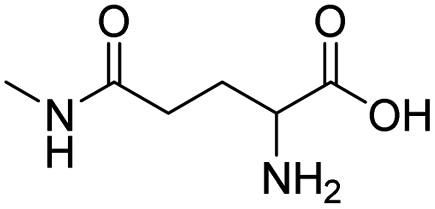
|
134 | |
| Hypoxia | 2-Nitroimidazole |
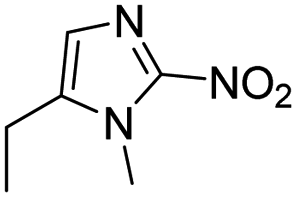
|
131 |
| Azobenzene |
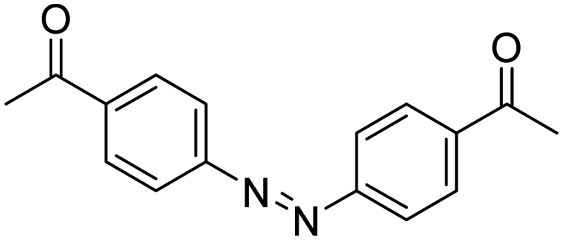
|
132 | |
| pH/ROS | Polyphenol–metal coordination |
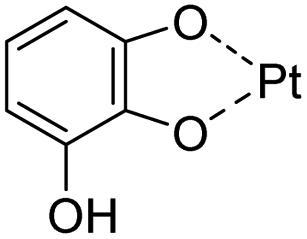
|
136 |
2. Concept and design of prodrug nanoassemblies
2.1 The prodrug concept
The concept of prodrugs was first defined by Adrien Albert in 1958, and has thus far played a transformative role in the development of contemporary anticancer drugs.38 Since then, various prodrug strategies have been used to address key barriers in anti-cancer drug development, such as undesirable physicochemical, pharmacokinetic (PK), and pharmacodynamic (PD) properties of potential compounds that have poor solubility and stability, low cell permeability, and severe adverse off-target cytotoxicity.19 As shown in Fig. 1A, prodrugs are bio-reversible derivatives of parent drugs that have little or no pharmacological activity, but can be subsequently transformed into active ones through one to two specific chemical or enzymatic steps. The inactive prodrugs are typically prepared by reversibly introducing a pro-moiety into the parent drug via a stimulus-responsive linker which could be tailored to directly respond to physical, chemical or biological stimuli (such as acidic pH, hypoxia, aberrant enzyme expression, and high oxidation and reduction states) present in the tumor microenvironment (TME).39–42 On the basis of this, stimulus-responsive prodrugs delivered to the TME may maintain their overall antitumor effects while possessing minimized off-target cytotoxicity towards normal tissues.Significantly, there are several prerequisites for a successful prodrug design: (1) parent drugs should have functional groups that are suitable for chemical modifications; (2) favorable pro-moieties that can improve the deficient ADMET properties of the parent drug; (3) a reversible linkage between the parent drug and the pro-moiety that allows stimuli-responsive controlled-release of drugs without formation of cytotoxic byproducts.43 Various functional groups in a parent drug, including carboxyl, hydroxyl, amine and carbonyl, can be reversibly transformed into the corresponding ester, carbonate, carbamate, amide, and oxime, respectively. The pro-moiety generally dictates the chemical identity and can improve the ADMET properties of the parent drug.44,45 For example, lipidation is an effective approach to enhance the membrane permeability of a hydrophilic drug.46 Conversely, the aqueous solubility of a hydrophobic drug may be improved by introducing an ionizable pro-moiety.47
2.2 Design of prodrug nanoassemblies
Amphiphilic prodrugs containing both hydrophilic and hydrophobic domains could self-assemble into homogeneous NPs in an appropriate solution.48,49 Notably, according to their molecular weights, amphiphilic prodrugs could be broadly divided into two categories, namely polymer–drug conjugates ((poly)-prodrug amphiphiles) and drug–drug conjugates (amphiphilized homo/heterodimeric prodrugs).50 As illustrated in Fig. 1C, the amphiphilic polymer–drug conjugates were synthesized by attaching single or multiple parent drugs onto carrier materials of synthetic (block polymers) or biological origin (peptides or oligonucleotides) via covalent stimulus-labile linkages.51 The resultant (poly)-prodrug amphiphiles exhibit a superior self-assembly ability to form into nano-prodrugs for mono- or combinatorial tumor therapy (Fig. 1C).52 In contrast, prodrug amphiphiles could be obtained by reversibly conjugating drugs to the terminus of carrier materials possessing an opposite solubility to their parent drugs.Due to the high content of polymers used, (poly)-prodrug amphiphile-based nanoassemblies still possess a low drug loading capacity. Although most of the currently FDA-approved nanomedicines use inert biomaterials as drug carriers, they still elicit significant adverse effects. Therefore, the development of carrier-free nano-prodrugs through the self-assembly of prodrugs themselves has become necessary. One example is shown in Fig. 1C, in which homo- or heterodimeric drug–drug conjugates were synthesized by conjugating two identical or different drug molecules via a biodegradable linker, respectively.53 The resultant drug–drug conjugate-based nanoassemblies possessed a high drug loading capacity capable of minimizing excipient-triggered adverse toxicity. Therefore, by taking advantage of the benefits in both prodrug design strategies and nanomedicine, these prodrug nanoassemblies could possess key properties including improved systemic stability, prolonged blood circulation, and passive/active targeting ability, as well as potential for application in theranostics and combination therapy. More importantly, by properly encoding suitable linker chemistries, the resulting self-assembled prodrugs could be tailored for tumor-specific temporal/spatial controlled-release of drugs in response to chemical or biological stimuli present at the tumor sites.54,55
2.3 Common methods for preparation of self-assembled prodrugs
Molecular assembly and kinetic trapping are the commonly used approaches to fabricate prodrug-based nanoassemblies (Fig. 2).56,57 Molecular self-assembly is a spontaneous organization of molecules or parts of molecules into stable and ordered aggregates under equilibrium conditions through non-covalent bonds, and without any guidance from outside forces (Fig. 2A).58 Such a self-assembly process is based on the balance of attractive and repulsive forces including π–π stacking, hydrogen bonding, van der Waals and Coulomb interactions, which occur under the premise that mobile molecules encounter each other in a solution.59 Thus, the structural features of self-assembled prodrugs can be tailored by adjusting the assembly conditions such as pH, temperature, and ionic strength. Kinetic trapping, also termed nanoprecipitation, is another method to prepare self-assembled nanostructures, in which the monomer (i.e. an amphiphilic prodrug conjugate) dissolved in a small amount of organic solvent is mixed with a large amount of aqueous solution under external forces (e.g. extensive stirring). Subsequent removal of the organic solvent leads to spontaneous formation of homogeneous nanostructures (Fig. 2B).60 During this process, the hydrophilic components form the outer shell and the hydrophobic components form the inner core of the resulting NPs. In contrast to NPs obtained from molecular assembly, nanostructures made by kinetic trapping have a disordered internal component forced together by hydrophobic–hydrophilic interactions.61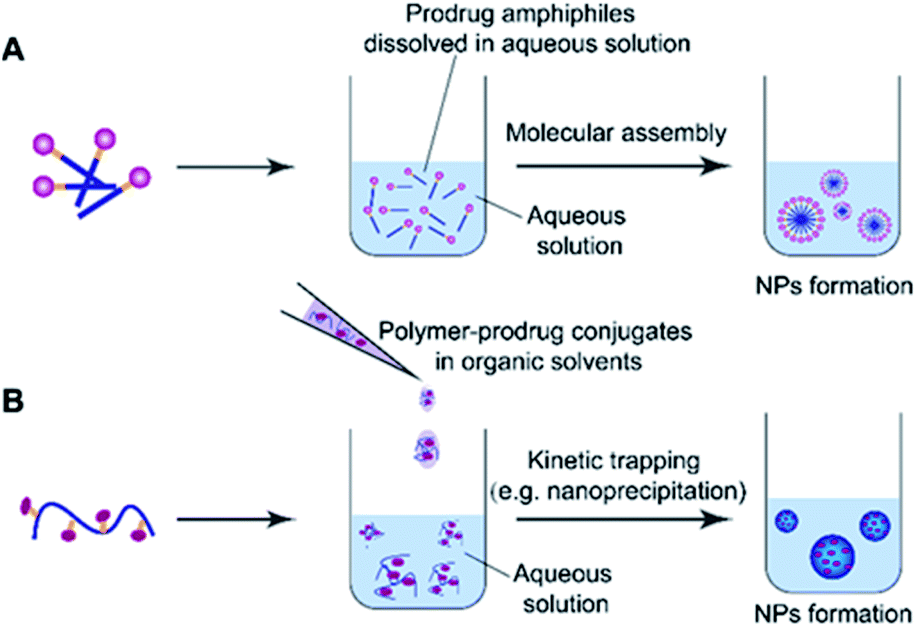 | ||
| Fig. 2 Schematic illustration of commonly used self-assembly methods for nano-prodrug preparation. (A) Nanoparticle (NP) formation induced by various non-covalent interactions including π–π stacking, hydrogen bonding, van der Waals and Coulomb interactions. (B) Nanoprecipitation-mediated NP formation by kinetic trapping with hydrophobic–hydrophilic interactions. | ||
3. pH-responsive nanoassemblies
Aberrant metabolism of tumor cells creates an acidic TME with pH 6.5–6.9, which is more acidic than normal physiological pH (∼7.4) in the blood and normal tissues.62,63 Furthermore, most NP-based drug delivery systems can enter into tumor cells via endocytosis and pass by the intracellular endosomes (pH = 5–6.5) and lysosomes (pH = 4.5–5), before being released into the less acidic cytosolic environment (pH = 7.4).64 Therefore, the significant pH difference that exists in extracellular and intracellular environments could be exploited as an inherent trigger for specific drug release at tumor sites, thus minimizing premature drug leakage during blood circulation or in normal tissues. In this section, we review several acid-sensitive linkers that were commonly used in the design of acid-labile nano-assembled prodrug conjugates, such as those based on the linker chemistries of hydrazines, imines and acetals.3.1 Polymer–drug conjugate-based nanoassemblies
The hydrazone bond is a commonly used moiety for pH-sensitive prodrug design.65 With the acidic TME in mind, Zhang and co-workers reported an acid-sensitive, dextran (DEX) polysaccharide-based amphiphilic poly-prodrug conjugate named DOM@DOX (i.e. DEX-POEGMA-b-PDOX) for improved chemotherapy (Table 1).66 As shown in Fig. 3A, to synthesize DOM@DOX with a bottlebrush-architecture, a dextran (DEX) polysaccharide was connected with hydrophilic poly(ethylene glycol)methyl ether methacrylate (POEGMA) through atom transfer radical polymerization (ATRP). After that, doxorubicin (DOX) was attached to the backbone of the obtained copolymer via the acid-labile hydrazone bond (Table 2), thus enabling pH-triggered drug release in the mildly acidic TME. In this study, dextrans of different molecular weights (Mn 1000 and Mn 5000) were used for poly-prodrug synthesis. The prepared amphiphilic dextran poly-prodrugs (DOM1000@DOX and DOM5000@DOX) were utilized to form prodrug micelles with hydrodynamic sizes of 37.56 and 49.26 nm in aqueous solution, respectively. The prepared nano-prodrugs exhibited prominent tumor inhibition and minimal systemic side effects. This approach was also explored by Chen and co-workers, who developed a carrier-free, pH-responsive nano-prodrug (PEG@D:siRNA) by co-self-assembly of PEGylated DOX (PEG-hyd-DOX) and a small interfering RNA (siRNA) against PD-L1 via cooperative π–π stacking and electrostatic interactions (Fig. 3B). In this study, the author demonstrated that PEG@D:siRNA could simultaneously induce immunogenic cell death (ICD) and suppress DOX-mediated upregulation of the immunosuppression pathway in targeted tumor cells, thus enabling efficient combinatorial chemo-immunotherapy (Table 1).67 To facilitate the pH-triggered drug release, the PEG-hyd-DOX was synthesized by connecting DOX with PEG through the acid-sensitive hydrazone bond (Table 2). It was shown that PEG@D:siRNA had a hydrodynamic size ranging from 25 nm to 45 nm and possessed a high cargo loading capacity of DOX (21.67%) and siRNA (4.13%), respectively. Upon successful entry into the targeted tumor cells, the DOX could be rapidly released due to the hydrolysis of hydrazone bonds inside acidic endosomes/lysosomes, leading to the eventual induction of ICD and stimulation of antitumor immune response. In the meantime, the released siRNA could inhibit the immunosuppressive gene PD-L1 to further strengthen the ICD-mediated antitumor immune response. The prepared PEG@D:siRNA NP that carried a PD-L1-targeting siRNA (named PEG@D:siPD-L1) could lead to an approximately 70% decrease of PD-L1 expression in CT26 cells. As expected, this nano-prodrug showed a superior tumor regression in two different tumor models (CT26 and Panc02). In a different study, Song et al. described a pH-sensitive micellar DOX prodrug for chemotherapy (Table 1).68 The nano-prodrug was constructed by self-assembly of an amphiphilic PEGylated DOX prodrug (PEG-Schiff-DOX) in aqueous solution (Fig. 3C). The PEG-Schiff-DOX having a low critical micelle concentration (CMC) value (0.018 mg mL−1) was synthesized by conjugating PEG with DOX through the acid-sensitive hydrazone bond (Table 2). These micellar prodrugs with an average z-size of 85 nm and a high drug loading capacity (∼38.1%) could maintain their structural integrity and avoid premature drug leakage under physiological conditions (pH = 7.4). Notably, the micelles could be disassembled inside acidic endosomes/lysosomes due to the cleavage of hydrazone bonds, thus leading to an efficient drug release (∼82.2%). The authors further showed that the DOX encapsulated in PEG-Schiff-DOX NPs could be programmed to achieve different drug release rates to maintain effective treatment over a longer period at tumor sites.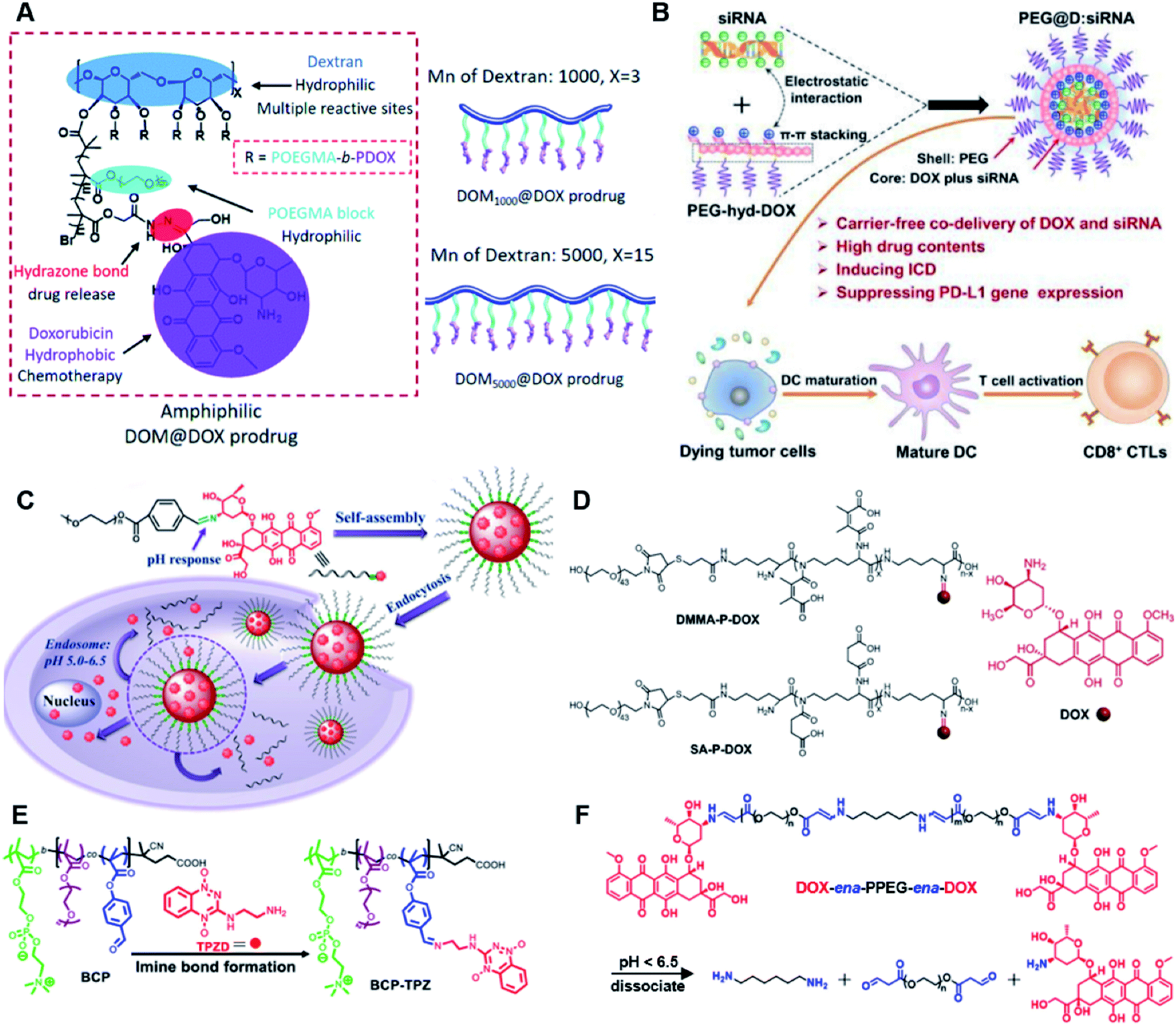 | ||
| Fig. 3 (A) Schematic illustration of the chemical structures of DOM@DOX and its self-assemblies. Reproduced from ref. 66 with permission from Royal Society of Chemistry, Copyright 2020. (B) Illustration of the fabrication of carrier-free, self-assembled prodrug (PEG@D:siRNA) to synergistically induce ICD and reverse tumor immunosuppression. Reproduced from ref. 67 with permission from Elsevier Ltd, Copyright 2020. (C) Synthesis of PEG-Schiff-DOX and the acid-sensitive intracellular drug release. Reproduced from ref. 68 with permission from Frontiers Media SA, Copyright 2021. (D) Schematic illustration of the preparation of DMMA-P-DOX and SA-P-DOX. Reproduced from ref. 70 with permission from Elsevier Ltd, Copyright 2020. (E) Synthesis of zwitterionic BCP-TPZ prodrug conjugates. Reproduced from ref. 71 with permission from American Chemical Society, Copyright 2021. (F) Chemical structure of acid-responsive DOX-ena-PPEG-ena-DOX and its drug activation mechanism. Reproduced from ref. 72 with permission from American Chemical Society, Copyright 2019. | ||
The imine bond is another promising linker for pH-triggered release of drugs due to its acid-labile properties.69 Recently, Guo et al. prepared a dual pH-responsive polymeric nano-prodrug (DMMA-P-DOX/LAP) with a charge-switchable ability for synergistic cancer therapy (Table 1).70 To synthesize 2,3-dimethylmaleic anhydride (DMMA)-P-DOX, as shown in Fig. 3D, DOX was first linked to the side chain of PEGylated ε-poly-L-lysine (EPLYS) via an imine bond to ensure a pH-triggered release of DOX (Table 2). SA-P-DOX was synthesized as a control. The CMC values of DMMA-P-DOX and SA-P-DOX were calculated to be 0.0069 mg mL−1 and 0.0057 mg mL−1, respectively. Such low CMC values enabled the corresponding nano-prodrugs to maintain an integral nanostructure during the blood circulation. The DMMA-P-DOX/LAP NPs were subsequently prepared by physical encapsulation of lapatinib (LAP), a dual tyrosine kinase inhibitor, within (DMMA-P-DOX)-based nanoassemblies. The hydrodynamic size of DMMA-P-DOX/LAP was determined to be 86.92 ± 1.82 nm, which was larger than those of DMMA-P-DOX (34.41 ± 2.97 nm) and SA-P-DOX (30.42 ± 4.11 nm). The positive charge of residual amino groups on the NP surface was shielded by modification of DMMA to further prolong its blood circulation time. Upon successful accumulation in the mildly acidic TME, the negative surface charge of the NP was reversed to positive, leading to an improved cell internalization and deep tumor penetration. The hydrolysis of imine bonds under low intracellular pH conditions could induce the disassembly of DMMA-P-DOX/LAP NPs, thus leading to the concurrent release of DOX and LAP in the cytosol for synergistic tumor therapy. Zhao et al. developed another zwitterionic block copolymer (BCP)-based micellar prodrug for pH-sensitive release of hypoxia-responsive prodrugs (Fig. 3E).71 The zwitterionic BCP-containing prodrug micelles were fabricated by self-assembly of PMPC-b-P(DEGMA-co-FPMA-g-TPZD) (BCP-TPZ), in which tirapazamine (TPZ) derivatives (named TPZD) were first conjugated onto PMPC-b-P(DEGMA-co-FPMA) through acid-labile imine bonds to facilitate pH-triggered drug release under acidic conditions (Table 2). By using pyrene as a fluorescence probe, the CMC values of BCP and BCP-TPZ were calculated to be 0.0608 and 0.0277 mg mL−1, respectively. The hydrodynamic size of BCP-TPZ micelles in PBS was determined to be 51–59 nm by DLS, which was larger than the size of BCP-only micelles (∼25 nm). The resulting micellar prodrugs maintained a good colloidal stability in PBS and DEME (with 10% FBS). In vitro TPZD release at different pH conditions of 7.4, 6.5 and 5.5 within 50 h was estimated to be 30%, 60% and 90%, respectively. As expected, the micellar BCP-TPZ exhibited at least 13.7-fold higher hypoxia-selective cytotoxicity against both HeLa and HepG2 cancer cells. In another study, Li and co-workers synthesized a pH-responsive polymeric prodrug (DOX-ena-PPEG-ena-DOX) that could self-assemble into homogeneous NPs with a good water solubility and biocompatibility (Fig. 3F).72 To obtain DOX-ena-PPEG-ena-DOX, the acid-sensitive alkynyl-terminated block polymer (A-P(PEG-alt-HMDA)-A) was synthesized by alternately connecting polyethylene glycol (PEG) with hexamethylenediamino (HMDA) via enamine bonds (-ena-). Subsequently, the corresponding polymer–prodrug conjugate (DOX-ena-PPEG-ena-DOX) was synthesized by attaching DOX to A-P(PEG-alt-HMDA)-A via amino–yne click chemistry. The resulting nano-prodrug had an average hydrodynamic size of 140 nm capable of causing ∼16.1% drug release at pH = 7.4 after 118 h of incubation. In contrast, approximately 85.0% of drug release was detected at pH = 5 (which mimicked the intracellular lysosome/endosome conditions), presumably caused by cleavage of the acid-labile enamine bond (Table 2).
Similarly, acetals or ketals are also promising linkers for the development of acid-responsive prodrugs.73 Mu and co-workers reported pH-sensitive polymeric paclitaxel (PTX) prodrugs (named PKPs) for ovarian cancer therapy (Table 1).74 As shown in Fig. 4A, in order to synthesize PKPs, the intermediate compound 2 containing acrylate groups was synthesized by linking PTX with compound 1via the acid-sensitive acetone-based acyclic ketal linker (Table 2). After that, compound 2 was subsequently conjugated with different lengths of poly(ethylene glycol) (PEG) to obtain PKPs, which formed into uniform micelles upon nanoprecipitation. The obtained micellar prodrug remained stable in the normal physiological environment but achieved accelerated drug release upon hydrolysis of the acid-labile bonds within the mildly acidic TME. Notably, the authors found that the molecular weight of PEG could affect the CMC values and the particle size of PKPs. The shorter PEGs could endow PKPs with favourable properties including smaller NP sizes, lower CMC values, prolonged circulation time, high accumulation at the tumor site and better antitumor activities. The particle sizes of various NPs (named PKP750, PKP1000, and PKP2000) determined by DLS were about 13, 22, and 48 nm, respectively. In addition, the author investigated the hydrolysis kinetics of PKPs at different pH conditions (7.4, 6.8 and 5.0). After 24 h incubation, release of PTX from PKP750, PKP1000, and PKP2000 NPs reached about 5.4%, 10.5%, and 11.4% at pH = 7.4, respectively (Fig. 4B). In contrast, higher degrees of PTX release were observed for the same PKPs (25.5%, 37.7% and 45.3%, respectively) at pH = 6.8. Finally, nearly complete release of PTX was observed for all three types of PKPs at pH = 5.0 after only 4 h incubation. In another study, Zhong et al. synthesized acid-activable gemcitabine (Gem)–polyketal conjugates (PKGems) by means of Michael addition polymerization of dithiol and diacryl-modified Gem.75 As shown in Fig. 4C, Gem was introduced onto a polyketal backbone via an acid-labile ketal linkage (Table 2), which facilitated the pH-triggered drug release under the acidic TME and intracellular endolysosomal conditions. The resulting hydrophobic PKGems were co-polymerized with mPEG-PDLLA to form a nano-prodrug (named GKNPs) that exhibited a good stability with minimal drug leakage at physiological pH = 7.4, but achieved a rapid drug release within the acidic TME. Consequently, these GKNPs showed a better antitumor effect than free gemcitabine in the ovarian tumor animal model implanted with A2780 cancer cells. Takemoto et al. reported a GEM-conjugated polymer (PGEM) for improved drug solubility and pharmacological activity of GEM.76 As shown in Fig. 4D, PGEM was synthesized by conjugating the native drug GEM onto the side chains of a poly(amino acid) through a cyclic acetal linkage. The obtained PGEM was able to self-assemble into a nano-prodrug that had a hydrodynamic size of 9.90 ± 0.97 nm in PBS. In comparison to Doxil (ca. 80 nm), the PGEM could achieve a deep homogeneous intratumor distribution in tumor tissues due to its smaller size. The PGEM system also showed a minimal drug leakage due to the stability of the 1,3-dioxane linkage under physiological conditions (pH = 7.4), thus causing negligible GEM-derived off-target effects. Upon entering into tumor cells via endocytosis, the encapsulated GEM was rapidly released due to hydrolysis of the 1,3-dioxane linkage in response to the acidic luminal pH in the endosomes/lysosomes. In addition, this PGEM system possessed a prolonged blood circulation time and exhibited a higher antitumor effect in two distinct pancreatic tumor models.
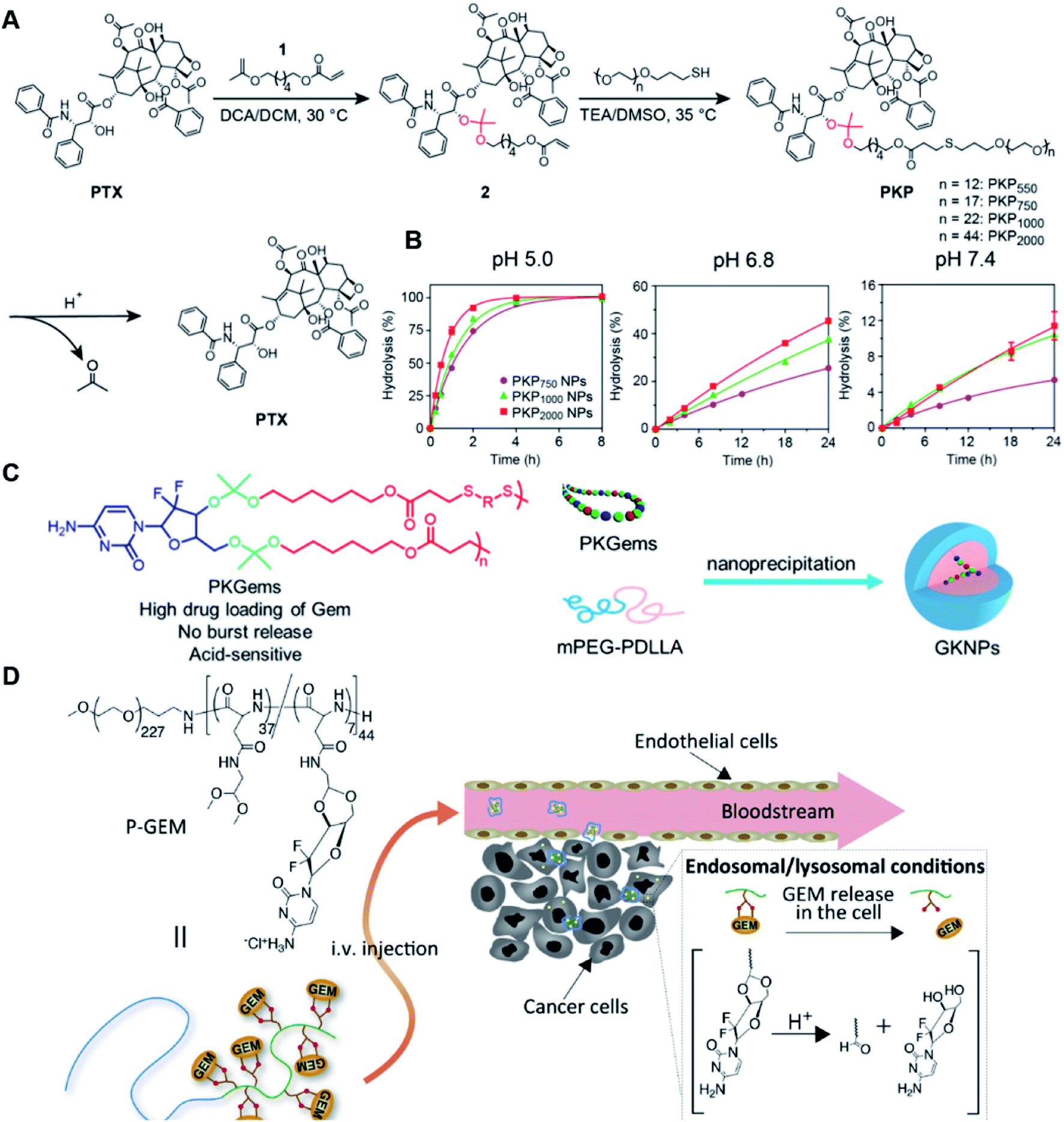 | ||
| Fig. 4 (A) Illustration of the design and self-assembly of pH-responsive PEGylated paclitaxel prodrugs (PKPs). (B) PTX release from PKP750, PKP1000 and PKP2000 NPs after 24 h incubation at pH 7.4, 6.8 and 5, respectively. Reproduced from ref. 74 with permission from Elsevier Ltd, Copyright 2020. (C) Molecular structures of pH-responsive gemcitabine (Gem)–polyketal conjugates (PKGems) and nanoparticle formation by nanoprecipitation. Reproduced from ref. 75 with permission from American Chemical Society, Copyright 2020. (D) Chemical design of polymer–GEM conjugates and acid-responsive drug release mechanism under endolysosomal conditions. Reproduced from ref. 76 with permission from Elsevier Ltd, Copyright 2020. | ||
The chelation between boronic acid and other reactive functional groups (such as hydroxyl or amino groups) could be used as a self-immolative linker for traceless release of some boric acid- or amine-containing drugs/proteins.77,78 Recently, Hu et al. designed a pH-activable dendrimeric bortezomib (BTZ) prodrug to improve the stability of BTZ during blood circulation and minimize its system toxicity.79 To prepare this dendrimeric BTZ prodrug, an amphiphilic PEGylated dendrimer linked with dopamine was synthesized by conjugating azide-functionalized polyethylene glycol with an alkyne-functionalized dendrimer, followed by attachment of dopamine (Fig. 5A). The resulting BTZ drug could be subsequently introduced onto the PEGylated dendrimer by virtue of the high reactivity between boric anhydride on BTZ and dopamine, leading to formation of reversible drug conjugates (Fig. 5B and Table 2). At low pH conditions, the conjugation between dopamine and BTZ was weakened, thus resulting in selective release of BTZ in the acidic TME. The obtained amphiphilic PEGylated dendrimer loaded with BTZ was next formulated into NPs (named c(RGDyK)-NPs/BTZ) with additional filling agents including c(RGDyK)-PEG3400-DSPE and PEG-PLGA. Transmission electron microscopy (TEM) images of the resulting NPs showed a size distribution of ∼100 nm. The encapsulation efficiency of BTZ in the c(RGDyK)-NPs/BTZ was estimated to be above 95%. The obtained c(RGDyK)-NPs/BTZ was shown to possess an improved serum stability and reduced system toxicity than the free BTZ. Due to the existence of c(RGDyK), this nano-prodrug could significantly improve the tumor-targeting ability. When compared to the PBS or free BTZ-treated group, the tumor growth in both NPs/BTZ- and c(RGDyK)-NPs/BTZ-treated groups was significantly suppressed. Notably, cRGDyK modification could further improve the therapeutic efficacy of the NPs/BTZ. More importantly, this design can be readily applied for preparation of similar nano-drugs from well-known chemotherapeutics such as ixazomib and cisplatin. In a similar study, Liu et al. developed an efficient method for preparation of pH-sensitive polymer–drug conjugates (P1-BTZ) based on the ultrafast and reversible boronic acid–salicylhydroxamate click reaction (Fig. 5C).80 To prepare P1-BTZ, the salicylhydroxamate-bearing polymer (P1) composed of salicylhyroxymate and PEG-methacrylate monomers was first synthesized through an initial RAFT co-polymerization. Due to the rapid condensation between boronic acid and salicylhydroxamate, the resultant P1 could be used for pH-responsive delivery of drugs that have an intrinsic boronic acid group. In this study, the FDA-approved bortezomib (BTZ) was used as a model drug to test the utility of such strategies. The condensation between boronic acid and salicylhydroxamate subsequently induced in situ self-assembly of the polymer–drug conjugate. The P1-BTZ conjugate, but not P1 alone, was shown to self-assemble into uniform NPs with a hydrodynamic size of about 50 nm. As expected, pH-dependent release of bortezomib from this nanoassembly was observed due to the reversibility of the boronic acid–salicylhydroxamate linkage under mildly acidic conditions (Fig. 5C). Similarly, P1 could also be applicable for drugs that were pre-linked with arylboronic acid by using a self-immolative linker. In another study, Ma and co-workers developed a biomimetic peptide (BTZ-PEP-RGD)-based nano-prodrug for pH-triggered release of BTZ selectively at tumor sites (Table 2).81 To obtain BTZ-PEP-RGD, as shown in Fig. 5D, the dye-labeled biomimetic PEP-RGD (FITC-(DOPA)4-G5-RGDS) was synthesized by inserting a non-bioactive quintuple glycine (G5) spacer between the cancer-targeting ligand RGDS and fluorescein isothiocyanate (FITC)-linked tetrapeptide (DOPA)4 bearing catechol groups. The catechol groups were used for dynamic conjugation of BTZ through formation of pH-sensitive boronic acid–catechol esters. The obtained BTZ-PEP-RGD could self-assemble into homogeneous NPs with a small size (173 ± 34 nm) and high drug-loading content (37.4 wt%). The resulting nano-prodrug showed a fast drug release in acidic endosomes/lysosomes (pH 5.0–6.0) due to the rapid dissociation of the catechol–BA linkage, and a significantly higher antitumor activity when compared to free drugs.
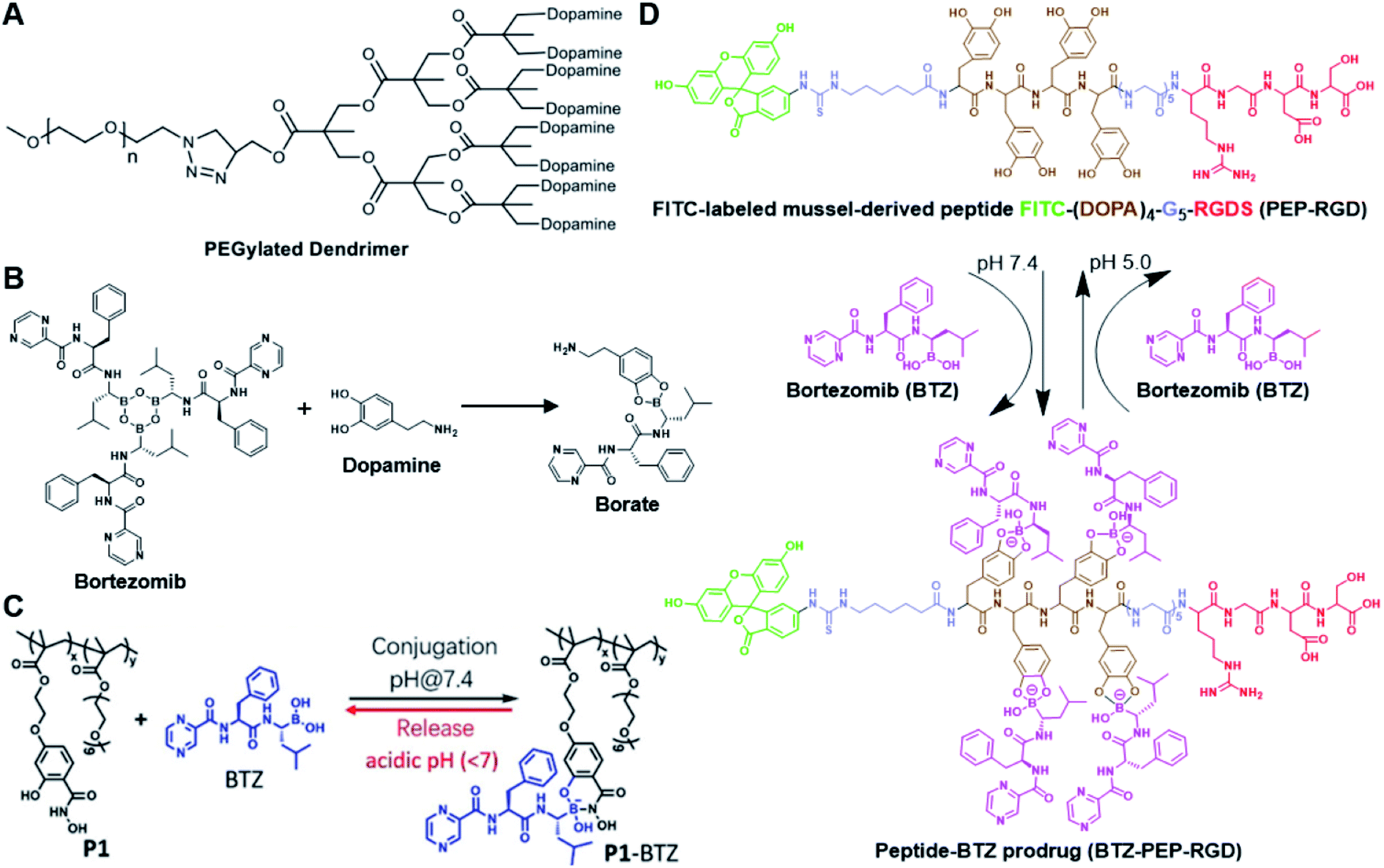 | ||
| Fig. 5 (A) Chemical structure of the dopamine-linked amphiphilic PEGylated dendrimer; (B) reaction between dopamine and boric acid on BTZ. Reproduced from ref. 79 with permission from Wiley-VCH, Copyright 2020. (C) Synthesis of the polymeric BTZ prodrug (P1-BTZ) and mechanism of pH-triggered drug release. Reproduced from ref. 80 with permission from Wiley-VCH, Copyright 2020. (D) Illustration of the pH-responsive dynamic conjugation between the mussel-derived cancer-targeting peptide and BTZ. Reproduced from ref. 81 with permission from American Chemical Society, Copyright 2019. | ||
3.2 Dimeric prodrug-based nanoassembly
Self-assembly of dimeric prodrugs has attracted much attention due to its high drug loading capacity and the less use of carrier materials. Recently, Li et al. reported a doxorubicin dimer-based nano-prodrug for pH-triggered drug release.82 As shown in Fig. 6A, doxorubicin dimer-based surfactants, DOX-ADH-DOX-PEG and ADH-(DOX-PEG)2, were synthesized by conjugating two molecules of DOX via an adipic acid dihydrazide (ADH) linker, followed by mono-PEGylation and bi-PEGylation, respectively. The resulting DOX-ADH-DOX-PEG and ADH-(DOX-PEG)2 could self-assemble into NPs with hydrodynamic sizes of 142–163 nm and 157–225 nm, respectively. The author found that the prodrug surfactant with mono-PEGylation had higher drug-loading capacity (51%) when compared to that with bi-PEGylation, which was probably due to the lower PEG content. The obtained PEGylated nano-prodrug showed a minimal drug leakage (7%) under normal physiological conditions (pH 7.4), but could release DOX completely in the tumor intracellular microenvironment (pH = 5.0). In another study, Li et al. synthesized a self-assembled homodimeric prodrug (D-DOXcar) for slow intracellular release of doxorubicin under acid conditions.83 The D-DOXcar was prepared by inserting an acid-sensitive carbamate linker (NC–NC) between two molecules of DOX (Fig. 6B). The NC–NC linker was synthesized by connecting 1,5-pentanediol with 4-nitrophenyl chloroformate (4-NPC). The obtained D-DOXcar self-assembled into NPs (D-DOXcar-NP) by nanoprecipitation. In this study, different nano-prodrugs (D-DOXcar-bNP and D-DOXcar-sNP) with different hydrodynamic sizes (180 nm and 95 nm, respectively) possessed a high drug-loading capacity of 86%. The author found that the pH-responsive slow drug release was dependent on the particle size and concentration. Larger-size DOXcar-NPs showed a slightly higher cumulative release than smaller-size NPs at the same concentration. Notably, higher cumulative drug release for D-DOXcar-sNP could be obtained at a lower concentration at pH 5.0 in PBS within 60 h. Similarly, Li et al. prepared another doxorubicin–doxorubicin conjugate (D-DOX)-based nanoassembly to minimize premature drug release and improve the antitumor effect (Fig. 6C).84 The acid-labile anhydride-linked D-DOXMAH was synthesized by amidation of DOX with different anhydrides (MAH, suc, and 2,3-dimethylmaleic anhydride (DMMAH)). The PEGylated D-DOXMAH was subsequently synthesized through a thiol–ene addition reaction. The obtained D-DOXMAH and amphiphilic PEGylated D-DOXMAH were shown to co-self-assemble into NPs (∼162 nm) at a feed ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. The obtained D-DOXMAH-S-PEG-based nanoassemblies with a high drug-loading capacity (51.2%) exhibited low premature drug leakage (∼4.5%) at pH = 7.4 but achieved 40.6% of cumulative DOX release under acidic intracellular tumor conditions.
:5. The obtained D-DOXMAH-S-PEG-based nanoassemblies with a high drug-loading capacity (51.2%) exhibited low premature drug leakage (∼4.5%) at pH = 7.4 but achieved 40.6% of cumulative DOX release under acidic intracellular tumor conditions.
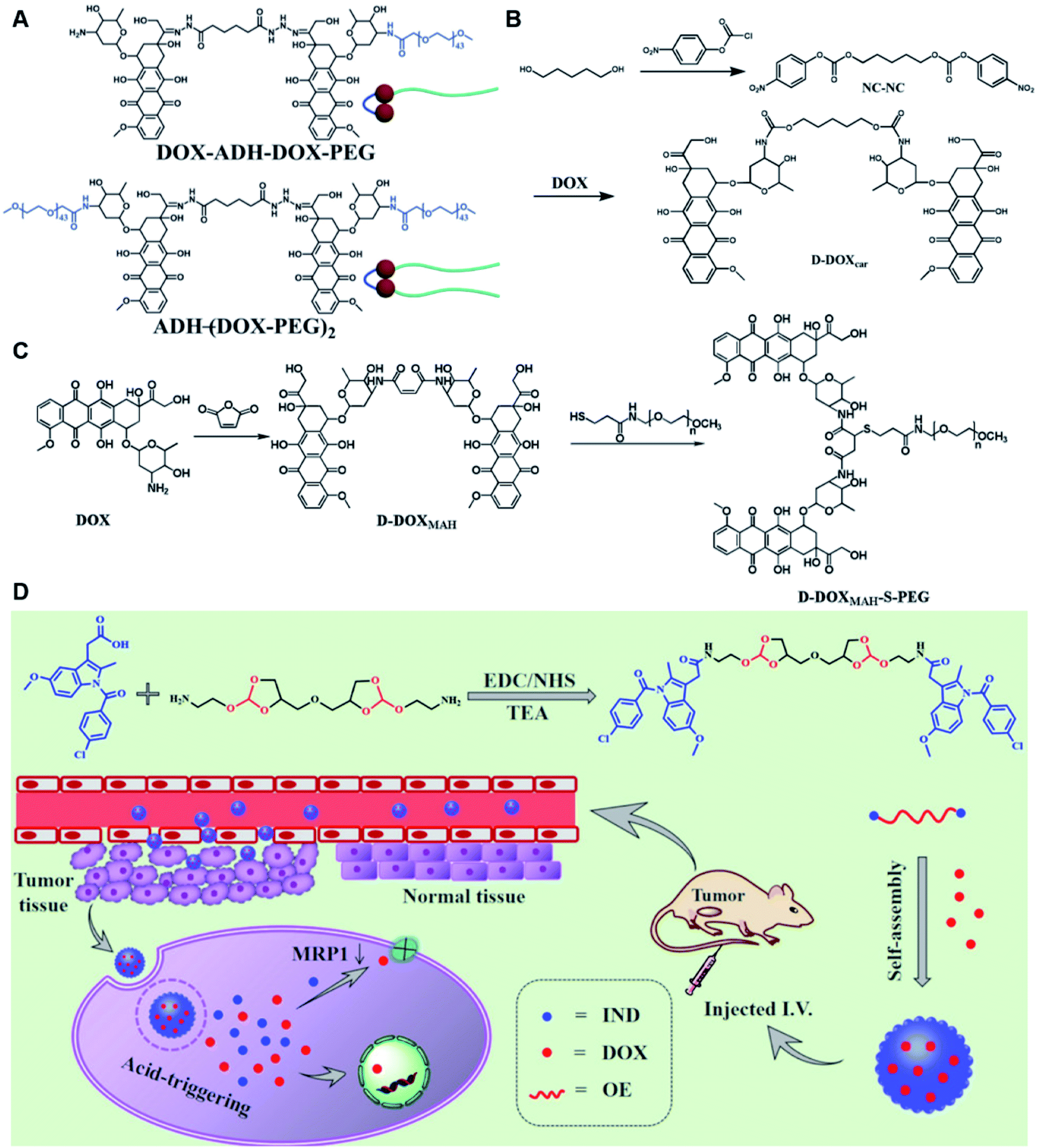 | ||
| Fig. 6 (A) Chemical structures of pH-responsive DOX-ADH-DOX-PEG and ADH-(DOX-PEG)2. Reproduced from ref. 82 with permission from WILEY-VCH, Copyright 2019. (B) Synthesis of the homodimeric doxorubicin prodrug (D-DOXcar). Reproduced from ref. 83 with permission from Elsevier Ltd, Copyright 2020. (C) Synthesis of D-DOXMAH and D-DOXMAH-S-PEG. Reproduced from ref. 84 with permission from Elsevier Ltd, Copyright 2020. (D) Chemical structure and the nano-assembly of the proposed ortho-ester-linked indomethacin (IND) dimer. Reproduced from ref. 85 with permission from American Chemical Society, Copyright 2019. | ||
Wang et al. reported an acid-responsive indomethacin (IND) dimeric nano-prodrug (named IND-OE/DOX) with physical encapsulation of doxorubicin for breast cancer therapy (Table 1).85 The dimeric IND prodrug was synthesized by conjugating two molecules of IND via an acid-labile ortho-ester linkage. The prepared IND dimers could form into homogeneous NPs by using an O/W single-emulsion solvent volatilization method (Fig. 6D). The hydrodynamic diameters of the obtained pH-sensitive NPs (named IND-OE/DOX) and pH-insensitive NPs (named IND-C12/DOX) were about 160–170 nm. The DOX drug-loading capacity of IND-OE/DOX and IND-C12/DOX was 11.39% and 9.43%, respectively. When compared to the pH-insensitive IND-C12/DOX, IND-OE/DOX showed a rapid degradation behaviour and accelerated drug release in mildly acidic environments. More than 85% DOX was released from IND-OE/DOX at pH = 5.0 within 48 h and about 15% of DOX was released at pH = 7.4. In contrast, about 20% DOX was released from IND-C12/DOX NPs after incubation at pH 5.0 or 7.4 within 4 days. Upon internalization by tumor cells, IND-OE/DOX exhibited an accelerated drug release inside the acidic endo-lysosomal system due to hydrolysis of the ortho ester. Notably, the released IND could reduce DOX efflux by P-gp down-regulation to enhance the chemotherapeutic effects. In vivo experiments confirmed that IND-OE/DOX showed synergistic antitumor properties when compared to free DOX.
4. ROS-responsive nanoassemblies
Reactive oxygen species (ROS), including superoxide (O2−), hydroxyl radicals (˙OH), hypochlorite ions (ClO−), hydrogen peroxide (H2O2) and singlet oxygen species (1O2), are by-products of normal cellular metabolism and play an essential role in maintaining various intracellular biological processes.86,87 It is well known that most tumor tissues/cells are in an oxidative stress state with higher levels of ROS when compared to normal tissues/cells due to their accelerated aerobic metabolism. Therefore, the elevated ROS levels in tumor tissues/cells could be utilized as endogenous stimuli for targeted drug release within the TME.88 To date, various ROS-responsive drug delivery systems have been developed on the basis of ROS-labile linkers, including thioethers, thioketals, boronic esters and others.894.1 Polymer–drug conjugate-based nanoassemblies
Due to the high ROS-producing state in tumor cells, Chu and co-workers described a ROS-responsive polymeric camptothecin (CPT) prodrug (MPEG-(TK-CPT)-PPa)-based nanoassembly for synergistic photodynamic therapy (PDT) and chemotherapy (Table 1).90 As shown in Fig. 7A, MPEG-(TK-CPT)-PPa was synthesized by introducing CPT into poly(ethylene glycol) methyl ether (MPEG) via the ROS-responsive thioketal (TK) linker (Table 2). A photosensitizer, pyropheophorbide-a (PPa), was attached to the same MPEG through lipid linkage. As an amphiphilic block copolymer, the resulting MPEG-(TK-CPT)-PPa self-assembled to form stable and uniform NPs that had an average hydrodynamic size of 43.6 ± 0.8 nm. During the formation of NP, the hydrophobic components (CPT and PPa) formed the inner core and the hydrophilic components (MPEG) formed the outer shell to prolong blood circulation time. Significantly, a 660 nm laser could be locally irradiated at tumor sites for 5 min (100 mW cm−2) under guidance of fluorescent PPa. Subsequently, ROS generated from PPa could elicit a substantial photodynamic therapeutic effect while also inducing TK linker cleavage and controlled release of CPT, thus achieving a combinatorial photodynamic and chemotherapy. In a similar study, Sun et al. developed another ROS-responsive polymeric prodrug (PTX-S-OA/PPa-PEG2k) with a core–shell structure for synergistic chemo-photodynamic therapy (Table 1).91 PTX-S-OA/PPa-PEG2k NPs were constructed by co-self-assembly of PEGylated PPa (PPa-PEG2k) and oleic acid (OA) linked PTX (PTX-S-OA), which formed an inner core and outer layer, respectively (Fig. 7B). The author found that this design could avoid the aggregation-caused quenching (ACQ) effect of the photosensitizer. The obtained nano-prodrug had an average hydrodynamic size of 75 nm and exhibited a superior colloidal stability in PBS when compared to non-PEGylated prodrug NPs. To ensure ROS-triggered drug release, PTX was coupled with OA via a ROS-responsive thioether linker (Table 2). Approximately 90% of PTX was successfully released after 6 h incubation under conditions of 10 mM H2O2. In contrast, lower degrees of PTX release were observed from ROS-insensitive PTX-OA/PPa-PEG2k NPs under the same conditions. Unlike passive encapsulation of PPa within the dense inner core of NPs, the outer layer PPa-PEG2k could avoid the aggregation-caused quenching (ACQ) effect of PPa, thus improving therapeutic PDT efficiency. Notably, the ROS generated by PPa under IR irradiation could induce effective PDT therapy while simultaneously inducing ROS-triggered release of PTX.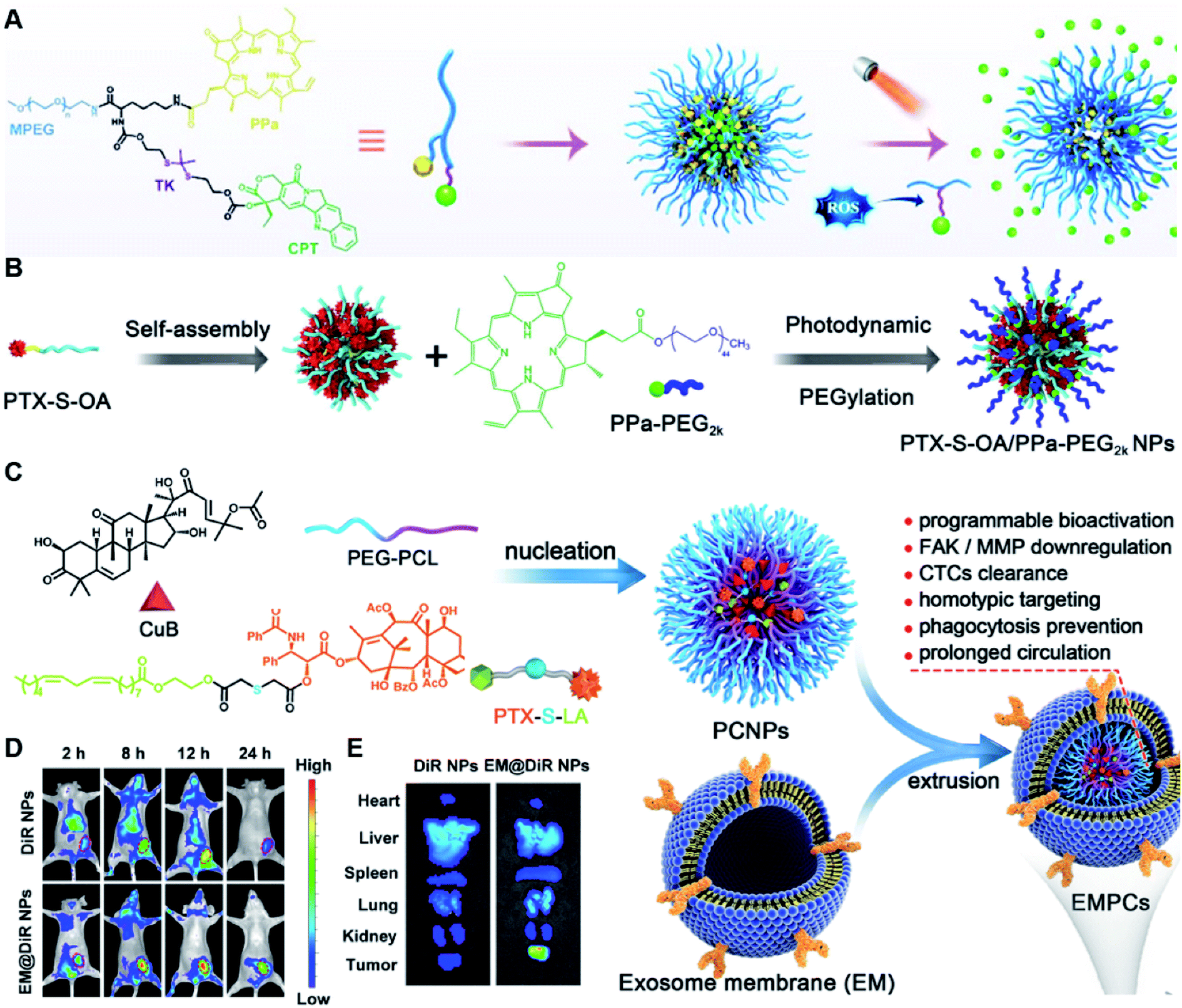 | ||
| Fig. 7 (A) Chemical design of MPEG-(TK-CPT)-PPa and its self-assembly for ROS-triggered CPT release and tumor therapy under laser irradiation. Reproduced from ref. 90 with permission from WILEY-VCH, Copyright 2020. (B) Schematic illustration of engineered PTX-S-OA/PPa-PEG2k NPs with photodynamic PEG coating for self-enhanced core–shell combination therapy. Reproduced from ref. 91 with permission from Elsevier Ltd, Copyright 2019. (C) Schematic representation of exosome-cloaked sequential-bioactivating paclitaxel prodrug nanoassemblies for cascade-amplified PTX chemotherapy. (D) In vivo fluorescence imaging of DiR NPs and EM@DiR NPs in orthotopic MDA-MB-231 tumor-bearing mice at 2, 8, 12, and 24 h post i.v. injection of NPs. (E) Ex vivo fluorescence imaging of DiR and EM@DiR NPs in the major organs at 24 h after NP injection. Reproduced from ref. 92 with permission from Elsevier Ltd, Copyright 2020. | ||
In another study, Wang et al. described an exosome mimetic sequential-bioactivation paclitaxel nano-prodrug (named EMPCs) that could inhibit breast cancer metastasis by recognizing circulating tumor cells (CTCs) in the bloodstream.92 To fabricate EMPCs, paclitaxel was attached to linoleic acid by using a ROS-responsive thioether linker, and the resulting PTX-S-LA was co-encapsulated with cucurbitacin B (CuB) within PEG-PCL polymeric micelles followed by cloaking with exosome membrane (EM) to achieve a homotypic tumor targeting capability (Fig. 7C). The prepared EMPCs with a spherical shape had an average hydrodynamic size of 100 nm capable of simultaneously targeting the primary tumor and capturing the CTCs during blood circulation. Upon successful entry into tumor cells, the released CuB not only inhibited the tumor metastasis by blocking the FAK/MMP signaling pathway, but also increased endogenous ROS levels to facilitate ROS-triggered PTX release. As shown in Fig. 7D and E, the fluorescence intensity of the exosome membrane-cloaked NPs (EM@DiR) showed an obvious increase at tumor sites from 2 h to 12 h post i.v. injection in MDA-MB-231 tumor-bearing mice, when compared to NPs without EM cloaking. The author demonstrated that the excellent tumor-targeting ability was caused by CD44-based homotypic targeting and CD47-mediated prolonged blood circulation time. When compared to control groups treated with saline or PCNPs that showed remarkable micro-metastases in the lungs, negligible metastatic nodules were observed in the lungs of mice upon treatment with EMPCs. Recently, Xia et al. reported an amphiphilic peptide–drug conjugate-based nanoassembly (named RGD-TK-Epo B) for tumor-targeted therapy.93 The RGD-TK-Epo B was synthesized by conjugating hydrophilic RGD peptide with hydrophobic cytotoxin epothilone B (Epo B) through the ROS-labile thioketal (TK) linker (Fig. 8A). Due to its inherent amphiphilic structure, the resulting RGD-TK-Epo B self-assembled into NPs (RECNs) with a uniform hydrodynamic size of 85.73 nm in an aqueous solution. The resulting RECNs had a good colloidal stability in PBS supplemented with 10% FBS and exhibited a specific targeting ability to tumor cells due to the high affinity of RGD for αvβ3 integrin overexpressed in tumor cells. Upon internalization by targeted tumor cells, Epo B was rapidly released as a result of cleavage in the thioketal bond under high intracellular levels of ROS. In vivo biodistribution studies showed that these RECNs exhibited excellent tumor-targeting ability after 8 h post i.v. injection. As expected, the RECNs presented an efficient antitumor effect in PC-3 tumor-bearing mice.
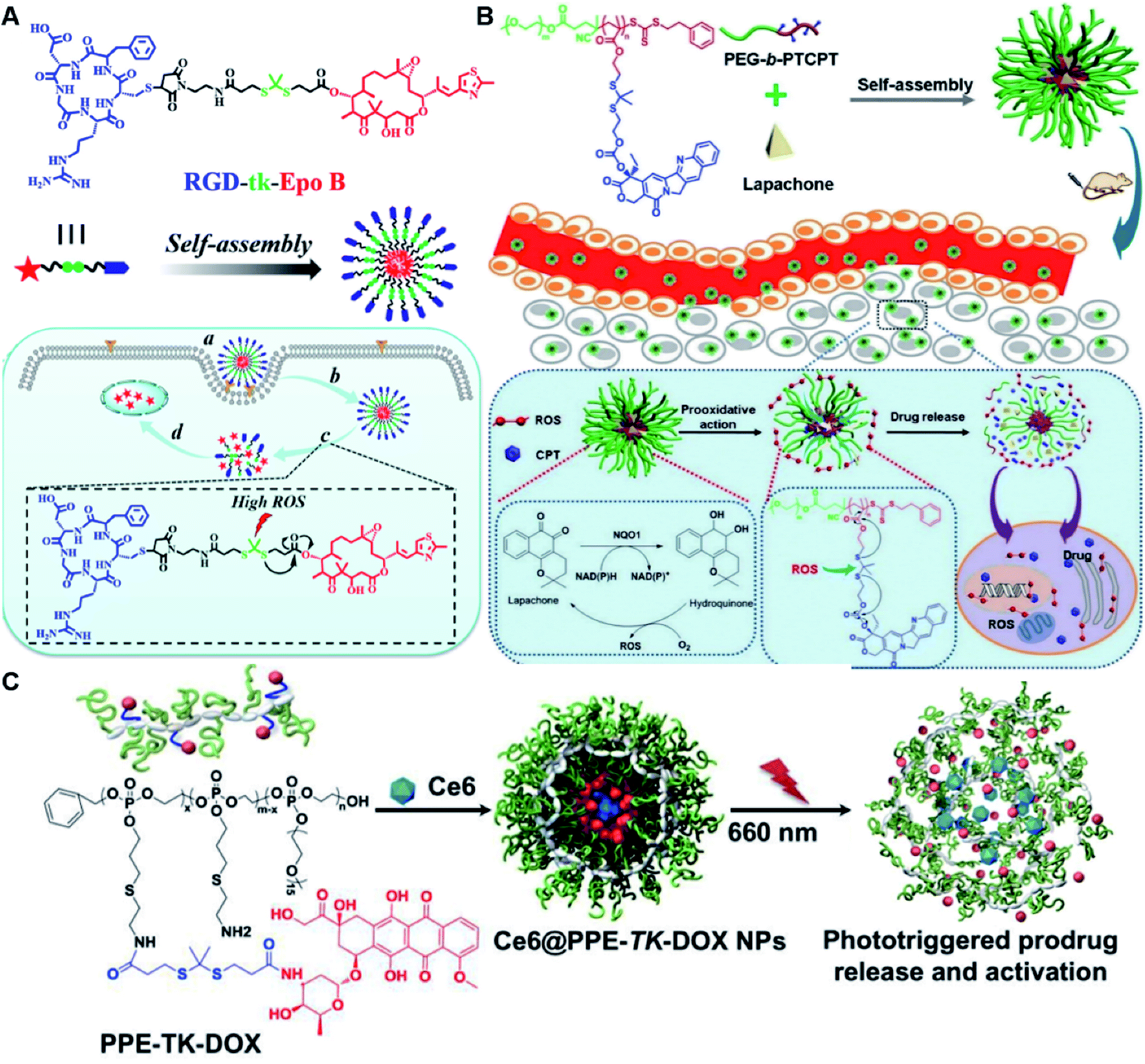 | ||
| Fig. 8 (A) Chemical structure of an RGD-tk-Epo B conjugate and its self-assembly for tumor-targeted drug delivery. (a and b) RGD-mediated tumor targeting and cell internalization of RECNs; (c) ROS-triggered release of Epo B from RECNs upon cleavage of the thioketal linker; (d) Epo B-induced cell apoptosis. Reproduced from ref. 93 with permission from American Chemical Society, Copyright 2020. (B) Schematic illustration of amphiphilic-block-copolymer Lapa@NPs with encapsulation of β-lapachone, which could catalyze the NAD(P)H-dependent increase in intracellular oxidative stress, thus enabling ROS-responsive drug release in tumor cells. Reproduced from ref. 94 with permission from Elsevier Ltd, Copyright 2019. (C) Chemical structure of PPE-TK-DOX and its self-assembly with encapsulation of photosensitizer Ce6 to enable ROS-triggered drug release under laser irradiation. Reproduced from ref. 95 with permission from Elsevier Ltd, Copyright 2019. | ||
Yin et al. synthesized an amphiphilic block copolymer prodrug (PEG-b-PTCPT) by conjugating camptothecin (CPT) to the backbones of PEG-linked polymerized methacrylate monomers via ROS-labile thioketal bonds (Table 2).94 The nano-prodrugs (Lapa@NPs) were prepared by encapsulation of β-lapachone within PEG-b-PTCPT-based micelles (Fig. 8B). The resulting Lapa@NPs with a hydrodynamic size of 48 nm exhibited good colloidal stability in serum-containing media for over 3 days. High drug-loading capacity (9%) and high encapsulation efficiency (98.5%) of Lapa in Lapa@NPs could be achieved when the weight ratio of polymer to Lapa was set at 10:1 during NP formation. Upon internalization to targeted tumor cells, β-lapachone could catalyze NAD(P)H to remarkably increase the intracellular ROS level, which concurrently amplified tumor oxidative stress to induce tumor cell apoptosis and ensure ROS-triggered drug release without additional laser irradiation.
In vitro drug release experiments showed that about 75% of the encapsulated CPT was released from the obtained nano-prodrugs under the conditions of 1 mM H2O2 in the presence of Fe2+ within 80 h. As expected, in vivo experiments showed that the Lapa@NP-treated group exhibited better antitumor effects than other groups, which may be attributed to combinatorial oxidation/chemotherapy. In another study, Pei and co-workers synthesized a polyphosphoester–doxorubicin conjugate (PPE-TK-DOX) by linking doxorubicin (DOX) to the side chains of polyphosphoesters (PPEs) through ROS-sensitive thioketal linkers (Fig. 8C).95 Upon co-self-assembly with photosensitizer Ce6, the resulting PPE-TK-DOX could form into NPs (Ce6@PPE-TK-DOX) in aqueous solution capable of avoiding premature drug leakage during blood circulation. The prepared Ce6@PPE-TK-DOX NPs had a hydrodynamic size of 73 nm and maintained a good colloidal stability with insignificant size changes in PBS supplemented with 10% FBS for over 7 days. Notably, DOX was successfully activated and locally released at tumor sites upon laser irradiation, resulting in minimized systemic side effects. Approximately 71.3% of the thioketal bond was cleaved under 660 nm laser irradiation for 40 min. The in vivo imaging data showed that the Ce6 fluorescence intensity at tumor sites reached a plateau at 4 h post i.v. injection of Ce6@PPE-TK-DOX. Ma et al. synthesized a ROS-activable aspirin–dextran (DEX) conjugate that could self-assemble into a polymeric prodrug (P3C-Asp) for TME regulation and enhanced tumor immunotherapy (Table 1).96 As shown in Fig. 9A, aspirin was attached to DEX via ROS-cleavable p-boronabenzyl ester. The resulting P3C-Asp with an average hydrodynamic size of 40 nm had a high drug-loading content (10 wt%). Upon successful entry into tumor cells, P3C-Asp NPs could be hydrolysed under elevated intracellular levels of ROS leading to the rapid release of deacetylated active aspirin (which could act as a COX-2 inhibitor) to inhibit the PGE2 secretion. Moreover, P3C-Asp could also enhance the infiltration of CD8+ T cells and M1 macrophages while inhibiting the infiltration of myeloid-derived suppressor cells (MDSC) in the TME. Consequently, this polymeric prodrug was capable of eradicating tumor cells in a murine colorectal cancer model upon combination treatment with anti-PD-1 antibodies.
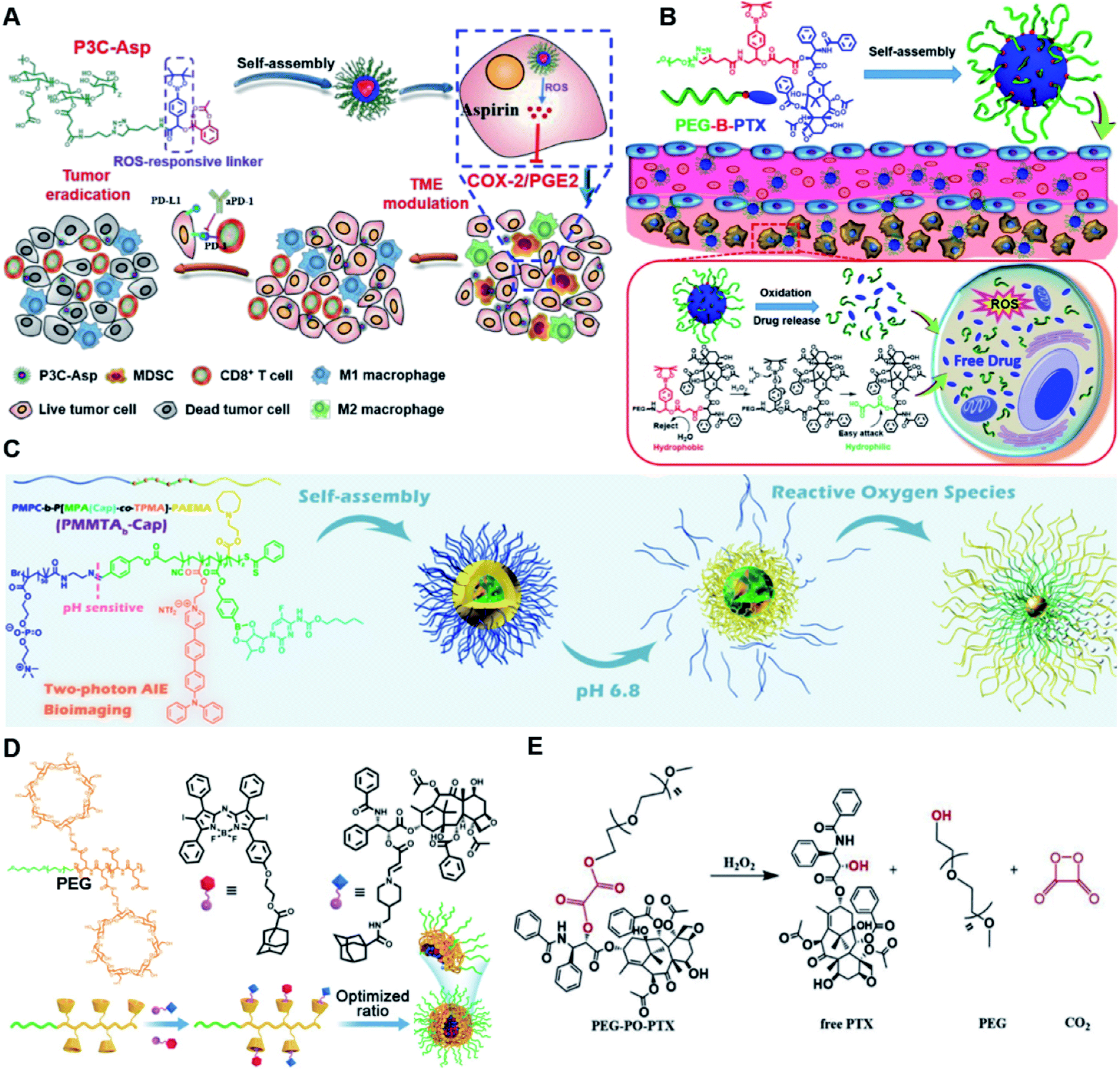 | ||
| Fig. 9 (A) Chemical structures of the ROS-responsive P3C-Asp prodrug and its self-assembly for TME regulation and enhanced tumor immunotherapy. Reproduced from ref. 96 with permission from Chinese Chemical Society, Copyright 2020. (B) Schematic representation of engineered PTX prodrug-based micelles (PEG-B-PTX) for ROS-triggered drug release in tumor therapy. Reproduced from ref. 97 with permission from Elsevier Ltd, Copyright 2020. (C) Chemical structure of PMPC-b-P[MPA(Cap)-co-TPMA]–PAEMA (PMMTAb-Cap) and its self-assembly for ROS-responsive drug release. Reproduced from ref. 98 with permission from American Chemical Society, Copyright 2019. (D) Chemical structures of PEG–PGA–β-CD, Ada–BODIPY, Ada–PTX and their nanoassemblies at an optimized ratio for combined chemo-dynamic therapy. Reproduced from ref. 99 with permission from Wiley-VCH, Copyright 2019. (E) Chemical design of PEG-PO-PTX and its drug release mechanism in the presence of H2O2. Reproduced from ref. 100 with permission from Elsevier Ltd, Copyright 2019. | ||
Dong and co-workers developed an amphiphilic paclitaxel (PTX) prodrug (PEG-B-PTX) by conjugating PTX to polyethylene glycol (PEG) via a p-(boronic ester) benzyl-based linker (Table 2).97 As shown in Fig. 9B, both ROS-sensitive PEG-B-PTX and ROS-insensitive PEG-H-PTX could form into micellar nano-prodrugs (∼50 nm) by self-assembly, providing NPs with a high PTX loading (13.3 wt%). The PEG-B-PTX micelles exhibited a good stability in physiological environments and exhibited a prolonged blood circulation time. In vitro experiments showed that negligible PTX (<5%) was released from PEG-B-PTX under physiological conditions (pH 7.4) even under acidic conditions (pH 5) without the addition of H2O2. In contrast, approximately 65% encapsulated PTX was released within 96 h upon addition of 200 μM of H2O2 at pH 7.4. Notably, accelerated drug release was observed in the presence of H2O2 (200 μM) at pH 5.0 which was approximately due to a stronger oxidation potential of H2O2 under acidic conditions. In another study, Ma et al. synthesized a hierarchical pH and ROS dual-responsive triblock polymeric prodrug (PMMTAb-Cap) that could self-assemble into homogeneous prodrug micelles for efficient cancer theranostics (Fig. 9C).98 The resulting nano-prodrugs possessed a core–shell structure and the outer block poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) was conjugated with a two-photon fluorophore (TP)-linked poly(2-azepane ethyl methacrylate) (PAEMA) through a pH-sensitive benzoyl imide linker (Table 2), and the Capecitabine (Cap) was introduced onto PAEMA via an ROS-responsive boron ester bond. The resulting PMMTAb-Cap prodrug micelles with a hydrodynamic size of 69 nm exhibited an excellent colloidal stability in PBS supplemented with 10% FBS. Upon accumulation in the mildly acidic TME (pH 6.8), the PMPC could be detached from the prodrug micelles due to the cleavage of imide bonds. Meanwhile, the inner PAEMA became electropositive and hydrophilic (from the original hydrophobic form), thus enabling a decrease in the micellar size and subsequently leading to the eventual enhancement in tissue penetration and endocytosis of these prodrug micelles. Upon successful internalization by targeted tumor cells, rapid drug release was observed under overexpressed intracellular ROS conditions. Notably, the two-photon fluorophore loading enabled these prodrug micelles to possess two-photon AIE bioimaging ability and therefore could be used as a potential theranostic drug delivery system, in which the co-delivery of photosensitizers and prodrugs at a precise ratio might have potential utilities to improve combination therapy.
In another study, Chen et al. reported a supramolecular nano-prodrug to co-deliver photosensitizers and ROS-responsive prodrugs at optimized ratios by using a novel host–guest strategy (Fig. 9D).99 To construct this nanosystem, a block copolymer (PEG–PGA–β-CD) was synthesized by conjugating β-CD to polyethylene glycol (PEG)-linked poly-L-glutamic acid (PGA). Aza-BODIPY and paclitaxel were modified with adamantane to serve as the PS and prodrug guest molecules, respectively. By virtue of the stable host–guest inclusion complex made by β-CD and the adamantane unit, the prodrug and PS could be easily grafted to PEG–PGA–β-CD with an optimized ratio. TEM images showed that Ada–PTX(60%)–BODIPY(40%)–PNs had a uniform and polymersome-like morphology with a hydrodynamic size of 100 nm. Upon NIR light irradiation, the ROS generated by the PS could enhance the release of active paclitaxel. Notably, a smaller NP size (∼68 nm) was observed after NIR irradiation for 30 min, indicating successful PTX release. IVIS imaging data showed that the control groups of Ada–BODIPY and Ada–BODIPY(100%)–PNs exhibited a similar tumor accumulation in tumor-bearing mice at 3 h post i.v. injection. However, the fluorescence intensity at tumor sites in the group treated with Ada–BODIPY(100%)–PNs was 6.5-fold higher in comparison to the Ada–BODIPY treatment group at 24 h post injection. These results indicated that the Ada–BODIPY(100%)–PNs with a prolonged blood circulation time could accumulate selectively in tumor regions. Notably, the mice treated with Ada–PTX(60%)–BODIPY(40%)–PNs exhibited a higher therapeutic efficiency with a lower dose of chemotherapeutic drug than other groups. Li et al. reported ROS-sensitive prodrug micelles for imaging-guided chemo-phototherapy (Fig. 9E).100 The prodrug micelles were fabricated by encapsulating free PTX into the co-self-assemblies of PEG-peroxalate ester-PTX (PEG-PO-PTX) and folate-PEG-Cypate. To achieve ROS-triggered drug release in tumor cells, PEG-PO-PTX was synthesized by conjugating PEG with PTX via an H2O2-labile peroxalate ester bond (Table 1). Folate was utilized to enable a specific tumor-targeting ability of the nano-prodrugs, as folate receptors (FR) are overexpressed in many cancer cells. The prepared nano-prodrugs had a hydrodynamic size of ∼117.6 nm capable of causing 74% PTX release in the presence of 100 μM H2O2 within 24 h. In contrast, less than 20% PTX was released in the absence of H2O2. Notably, the prodrug micelles could maintain a good colloidal stability in PBS with no obvious changes in size for over 1 week. Upon successful internalization into tumor cells which had high levels of intracellular H2O2, the micelles underwent successful disassembly to release both free and conjugated PTX as a result of cleavage in the H2O2-labile peroxalate ester bond. Cypate was used as a photosensitive/photothermal agent to enable optical imaging-guided combinatorial phototherapy under laser irradiation.
4.2 Drug–drug conjugate-based nanoassemblies
Recently, Zhang and co-workers reported a ROS-sensitive photosensitizer-driven nanoassembly (CTX-S-CTX/PPa) for combinatorial chemo-photodynamic therapy (Table 1).101 A homodimeric cabazitaxel (CTX) prodrug (CTX-S-CTX) was synthesized by conjugating two molecules of CTX through an ROS-labile single thioether bond (Table 2). CTX-S-CTX itself failed to form NPs due to its highly symmetric structure, while it was able to co-assemble with PPa into NPs due to multiple intermolecular forces including hydrogen bonds, hydrophobic interactions, π–π stacking, and π–cation interactions (Fig. 10A). Moreover, a PEGylated nanoassembly (pCTX-S-CTX/PPa) with prolonged blood circulation time and a minimized ACQ effect from PPa was next obtained by inserting 20% (w/w) of DSPE-PEG2K into this nanostructure. The hydrodynamic size of the non-PEGylated control NP (CTX-S-CTX/PPa) was about 79 nm and increased to 88 nm after PEGylation. The pCTX-SCTX/PPa exhibited a good colloidal stability as the PEGylation could significantly increase resistance to salting out. Under NIR laser irradiation, ROS generated by PPa could oxidize the thioester bond in CTX-S-CTX and subsequently disassembly of NPs leading to 80% CTX release within 24 h in the presence of 1 mM H2O2 (Fig. 10B). In a similar study, Luo and coworkers reported a heterotypic dimer-based nanoassembly for combinatorial chemo-photodynamic therapy (Table 1).102 To achieve ROS-triggered drug release, the heterotypic dimer was synthesized by coupling PTX with PPa via ROS-labile thioether bonds (Fig. 10C). The obtained heterotypic dimer could form into NPs in deionized water followed by inserting DSPE-PEG2K on the surface to finish the PEGylation. The resultant nano-prodrug (PPa-S-PTX/DSPE-PEG2k NPs) had an average diameter of 90 nm and possessed an ultrahigh drug loading capacity for both PTX (44.2 wt%) and PPa (27.6 wt%). Upon NIR irradiation, the ROS generated from PPa could not only enable the PDT therapy but also facilitated the PTX release within tumor cells due to the cleavage of thioether bonds. In vitro drug release experiments showed that over 90% PTX was released from the nano-prodrug within 8 h under the conditions of 10 mM H2O2. Significantly, the PTX release could lead to the disassembly of the NP to effectively alleviate the ACQ effect of PPa, thus achieving a synergistic chemo-photodynamic antitumor therapy.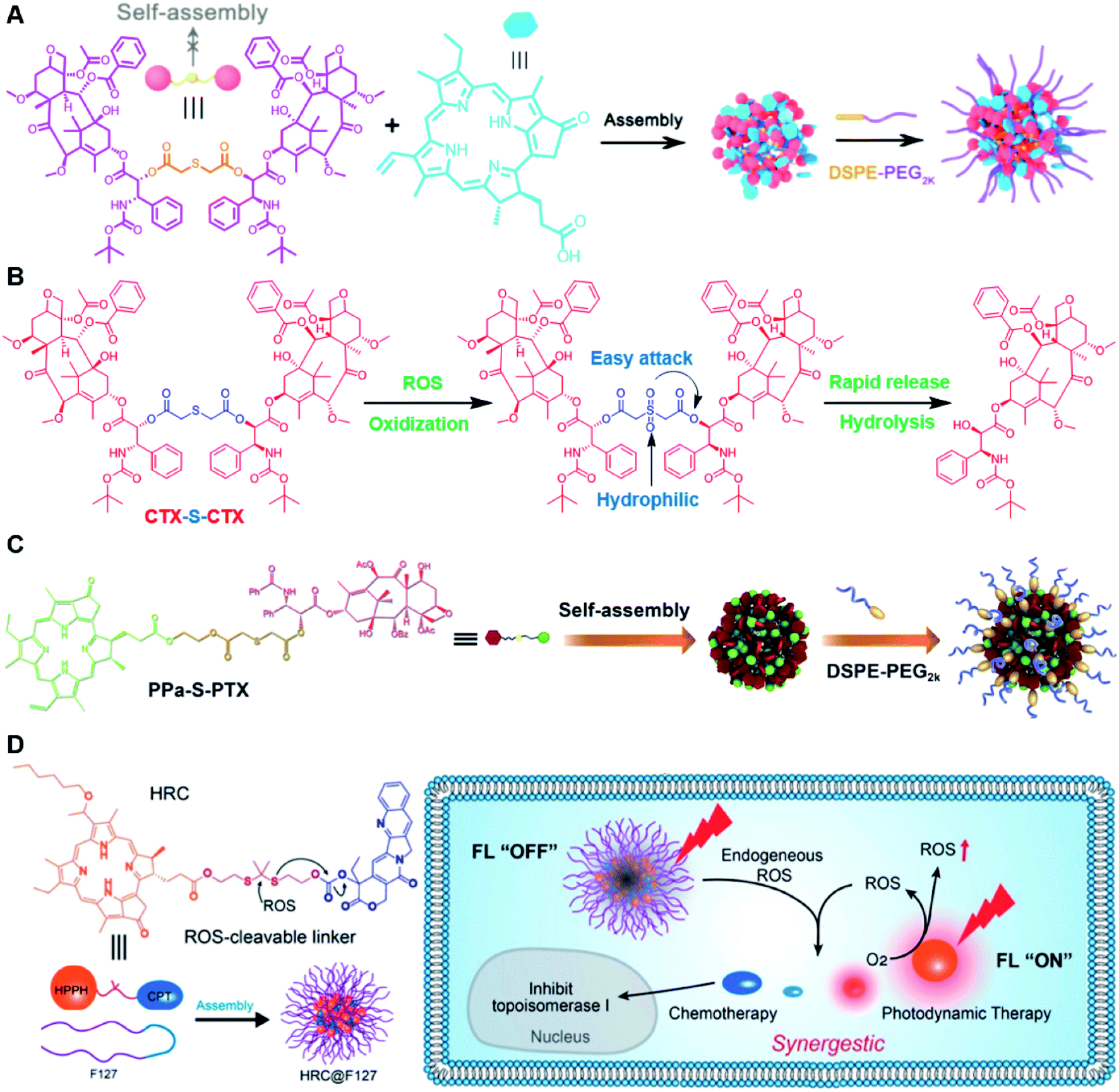 | ||
| Fig. 10 (A) Schematic illustration of PEGylated PPa-driven nanoassembly of CTX-S-CTX. (B) CTX activation and release mechanism from CTX-S-CTX. Reproduced from ref. 101 with permission from Ivyspring International Publisher, Copyright 2021. (C) Schematic illustration of the construction of PPa-S-PTX-based nanoassemblies for synergistic photo-chemotherapy. Reproduced from ref. 102 with permission from Elsevier Ltd, Copyright 2019. (D) Molecular structures of the ROS-responsive HRC prodrug and the formation of nanoparticles by encapsulating HRC into F127 polymeric micelles for cancer imaging and combinatorial chemo-photodynamic therapy. Reproduced from ref. 103 with permission from American Chemical Society, Copyright 2020. | ||
In another study, Jiang et al. developed a ROS-activable nanoprodrug (HRC@F127) for combined chemo-photodynamic therapy (Table 1).103 As shown in Fig. 10D, the HRC@F127 was prepared by encapsulating the heterodimeric prodrug (HRC) in the F127-based nanoassemblies. To achieve ROS triggered drug release, the HRC was synthesized by linking camptothecin (CPT) with photosensitizer 2-(1-hexyloxyethyl)-2-devinylpyropheophorbide-a (HPPH) through a thioketal linker (Table 2). The obtained HRC@F127 (23 ± 5 nm) with a high drug loading efficiency (>95%) could keep a good colloidal stability with minimal premature drug leakage during the blood circulation. Notably, the fluorescence of HPPH in HRC@F127 could be quenched due to the existence of strong π–π stacking of HPPH. Upon successful entry into tumor cells, the HPPH and CPT could be rapidly released from HRC@F127 NPs in the presence of high endogenous ROS levels, thus lighting up the tumor cells and achieving an efficient combinatorial chemo-photodynamic therapy. High fluorescence intensity was observed in HRC@F127 NP-treated HCT116 cells, but the ROS-unresponsive control group (HCC@F127)-treated tumor cells showed a negligible fluorescence, indicating the ROS-triggered thioketal bond cleavage and HPPH release from HRC@F127 NPs. To study the in vivo biodistribution of this nano-prodrug, the control NPs (HPPH@F127) were prepared by encapsulating HPPH into the F127 NPs. Both HPPH@F127 and HRC@F127 NPs could specifically accumulate in tumor tissues via the EPR effect, while negligible PET signals were detected in free HPPH molecule-treated mice, which was probably due to the fast clearance of free HPPH during blood circulation. The HRC@F127 treated group exhibited higher PET signals than the control group, which could be probably attributed to lower premature drug leakage during blood circulation due to the higher hydrophobicity of the HRC prodrug.
5. GSH-responsive nanoassemblies
Glutathione (GSH) is a normal byproduct in living cells, which has a significant influence on the several cellular processes including antioxidant, cell differentiation, proliferation, and apoptosis.104,105 The concentration of cytosolic GSH is found to be approximately 100–1000 times higher than that in the extracellular matrixes or human blood circulation.106 As most tumor cells are often in a high oxidative stress state, GSH as an antioxidant is overexpressed in some tumor cells with about 4 times higher (2–10 mM) than normal cells, which could be exploited as an internal stimuli for cancer-specific drug delivery.1075.1 Polymer–drug conjugate-based nanoassemblies
Specifically targeting the sub-organelles in tumor cells such as mitochondria is playing an increasingly important role in tumor therapy.108 In a recent study, Xiao et al. reported a new mitochondrion-targeting peptide (MTP) with cell-penetrating ability.109 Based on this MTP, a mitochondrion-targeting prodrug (Fig. 11A) was synthesized by inserting a disulfide linker between DOX and biotin (a tumor-targeting ligand)-tagged MTP to achieve mitochondrion-targeting properties (Table 2). These prodrug conjugates showed significant specific cytotoxicity to HeLa cells, but no obvious cytotoxicity to normal Chinese hamster ovary (CHO) cells. The resulting peptide–drug conjugates could self-assemble into NPs with a hydrodynamic size of 90 nm upon gentle stirring in aqueous solution. The obtained prodrug nanoassemblies possessed a high mitochondrion-targeting ability due to the appropriate balance between lipophilicity and positive charges in the peptide. DOX could be selectively delivered into mitochondria and activated in the presence of abundant endogenous GSH, thus leading to cell apoptosis via mitochondrial depolarization by decreasing matrix metalloproteinases (MMP). In another study, Li et al. reported a redox-activable polyprodrug nanosystem (named polyHCPT) for targeted co-delivery of Bcl-2 siRNA (siBcl-2) and combinatorial tumor therapy (Table 1).110 As shown in Fig. 11B, this nano-prodrug was fabricated by co-self-assembly of the hydrophobic 10-hydroxycamptothecin (HCPT)-based polyHCPT and cationic lipid (G0-C14)/siRNA complexes followed by cloaking with amphiphilic lipid-poly(ethylene glycol) (lipid-PEG) on the surface of the resulting NP. During NP formation, polyHCPT formed the inner core and the amphiphilic lipid-poly(ethylene glycol) (lipid-PEG) formed the outer shell. In addition, lactobionic acid (LA) was modified at the surface of the NP to improve the hepatoma-targeting ability. The obtained nano-prodrug had an average hydrodynamic size of 85 nm capable of causing 80% release of siRNA and 90% release of HCPT within 24 h in the presence of 10 mM GSH. Notably, the nano-prodrug changed from spherical to amorphous aggregates after incubation in PBS solution containing 10 mM GSH. Upon successful entry into tumor cells, both HCPT and siBcl-2 could be rapidly released due to the cleavage of disulfide bonds in polyHCPT at high intracellular concentrations of GSH. The released CPT and siBcl-2 could induce cell apoptosis and concurrently inhibit Bcl-2-mediated anti-apoptotic pathways, eventually leading to a highly efficient combinatorial therapy. In another study, Lu et al. developed a GSH-responsive blood–brain-barrier (BBB)-penetrating prodrug micelle (CPT-S-S-PEG-iRGD@IR780) for combinatorial chemo-photodynamic therapy against glioma (Table 1).111 The CPT-S-S-PEG-iRGD@IR780 was constructed by encapsulating IR780 in CPT-S-S-PEG-iRGD-based nanoassemblies. As shown in Fig. 11C, CPT-S-S-PEG-iRGD was synthesized by introducing CPT into polyethylene glycol (PEG) via a disulfide bond, followed by surface modification with iRGD peptide at the terminus of PEG. The hydrodynamic size of the resulting CPD@IR780 was determined to be 140.68 ± 8.53 nm. In vitro drug release experiments showed that around 22% and 60% of CPT was released from the prodrug micelles in the presence of 2 mM and 10 mM GSH, respectively. In contrast, negligible CPT leakage (<5%) was observed in the absence of GSH. Notably, the amount of ROS generated by CPD@IR780 micelles in tumor cells was higher than that of free IR780 under NIR laser irradiation, indicating that IR780 could be efficiently delivered into tumor cells by these micelles. In vitro BBB-crossing experiments revealed that these iRGD-modified prodrug micelles exhibited a stronger BBB-penetrating ability. Similarly, in vivo biodistribution experiments indicated that CPD@IR780 exhibited an excellent glioma-targeting ability when compared to CPC@IR780, which was attributed to the αvβ integrin and neuropilin-1-mediated ligand transportation. As expected, CPD@IR780 micelles could significantly prolong the median survival of the orthotopic glioma model.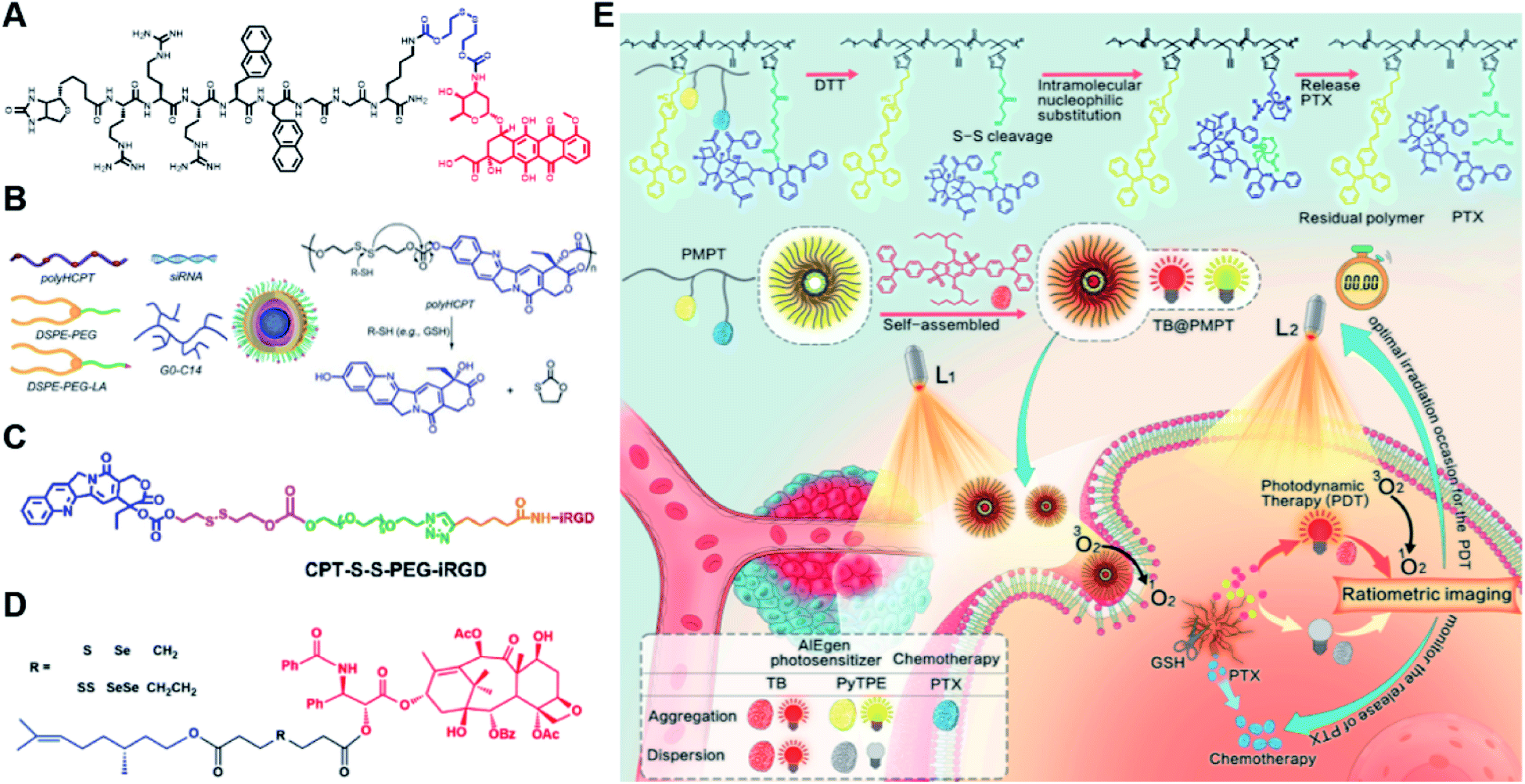 | ||
| Fig. 11 (A) Chemical structure of mitochondrion-targeting prodrug–peptide conjugates. Reproduced from ref. 109 with permission from John Wiley & Sons, Inc., Copyright 2021. (B) Illustration of the engineered redox-sensitive polyHCPT-based NPs and redox-responsive drug release mechanism. Reproduced from ref. 110 with permission from Elsevier Ltd, Copyright 2020. (C) Chemical structure of CPT-S-S-PEG-iRGD. Reproduced from ref. 111 with permission from Elsevier Ltd, Copyright 2020. (D) Schematic illustration of PTX–CIT conjugates linked by different chemical bonds (sulfur/selenium/carbon). Reproduced from ref. 113 with permission from Springer Nature, Copyright 2019. (E) Chemical structure of the polymeric PMPT prodrug and its self-assembly with encapsulation of two AIE photosensitizers (PyTPE and TB) for imaging-guided PDT and drug release. Reproduced from ref. 114 with permission from American Chemical Society, Copyright 2021. | ||
The diselenide bond (Se–Se), a ROS-responsive linker, was also used in the redox-responsive drug delivery system. The bond dissociation energy (172 kJ mol−1) of Se–Se is lower than the disulfide bond (240 kJ mol−1).112 Thus, the diselenide linker has a greater potential than the disulfide linker for redox-responsive drug release in tumor cells. In a recent study, Sun et al. synthesized six paclitaxel–citronellol (PTX–CIT) conjugates to investigate the effects of various chemical bonds (sulfur/selenium/carbon) and bond angles/dihedral angles on self-assembly performance, drug release, cytotoxicity, stability, and pharmacokinetics (Fig. 11D).113 The hydrophobic PTX–CIT conjugates could form into uniform nano-prodrugs through one-step nanoprecipitation followed by PEGylation with DSPE-PEG2K to obtain better colloidal stability and pharmacokinetic behavior. The obtained PEGylated nano-prodrugs had an average hydrodynamic size of ∼90 nm and high drug loading content (>50 wt%). In this study, the authors found that the thioether bonds and selenoether bonds showed much higher oxidation sensitivity than reduction sensitivity. As expected, the diselenide bond-based nano-prodrugs exhibited the most promising antitumor activity compared to other responsive linkers. The superior antitumor effect was probably attributed to several advantages of these nano-prodrugs including the good colloidal stability, prolonged blood circulation time, preferential tumor distribution, and efficient drug release. The selenoether/diselenide bonds enabled reactive oxygen species production, which could boost the cytotoxicity of these prodrugs. This study provides a strategy for rational design of redox-responsive prodrug delivery systems. In another study, Yi et al. reported a self-guiding prodrug micelle (TB@PMPT) for improved chemo-photodynamic therapy (Table 1).114 As shown in Fig. 11E, TB@PMPT was prepared by co-self-assembly of a polymeric prodrug (PEG-b-PMPMC-g-PTX-g-PyTPE, PMPT) with physical encapsulation of another AIE photosensitizer TPA-BDTP (TB). The PMPT was synthesized by simultaneously conjugating the AIE photosensitizer PyTPE and reduction-sensitive PTX prodrug (PTXSS-N3) onto the backbone of an amphiphilic polycarbonate. The obtained TB@PMPT micelles could emit yellow and red fluorescence from PyTPE and TB, respectively. Upon accumulation in tumor tissues, the PyTPE and TB generated ROS on the first light irradiation and subsequently induced lipid peroxidation and increased the permeability of the cell membrane, leading to enhanced cell internalization of TB@PMPT micelles. The cleavage of the disulfide bond under the conditions of high intracellular GSH leads to the rapid release of PTX. Notably, PTX release could improve the hydrophilicity of the residual amphiphilic polymer and increase the dispersion of PyTPE in an aqueous solution capable of causing fluorescence ratio transformation as TPA-BDTP (TB) remained aggregated after PTX release. The fluorescence ratio transformation could be employed as a ratiometric fluorescence probe to guide the occasion for second irradiation to achieve PDT, as well as for drug release monitoring.
5.2 Drug–drug conjugate-based nanoassemblies
In a recent study, Yang et al. synthesized various homodimeric doxorubicin prodrugs based on different GSH-responsive linkers including thioether/disulfide/trisulfide bonds, which were termed as DOX-S-DOX (DSD), DOX-SS-DOX (DSSD), and DOX-SSS-DOX (DSSSD), respectively (Fig. 12A).115 DSD or DSSD alone failed to form stable NPs in aqueous solution as they gradually precipitated from the initial transparent red solution. In contrast, DSSSD could form spherical NPs that had an average hydrodynamic size of 150 nm and a good colloidal stability without precipitation within 48 h. By co-assembly with DSPE-PEG2K, DSSD and DSSSD could form into stable NPs that did not precipitate within 24 h, while DSD still failed to self-assemble into stable NPs even with the help of DSPE-PEG2K. The resulting trisulfide bond-based nano-prodrugs were more sensitive to GSH, thereby enabling ultrahigh tumor selectivity in cancer cells as compared to normal cells. Notably, trisulfide bond-based dimeric nano-prodrugs (DSSSD) had a higher drug loading capacity (67.24%, w/w) capable of causing 25% DOX-SH release from DSSSD within 30 min under the conditions of 0.5 mM GSH, and 68% DOX-SH release within 1 h in the presence of 1 mM GSH (Fig. 12B and C). In contrast, slower DOX-SH release from DSSD was observed under the same conditions. In addition, DSSSD exhibited higher tumor targeting ability over disulfide and thioether bond-based NPs. As expected, DSSSD NPs exhibited a superior antitumor effect with almost no growth in tumor volume to the conventional disulfide bond-based nano-prodrugs. In another study, Xie et al. developed a GSH responsive nano-prodrug (CEL@G-SS-NIR) for tumor theranostics (Table 1).116 The CEL@G-SS-NIR was prepared by physical encapsulation of celastrol (a serine threonine protein kinase inhibitor) in the gefitinib (a tyrosine kinase inhibitor) prodrug (G-SS-NIR)-based nanoassemblies (Fig. 12D). The G-SS-NIR was synthesized by linking gefitinib and a near-infrared (NIR) dye with disulfide bonds (Table 2). The hydrodynamic size of CEL@G-SS-NIR with a high drug loading content of celastrol (42 ± 2 wt%) was determined to be 119 ± 6 nm. Nearly 90% of gefitinib and celastrol was released from CEL@G-SS-NIR NPs in the presence of varied quantities of GSH. Upon successful entry into tumor cells, the nano-prodrug was disassembled in the presence of abundant endogenous GSH causing the rapid release of two drugs that could simultaneously inhibit the phosphorylation of upstream EGFR and downstream Akt in the EGFR signaling pathway. Importantly, the released dyes could be used for fluorescence and optoacoustic imaging to monitor the drug release in vivo. Obvious fluorescence was observed in the lung region in the orthotopic A549-luc tumor-bearing mice at 1 h post injection of CEL@G-SS-NIR, and the fluorescence showed a progressive enhancement in the lung region within 12 h. In contrast, negligible fluorescence was observed in the NIR-OH treated group. As expected, CEL@G-SS-NIR exhibited a superior antitumor efficiency in both PC-9 and A549 tumor-bearing mice.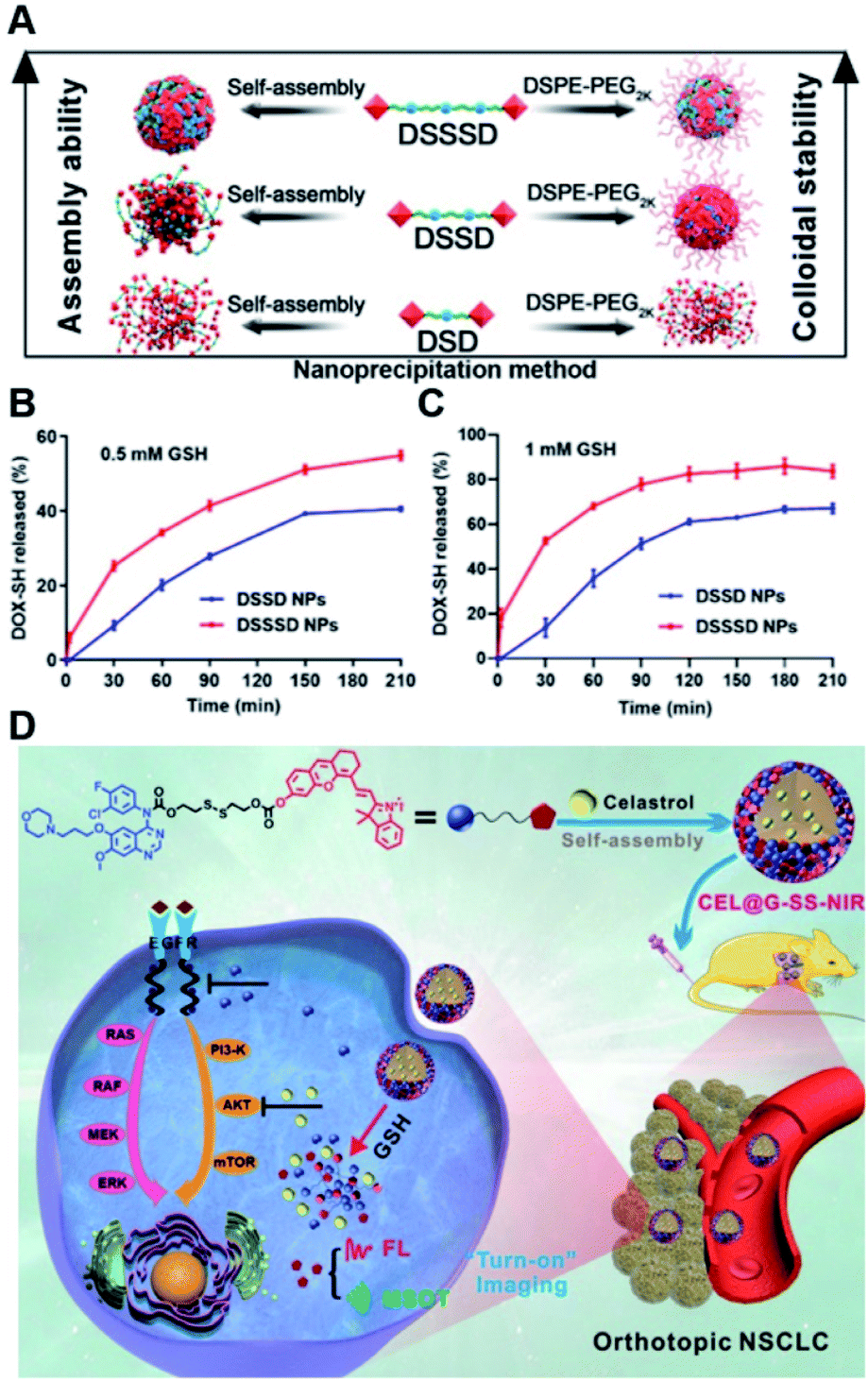 | ||
| Fig. 12 (A) Nano-prodrug constructed by self-assembly of DSD, DSSD and DSSSD with or without DSPE-PEG2K. In vitro DOX-SH release from DSSD and DSSSD in (B) 0.5 mM or (C) 1 mM GSH. Reproduced from ref. 115 with permission from American Association for the Advancement of Science, Copyright 2020. (D) Chemical structure of a gefitinib linked near-infrared dye and its self-assembly with encapsulation of celastrol for tumor therapy. Reproduced from ref. 116 with permission from Wiley-VCH, Copyright 2019. | ||
6. Enzyme-responsive nanoassemblies
Enzymes are involved in various key biological and metabolic processes in living cells and the aberrant enzyme expression is often found in several pathological situations, especially in cancer.117 For example, cathepsin B and matrix metalloproteinases (MMP) are overexpressed in several tumors, which play an important role in tumor progression, invasion, and metastasis.118,119 Such tumor associated enzyme dysregulation could be exploited as endogenous triggers for construction of the enzyme-responsive drug delivery system to achieve specific drug activation and release in tumor tissues.120,121 Various enzyme-labile bonds (specific esters or peptide bonds) have been exploited so far for tumor theranostics, especially in the development of prodrug-based nanomedicines.6.1 Polymer–drug conjugate-based nanoassemblies
In a recent study, Wang et al. reported an enzyme-responsive, amphiphilic polymeric imidazoquinoline TLR7/8 agonist prodrug (PEG5k-GL2-IMDQ) for immune-stimulatory drug delivery (Table 1).122 As shown in Fig. 13A, the PEG5k-GL2-IMDQ was synthesized by inserting the hydrophobic enzyme-responsive and self-immolative linker between the imidazoquinoline TLR7/8 agonist and the hydrophilic PEG. The enzyme responsive linker was composed of β-glucuronidase (β-GUS)-sensitive glucuronide capped benzyl carbamate residues and ester bonds (Table 2). The obtained amphiphilic polymeric prodrug could self-assemble into vesicular polymer vesicles (PEG5k-GL2-IMDQ) in aqueous solution by means of nanoprecipitation. The resultant PEG5k-GL2-IMDQ with a hydrodynamic size of 200 nm could disintegrate specifically and achieve a rapid drug release, which was approximately attributed to the cleavage of the ester bond and glucuronide under conditions of the overexpressed esterases and β-GUS in tumor cells, respectively. The residual linker was prone to being self-immolative, thus enabling the release of IMDQ in native form. In vitro cumulative drug release experiments showed that minimal IMDQ release was observed in the absence of enzymes. In contrast, the PEG5k-GL2-IMDQ could achieve a sustained drug release and reached a plateau in the presence of enzymes after 4 days. Thus, this design could avoid the severe systemic side effects caused by drug leakage during blood circulation. Significantly, this nano-prodrug could provoke a strong and long-lasting immune stimulation in draining lymphoid tissues avoiding systemic inflammation. In addition, PEG5k-GL2-IMDQ could also promote the recruitment and maturation of dendritic cells (DCs) in lymph nodes. In another study, Mou and co-workers reported “chemogene”-based nanoassemblies for combinatorial chemo and gene therapy to reverse drug resistance (Table 2).123 As shown in Fig. 13B, the chemogene was synthesized by incorporating a nucleoside analog antitumor drug (floxuridine (F)) in the antisense sequence which took the place of thymine (T). The block copolymer poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG-b-PCL) conjugated chemogene had a very low CMC value (∼8.37 μg mL−1) and could self-assemble into spherical nucleic acid (SNA)-like NPs for co-delivering the chemodrugs and B-cell lymphoma 2 (Bcl-2) protein-targeted antisense oligonucleotides. The obtained two-in-one NPs termed SNA(F-Bcl-2ASO) with PCL polymerization of ∼28, 68, and 120 have a hydrodynamic size of approximately 17.5, 24.0, and 40.7 nm, respectively. The SNA (F-Bcl-2 ASO) exhibited an excellent cellular uptake efficiency compared to the naked F-Bcl-2 ASO. Upon successful entry into tumor cells, the released F-Bcl-2 ASO could hybridize with Bcl-2 mRNA and subsequently recruited the RNase H to digest the target mRNA, thus enabling the relative gene downregulation, and the chemogenes could release toxic floxuridine upon cleavage by DNase II in tumor cells. Notably, about 54% knockdown of Bcl-2 mRNA was observed in drug-resistant BEL-7402 cells. In contrast, the naked F-Bcl-2 ASO failed to downregulate the Bcl-2 mRNA due to its low cell internalization efficiency. As expected, SNA (F-Bcl-2 ASO) exhibited an excellent antitumor efficacy against subcutaneous and orthotopic drug-resistant BEL-7402 tumors compared to the free floxuridine combined with naked F-Bcl-2 ASO. This reported chemogene strategy could be applied to other therapeutic nucleic acids such as microRNA, siRNA, and small hairpin RNA.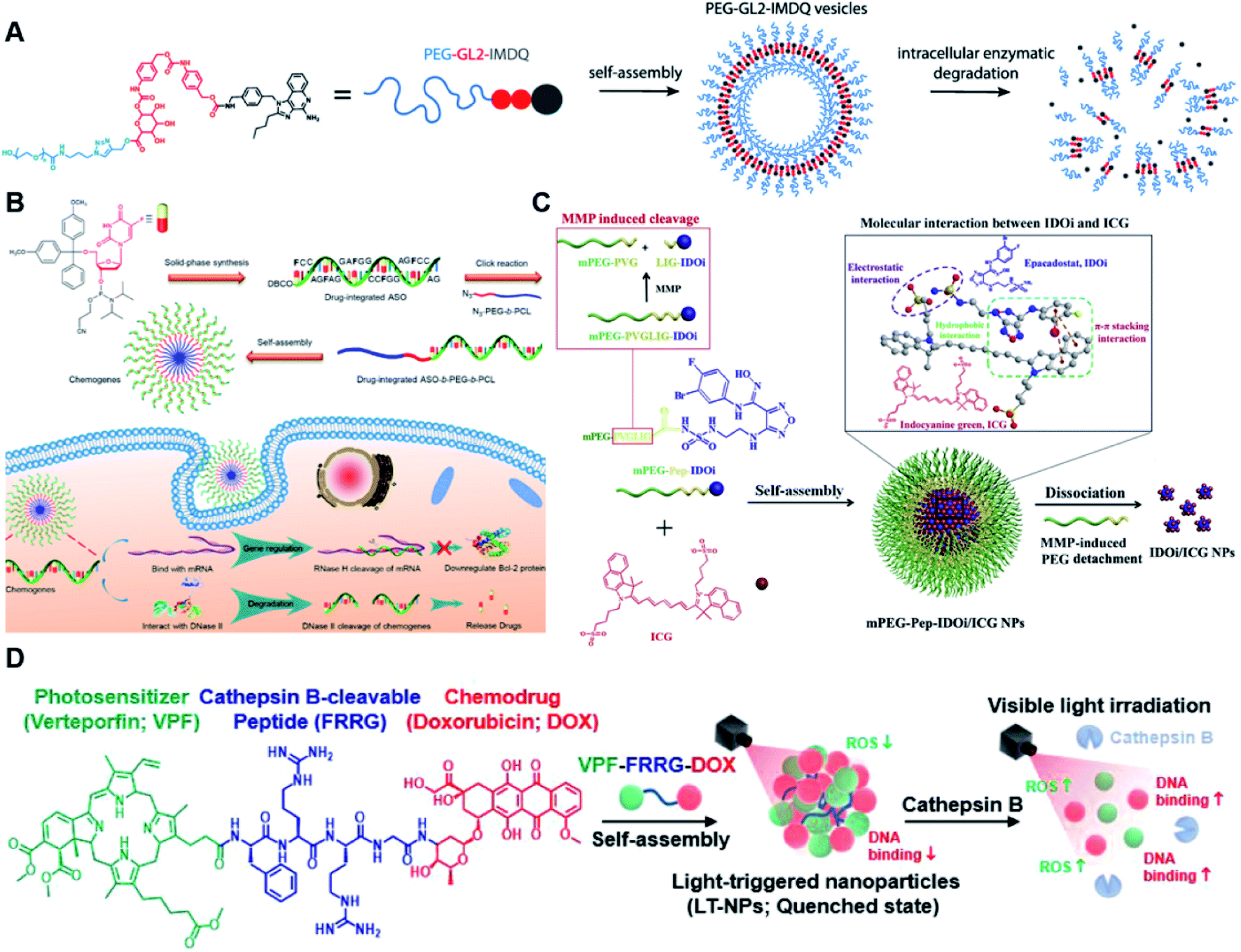 | ||
| Fig. 13 (A) Chemical design of the amphiphilic imidazoquinoline (IMDQ) prodrug and its self-assembly into vesicular nanostructures. Reproduced from ref. 122 with permission from American Chemical Society, Copyright 2020. (B) Illustration of the F-integrated ASO-b-PEG-b-PCL triblock copolymer and its self-assembly to reverse chemoresistance. Reproduced from ref. 123 with permission from American Chemical Society, Copyright 2021. (C) Preparation of TME-responsive nano-prodrugs by co-self-assembly of a photothermal agent and IDO inhibitor for cancer immunotherapy. Reproduced from ref. 125 with permission from Elsevier Ltd, Copyright 2020. (D) Chemical structures of a cathepsin B-responsive prodrug and its self-assemblies for combinatorial chemo-photodynamic therapy to potentiate the effective checkpoint blockade-based tumor immunotherapy. Reproduced from ref. 126 with permission from American Chemical Society, Copyright 2021. | ||
Luo et al. synthesized another enzyme-responsive dendritic polymer–paclitaxel prodrug conjugate (POEGMA-GFLG-PTX), in which PTX was conjugated onto poly[oligo(ethylene glycol) methyl ether methacrylate] (polyOEGMA) via a cathepsin B-responsive tetra-peptide GFLG linkage (Table 2).124 The resulting POEGMA-GFLG-PTX had a low CMC value of 0.2 μg mL−1, which could self-assemble into NPs with a hydrophobic core for encapsulation of T1 (an imidazole derivative) and pyropheophorbide a. The average hydrodynamic size of the obtained nano-prodrug was about 163.1 ± 13.0 nm. Both cargoes (T1 and PPa) in this nanoassembly had a high encapsulation efficiency of over 60%. In this study, the T1 with two-photon (TP) absorption and high energy-transfer efficiency was exploited to enhance the two-photon photodynamic therapy. Upon successful internalization by tumor cells, PTX was rapidly released due to the cleavage of GFLG peptide in the presence of intracellular cathepsin B. The resulting nano-prodrug could achieve enhanced antitumor efficiency in 4T1 xenograft mice through combinatorial chemotherapy and two-photon photodynamic therapy. In another study, Liu and co-workers reported a tumor microenvironment-responsive nano-prodrug for improved photo-immunotherapy (Table 1).125 As shown in Fig. 13C, the MMP-2 responsive peptide sequence PVGLIG was introduced as a linker to conjugate the indoleamine 2,3-dioxygenase (IDO) inhibitor (IDOi, Epacadostat) with PEG (Table 2). The PEGylated IDOi and photosensitizer (ICG) could co-self-assemble into uniform NPs (mPEG-Pep-IDOi/ICG NPs) via intermolecular interactions. As a result of the peptide cleavage by MMP-2 in the TME, the resulting nano-prodrug with a large initial size (∼140 nm) could transform to smaller-sized (<40 nm) drug–drug complexes that could enhance the drug penetration in tumor tissues. The mPEG-Pep-IDOi/ICG NPs exhibited an enhanced release of both IDOi and ICG under conditions of MMP-2 when compared to the mPEG-IDOi/ICG NPs under the same conditions. Upon NIR laser irradiation, the ICG mediated phototherapy could kill tumor cells and evoke an in situ antitumor immune response to facilitate IDO-mediated immunosuppression. The author found that the phototherapy mediated by mPEG-Pep-IDOi/ICG NPs could improve the expression of the co-stimulatory molecules (CD80 and CD86) on dendritic cells (DCs). Moreover, the mPEG-Pep-IDOi/ICG NPs could promote DC maturation in the tumor site probably due to their improved tumor specific accumulation and tumor cell internalization ability. Notably, this MMP-2-responsive nano-prodrug could synergistically enhance the tumor immunotherapy in combination with the PD-L1 checkpoint blockade, leading to a substantial antitumor effect on both primary and abscopal tumors.
6.2 Drug–drug conjugate-based nanoassemblies
Recently, Choi et al. reported light-triggered hetero-drug–drug conjugate-based nanoassemblies (LT-NPs) for synergistic chemo-photodynamic therapy to enhance the checkpoint blockade immunotherapy (Table 1).126 The amphiphilic heterodimeric prodrug (VPF-FRRG-DOX) was synthesized by linking the photosensitizer (verteporfin; VPF) with the doxorubicin (DOX) via cathepsin B-specific cleavable peptide (FRRG). The obtained VPF-FRRG-DOX could self-assemble into LT-NPs without additional helper materials through intermolecular π–π stacking and hydrophobic interactions (Fig. 13D). The obtained LT-NPs had an average size of 87.12 ± 3.95 nm and high drug loading efficiency (>70%). The LT-NPs showed minimized side effects due to the accelerated drug release of DOX and VPF in cathepsin B-overexpressed tumor cells rather than the normal cells that have a lower cathepsin B expression. Upon laser irradiation, the DOX and VPF could be released for combined chemo-photodynamic therapy (Table 1) that elicited immunogenic cell death (ICD) by activating the damage-associated molecular pattern (DAMP) signals, thereby leading to dendritic cell (DC) maturation and cytotoxic T lymphocyte activation. Strong antitumor immune responses were observed by combinatorial treatment with anti-PD-L1 antibodies that not only led to complete primary tumor regression with local light irradiation treatment, but also prevented the progression of distant pulmonary metastatic tumors and rechallenged tumors.7. Hypoxia-responsive nanoassemblies
Hypoxia is a common pathological feature in most solid tumors.127 The highly abnormal and defective tumor microvasculature results in the impaired microcirculation and subsequently limited the transport and diffusion of oxygen in the solid TME.128,129 Thus, the cancer cells within the interior tumor tissues are deprived of adequate oxygen supply. Therefore, hypoxia could be exploited as an endogenous stimulus for tumor-specific controlled drug release by using different hypoxia-responsive linkers.130 Various hypoxia-responsive drug delivery systems have been explored for tumor targeted drug delivery.7.1 Polymer–drug conjugate-based nanoassemblies
In a recent study, Cui et al. developed a semiconducting polymer-based nano-prodrug (SPNpd) for synergistic chemo-photodynamic therapy (Table 1).131 As shown in Fig. 14A, SPNpd was prepared by self-assembly of the amphiphilic PEGylated semiconducting polymer brush, in which the bromoisophosphoramide mustard intermediate (IPM-Br) was conjugated onto the light-responsive photodynamic backbone through hypoxia-cleavable linkers (Table 2). TEM images showed that the resulting SPNpd and drug-free control NPs (SPNc) had a uniform spherical morphology with an average size of ∼30 nm and could maintain a good colloidal stability in PBS for over 20 days. Upon NIR irradiation, the SPNpd could generate singlet oxygen (1O2) for photodynamic therapy. Meanwhile, the photoirradiation-depleted oxygen exacerbated hypoxia in the TME, which facilitated the rapid release of IPM-Br under catalysis of nitroreductase, thereby enabling the synergistic chemo-photodynamic therapy. Notably, there was no difference in the cell viability of SPNc (45.7%) and SPNpd (43.7%) treated cells upon light irradiation, which indicated that the prodrug was not activated in normoxia. However, the cell viability of SPNpd-treated cells decreased to 34.1% under hypoxic conditions without light irradiation, which was 2.5-fold lower than that of the SPNpd-treated cells under normoxic conditions (84.8%), indicating the activation of the prodrug under hypoxic conditions. This study describes the first hypoxia-activable phototherapeutic polymeric prodrug system that had a high potential for efficient tumor therapy. | ||
| Fig. 14 (A) Schematic illustration of a hypoxia-activable semiconducting polymeric nano-prodrug for synergistic chemo-photodynamic therapy. Reproduced from ref. 131 with permission from Wiley-VCH, Copyright 2018. (B) Chemical structures of a hypoxia-responsive nano-prodrug (Ce6/PTX2-Azo) and its self-assembly for synergistic photodynamic-chemotherapy. Reproduced from ref. 132 with permission from Wiley-VCH., Copyright 2020. | ||
7.2 Drug–drug conjugate-based nanoassemblies
Recently, Zhou et al. reported a hypoxia-activable nano-prodrug (Ce6/PTX2-Azo) for synergistic photodynamic-chemotherapy (Table 1).132 As shown in Fig. 14B, the Ce6/PTX2-Azo was prepared by co-self-assembly of the hypoxia-responsive dimeric prodrug (PTX2-Azo) and photosensitizer chlorin e6 (Ce6) decorated peptide copolymer. The PTX2-Azo was synthesized by inserting the azobenzene (Azo) linker between two molecules of PTX. The hydrodynamic sizes of the prepared Ce6/PTX2-Azo NPs, Ce6 NPs, and PTX2-Azo NPs were determined to be 35 ± 5 nm, 45 ± 10 nm and 75 ± 12 nm, respectively. The sodium dithionite (Na2S2O4) solution was used to mimic hypoxic conditions to study the in vitro drug release of the nano-prodrugs. Interestingly, an increased concentration of Na2S2O4 led to an increased PTX release and the yellow NP solution became transparent during the drug release process. Upon successful internalization by tumor cells, Ce6 could generate ROS to induce cell apoptosis. Notably, the ROS generation could consume O2 to aggravate hypoxia to promote PTX release, thus enabling a synergistic chemo-photodynamic antitumor effect. As expected, enhanced tumor inhibition was observed in the Ce6/PTX2-Azo NP + L-treated group, which was found to be better than the single PDT or chemotherapeutic effect (Fig. 14B).8. Multiple stimulus-responsive nanoassemblies
Self-assembled prodrugs could be designed for hierarchical drug delivery in response to dual or multiple stimuli that could significantly improve the selectivity and efficiency of drug release in the tumor site.133 In previous sections, most of the above-mentioned prodrug nanoassemblies were constructed based on the degradable linkers in response to single stimuli including the acidic TME/lysosome conditions and intracellular oxidative/reductive environment. However, some normal cells possess similar acidic and oxidative/reductive environments that could lead to potential off-target drug activation. Therefore, single stimulus-responsive drug release may be inadequate.8.1 Polymer–drug conjugate-based nanoassemblies
Limited tumor penetration and diffusion of nanomedicine creates obstacles for effective anticancer drug delivery. To overcome this barrier, Zhou et al. reported a γ-glutamyl transpeptidase (GGT) and GSH dual-responsive zwitterionic polymer–camptothecin conjugate for enhanced drug penetration and cancer therapy (Table 1).134 It should be noted that the hydrolysis of γ-glutamylamides is structure-dependent. For example, the γ-glutamylamides of glutathione (GSH) and its ophthalmic acid analogue could be easily cleaved by GGT, while GGT is much less active towards L-γ-glutamylglycine. In this study, two different polymer drug conjugates PBEAGA-CPT and PEAGA-CPT were synthesized by introducing camptothecin (CPT) to the 2-(L-γ-glutamyl-L-α-aminobutyrylamino)ethyl acrylamide (BEAGA) and 2-(L-γ-glutamylamino)ethyl acrylamide (EAGA) via disulfide bonds, respectively (Fig. 15A). The resulting PBEAGA18-CPT5 could self-assemble into homogeneous NPs that had a hydrodynamic size of 9 nm. The zeta potential (ζ) of PBEAGA18-CPT5 became positive within 15 h under conditions of 10 U ml−1 GGT, while PEAGA18-CPT5 still remained negatively charged even after 48 h under the same conditions. The zeta potential (ζ) of both PBEAGA18-CPT5 and PEAGA18-CPT5 remained negative even after 48 h incubation in the presence of the normal blood level of GGT (0.05 U ml−1), thereby minimizing the side effects. Upon arriving at the tumor blood vessels or extravasating into the tumor interstitium, the γ-glutamyl moieties were cleaved by overexpressed γ-glutamyl transpeptidase on endothelial cell membranes or tumor cell membranes, leading to the exposure of the positively charged primary amines. Significantly, the positively charged amine could enhance the trans-endothelial and transcellular transport of the nanomedicines to enable homogeneous drug distribution in tumor tissues. CPT was released in tumor cells under highly abundant endogenous GSH, thus realizing an improved antitumor efficiency. In another study, Dong et al. reported a polymeric prodrug, named CPT-ss-poly(BYP-hyd-DOX-co-EEP), for pH/redox dual responsive delivery of camptothecin (CPT) and doxorubicin (DOX).135 As shown in Fig. 15B, the CPT-ss-poly(BYP-hyd-DOX-co-EEP) was synthesized by a CPT derivative (CPT-ss-OH)-mediated random ring-opening polymerization (ROP) of two cyclic phosphate monomers (BYP and EEP) followed by introducing the DOX onto the resultant PBYP via the acid-labile hydrazone linker (–hyd–, Table 2). The resulting amphiphilic polymeric prodrug could form NPs with core–shell structures, in which the hydrophobic PBYP segment containing DOX and CPT formed the core and the hydrophilic PEEP parts formed the outer shell. The hydrodynamic size of the obtained nano-prodrug was determined to be 90 nm. Notably, the CPT-ss-poly(BYP-hyd-DOX-co-EEP) NPs could disassemble due to the degradation of the polyphosphate chain under the conditions of phosphodiesterase I. At the same time, the cleavage of the redox-sensitive disulfide carbonate linker and acid-sensitive hydrazone bond under reductive and acidic conditions could cause the rapid release of CPT and DOX, respectively. | ||
| Fig. 15 (A) Chemical design of GGT-activable cationizing PBEAGA-CPT and the non-GGT-activable PEAGA-CPT, and the mechanism for GGT-catalysed γ-glutamylamide hydrolysis. Reproduced from ref. 134 with permission from Springer Nature, Copyright 2019. (B) Molecular structure of the redox/pH dual-responsive CPT-ss-poly(BYP-hyd-DOX-co-EEP). Reproduced from ref. 135 with permission from American Chemical Society, Copyright 2019. (C) Schematic illustration of the PEG-GAx/Pt formation through coordination of the polyphenol and CDDP, and the mechanism for intracellular dual-responsive drug release. Reproduced from ref. 136 with permission from Elsevier Ltd, Copyright 2020. (D) Molecular structures of mPEG-Phe-TK-Phe-hyd-DOX and its self-assemblies for tumor therapy. Reproduced from ref. 137 with permission from Elsevier Ltd, Copyright 2021. (E) Chemical structure of diblock pDHPMA-DOX. Reproduced from ref. 138 with permission from Elsevier Ltd, Copyright 2019. | ||
In another study, Xiang et al. developed a novel polyphenol–cisplatin complexation-based core–shell structured nano-prodrug (PEG-GAx/Pt) to achieve pH and ROS-responsive drug delivery (Table 1).136 To prepare the PEG-GAx/Pt, methoxyl-PEG terminated with one (PEG-GA) or two (PEG-GA2) gallic acid moieties was complexed with cisplatin (CDDP) by using the polyphenol–metal coordination method (Fig. 15C). When the molar ratio of CDDP to GA (MPt:MGA) was set to 1:1 and 10:1, the resulting PEG-GA/Pt and PEG-GA2/Pt could self-assemble into homogeneous NPs that have a volume size of 80–100 nm and 110–130 nm, respectively. The obtained NPs had a good colloidal stability with no obvious size changes in PBS supplemented with 5%, 10%, or 50% FBS for 72 h at 37 °C. The loading content of CDDP in PEG-GA and PEG-GA2 was determined to be about 17.7% to 29.8%, respectively. In vitro drug release experiments showed that about 47% and 59% of Pt was released from PEG-GA/Pt and PEG-GA2/Pt NPs at pH 5.0 within 48 h, respectively. However, <20% Pt drug was released from both PEG-GA/Pt and PEG-GA2/Pt at pH 7.4. The pH-triggered Pt release was attributed to the weakened coordination between polyphenols and Pt(II) under acidic conditions.
Notably, the addition of H2O2 could promote drug release due to the oxidation of galloyl groups. The PEG-GAx/Pt NPs exhibited prolonged blood circulation time and enhanced tumor accumulation, thus increasing antitumor efficiency, and minimizing toxicity. In another study, Xu et al. synthesized a ROS/pH dual-responsive polymer–DOX conjugate (mPEG-Phe-TK-Phe-hyd-DOX) for efficient drug release and improved antitumor efficiency (Fig. 15D).137 In this study, the acylhydrazine linker (ahy-Phe-TK-Phe-ahy) containing ROS-sensitive thioketal (TK) bonds was inserted between the mPEG and DOX, in which DOX was conjugated onto the ahy-Phe-TK-Phe-ahy via acid-sensitive hydrazone bonds (Table 2). The CMC value of mPEG-Phe-TK-Phe-hyd-DOX was calculated to be 3.79 μg mL−1. The hydrophobic phenylalanine moieties (Phe-TK-Phe) could facilitate the self-assembly of the mPEG-Phe-TK-Phe-hyd-DOX to uniform nanostructures with a hydrodynamic size of 41.32 nm. The resulting prodrug micelles had a high drug loading content (11.2%) and good colloidal stability due to the π–π interactions of prodrug molecules, which could minimize the drug leakages during blood circulation. In vitro cytotoxicity experiments showed that the IC50 of mPEG-Phe-TK-Phe-hyd-DOX was about 5.50 μg mL−1 against HeLa cells, which was lower than those of mPEG-hyd-DOX (19.2 μg mL−1) and DOX/mPEG-ahy-Phe-TK-Phe-ahy (35.4 μg mL−1). As expected, the ROS/pH-responsive nano-prodrugs exhibited a better antitumor efficacy than the ROS-inert control nano-prodrug. In a similar study, Chen et al. reported an enzyme/pH dual sensitive polymer–doxorubicin conjugate (diblock pDHPMA-DOX) for improved antitumor efficiency of the chemotherapeutic drugs and minimized systemic side effects (Table 1).138 To obtain the diblock pDHPMA-DOX, the DOX was attached onto the GFLG peptide-functionalized diblock copolymer through acid-labile hydrazone bonds (Table 2), in which the N-(1,3-dihydroxypropan-2-yl) methacrylamide (DHPMA) copolymer was synthesized by RAFT polymerization (Fig. 15E). The diblock pDHPMA-DOX could self-aggregate into nanoassemblies that had an average hydrodynamic size of 21 ± 2.2 nm capable of causing ∼82% and ∼80% DOX release at pH 5.0 within 10 h in the presence of cathepsin B or no cathepsin B, respectively. In contrast, a negligible amount of DOX (∼5%) was released from the nano-prodrug at pH 7.4 with or without addition of cathepsin B. The diblock pDHPMA-DOX NPs could maintain a good colloidal stability with no significant aggregation/degradation within 24 h in PBS supplemented with 50% FBS. Upon successful internalization by tumor cells, the diblock copolymer-based NPs (90 kDa) were degraded into small fragments that had smaller molecular weight (45 kDa) as a result of the cleavage of GFLG peptide under conditions of lysosomal cathepsin B. In vivo pharmacokinetics experiments revealed that the diblock pDHPMA-DOX NPs had a prolonged blood circulation time of 9.8 h, which was longer than that of free DOX (3.7 h). As expected, the diblock pDHPMA-DOX NPs displayed an enhanced antitumor efficiency for 4T1 xenograft tumors.
8.2 Drug–drug conjugate-based nanoassemblies
In a recent study, Zhu et al. described precise PTX–chemogene conjugate-based and carrier-free nanoassemblies for combinatorial therapy to overcome drug resistance (Table 1).139 To obtain the drug–chemogene conjugates, the PTX dimer was synthesized first by connecting two molecules of PTX through a fluorescent and azide-containing dithiomaleimide (DTM) linker (Table 2). After that, the floxuridine (FdU)-integrated antisense oligonucleotides (i.e. chemogene) were subsequently conjugated with PTX dimers via click chemistry (Fig. 16A). The resulting PTX–chemogene conjugates having a low CMC value (7.5 μM) could self-assemble into spherical nucleic acid (SNA)-like prodrug micelles with an average size of 81 nm. Notably, the PTX–chemogene-based prodrug micelles could simultaneously co-deliver three different kinds of drugs to avoid dramatic pharmacokinetic differences, thus maximizing the combination therapy efficiencies. Upon successful entry into tumor cells, the antisense oligonucleotides could downregulate the P-glycoprotein (P-gp) expression followed by simultaneously releasing fluoride monophosphate (FdUMP) and PTX under DNase II degradation and intracellular GSH. In vitro qRT-PCR analysis experiments showed that about 67% of P-gp mRNA was downregulated in HeLa/PTX cells after treatment with PTX-chemogene-based nanoassemblies, which was higher than that of the naked ASO (20%). Notably, the PTX-chemogene has a lower IC50 value (0.06 μM) against HeLa cells than the control group (PTX-ASO). As expected, PTX-chemogene exhibited a higher inhibition rate (∼80%) against drug-resistant tumors. In another study, Xia et al. synthesized different linker (sulfur, selenide, or telluride)-bridged paclitaxel dimers that could form into homogeneous NPs (PTX2-R NPs, R = S, Se and Te) for programmed drug release (Fig. 16A).140 The obtained NPs (PTX2-R NPs, R = S, Se and Te) had a hydrodynamic size of 179.9 nm, 187.5 nm, and 204.9 nm, respectively. The obtained PTX2-R NPs with homogeneous spherical structures had a high drug loading capacity (>84 wt%) and could maintain a good stability in water for one week with minor size changes. The in vitro drug release rates of PTX2-S, PTX2-Se, and PTX2-Te dimers were evaluated under the same conditions of H2O2 and DTT, and the results showed that the PTX2-Te dimer could be almost completely oxidized when incubated in 1 mM H2O2 for 1 h. In contrast, only 10% of PTX2-S and 50% of PTX2-Se dimer was oxidized under the same conditions. Similarly, 95% of PTX2-Te was degraded in the presence of 10 mM DTT in 24 h, which was faster than the degradation rate of PTX2-S and PTX2-Se (<20%). These results showed that the tellurium-bridged paclitaxel dimer presents a preferable GSH and ROS dual-responsive capability when compared with the sulfur and selenium-bridged paclitaxel dimeric prodrugs. The in vitro cytotoxicity experiments showed that PTX2-Te NPs exhibited a highly selective cytotoxicity against tumor cells. | ||
| Fig. 16 (A) Chemical design and synthetic routes of the PTX-chemogene conjugate and its self-assembly for combination therapy to overcome drug resistance. Reproduced from ref. 139 with permission from Wiley-VCH, Copyright 2020. (B) Schematic representation of sulfur/selenium/tellurium-bridged homodimeric prodrugs and their intracellular ROS/GSH dual-responsive drug release. (C) HPLC analysis of PTX2-S, PTX2-Se and PTX2-Te dimers in the presence of 1 mM H2O2 or 10 mM DTT for 24 h at 37 °C. Reproduced from ref. 140 with permission from Elsevier Ltd, Copyright 2020. | ||
9. Polymer–drug conjugate-based nano-prodrugs in clinical translation
Polymer–drug conjugates have long been used for drug delivery due to their advantages, including enhanced drug solubility, improved pharmacokinetics, controlled drug release ability, etc.141 Since the first polymer–enzyme conjugate drug (Adagen) obtained its market approval, a remarkable growth of polymer–drug conjugates in the field of drug delivery was observed, and many polymer–drug conjugates that could be easily prepared have already entered the market or are in different stages of clinical trials.142,143 This section will summarize some representative polymer–drug conjugate-based nanomedicines that are in clinical development.NC-6004, a polymeric cisplatin nanoparticle (∼30 nm), was constructed by encapsulating cisplatin into the micelles through the formation of a polymer–metal complex between polyethylene glycol-poly(glutamic acid) block copolymers (PEG-P(Glu)) and cisplatin (Table 2).144,145 Compared to free cisplatin that could be excreted rapidly from the human body, NC-6004 could significantly extend the blood circulation time of cisplatin as the outer PEG shell on the surface of the micelles could minimize its clearance by the reticuloendothelial systems.146 Furthermore, NC-6004 with a sustained drug release ability has lower toxicity than the native cisplatin.147 In a phase Ib/II trial study, NC-6004 plus gemcitabine exhibited a greater anti-tumor activity and no clinically significant neuro-, oto-, or nephrotoxicity. NC-6300, another clinical candidate, was prepared by conjugating epirubicin to PEG-b-poly(aspartic acid) through hydrazone linkages.148 The resulting polymer–epirubicin conjugates could form into micelles that could keep stable during blood circulation but achieve fast release of drug in acidic environments (e.g. endosomal and lysosomal).149 Notably, NC-6300 could significantly minimize the system side-effects so that higher doses of drug could be used through micellar therapy.148 Similarly, NK911 is a doxorubicin-loaded PEG-b-poly(aspartic acid) copolymer-based prodrug micelle (∼40 nm), in which the doxorubicin is both physically encapsulated in the micelle and conjugated to the PEG-b-poly(aspartic acid) copolymer via amide bonds.150 NK911 exhibited a prolonged blood circulation time and a larger area under the plasma drug concentration–time curve (AUC) than native doxorubicin in a phase I clinical trial.150 CriPec® Docetaxel (CPC634) consists of core-cross linked micellar nanoparticles with covalently entrapped docetaxel. CPC634 entered the first-in-human study and finished its phase I testing in 2018. The CPC634 exhibited an improved pharmacokinetics and therapeutic index in patients with advanced solid tumors.151 The polymeric nanoassemblies CRLX101 and CRLX301 are constructed by self-assembly of cyclodextrin–drug conjugates, which were synthesized by covalently conjugating cyclodextrin-PEG copolymer with camptothecin and docetaxel, respectively.152,153 CRLX301 exhibited a controlled drug release ability and slower clearance than free docetaxel in a phase I/IIa study.154 In a randomized phase II trial, CRLX101 plus bevacizumab had no discernible increase in progression-free survival (PFS) when compared to the standard of care (SOC) in metastatic renal cell carcinoma.155
10. Summary and perspectives
Self-assembled prodrugs, by integration of prodrug design and biomedical nanotechnology, offer a great deal of versatility in various drug delivery systems for controlled release of anti-cancer drugs. In this review, we have comprehensively summarized recent advances of single/dual-stimulus-responsive self-assembled prodrugs for tumor chemotherapy, phototherapy, immunotherapy, theranostics and combinatorial therapy, by covering different conjugation strategies (polymer–drug conjugates, drug–drug conjugates, etc.) in the preparation of nano-prodrugs. Significantly, we provided details of various linker chemistries that allowed self-assembly of amphiphilic prodrugs to achieve temporal/spatial controlled-release of drugs. Notably, the specific pathophysiological characteristics in tumor sites including hypoxia, aberrant enzymes, low pH, high redox and oxidative stress state have inspired the development of various prodrug chemistries for controlled drug release at tumor sites. Self-assembled prodrugs have many advantages over their small-molecule drug/prodrug counterparts, including improved ADMET properties, prolonged blood circulation time, active/passive tumor targeting ability, spatial/temporal controlled-release of drugs, combinatorial therapy, and others.Despite obvious benefits afforded by self-assembled prodrug strategies, there are still several challenges that need to be circumvented before their successful clinical translation. First, the CMC value of amphiphilic prodrugs should be as low as possible, as it is an important factor that affects the systemic stability of prodrug micelles during blood circulation. Second, the non-homogeneous nano-prodrug activation and distribution still exists due to the heterogeneous nature of tumor tissues, thereby leading to the suboptimal therapeutic efficacy. Therefore, exogenous physical stimuli (light, temperature, ultrasound, X-ray, etc.) plus endogenous stimuli (pH, GSH, ROS, enzyme, and hypoxia) could thus be exploited for development of multi-responsive drug delivery systems. Third, the potential systemic cytotoxicity of the carrier materials still poses challenges that restrict the clinical translation of the polymeric nanoparticles. Thus, materials with high biodegradability should be well explored, or carrier-free drug delivery systems could be developed. Fourth, it is difficult to avoid the batch-to-batch variations of polymer–drug conjugation limiting its clinical translation. That is why the design of prodrug nanoassemblies should be as simple as possible to facilitate easy preparation. Finally, there is an urgent need to develop novel prodrug chemistry that allows more efficient and specific drug release at tumor sites. For example, we can try to develop self-immolative linker-based combinatorial chemistry with a cascade reaction ability to improve the stimulus-responsive drug release ability. Nevertheless, self-assembled prodrug designs have great potential in tumor-targeted drug delivery systems, and we look forward to seeing more clinical translation in the coming years.
Author contributions
Xiao Dong and Shao Q. Yao conceived the idea of the review and supervised this work. Xiao Dong collected references and prepared the initial draft; Rajeev K. Brahma contributed to the graphic design; Chao Fang performed the analysis with discussions and modified the images; Xiao Dong and Shao Q. Yao edited the manuscript; all authors participated in the discussion of the draft and gave approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the GSK-EDB Trust Fund (R-143-000-688-592), the Synthetic Biology Research & Development Programme (SBP) of the National Research Foundation (SBP-P4 and SBP-P8) of Singapore, Fundamental Research Funds for the Central Universities, Yangfan Project of Shanghai Science and Technology Commission (No. 22YF1427500), and Development Scholarship for Outstanding Doctoral Graduates of Shanghai Jiaotong University.Notes and references
- J. Ferlay, M. Colombet, I. Soerjomataram, D. M. Parkin, M. Pineros, A. Znaor and F. Bray, Int. J. Cancer, 2021, 149, 778 CrossRef CAS PubMed.
- M. J. Markham, K. Wachter, N. Agarwal, M. M. Bertagnolli, S. M. Chang, W. Dale, C. S. M. Diefenbach, C. Rodriguez-Galindo, D. J. George, T. D. Gilligan, R. D. Harvey, M. L. Johnson, R. J. Kimple, M. A. Knoll, N. LoConte, R. G. Maki, J. L. Meisel, J. A. Meyerhardt, N. A. Pennell, G. B. Rocque, M. S. Sabel, R. L. Schilsky, B. J. Schneider, W. D. Tap, R. G. Uzzo and S. N. Westin, J. Clin. Oncol., 2020, 38, 1081 CrossRef PubMed.
- V. Schirrmacher, Internet J. Oncol., 2019, 54, 407–419 CAS.
- A. G. Waks and E. P. Winer, JAMA, 2019, 321, 288–300 CrossRef CAS PubMed.
- E. C. Ko, D. Raben and S. C. Formenti, Clin. Cancer Res., 2018, 24, 5792–5806 CrossRef CAS PubMed.
- S. H. Yun and S. J. J. Kwok, Nat. Biomed. Eng., 2017, 1, 0008 CrossRef CAS PubMed.
- S. Mohanty, R. Rajaram, K. Y. Bilimoria, R. Salem, T. M. Pawlik and D. J. Bentrem, Eur. J. Surg. Oncol., 2016, 113, 175–180 CrossRef PubMed.
- C. E. Humber, J. F. Tierney, R. P. Symonds, M. Collingwood, J. Kirwan, C. Williams and J. A. Green, Ann. Oncol., 2007, 18, 409–420 CrossRef CAS PubMed.
- A. M. Vargason, A. C. Anselmo and S. Mitragotri, Nat. Biomed. Eng., 2021, 5, 951–967 CrossRef PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 7 CrossRef PubMed.
- P. Y. Muller and M. N. Milton, Nat. Rev. Drug Discovery, 2012, 11, 751–761 CrossRef CAS PubMed.
- R. Walther, J. Rautio and A. N. Zelikin, Adv. Drug Delivery Rev., 2017, 118, 65–77 CrossRef CAS PubMed.
- B. Du, D. Li, J. Wang and E. Wang, Adv. Drug Delivery Rev., 2017, 118, 78–93 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H. S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- A. Albert, Nature, 1958, 182, 421–423 CrossRef CAS PubMed.
- S. Sheikh, D. Ernst and A. Keating, Mol. Ther., 2021, 29, 1716–1728 CrossRef CAS PubMed.
- X. Xia, Y. Zhou and H. Gao, Chem. Commun., 2021, 57, 8842–8855 RSC.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- A. Kakkar, G. Traverso, O. C. Farokhzad, R. Weissleder and R. Langer, Nat. Rev. Chem., 2017, 1, 0063 CrossRef CAS PubMed.
- W. Mu, Q. Chu, Y. Liu and N. Zhang, Nanomicro Lett., 2020, 12, 142 CAS.
- S. S. Liew, J. Zhou, L. Li and S. Q. Yao, Chem. Commun., 2021, 57, 3215–3218 RSC.
- P. Yuan, X. Mao, X. Wu, S. S. Liew, L. Li and S. Q. Yao, Angew. Chem., Int. Ed., 2019, 58, 7657–7661 CrossRef CAS PubMed.
- X. Dong, L. L. Mu, X. L. Liu, H. Zhu, S. C. Yang, X. Lai, H. J. Liu, H. Y. Feng, Q. Lu and B. B. S. Zhou, Adv. Funct. Mater., 2020, 30, 2000309 CrossRef CAS.
- F. Moradi Kashkooli, M. Soltani and M. Souri, J. Controlled Release, 2020, 327, 316–349 CrossRef CAS PubMed.
- M. T. Manzari, Y. Shamay, H. Kiguchi, N. Rosen, M. Scaltriti and D. A. Heller, Nat. Rev. Mater., 2021, 6, 351–370 CrossRef CAS PubMed.
- W. Yu, R. Liu, Y. Zhou and H. Gao, ACS Cent. Sci., 2020, 6, 100–116 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- S. Raj, S. Khurana, R. Choudhari, K. K. Kesari, M. A. Kamal, N. Garg, J. Ruokolainen, B. C. Das and D. Kumar, Semin. Cancer Biol., 2021, 69, 166–177 CrossRef CAS PubMed.
- A. C. Marques, P. J. Costa, S. Velho and M. H. Amaral, J. Controlled Release, 2020, 320, 180–200 CrossRef CAS PubMed.
- X. Dong, H.-J. Liu, H.-Y. Feng, S.-C. Yang, X.-L. Liu, X. Lai, Q. Lu, J. F. Lovell, H.-Z. Chen and C. Fang, Nano Lett., 2019, 19, 997–1008 CrossRef CAS PubMed.
- G. Li, B. Sun, Y. Li, C. Luo, Z. He and J. Sun, Small, 2021, 17, 2101460 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev., 2016, 1, 16014 CAS.
- L. P. Ganipineni, F. Danhier and V. Préat, J. Controlled Release, 2018, 281, 42–57 CrossRef CAS PubMed.
- D. Dutta, W. Ke, L. Xi, W. Yin, M. Zhou and Z. Ge, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1585 CAS.
- V. Taresco, C. Alexander, N. Singh and A. K. Pearce, Adv. Ther., 2018, 1, 1800030 CrossRef.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Chem. Soc. Rev., 2017, 46, 6638–6663 RSC.
- C. Souza, D. S. Pellosi and A. C. Tedesco, Expert Rev. Anticancer Ther., 2019, 19, 483–502 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- R. Kumari, D. Sunil and R. S. Ningthoujam, J. Controlled Release, 2020, 319, 135–156 CrossRef CAS PubMed.
- X. Ji, Z. Pan, B. Yu, L. K. De La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC.
- S. P. Vaidya and M. Patra, ChemBioChem, 2021, 22, 2998–3000 CrossRef CAS PubMed.
- A. Nguyen, R. Bottger and S. D. Li, Biomaterials, 2021, 275, 120955 CrossRef CAS PubMed.
- A. Najjar, A. Najjar and R. Karaman, Molecules, 2020, 25, 884 CrossRef CAS PubMed.
- B. Sun, C. Luo, W. Cui, J. Sun and Z. He, J. Controlled Release, 2017, 264, 145–159 CrossRef CAS PubMed.
- F. Olim, A. R. Neves, M. Vieira, H. Tomás and R. Sheng, Eur. J. Lipid Sci. Technol., 2021, 123, 2000337 CrossRef CAS.
- B. M. A. Sanches and E. I. Ferreira, Int. J. Pharm., 2019, 568, 118498 CrossRef CAS PubMed.
- Z. Wang, J. Chen, N. Little and J. Lu, Int. J. Pharm., 2020, 111, 20–28 CAS.
- A. Xie, S. Hanif, J. Ouyang, Z. Tang, N. Kong, N. Y. Kim, B. Qi, D. Patel, B. Shi and W. Tao, EBioMedicine, 2020, 56, 102821 CrossRef PubMed.
- J. Xiang, X. Liu, G. Yuan, R. Zhang, Q. Zhou, T. Xie and Y. Shen, Adv. Drug Delivery Rev., 2021, 179, 114027 CrossRef CAS PubMed.
- B. Yang, J. Gao, Q. Pei, H. Xu and H. Yu, Adv. Sci., 2020, 7, 2002365 CrossRef CAS PubMed.
- Y. Zhang, H. Cui, R. Zhang, H. Zhang and W. Huang, Adv. Sci., 2021, 8, 2101454 CrossRef CAS PubMed.
- S. Li, X. Shan, Y. Wang, Q. Chen, J. Sun, Z. He, B. Sun and C. Luo, J. Controlled Release, 2020, 326, 510–522 CrossRef CAS PubMed.
- K. J. Chen, A. J. Plaunt, F. G. Leifer, J. Y. Kang and D. Cipolla, Eur. J. Pharm. Biopharm., 2021, 165, 219–243 CrossRef CAS PubMed.
- M. R. Horsman and P. Vaupel, Front. Oncol., 2016, 6, 66 Search PubMed.
- H. Su, J. M. Koo and H. Cui, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- W. Ma, A. G. Cheetham and H. Cui, Nano Today, 2016, 11, 13–30 CrossRef CAS PubMed.
- R. J. Thibault and V. M. Rotello, Molecular Self-Assembly, Encyclopedia of Polymer Science and Technology, 2004, DOI:10.1002/0471440264.pst504.
- K. J. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- Y. Yan, J. Huang and B. Z. Tang, Chem. Commun., 2016, 52, 11870–11884 RSC.
- U. Bilati, E. Allémann and E. Doelker, Eur. J. Pharm. Sci., 2005, 24, 67–75 CrossRef CAS PubMed.
- V. Estrella, T. Chen, M. Lloyd, J. Wojtkowiak, H. H. Cornnell, A. Ibrahim-Hashim, K. Bailey, Y. Balagurunathan, J. M. Rothberg, B. F. Sloane, J. Johnson, R. A. Gatenby and R. J. Gillies, Cancer Res., 2013, 73, 1524–1535 CrossRef CAS PubMed.
- D. Neri and C. T. Supuran, Nat. Rev. Drug Discovery, 2011, 10, 767–777 CrossRef CAS PubMed.
- E. Boedtkjer and S. F. Pedersen, Annu. Rev. Physiol., 2020, 82, 103–126 CrossRef CAS PubMed.
- S. J. Sonawane, R. S. Kalhapure and T. Govender, Eur. J. Pharm. Sci., 2017, 99, 45–65 CrossRef CAS PubMed.
- T. Zhang, Y. Wang, X. Ma, C. Hou, S. Lv, D. Jia, Y. Lu, P. Xue, Y. Kang and Z. Xu, Biomater. Sci., 2020, 8, 473–484 RSC.
- S. Chen, D. Li, X. Du, X. He, M. Huang, Y. Wang, X. Yang and J. Wang, Nano Today, 2020, 35, 100924 CrossRef CAS.
- J. Song, B. Xu, H. Yao, X. Lu, Y. Tan, B. Wang, X. Wang and Z. Yang, Front. Oncol., 2021, 11, 656717 CrossRef PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- Z. Guo, J. Sui, M. Ma, J. Hu, Y. Sun, L. Yang, Y. Fan and X. Zhang, J. Controlled Release, 2020, 326, 350–364 CrossRef CAS PubMed.
- J. Zhao, Y.-Y. Peng, D. Diaz-Dussan, J. White, W. Duan, L. Kong, R. Narain and X. Hao, Mol. Pharm., 2021 DOI:10.1021/acs.molpharmaceut.1c00518.
- D. Li, Y. Song, J. He, M. Zhang and P. Ni, ACS Biomater. Sci. Eng., 2019, 5, 2307–2315 CrossRef CAS PubMed.
- A. M. Wenthe and E. H. Cordes, J. Am. Chem. Soc., 1965, 87, 3173–3180 CrossRef CAS.
- J. Mu, H. Zhong, H. Zou, T. Liu, N. Yu, X. Zhang, Z. Xu, Z. Chen and S. Guo, J. Controlled Release, 2020, 326, 265–275 CrossRef CAS PubMed.
- H. Zhong, J. Mu, Y. Du, Z. Xu, Y. Xu, N. Yu, S. Zhang and S. Guo, Biomacromolecules, 2020, 21, 803–814 CrossRef CAS PubMed.
- H. Takemoto, T. Inaba, T. Nomoto, M. Matsui, X. Liu, M. Toyoda, Y. Honda, K. Taniwaki, N. Yamada, J. Kim, K. Tomoda and N. Nishiyama, Biomaterials, 2020, 235, 119804 CrossRef CAS PubMed.
- K. A. Andersen, T. P. Smith, J. E. Lomax and R. T. Raines, ACS Chem. Biol., 2016, 11, 319–323 CrossRef CAS PubMed.
- H. Maslah, C. Skarbek, S. Pethe and R. Labruère, Eur. J. Med. Chem., 2020, 207, 112670 CrossRef CAS PubMed.
- X. Hu, Z. Chai, L. Lu, H. Ruan, R. Wang, C. Zhan, C. Xie, J. Pan, M. Liu, H. Wang and W. Lu, Adv. Funct. Mater., 2019, 29, 1807941 CrossRef.
- B. Liu, R. Wu, S. Gong, H. Xiao and S. Thayumanavan, Angew. Chem., Int. Ed., 2020, 59, 15135–15140 CrossRef CAS PubMed.
- Y. Ma, P. He, X. Tian, G. Liu, X. Zeng and G. Pan, ACS Appl. Mater. Interfaces, 2019, 11, 23948–23956 CrossRef CAS PubMed.
- J. Li and P. Liu, Part. Part. Syst. Charact., 2019, 36, 1900113 CrossRef.
- J. Li, X. Li and P. Liu, Colloids Surf., B, 2020, 185, 110608 CrossRef CAS PubMed.
- J. Li, X. Li, M. Pei and P. Liu, Colloids Surf., B, 2020, 192, 111064 CrossRef CAS PubMed.
- X. Wang, X. Cheng, L. He, X. Zeng, Y. Zheng and R. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 28597–28609 CrossRef CAS PubMed.
- C. R. Reczek and N. S. Chandel, Annu. Rev. Cancer Biol., 2017, 1, 79–98 CrossRef.
- V. J. Thannickal and B. L. Fanburg, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2000, 279, L1005–L1028 CrossRef CAS PubMed.
- H. Pelicano, D. Carney and P. Huang, Drug Resistance Updates, 2004, 7, 97–110 CrossRef CAS PubMed.
- G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef PubMed.
- B. Chu, Y. Qu, X. He, Y. Hao, C. Yang, Y. Yang, D. Hu, F. Wang and Z. Qian, Adv. Funct. Mater., 2020, 30, 2005918 CrossRef CAS.
- B. Sun, Y. Chen, H. Yu, C. Wang, X. Zhang, H. Zhao, Q. Chen, Z. He, C. Luo and J. Sun, Acta Biomater., 2019, 92, 219–228 CrossRef CAS PubMed.
- K. Wang, H. Ye, X. Zhang, X. Wang, B. Yang, C. Luo, Z. Zhao, J. Zhao, Q. Lu, H. Zhang, Q. Kan, Y. Wang, Z. He and J. Sun, Biomaterials, 2020, 257, 120224 CrossRef CAS PubMed.
- X. Xia, X. Yang, P. Huang and D. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 18301–18308 CrossRef CAS PubMed.
- W. Yin, W. Ke, W. Chen, L. Xi, Q. Zhou, J. F. Mukerabigwi and Z. Ge, Biomaterials, 2019, 195, 63–74 CrossRef CAS PubMed.
- P. Pei, C. Sun, W. Tao, J. Li, X. Yang and J. Wang, Biomaterials, 2019, 188, 74–82 CrossRef CAS PubMed.
- S. Ma, W. Song, Y. Xu, X. Si, Y. Zhang, Z. Tang and X. Chen, CCS Chem., 2020, 2, 390–400 CrossRef CAS.
- C. Dong, Q. Zhou, J. Xiang, F. Liu, Z. Zhou and Y. Shen, J. Controlled Release, 2020, 321, 529–539 CrossRef CAS PubMed.
- B. Ma, W. Zhuang, H. Xu, G. Li and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 47259–47269 CrossRef CAS PubMed.
- H. Chen, X. Zeng, H. P. Tham, S. Z. F. Phua, W. Cheng, W. Zeng, H. Shi, L. Mei and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 7641–7646 CrossRef CAS PubMed.
- X. Li, C. Zhang, Q. Zheng and X. Shi, Colloids Surf., B, 2019, 179, 218–225 CrossRef CAS PubMed.
- S. Zhang, Z. Wang, Z. Kong, Y. Wang, X. Zhang, B. Sun, H. Zhang, Q. Kan, Z. He, C. Luo and J. Sun, Theranostics, 2021, 11, 6019–6032 CrossRef CAS PubMed.
- C. Luo, B. Sun, C. Wang, X. Zhang, Y. Chen, Q. Chen, H. Yu, H. Zhao, M. Sun, Z. Li, H. Zhang, Q. Kan, Y. Wang, Z. He and J. Sun, J. Controlled Release, 2019, 302, 79–89 CrossRef CAS PubMed.
- M. Jiang, J. Mu, O. Jacobson, Z. Wang, L. He, F. Zhang, W. Yang, Q. Lin, Z. Zhou, Y. Ma, J. Lin, J. Qu, P. Huang and X. Chen, ACS Nano, 2020, 14, 16875–16886 CrossRef CAS PubMed.
- J. A. Cook, D. Gius, D. A. Wink, M. C. Krishna, A. Russo and J. B. Mitchell, Radiat. Oncol., 2004, 14, 259–266 CrossRef PubMed.
- K.-i. Matsumoto, F. Hyodo, A. Matsumoto, A. P. Koretsky, A. L. Sowers, J. B. Mitchell and M. C. Krishna, Clin. Cancer Res., 2006, 12, 2455 CrossRef CAS PubMed.
- L. Chaiswing and T. D. Oberley, Antioxid. Redox Signaling, 2009, 13, 449–465 CrossRef PubMed.
- X. Guo, Y. Cheng, X. Zhao, Y. Luo, J. Chen and W.-E. Yuan, J. Nanobiotechnol., 2018, 16, 74 CrossRef PubMed.
- X. Guo, N. Yang, W. Ji, H. Zhang, X. Dong, Z. Zhou, L. Li, H.-M. Shen, S. Q. Yao and W. Huang, Adv. Mater., 2021, 33, 2007778 CrossRef CAS PubMed.
- Q. Xiao, X. Dong, F. Yang, S. Zhou, M. Xiang, L. Lou, S. Q. Yao and L. Gao, Eur. J. Inorg. Chem., 2021, 27, 14721–14729 CAS.
- S. Li, P. E. Saw, C. Lin, Y. Nie, W. Tao, O. C. Farokhzad, L. Zhang and X. Xu, Biomaterials, 2020, 234, 119760 CrossRef CAS PubMed.
- L. Lu, X. Zhao, T. Fu, K. Li, Y. He, Z. Luo, L. Dai, R. Zeng and K. Cai, Biomaterials, 2020, 230, 119666 CrossRef CAS PubMed.
- N. K. Kildahl, J. Chem. Educ., 1995, 72, 423 CrossRef CAS.
- B. Sun, C. Luo, X. Zhang, M. Guo, M. Sun, H. Yu, Q. Chen, W. Yang, M. Wang, S. Zuo, P. Chen, Q. Kan, H. Zhang, Y. Wang, Z. He and J. Sun, Nat. Commun., 2019, 10, 3211 CrossRef PubMed.
- X. Yi, J. J. Hu, J. Dai, X. Lou, Z. Zhao, F. Xia and B. Z. Tang, ACS Nano, 2021, 15, 3026–3037 CrossRef CAS PubMed.
- Y. Yang, B. Sun, S. Zuo, X. Li, S. Zhou, L. Li, C. Luo, H. Liu, M. Cheng and Y. Wang, Sci. Adv., 2020, 6, eabc1725 CrossRef CAS PubMed.
- X. Xie, C. Zhan, J. Wang, F. Zeng and S. Wu, Small, 2020, 16, e2003451 CrossRef PubMed.
- N. Mitsiades, C. C. Sung, N. Schultz, D. C. Danila, B. He, V. K. Eedunuri, M. Fleisher, C. Sander, C. L. Sawyers and H. I. Scher, Cancer Res., 2012, 72, 6142–6152 CrossRef CAS PubMed.
- O. C. Olson and J. A. Joyce, Nat. Rev. Drug Discovery, 2015, 15, 712–729 CrossRef CAS PubMed.
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS PubMed.
- J. Mu, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2018, 47, 5554–5573 RSC.
- Y. Gu, Y. Zhong, F. Meng, R. Cheng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 2772–2780 CrossRef CAS PubMed.
- B. Wang, S. Van Herck, Y. Chen, X. Bai, Z. Zhong, K. Deswarte, B. N. Lambrecht, N. N. Sanders, S. Lienenklaus, H. W. Scheeren, S. A. David, F. Kiessling, T. Lammers, B. G. De Geest and Y. Shi, J. Am. Chem. Soc., 2020, 142, 12133–12139 CrossRef CAS PubMed.
- Q. Mou, Y. Ma, F. Ding, X. Gao, D. Yan, X. Zhu and C. Zhang, J. Am. Chem. Soc., 2019, 141, 6955–6966 CrossRef CAS PubMed.
- L. Luo, Z. Yin, Y. Qi, S. Liu, Y. Yi, X. Tian, Y. Wu, D. Zhong, Z. Gu, H. Zhang and K. Luo, Appl. Mater. Today, 2021, 23, 100996 CrossRef.
- Y. Liu, Y. Lu, X. Zhu, C. Li, M. Yan, J. Pan and G. Ma, Biomaterials, 2020, 242, 119933 CrossRef CAS PubMed.
- J. Choi, M. K. Shim, S. Yang, H. S. Hwang, H. Cho, J. Kim, W. S. Yun, Y. Moon, J. Kim, H. Y. Yoon and K. Kim, ACS Nano, 2021, 15, 12086–12098 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- G. Helmlinger, F. Yuan, M. Dellian and R. K. Jain, Nat. Med., 1997, 3, 177–182 CrossRef CAS PubMed.
- A. Patel and S. Sant, Biotechnol. Adv., 2016, 34, 803–812 CrossRef CAS PubMed.
- A. Sharma, J. F. Arambula, S. Koo, R. Kumar, H. Singh, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2019, 48, 771–813 RSC.
- D. Cui, J. Huang, X. Zhen, J. Li, Y. Jiang and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 5920–5924 CrossRef CAS PubMed.
- S. Zhou, X. Hu, R. Xia, S. Liu, Q. Pei, G. Chen, Z. Xie and X. Jing, Angew. Chem., Int. Ed., 2020, 59, 23198–23205 CrossRef CAS PubMed.
- R. Jia, L. Teng, L. Gao, T. Su, L. Fu, Z. Qiu and Y. Bi, Int. J. Nanomed., 2021, 16, 1525–1551 CrossRef PubMed.
- Q. Zhou, S. Shao, J. Wang, C. Xu, J. Xiang, Y. Piao, Z. Zhou, Q. Yu, J. Tang, X. Liu, Z. Gan, R. Mo, Z. Gu and Y. Shen, Nat. Nanotechnol., 2019, 14, 799–809 CrossRef CAS PubMed.
- S. Dong, Y. Sun, J. Liu, L. Li, J. He, M. Zhang and P. Ni, ACS Appl. Mater. Interfaces, 2019, 11, 8740–8748 CrossRef CAS PubMed.
- J. Xiang, Y. Li, Y. Zhang, G. Wang, H. Xu, Z. Zhou, J. Tang and Y. Shen, J. Controlled Release, 2021, 330, 992–1003 CrossRef CAS PubMed.
- C. Xu, L. Xu, R. Han, Y. Zhu and J. Zhang, Colloids Surf., B, 2021, 201, 111632 CrossRef CAS PubMed.
- K. Chen, H. Cai, H. Zhang, H. Zhu, Z. Gu, Q. Gong and K. Luo, Acta Biomater., 2019, 84, 339–355 CrossRef CAS PubMed.
- L. Zhu, Y. Guo, Q. Qian, D. Yan, Y. Li, X. Zhu and C. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17944–17950 CrossRef CAS PubMed.
- R. Xia, Q. Pei, J. Wang, Z. Wang, X. Hu and Z. Xie, J. Colloid Interface Sci., 2020, 580, 785–793 CrossRef CAS PubMed.
- S. Alven, X. Nqoro, B. Buyana and B. A. Aderibigbe, Pharmaceutics, 2020, 12, 406 CrossRef CAS PubMed.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- R. B. Greenwald, Y. H. Choe, J. McGuire and C. D. Conover, Adv. Drug Delivery Rev., 2003, 55, 217–250 CrossRef CAS PubMed.
- R. Plummer, R. H. Wilson, H. Calvert, A. V. Boddy, M. Griffin, J. Sludden, M. J. Tilby, M. Eatock, D. G. Pearson, C. J. Ottley, Y. Matsumura, K. Kataoka and T. Nishiya, Br. J. Cancer, 2011, 104, 593–598 CrossRef CAS PubMed.
- V. Subbiah, J. E. Grilley-Olson, A. J. Combest, N. Sharma, R. H. Tran, I. Bobe, A. Osada, K. Takahashi, J. Balkissoon, A. Camp, A. Masada, D. J. Reitsma and L. A. Bazhenova, Clin. Cancer Res., 2018, 24, 43–51 CrossRef CAS PubMed.
- N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamoto, Y. Kato, Y. Sugiyama, K. Nishio, Y. Matsumura and K. Kataoka, Cancer Res., 2003, 63, 8977–8983 CAS.
- H. Uchino, Y. Matsumura, T. Negishi, F. Koizumi, T. Hayashi, T. Honda, N. Nishiyama, K. Kataoka, S. Naito and T. Kakizoe, Br. J. Cancer, 2005, 93, 678–687 CrossRef CAS PubMed.
- H. Mukai, T. Kogawa, N. Matsubara, Y. Naito, M. Sasaki and A. Hosono, Invest. New Drugs, 2017, 35, 307–314 CrossRef CAS PubMed.
- A. Takahashi, Y. Yamamoto, M. Yasunaga, Y. Koga, J. Kuroda, M. Takigahira, M. Harada, H. Saito, T. Hayashi, Y. Kato, T. Kinoshita, N. Ohkohchi, I. Hyodo and Y. Matsumura, Cancer Sci., 2013, 104, 920–925 CrossRef CAS PubMed.
- Y. Matsumura, T. Hamaguchi, T. Ura, K. Muro, Y. Yamada, Y. Shimada, K. Shirao, T. Okusaka, H. Ueno, M. Ikeda and N. Watanabe, Br. J. Cancer, 2004, 91, 1775–1781 CrossRef CAS PubMed.
- F. Atrafi, H. Dumez, R. H. J. Mathijssen, C. W. Menke van der Houven van Oordt, C. J. F. Rijcken, R. Hanssen, F. Eskens and P. Schöffski, J. Controlled Release, 2020, 325, 191–197 CrossRef CAS PubMed.
- C. Young, T. Schluep, J. Hwang and S. Eliasof, Curr. Bioact. Compd., 2011, 7, 8–14 CrossRef CAS PubMed.
- E. Mukhtar, D. Worroll, G. Galletti, S. Schuster, S. A. Piha-Paul and P. Giannakakou, Cancer Drug Resist., 2020, 3, 636–646 CAS.
- S. A. Piha-Paul, K. Z. Thein, P. De Souza, R. Kefford, T. Gangadhar, C. Smith, S. Schuster, W. C. Zamboni, C. E. Dees and B. Markman, Invest. New Drugs, 2021, 39, 1047–1056 CrossRef CAS PubMed.
- M. H. Voss, A. Hussain, N. Vogelzang, J. L. Lee, B. Keam, S. Y. Rha, U. Vaishampayan, W. B. Harris, S. Richey, J. M. Randall, D. Shaffer, A. Cohn, T. Crowell, J. Li, A. Senderowicz, E. Stone, R. Figlin, R. J. Motzer, N. B. Haas and T. Hutson, Ann. Oncol., 2017, 28, 2754–2760 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |