
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of amino acid derivatives of substituted benzimidazoles and pyrazoles as Sirt1 inhibitors†
Nikil Purushotham‡
a,
Mrityunjay Singh‡ bc,
Bugga Paramesha‡b,
Vasantha Kumara,
Sharad Wakodec,
Sanjay K. Banerjee§
*b,
Boja Poojary*a and
Shailendra Asthana*b
bc,
Bugga Paramesha‡b,
Vasantha Kumara,
Sharad Wakodec,
Sanjay K. Banerjee§
*b,
Boja Poojary*a and
Shailendra Asthana*b
aDepartment of Studies in Chemistry, Mangalore University, Mangalagangotri, Karnataka-574 199, India. E-mail: bojapoojary@gmail.com; Tel: +91 9686940403
bTranslational Health Science and Technology Institute (THSTI), NCR Biotech Science Cluster, Faridabad, Haryana-121001, India. E-mail: skbanerjee@thsti.res.in; sasthana@thsti.res.in; Tel: +91 1292876475 Tel: +91 1292876489 Tel: +91 8447568689
cDelhi Institute of Pharmaceutical Sciences and Research, DPSR University, M.B Road, Pushp Vihar, Sector 3, New Delhi 110017, India
First published on 30th January 2022
Abstract
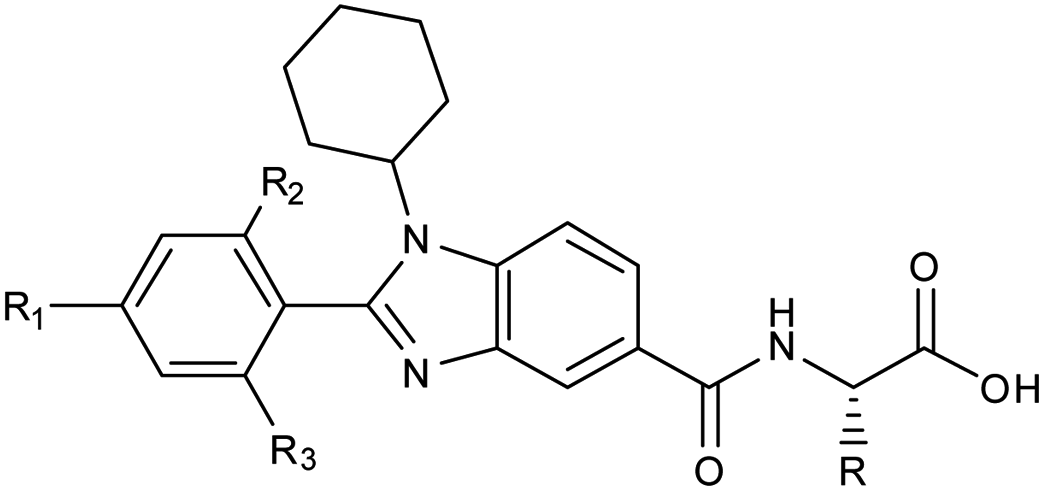
Owing to its presence in several biological processes, Sirt1 acts as a potential therapeutic target for many diseases. Here, we report the structure-based designing and synthesis of two distinct series of novel Sirt1 inhibitors, benzimidazole mono-peptides and amino-acid derived 5-pyrazolyl methylidene rhodanine carboxylic acid. The compounds were evaluated for in vitro enzyme-based and cell-based Sirt1 inhibition assay, and cytotoxic-activity in both liver and breast cancer cells. The tryptophan conjugates i.e. 13h (IC50 = 0.66 μM, ΔGbind = −1.1 kcal mol−1) and 7d (IC50 = 0.77 μM, ΔGbind = −4.4 kcal mol−1) demonstrated the maximum efficacy to inhibit Sirt1. The MD simulation unveiled that electrostatic complementarity at the substrate-binding-site through a novel motif “SLxVxP(V/F)A” could be a cause of increased Sirt1 inhibition by 13h and 13l over Sirt2 in cell-based assay, as compared to the control Ex527 and 7d. Finally, this study highlights novel molecules 7d and 13h, along with a new key hot-spot in Sirt1, which could be used as a starting lead to design more potent and selective sirtuin inhibitors as a potential anticancer molecule.
Introduction
Epigenetic regulation is a dynamic and reversible process, which can contribute to a broad range of human diseases that include metabolic and neurological diseases, inflammation and cancer, etc.1 In the recent past decade, human homologs of yeast silent information regulators 2 (Sir2), class-III lysine deacetylases, also called sirtuins that use NAD+ as the co-factor to catalyze the deacetylation,2 have emerged as targets for several diseases along with cancer chemotherapy.3,4 A total of seven Sir2 homologs (Sirt1–7) are found in humans, in which Sirt1, Sirt6 and Sirt7 are predominantly found in the nucleus, Sirt2 in the cytoplasm and Sirt3, Sirt4 and Sirt5 in mitochondria.5 All sirtuins have highly conserved NAD-binding, and catalytic core domains with distinct N- and C-terminal extensions.6–8 The length of dissimilar N- and C-terminals varies according to the type of binding partners, and their subcellular localization.6–8 Among all sirtuins, Sirt1's inhibition has gained more attention in recent years owing to its role in cancer,3 rheumatoid arthritis,9,10 HIV10,11 and autophagy.12The role of Sirt1 in cancer begins from the finding that it deacetylates p53 and E2F1, and finally inhibits the apoptosis via modulation of their transcriptional activity.13 Indeed, the role of Sirt1 in cancer is complex and depends on the phases and type of cancer; for example, in some types of cancers, it promotes tumorigenesis,14 while in others such as colon cancer, it acts as a tumor suppressor.14–17 Overexpression of Sirt1 in pancreatic ductal adenocarcinoma,17 and hepatocellular carcinoma (HCC)18 was associated with metastasis and chemoresistance. Though the mechanism is not yet known, more exploration is underway to find the role of Sirt1 inhibition in cancer progression.19,20
Even though sirtuins' biochemistry has been extensively studied, and several Sirt2 inhibitors (Sirt2 selective) such as pyridine containing inhibitor (1),21,22 tryptophan-containing Javamide-II (2),23 2,3-disubstituted benzimidazole-5-carboxylates (3),24 SirReal (4),25 a compound containing substituted 2-aminothiazole-carboxamide linked to a pyrimidine moiety, thienopyrimidinones (5),26 carboxamide containing AEM2 (6)27 and AGK2 (7)28 have been identified and have been shown to inhibit the growth of cancer cells, relatively few studies have examined the anti-proliferative effect of Sirt1 inhibitors on cancer cells29 (Fig. 1). Moreover, Sirt1 inhibitors such as 1,2,3,4 tetrahydrocarbazole Ex527 (8, Selisistat),30 pyrazole (9),31 sirtinol, cambinol, 4bb32 and MHY2256 (ref. 33) are also reported to trigger growth arrest, apoptosis or senescence in tumor cells34–39 (Fig. 1). Recently, some Sirt1 inhibitors are reported in adjuvant therapy for the treatment of paclitaxel-resistant human cervical cancer40 and cisplatin-resistant endometrial carcinoma41 and other cancers;42 thus, these findings have opened up a new avenue to explore the role of Sirt1 inhibitors as a therapy for the treatment of cancer cells.32,43,44
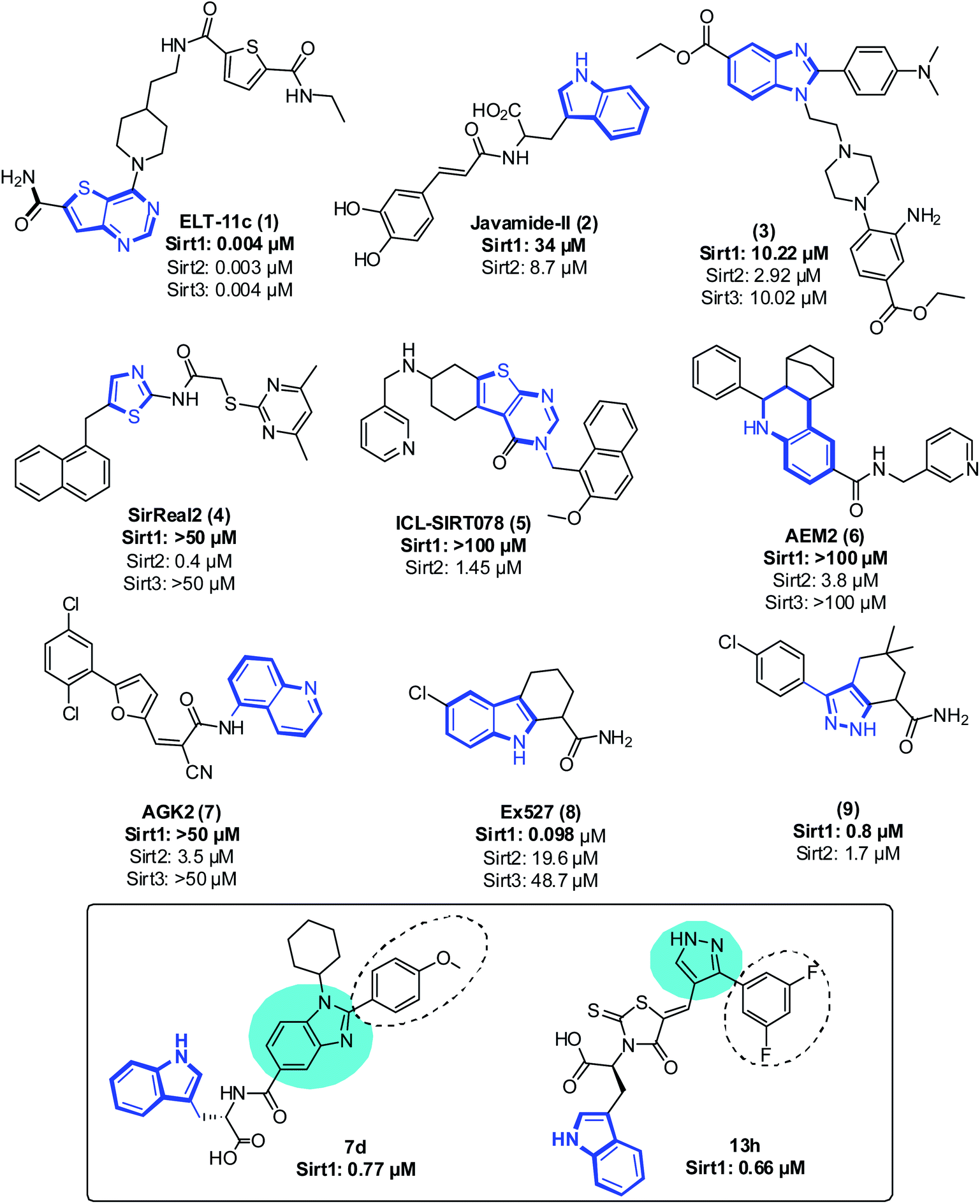 | ||
| Fig. 1 Representative sirtuin inhibitor structures with their IC50 values. The area shown in dark blue lines reflects the ligand's moiety bound at pocket-C. The most promising candidates from both the series are displayed in the box. The circles in cyan color and dotted line in black represent the core moiety and regions of selectivity, respectively. | ||
As a follow-up to our previous studies that observed differential selectivity patterns of pocket-C and extended pocket-C in Sirt1, Sirt2 and Sirt3,45,46 in this study, in addition to pocket-C, the substrate binding site was taken into account when designing a Sirt1 inhibitor. Here, we are reporting the structure-based designing47–50 and synthesis of 1,2-disubstituted benzimidazole mono-peptides via a one-pot reductive cyclization method and rhodanine acid conjugated pyrazoles via Knoevenagel condensation reactions as a Sirt1 inhibitor. The potent Sirt1 inhibitor Ex527 (6-membered ring) was used as a control for designing and comparative biological activity. The binding-pose-metadynamics (bpMD) followed by molecular dynamics (MD) simulations, per-residue energy contribution and thermodynamic profiling was performed to establish the structure–activity-relationship (SAR). The bpMD was implemented to predict the most stable binding pose of synthesized compounds. Furthermore, the Sirt1 inhibitory potential was investigated in enzyme- and cell-based assay systems and also correlated with their anti-cancer activity in two different cell lines. Finally, the electrostatic complementarity analysis was carried out to unveil the plausible mechanism of selective inhibition of Sirt1 via inhibitors 13d, 13h and 13l.
Results and discussion
Rationale for designing
Accurate prediction of binding-site and binding-mode in protein is challenging; however, identification of accurate binding mode inside the binding site is the prerequisite for the designing of molecules. Since the crystal structure pose of Sirt1 was available with Ex527* (7-membered ring), an analog of Ex527, it belongs to the same chemical class and has nearly the same binding affinity and IC50 values (IC50_Ex527 = 0.098 μM, IC50_4I5 = 0.124 μM); therefore, the protein structure PDB-id “4ZZI” was selected as a reference for model building, virtual screening, docking and MD simulation studies.30,51 Initially, focused docking and Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) calculations were performed for Ex527 and Ex527*, by using the center of mass of Ex527* as a grid center to establish the benchmark. It was reported that the inhibitor binding site in sirtuins is flexible and dynamic in nature;25 therefore, we performed the MD simulations of crystal-pose (Sirt1-Ex527*) and docked-pose (Sirt1-Ex527) complexes to get the exact binding modes of Ex527. The overlaid structures (the average structure extracted from the last 80 ns of total 200 ns MD simulation of each) had shown that both Ex527 and Ex527* were bound at the same place with a root mean square deviation (RMSD) of 0.98 Å (Fig. S1A†), nested deep within the active site, and primarily interacted through pi–pi, hydrophobic and hydrogen bond interactions (Fig. S1B†). The lowest MM-GBSA (ΔGbind) and docking scores among Ex527 and Ex527* were −60.00 kcal mol−1 and −6.0, respectively (Fig. S1B†). Therefore, we took these values as a cut-off for the selection of compounds through virtual screening (VS) and ligand designing. We have performed VS of our in-house curated library of 1.0 million e-molecules through the VS workflow module in Maestro 2017-2. After applying all filters available like QikProp, Lipinski's rule, and the reactive functional group, the VS workflow filtered 1562 molecules which have a dock-score < −10.0 kcal mol−1. Based on docking and MM-GBSA scores, the top three molecules were picked for further ligand designing and pocket optimization (Table S1†). The structural alignment of VS's top three molecules was further explored and two crucial pieces of information were obtained: first, the possibility of ligand expansion, i.e. the ligand further can be designed by considering three additional pockets D1, D2 and substrate binding site (pocket-A), along with pocket-C, when compared to the control Ex527*, and second, the role of the aromatic moiety for interaction at all the three sites, viz. substrate binding site, pocket-B and pocket-C (Fig. 2A and S1B†).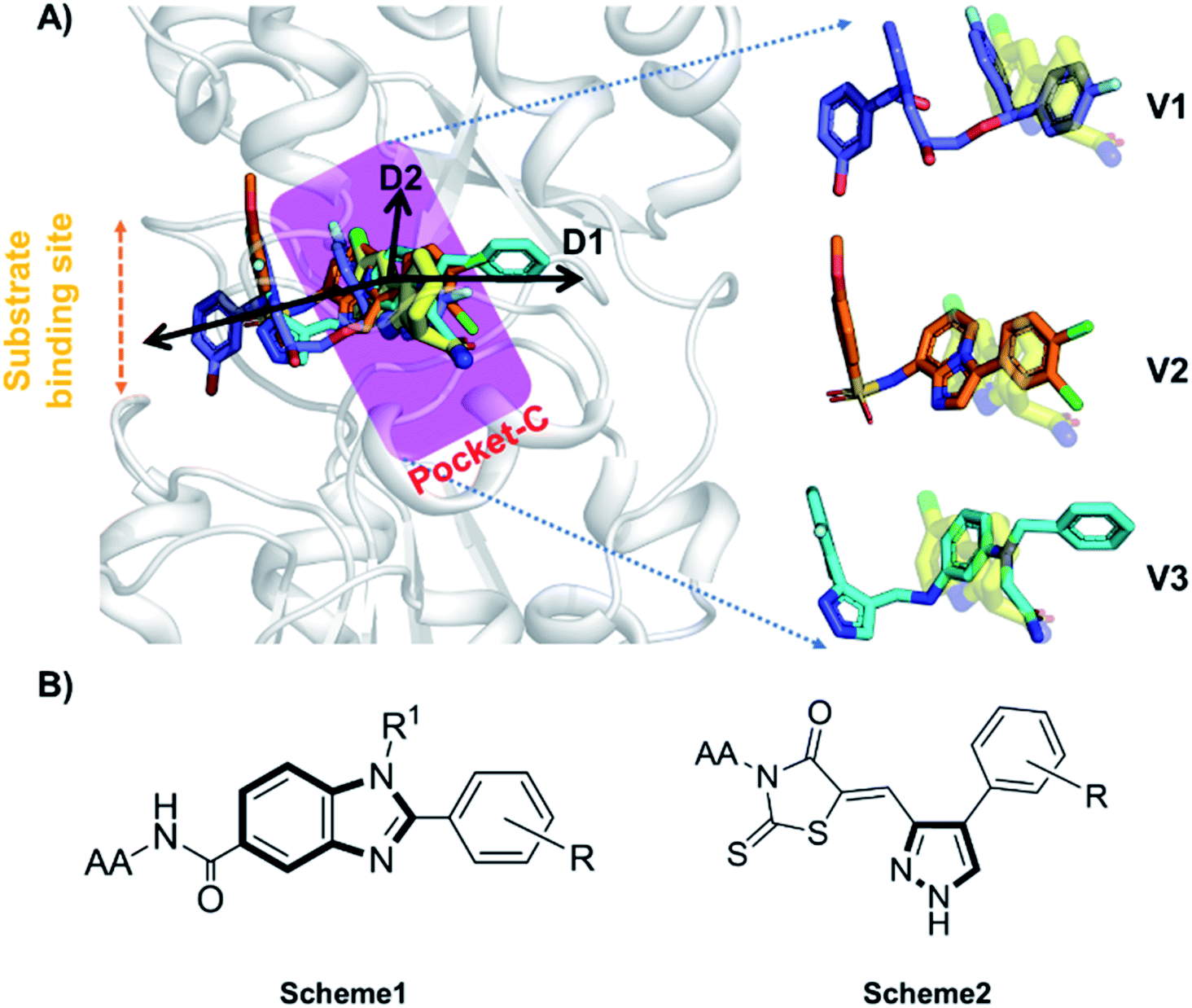 | ||
| Fig. 2 The rationale for compound designing. (A) The structural alignment of docking poses of top three molecules of VS (1.0 million in-house e-compounds) with the crystal structure PDB id. 4I5I. (B) The structure of the “chemical-core” identified for designing the ligands of Schemes 1 and 2. | ||
Moreover, the structural study of Sirt1–3's inhibitors revealed the importance of the aromatic heterocyclic moiety for interaction in pocket-C, the region necessary for inhibition of Sirt1 as reported in the crystal structure 4I5I (Fig. 1). The residues F273 and Y280 formed an aromatic zone at pocket-C (Fig. S1B†) and played a significant role in the stability of Ex527. Similarly, at pocket-B residue H363, and F414 form an aromatic cationic patch (Fig. S1B†) that appears to be more suitable for interaction with heteroaromatic groups such as pyrazoles and benzimidazole due to their pi–pi and pi–cationic interactions. Furthermore, these findings were validated by the molecule V3 (Fig. 2), in which the pyrazole moiety is located at pocket-B and forms a good interaction with residue H363. All of these findings led us to choose benzimidazole and pyrazole as the core structures. In continuation of our previous study on Sirt1–3 where we discussed the amino acid conservation of pocket-C and its role in differential dynamics and selectivity of Ex527*,45 here in this study we explored regions, specifically substrate binding site along with pocket-C for Sirt1-inhibitor designing.
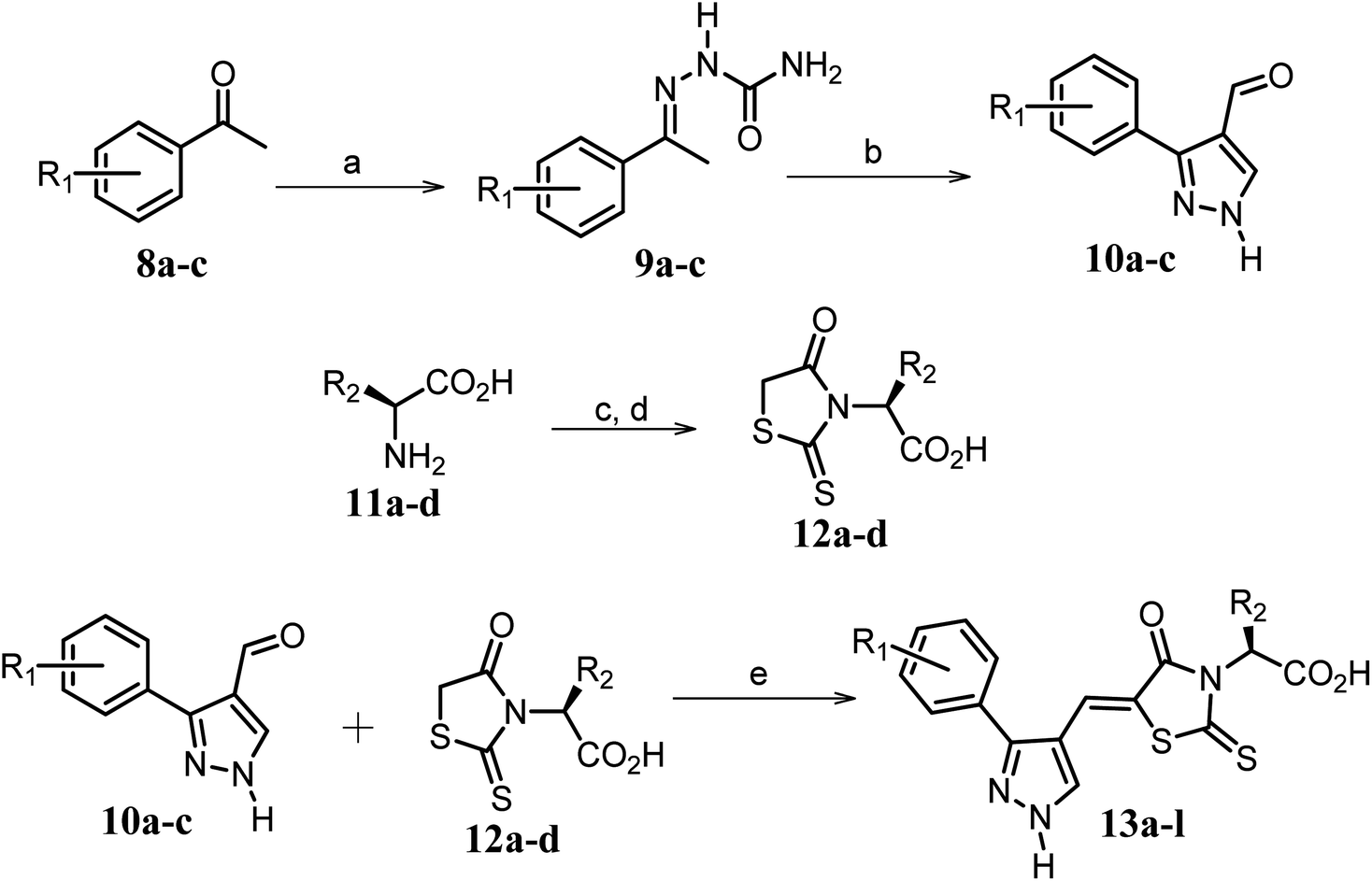
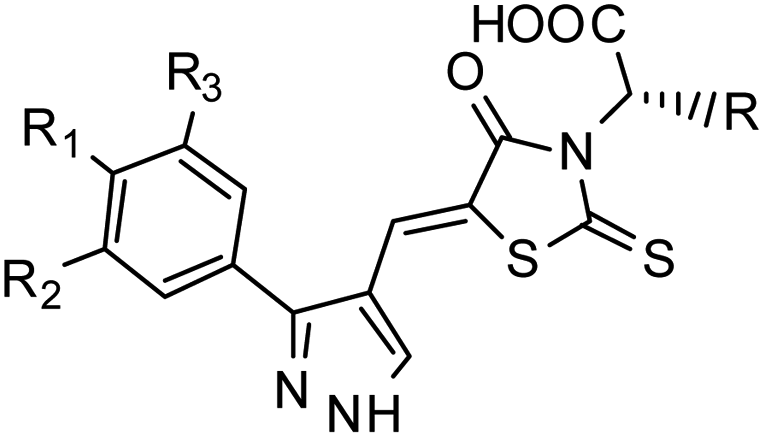
Furthermore, in the recent developments in drug design, strategically conjugating amino acid fragments to bioactive heterocycles has proved to enhance desirable pharmacological features such as low toxicity, high bioavailability, stability and cell permeability with modest potency due to its biocompatibility.52 After getting this initial information, a total of twenty-four amino acid–heterocycle conjugates were designed and synthesized using a combination of different amino acids on two types of heterocyclic scaffolds, namely benzimidazole and pyrazole (Fig. 2B). Out of the two series, series1 consisted of twelve substituted benzimidazole monopeptides derived from four amino acids viz. alanine, valine, leucine and tryptophan (Scheme 1 and Table 1), whereas series2 consisted of twelve substituted pyrazolyl methylidenes of rhodanine carboxylic acids derived from four amino acids viz. glycine, alanine, phenylalanine and tryptophan (Scheme 2 and Table 2).
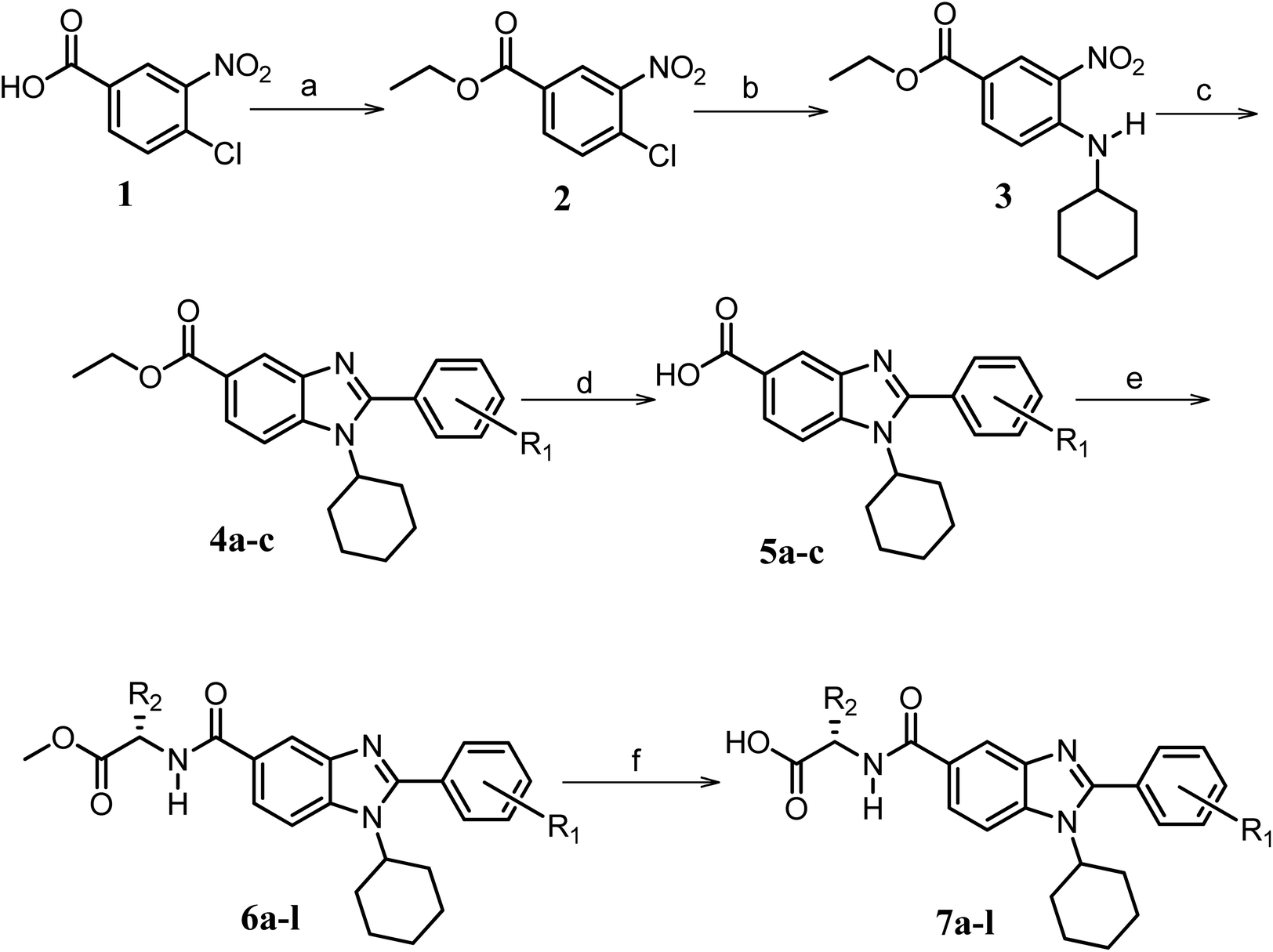 | ||
Scheme 1 Synthetic strategy for the benzimidazole monopeptides (7a–l). aReagents and conditions: (a) EtOH, conc. H2SO4 (catalytic), reflux; (b) cyclohexylamine (2.5 equiv.), TEA (3.0 equiv.), THF, r.t.; (c) substituted benzaldehyde (1 equiv.), Na2S2O4 (3 equiv.), DMSO, 90 °C; (d) NaOH (1.1 equiv.), water, reflux; (e) amino acid methyl ester hydrochloride (1 equiv.), NMM (2.5 equiv.), TBTU (1.25 equiv.), DMF, r.t.; (f) LiOH·H2O (1 equiv.), water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (2:1), 0 °C. :THF (2:1), 0 °C.  = 4-F, 4-OCH3, 2-Cl-6-F; = 4-F, 4-OCH3, 2-Cl-6-F; 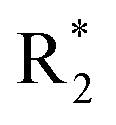 = CH3, CH(CH3)2, CH2–CH(CH3)2, 3-indolylmethyl. * Further for the substitution pattern see Table 1. = CH3, CH(CH3)2, CH2–CH(CH3)2, 3-indolylmethyl. * Further for the substitution pattern see Table 1. | ||
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | R | R1 | R2/R3 | Mol. mass | Static docking Sirt1 (kcal mol−1) | IF guided docking Sirt1 | % inhibition at 10 μM Sirt1 | |
| Dock score (kcal mol−1) | ΔGbind (kcal mol−1) | |||||||
| a The italicized rows represent the ligands chosen for further studies. | ||||||||
| 7a | 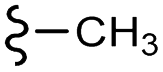 |
–OMe | –H | 421 | −9.23 | −6.26 | −58.96 | 88.03 |
| 7b | 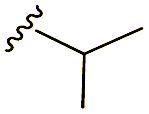 |
–OMe | –H | 450 | −7.98 | −7.06 | −59.49 | 88.18 |
| 7c | 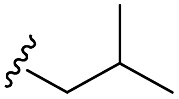 |
–OMe | –H | 464 | −9.15 | −6.68 | −58.85 | 86.34 |
| 7d | 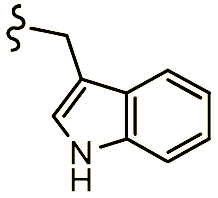 |
–OMe | –H | 537 | −8.81 | −6.70 | −72.08 | 89.99 |
| 7e |  |
–F | –H | 409 | −11.47 | −6.51 | −54.45 | <30 |
| 7f |  |
–F | –H | 437 | −9.32 | −7.40 | −54.66 | <30 |
| 7g |  |
–F | –H | 451 | −8.76 | −6.08 | −58.20 | 80.82 |
| 7h | 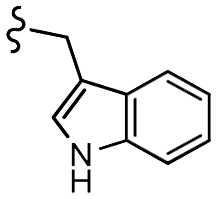 |
–F | –H | 525 | −8.67 | −5.84 | −61.60 | 86.83 |
| 7i | 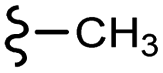 |
–H | –Cl/F | 472 | −10.70 | −7.08 | −49.04 | <30 |
| 7j | 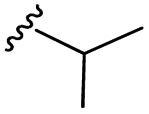 |
–H | –Cl/F | 486 | −9.71 | −5.33 | −33.49 | <30 |
| 7k | 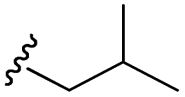 |
–H | –Cl/F | 559 | −8.49 | −5.61 | −52.97 | <30 |
| 7l | 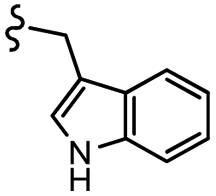 |
–H | –Cl/F | 559 | −8.12 | −6.61 | −56.97 | <30 |
| Control | Ex527 | −7.78 | −63.58 | 97.73 | ||||
 | ||
Scheme 2 Synthetic strategy for the preparation of pyrazole conjugated rhodanine carboxylic acids (13a–l). aReagents and conditions: (a) semicarbazide hydrochloride (1.1 equiv.), NaOAc (1.3 equiv.), EtOH, reflux; (b) POCl3 (10 equiv.), DMF, reflux; (c) CS2 (1.2 equiv.), KOH (1 equiv.), water, r.t.; (d) potassium chloroacetate (1 equiv.), 2 N HCl until pH 2, 90 °C; (e) β-alanine (2 equiv.), AcOH, reflux.  = 4-OCH3, 4-NO2, 3,5-F2; = 4-OCH3, 4-NO2, 3,5-F2; 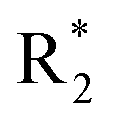 = H, CH3, CH2–C6H5, 3-indolylmethyl. * Further for the substitution pattern see Table 2. = H, CH3, CH2–C6H5, 3-indolylmethyl. * Further for the substitution pattern see Table 2. | ||
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | R | R1 | R2/R3 | Mol. mass | Static docking Sirt1 (kcal mol−1) | IF guided docking Sirt1 | % inhibition at 10 μM Sirt1 | |
| Dock score (kcal mol−1) | ΔGbind (kcal mol−1) | |||||||
| a The italicized rows represent the ligands chosen for further studies. | ||||||||
| 13a | –H | –OMe | –H | 375.42 | −7.42 | −6.54 | −47.19 | <30 |
| 13b | 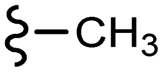 |
–OMe | –H | 389.44 | −7.64 | −6.18 | −46.74 | <30 |
| 13c | 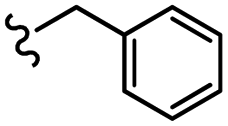 |
–OMe | –H | 465.08 | −5.81 | −7.44 | −40.64 | <30 |
| 13d | 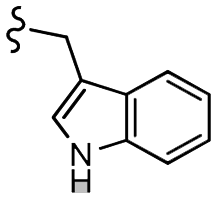 |
–OMe | –H | 504.58 | −6.90 | −7.26 | −63.40 | 90.64 |
| 13e | –H | –H | –F | 381.37 | −7.50 | −6.36 | −48.85 | <30 |
| 13f | 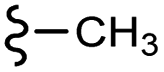 |
–H | –F | 395.40 | −7.22 | −6.51 | −48.22 | <30 |
| 13g | 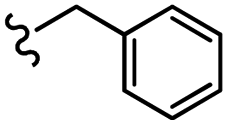 |
–H | –F | 471.50 | −5.05 | −7.42 | −52.96 | <30 |
| 13h | 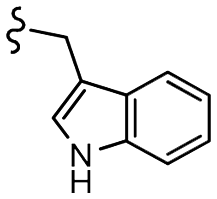 |
–H | –F | 510.53 | −7.88 | −7.21 | −60.23 | 89.15 |
| 13i | –H | –NO2 | –H | 390.39 | −7.18 | −6.97 | −59.30 | <30 |
| 13j | 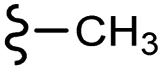 |
–NO2 | –H | 404.42 | −4.52 | −7.70 | −53.11 | <30 |
| 13k | 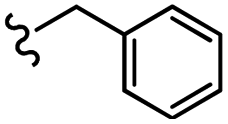 |
–NO2 | –H | 480.51 | −3.68 | −6.27 | −56.01 | <30 |
| 13l | 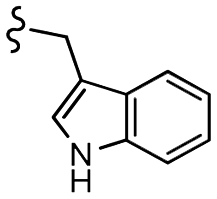 |
–NO2 | –H | 519.55 | −5.50 | −7.09 | −70.88 | 89.64 |
| Control | Ex527 | −7.78 | −63.58 | 97.73 | ||||
Chemistry
The synthetic strategy of novel benzimidazole monopeptides is depicted in Scheme 1. Initially, ethyl 4-chloro-3-nitrobenzoate (2) was prepared by esterification of 4-chloro-3-nitrobenzoic acid (1) in refluxing ethanol with a catalytic amount of sulphuric acid. The resulting ester was subjected to nucleophilic aromatic substitution with cyclohexylamine in THF using triethylamine as the base at room temperature to obtain ethyl 4-cyclohexylamino-3-nitrobenzoate (3).53 Successively, the benzimidazole core was accomplished by sodium dithionite assisted reductive cyclization of ethyl 4-cyclohexylamino-3-nitrobenzoate (3) with substituted benzaldehydes in DMSO at 90 °C. The benzimidazole esters (4a–c) were then hydrolyzed to the corresponding carboxylic acids (5a–c) in refluxing aqueous sodium hydroxide solution. These benzimidazole carboxylic acids, when coupled with various amino acid methyl ester hydrochlorides, in the presence of N-methyl morpholine using TBTU as the coupling agent in DMF media, furnished ester protected benzimidazole monopeptides (6a–l). These monopeptide esters were hydrolyzed to obtain the target benzimidazole monopeptides (7a–l) using lithium hydroxide monohydrate in THF–water mixture at 0 °C.The synthetic strategy for the rhodanine carboxylic acid conjugated pyrazoles is depicted in Scheme 2. In order to synthesize the final compounds, the two key scaffolds (i) pyrazole aldehyde and (ii) rhodanine acids were initially prepared separately. For the preparation of 3-substituted-1H-pyrazole-4-carboxaldehydes,54 appropriately substituted acetophenones (8a–c) were heated with semicarbazide hydrochloride in the presence of sodium acetate in acetic acid to obtain the corresponding semicarbazones (9a–c). These on cyclization with phosphorous oxychloride via Vilsmeier–Haack reaction furnished 3-substituted-1H-pyrazole-4-carboxaldehydes (10a–c).55 For the preparation of rhodanine acetic acids (12a–d), suitable amino acids (11a–d) were initially dissolved in an aqueous solution of potassium hydroxide and treated with carbon disulfide to obtain the corresponding potassium salt of dithiocarbamates. These on treatment with potassium chloroacetate, followed by heating with 2 N HCl solution, yielded the 2-(4-oxo-2-thioxothiazolidin-3-yl)-amino acids (12a–d).56 Finally, the two key scaffolds were clubbed together by means of Knoevenagel condensation using beta-alanine as the catalyst57 to accomplish the target compounds (13a–l) in good yield.
Biological evaluation
| Comp. | % Inhibition | Sirt1 IC50 (μM) | |
|---|---|---|---|
| Sirt1 | Sirt2 + Sirt3 | ||
| a The italicized rows show compounds with the highest Sirt1 inhibition. nd = not determined.b Biological assay control or standard compound i.e. Ex527. | |||
| 7a | <1 | nd | nd |
| 7b | <1 | nd | nd |
| 7c | 3.24 ± 0.27 | 5.21 ± 0.78 | nd |
| 7d | 38.33 ± 0.37 | 19.89 ± 0.89 | 0.77 ± 0.04 |
| 7g | <1 | nd | nd |
| 7h | <1 | nd | nd |
| 13d | 36.34 ± 0.99 | 6.04 ± 0.91 | 0.71 ± 0.03 |
| 13h | 48.54 ± 0.91 | 9.53 ± 0.65 | 0.66 ± 0.02 |
| 13l | 30.06 ± 0.23 | 9.47 ± 0.92 | 0.73 ± 0.06 |
| Ex527b | 33.33 ± 0.64 | 15.21 ± 0.99 | 0.60 ± 0.02 |
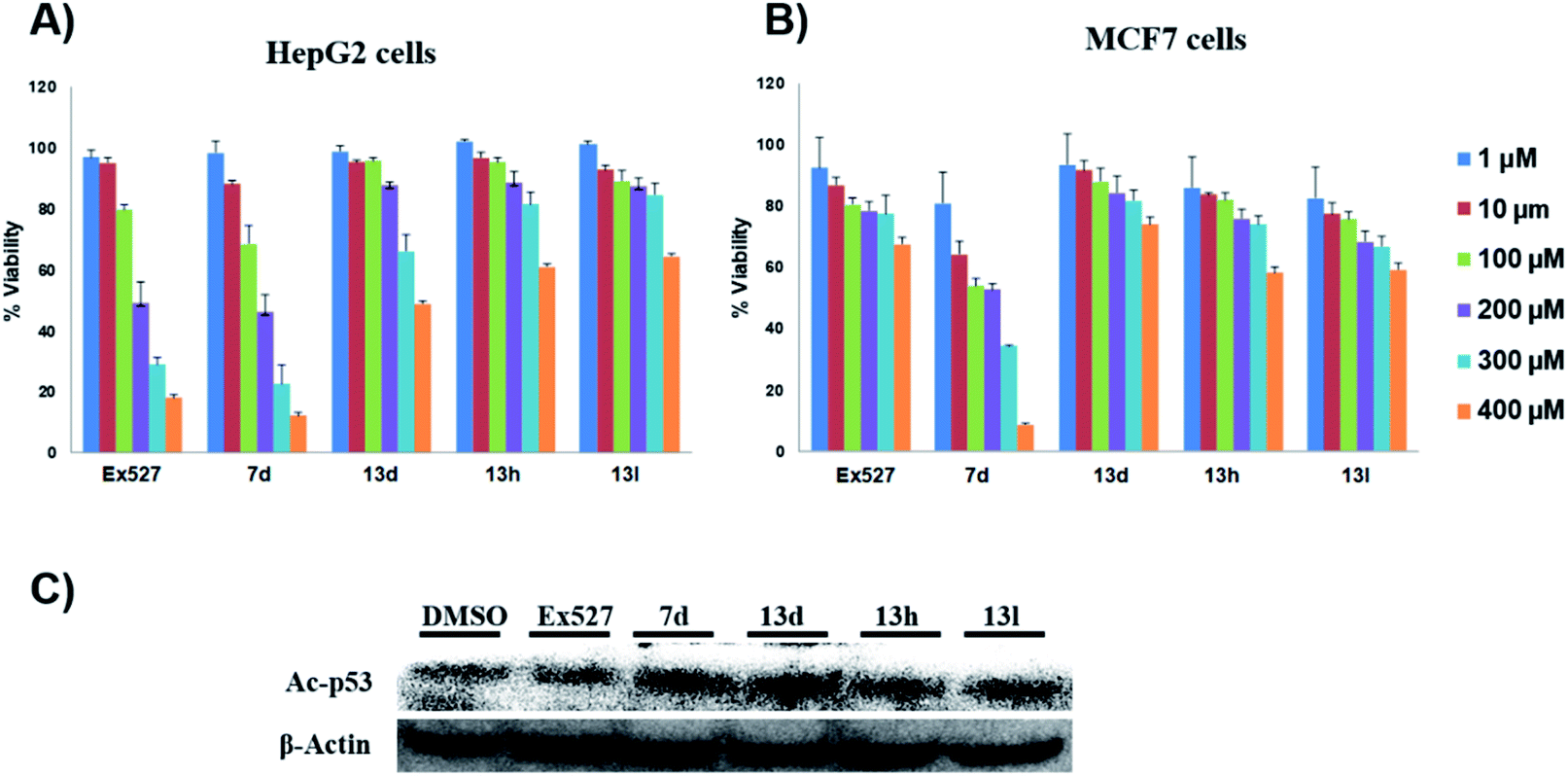 | ||
| Fig. 3 In vitro evaluation of computationally promising compounds. (A and B) The % viability of cancer cells in the presence of test compounds at different concentrations. (A) Effect of test compounds on HepG2 cell viability. (B) Effect of test compounds on MCF7 cell viability, n = 3, and the results were represented as the mean of 3 independent experiments. (C) Immune blotting shows the effect of control and test compounds on the protein levels of Ac-p53 in HepG2 cells. | ||
These assays' outcome helps to decipher that 13d, 13h and 13l standout as a Sirt1 selective molecule in comparison with Ex527. As compound 7d inhibits Sirt1 along with Sirt2 and decreased cell viability significantly higher than 13h and 13d similar to Ex527, it can be explored further as an anticancer molecule (Fig. 3). Overall, the biological evaluation studies suggested that both Sirt1 and Sirt2 inhibition is a prerequisite property of an anticancer molecule.
Computational studies
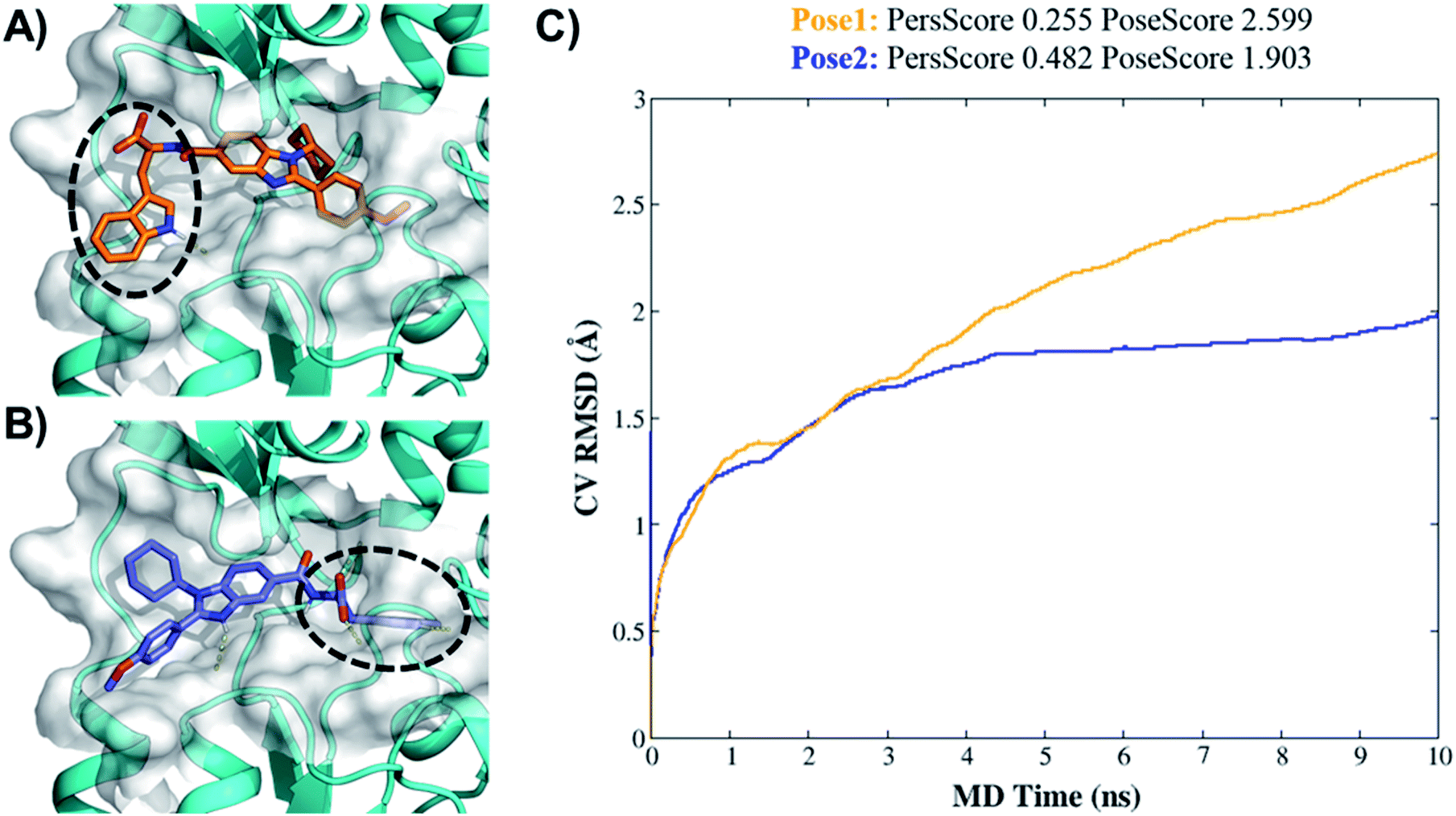 | ||
| Fig. 4 Binding pose metadynamics to identify the most stable orientation. (A) Pose1 of 7d (initial pose). (B) Pose2 of 7d (IFD-pose). (C) The average RMSD of 7d during the 10 × 10 ns metadynamics runs in pose1 (orange line) and pose2 (blue line). | ||
Higher PersScore values are equivalent to higher ligand stability. Since pose1 has a higher PoseScore 2.599 this indicated higher ligand displacement in position and orientation relative to the initial position compared to pose2 (PoseScore = 1.903). Furthermore, in pose1, only one HB remained stable throughout the trajectory, resulting in a lower PersScore of 0.255. In pose2, PresScore 0.482, 4 HBs remained stable for up to 60% of the simulation time, indicating that 7d was more stable in pose2 than pose1. Finally, CompScore gives an overall assessment of the stability of systems through the formula “CompScore = PoseScore − 5 × PersScore”. The lower the CompScore the more robust the complex. The results of bpMD revealed that pose2, with a CompScore of 0.103, was significantly more stable and accurate than pose1 with a CompScore of 2.089 (Fig. 4C). Furthermore, in the view of the result of bpMD, rescoring and guided docking were performed for each molecule with consideration of protein flexibility up to 4.0 Å to get the most likely orientation of each compound (Fig. S3;† Tables 1 and 2). As reflected from Fig. S3,† a total number of 9 molecules (7a, 7b, 7c, 7d, 13d, 13h and 13l) in Sirt1, only one molecule (7d) in Sirt2 and none of the molecules in Sirt3 cross the cut-off values. Although the outcomes of docking results matched well with the results of the biological assays, in cell-based assay only four molecules 7d, 13d, 13h and 13l showed good results. Further to investigate this change in the activity pattern of ligands in cell-based assay and to prepare SAR of molecules, the MD simulations were performed.
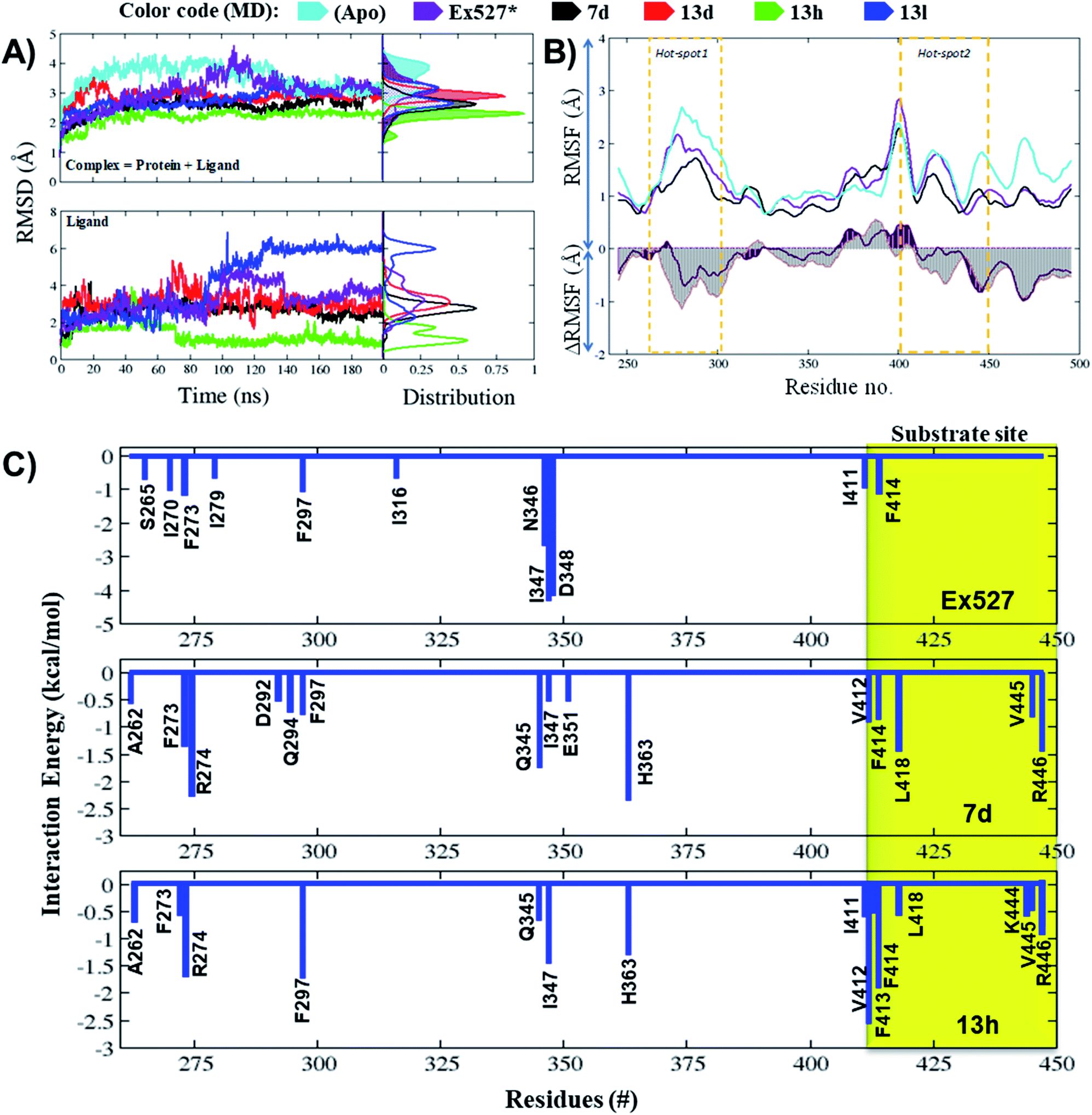 | ||
| Fig. 5 Molecular dynamics simulations reflect the significant changes in ligand behavioral patterns. (A and B) MD trajectory analysis of apo Sirt1 and protein–ligand complexes of 7d, 13d, 13h and 13l. (A) The root mean square deviation (RMSD) of protein (all backbone atoms) and ligand in coordinates as a function of the simulation time. (B) The root mean square fluctuation (RMSF) and ΔRMSF of C-alpha atoms of MD systems, Sirt1-apo, Sirt1-Ex527* and Sirt1-7d. (C) Per-residue energy decomposition of Ex527*, 7d and 13d. The residue which reflected the major contribution in binding free energies (ΔGpbsa ≤ −0.5 kcal mol−1) is shown in the graphs. The region highlighted with yellow color background belongs to the substrate-binding site. | ||
The average RMSD of complex systems of Sirt1 with compounds 7d, 13d, 13h and 13l was 2.5 Å, 2.8 Å, 2.1 Å and 2.8 Å, respectively. Since complex 13h was the most stable complex among all, it had shown the lowest, smooth and unimodal RMSD distribution. Although complexes 13d and 13l both had nearly the same average RMSD, which was highest among complex systems (7d, 13d, 13h, and 13l), the ligand RMSD distribution plot for 13l was bimodal, while that of 13d is unimodal. The bimodal RMSD distribution showed higher fluctuation than unimodal and since complex 13l instability increased after 100 ns from 2.5 Å to 3.0 Å, 13d was found to be more stable. In cell-based assay the biological inhibitory activity pattern was 13h > 7d > 13d > 13l, that agrees well with MD outcomes (Fig. 5A, MD stability, 13h > Ex527 ∼ 7d > 13d > 13l).
| Energy component | Average energy in kcal mol−1 | ||
|---|---|---|---|
| Ex527 | 7d | 13h | |
| ΔEint | 0 | 0 | 0 |
| ΔEvdW | −31.31 ± 0.33 | −54.88 ± 0.58 | −43.77 ± 0.42 |
| ΔEele | −22.13 ± 0.51 | −86.63 ± 1.30 | −82.11 ± 1.20 |
| ΔEpb_solv | 32.76 ± 0.47 | 119.88 ± 1.58 | 102.44 ± 1.08 |
| ΔEnp_solv | −2.97 ± 0.01 | −5.96 ± 0.03 | −4.49 ± 0.02 |
| ΔGgas | −53.65 | −141.51 ± 1.55 | −125.88 ± 1.30 |
| ΔGsolv | −29.79 ± 0.47 | 113.91 ± 1.56 | 97.95 ± 1.07 |
| ΔH | −23.66 ± 0.37 | −27.60 ± 0.78 | −27.93 ± 0.67 |
| T × ΔS | −18.80 ± 0.85 | −26.70 ± 0.92 | −23.50 ± 2.36 |
| ΔGbind_pbsa (kcal mol−1) | −4.86 | −1.10 | −4.43 |
Docking is a static technique in which the ligand was directly docked over the protein without considering the solvation pattern. Furthermore, the prime tool of Schrodinger calculated binding energy only through the MM-GBSA protocol. We initially filtered our molecules solely based on the docking and MM-GBSA scores provided by the Schrodinger prime tools. However, to obtain absolute affinity and more conclusive results of filtered compounds, the MM-PBSA calculation was carried out with real-time MD simulations.
The total MM-PBSA binding free energies (ΔGbind_pbsa) for Ex527, 7d and 13h were −4.86 kcal mol−1, −1.10 kcal mol−1 and −4.43 kcal mol−1, respectively (Table 4). The outcomes of ΔGbind_pbsa clearly followed the trends of IC50 values. The per-residue energy decomposition claimed that in Ex527, a total of 11 residues (S265, I270, F273, I279, F297, I316, N346, I347, D348 and I411, F414) reflected major contribution in binding free energies (ΔGpbsa ≤ −0.5 kcal mol−1), and among them, I347, N346 and D348 contributed highest (Fig. 5C). However, in the case of compounds 7d and 13h, around 15 residues showed major contribution to binding free energy (ΔGpbsa ≤ −0.5 kcal mol−1) (Fig. 5C). In 7d, residues A262, F273, R274, D292, Q294, F297, Q345, I347, E351, H363, V412, F414, L418, V445 and R446 reflected major contribution in binding free energy and among them, F273, H363 from pockets-B and -C, and L418 and R446 from hot-spot2 contributed highest (Fig. 5C). Similarly, in the case of 13h, residues A262, F273, R274, F297, Q345, I347, H363, I411, V412, F413, F414, L418, K444, V445 and R446 showed major contribution and among them, F297, I347, H363 form pockets-B and -C and V412, F413 and F414 from substrate binding site contributed highest (Fig. 5C).
We observed that in Ex527 all 10 residues that played a key role in interaction belong to pockets-B and -C only and no interaction was observed with residues at the substrate binding site except F414, while in the cases of 7d, 13d, 13h and 13l both, the residues from all three sites, pockets-B and -C and substrate binding sites, played a major role in stability (Fig. 6A). Further to validate the outcomes of energy decomposition, we also performed interaction fraction analysis of whole 200 ns MD simulation (Fig. 6B). The results of residue-wise interaction analysis revealed that, while 7d and 13h had the most interaction at the NAD+ and pocket-C sites, 13h had more interaction at the substrate site.
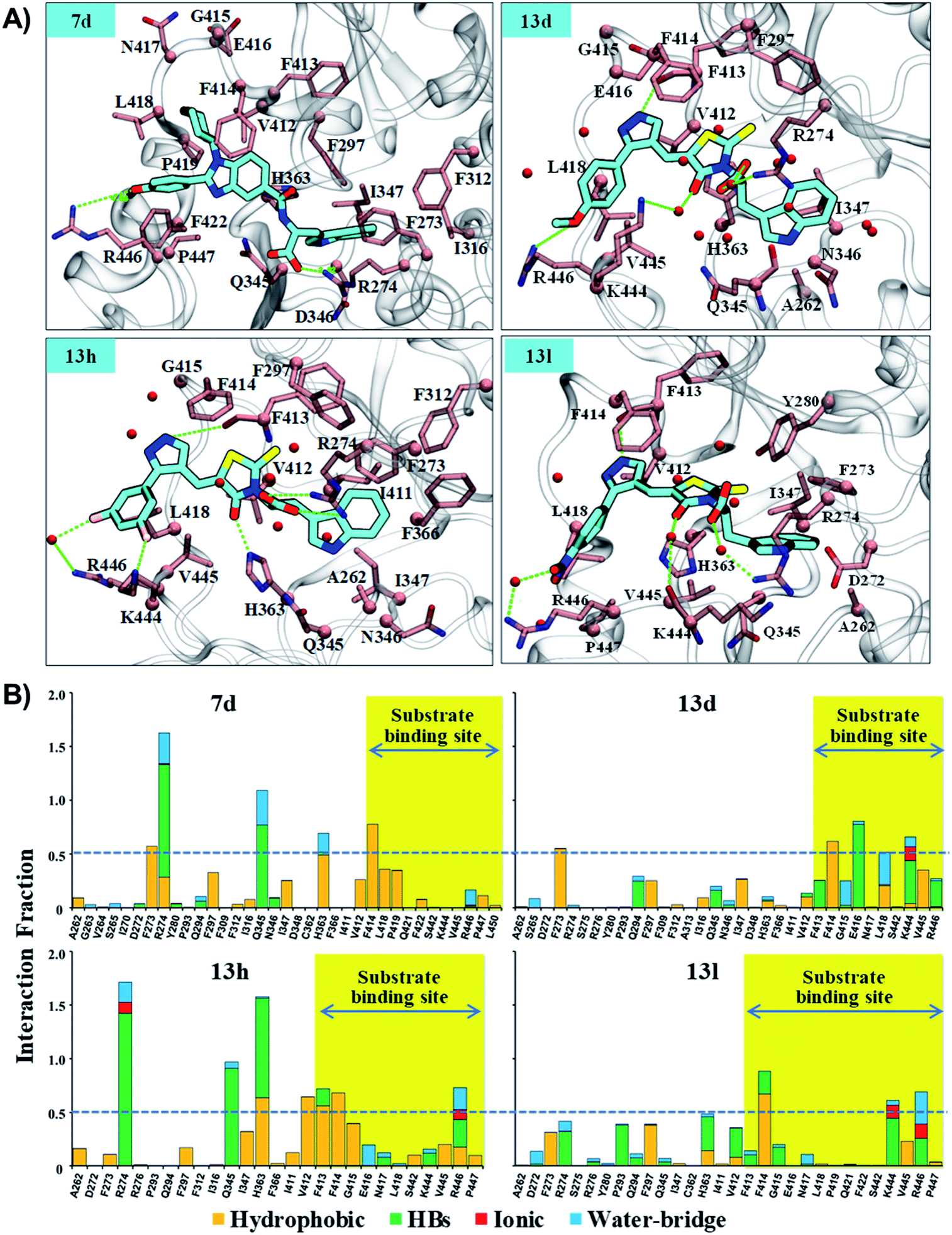 | ||
| Fig. 6 The residue interaction analysis of ligands 7d, 13d, 13h and 13l. (A) The interaction map of 7d, 13d, 13h, and 13l. The ligands 7d, 13d, 13h and 13l are shown in “stick” representation in color-type “element” with “C” in fluorescent cyan. The key residue within 3.5 Å cutoff from ligands are highlighted in color-type “element” with “C” in pink color. The C-alpha atom and side chain are shown in “sphere” and “stick” representation, respectively. Backbone atoms are shown for only those residues which are involved in HB interaction. Backbone atoms are shown in stick representation. Water (oxygen atom) within 2.5 Å from the ligand is shown in sphere representation in red color. HBs are shown with a dotted line and green color. (B) The residue interaction fraction analysis of complex systems, control (Sirt1-Ex527*), 7d (Sirt1-7d), 13d (Sirt1-13d), 13h (Sirt1-13h) and 13l (Sirt1-13l) throughout 200 ns MD simulation. The dotted line represents a cut-off value of 0.5, which indicated that the specific interaction is maintained ≥50% of the total simulation time. | ||
Establishment of structure–activity relationship (SAR)
As reflected from the 2d interaction graph in Fig. S4,† the inhibitors Ex527 (docked complex) and Ex527-analogue (Ex527*, co-crystal, PDB id: 4I5I) both bind into the pocket-C and share high conservity in the interaction pattern. The pocket-C is made up of two types of residues hydrophobic and polar. The residues F273, Y280, and I347 cover it from the front- and side-faces and are involved in pi–pi, pi–cation and hydrophobic interactions. The residues Q345, N346 and D348 form the base of pocket-C and establish the polar electrostatic interactions (Fig. S1B†). Both types of interactions appeared to be necessary for the inhibitory activity of Ex527, as reflected from the per-residue energy decomposition and residue interaction fraction analysis graph (Fig. 5C and S5†). The residue interaction fraction analysis graph revealed that three key residues F273, I347 and D348 are crucial for interaction with Ex527* at pocket-C, and maintained the interaction for 75%, 100% and 125% of total simulation time, respectively (Fig. S5,† stable MD pose).
The results of per-residue energy decomposition and interaction fraction analysis matched well with the outcome of RMSF, and revealed that 7d, 13d, 13h and 13l have an additional interaction zone, the substrate site (pocket-A, hot-spot2) when compared to Ex527. We observed that at the substrate site most of the interactions are electrostatic (HBs, water bridges and ionic) and dominate the overall interaction contribution in 13d and 13l. As shown in Fig. 5B and 6A, the substrate-binding site residues (412–419 and 442–449) formed hot-spot2. The aromatic residues at hot-spot2, F413 and F414 formed pi–pi or pi–cation interactions with the benzimidazole moiety of 7d and pyrazole and rhodanine moiety of 13d, 13h and 13l; residues L418 and V445 formed hydrophobic contact with the R1 substituted phenyl moiety, and the small substitution of 4-OMe/4-NO2/2-Cl-6-F at the R1-position was found to be good for the stability of compounds and formed polar, water bridge, or direct HB contact with basic residues K444 and R446 (Fig. 6B). In addition, the study of the interaction fraction (Fig. 6B) showed that compounds 13d, 13h and 13l experienced unusually higher polar, ionic and water–bridge interactions at the hot-spot2 compared to 7d, which may be an explanation for comparatively higher Sirt1 selectivity of 13d, 13h and 13l. Moreover, Fig. 6A reflected that in all three 13d, 13h and 13l, residues R274 and Y280 formed either direct HB or water bridge contact with the carboxylic acid moiety at pocket-B, which was missing in the case of 7d due to structural orientation, which could be a reason for the higher affinity of 13d, 13h and 13l. These observations are consistent with the Sirt1 selectivity pattern of compounds of the pyrazole group viz. 13h and benzimidazole group 7d in biological assay.
The structural analysis of docked complexes of 7d, 13d, 13h and 13l reflected that in both benzimidazole and pyrazole classes of compounds, the –R1 substitution localized toward the substrate-binding site and the substitution –R2 is protruded towards the pocket-C (Fig. 6). Similarly, when we superimposed the best docked poses of all the compounds of the benzimidazole monopeptide class (Table 1), which had 4-OMe at –R1 substitution, with complex 7d, we found that the polar aromatic indole moiety which appeared to be crucial for the interaction was missing in other compounds (Fig. S6A†), which could be a possible reason for the loss of their inhibitory activity. Similarly, as shown in Table 1 three compounds 7d, 7h and 7l from the benzimidazole class had an indole group at the –R2 position that was required for the inhibition of Sirt1 biological activity as predicted above, but among them, only 7d was found to be active in cell-based assay (Table 3). We observed that compounds 7d, 7h and 7l only differ in substitution at the R1 position (Fig. S6B† and Table 1), and compound 7d which had 4-OMe at the R1 position showed good inhibition of Sirt1 catalytic activity. In the case of compound 7l, R1 substitution was “2-Cl”, which was at the ortho position, and –Cl substitution at the ortho position formed steric clash with residue L418 and decreased the stability of the compound, revealed in MD analysis (Fig. S6B and C†). Similarly, in the case of compounds 7h the –R1 substitution was 4-F (Fig. S6B†). During MD simulation, it was found that compound 7h was comparatively less stable than 7d (Fig. S6C†). Interestingly, as shown in Fig. 6 the upper hydrophobic groove which is flexible and made by residues V412 to P419 makes a flexible groove in which the phenyl moiety of 7d was trapped completely, providing the gain in binding affinity. This groove is a narrow hydrophobic pocket, which could be an area of future exploration to find additional classes of selective Sirt1 inhibitors. Also, the compounds 13h and 13d gain additional interaction in the form of stable HBs, water bridges, or polar contacts with basic residues K444 and R446 (Fig. 6), which appears to be a peculiar selectivity feature as K444 and R446 are present in Sirt1 only (in place of K/R it is Q/E in Sirt2, respectively). In addition, while compounds 7d, 13d, 13h and 13l explored additional interactions at the Sirt1 substrate-binding site, one primary interaction with residue D348 was missing unlike Ex527*, and any substitution at pocket-C with a group like polar basic amine may further enhance the potency of the molecule along with Sirt1 selectivity which can be explored in the future.
Possible mechanism of selectivity
The electrostatic interactions play a critical role in ligand binding, as protein has a specific electrostatic environment that the ligand needs for binding. We compared the Sirt1–3 binding sites in terms of electrostatic surface potential (ESP), as the ESP is an important key factor at the region of substrate binding site and played a key role in the Sirt1 selectivity and binding of 7d (Fig. 7). As shown in Fig. 7C and D, the ESP of 7d is complementarity to the ESP of the binding site and it favors the binding of 7d with Sirt1. Furthermore, at the substrate-binding site, we observed that the residues 442–449 (Sirt1) have positive ESP (in blue), while residues 263–270 (Sirt2) and 321–328 (Sirt3) have negative ESP (in red). Since ligands 7d, 13d, 13h and 13l have negative electrostatic potential near the substrate binding site, the ESP of the protein–ligand complex system in Sirt1 reflected complete ESP complementarity that is favorable for binding (Fig. 7E), while contradictory, unfavorable ESP was observed in the case of Sirt2 (Fig. 7F) and Sirt3 (Fig. 7G) which repel each other and is possibly a root cause of decreased affinity of 7d, 13d, 13h and 13l in Sirt2 and Sirt3. We observed that the lower panel of substrate binding sites among Sirt1–3 has a binding motif “SLxVxP(V/F)A”, and the variable residues (x) in Sirt1 are K444 and R446 and have shown substantial interaction with compounds (Fig. 7A and B). Since both the residues K444 and R446 are basic and have positive ESP, they serve as a selectivity hot-spot for interaction with compounds. In the cases of Sirt2 and Sirt3, the counterpart residues are glutamine (Q265 and Q267) and glutamic acid (E321 and E325), respectively, which make ESP negative (Fig. 7). This remarkable difference in ESP behavior at the substrate binding site in Sirt1–3 reflected that the compounds with negative ESP at the substrate binding site can be more Sirt1 selective.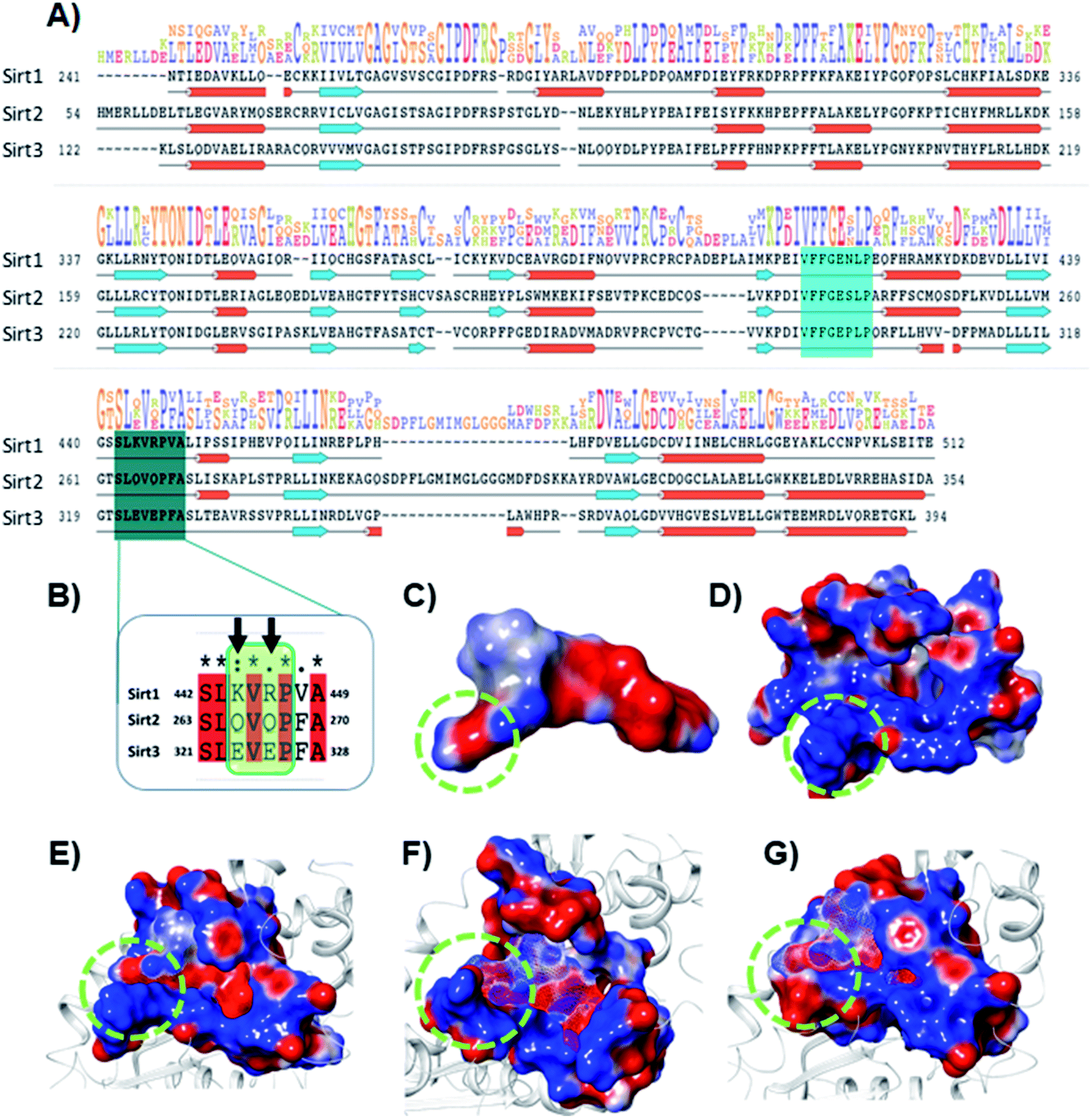 | ||
| Fig. 7 Selectivity at the substrate binding site explored at the sequence and structural level. (A) The sequence alignment of Sirt1, Sirt2 and Sirt3 PDB id. 4I5I, 4RMG and 4JSR, respectively. The region highlighted in cyan and deep cyan color represents the substrate-binding site. (B) The enlarged view of the sequence alignment of lower panel of substrate binding zone of Sirt1–3 (green color box). The arrow sign marked the residues K444 and R446 that played a key role in Sirt1 selectivity of 7d via polar electrostatic interaction. (C and D) Electrostatic surface view of ligand 7d (C) and Sirt1 binding cavity (D). (E–G) The electrostatic surface potential (ESP) complementarity analysis of 7d with Sirt1 (E), Sirt2 (F) and Sirt3 (G). Protein is shown in the solid surface, while 7d is shown in the mesh surface. The blue and red color surface represents positive and negative potential, respectively. The region highlighted with a green dotted circle highlights the selectivity zone at the substrate-binding site. | ||
Conclusion
In summary, from virtual screening (1.0 million in-house e-compounds) and structure-based approaches, two series of novel compounds were designed, synthesized and evaluated in enzyme- and cell-based assays. The bpMD, MD simulations, MM-GBSA and MM-PBSA were conducted to explore Sirt1's structural dynamics and develop comprehensive molecular SAR. The IFD docking and MM-GBSA calculations of the compounds along with in vitro enzymatic and cell-based assays revealed that the tryptophan conjugates of both the scaffolds were potent and selective in inhibiting Sirt1 enzyme over the rest of the homologs at micromolar potency, comparable to the known inhibitor, Ex527. The residue interaction fraction analysis and MM-PBSA studies illustrated the need for the compounds to possess an aromatic moiety with the attached polar group that can interact with pocket-C to exhibit inhibitory action; another aromatic ring at the substrate-binding site with small substitutions such as methoxy, nitro, or di-fluoro on the phenyl ring can further improve the stability and selectivity of the enzyme–inhibitor complex. The HB interaction with D348 appeared promising at pocket-C interaction and any substitution with polar basic amine like groups at pocket-C may further enhance the potency of the molecule which can be explored in the future. This study also highlighted the importance of IFD and binding pose metadynamics in the designing of ligands for the flexible and solvent-exposed binding site over conventional docking. We also explore the possible mechanism of improved Sirt1 selectivity. We found that the improved Sirt1 selectivity of our compounds is due to the presence of strong electrostatic interactions with the two basic residues K444 and R446 of the “SLxVxP(V/F)A” motif at the lower cleft of the substrate binding site, which favors the interactions with electronegative atoms like –O and –F via formation of multiple hydrogen bonds, water bridge and polar contacts. Finally, the experimental validation via in vitro enzyme- and cell-based assays confirmed that 7d, 13d, 13h, and 13l compounds were effective to inhibit Sirt1 similar to Ex527. In a nutshell, this study illustrated the role of substrate binding sites and electrostatic complementarity in Sirt1–3 and will pave the way for designing of more potent and Sirt1–3 specific inhibitors in future.Experimental section
Chemistry
All precursor chemicals and solvents were procured from Spectrochem Pvt. Ltd (India) and Sigma-Aldrich (India) via commercial vendors in suitable grades and used without further purification. The reactions were conducted with a guard tube containing calcium chloride attached to the reaction flask. The open capillary method was used to determine the uncorrected melting points. A Shimadzu FT-IR 157 spectrometer was used to record the IR spectra. A Bruker Advance II – 400 spectrometer was used to record NMR spectra at 400 MHz for 1H and at 100 MHz for 13C nuclei respectively. Tetramethylsilane (TMS) served as an internal standard. The values of coupling constants (J) and the chemical shifts (δ) are expressed in Hertz and parts per million (ppm) respectively. An Agilent Technology LC-mass spectrometer with ESI ionization was used to record the mass spectra. Elemental analysis was conducted in a CHNS Elementar Vario EL III. Thin-layer chromatography (TLC) was carried out on a silica-coated aluminum sheet (silica gel 60F254) to monitor the reaction progress and purity of the compounds using ethyl acetate and hexane solvent systems as the mobile phase and visualized under UV light at 254 nm. Column chromatography was performed with 60–120 mesh silica gel. Further for the detailed information please see the ESI file (page no. S10 to S27†).Biological evaluation
In silico computational methods
The overall computational protocol implemented in this analysis is demonstrated in Fig. S7.† All calculations were performed, on a Linux Centos 7.0 based workstation (RAM 132 GB, CPU-core 8, GPU-card 1 GTX1080) and Linux Centos 6.9 based server (RAM 64 GB, CPU(S) 64, GPU(S) 2 Tesla-P100), by using the Maestro release 2017–2 graphical user interface (GUI) of the Schrodinger software suite,66 VMD 1.9.3,67 ChemDraw Professional 15.1.68 Desmond3.7 (ref. 69) and Amber 2016.70P for all compounds were calculated using ChemDraw.Subsequently, with the help of Glide (Glide, Maestro 11.2.014, Schrödinger LLC), receptor grids of these complexes were generated. A virtual grid box of volume 20 × 20 × 20 Å region in space centered at the original ligand of the complex structures was considered, generating a receptor grid. For the other parameters, the default values were assigned.75
Furthermore, the per-residue energy decomposition of the MD system was calculated through the MM-PBSA protocol, implemented in Amber 2016, over the most stable pose extracted from the equilibrated trajectory of last 50 ns of MD simulation.45,78 A total of 200 snapshots were extracted from the equilibrated trajectory of each system and binding energy was calculated through the MM-PBSA.py script implemented in Amber.
ΔGbind is the total sum of difference in minimized energy (ΔEMM), difference in solvation energy (ΔGsolv), and difference in surface area energy.
| ΔGbind = ΔEMM + ΔGsolv + ΔGSA |
| ΔEMM = Emin(complex) − (Emin(protein) + Emin(ligand)) |
| ΔGsolv = Gsolv(complex) − (Gsolv(protein) + Gsolv(ligand)) |
| ΔGSA = GSA(complex) − (GSA(protein) + GSA(ligand)) |
000 frames.80Abbreviations
| Sirt1–3 | Sirt1, Sirt2 and Sirt3 |
| Sirt1 | NAD-dependent protein deacetylase sirtuin-1, silent information regulator2 homolog 1 |
| Sirt2 | NAD-dependent protein deacetylase sirtuin-2, silent information regulator2 homolog 2 |
| Sirt3 | NAD-dependent protein deacetylase sirtuin-3, silent information regulator2 homolog 3 |
| MD simulation | Molecular dynamics simulation |
| HepG2 cells | Human hepatoma G2, human liver cancer cell |
| MCF cells | Michigan cancer foundation-7, breast cancer cells |
| IC50 | Half maximal inhibitory concentration |
| HBs | Hydrogen bonds |
| ESP | Electrostatic surface potential |
| MM-GBSA | Molecular mechanics-generalized Born surface area |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| Ex527* | Ex527 analogue with a 7-membered ring |
Author contributions
Shailendra Asthana conceived the idea and designed the work. Nikil Purushotham and Boja Poojary performed chemical synthesis, Mrityunjay Singh and Shailendra Asthana performed and supervised computational designing and molecular modeling studies, and Bugga Paramesha and Sanjay K. Banerjee performed and supervised experimental assays. All authors wrote this paper and have approved the final version of the manuscript.Conflicts of interest
The authors declare that they do not have any conflicts of interest. The authors declare no competing financial interest.Acknowledgements
Nikil Purushotham is thankful to DST New Delhi for the INSPIRE fellowship. Mrityunjay Singh is thankful to Council of Scientific & Industrial Research (CSIR), New Delhi, for awarding Senior Research Fellowship (grant no. 09/1049(0023)/017/EMR-1), and Bugga Paramesha is thankful to the Council of Scientific & Industrial Research (CSIR), New Delhi, for awarding Junior Research Fellowship. Boja Poojary is thankful to UGC SAP for financial assistance. Sanjay K Banerjee and Shailendra Asthana are thankful to THSTI for providing the necessary support and core funding. We are also thankful to the NMR facility at THSTI for assisting us in generating compounds' 1H and 13C NMR spectra.References
- W. W. Hsu, B. Wu and W. R. Liu, ACS Chem. Biol., 2016, 11, 792–799 CrossRef CAS PubMed.
- S. Michan and D. Sinclair, Biochem. J., 2007, 404, 1–13 CrossRef CAS PubMed.
- T. Liu, P. Y. Liu and G. M. Marshall, Cancer Res., 2009, 69, 1702–1705 CrossRef CAS PubMed.
- S. M. Jeong and M. C. Haigis, Mol. Cells, 2015, 38, 750–758 CrossRef CAS PubMed.
- R. A. Frye, Biochem. Biophys. Res. Commun., 2000, 273, 793–798 CrossRef CAS PubMed.
- H. Yuan and R. Marmorstein, J. Biol. Chem., 2012, 287, 42428–42435 CrossRef CAS PubMed.
- M. C. Haigis and D. A. Sinclair, Annu. Rev. Pathol., 2010, 5, 253–295 CrossRef CAS PubMed.
- C. Sebastian, F. K. Satterstrom, M. C. Haigis and R. Mostoslavsky, J. Biol. Chem., 2012, 287, 42444–42452 CrossRef CAS PubMed.
- F. Niederer, C. Ospelt, F. Brentano, M. O. Hottiger, R. E. Gay, S. Gay, M. Detmar and D. Kyburz, Ann. Rheum. Dis., 2011, 70, 1866–1873 CrossRef CAS PubMed.
- A. Engler, C. Tange, M. Frank-Bertoncelj, R. E. Gay, S. Gay and C. Ospelt, J. Mol. Med., 2016, 94, 173–182 CrossRef CAS PubMed.
- M. R. Pinzone, B. Cacopardo, F. Condorelli, M. Di Rosa and G. Nunnari, Curr. Drug Targets, 2013, 14, 648–652 CrossRef CAS PubMed.
- E. Morselli, M. C. Maiuri, M. Markaki, E. Megalou, A. Pasparaki, K. Palikaras, A. Criollo, L. Galluzzi, S. A. Malik, I. Vitale, M. Michaud, F. Madeo, N. Tavernarakis and G. Kroemer, Cell Death Dis., 2010, 1, e10 CrossRef CAS PubMed.
- J. Luo, A. Y. Nikolaev, S.-i. Imai, D. Chen, F. Su, A. Shiloh, L. Guarente and W. Gu, Cell, 2001, 107, 137–148 CrossRef CAS PubMed.
- H. C. Chen, Y. M. Jeng, R. H. Yuan, H. C. Hsu and Y. L. Chen, Ann. Surg. Oncol., 2012, 19, 2011–2019 CrossRef PubMed.
- D. K. Alves-Fernandes and M. G. Jasiulionis, Int. J. Mol. Sci., 2019, 20, 3153 CrossRef CAS PubMed.
- V. Carafa, L. Altucci and A. Nebbioso, Front. Pharmacol., 2019, 10, 38 CrossRef CAS PubMed.
- S. Li, H. Hong, H. Lv, G. Wu and Z. Wang, Med. Sci. Monit., 2016, 22, 1593–1600 CrossRef CAS PubMed.
- M. Farcas, A. A. Gavrea, D. Gulei, C. Ionescu, A. Irimie, C. S. Catana and I. Berindan-Neagoe, Front. Nutr., 2019, 6, 148 CrossRef PubMed.
- Z. Wang and W. Chen, Genes Cancer, 2013, 4, 82–90 CrossRef CAS PubMed.
- K. Kojima, R. Ohhashi, Y. Fujita, N. Hamada, Y. Akao, Y. Nozawa, T. Deguchi and M. Ito, Biochem. Biophys. Res. Commun., 2008, 373, 423–428 CrossRef CAS PubMed.
- H. Dai, A. W. Case, T. V. Riera, T. Considine, J. E. Lee, Y. Hamuro, H. Zhao, Y. Jiang, S. M. Sweitzer, B. Pietrak, B. Schwartz, C. A. Blum, J. S. Disch, R. Caldwell, B. Szczepankiewicz, C. Oalmann, P. Yee Ng, B. H. White, R. Casaubon, R. Narayan, K. Koppetsch, F. Bourbonais, B. Wu, J. Wang, D. Qian, F. Jiang, C. Mao, M. Wang, E. Hu, J. C. Wu, R. B. Perni, G. P. Vlasuk and J. L. Ellis, Nat. Commun., 2015, 6, 7645 CrossRef PubMed.
- J. S. Disch, G. Evindar, C. H. Chiu, C. A. Blum, H. Dai, L. Jin, E. Schuman, K. E. Lind, S. L. Belyanskaya, J. Deng, F. Coppo, L. Aquilani, T. L. Graybill, J. W. Cuozzo, S. Lavu, C. Mao, G. P. Vlasuk and R. B. Perni, J. Med. Chem., 2013, 56, 3666–3679 CrossRef CAS PubMed.
- J. B. Park, PLoS One, 2016, 11, e0150392 CrossRef PubMed.
- Y. Yoon, H. Osman and T. Choon, MedChemComm, 2016, 7, 2094–2099 RSC.
- T. Rumpf, M. Schiedel, B. Karaman, C. Roessler, B. J. North, A. Lehotzky, J. Olah, K. I. Ladwein, K. Schmidtkunz, M. Gajer, M. Pannek, C. Steegborn, D. A. Sinclair, S. Gerhardt, J. Ovadi, M. Schutkowski, W. Sippl, O. Einsle and M. Jung, Nat. Commun., 2015, 6, 6263 CrossRef CAS PubMed.
- P. Di Fruscia, E. Zacharioudakis, C. Liu, S. Moniot, S. Laohasinnarong, M. Khongkow, I. F. Harrison, K. Koltsida, C. R. Reynolds, K. Schmidtkunz, M. Jung, K. L. Chapman, C. Steegborn, D. T. Dexter, M. J. Sternberg, E. W. Lam and M. J. Fuchter, ChemMedChem, 2015, 10, 69–82 CrossRef CAS PubMed.
- G. Hoffmann, F. Breitenbucher, M. Schuler and A. E. Ehrenhofer-Murray, J. Biol. Chem., 2014, 289, 5208–5216 CrossRef CAS PubMed.
- T. F. Outeiro, E. Kontopoulos, S. M. Altmann, I. Kufareva, K. E. Strathearn, A. M. Amore, C. B. Volk, M. M. Maxwell, J. C. Rochet, P. J. McLean, A. B. Young, R. Abagyan, M. B. Feany, B. T. Hyman and A. G. Kazantsev, Science, 2007, 317, 516–519 CrossRef CAS PubMed.
- M. C. Haigis and L. P. Guarente, Genes Dev., 2006, 20, 2913–2921 CrossRef CAS PubMed.
- A. D. Napper, J. Hixon, T. McDonagh, K. Keavey, J. F. Pons, J. Barker, W. T. Yau, P. Amouzegh, A. Flegg, E. Hamelin, R. J. Thomas, M. Kates, S. Jones, M. A. Navia, J. O. Saunders, P. S. DiStefano and R. Curtis, J. Med. Chem., 2005, 48, 8045–8054 CrossRef CAS PubMed.
- E. Therrien, G. Larouche, N. Nguyen, J. Rahil, A. M. Lemieux, Z. Li, M. Fournel, T. P. Yan, A. J. Landry, S. Lefebvre, J. J. Wang, K. MacBeth, C. Heise, A. Nguyen, J. M. Besterman, R. Deziel and A. Wahhab, Bioorg. Med. Chem. Lett., 2015, 25, 2514–2518 CrossRef CAS PubMed.
- A. Ghosh, A. Sengupta, G. P. K. Seerapu, A. Nakhi, E. V. V. Shivaji Ramarao, N. Bung, G. Bulusu, M. Pal and D. Haldar, Biochem. Biophys. Res. Commun., 2017, 488, 562–569 CrossRef CAS PubMed.
- M. J. Kim, Y. J. Kang, B. Sung, J. Y. Jang, Y. R. Ahn, H. J. Oh, H. Choi, I. Choi, E. Im, H. R. Moon, H. Y. Chung and N. D. Kim, Biomol. Ther., 2020, 28, 561–568 CrossRef PubMed.
- H. Ota, E. Tokunaga, K. Chang, M. Hikasa, K. Iijima, M. Eto, K. Kozaki, M. Akishita, Y. Ouchi and M. Kaneki, Oncogene, 2006, 25, 176–185 CrossRef CAS PubMed.
- Y. Cen, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1635–1644 CrossRef CAS PubMed.
- S. Portmann, R. Fahrner, A. Lechleiter, A. Keogh, S. Overney, A. Laemmle, K. Mikami, M. Montani, M. P. Tschan, D. Candinas and D. Stroka, Mol. Cancer Ther., 2013, 12, 499–508 CrossRef CAS PubMed.
- T. Wang, X. Li and S. L. Sun, Anticancer Drugs, 2020, 31, 19–26 CrossRef CAS PubMed.
- N. Panathur, N. Gokhale, U. Dalimba, P. V. Koushik, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2015, 25, 2768–2772 CrossRef CAS PubMed.
- M. Spinck, M. Bischoff, P. Lampe, F.-J. Meyer-Almes, S. Sievers and H. Neumann, J. Med. Chem., 2021, 64, 5838–5849 CrossRef CAS PubMed.
- X. Xia and X. Zhou, Cell. Mol. Biol., 2018, 64, 36–41 CrossRef.
- R. Asaka, T. Miyamoto, Y. Yamada, H. Ando, D. H. Mvunta, H. Kobara and T. Shiozawa, Lab. Invest., 2015, 95, 1363–1373 CrossRef CAS PubMed.
- H. K. Choi, K. B. Cho, N. T. Phuong, C. Y. Han, H. K. Han, T. T. Hien, H. S. Choi and K. W. Kang, Mol. Pharm., 2013, 10, 2517–2527 CrossRef CAS PubMed.
- I. H. Tae, E. Y. Park, P. Dey, J. Y. Son, S.-Y. Lee, J. H. Jung, S. Saloni, M.-H. Kim and H. S. Kim, Int. J. Oncol., 2018, 53, 2518–2530 CAS.
- N. Wossner, Z. Alhalabi, J. Gonzalez, S. Swyter, J. Gan, K. Schmidtkunz, L. Zhang, A. Vaquero, H. Ovaa, O. Einsle, W. Sippl and M. Jung, Front. Oncol., 2020, 10, 657 CrossRef PubMed.
- M. Singh, M. Srivastava, S. R. Wakode and S. Asthana, J. Chem. Inf. Model., 2021, 61, 1105–1124 CrossRef CAS PubMed.
- M. Singh, M. Srivastava, N. Purushotham, B. Paramesha, S. R. Wakode, B. Poojary, S. K. Banerjee and S. Asthana, Biophys. J., 2020, 118, 207a CrossRef.
- U. Uciechowska, J. Schemies, R. C. Neugebauer, E. M. Huda, M. L. Schmitt, R. Meier, E. Verdin, M. Jung and W. Sippl, ChemMedChem, 2008, 3, 1965–1976 CrossRef CAS PubMed.
- P. Kokkonen, T. Kokkola, T. Suuronen, A. Poso, E. Jarho and M. Lahtela-Kakkonen, Eur. J. Pharm. Sci., 2015, 76, 27–32 CrossRef CAS PubMed.
- G. Eren, A. Bruno, S. Guntekin-Ergun, R. Cetin-Atalay, F. Ozgencil, Y. Ozkan, M. Gozelle, S. G. Kaya and G. Costantino, J. Mol. Graphics Modell., 2019, 89, 60–73 CrossRef CAS PubMed.
- R. C. Neugebauer, U. Uchiechowska, R. Meier, H. Hruby, V. Valkov, E. Verdin, W. Sippl and M. Jung, J. Med. Chem., 2008, 51, 1203–1213 CrossRef CAS PubMed.
- X. Zhao, D. Allison, B. Condon, F. Zhang, T. Gheyi, A. Zhang, S. Ashok, M. Russell, I. MacEwan, Y. Qian, J. A. Jamison and J. G. Luz, J. Med. Chem., 2013, 56, 963–969 CrossRef CAS PubMed.
- M. Wang, K. P. Rakesh, J. Leng, W. Y. Fang, L. Ravindar, D. Channe Gowda and H. L. Qin, Bioorg. Chem., 2018, 76, 113–129 CrossRef CAS PubMed.
- S. Oda, H. Shimizu, Y. Aoyama, T. Ueki, S. Shimizu, H. Osato and Y. Takeuchi, Org. Process Res. Dev., 2011, 16, 96–101 CrossRef.
- A. M. Vijesh, A. M. Isloor, S. K. Peethambar, K. N. Shivananda, T. Arulmoli and N. A. Isloor, Eur. J. Med. Chem., 2011, 46, 5591–5597 CrossRef CAS PubMed.
- A. V. Lebedev, A. B. Lebedeva, V. D. Sheludyakov, E. A. Kovaleva, O. L. Ustinova and I. B. Kozhevnikov, Russ. J. Gen. Chem., 2005, 75, 782–789 CrossRef CAS.
- L. Slepikas, G. Chiriano, R. Perozzo, S. Tardy, A. Kranjc, O. Patthey-Vuadens, H. Ouertatani-Sakouhi, S. Kicka, C. F. Harrison, T. Scrignari, K. Perron, H. Hilbi, T. Soldati, P. Cosson, E. Tarasevicius and L. Scapozza, J. Med. Chem., 2016, 59, 10917–10928 CrossRef CAS PubMed.
- F. S. Prout, J. Org. Chem., 2002, 18, 928–933 CrossRef.
- B. Peck, C. Y. Chen, K. K. Ho, P. Di Fruscia, S. S. Myatt, R. C. Coombes, M. J. Fuchter, C. D. Hsiao and E. W. Lam, Mol. Cancer Ther., 2010, 9, 844–855 CrossRef CAS PubMed.
- J. M. Solomon, R. Pasupuleti, L. Xu, T. McDonagh, R. Curtis, P. S. DiStefano and L. J. Huber, Mol. Cell. Biol., 2006, 26, 28–38 CrossRef CAS PubMed.
- S. S. Mahajan, M. Scian, S. Sripathy, J. Posakony, U. Lao, T. K. Loe, V. Leko, A. Thalhofer, A. D. Schuler, A. Bedalov and J. A. Simon, J. Med. Chem., 2014, 57, 3283–3294 CrossRef CAS PubMed.
- S. M. Reed and D. E. Quelle, Cancers, 2014, 7, 30–69 CrossRef PubMed.
- J. Yi and J. Luo, Biochim. Biophys. Acta, 2010, 1804, 1684–1689 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS PubMed.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. H. Zhang and T. Hou, Chem. Rev., 2019, 119, 9478–9508 CrossRef CAS PubMed.
- P. Mellini, Y. Itoh, E. E. Elboray, H. Tsumoto, Y. Li, M. Suzuki, Y. Takahashi, T. Tojo, T. Kurohara, Y. Miyake, Y. Miura, Y. Kitao, M. Kotoku, T. Iida and T. Suzuki, J. Med. Chem., 2019, 62, 5844–5862 CrossRef CAS PubMed.
- Schrödinger LLC, Schrödinger Release 2017-2: Maestro. Schrödinger LLC, New York, 2017 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
- N. Mills, J. Am. Chem. Soc., 2006, 128, 13649–13650 CrossRef CAS.
- Schrödinger Release, Maestro-Desmond Interoperability Tools, Version, 3, DE Shaw Research, New York, NY, 2014 Search PubMed.
- D. A. Case, D. Cerutti, T. Cheatham, T. Darden, R. Duke, T. Giese, H. Gohlke, A. Goetz, D. Greene and N. Homeyer, Amber 2016, University of California, San Francisco, 2016 Search PubMed.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS PubMed.
- M. P. Repasky, M. Shelley and R. A. Friesner, Curr. Protoc. Bioinf., 2007, 18, 811–1218 Search PubMed.
- S. Asthana, P. Zucca, A. V. Vargiu, E. Sanjust, P. Ruggerone and A. Rescigno, J. Agric. Food Chem., 2015, 63, 7236–7244 CrossRef CAS PubMed.
- S. Mattapally, M. Singh, K. S. Murthy, S. Asthana and S. K. Banerjee, Oncotarget, 2018, 9, 13713–13732 CrossRef PubMed.
- L. Mittal, A. Kumari, C. Suri, S. Bhattacharya and S. Asthana, J. Biomol. Struct. Dyn., 2020, 38, 1612–1625 CrossRef CAS PubMed.
- A. J. Clark, P. Tiwary, K. Borrelli, S. Feng, E. B. Miller, R. Abel, R. A. Friesner and B. J. Berne, J. Chem. Theory Comput., 2016, 12, 2990–2998 CrossRef CAS PubMed.
- J. Li, R. Abel, K. Zhu, Y. Cao, S. Zhao and R. A. Friesner, Proteins, 2011, 79, 2794–2812 CrossRef CAS PubMed.
- L. Mittal, M. Srivastava and S. Asthana, J. Phys. Chem. B, 2019, 123, 6150–6160 CrossRef CAS PubMed.
- M. Srivastava, C. Suri, M. Singh, R. Mathur and S. Asthana, Oncotarget, 2018, 9, 34289 CrossRef PubMed.
- L. Subramanian, S. Maghajothi, M. Singh, K. Kesh, A. Kalyani, S. Sharma, M. Khullar, S. M. Victor, S. Swarnakar, S. Asthana, A. S. Mullasari and N. R. Mahapatra, Hypertension, 2019, 74, 1448–1459 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S8. See DOI: 10.1039/d1ra06149f |
| ‡ Nikil Purushotham, Mrityunjay Singh and Bugga Paramesha contribute equally to this work. |
| § Present address: Department of Biotechnology, National Institute of Pharmaceutical Education and Research (NIPER), Guwahati-781101, India. |
| This journal is © The Royal Society of Chemistry 2022 |