
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide cyclisation promoted by supramolecular complex formation†
Arnout P. T.
Hartendorp
,
Felix J.
de Zwart
 ,
Hans
Bieräugel
,
Bas de
Bruin
,
Joost N. H.
Reek
and
Jan H.
van Maarseveen
*
,
Hans
Bieräugel
,
Bas de
Bruin
,
Joost N. H.
Reek
and
Jan H.
van Maarseveen
*
Van‘t Hoff Institute for Molecular Sciences (HIMS), Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.h.vanmaarseveen@uva.nl
First published on 16th December 2021
Abstract
Phenol ester activated dipeptides that are reluctant to ring-close have been cyclised with the aid of sterically shielding metallo-porphyrins avoiding unwanted intermolecular reactions. The binding of ZnTPP to the dipyridine-functionalised activating phenolic ester was studied by NMR titrations and modelling. Staudinger-mediated cyclisations in the presence of ZnTPP increased the yield of the cyclic dipeptide from 16% to 40%.
Cyclic peptides are widespread in nature. Bacteria and plants use cyclic peptides for protection against a plethora of invaders. As a result, cyclic peptides constitute an important molecular class in drug discovery. In 2017, over 40 cyclic peptide-based drugs were in clinical use.1 Most of these peptide drugs consist of 5–14 amino acids.2 There are still challenges in the field of drug research and arguably the two most important ones are oral availability and cell membrane permeability.1,3 To overcome these potential problems, lowering the molecular weight within the range of Lipinski's rules4 (MW < 500) is a prerequisite. However, decreasing the ring size of cyclic peptides poses a synthetic challenge.
The synthesis of cyclic seven-membered dipeptides is difficult due to the low concentration of the productive (cisoid) conformation of the linear precursor. For this, the predominantly transoid-configured amide linkage has to adopt a cisoid conformation to enable cyclisation.5 Because this equilibrium lies far to the transoid side, intermolecular reactions take precedence over the desired unimolecular cyclisation of the monomer. In order to decrease the unwanted intermolecular reactions, we have previously used, next to covalent templates providing lactams via ring-contraction strategies, cavities within carbosilane dendrimers to shield the dipeptides from the bulk solution and prevent intermolecular side reactions.6 However, the required precursors for such lacatamisation reactions are tedious to prepare. As such, we wondered if similar site-isolation effects can be achieved by using simpler building blocks. In this context, we realised that a facile way to shield the peptide from other peptides may be to utilise the known dipyridine backbones that are typically used as building block for the self-assembly of well-defined supramolecular structures.7–11 As compared to the lengthy covalent multistep synthesis of the carbosilane dendrimers, supramolecular self-assembly is an interesting tool that enables access to large and/or complex structures.
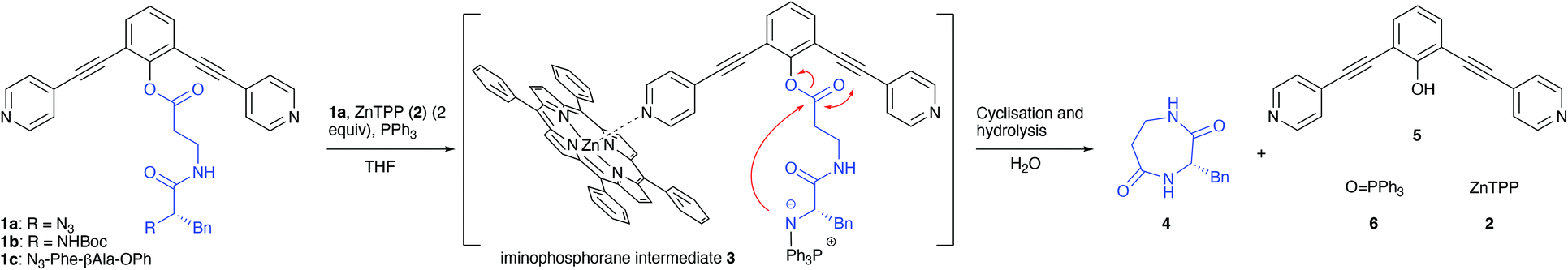
As outlined in Fig. 1, our strategy is based on peptide phenolic esters 1a,b of which the phenyl group is functionalised with two 2,6-alkyne tethered 4-pyridines.13 Metallo-porphyrins, e.g. 5,10,15,20-tetraphenyl-21H,23H-porphine zinc (ZnTPP, 2) can coordinate to the pyridine-moieties and, while doing so, sterically shield the mildly activated phenolic-ester linkage.14 As a result, the ester is confined within a small cavity defined by the porphyrins associated to the pyridyl functions, which should lead to a favourable intramolecular reaction compared to intermolecular processes that eventually lead to oligomeric (cyclisation) products. As a target that is reluctant to ring-closure seven-membered 1,4-diazepane-2,5-dione c[βAla-Phe] 4 was chosen. The nucleophilic amine terminus of the linear precursor was liberated both via a Staudinger reaction from N3-Phe-βAla-OAr (1a) or after protolytic removal of the Boc-protective group at BocHN-Phe-βAla-OAr (1b). As a reference compound also N3-Phe-βAla-OPh (1c), thus lacking the metallo-porphyrin coordination sites, was included in this study.12
 | ||
| Fig. 1 The concept of site-isolation of an activated peptide by self-assembly by porphyrin-pyridyl coordination to form a cavity, possibly leading to more efficient formation of cyclic dipeptide. | ||
For the synthesis and characterisation (1H NMR, 13C NMR and HR-MS) of cyclisation precursors 1a–c we refer to the ESI.† With the building blocks in hand we needed to establish whether the proposed self-assembled structures can form indeed. Based on previous research on template-ligand approaches towards confined catalysts,15–17 we anticipated that just mixing the new building block with Zn porphyrin 2 should lead to the proposed 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex. In this complex, the reactive phenolic ester is well buried within the cavity defined by the porphyrins, thus hampering unwanted intermolecular reactions.
:1 complex. In this complex, the reactive phenolic ester is well buried within the cavity defined by the porphyrins, thus hampering unwanted intermolecular reactions.
The binding event of ZnTPP (2a) to the dipyridine backbone was evaluated by 1H NMR spectroscopy using THF-d8 as the solvent as will be the case for the cyclisation reactions. For determining of the binding constant, 2,6-bis(pyridin-4-ylethynyl)phenyl acetate (7) was chosen of which the concentration was kept constant at 8.3 mM, while gradually increasing the amount of ZnTPP. The coordination of the metallo-porphyrins to the pyridines in THF-d8 is evident from the typical upfield shifts in the 1H NMR of the pyridyl protons, with the signals of the protons ortho with respect to the nitrogen atoms shifting the most (see Fig. S1†). These NMR data, was used to construct binding curves that were fitted to calculate the binding constants under these conditions using Bindfit v0.5 (accessed through http://app.supramolecular.org/bindfit). As there should be no electronic communication between the two binding sites on the backbone, the data were fitted to a non-cooperative 2:1 fit, which assumes that the binding sites are identical and thus the apparent binding of the second porphyrin is four times as small (K1/K2 = 4) which is purely a statistical factor.18 The found binding constant of 2 × 102 M−1 is in line with reported association constants in the range of 102–104 M−1 of pyridine to ZnTPP in various non-coordinating solvents.19–24 This binding constant indicates that for the exclusive formation of the 2:1 complex high concentrations of ZnTPP are required, and that under the typical reaction conditions used for the cyclisation step the 1:1 complex is the dominant species. Although we aimed for complete encapsulation, we decided to continue as the concept may work also for the 1:1 complex as this already provides substantial steric hindrance prohibiting intermolecular reactions (see Fig. 2).
 | ||
| Fig. 2 3 coordinated to ZnTPP in stick (left) and space-filling (right) representation. | ||
With these results in hand, we set out to perform the cyclisation reactions (see Table 1). These lactamisations were carried out in THF-d8 as the solvent and using 1,3,5-trimethoxy benzene as an internal standard to allow quantification of seven membered ring product formation by 1H NMR.
|
|
|||
|---|---|---|---|
| Entry | Precursor | Porphyrin (equiv.) | Yield of 4 |
| Conditions: dipeptide (0.017 mM), PPh3 (1.1 equiv.) and porphyrin (see table) were dissolved in THF-d8 and heated in a microwave (max. 250 W) at 80 °C. After five hours, 60 μl H2O was added and the mixture heated for one additional hour at 80 °C. The yield was calculated based on 1H NMR analysis using 1,3,5-trimethoxy benzene as an internal standard.a Single reaction performed.b Reaction performed in duplo.c Reaction performed in triplo.d The amine group was liberated by treatment with TFA followed by removal of the volatiles and subsequent DiPEA addition.e The mixture was heated at 80 °C in an NMR tube in a sand bath for 20 hours. H2O was present at the start of the reaction. | |||
| 1 | 1c | — | 0%a |
| 2 | 1c | ZnTPP (3) | 0%b |
| 3 | 1a | — | 16 ± 7%c |
| 4 | 1a | ZnTPP (0.05) | 15 ± 4%b |
| 5 | 1a | ZnTPP (0.5) | 23 ± 5%b |
| 6 | 1a | ZnTPP (3) | 40 ± 7%c |
| 7 | 1b | — | Tracea |
| 8 | 1b | ZnTPP (3) | Tracea |
| 9 | 1a | ZnTPP (3) | 12%a,e |

The linear peptide precursor was mixed with triphenylphosphine and three equivalents of ZnTPP, followed by heating in a microwave at 80 °C for five hours. After this, an aliquot of 60 μl of H2O was added and the mixture was heated for an additional hour at 80 °C to liberate the product and triphenylphosphine oxide. In the first set of experiments, cyclisation of N3-βAla-Phe-OAr (1a) was compared to the pyridine-free compound N3-βAla-Phe-OPh (1c) as a reference as it is unable to bind porphyrins. The reactions were carried out both in the presence and absence of porphyrins to allow evaluation of the cavity effect (entries 1–4). When starting from pyridine-free ester 1c, the only observed products were H2N-βAla-Phe-OPh (loss of N2), Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-βAla-Phe-OPh (iminophosphorane intermediate), triphenylphosphine oxide (6) and one unidentified compound that did not ionise, preventing its characterisation by MS. If the sample was left to stand longer, the peaks of cyclisation precursor H2N-βAla-Phe-OPh and triphenylphosphine oxide remain. Thus, no formation of cyclic peptide 4 was observed when 1c was subjected to the reaction conditions, regardless of the presence of ZnTPP. Interestingly, when dipyridine 1a was subjected to the reaction conditions, yields of 16 ± 7% of 4 were already obtained in the absence of ZnTPP (entry 3). This shows that in 1c without ZnTPP the two ortho acetylene-pyridine groups already increase the sterics around the ester bond considerably, hampering unwanted intermolecular reactions. Another explanation is that the ortho groups lead to a change in conformation and preorganise the molecule to favour an intramolecular reaction. In the presence of ZnTPP (2a) the yield of the cyclised product 4 was significantly improved and increases with the amount of ZnTPP. In the presence of three equiv. of ZnTPP, a maximum yield of 40 ± 7% (entries 4–6) was obtained. Due to its limited solubility, higher concentrations of ZnTPP could not be achieved.
N-βAla-Phe-OPh (iminophosphorane intermediate), triphenylphosphine oxide (6) and one unidentified compound that did not ionise, preventing its characterisation by MS. If the sample was left to stand longer, the peaks of cyclisation precursor H2N-βAla-Phe-OPh and triphenylphosphine oxide remain. Thus, no formation of cyclic peptide 4 was observed when 1c was subjected to the reaction conditions, regardless of the presence of ZnTPP. Interestingly, when dipyridine 1a was subjected to the reaction conditions, yields of 16 ± 7% of 4 were already obtained in the absence of ZnTPP (entry 3). This shows that in 1c without ZnTPP the two ortho acetylene-pyridine groups already increase the sterics around the ester bond considerably, hampering unwanted intermolecular reactions. Another explanation is that the ortho groups lead to a change in conformation and preorganise the molecule to favour an intramolecular reaction. In the presence of ZnTPP (2a) the yield of the cyclised product 4 was significantly improved and increases with the amount of ZnTPP. In the presence of three equiv. of ZnTPP, a maximum yield of 40 ± 7% (entries 4–6) was obtained. Due to its limited solubility, higher concentrations of ZnTPP could not be achieved.
Finally, the Boc-protected cyclisation precursor 1b was employed to determine whether primary amines could also directly undergo cyclisation. The Boc-group was removed prior to the reaction by treatment with TFA, followed by removal of the volatiles. After the addition of DiPEA the nucleophilic amine was liberated from the salt. After prolonged reaction times, monitored by 1H NMR, only traces of 4 were detected regardless the presence of ZnTPP (entries 7 and 8, respectively). The low yield found in the experiment described in entry 8 (ZnTPP present) shed some light on the mechanism of the reaction. Obviously, product formation does not occur via the free amine. If this were the case, the yields found for the experiments of entries 6 and 8 would be comparable. The difference might be explained by the high nucleophilicity of the negatively charged N-atom in the iminophosphorane intermediate 3 (see Fig. 1) as compared to the neutral amine as obtained from 1b (Fig. 3).25
 | ||
| Fig. 3 Schematic representation of calculated structures of cisoid isomers of iminophosphorane intermediates 3 and 3-ZnTPP (Turbomole, BP86, def2-TZVP) with N–C distances in red. | ||
When the reaction was carried out in a closed vessel under heating at 80 °C, no product formation was observed after five hours. After allowing the reaction to stand for 20 hours at that temperature, lactam 4 was obtained in 12% yield (entry 9). While the usage of microwave conditions provided better results, the reaction is also possible under solvothermal conditions. Several reactions (entries 3, 6 and 8) were scaled up to a preparative scale. After isolation of the cyclic dipeptides by column chromatography, similar yields were obtained as for the experiments that were carried out on an analytical scale and quantified by integration of the NMR signals.
As the use of the Zn porphyrin template leads to a small but significant increase in the yield of cyclisation product 4, a qualitative DFT investigation into the conformers of this assembly was deemed useful. From previous investigations it is known that the step preceding cyclisation of α,β-dipeptides is isomerisation of the amide bond into a cisoid conformation. Isomerisation of the amide bond in iminophosphorane 3 from a transoid to cisoid conformation decreases the N–C (iminophosphorane nitrogen to ester carbon) distance from 6.51 Å to 6.18 Å (see ESI† for full computational details). The cisoid isomer is lower in energy by 5.1 kcal mol−1, presumably due to the release of strain between the ligand backbone and the bulky triphenylphosphine, possibly explaining why an appreciable yield is obtained even without ZnTPP. Upon investigation of the transoid and cisoid conformers of the 3-ZnTPP adduct, stark geometric differences were found (ESI S17†). Isomerisation of the amide bond in 3-ZnTPP causes the peptide chain to retract into the porphyrin-backbone cavity, leading to a decrease in the N–C distance from 6.38 Å to 4.07 Å, accompanied by an energetic penalty of 7.2 kcal mol−1. The steric bulk of the porphyrin cavity causes the preorganisation of the peptide for cyclisation by bringing the chain ends closer together, which could be responsible for the observed increase in yield. Further inspection revealed a close amide NH–Npyridine distance of 2.62 Å, leading us to hypothesise that future research efforts could be directed at enhancing this preorganisation effect by placing a strong hydrogen bond acceptor in the porphyrin cavity, stabilising the cis-3-ZnTPP conformation. Furthermore, the steric bulk of the porphyrin coordination also prevents dimerisation, as is reflected in the decrease of the accessible surface area on the ester carbon from 121 Å2 to 85 Å2 upon coordination of ZnTPP.
In summary, cyclisation reactions have been carried out starting from N3-βAla-Phe-OPh (1c) and the analogous dipyridine-functionalised α,β-dipeptide 1a. Cyclisation of dipyridine-functionalised α,β-dipeptide 1b featuring a Boc-protected N-terminal amine instead of the azide only gave traces of the 7-membered lactam. The cyclisation reactions have been carried out in the presence of metallo-porphyrins with the aim to effectuate site-isolation of the activated ester moiety. By starting from model peptide 1c, which lacks the coordinating pyridines, no cyclic peptide was formed after phosphine-mediated azide reduction. In the absence of ZnTPP, the dipyridine-functionalised α,β-dipeptide 1b gave 16% of the 7-membered lactam. By binding of ZnTPP to the dipyridine backbone, the yield could be more than doubled to 40 ± 7%. Binding of ZnTPP to the dipyridine backbone was studied by modelling and NMR titrations. Based on the results it is likely that the beneficial effect in yield can be attributed to coordination of a single ZnTPP molecule to one side of the dipyridine backbone, that causes preorganisation of the peptide for cyclisation.
This proof-of-principle study shows that supramolecular chemistry can be used to easily introduce steric bulk by self-assembly which favours cyclisation by hampering intermolecular reaction pathways or inducing conformational changes to preorganise the molecule and facilitate intramolecular reaction. Further optimisation of the building blocks changing the number of pyridine functions and the location with respect to the peptide chain as well as the zinc(II)porphyrin building blocks may lead to further increased yields. As it is likely that the 1:1 complex provides the productive pathway for cyclisation, it is also possible to lower the molecular weight of the auxiliary by using a monotopic pyridine backbone. Considering the ease of mix-and-react, the facile synthesis of libraries of small and strained lactams could be envisioned.
Author contributions
A. H., J. R. and J. v. M. proposed the research. Experiments were conducted by A. H. and H. B. F. d. Z., A. H. and B. d. B. carried out the computational studies. The manuscript was prepared by J. v. M., A. H., F. d. Z. and J. R.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by NWO Chemical Sciences TOP-PUNT Grant ‘Catalysis in Confined Spaces’ 718.015.004.References
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- D. Gang, D. Kim and H.-S. Park, Genes, 2018, 9, 557 CrossRef PubMed.
- D. S. Nielsen, N. E. Shepherd, W. Xu, A. J. Lucke, M. J. Stoermer and D. P. Fairlie, Chem. Rev., 2017, 117, 8094–8128 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- (a) J. S. Davies, J. Pept. Sci., 2003, 9, 471–501 CrossRef CAS PubMed; (b) V. Sarojini, A. J. Cameron, K. G. Varnava, W. A. Denny and G. Sanjayan, Chem. Rev., 2019, 119, 10318–10359 CrossRef CAS.
- A. Amore, R. Van Heerbeek, N. Zeep, J. Van Esch, J. N. H. Reek, H. Hiemstra and J. H. Van Maarseveen, J. Org. Chem., 2006, 71, 1851–1860 CrossRef CAS.
- S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Adv. Synth. Catal., 2016, 358, 1509–1518 CrossRef CAS.
- S. Gonell and J. N. H. Reek, ChemCatChem, 2019, 11, 1458–1464 CrossRef CAS PubMed.
- S. Gonell, X. Caumes, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Chem. Sci., 2019, 10, 1316–1321 RSC.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- K. Harris, Q. Sun, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 12497–12499 CrossRef CAS PubMed.
- O. David, W. J. N. Meester, H. Bieräugel, H. E. Schoemaker, H. Hiemstra and J. H. van Maarseveen, Angew. Chem., Int. Ed., 2003, 42, 4373–4375 CrossRef CAS PubMed.
- M. Kawano, S. Sato, M. Fujita, K. Suzuki, M. Kawano, S. Sato and M. Fujita, J. Am. Chem. Soc., 2007, 129, 10652–10653 CrossRef PubMed.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476–476 CrossRef CAS.
- V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., 2001, 113, 4401–4404 CrossRef.
- V. F. Slagt, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 1526–1536 CrossRef CAS PubMed.
- V. Bocokić, A. Kalkan, M. Lutz, A. L. Spek, D. T. Gryko and J. N. H. Reek, Nat. Commun., 2013, 4, 2670 CrossRef PubMed.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- A. Satake and Y. Kobuke, Tetrahedron, 2005, 61, 13–41 CrossRef CAS.
- S. J. Cole, G. C. Curthoys, E. A. Magnusson and J. N. Phillips, Inorg. Chem., 1972, 11, 1024–1028 CrossRef CAS.
- M. Nappa and J. S. Valentine, J. Am. Chem. Soc., 1978, 100, 5075–5080 CrossRef CAS.
- G. C. Vogel and J. R. Stahlbush, Inorg. Chem., 1977, 16, 950–953 CrossRef CAS.
- P. Bhyrappa, V. Krishnan and M. Nethaji, J. Chem. Soc., Dalton Trans., 1993, 1901 RSC.
- L. Favereau, A. Cnossen, J. B. Kelber, J. Q. Gong, R. M. Oetterli, J. Cremers, L. M. Herz and H. L. Anderson, J. Am. Chem. Soc., 2015, 137, 14256–14259 CrossRef CAS PubMed.
- P. N. M. Botman, O. David, A. Amore, J. Dinkelaar, M. T. Vlaar, K. Goubitz, J. Fraanje, H. Schenk, H. Hiemstra and J. H. Van Maarseveen, Angew. Chem., Int. Ed., 2004, 43, 3471–3473 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob02309h |
| This journal is © The Royal Society of Chemistry 2022 |