
Engineering the oxygen vacancies of rocksalt-type high-entropy oxides for enhanced electrocatalysis†
Yaohang
Gu
a,
Ateer
Bao
a,
Xuanyu
Wang
a,
Yizhen
Chen
b,
Liang
Dong
cd,
Xin
Liu
ac,
Haijun
Pan
*c,
Ying
Li
e and
Xiwei
Qi
 *f
*f
aSchool of Materials Science and Engineering, Northeastern University, Shenyang 110189, China
bHefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, China
cSchool of Resources and Materials, Northeastern University at Qinhuangdao, Qinhuangdao 066004, China. E-mail: panhaijun@neuq.edu.cn
dKey Laboratory of Dielectric and Electrolyte Functional Material Hebei Province, Northeastern University at Qinhuangdao, Qinhuangdao 066004, China
eSchool of Science, Inner Mongolia University of Science and Technology, Baotou 014010, China
fCollege of Metallurgy and Energy, North China of Science and Technology, Tangshan 063210, China. E-mail: qxw@mail.neuq.edu.cn
First published on 7th December 2021
Abstract
High-entropy oxides (HEOs), a class of compounds that include five or more elemental species, have gained increasing attraction for their capability of optimizing the target properties. To date, even though some high-entropy oxides have been successfully prepared, their applications still need to be explored. In the present study, a lithium-manipulation strategy for constructing transition metal oxides (LTM) via a modified solid-state method was investigated. The as-synthesized LTM contained six highly dispersed metal species (Li, Fe, Co, Ni, Cu, Zn) and demonstrated a rocksalt-type structure. Besides, with the introduction of Li, more oxygen vacancies were produced which was also accompanied by shrinking of the lattice constant. When the molar ratio of Li was equal to the other TM cations (LTM16.7), the electrical conductivity was greatly enhanced by a factor of 10 times. Moreover, LTM16.7 achieved the best HER (η = 207 mV at 10 mA cm−2) and OER performances (η = 347 mV at 10 mA cm−2) with elevated electrical conductivity. To facilitate further design of this new kind of materials, we also conducted DFT calculations and elemental alternation experiments, which showed that Fe acted as electrocatalytic sites in this HEOs system. This Li-incorporation strategy opens a new way to understand and modify defect-related HEOs.
Introduction
High-entropy oxides (HEOs), derived from the concept of high-entropy alloys (HEAs), belong to a new class of solid solutions that have aroused enormous attention due to their unique physical properties.1–7 Over the past few years, the concept of high entropy (HE) has been extended to energy storage,8–12 superionic conductivity13,14 and catalysis.15–17 With five or more equimolar components in the one single-phase, HEOs can create a large configurational entropy, which results in a minimal Gibbs free energy (G = H TS) and improvement of the solubility of a great number of species.2 This indicates that the target properties of HEOs can be tuned by the multiple selections; however, the physical properties cannot be easily predicted from composite elements or the complex entropic structure. According to recent studies on HE compounds, including metal carbides,18,19 nitrides10 and oxides,2,8,9 a uniform distribution of transition metal (TM) or rare earth (RE) atoms have been successfully synthesized, showing the solubility merits in long-range ordering.9,20Given that the inherent surface complexity of HEOs, constructing multielement-type oxides is of great interest for applying HEOs as electrocatalysts. Owing to the exposure of unique active binding sites, HEOs can not only achieve a nearly continuous adsorption energy level but can also promote the regulation of various properties simultaneously.15,21 Recent progress in entropy-stabilized system has involved the choice of 3d transition metal elements (such as Fe, Co, Ni, Cu, Zn) as the main source owing to their superior electrochemical property.13,14,22,23 One common consideration for this is that the 3d electronic structure can be modulated by manufacturing multimetal oxides, followed by an enhanced electrocatalytic activity.24 Furthermore, electrical conductivity is considered to be another necessary factor for electrocatalysts and determines the charge transfer during the catalysis process.25,26 High conductivity guarantees a rapid electron transport between the current collector and electrolyte, and as a result, the electrocatalyst will have a lower overpotential and higher energy efficiency, as has been demonstrated in previous studies.25–28 Based on the understanding of these effects, designing an advanced electrocatalyst derived from an HE strategy should lead to improved electrical conductivity and electrochemical activity.
Here, we develop several (FeCoNiCuZn)O-based TM-HEOs by incorporating Li into the matrix lattice to form a pure rocksalt structure (LTM). New results on the electrochemical properties are presented herein, in which the HE concept enabled us to build a new type of multifunctional material with hydrogen evolution reaction (HER), oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) performance. It was also found that increasing the amount of latticed Li brought about more oxygen vacancies, while the disappearance of ferromagnetism also revealed the interaction between the oxygen vacancies and 3d electronic structure. Consequently, the concentration of oxygen vacancies together with the electrical conductivity could be optimized by Li, in which the defect-engineered electronic structure and electrical properties led to an elevated reaction rate in electrocatalytic processes. When the lithium was in an equiatomic concentration, the LTM demonstrated the best electrocatalytic performance. Moreover, the reduced entropy through removing one single element could hardly maintain the single structure, causing a degradation of the catalysis activity, allowing the deduction that the catalysis performance was determined by an entropy-stabilization effect. Finally, density functional theory (DFT) calculations were performed and showed that Fe acts as the catalytic centre, which proves the results from the elemental alternation experiments. As further proof of the active centre, we also successfully synthesized Mg-replacement HEOs with the same rocksalt structure, in which only the substitution of Fe weakened the electrocatalytic activity. Finally, for further proof that Li was the only choice among the alkali metals, Na- and K-manipulated HEOs (NaFeCoNiCuZnO and KFeCoNiCuZnO) were prepared with the same method.
Results and discussion
All three HEOs with different Li amounts were successfully synthesized via wet-soaking treatment as shown in Fig. 1a, in which the overview scanning electron microscopy (SEM) image in Fig. S1† revealed that the particle size of the HEOs were distributed from nanometres to micrometres. The different lithium amounts did not change the morphology of the products. According to previous studies, the appearance of oxygen vacancies is usually accompanied with lattice variations.29,30 In this work, the effect of the lithium incorporation was verified by powder X-ray diffraction (XRD), as seen in Fig. 1b. Also, the corresponding Rietveld refinement yielded values for the lattice constant a of 4.2415, 4.2354 and 4.2032 Å for LTM0, LTM12 and LTM16.7, respectively. Besides, the Rietveld refinement lattice parameters were also verified by the lattice fringes in the HRTEM images in Fig. S2 and S3,† which were close to the XRD results. Interestingly, as the lithium content increased, the intensity of the oxygen vacancy signal increased in the electron paramagnetic resonance (EPR) spectroscopy analysis, as seen in Fig. 1c. In addition, a great amount of experimental and theoretical results demonstrated that oxygen vacancies play a significant role in the origin of ferromagnetism.31,32 As the Li content increased, the ferromagnetism weakened gradually until it disappeared for LTM16.7, as shown in Fig. S4.† The disappearance of ferromagnetism resulted from the reduction of the magnetic interaction hopping integration between the TM atoms, owing to the presence of more oxygen vacancies.33 By tuning the external electrons at the 3d orbitals of the TM species, these defects could steer the conductivity of the material, and thus the electrochemical activity, which is discussed below.33,34 As an ordered or disordered rocksalt-type oxide, the configurational space usually depends largely on the stoichiometric lithium.9,35,36 From previous studies, lithium insertion usually leads to a shrinking of the TM oxides lattice parameters, which is the key factor leading to oxygen vacancies.37–41 From the X-ray photoelectron spectroscopy (XPS) analysis in Fig. 1d, there was a negligible difference in the O 1s peak at 531.3 eV, which, combined with the EPR spectroscopy results, indicated that the variation of oxygen vacancies for the LTM series sample mainly came from the bulk lattice.42–46 Moreover, the binding energy of Fe species shifted to lower energies with the increase in lithium, as can be seen from Fig. S5,† indicating the transportation of electrons bonded to oxygen vacancies to the Fe atoms.47,48 Also, two fitted peaks in the Co 2p3/2 spectra showed the Co3+/Co2+ ratio at the surface area decreased from 3.25 for LTM0 to 2.35 for LTM12 and 2.26 for LTM16.7, respectively, indicating the movement of electrons to Co after Li incorporation. However, the variations of the Co valence state between LTM12 and LTM16.7 remained almost unchanged. The alternation of the electron density of Co and Fe demonstrated another proof of the defect changes in the LTM structure. By contrast, the XPS spectra of other metal species (Ni, Cu, and Zn) remained unchanged, in which all of them existed in the +2 valence state. As the rocksalt structure was M2+O2−, there must be lattice defects for the coexistence of Fe3+ and Co3+ according to the valence neutralization rule. As a result, it is reasonable to obtain controllable lattice oxygen defects for this HEOs system and to manipulate the amount of Li+. Besides, the XPS results showed that the excessive electrons produced by oxygen vacancies only accumulated towards Fe when the Li content reached 16.7%.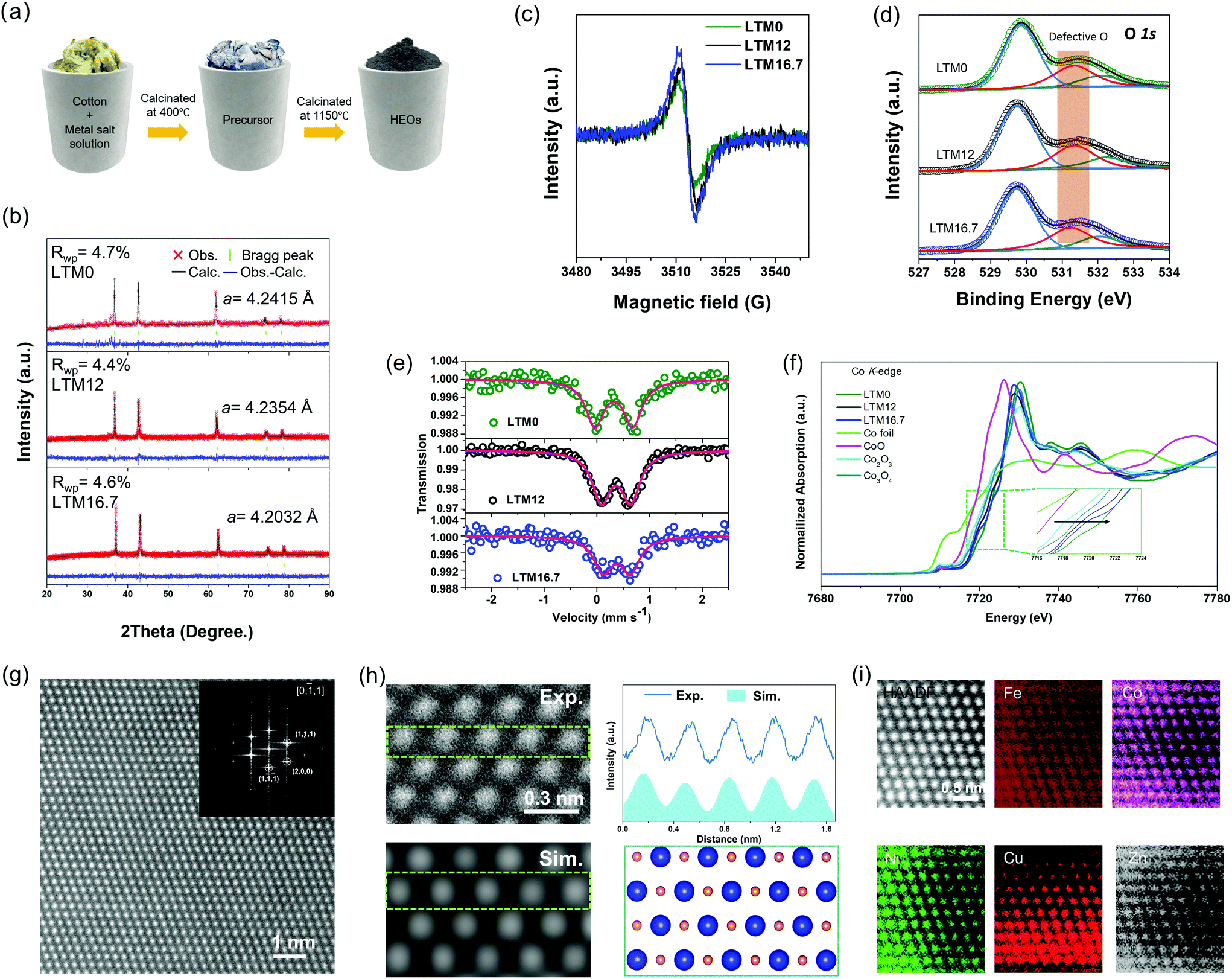 | ||
| Fig. 1 (a) Schematic illustration of the cotton-modified solid-state calcination process. (b) Powder XRD patterns and corresponding Rietveld refinement results. (c) EPR spectra, which present the existence of oxygen vacancies with g = 2.003. (d) XPS profiles of the O 1s region for each LTM sample. The orange part indicates the oxygen vacancies signal near the surface. (e) 57Fe Mössbauer spectrum for each LTM sample. (f) XANES spectra of LTM series samples and reference samples at the Co K-edge. (g) Atomic-resolution HAADF-STEM image and corresponding FFT results (inset). Scale bar is 1 nm. (h) Enlarged view of the region in (g), corresponding to the atomic model of the LTM16.7 surface (the blue spheres represent metal atoms and orange spheres represent oxygen atoms) with the simulation HAADF-STEM image and intensity profile analysis of atoms marked in the green frame. Scale bar is 0.3 nm. (i) HAADF-STEM image and corresponding STEM-EDS analysis. Scale bar is 0.5 nm. | ||
To further verify the XPS data, the 57Fe Mössbauer spectroscopy was performed for LTM0, LTM12 and LTM16.7 at room temperature, as can be seen in Fig. 1e. Each Mössbauer spectrum was fitted by one set of doublets using Lorentzian line shapes, showing that Fe existed at one single structure site. The isomer shift (IS) of 57Fe reflected information on the oxidation state of iron, in which the reduction of the iron oxidation state leads to a higher isomer shift.49 The gradually increased IS values of 0.321, 0.362 and 0.375 mm s−1 in Table S1† for LTM0, LTM12 and LTM16.7 were consistent with the XPS results, suggesting the presence of high-spin trivalent iron oxides in the polyhedral structure.49–52 Our following dataset with quadrupole splitting (QS) enabled us to identify the distortion of Fe3+ sites. After mixing lithium into the system, the apparently decreased QS value showed that lithium could release the distorted environment adjacent to Fe3+ ions.51,53 Except for the above investigations of Fe, Co is another possible electron acceptor according to the XPS results. Necessarily, analysis of the oxidation states of Co cations was performed by XANES analysis. Fig. 1f presents the Co K-edge spectra of the LTM series samples together with Co foil, CoO, Co2O3 and Co3O4. According to the edge positions, it is obvious that the valence of Co cations in the LTM system was close to Co3+. Even though Co was shifted to a lower oxidation state as with the incorporation of Li, LTM16.7 was still higher than that of LTM12, which matched well with the XPS results. We can attribute the unusual changes of oxidation states on Co3+ to the gradual reduction of Fe3+, which resulted from variations in the electron density. To better understand the fine structural environment of Co in the high-entropy system, extended-X-ray absorption fine-structure (EXAFS) analysis was performed, as shown in Fig. S6.† The Fourier-transformed data showed notable peaks at 1.46 and 2.42 Å, which were contributed from Co–O and Co–Co bonds with the same location, in contrast to the Co3O4 reference.
The crystal structure of the Li (16.7%)-incorporated TM-HEOs (LTM16.7) was found to be monocrystalline, as was demonstrated by the transmission electron microscopy (TEM) and selected area electron diffraction (SAED) analyses in Fig. S3,† where the diffraction spots of the [2,0,0] and [1,1,1] facets could be indexed, indicating the main exposed [0,−1,1] facet. The corresponding zone in the high-resolution TEM (HRTEM) image of the as-prepared LTM16.7 showed a normal structure, which further confirmed the single-crystalline structure. Besides, the fast Fourier transform (FFT) analysis of the HRTEM image of the as-prepared LTM16.7 showed the [0,1,1] zone, which agreed well with the SAED results.54 To further clarify the precise atomic structure of the LTM16.7 lattice, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were obtained. Based on the results of HRTEM, LTM16.7 in Fig. 1g still demonstrated a smooth surface with high crystallinity along the [0,1,1] zone axis. The corresponding FFT pattern proved the consistency at the atomic level and the lattice constant a as well. Scanning transmission electron microscopy (STEM)/energy-dispersive spectroscopy (EDS) were applied to examine the distribution of each element in the oxides. Besides, HAADF-STEM simulations were conducted to compare our experiment images. The upper left image in Fig. 1h is an enlarged view of the image from Fig. 1g, where it can be seen the atoms were uniformly dispersed. Accordingly, we constructed an atomic model with the [0,1,1] view in the lower-right panel of Fig. 1h, upon which the simulation result is exhibited in the lower left. It is obvious that the contrast distribution matched well with the experimental HAADF-STEM image, indicating the accuracy of the calculation models. Fig. 1i reveals the STEM/EDS mapping of one typical area of the as-prepared LTM16.7, exhibiting that the corresponding TM species were uniformly distributed in the atomic range. Not only in the small area, but the relatively large particles with the size of nanometres and micrometres showed the same result as in the EDS mapping characterization (Fig. S1 and S3†).
To further investigate the effects of the defect sites, the water splitting performance of the as-synthesized HEOs with different Li contents was evaluated using a standard three-electrode system in 1.0 M KOH electrolyte. The catalyst was cast onto carbon fibre paper (CFP) with a total mass loading of ∼2 mg cm−2. Fig. 2a compares the linear sweep voltammetry (LSV) curves of LTM0, LTM12 and LTM16.7 at a slow scan rate of 2 mV s−1 to eliminate the current caused by the capacitance behaviour after 20 cyclic voltammetric scans for OER. IR correction was applied to the original data to show the intrinsic trend of the as-prepared HEOs. Here, LTM16.7 displayed the lowest overpotential (η = 207 mV) to reach 10 mA cm−2 in comparison with LTM0, LTM12 and the CFP substrate. In particular, LTM16.7 acquired a current density of ∼88 mA cm−2, which was 7.3 and 4.2 times that of LTM0 and LTM12, respectively (Fig. S7†). To gain further insights into this series of materials, Fig. 2b shows that the Tafel slope of LTM16.7 (79.4 mV dec−1) was lower than that of LTM0 (79.6 mV dec−1) and LTM12 (85.3 mV dec−1). Except for the HER, the OER polarization curves were examined to explore the overall water splitting ability in 1.0 M KOH as well. As shown in Fig. 2c, after manipulating the lithium amount in the HEOs matrix gradually, the overpotential decreased to the lowest value at 10 mA cm−2 (η = 347 mV) compared with the other samples. Meanwhile, the current density at 1.6 V vs. RHE for LTM16.7 could reach 54 mA cm−2, which was much higher than that for the other LTM series samples (Fig. S7†). Further, LTM16.7 possessed the lowest slope of 53.8 mV dec−1 from the corresponding Tafel plots (Fig. 2d), illustrating the important role of oxygen vacancies in boosting the electrocatalytic kinetics of OER and HER.55 In addition, the electrochemical surface areas (ECSAs) of the working electrode were determined by measuring the electrochemical double-layer capacitances (EDLCs, Cdl), which represented the effective active sites of the LTM catalysts. As shown in Fig. S8,† the ECSAs of LTM rose from 26.6 mF cm−2 (LTM0), to 29.2 mF cm−2 (LTM12) to 38.6 mF cm−2 (LTM16.7). However, the morphology was almost the same among the LTM series of catalysts as discussed above, indicating that it was the active surface area and more exposed active sites that led to the higher ECSAs results.25,27 As a consequence, the inner property of the catalysts must play a significant role in the electrochemical reactions. Besides the catalytic performance, the durability of LTM16.7 was probed under a static overpotential, in which a stable current was observed for 20 h operation without apparent attenuation less than 5% (Fig. S9†). The stability of LTM16.7 to served as a bifunctional electrocatalyst for overall water splitting was tested using a two-electrode system with 1.0 M KOH electrolyte. With a cell voltage of 1.89 V, the current density reached 30 mA cm−2 with only a little degradation (8.4%) after an even longer period of 40 h test. The SEM image in Fig. S10† for LTM16.7 serving as both the anode and cathode after the stability test showed a negligible change of the morphology. Also, a detailed summary of the HER and OER performance with similar electrocatalysts is presented in Tables S2 and S3,† respectively.
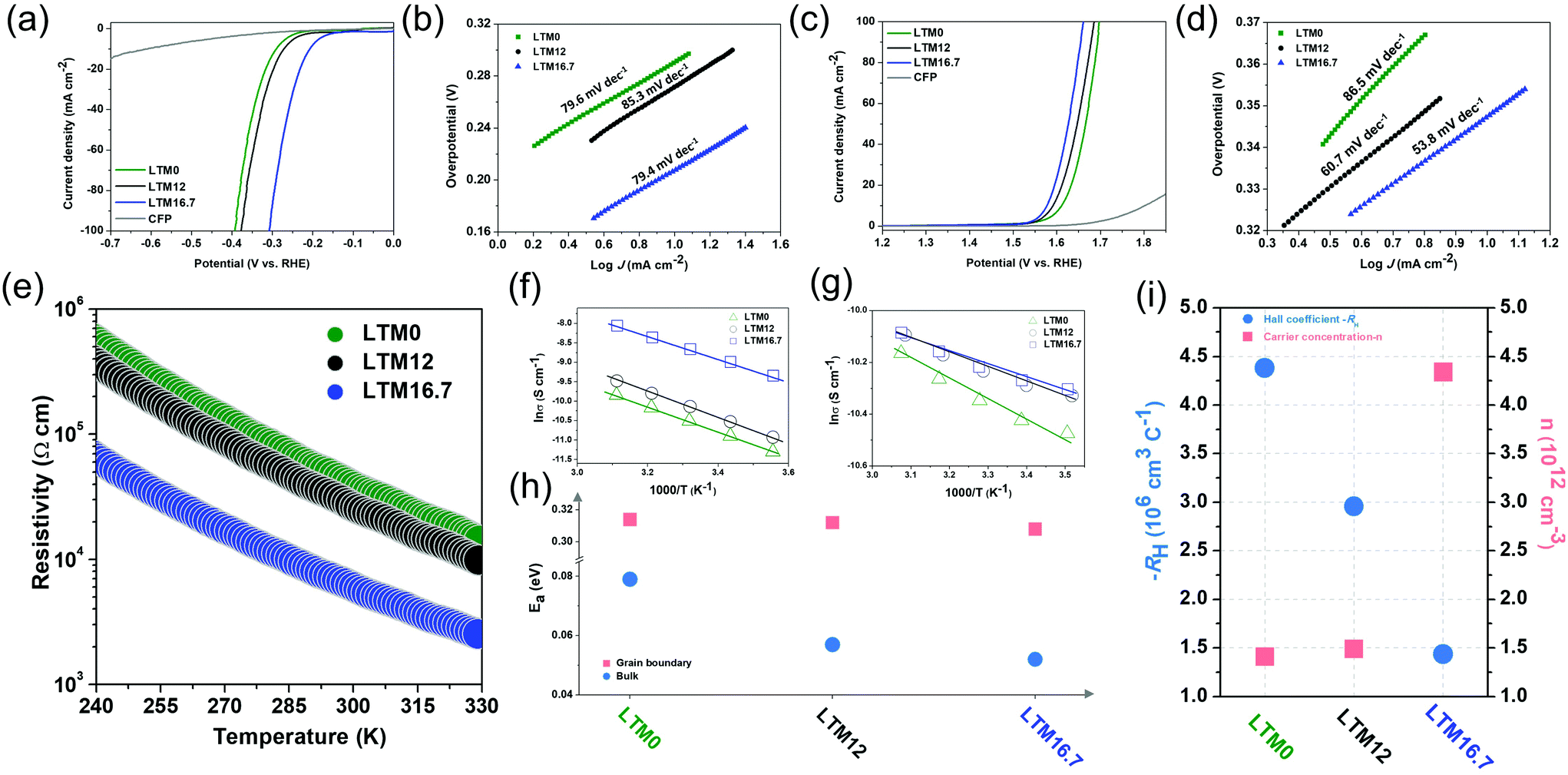 | ||
| Fig. 2 (a), (b) HER polarization curves and Tafel plots. (c), (d) OER polarization curves and Tafel plots. (e) Temperature-dependent (ρ–T) electrical resistivity test. (f), (g) The log(1/ρ) vs. 1000/T plots under low frequency and high frequency, respectively. (h) Corresponding activation energy (Ea) values obtained from (f) and (g). (i) Hall coefficient (RH) and carrier concentration (n) plots. | ||
The oxygen reduction reaction (ORR) is another important electrocatalytic reaction, which was assessed by conducting rotating disc electrode (RDE) experiments in O2-saturated 0.1 M KOH solution. The LSV curves of the LTM series of catalysts on the glassy carbon (GC) electrode under 1600 rpm rotation speed at a scan rate of 5 mV s−1 are exhibited in Fig. S11.† Here, LTM16.7 had a higher limiting current density even though their onset overpotential stayed the same. To further detect the ORR kinetics and catalytic mechanisms of the catalysts, RDE tests were performed at different rotation speeds (100–3600 rpm). As is well known, the ORR process can be divided into 2-electron (H2O + O2 + 4e− = 4OH−) or 4-electron (H2O + O2 + 2e− = OH− + HO2−, H2O + HO2− + 2e− = 3OH−) pathways, which can be calculated by the Koutecky–Levich (K–L) plots. The electron-transfer number (n) for LTM0, LTM12 and LTM16.7 at 0.3 V was 2.24, 2.85 and 3.21, respectively, which meant that the 4-electron way dominated the ORR procedure when there were more lithium atoms in the lattice. This is further evidence that the electrical conductivity from the defected structure could promote charge transfer for electrochemical reactions.56,57 In general, the electrochemical performance is enhanced following the increase in the conductivity, which can be easily tuned by manipulating the concentration of oxygen vacancies in the bulk.
For investigating the influence of lattice defects on the conductivity, measurements of the temperature-dependent resistivity (ρ–T) of LTM0, LTM12 and LTM16.7 were introduced. According to Fig. 2e, the electrical resistivity of the oxides all decreased with the elevation in temperature, exhibiting dρ/dT < 0. This tendency indicated a typical semiconductor behaviour. The resistivity was reduced sharply as Li increased, showing a promoted charge-transfer efficiency. Especially for LTM16.7, the resistivity decreased dramatically by a factor of 10 times compared with LTM0 and LTM12 in the room temperature range. Furthermore, the charge-transfer mechanism in different frequency regions could be explained by the grain boundary barrier layer capacitance (IBLC) model, which originated from impedance spectroscopy, as illustrated in Fig. 2f, g and Fig. S12.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 58 The Z plots in Fig. S12† presented the bulk and grain boundary arcs located in the high frequency (1 MHz) and low frequency (100 Hz) areas, respectively. Both the corresponding conductivity data (σ, σ = ρ−1) fitted well with the Arrhenius law. The derived activation energy (Ea) stayed almost unchanged with the values of 0.314, 0.312 and 0.308 eV for LTM0, LTM12 and LTM16.7 under the low frequency condition, indicating that there were no variations with the conductivity at the grain boundary. However, the activation energy values at the high frequency dropped from 0.079 eV to 0.057 eV and 0.052 eV for LTM0, LTM12 and LTM16.7, respectively, which manifested the greater conductive character of the bulk part (Fig. 2h). Hall coefficient (RH) measurements were conducted to reveal the charge-transfer properties of the HEOs in Fig. 2i. In addition, the activation energy results corresponded nicely with the above EPR and XPS results, showing further evidence that the oxygen vacancies in the bulk in the LTM series materials have a significant influence on the electrical conductivity. The negative RH of all the samples showed that the electrons were the charge carriers (n-type), further explaining the potentials for the electrocatalysts. The carrier concentration (n) obtained from the Hall coefficient for LTM16.7 showing an increasement of 3 times verified the improved electrical conductivity. Besides, the charge-transfer resistance (Rct) values for LTM0, LTM12 and LTM16.7 from the semicircles in the Nyquist plots also corresponded well with the electrical conductivity results (Fig. S13†).
58 The Z plots in Fig. S12† presented the bulk and grain boundary arcs located in the high frequency (1 MHz) and low frequency (100 Hz) areas, respectively. Both the corresponding conductivity data (σ, σ = ρ−1) fitted well with the Arrhenius law. The derived activation energy (Ea) stayed almost unchanged with the values of 0.314, 0.312 and 0.308 eV for LTM0, LTM12 and LTM16.7 under the low frequency condition, indicating that there were no variations with the conductivity at the grain boundary. However, the activation energy values at the high frequency dropped from 0.079 eV to 0.057 eV and 0.052 eV for LTM0, LTM12 and LTM16.7, respectively, which manifested the greater conductive character of the bulk part (Fig. 2h). Hall coefficient (RH) measurements were conducted to reveal the charge-transfer properties of the HEOs in Fig. 2i. In addition, the activation energy results corresponded nicely with the above EPR and XPS results, showing further evidence that the oxygen vacancies in the bulk in the LTM series materials have a significant influence on the electrical conductivity. The negative RH of all the samples showed that the electrons were the charge carriers (n-type), further explaining the potentials for the electrocatalysts. The carrier concentration (n) obtained from the Hall coefficient for LTM16.7 showing an increasement of 3 times verified the improved electrical conductivity. Besides, the charge-transfer resistance (Rct) values for LTM0, LTM12 and LTM16.7 from the semicircles in the Nyquist plots also corresponded well with the electrical conductivity results (Fig. S13†).
Given the great number of cation species in the LTM, a composition experiment was carried out to exclude specific elements and then to figure out the resulting differences with the electrocatalytic behaviour, thus identifying the electrochemical roles for the corresponding species. The removal of one of the species in the 5-TM cations system led to a reduction in the configurational entropy from ∼1.79R to ∼1.61R, as treated under the same annealing process compared with the 6 component HEOs. However, it is difficult for the one-cation excluded oxides to form one single phase even though their main structure still belonged to FCC (Fig. 3a). Fig. 3b–e contrast the electrochemical performance of LTM16.7 compared with the other 5-cation systems, as it was anticipated that the overpotential of each element-removed catalyst would exhibit a different degree of degradation. The overpotential of the 5-cation system at 10 mA cm−2 for the HER and OER polarization curves are shown in Fig. S14,† Fe should play the most important role in the HER reactions. For the OER process (Fig. 3d), Fe seemed to be the most important element as well, followed by Co, Zn and Ni. In addition, the Tafel slope of the oxides with low entropy in Fig. 3c and e also showed their low electrochemical activity. The elemental exclusion result for the ORR process was not affected so much as the above reaction; however, like for the OER process, the performance of oxides without Fe, Co and Ni receded more than the others (Fig. S15†). The different results for each kind of catalytic reaction in the element-removal experiments confirmed that the TM species constructed an entropy-stabilized system, and Fe was the most important electrocatalytic active site among them. As is well known, reversibility is a necessary factor for entropy-driven transitions.2,59 In order to test the reversibility of LTM16.7, the precursor powder was put into a muffle furnace calcinated from 800 °C to 1150 °C with 50 °C increments. Fig. 3f shows the XRD patterns with the phase-transformation process. It could be found that only when the temperature reached 1150 °C did the single rocksalt structure occur. When the as-prepared LTM16.7 was sintered back to 800 °C again, another spinel phase appeared. But when we treated the sample under 1150 °C again, it was found that the as-prepared material became single phase again. The corresponding electrochemical performance also degraded when LTM16.7 was sintered under 800 °C. However, when the calcination temperature went back to 1150 °C, the catalytic activity was enhanced again, as is shown in Fig. 3g, h and S16. The results of the reversibility experiments indicated that the entropy-stabilized system should be of great importance for electrochemical reactions.
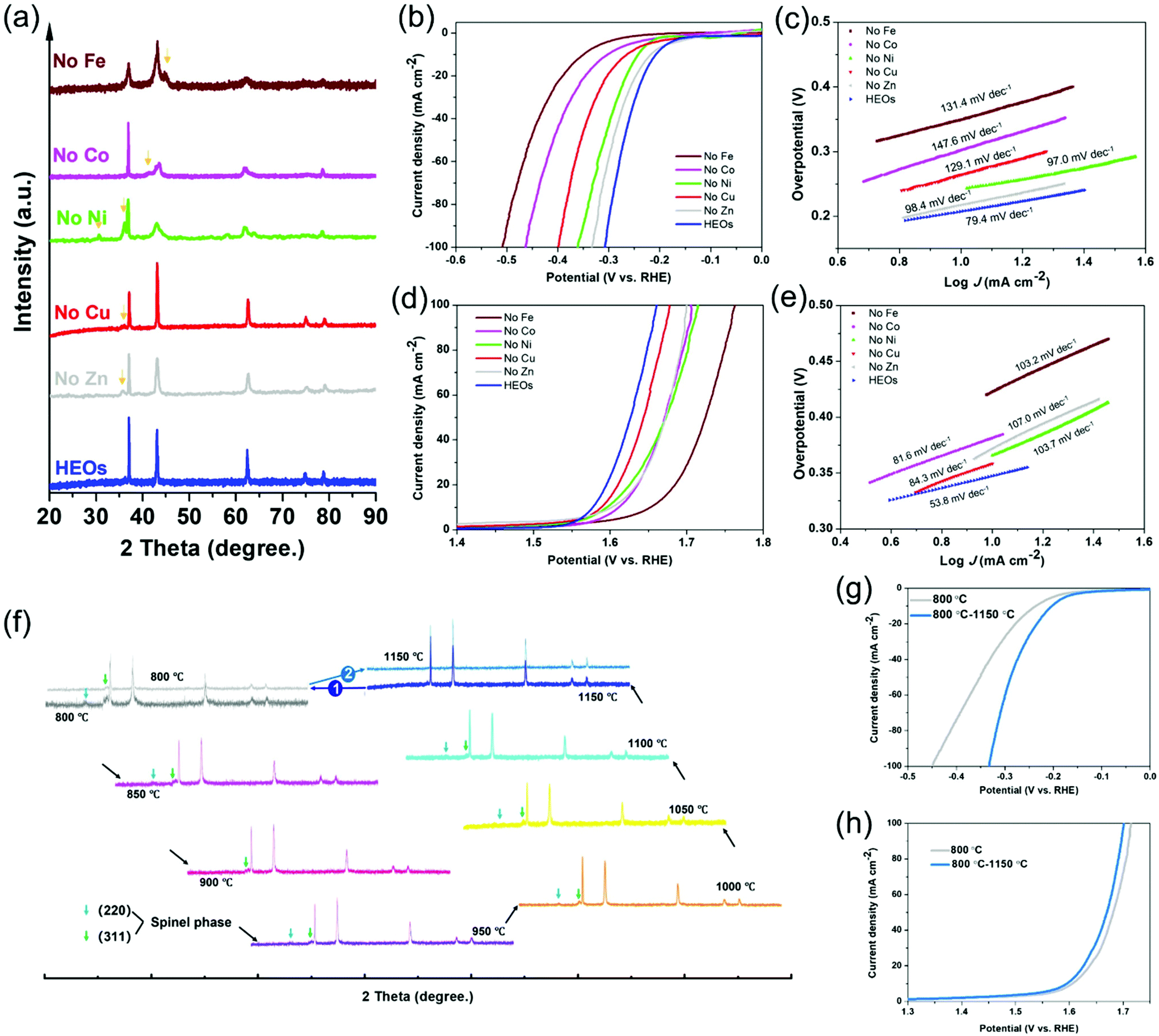 | ||
| Fig. 3 (a). XRD patterns for the composition alternation experiments. An individual component is removed from the main composites, where the yellow arrow marks the peaks from the other crystal phases. The corresponding samples for (b) HER polarization curves and (c) Tafel plots. (d) OER polarization curves and (e) Tafel plots. (f) XRD patterns for the reversibility of high-entropy oxides. (g) HER and (h) OER polarization curves of LTM16.7 sintered under 800 °C and then back to 1150 °C catalysts. | ||
To fully rationalize the inherent merits of the electrocatalytic activity and to determine each active site in the HEOs, a first-principles density functional theory (DFT) study was carried out to evaluate the whole HER and OER processes with the kinetics or thermodynamic details at the atomic level. Since the face-centred cubic structure of LTM16.7 was oriented by the [0,1,1] facet (as exhibited by the TEM results in Fig. 1 and S3†), we compared the reaction process of each metal element on the [0,1,1] crystal plane, as demonstrated in Fig. 4a. The identification of the active sites of the HER process and the corresponding calculated free-energy diagram are summarized in Fig. 4b. It was obvious that Ni, Cu, Zn, and Li found it harder to absorb H* in the first step, especially considering the adsorption free energy for Li (2.38 eV) and Cu (1.22 eV), which were much higher than that of Zn (0.48 eV) and Co (0.6 eV), indicating that all these elements hardly acted as absorption sites for HER. However, for Fe and Ni, the binding energy varied significantly to −0.39 eV and −0.24 eV, respectively, suggesting their dominating role. It should be noted that the binding energy for the OH* group can facilitate the water disassociation except for the H* binding ability during the Volmer step.60,61 Fe and Ni occupied the lowest free energy, which corresponded well with the H* adsorption energy. The experiments about the element-removal results as well as theoretical calculations revealed that Fe was the most important active sites for the HER process. For simplicity, in considering the corresponding catalytic process, we only depict the central active site (dark red atom in the inset graph), in which the general process on the [0,1,1] surface comprised hydrogen adsorption and electrochemical desorption. In addition, the optimized structure of HEOs was also drawn for the purpose of computation (Fig. S17†).
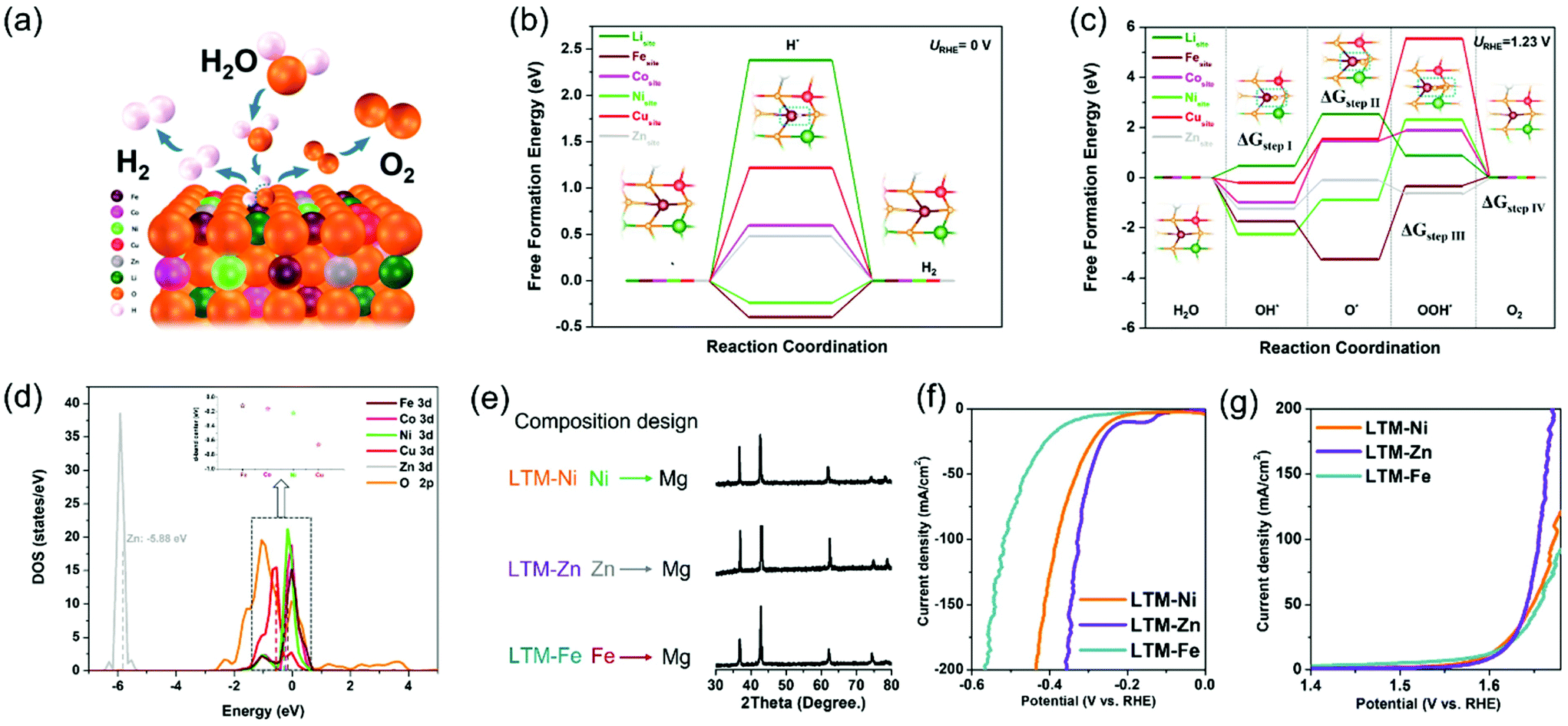 | ||
| Fig. 4 (a) Illustration of the mechanism for the electrocatalytic process under alkaline conditions. Free-energy diagrams on the surface of LTM16.7 for (b) HER and (c) OER processes, respectively. (d) Calculated DOS of LTM16.7. (e) Demonstration of Mg-substitution composition design for the entropy-stabilized system and corresponding XRD pattern. The as-prepared new entropy-stabilized materials with (f) HER and (g) OER polarization curves for the purpose of clarifying the active sites. | ||
Similarly, the free-energy calculations of each metal species in the HEOs for OER were also performed. As demonstrated in Fig. 4c, the complete pathways contained intermediates with O*, OH* and OOH*. For Li, Co and Zn, the rate-determining step was the formation of an O* group from an OH* group (ΔGstepII), which was determined by the Gibbs free energy of these two intermediates. However, the other metal elements (Fe, Ni, and Cu) were determined by the process occurring from the O* group to OOH* group (ΔGstepIII). Moreover, compared with Li (2.06 eV) and Co (2.44 eV), Zn showed a lower free energy (1.13 eV) for the rate-determining step (ΔGstepII), while Fe demonstrated 2.92 eV for the ΔGstepIII, lower than that of Ni (3.18 eV) and Cu (4.00 eV). The data for the rate-determining step revealed that HEOs without Cu did not degrade so much as the other species, because they needed to overcome a really large energy barrier.62 Combined with the data from the H* adsorption energy, Fe and Ni should be essential in the OER process for their kinetically favourable merits. Unlike the calculation results in Fig. 3, Fe was the most important element according to the elemental elimination experiments. In summary, the participation of the active sites in the electrocatalytic reaction should be another key factor in the electrocatalysis process.
To further illustrate the free-energy results, the density of states (DOS) near the Fermi level (Ef) was performed to compare the d-band centre of each TM species in Fig. 4d, in which Fe showed the nearest location (−0.12 eV) from Ef, indicating the adsorption of H2O during the electrocatalytic reaction process decided the participation of Fe with high priority.63,64 In addition, the DOS of the O 2p curve dominated the conduction band minimum compared with the total DOS of LTM16.7 (Fig. S18†), which showed that the electrons in the O 2p orbit should play a key factor in the charge-transfer process. As a result, this is further evidence showing how the oxygen vacancy affects the electrical conductivity as well as the electrocatalysis. Based on the calculation results and composition alternation experiments, it can be inferred that keeping Fe in the parent phase could still achieve a similar performance with that of LTM16.7. Since an entropy-stabilization system should be necessary for the enrolment of a charge-transfer process, and combined with the above reversibility results, we substituted Ni, Zn and Fe with the electrochemically inactive Mg element to test our hypothesis. All the Mg-replaced samples showed one single rocksalt formation due to the entropy-stabilized driving force (Fig. 4e). More importantly, it was obvious that the displacement of Fe will hinder the HER and OER performance, as shown in Fig. 4f and g. However, the substitution of Ni and Zn did not show any apparent degradation, which could further explain the above deduction.
Conclusions
In summary, we successfully prepared Li-manipulated transition metal high-entropy oxides via a wet soaking-assisted modified solid-state strategy. Six metal species, including Li, Fe, Co, Ni, Cu, Zn, were highly distributed in the as-prepared LTM and demonstrated a single rocksalt structure. When the Li content was varied, the amount of oxygen vacancies changed following the variations of the lattice constant. Moreover, higher electrical conductivity was usually followed up with more oxygen vacancies, which could facilitate the electrocatalytic activities of the LTM series catalysts as well. According to the DFT calculations and experimental studies, Fe should be the most significant catalytic sites for the catalytic performance in the entropy-stabilized system. This work not only opens a new door for the large-scale production of high-entropy oxides, but also provides a theoretical basis for enhancing the electrocatalytic performance of TM-based HEOs.Author contributions
Y. G. and X. Q. proposed the project, designed the experiments, and prepared the draft manuscript; Y. G. and A. B. performed the experiments; X. W., Y. C. and L. D. assisted in analyzing the experimental data; X. L. performed Mössbauer test and analysis; H. P. performed the DFT calculation; X. Q. supervised the whole project; Y. L. provided partial financial support and EPR test. Y. G. and A. B. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Natural Science Foundation of China (No. 51972048 and No. 11775226), Fundamental Research Funds for the Central Universities (N2023029), and the Natural Science Foundation of Inner Mongolia (No. 2020MS05038).Notes and references
- B. Cantor, I. T. H. Chang, P. Knight and A. J. B. Vincent, Mater. Sci. Eng., A, 2004, 375–377, 213–218 CrossRef
.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J. P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS PubMed
- R.-Z. Zhang and M. J. Reece, J. Mater. Chem. A, 2019, 7, 22148–22162 RSC
- S. Jiang, T. Hu, J. Gild, N. Zhou, J. Nie, M. Qin, T. Harrington, K. Vecchio and J. Luo, Scr. Mater., 2018, 142, 116–120 CrossRef CAS
- J.-W. Yeh, S.-K. Chen, S.-J. Lin, J.-Y. Gan, T.-S. Chin, T.-T. Shun, C.-H. Tsau and S.-Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS
- C. Oses, C. Toher and S. Curtarolo, Nat. Rev. Mater., 2020, 5, 295–309 CrossRef CAS
- H. Xiang, Y. Xing, F.-z. Dai, H. Wang, L. Su, L. Miao, G. Zhang, Y. Wang, X. Qi, L. Yao, H. Wang, B. Zhao, J. Li and Y. Zhou, J. Adv. Ceram., 2021, 10, 385–441 CrossRef CAS
- J. Yan, D. Wang, X. Zhang, J. Li and X. Qi, J. Mater. Sci., 2020, 55, 6942–6951 CrossRef CAS
- Z. Lun, B. Ouyang, D. H. Kwon, Y. Ha, E. E. Foley, T. Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clement, H. Ji and G. Ceder, Nat. Mater., 2020, 20, 214–221 CrossRef PubMed
- T. Jin, X. Sang, R. R. Unocic, R. T. Kinch, X. Liu, J. Hu, H. Liu and S. Dai, Adv. Mater., 2018, 30, 1707512 CrossRef PubMed
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, 1806236 CrossRef PubMed
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kubel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef PubMed
- D. Bérardan, S. Franger, A. K. Meena and N. Dragoe, J. Mater. Chem. A, 2016, 4, 9536–9541 RSC
- M. Gazda, T. Miruszewski, D. Jaworski, A. Mielewczyk-Gryń, W. Skubida, S. Wachowski, P. Winiarz, K. Dzierzgowski, M. Łapiński, I. Szpunar and E. Dzik, ACS Mater. Lett., 2020, 2, 1315–1321 CrossRef CAS
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3, 834–845 CrossRef CAS
- P. Xie, Y. Yao, Z. Huang, Z. Liu, J. Zhang, T. Li, G. Wang, R. Shahbazian-Yassar, L. Hu and C. Wang, Nat. Commun., 2019, 10, 4011 CrossRef PubMed
- D. Feng, Y. Dong, L. Zhang, X. Ge, W. Zhang, S. Dai and Z. A. Qiao, Angew. Chem., Int. Ed., 2020, 59, 19503–19509 CrossRef CAS PubMed
- T. J. Harrington, J. Gild, P. Sarker, C. Toher, C. M. Rost, O. F. Dippo, C. McElfresh, K. Kaufmann, E. Marin, L. Borowski, P. E. Hopkins, J. Luo, S. Curtarolo, D. W. Brenner and K. S. Vecchio, Acta Mater., 2019, 166, 271–280 CrossRef CAS
- T. Wang, F. Okejiri, Z. A. Qiao and S. Dai, Adv. Mater., 2020, 32, 2002475 CrossRef PubMed
- Y. Yao, Z. Huang, L. A. Hughes, J. Gao, T. Li, D. Morris, S. E. Zeltmann, B. H. Savitzky, C. Ophus, Y. Z. Finfrock, Q. Dong, M. Jiao, Y. Mao, M. Chi, P. Zhang, J. Li, A. M. Minor, R. Shahbazian-Yassar and L. Hu, Matter, 2021, 4, 1–14 CrossRef
- G. Zhang, K. Ming, J. Kang, Q. Huang, Z. Zhang, X. Zheng and X. Bi, Electrochim. Acta, 2018, 279, 19–23 CrossRef CAS
- A.-L. Wang, H.-C. Wan, H. Xu, Y.-X. Tong and G.-R. Li, Electrochim. Acta, 2014, 127, 448–453 CrossRef CAS
- X. Yan, Y. Jia and X. Yao, Small Struct., 2021, 2, 2000067 CrossRef
- B. Zhang, X. Zheng, O. Voznyy, R. Comin and M. Bajdich, Science, 2016, 352, 333–337 CrossRef CAS PubMed
- X. Zhao, X. Li, Y. Yan, Y. Xing, S. Lu, L. Zhao, S. Zhou, Z. Peng and J. Zeng, Appl. Catal., B, 2018, 236, 569–575 CrossRef CAS
- K. Xu, H. Ding, H. Lv, P. Chen, X. Lu, H. Cheng, T. Zhou, S. Liu, X. Wu, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 3326–3332 CrossRef CAS PubMed
- X. Zhao, H. Zhang, Y. Yan, J. Cao, X. Li, S. Zhou, Z. Peng and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 328–332 CrossRef CAS PubMed
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8846–8851 CrossRef
- Y. Shin, K.-Y. Doh, S. H. Kim, J. H. Lee, H. Bae, S.-J. Song and D. Lee, J. Mater. Chem. A, 2020, 8, 4784–4789 RSC
- X. Wu, J. Walter, T. Feng, J. Zhu, H. Zheng, J. F. Mitchell, N. Biškup, M. Varela, X. Ruan, C. Leighton and X. Wang, Adv. Funct. Mater., 2017, 27, 1704233 CrossRef
- S. Jaiswar and K. D. Mandal, J. Phys. Chem. C, 2017, 121, 19586–19601 CrossRef CAS
- N. Biškup, J. Salafranca, V. Mehta, M. P. Oxley, Y. Suzuki, S. J. Pennycook, S. T. Pantelides and M. Varela, Phys. Rev. Lett., 2014, 112, 087202 CrossRef
- Q. Zhang, Z. F. Xu, L. F. Wang, S. H. Gao and S. J. Yuan, J. Alloys Compd., 2015, 649, 1151–1155 CrossRef CAS
- Z. Zhuang, Y. Li, Y. Li, J. Huang, B. Wei, R. Sun, Y. Ren, J. Ding, J. Zhu, Z. Lang, L. V. Moskaleva, C. He, Y. Wang, Z. Wang, D. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 1016–1028 RSC
- A. Urban, J. Lee and G. Ceder, Adv. Energy Mater., 2014, 4, 1400478 CrossRef
- Y. Xie, M. Saubanère and M. L. Doublet, Energy Environ. Sci., 2017, 10, 266–274 RSC
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y. S. Liu, K. Edstrom, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed
- W. S. Yoon, M. Balasubramanian, K. Y. Chung, X. Q. Yang, J. McBreen, C. P. Grey and D. A. Fischer, J. Am. Chem. Soc., 2005, 127, 17479–17487 CrossRef CAS PubMed
- F. Li, X. Zhang, J. Lin, J. Ma, S. Zhang and G. Yang, J. Phys. Chem. C, 2019, 123, 23403–23409 CrossRef CAS
- C. R. Fell, D. Qian, K. J. Carroll, M. Chi, J. L. Jones and Y. S. Meng, Chem. Mater., 2013, 25, 1621–1629 CrossRef CAS
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. N. Waterhouse, W. Huang and T. Zhang, Adv. Energy Mater., 2017, 7, 1700005 CrossRef
- Z. Cai, Y. Bi, E. Hu, W. Liu, N. Dwarica, Y. Tian, X. Li, Y. Kuang, Y. Li, X.-Q. Yang, H. Wang and X. Sun, Adv. Energy Mater., 2018, 8, 1701694 CrossRef
- G. Wu, G. Zhao, J. Sun, X. Cao, Y. He, J. Feng and D. Li, J. Catal., 2019, 377, 271–282 CrossRef CAS
- Y. Zheng, R. Zhang, L. Zhang, Q. Gu and Z. A. Qiao, Angew. Chem., Int. Ed., 2021, 60, 4774–4781 CrossRef CAS PubMed
- R. Zhang, Z. Liu, T. N. Gao, L. Zhang, Y. Zheng, J. Zhang, L. Zhang and Z. A. Qiao, Angew. Chem., Int. Ed., 2021, 60, 24299–24305 CrossRef CAS PubMed
- H. Xiong, H. Zhou, G. Sun, Z. Liu, L. Zhang, L. Zhang, F. Du, Z. A. Qiao and S. Dai, Angew. Chem., Int. Ed., 2020, 59, 11053–11060 CrossRef CAS PubMed
- H. Li, S. Zhang and Q. Zhong, J. Colloid Interface Sci., 2013, 402, 190–195 CrossRef CAS PubMed
- Y. Li, C. Wang, H. Zheng, F. Wan, F. Yu, X. Zhang and Y. Liu, Appl. Surf. Sci., 2017, 391, 654–661 CrossRef CAS
- F. Menil, J. Phys. Chem. Solids, 1985, 46, 763–789 CrossRef CAS
- L. Ma, R. K. Brow and A. Choudhury, J. Non-Cryst. Solids, 2014, 402, 64–73 CrossRef CAS
- M. Tabuchi, A. Nakashima, H. Shigemura, K. Ado, H. Kobayashi, H. Sakaebe, K. Tatsumi, H. Kageyama, T. Nakamura and R. Kanno, J. Mater. Chem., 2003, 13, 1747–1757 RSC
- C. Delmas, G. Prado, A. Rougier, E. Suard and L. Fournès, Solid State Ionics, 2000, 135, 71–79 CrossRef CAS
- E. McCalla, M. T. Sougrati, G. Rousse, E. J. Berg, A. Abakumov, N. Recham, K. Ramesha, M. Sathiya, R. Dominko, G. Van Tendeloo, P. Novak and J. M. Tarascon, J. Am. Chem. Soc., 2015, 137, 4804–4814 CrossRef CAS PubMed
- H. Xiong, G. Sun, Z. Liu, L. Zhang, L. Li, W. Zhang, F. Du and Z. A. Qiao, Angew. Chem., Int. Ed., 2021, 60, 10334–10341 CrossRef CAS PubMed
- L. Ji, X. Peng and Z. Wang, Trans. Tianjin Univ., 2020, 26, 373–381 CrossRef CAS
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. Waterhouse, L. Z. Wu, C. H. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517–6524 CrossRef CAS PubMed
- D. C. Sinclair, T. B. Adams, F. D. Morrison and A. R. West, Appl. Phys. Lett., 2002, 80, 2153–2155 CrossRef CAS
- S. A. Jones, J. Fenerty and J. Pearce, Thermochim. Acta, 1987, 114, 61–66 CrossRef CAS
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed
- W. Luc, Z. Jiang, J. G. Chen and F. Jiao, ACS Catal., 2018, 8, 9327–9333 CrossRef CAS
- X. Yan, C. Chen, Y. Wu, S. Liu, Y. Chen, R. Feng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 6638–6645 RSC
- M. Zhou, H. Li, A. Long, B. Zhou, F. Lu, F. Zhang, F. Zhan, Z. Zhang, W. Xie, X. Zeng, D. Yi and X. Wang, Adv. Energy Mater., 2021, 11, 2101789 CrossRef CAS
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbruster, A. Knop-Gericke and R. Schlogl, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr07000b |
| This journal is © The Royal Society of Chemistry 2022 |