
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceValorisation of phenols to coumarins through one-pot palladium-catalysed double C–H functionalizations†
Giulia
Brufani‡
ac,
Federica
Valentini‡
ab,
Flavio
Sabatelli
a,
Benedetta
Di Erasmo
a,
Anastasiia M.
Afanasenko
 c,
Chao-Jun
Li
*c and
Luigi
Vaccaro
*a
c,
Chao-Jun
Li
*c and
Luigi
Vaccaro
*a
aLaboratory of Green S.O.C. – Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123, Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: https://greensoc.chm.unipg.it/
bConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali (INSTM), Via Giusti, 9, 50121 Firenze, Italy
cDepartment of Chemistry, and FQRNT Centre for Green Chemistry and Catalysis – McGill University, 801 Sherbrooke Street West, Montreal, QC H3A0B8, Canada
First published on 28th October 2022
Abstract
Phenols’ valorisation via step- and atom-efficient protocols is of great importance for the transition to sustainable chemical production. Among the transformation of phenols, C–H functionalization is limited to a few examples with homogeneous metal-based catalysts. Herein, a robust one-pot approach for constructing C8 alkyl substituted coumarins starting from simple phenols through the palladium-catalysed double C–H functionalizations is presented. In such a combined protocol, a Pd/C was used as a heterogeneous catalyst for the first time. The developed methodology enables the synthesis of structurally diverse coumarins, including natural products, in good isolated yields using potentially biomass-derived phenols. Several TEM and XPS measurements combined with elemental analysis were conducted in order to gain insight into the nature of the active catalyst and the factors affecting its reactivity.
Introduction
The coumarin skeleton, belonging to the class of benzopyrones, is a common structural scaffold (Fig. 1A) in natural products,1 active pharmaceutical ingredients (API),2 pesticides,3 food additives,4 and cosmetics.5 Prenylated coumarin compounds, such as osthole and its derivatives, are a target of high interest for inflammatory, oncological diseases, and diabetes.6 Owing to their importance, numerous synthetic pathways have been developed for the construction of coumarins, in particular, via Knoevenagel or Pechmann condensations.7 However, these conventional methodologies require stoichiometric amounts of strong Lewis or Brønsted acid additives and harsh reaction conditions, which subsequently leads to considerable waste formation, as well as poor atom economy. The multistep protocols for the synthesis of osthole-like derivatives (Fig. 1B),8 coumarins functionalized at the C8 position with an aliphatic chain, frequently involve the use of toxic reagents and non-recyclable catalysts that impose restrictions on its practical utilization.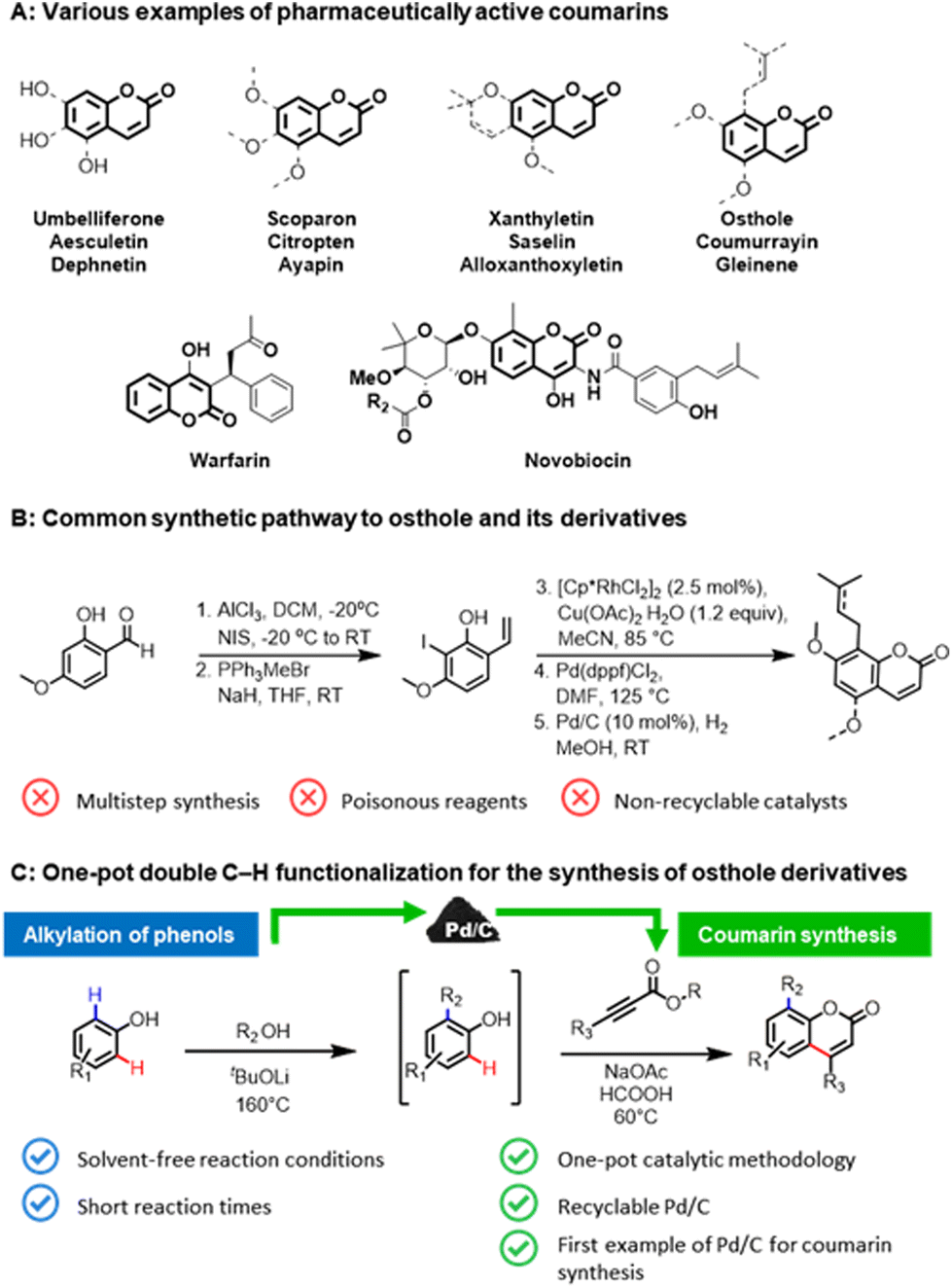 | ||
| Fig. 1 An overview of coumarin production. (A) Representative examples of pharmaceutically and biologically active compounds of interest to this work. (B) Multistep common synthetic pathway to osthole and its derivates. (C) One-pot double C–H functionalizations of phenols to osthole derivatives over a Pd/C catalyst. | ||
An alternative pathway for constructing prenylated coumarins would be a double C–H functionalization9 of phenols (Fig. 1C). In this way, the aliphatic chain will be introduced at the C8 position via an ortho-alkylation of phenols with alcohols,10 and then the resulting intermediate will be directly utilized in the formation of the core coumarin moiety via the second C–H functionalization of the formed phenolic derivatives with alkynoates. Such a one-pot strategy will enable the rapid generation of molecular complexity in a step- and atom-efficient way employing potentially bio-derived phenols,11 and thus pave a highway toward the sustainable production of pharmaceutically-relevant coumarins directly from renewable feedstocks. Surprisingly, this approach has not been realized to date.
Despite the remarkable achievements over the past decades in the synthesis of coumarins through the C–H functionalization of simple phenolic compounds using homogeneous catalyst systems,12 methodology development in this field has generally overlooked the use of heterogeneous catalysts in such transformation. Reported protocols are limited to the use of non-recyclable noble metal catalysts, making them irrelevant in the above-proposed one-pot approach. In this context, heterogeneous catalysts13 can be a promising alternative to overcome the principal drawback of the existing protocols and to carry out the overall coumarin synthesis process in line with the main principles of green chemistry.14
Thus, with an increasing demand for sustainable synthesis of biologically active coumarins and in continuation of our interest in formic acid-assisted valorisation15 of phenols and heterogeneous C–H functionalization processes,16 we report herein the first heterogeneous palladium-based catalyst system for the synthesis of coumarins and its prenylated derivatives. At the core of this one-pot methodology is the use of commercially available Pd/C in both steps, namely the ortho-alkylation of phenols with alcohols under solvent-free reaction conditions and the coumarin skeleton formation using formic acid as a green reaction medium. Our attention was particularly focused on the recycling studies of the heterogeneous catalyst and the investigation of the reaction mechanism.
Results and discussion
We started our investigation by optimizing the synthesis of coumarins from phenols using a heterogeneous catalyst system in formic acid as the reaction medium. Initially, the reactions between 3,5-dimethoxyphenol (1a) and ethyl propiolate (2a) in the presence of different palladium-based catalysts were conducted (Table S1 – section 3, in ESI†). Considering the importance of Pd0 in the catalytic cycle previously proposed by Trost and Toste,12b we chose to investigate two different commercially available heterogeneous catalysts, Pd/C and Pd/Al2O3 in presence of 20 mol% of NaOAc as a base co-catalyst using formic acid as both the reaction medium and reducing agent at 60 °C. Gratifyingly, by replacing the homogeneous Pd(OAc)2 catalyst with Pd/C under the selected reaction conditions, 88% of 5,7-dimethoxy coumarin (3a) was obtained within 16 hours (entry 3, Table S1 – section 3.1, in ESI†). When the reaction time was increased to 24 hours, both Pd/C and Pd/Al2O3 afforded quantitative conversion of 1a (entries 4 and 6, Table S1 – section 3.1, in ESI†). Having chosen Pd/C for further studies, the selection of a suitable reaction medium, varying the ratio between formic acid and water as well as employing a mixture of formic acid and organic solvents, was carried out (Table 1). With a gradual increase in the percentage of water in the formic acid medium, a decrease in conversion was observed (entries 1–6, Table 1). This is probably due to the faster release of molecular hydrogen from the aqueous formic acid medium catalysed by Pd/C,17 compared to the formation of cinnamate ester intermediate. When toluene was used as a co-solvent in the C–H functionalization of 1a, its complete conversion to 3a was observed (entry 8, Table 1). However, the phase separation of this solvent mixture led us to select 98% formic acid as the best reaction medium. The reaction did not proceed using cyclopentylmethylether (CPME) as a co-solvent (entry 7, Table 1). When acetic acid was used as reaction medium, only traces amount of product 3a was detected (Table 1, entry 9), further confirming the key role of formic acid also as reducing agent.12b|
|
||||
|---|---|---|---|---|
| Entry | Reaction medium | Conc. (m) | Time (h) | Conv.b (%) |
| a 1a (0.3 mmol); 2a (2 equiv.), NaOAc (20 mol%), Pd/C (10 mol%), under argon, 60 °C. b Conversion determined by GC analysis; the remaining materials are unreacted 1a. c 1a (1 mmol). | ||||
| 1 | HCOOH 98% | 0.1 | 24 | >99 |
| 2c | HCOOH 98% | 0.15 | 24 | >99 |
| 3 | HCOOH 98% | 0.1 | 2 | 77 |
| 4 | HCOOH 85% | 0.1 | 2 | 54 |
| 5 | HCOOH 50% | 0.1 | 2 | <5 |
| 6 | HCOOH 50% | 0.1 | 24 | 45 |
| 7 | HCOOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CPME (1:1) :CPME (1:1) |
0.1 | 24 | <5 |
| 8 | HCOOH:toluene (1:1) |
0.1 | 24 | >99 |
| 9 | CH3COOH | 0.1 | 24 | Traces |
To better understand the mechanism of such a heterogeneous catalytic process, we decided to investigate the role of the base co-catalyst. To our surprise, we observed a complete conversion of 1a in 2 hours upon removal of the NaOAc base co-catalyst. Next, the effect of counterion size and loading of the base co-catalyst were evaluated (Table 2). A decrease in conversion was observed with an increase in the size of the counterion, which suggests that the acetate is coordinated with Pd. The coordination effect of acetate is also consistent with the complete conversion obtained without NaOAc. Replacing the sodium acetate with another base such as NaOH inhibited the reaction significantly (entry 8, Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Base co-catalyst | Mol% | Time (h) | Conv.b (%) |
| a 1a (0.3 mmol); 2a (2 eq.), HCOOH 98% (0.1 M), Pd/C (10 mol%), under argon, 60 °C, 2 h. b Conversion determined by GC analysis; the remaining materials are unreacted 1a. | ||||
| 1 | –– | –– | 2 | 99 |
| 2 | NaOAc | 5 | 2 | 80 |
| 3 | NaOAc | 10 | 2 | 73 |
| 4 | NaOAc | 20 | 2 | 77 |
| 5 | NaOAc | 100 | 2 | 45 |
| 6 | NH4OAc | 20 | 2 | 68 |
| 7 | TBAAc | 20 | 2 | 50 |
| 8 | NaOH | 20 | 2 | 20 |
Taking into account our findings on the role of the base co-catalyst, kinetic and recycling tests were carried out for the catalyst system in the absence of NaOAc. Regarding the kinetic test, without NaOAc, the reaction reached a plateau faster compared to the process with NaOAc. The latter required 12 hours to reach a plateau and 24 hours to complete conversion (Fig. 2a).
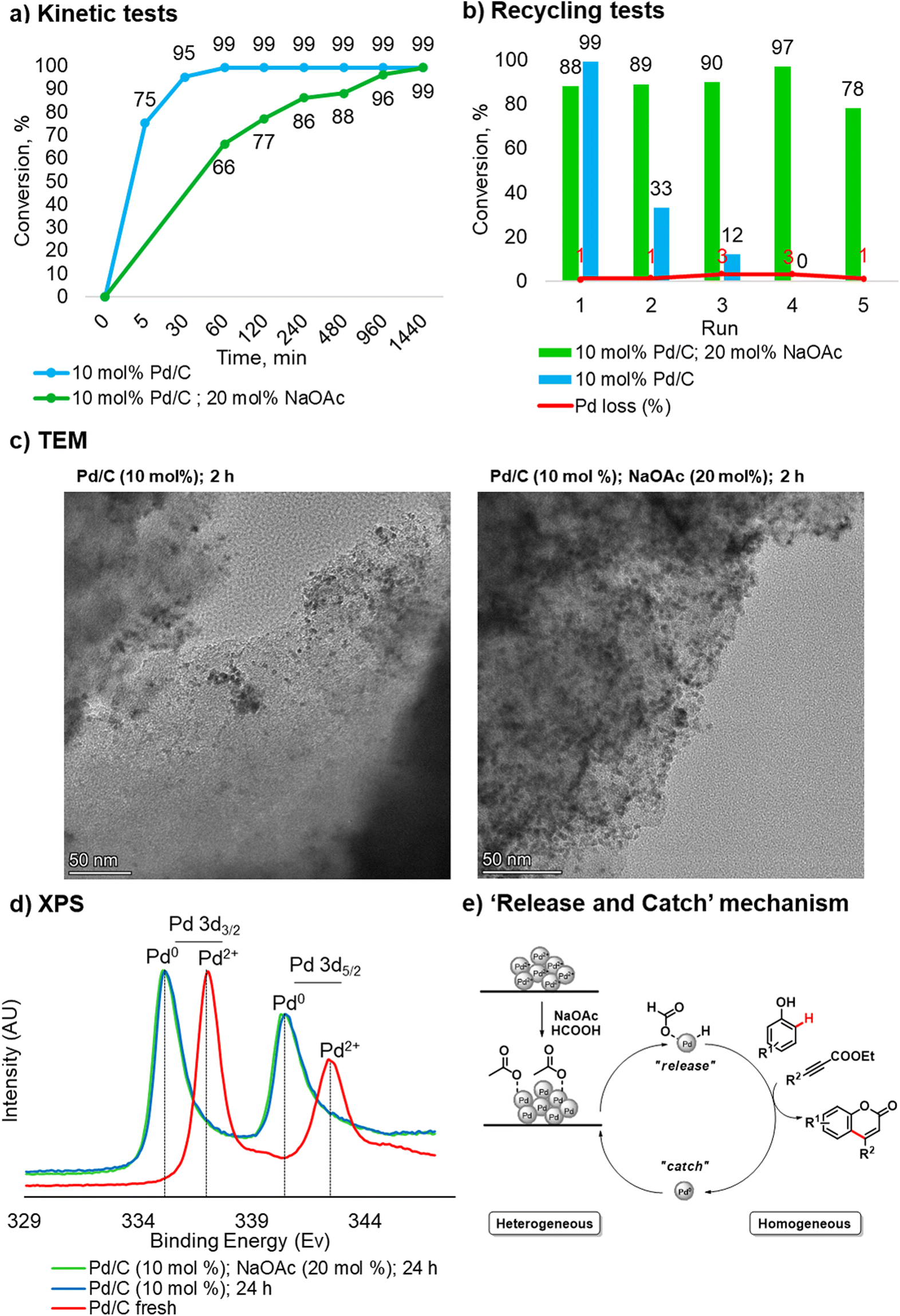 | ||
| Fig. 2 Catalytic studies and mechanistic investigation. (A) Kinetic tests, conducted with 10 mol% of Pd/C in the presence/absence of 20 mol% of NaOAc. (B) Recycling tests of 10 mol% of Pd/C, conducted with 10 mol% in the presence of 20 mol% of NaOAc after 24 hours and the absence of the additive after 2 hours. (C) TEM images: sample without 20 mol% of NaOAc after 2 hours (left). Sample with 20 mol% of NaOAc after 2 hours (right). (D) XPS analyses. (E) General representation of ‘release and catch’ mechanism. | ||
However, upon recovery and reuse of the Pd/C catalyst used in the C–H functionalization in the absence of NaOAc, a significant drop in conversion was observed in the second run (Fig. 2b). These results highlight the crucial role of NaOAc in this transformation and suggest that the process may go through a “release and catch” mechanism.18 In this scenario, the acetate stabilizes the Pd species in the solution and prevents aggregation in the “catch” step (Fig. 2e). Indeed, without NaOAc, the stabilization effect is lost, and although the reaction proceeds faster than in the protocol using a base co-catalyst, Pd in solution leads to the formation of catalytically inactive aggregates.
To confirm the proposed mechanism, TEM analyses and measurements of the Pd content in solution in the case of incomplete conversion were carried out. Regarding the measurement of Pd in solution, since the success of the protocol is strictly related to the pressure generated inside the reactor, it was impossible to perform a hot filtration test. As such, a leaching analysis was conducted after filtering the hot mixture at a phenol conversion of 75%. In accordance with previous kinetic data, the protocol in the absence of NaOAc was filtered after 5 min, and the reaction carried out in presence of NaOAc was filtered after 120 min. In the first case, the loss of palladium in the crude reaction mixture was 18%, and in the second case, 40%. By comparing these results with the leaching values obtained during the recycling tests, the return of Pd to the support at the end of the process can be estimated. Indeed, the total loss of Pd over 5 consecutive runs (9%) and the values measured by filtering the hot mixture confirm a “release and catch” mechanism.18 The differences in Pd leaching found when comparing two different protocols are mainly due to the reaction time required to reach 75% conversion of phenol 1a.
TEM analyses were performed on Pd/C samples obtained after 2 hours and 24 hours in the presence/absence of NaOAc (20 mol%). The qualitative analysis of the samples revealed the formation of aggregates and a non-uniform distribution of the Pd particles without NaOAc already after 2 hours (Fig. 2c), and after 24 hours large aggregation of Pd is clearly visible (see more details in section 3.3, in ESI†). On the other hand, a better distribution of Pd particles can be noted in the sample with NaOAc. The TEM analysis of Pd/C obtained after three consecutive recycling cycles without NaOAc (20 mol%) shows the formation of inactive Pd aggregates. It is noteworthy that when comparing the latter with the catalyst recycled 5 times in the presence of NaOAc (20 mol%), degradation of the support takes place. These experimental findings additionally confirm the key role of NaOAc: the coordination of acetate with Pd prevents the formation of the metal aggregates and increases the lifetime of the heterogeneous catalyst, allowing it to be recycled.
XPS analyses confirmed that the oxidation state of Pd at the end of the reaction is Pd0, regardless of whether an additive was used or not. The latter is consistent with the mechanism proposed by Trost and Toste12b and was expected under such highly reducing reaction conditions (Fig. 2d).
A comparison of the TON and TOF values of heterogeneous Pd/C with homogeneous catalyst systems used in previous literature works,12b,c,e was conducted. The TON value of 41, obtained considering 5 consecutive runs, is greater than the value obtained for the currently known homogeneous systems (6.2–32.3); the TOF values are comparable for all systems (see section 3.4 for more details, in ESI†). In addition, the E-factor value19 of our protocol was also compared with those of the processes reported in the literature (see section 4 in ESI for more details†).12b,c,e With our optimized conditions the waste associated to the coumarins synthesis was reduced up to 56% (as confirmed by the E-factor values).
Next, the scope and limitations of the newly established protocol were explored. A wide range of phenols was efficiently coupled with variously functionalized alkynoates under the optimized reaction conditions (Scheme 1). Notably, the reaction requires a strong activation of the aromatic ring of the phenol, therefore the role of the electron-donating substituent is crucial.12b Monosubstituted methoxy and methyl phenols did not lead to a significant conversion (see section 3.5, in ESI for further details†). We focused the study towards the synthesis of target coumarins of relevance as natural products.
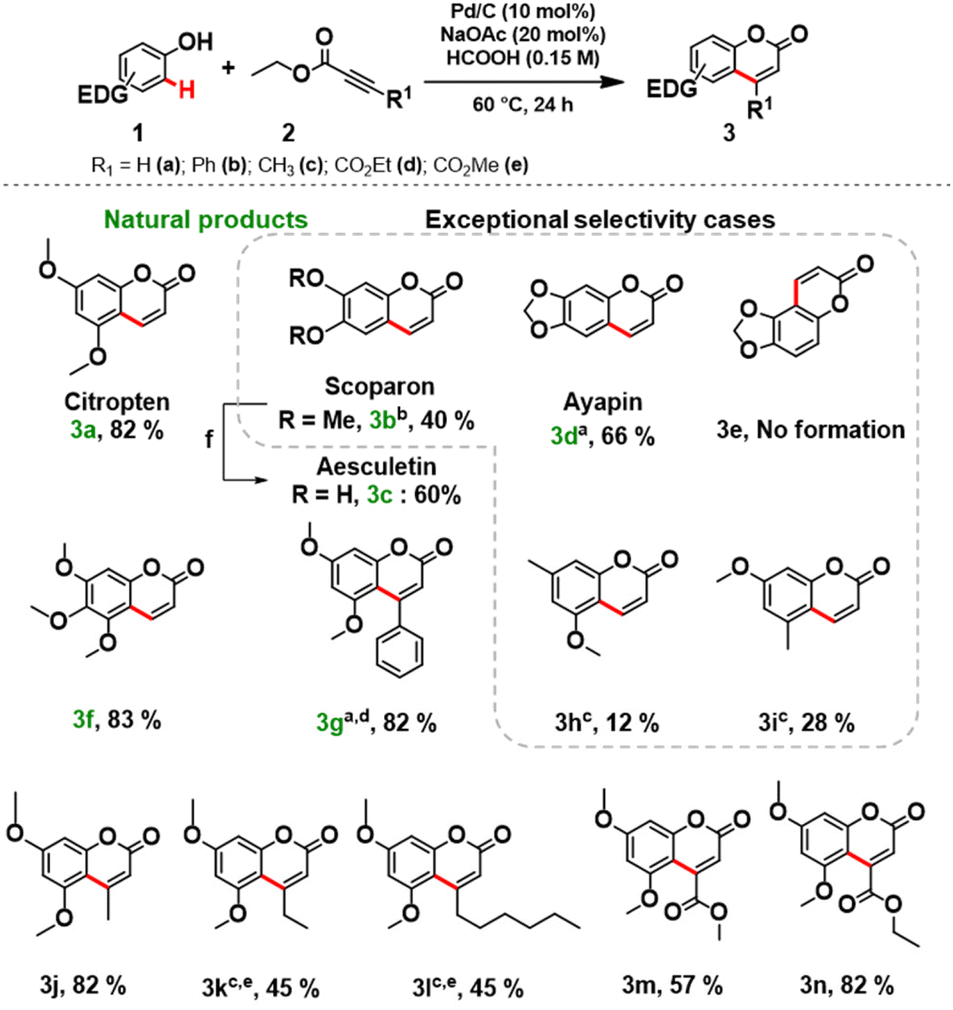 | ||
| Scheme 1 Coumarins synthetized via C–H functionalization of phenols with alkynoates using Pd/C as a catalyst. Reaction conditions: 1 (1 mmol), 2 (2 equiv.), Pd/C (10 mol%), NaOAc (20 mol%), HCOOH (0.15 M), 60 °C and 24 hours. a80 °C and 24 hours. b80 °C and 48 hours. c90 °C 48 hours. d2 (3 equiv.). e1 (0.3 mmol), HCOOH (0.1 M); fCH3CN 0.8 M, Al powder (4.3 equiv.), I2 (1.7 equiv.), after 24 hours at 80 °C under reflux quenched with HCl (2 M, 1 mL). | ||
3,5-Dimethoxyphenol (1a), 3,4-dimethoxyphenol (1b), sesamol (1c), and 3,4,5-trimethoxyphenols (1d) smoothly reacted with ethyl propiolate (2a) affording the desired natural products (3a–g) in good isolated yields (Scheme 1). Remarkably, aesculetin (3c) was readily obtained without any further purification procedures by cleaving the methyl ethers of scoparon (3b) under acidic conditions.
Replacement of 2a with methyl propiolate gave comparable results for isolated coumarins, suggesting that the alkynoates do not interfere with the protocol. To access C3-functionalized coumarins, various alkynoates were successfully applied in the synthesis of products 3j–m (Scheme 1). An excellent isolated yield (82%) of 3g was obtained by increasing the amount (3 equiv.) of methyl phenylpropiolate (2b) at a slightly elevated temperature (80 °C). Unfortunately, the reactions of phenylpropiolic acid with phenols 1a and 1c did not occur, and the phenols were recovered unreacted.
To our great delight, the coupling of 1f with 2a under the optimized reaction conditions afforded a key intermediate 3o for the synthesis of alloxanthoxyletin natural product (Scheme 2). The regioselectivity of this Pd-catalysed heterogeneous process is in line with the results previously obtained by Trost and Toste.12b Indeed, the C–H functionalization process favors the reaction at the C8 position, product 3o was obtained with exceptional selectivity, while the product resulting from the reaction at the C6-position 3p was not detected in the reaction mixture (see section 3.6, in ESI†).
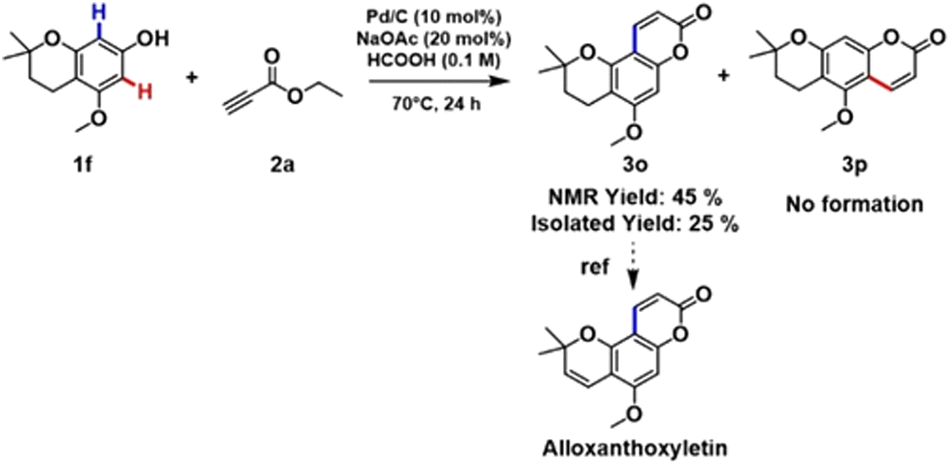 | ||
| Scheme 2 Synthesis of alloxanthoxyletin intermediate employing our developed methodology. | ||
In order to prove the feasibility of our newly developed methodology and to extend the scope of natural coumarin products, we turned our attention to the synthesis of osthole-like derivatives. We disclosed a one-pot double C–H functionalizations of phenols to access coumarins alkylated at the C8 position using Pd/C as a universal catalyst. The process consists of two steps: ortho-alkylation of phenols with alcohols followed by the C–H functionalization of the alkylated phenolic intermediates with alkynoates (Scheme 3).
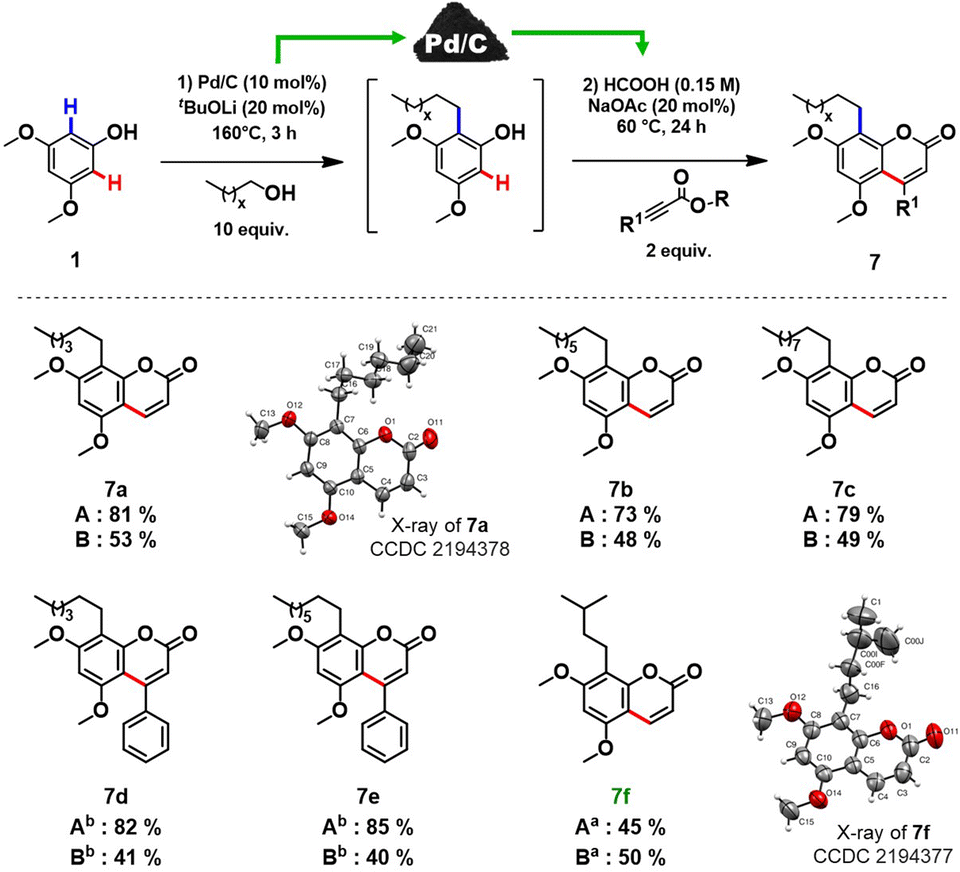 | ||
| Scheme 3 Coumarins synthesized via double C–H functionalizations of phenols using Pd/C as a catalyst. Method A: Coumarins synthesized via a two-step double C–H functionalizations of phenols using Pd/C. Method B: Coumarins synthesized via one-pot double C–H functionalizations of phenols using Pd/C. Reaction conditions 1st step: 1 (1 mmol), alcohol (10 equiv.), Pd/C (10 mol%), tBuOLi (20 mol%), 160 °C, 3 hours. Reaction conditions 2nd step: 2 (2 equiv.), NaOAc (20 mol%), HCOOH (0.15 M), 60 °C, 24 hours. a1 (0.3 mmol), HCOOH (0.1 M). b2 (3 equiv.). | ||
Aiming to design a robust sustainable methodology for the synthesis of coumarins, we reoptimized the reaction conditions of our previously reported protocol for ortho-alkylation of phenols catalysed by Pd/C in toluene.10a As such, the ‘first’ formal step of the reaction can be completed in 3 hours under solvent-free conditions (see section 3.2, in ESI† for further details), delivering the target ortho-alkylated product with exceptional selectivity. At the ‘second’ step, employing the same Pd/C as in the ‘first’ step, the formed intermediate without additional isolation is coupled with the alkynoate using formic acid as a green reaction medium to afford the target C8 alkyl substituted coumarins. Under the established reaction conditions, structurally diverse coumarins were synthesised in a remarkably simple manner (Scheme 3) affording reasonable overall isolated yields (7a–f, 40–53%). The overall yields of the target coumarins obtained by the one-pot approach (Scheme 3 – Method B) were comparable to that obtained by the two-step methodology (Scheme 3 – Method A). Gratifyingly, natural product 7f, the hydrogenated derivative of osthole, can be obtained in 50% overall isolated yield starting from a simple 3,5-dimethoxyphenol 1a. The structures of 7a and 7f were further confirmed by X-ray diffraction studies (see section 5, in ESI†).
Moreover, if compared with other multi-step procedure (Fig. 1B), our newly defined one-pot procedure for the synthesis of osthole derivates allowed a significant improvement in terms of atom economy, from 32% to 81%.
Conclusion
In summary, we have developed a general methodology based on a palladium-catalysed double C–H functionalizations of activated phenols for a one-pot synthesis of structurally diverse coumarins. The described protocol is remarkably easy to implement, demonstrates high selectivity, a broad scope, and ideally follows the twelve principles of green chemistry. Furthermore, this method enables the synthesis of biologically active products such as scoparon, aesculetin, ayapin, citropten as well as osthole-like derivatives employing potentially biomass-derived phenols. In order to gain further insight into the nature of the active catalyst system and the influence of reaction parameters on the catalyst morphology, a series of TEM and XPS measurements combined with elemental analysis and leaching experiments were performed. These experiments revealed the stabilizing role of the NaOAc for the Pd species in solution and suggested a ‘release and catch’ reaction mechanism. Notably, despite the strongly acidic conditions, Pd/C was efficiently recycled for five consecutive runs with negligible loss in conversion and metal leaching in the solution. To the best of our knowledge, this is the first example of using a heterogeneous catalyst system for the valorisation of phenols to C8 alkyl substituted coumarins via double C–H functionalizations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Università degli Studi di Perugia and MUR are acknowledged for financial support for the project AMIS, through the program “Dipartimenti di Eccellenza – 2018–2022”. C.-J. Li thanks NSERC, CFI, FQRNT, and Canada Research Chairs program for financial support. We are thankful to Dr. Alexander Wahba for the help with HRMS measurements, Dr. Hojatollah Vali for the help with TEM analysis, Dr. Hatem Titi for single crystal XRD measurements, Dr. Hui Su for the help with XPS measurements.References
- (a) Coumarins: Biology, Applications and Mode of Action. ed. R. O'Kennedy, R. D. Thornes, Wiley & Sons Ltd, 1997. Search PubMed; (b) R. D. H. Murray, in Prog. Chem. Org. Nat. Prod ed. A.D. Kinghorn, H. Falk, S. Gibbons, Y. Asakawa, J.-K. Liu, V.M. Dirsch, Springer-Verlag, Wien, 1997, pp. 2–105 Search PubMed.
- (a) S. Emami and S. Dadashpour, Eur. J. Med. Chem., 2015, 102, 611–630 CrossRef CAS PubMed; (b) T. Al-Warhi, A. Sabt, E. B. Elkaeed and W. M. Eldehna, Bioorg. Chem., 2020, 103, 104163 CrossRef CAS PubMed; (c) A. Detsi, C. Kontogiorgis and D. Hadjipavlou-Litina, Expert Opin. Ther. Pat. 2017, 27, 1201–1226 Search PubMed.
- J. Chen, Y. Yu, S. Li and W. Ding, Molecules, 2016, 21, 1501 CrossRef PubMed.
- S. Krüger, L. Winheim and G. E. Morlock, Food Chem., 2018, 239, 1182–1191 CrossRef PubMed.
- C. Stiefel, T. Schubert and G. E. Morlock, ACS Omega, 2017, 2, 5242–5250 CrossRef CAS PubMed.
- (a) T. T. Lin, Y. Y. Huang, G. H. Tang, Z. B. Cheng, X. Liu, H. B. Luo and S. Yin, J. Nat. Prod., 2014, 77, 955–962 CrossRef CAS PubMed; (b) Y. P. Liu, G. Yan, Y. T. Xie, T. C. Lin, W. Zhang, J. Li, Y. J. Wu, J. Y. Zhou and Y. H. Fu, Bioorg. Chem., 2020, 97, 103699 CrossRef CAS PubMed.
- (a) S. M. Setena and N. M. Shah, Chem. Rev., 1945, 36, 1–62 CrossRef; (b) M. Molnar, M. Lončarić and M. Kovač, Curr. Org. Chem., 2020, 24, 4–43 CrossRef CAS.
- M. Sun, M. Sun and J. Zhang, Med. Chem. Res., 2021, 30, 1767–1794 CrossRef CAS PubMed.
- For reviews of C–H activation: (a) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (b) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed; (c) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (d) C. Yu, J. Sanjosé-Orduna, F. W. Patureau and M. H. Pérez-Temprano, Chem. Soc. Rev., 2020, 49, 1643–1652 RSC; (e) P. Gandeepan, N. Kaplaneris, S. Santoro, L. Vaccaro and L. Ackermann, ACS Sustainable Chem. Eng., 2019, 7, 8023–8040 CrossRef CAS; (f) T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends Chem., 2019, 1, 63–76 CrossRef CAS; (g) T. Gensch, M. J. James, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 2296–2306 CrossRef CAS PubMed; (h) S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601–1612 RSC; (i) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (j) R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS PubMed; (k) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed; (l) I. Choi, J. Struwe and L. Ackermann, Photochem. Photobiol. Sci., 2021, 20, 1563–1572 CrossRef CAS PubMed; (m) C.-J. Li, Acc. Chem. Res., 2009, 335–344 CrossRef PubMed.
- (a) J. Yu, C.-J. Li and H. Zeng, Angew. Chem., Int. Ed., 2021, 60, 4043–4048 CrossRef CAS PubMed; (b) S. W. Youn and C. G. Cho, Org. Biomol. Chem., 2021, 19, 5028–5047 RSC.
- The phenols used in this protocol that could potentially be obtained from biomass: (a) B. F. De Simon, E. Esteruelas, À. M. Muñoz, E. Cadahia and M. Sanz, J. Agric. Food Chem., 2009, 57, 3217–3227 CrossRef PubMed; (b) P. Picart, C. Müller, J. Mottweiler, L. Wiermans, C. Bolm, P. D. De Marfa and A. Schallmey, ChemSusChem, 2014, 7, 3164–3171 CrossRef CAS PubMed; (c) J. F. Yang, C. H. Yang, M. T. Liang, Z. J. Gao, Y. W. Wu and L. Y. Chuang, Molecules, 2016, 21, 1–10 Search PubMed.
- For examples of C–H functionalization of simple phenolic compounds using homogeneous catalyst systems: (a) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1996, 118, 6305–6306 CrossRef CAS; (b) B. M. Trost, F. D. Toste and K. Greenman, J. Am. Chem. Soc., 2003, 125, 4518–4526 CrossRef CAS PubMed; (c) M. Kotani, K. Yamamoto, J. Oyamada, Y. Fujiwara and T. Kitamura, Synthesis, 2004, 1466–1470 CAS; (d) T. Kitamura, K. Yamamoto, M. Kotani, J. Oyamada, C. Jia and Y. Fujiwara, Bull. Chem. Soc. Jpn., 2003, 76, 1889–1895 CrossRef CAS; (e) U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., 2013, 125, 12901–12905 CrossRef.
- S. Santoro, S. I. Kozhushkov, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 3471–3493 RSC.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed; (b) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC; (c) H. C. Erythropel, J. B. Zimmerman, T. M. De Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- (a) V. R. Jumde, E. Petricci, C. Petrucci, N. Santillo, M. Taddei and L. Vaccaro, Org. Lett., 2015, 17, 3990–3993 CrossRef CAS PubMed; (b) Z. Chen, H. Zeng, H. Gong, H. Wang and C.-J. Li, Chem. Sci., 2015, 6, 4174–4178 RSC; (c) Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C.-J. Li, Angew. Chem., Int. Ed., 2015, 54, 14487–14491 CrossRef CAS PubMed; (d) F. Valentini, N. Santillo, C. Petrucci, D. Lanari, E. Petricci, M. Taddei and L. Vaccaro, ChemCatChem, 2018, 10, 1277–1281 CrossRef CAS; (e) A. Dominguez-Huerta, I. Perepichka and C.-J. Li, ChemSusChem, 2019, 12, 2999–3002 CrossRef CAS PubMed; (f) Z. Qiu, H. Zeng and C.-J. Li, Acc. Chem. Res., 2020, 53, 2395–2413 CrossRef CAS PubMed.
- (a) F. Ferlin, I. Anastasiou, N. Salameh, T. Miyakoshi, O. Baudoin and L. Vaccaro, ChemSusChem, 2022, 15, e202102736 CrossRef CAS PubMed; (b) N. Salameh, I. Anastasiou, F. Ferlin, F. Minio, S. Chen, S. Santoro, P. Liu, Y. Gu and L. Vaccaro, Mol. Catal., 2022, 522, 112211 CrossRef CAS; (c) D. Sciosci, F. Valentini, F. Ferlin, S. Chen, Y. Gu, O. Piermatti and L. Vaccaro, Green Chem., 2020, 22, 6560–6566 RSC; (d) F. Ferlin, I. Anastasiou, L. Carpisassi and L. Vaccaro, Green Chem., 2021, 23, 6576–6582 RSC; (e) N. Salameh, F. Ferlin, F. Valentini, I. Anastasiou and L. Vaccaro, ACS Sustainable Chem. Eng., 2022, 10, 3766–3776 CrossRef CAS.
- (a) Y. Kim and D. H. Kim, Appl. Catal., B, 2019, 244, 684–693 CrossRef CAS; (b) A. Kosider, D. Blaumeiser, S. Schötz, P. Preuster, A. Bösmann, P. Wasserscheid, J. Libuda and T. Bauer, Catal. Sci. Technol., 2021, 11, 4259–4271 RSC.
- M. Gruttadauria, F. Giacalone and R. Noto, Green Chem., 2013, 15, 2608–2618 RSC.
- J. Andraos and A. Hent, J. Chem. Educ., 2015, 92, 1820–1830 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03579k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |