
Difunctionalization of unactivated olefins via selective electrochemical chlorosulfuration or chlorosulfoxidation†
Pan
Zhou‡
,
Kaikai
Niu‡
,
Hongjian
Song
 *,
Yuxiu
Liu
* and
Qingmin
Wang
*
*,
Yuxiu
Liu
* and
Qingmin
Wang
*
State Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, College of Chemistry, Frontiers Science Center for New Organic Matter, Nankai University, Tianjin 300071, China. E-mail: songhongjian@nankai.edu.cn; liuyuxiu@nankai.edu.cn; wangqm@nankai.edu.cn
First published on 13th July 2022
Abstract
Direct difunctionalization of olefins—which are the most commonly used compounds in organic synthesis—is a powerful tool for rapid formation of structurally complex building blocks from readily available starting materials. However, difunctionalization of unactivated olefins remains a formidable challenge. Herein, we describe a green, cost-effective, and eco-friendly electrochemical protocol for selective chlorosulfuration or chlorosulfoxidation of unactivated alkenes. This electrochemical method creates the potential possibility for the preparation of high-value-added products in a more sustainable and greener manner.
Sulfide and sulfoxide motifs are prevalent in drug molecules and natural products (Scheme 1a),1,2 and compounds containing these motifs are commonly used as ligands for metal catalysts.3,4 In addition, olefins are useful building blocks for organic synthesis, and olefin difunctionalization has emerged as an efficient method for construction of the ubiquitous scaffold.5–9 Therefore, methods for the formation of C–S bonds by means of reactions involving olefins are of interest to synthetic chemists. In particular, sulfuration of olefins is an ideal strategy for introducing C–S bonds into molecules.10–14 Despite the importance of these methods, we have tried to eliminate the drawbacks such as the use of expensive and unavailable catalysts15–17 or oxidants,18,19 restricted use to activated olefins, the challenge of over-oxidation, and the poor agreement with green chemistry principles that have been reported in previous works (Scheme 1b). Therefore, a more green, cost-effective, and eco-friendly method for sulfuration of olefins and subsequent sulfide oxidation would be highly desirable.
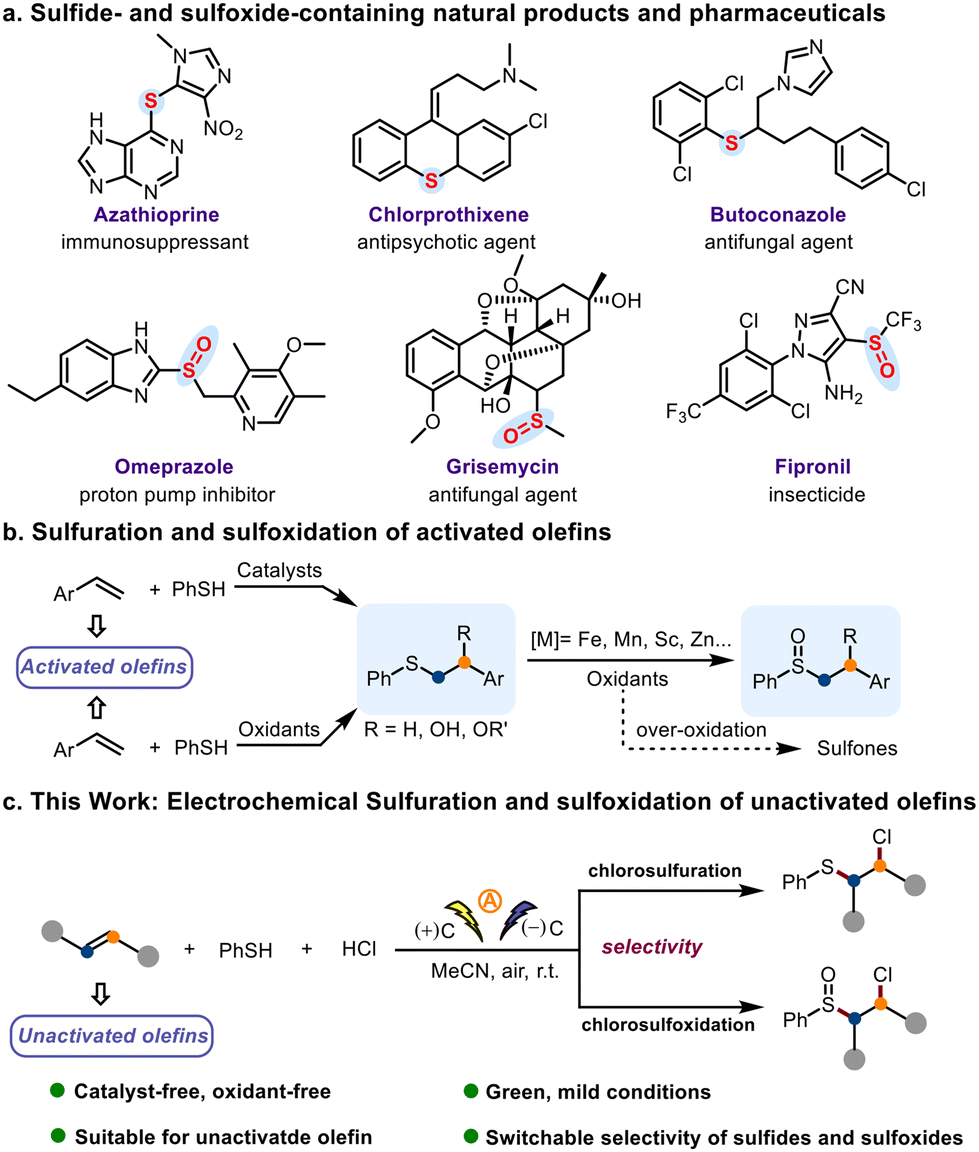 | ||
| Scheme 1 Representative sulfide- and sulfoxide-containing natural products and pharmaceuticals and methods for olefin difunctionalization. | ||
In 1998, Anastas and Warner defined the “12 Principles of Green Chemistry” as “design rules” to make chemical processes more sustainable.20–25 Electrochemistry is considered to be a green and sustainable technology since it could avoid the use of chemical oxidants or reductants and does not generate reagent waste.26–29 Electrochemical difunctionalization of olefins is emerging as a strategy for forging C–S bonds.30,31 We envisioned that electrochemical oxidation of thiophenols could be controlled by varying the operating voltage and reaction time and that the resulting thiyl radicals could be used for selective sulfuration and sulfoxidation of unactivated olefins.
Indeed, we herein report that we have successfully developed an electrochemical protocol for selective chlorosulfuration or chlorosulfoxidation of unactivated olefins by means of reactions with thiophenols and HCl (Scheme 1c). The significant features of this procedure for the preparation of sulfides include the use of simple graphite as electrodes, low cost, mild conditions, and environment friendliness, which all together make this approach in line with the 12 principles of green chemistry.
We began by selecting cyclohexene (1a) and p-toluenethiol (2a) as model substrates. In an undivided cell with graphite electrodes, constant-current electrolysis of 1a and 2a for 10 h at room temperature in MeCN containing nBu4NBF4 as the electrolyte and concentrated HCl as a chlorine source gave the desired sulfide 3a in 74% isolated yield (Table 1, entry 1). Replacement of MeCN with other solvents led to diminished yields (entries 2–6). Evaluation of various chlorine sources revealed that HCl was optimal (entries 7–9), which indicates that water in the HCl may have participated in the reaction. The similarity of the yields from the reaction under air (entry 1) and a reaction under O2 (entry 10) suggests that sulfide 3a was not oxidized by O2. Strikingly, when the reaction time was increased to 22 h, sulfoxide product 4a was obtained in 81% isolated yield (entry 11). Electricity was indispensable for the reaction (entry 12).
|
|
||
|---|---|---|
| Entry | Variation from standard conditionsa | Yields of 3a/4ab (%) |
| a Standard conditions: 1a (0.5 mmol), 2a (1.0 mmol), nBu4NBF4 (1.0 mmol), MeCN (5 mL), an undivided cell with graphite electrodes (each 1.0 × 1.0 cm2), room temperature (r.t.), 10 mA, 10 h. b Yields were determined by 1H NMR spectroscopy with CH2Br2 as an internal standard. c Isolated yield. | ||
| 1 | None | 80 (74c)/12 |
| 2 | DCM as the solvent | 66/0 |
| 3 | DMF as the solvent | 0/57 |
| 4 | DMSO as the solvent | 0/0 |
| 5 | DCE as the solvent | 64/0 |
| 6 | DMA as the solvent | 0/0 |
| 7 | NaCl instead of HCl | 5/0 |
| 8 | KCl instead of HCl | 2/0 |
| 9 | LiCl instead of HCl | 12/0 |
| 10 | O2 | 82/10 |
| 11 | 22 h | 0/87 (81c) |
| 12 | No electric current | 0 |
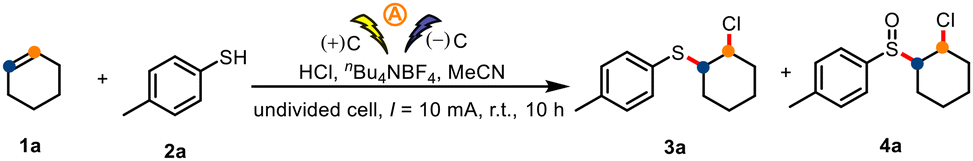
Using the optimized conditions, we turned our attention to exploring the generality of the protocol. First, we evaluated various thiophenols 2 in the electrochemical chlorosulfuration reactions of cyclohexene (1a, Scheme 2). Thiophenols with a para, ortho, or dimethyl group delivered the corresponding products 3a–3c, respectively. In addition, thiophenols bearing a variety of para substituents (tert-butyl, halogens, trifluoromethyl, methoxy) were suitable for the reaction, giving products 3d–3i.
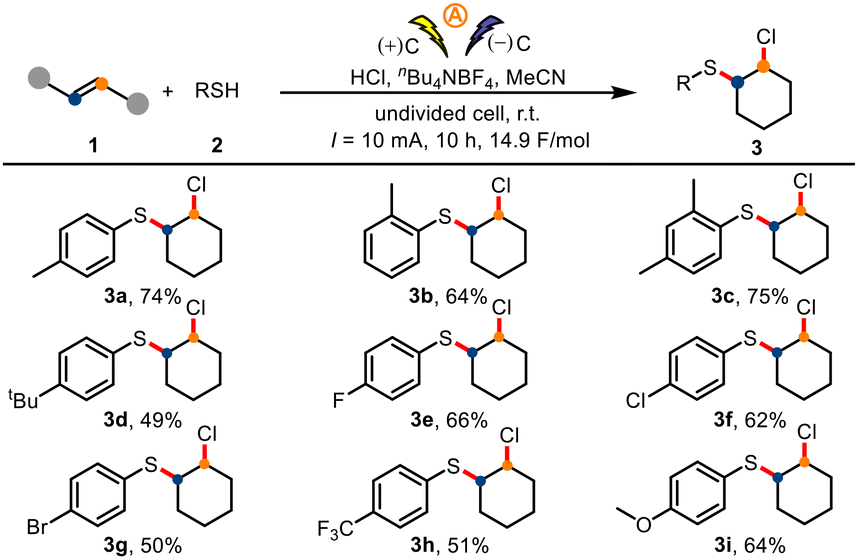 | ||
| Scheme 2 Substrate scope of chlorosulfuration. Standard chlorosulfuration conditions: 1 (0.5 mmol), 2 (1.0 mmol), nBu4NBF4 (1.0 mmol), MeCN (5 mL), an undivided cell with graphite electrodes (each 1.0 × 1.0 cm2), room temperature (r.t.), 10 mA, 10 h. | ||
We also investigated the substrate scope for direct preparation of sulfoxides from unactivated olefins and thiophenols by means of our electrochemical protocol (Scheme 3). Thiophenols with a para, ortho, or meta methyl group, along with dimethyl- and tert-butyl-substituted thiophenols, afforded the corresponding products (4a–4e) in 63–88% yields. Halogen atoms (F, Cl, Br; 4f–4i), electron-withdrawing groups (CN, CF3; 4j and 4k), and an electron-donating group (OMe, 4l) were also tolerated. 2-Naphthiophenol gave target 4m in 39% yield. Gratifyingly, phenylselenol also underwent the reaction under the standard conditions to give 4n, albeit in only 21% yield. Next, we probed the difunctionalization of various unactivated alkenes 1 in reactions with p-toluenethiol (2a). As shown in Scheme 3, symmetrical cycloalkenes—including cyclopentene, cycloheptene, cyclooctene, and norbornene—delivered the corresponding products (4o–4t) in 51–73% yields. A diene also underwent the desired difunctionalization reaction, giving 41% yield of 4u, the product of functionalization of only one double bond. Activated olefins (4v, 4w) were competent reaction partners (see the ESI†).
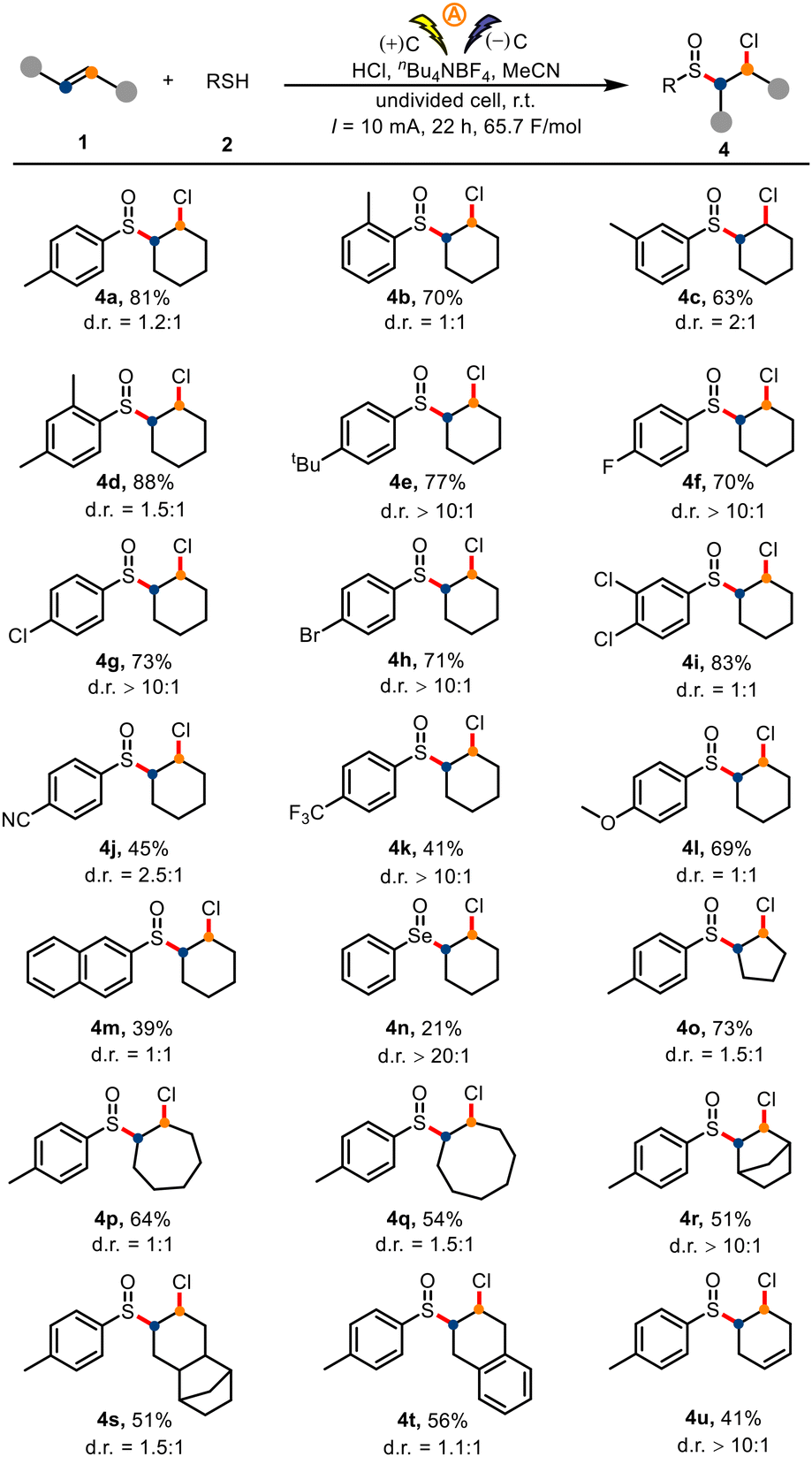 | ||
| Scheme 3 Substrate scope of chlorosulfoxidation. Standard chlorosulfoxidation conditions: 1 (0.5 mmol), 2 (1.0 mmol), nBu4NBF4 (1.0 mmol), MeCN (5 mL), an undivided cell with graphite electrodes (each 1.0 × 1.0 cm2), room temperature (r.t.), 10 mA, 22 h. | ||
To study the reaction mechanism, we carried out a series of control experiments. When the radical inhibitor 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 1.0 mmol) was added to a reaction mixture containing 1a (0.5 mmol) and 2a (1.0 mmol) under otherwise standard conditions, the desired transformation shut down completely, and radical-trapping compound 5 was detected by high-resolution mass spectrometry (Scheme 4a). This result suggests that radical intermediates were involved in the reaction. Upon reaction for only 3 h under otherwise standard conditions, the homocoupling product bis-(4-methylphenyl)disulfide (6) was obtained in 60% isolated yield (Scheme 4b). When p-toluenethiol (2a) was replaced by 6, product 4a was obtained in 82% yield (Scheme 4c). These results indicate that thiyl radicals were produced under these electrochemical conditions. Moreover, when H218O (1.0 mL) was present under otherwise standard conditions, we obtained 4a and 18O-labeled compound 7, which means that the oxygen atom in product 4a came from H2O present in the concentrated HCl rather than from O2 (Scheme 4d).
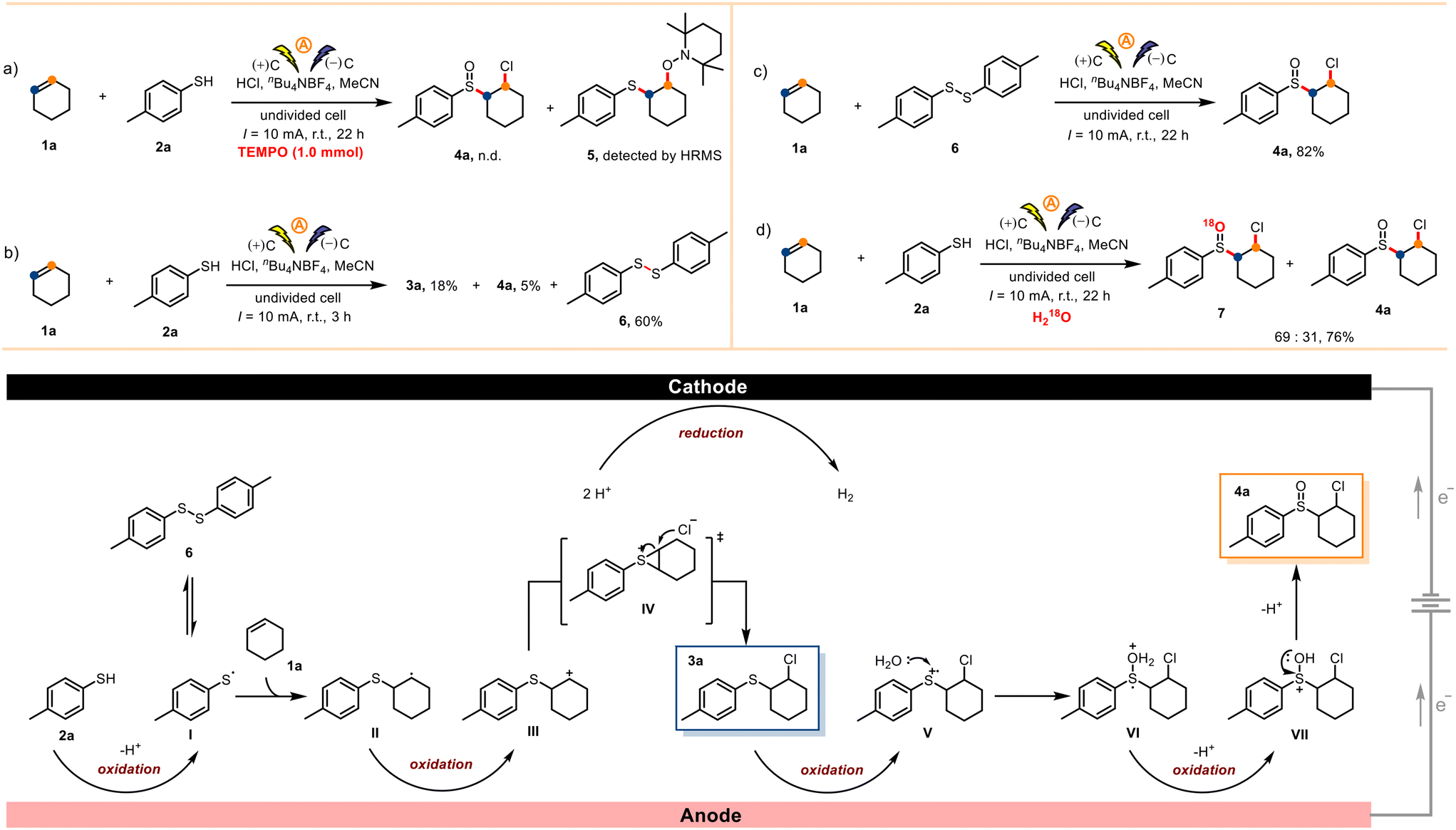 | ||
| Scheme 4 Mechanistic studies and the proposed mechanism. | ||
On the basis of the above-described results, a proposed mechanism is depicted in Scheme 4. First, electrochemical oxidation of p-toluenethiol (2a) at the anode generates thiyl radical I, which undergoes radical addition with cyclohexene (1a) to afford carbon radical II. Radical II is oxidized at the anode to produce carbocation III, which can also exist as a sulfonium intermediate (IV). Nucleophilic attack on IV by a chloride ion generates sulfide 3a, which can be oxidized at the anode to cationic radical V. Cationic radical V is trapped by H2O in HCl to afford oxonium ion VI.32 Finally, VI is oxidized to sulphion VII at the anode, and subsequent deprotonation generates sulfoxide product 4a.
Conclusions
We introduced a combination of simple graphite electrodes, mild conditions, and an environmentally friendly method for the selective preparation of sulfides or sulfoxides via unactivated olefins and thiophenols through a green electrochemical protocol. Avoiding the use of chemical oxidants or reductants, we chose electrochemistry as a green, environmentally friendly and economical transformation. We also expect this green method to be attractive for academic research as well as industrial applications to prepare more high-value products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21732002, 22077071) and the Frontiers Science Center for New Organic Matter, Nankai University (No. 63181206) for generous financial support for our programs.Notes and references
- C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed.
- J. Zhao and X. Jiang, Chin. Chem. Lett., 2018, 29, 1079–1087 CrossRef CAS.
- L. H. Smith, S. C. Coote, H. F. Sneddon and D. J. Procter, Angew. Chem., Int. Ed., 2010, 49, 5832–5844 CrossRef CAS PubMed.
- J. B. Sweeney, Angew. Chem., Int. Ed., 2016, 55, 13928–13928 CrossRef CAS.
- Y. C. Wu, Y. T. Xiao, Y. Z. Yang, R. J. Song and J. H. Li, ChemCatChem, 2020, 12, 5312–5329 CrossRef CAS.
- S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327–1339 CAS.
- X. Cheng, X. Liu, S. Wang, Y. Hu, B. Hu, A. Lei and J. Li, Nat. Commun., 2021, 12, 4366 CrossRef CAS PubMed.
- F.-H. Cui, Y. Hua, Y.-M. Lin, J. Fei, L.-H. Gao, X. Zhao and H. Xia, J. Am. Chem. Soc., 2022, 144, 2301–2310 CrossRef CAS PubMed.
- D.-M. Yan, S.-H. Xu, H. Qian, P.-P. Gao, M.-H. Bi, W.-J. Xiao and J.-R. Chen, ACS Catal., 2022, 12, 3279–3285 CrossRef CAS.
- S.-F. Zhou, X. Pan, Z.-H. Zhou, A. Shoberu and J.-P. Zou, J. Org. Chem., 2015, 80, 3682–3687 CrossRef CAS PubMed.
- C. Huo, Y. Wang, Y. Yuan, F. Chen and J. Tang, Chem. Commun., 2016, 52, 7233–7236 RSC.
- K. Surendra, N. S. Krishnaveni, R. Sridhar and K. R. Rao, J. Org. Chem., 2006, 71, 5819–5821 CrossRef CAS PubMed.
- H. Xi, B. Deng, Z. Zong, S. Lu and Z. Li, Org. Lett., 2015, 17, 1180–1183 CrossRef CAS PubMed.
- K. Choudhuri, A. Mandal and P. Mal, Chem. Commun., 2018, 54, 3759–3762 RSC.
- S. Chatterjee and T. K. Paine, Angew. Chem., Int. Ed., 2015, 54, 9338–9342 CrossRef CAS PubMed.
- Y. Li, S. A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499–13506 CrossRef CAS PubMed.
- K.-J. Liu, Z. Wang, L.-H. Lu, J.-Y. Chen, F. Zeng, Y.-W. Lin, Z. Cao, X. Yu and W.-M. He, Green Chem., 2021, 23, 496–500 RSC.
- C. G. Overberger and R. W. Cummins, J. Am. Chem. Soc., 1953, 75, 4250–4254 CrossRef CAS.
- B. Yu, A.-H. Liu, L.-N. He, B. Li, Z.-F. Diao and Y.-N. Li, Green Chem., 2012, 14, 957–962 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CAS.
- B. Zhang, Y. Gao, Y. Hioki, M. S. Oderinde, J. X. Qiao, K. X. Rodriguez, H. J. Zhang, Y. Kawamata and P. S. Baran, Nature, 2022, 606, 313–318 CrossRef CAS PubMed.
- G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef CAS.
- Y. Yuan, Y. Chen, S. Tang, Z. Huang and A. Lei, Sci. Adv., 2018, 4, eaat5312 CrossRef CAS PubMed.
- Y. Wang, L. Deng, H. Mei, B. Du, J. Han and Y. Pan, Green Chem., 2018, 20, 3444–3449 RSC.
- H. Wang, M. Yu, P. Zhang, H. Wan, H. Cong and A. Lei, Sci. Bull., 2022, 67, 79–84 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02134j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |