
Hydrodeoxygenation of potential platform chemicals derived from biomass to fuels and chemicals†
Keiichi
Tomishige
 *,
Mizuho
Yabushita
,
Ji
Cao
and
Yoshinao
Nakagawa
*,
Mizuho
Yabushita
,
Ji
Cao
and
Yoshinao
Nakagawa
Department of Applied Chemistry, School of Engineering, Tohoku University, 6-6-07 Aoba, Aramaki, Aoba-ku, Sendai, Miyagi 980-8579, Japan. E-mail: tomi@erec.che.tohoku.ac.jp
First published on 23rd June 2022
Abstract
Biomass valorization has emerged as a promising and sustainable means of accommodating value-added chemicals. To produce such chemicals, various transformation methods of the top biomass-derived platform chemicals nominated by the U.S. Department of Energy (DOE) have been extensively investigated in recent decades. However, as represented by 5-hydroxymethylfurfural, which is one of the most widely investigated top platform chemicals, their use is still under development due to difficulties in their production, handling, and selective transformation. In this context, revisiting the U.S. DOE's lists and refocusing on other potent platform chemicals are beneficial for this research field. This review introduces such platform chemicals, viz., levoglucosan (+levoglucosenone), alkyl glycosides, glucaric acid, erythritol (+1,4-anhydroerythritol), and N-acetylglucosamine. Hydrodeoxygenation to remove oxygenated functionalities partially or completely using hydrogen as a reductant is a promising means of upgrading these compounds to fuels and useful chemicals, whose production is difficult from conventional top platform chemicals. Herein, a variety of catalytic systems to achieve the selective hydrodeoxygenation of the compounds introduced above are summarized with a description of their features, including catalytic performance, active sites, and mechanism.
 Keiichi Tomishige | Keiichi Tomishige received his B.S., M.S., and Ph.D. from the Graduate School of Science, Department of Chemistry, The University of Tokyo, under the guidance of Prof. Y. Iwasawa. During his Ph.D. course in 1994, he moved to the Graduate School of Engineering, The University of Tokyo, as a research associate and worked with Prof. K. Fujimoto. In 1998, he became a lecturer, and then he moved to the Institute of Materials Science, University of Tsukuba, as a lecturer in 2001. Since 2004 he has been an associate professor at the Graduate School of Pure and Applied Sciences, University of Tsukuba. Since 2010 he has been a professor at the School of Engineering, Tohoku University. His research interests are the development of heterogeneous catalysts for (1) the production of biomass-derived chemicals, (2) the non-reductive conversion of CO2 with alcohols/amines into organic carbonates, carbamates, and ureas, and (3) the reforming of hydrocarbons such as biomass tar and methane. He received a Catalysis Society of Japan Award (Academic field) in 2021. He is a member of the editorial boards of Green Chemistry (since Aug., 2017) and Applied Catalysis A (since Apr., 2009), a member of the international advisory boards of ChemSusChem (since Jan., 2015) and ChemCatChem (since Jan., 2021), a member of the editorial advisory board of ACS Omega (since Jan., 2018), and a member of the editorial boards of Catalysis Surveys from Asia (since Oct., 2021), Sustainable Chemistry for Climate Action (since Jan., 2022) and Current Opinion in Chemical Engineering (since Jan., 2022). |
 Mizuho Yabushita | Mizuho Yabushita received his Ph.D. in 2015 from the Graduate School of Chemical Sciences and Engineering, Hokkaido University under the guidance of Prof. A. Fukuoka. From 2015 to 2018, he worked as a postdoctoral fellow at the University of California, Berkeley, with Prof. A. Katz and at Hokkaido University with Prof. A. Fukuoka. After two years as an assistant professor in Prof. A. Muramatsu's group at Tohoku University, he moved to the research group led by Prof. K. Tomishige as an assistant professor. His current research interests are the catalytic conversion of biomass-derived compounds and the synthesis and application of zeolites. |
 Ji Cao | Ji Cao is an assistant professor at the School of Engineering, Tohoku University. She received her B.E degree in 2014 from Taiyuan University of Technology, and her M.ScT. degree in 2016 from Hirosaki University. She obtained her Ph.D. in 2020 from the Graduate School of Engineering at Tohoku University under the guidance of Prof. K. Tomishige. Her current research focuses on the development of heterogeneous catalysts for the conversion of biomass-related compounds into value-added chemicals. |
 Yoshinao Nakagawa | Yoshinao Nakagawa received his Ph.D. in 2005 from the Graduate School of Engineering, The University of Tokyo, under the guidance of Prof. N. Mizuno. After 4 years of postdoctoral research in the same group, he joined the research group of Keiichi Tomishige at the University of Tsukuba. He moved to Tohoku University and became an assistant professor in 2010. Since 2013, he has been an associate professor. His current research interests are catalytic oxidations and reductions of bio-related chemicals. |
1. Introduction
Decreasing CO2 emission is one of the urgent tasks for the prevention of global warming. Much attention has been recently paid to the concept of “carbon recycling”, which can include CO2 conversion to fuels and chemicals by renewable energies.1–3 A typical method for CO2 conversion is the reduction of CO2 with renewable hydrogen to CO, formic acid, formaldehyde, methanol, methane, ethanol, longer chain hydrocarbons, and so on. CO2 contains carbon atoms with the highest oxidation state, meaning that a large amount of renewable hydrogen is required for CO2 reduction to hydrocarbons. The availability of renewable hydrogen influences the feasibility of these CO2 hydrogenation processes significantly. A variety of useful oxygenates have been produced by the oxidation and/or hydration of hydrocarbons in the petroleum refining industry. It is thought that the production of such oxygenates by the oxidation/hydration of hydrocarbons from CO2 reduction is not energy-efficient. Oxygenates can be produced by the reduction of biomass-derived platform chemicals with renewable hydrogen because the oxygen content of biomass-derived platform chemicals is clearly higher than that of oxygenates produced in the petrochemical industry. The reduction of biomass-derived platform chemicals is more favorable than CO2 hydrogenation because a lower amount of H2 is necessary for the former case. The more energy efficient production of value-added chemicals via biomass-derived platform chemicals than those via CO2-derived hydrocarbons will maintain the importance of biomass utilization.Two famous lists of biomass-derived platform chemicals were issued by the U.S. Department of Energy (DOE) in 2004 and 2010, and the newer one was published in this journal.4,5 The older list was made by the screening of various compounds that can be obtained from carbohydrates. Twenty-eight carbohydrate-derived compounds, many of which were fermentation products, were selected, and the combination of these compounds with CO and synthesis gas (syngas) gave the “Top 30” candidates. Further refinement gave the “Top 15” candidates. They are sometimes also called the “Top 12” by combining three C4 dicarboxylic acids (succinic acid, malic acid, and fumaric acid) and C5 sugar alcohols (xylitol and arabinitol) into their respective groups, and they are even colloquially called the “Top 10”. The newer list was re-compiled from compounds in a wider range, including those derived from vegetable oil and algae. The manufacturing cost and availability were more focused on for the newer list, and compounds synthesized by chemical processes, rather than fermentation, tended to be selected. Numerous studies on the synthesis and conversion of these platform chemicals have been carried out in this decade on the basis of these lists.6 The compounds introduced as top biomass-derived platform chemicals in the two lists are shown in Table 1.
| Compound name | Chemical formula | Feedstock, type of production processa | Carbon number | DOE report in 2004 b | Green Chem. in 2010c | This review | |
|---|---|---|---|---|---|---|---|
| “Top 12 (or 15)” | “Top 30” | ||||||
| a B: Produced by biological or biochemical processes such as fermentation; C: produced by chemical or physical processes. b Ref. 4. c Ref. 5. d Carbon atoms in the alkyl or acetyl group are not included. | |||||||
| Carbon monoxide, syngas | Lignocellulose, C | 1 | — | X | — | — | |
| Ethanol |
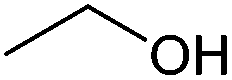
|
Sugars, B | 2 | — | — | X | — |
| Glycerol |
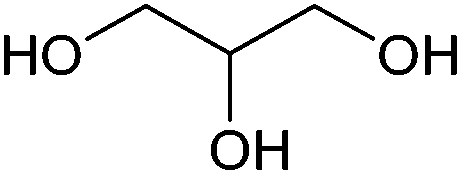
|
Oil/fat, C | 3 | X | X | X | — |
| 3-Hydroxypropionic acid |
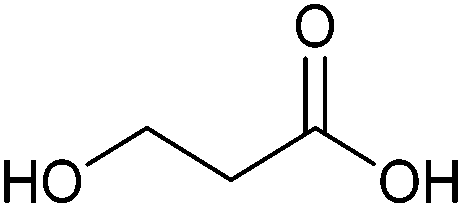
|
Sugars, B | 3 | X | X | X | — |
| Lactic acid |
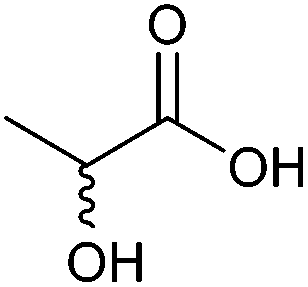
|
Sugars, B | 3 | — | X | X | — |
| Malonic acid |
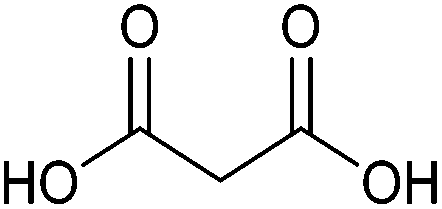
|
Sugars, B | 3 | — | X | – | — |
| Propionic acid |

|
Sugars, B | 3 | — | X | – | — |
| Serine |

|
Sugars, B | 3 | — | X | – | — |
| Succinic acid |
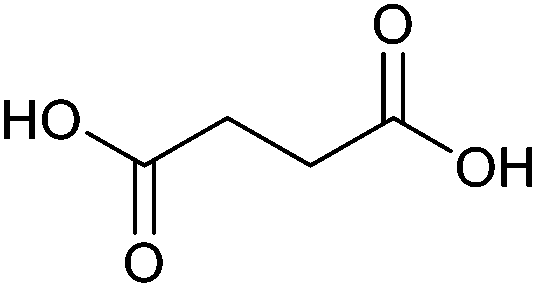
|
Sugars, B | 4 | X | X | X | — |
| Malic acid |
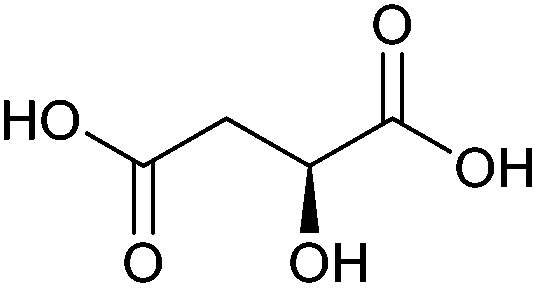
|
Sugars, B | 4 | X | X | — | — |
| Fumaric acid |
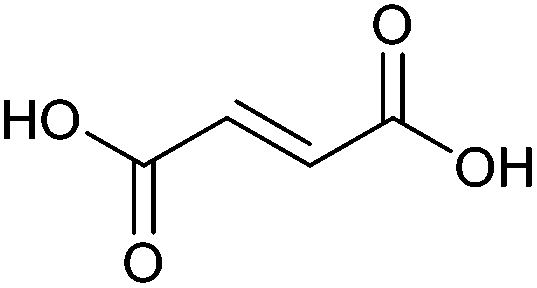
|
Sugars, B | 4 | X | X | — | — |
| Aspartic acid |
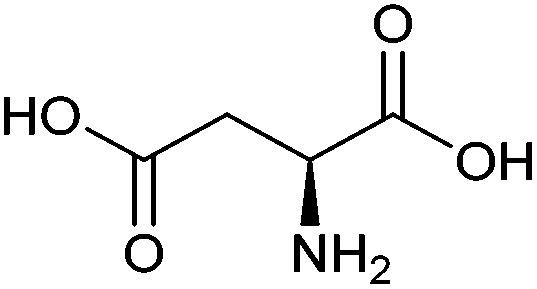
|
Sugars, B | 4 | X | X | — | — |
| Threonine |
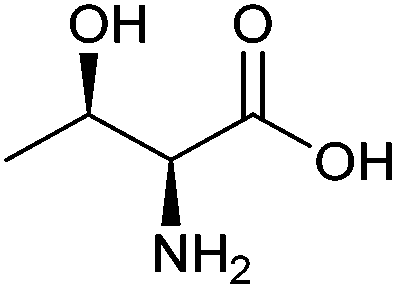
|
Sugars, B | 4 | — | X | — | — |
| Acetoin |
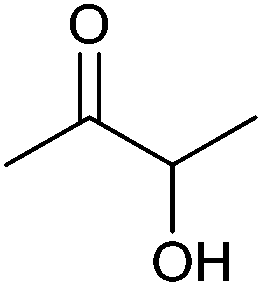
|
Sugars, B | 4 | — | X | — | — |
| 3-Hydroxybutyrolactone |
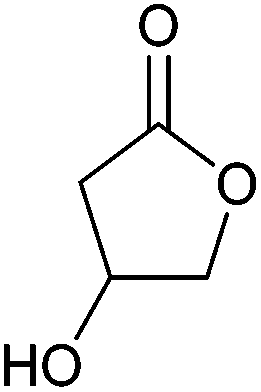
|
Sugars, B | 4 | X | X | — | — |
| Erythritol |

|
Sugars, B | 4 | — | — | — | X |
| Furfural |
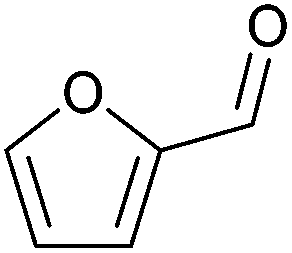
|
Hemicellulose, C | 5 | — | X | X | – |
| Itaconic acid |
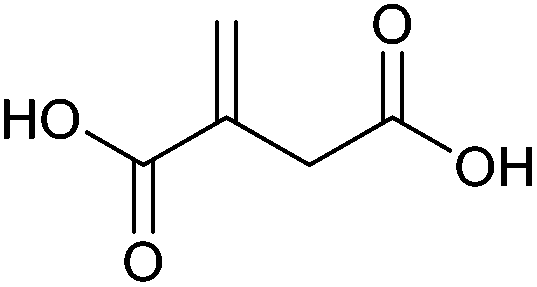
|
Sugars, B | 5 | X | X | — | — |
| Levulinic acid |
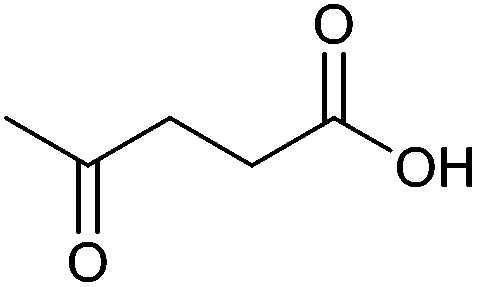
|
Sugars, C | 5 | X | X | X | – |
| Glutamic acid |
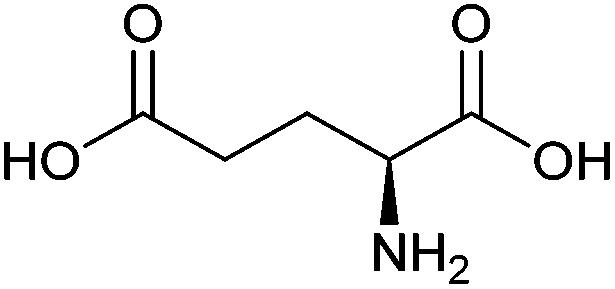
|
Sugars, B | 5 | X | X | — | — |
| Proline |
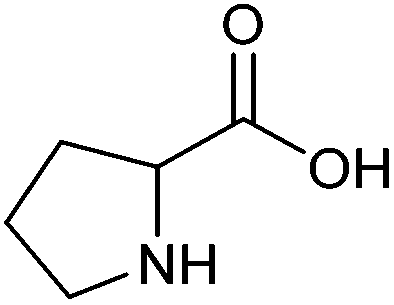
|
Sugars, B | 5 | — | X | — | — |
| Xylitol and arabinitol |
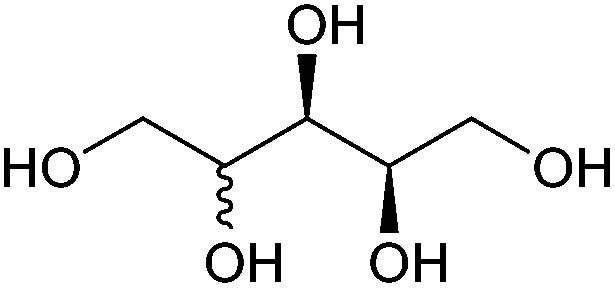
|
Hemicellulose, C | 5 | X | X | X | — |
| Xylonic acid |
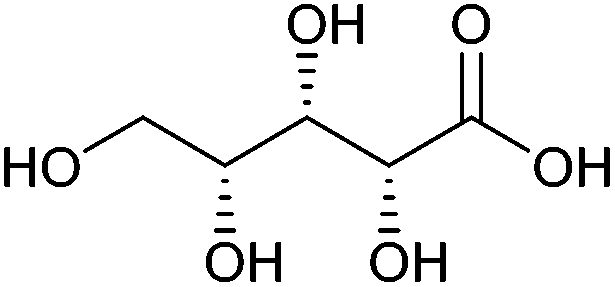
|
Hemicellulose, C | 5 | — | X | — | — |
| Isoprene |
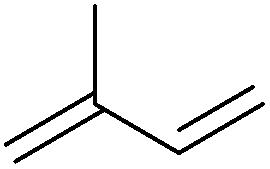
|
Sugars, B | 5 | — | — | X | — |
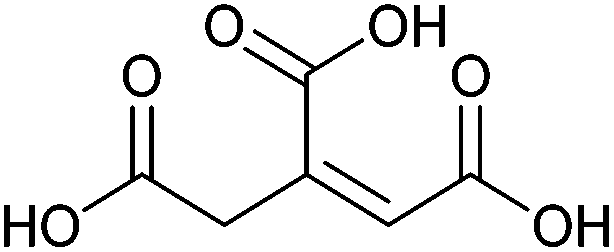
| Citric acid, aconitic acid |
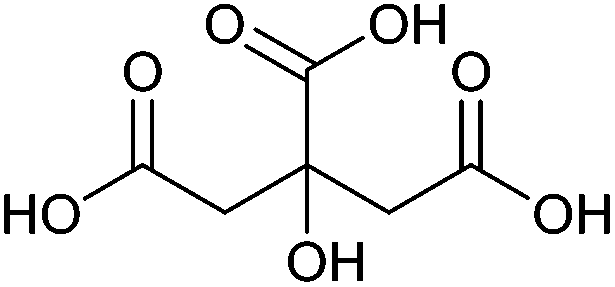
|
Sugars, B | 6 | — | X | — | — |
|
|||||||
| Lysine |
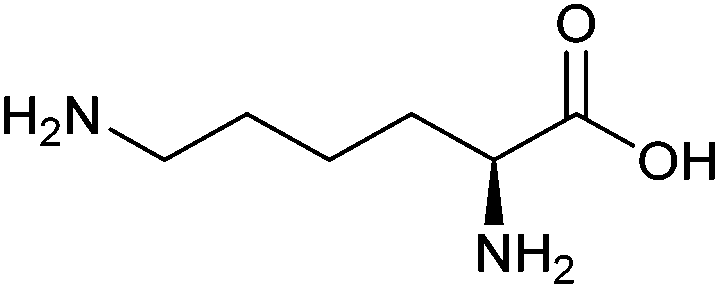
|
Sugars, B | 6 | — | X | — | — |
| 5-Hydroxymethylfurfural (HMF) |
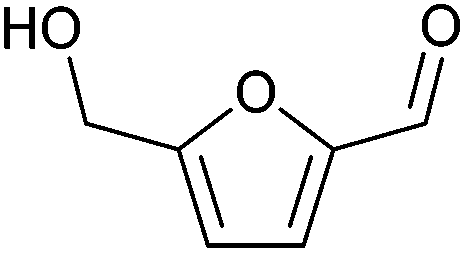
|
Sugars, C | 6 | — | — | X | — |
| 2,5-Furandicarboxylic acid (FDCA) |
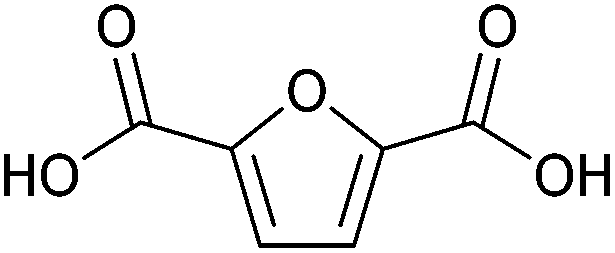
|
Sugars, C | 6 | X | X | X | — |
| Sorbitol |
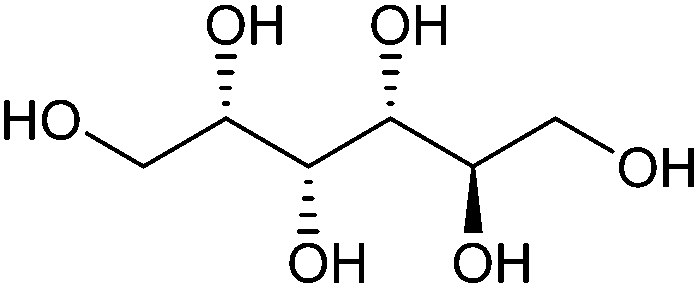
|
Sugars, C | 6 | X | X | X | — |
| Glucaric acid |
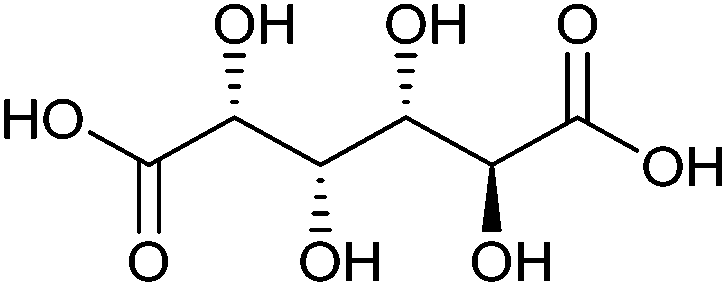
|
Sugars, C | 6 | X | X | — | X |
| Levoglucosan |
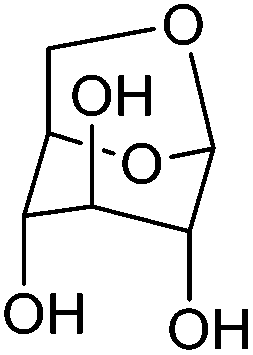
|
Cellulose, C | 6 | — | X | — | X |
| Alkyl glycosides |
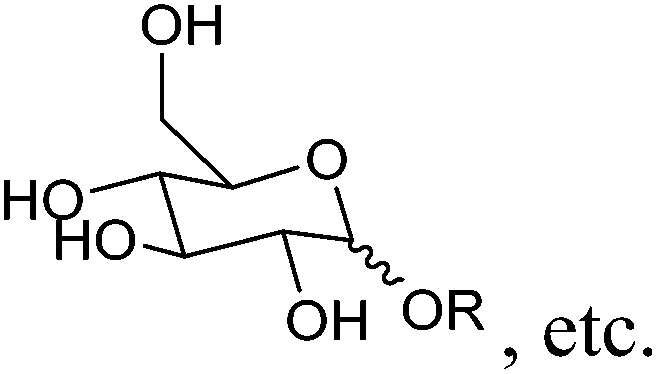
|
Sugars, C | 6d | — | — | — | X |
| N-Acetylglucosamine |
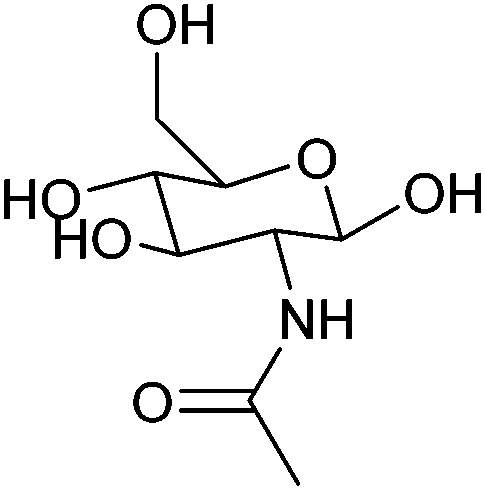
|
Chitin, C | 6d | — | — | — | X |
| Heavy biohydrocarbons |
|
Sugars, B or algae, C | ≥15 | — | — | X | — |
As the sole C2 platform chemical, ethanol can be easily converted into ethylene, meaning that ethanol can play similar roles to ethylene as an intermediate in petrochemical production. Glycerol can give a variety of C3 chemicals such as propanols, propanediols, and acrolein, suggesting that glycerol will be able to act as a substitute for propylene in the petrochemical industry. It is expected that C2 and C3 chemicals can be sufficiently supplied from these present platform chemicals. For C4 platform chemicals, various compounds were introduced in the older list;4 however, only one compound, succinic acid, remained a top platform chemical in the newer list.5 This is because the fermentative production of these C4 platform chemicals is inefficient and costly. The petrochemical industry finds it difficult to produce a C5 platform chemical, and thus, the accessibility of C5 platform chemicals is the advantage of the biomass refinery because of hemicellulose and natural rubber. One of the major strategies for the utilization of such C5 platform chemicals relies on furfural and its derivatives.7–11 In the petrochemical industry, a variety of C6 aliphatic and aromatic chemicals are produced mainly via benzene. Because of the importance of many C6 compounds in the chemical industry, biomass-derived platform chemicals for C6 compounds should be diverse with respect to functionality and feedstock, and we propose several new C6 platform chemicals.
A biomass-derived candidate for a C6 platform chemical with wide applicability is 5-hydroxymethylfurfural (HMF). HMF has been recognized as one of the most typical lignocellulosic biomass-derived platform chemicals, since this compound can be derivatized into valuable chemicals including 2,5-dimethylfuran (diesel fuel) and 2,5-furandicarboxylic acid (FDCA; building block of polyethylene furanoate),12–15 the latter of which is also listed in the top bio-based chemicals derived from sugars by the U.S. DOE along with HMF (Table 1).4,5 However, regardless of their attractive applications, the mass production of these furanic compounds inevitably faces serious and fundamental obstacles arising from the chemical nature of HMF, namely its reactive formyl and hydroxymethyl groups (Fig. 1), as described in a previous essay by Galkin and Ananikov.16 A well-known issue is the difficulty in the selective production of HMF from biomass-derived sugars. The most attractive substrate for synthesizing HMF is glucose, which is the most accessible monosaccharide in nature and typically undergoes a two-step transformation to yield HMF (isomerization of glucose to fructose catalyzed by a base or Lewis acid and subsequent dehydration of fructose to HMF in the presence of acids).12 However, due to the presence of reactive functional groups (formyl and hydroxymethyl groups) in both the substrate and HMF molecules, this reaction always suffers from side reactions, as exemplified by the intermolecular condensation of HMF and/or reactive intermediate compounds to produce insoluble humins and the hydrolysis of HMF to levulinic acid and formic acid.17–19 Low substrate concentration conditions and biphasic systems comprising water, sometimes containing salts, which are required for the dissolution of sugar substrates, and organic solvent, which extracts and stabilizes produced HMF, are effective for the suppression of undesired side reactions.20–22 However, the dilution of HMF and the use of additional solvents and additives require energy-consuming condensation, separation, and/or purification steps. Such additional steps make the entire HMF production process more complicated and costlier. What is worse, HMF degrades spontaneously to be polymerized even during storage.23 Therefore, to establish efficient commercial plants for manufacturing HMF derivatives, these fundamental limitations need to be solved. The protection of the reactive groups of HMF, e.g., acetalization of the formyl group by 1,3-propanediol,24,25 silylation of the hydroxymethyl group by the tert-butyldiphenylsilyl group,23 or etherification of the hydroxymethyl group,26 should be a promising approach (Fig. 1) and is in progress. More detailed protection strategies were summarized in a very recent review.27 Another issue for handling HMF arises from the negative effect of trace impurities on the catalytic conversions of HMF.28 HMF synthesis typically uses unconventional solvents such as dimethyl sulfoxide (DMSO), and the presence of trace DMSO greatly affects the hydrogenation of HMF.29
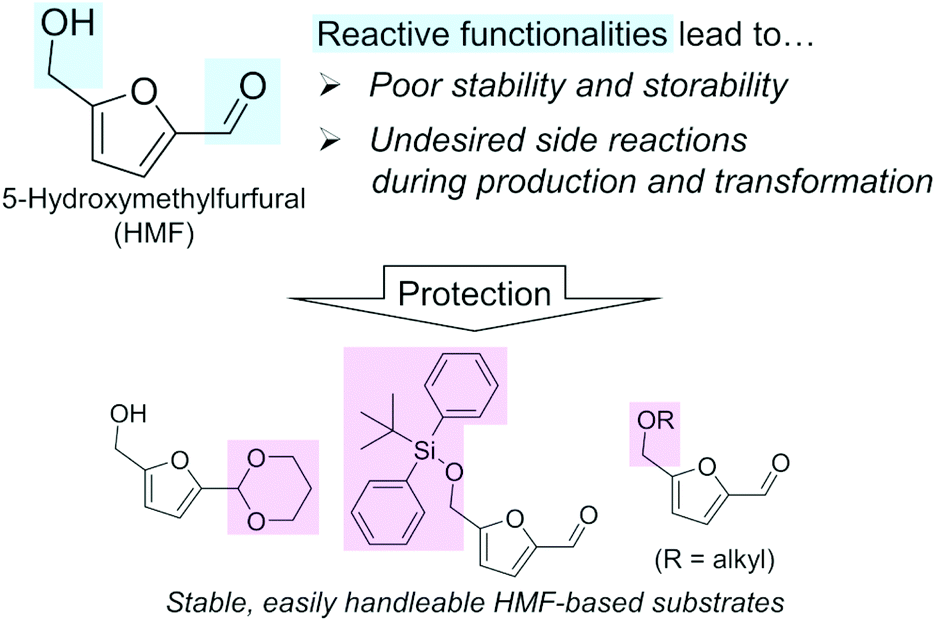 | ||
| Fig. 1 Origins of the difficulty in using HMF as a platform chemical and promising approaches for its protection. | ||
As mentioned above, some difficulties of HMF operation arise from the presence of the formyl group; this is common when handling sugar compounds. It is known that formyl groups can be protected by acetal formation. Sugars are usually present as cyclic hemiacetals through the intramolecular addition reaction of the OH group at the 4- or 5-position to the formyl group (Fig. 2). Further reaction of the hemiacetal with another OH group in the sugar molecule gives an acetal. A representative of such an acetal is levoglucosan, which is the acetal of β-glucose cyclized between the 1- and 6-positions via an O atom. Another type of acetal is that of external alcohols, alkyl glycosides. Here, we discuss the potential of these acetals (levoglucosan: section 2; alkyl glycosides: section 3) as C6 platform chemicals.
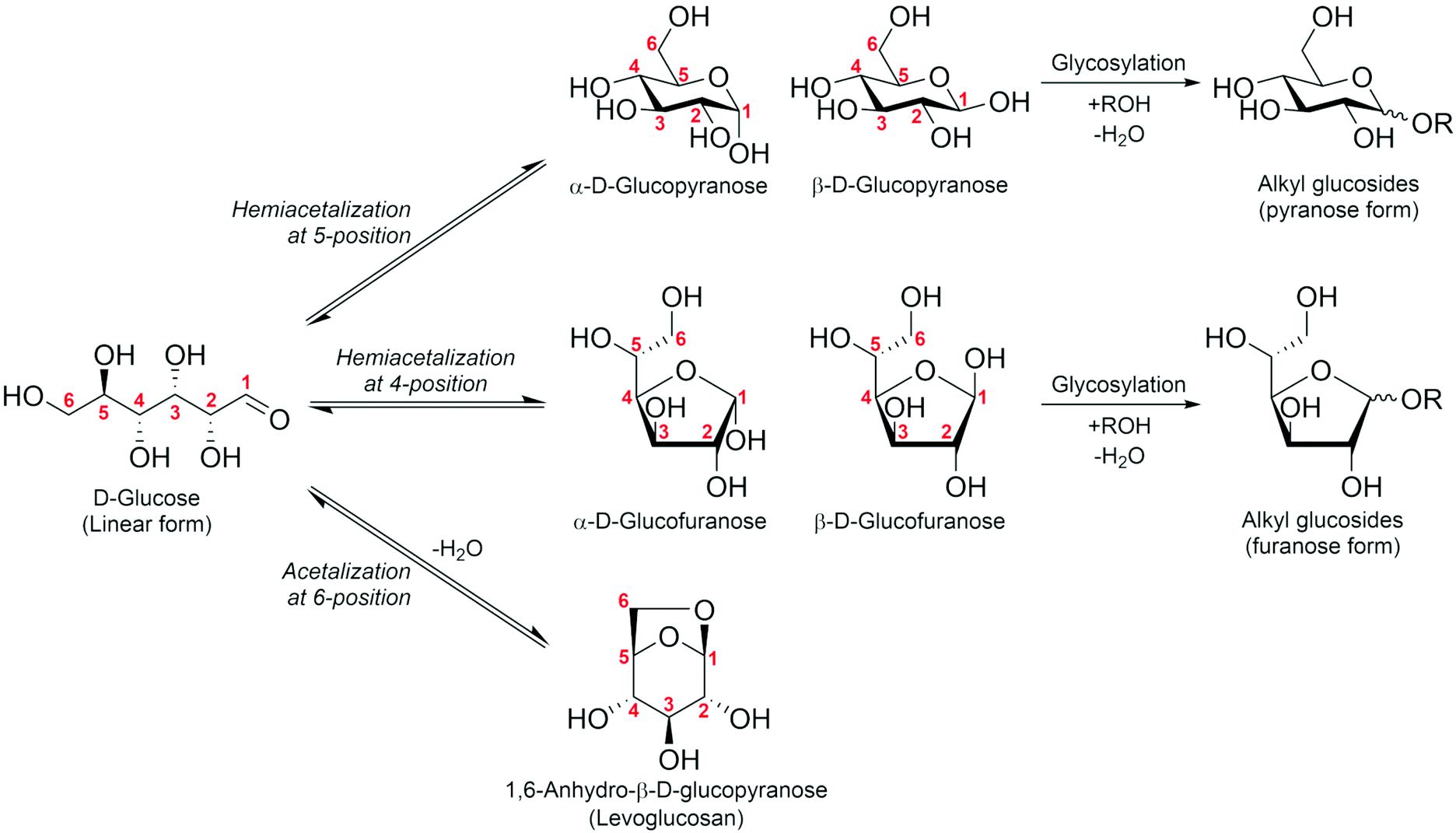 | ||
| Fig. 2 Hemiacetalization and acetalization of sugar compounds, as exemplified by D-glucose. | ||
Carboxylic groups can be formed via simple oxidation of formyl and primary hydroxy groups. The reactivity of the carboxylic group is not as high as that of the formyl group, leading to the ease of handling such compounds containing a carboxylic group(s). Therefore, sugar acids can be also considered as C6 platform chemicals (section 4).
Another problem of the platform-chemical list from 20104 is that C4 platform chemicals are limited to dicarboxylic acids, and the most easily accessible one is succinic acid. This is connected to the difficulty in functionalizing the 2-position. Syntheses of C4 chemicals from C5 platform chemicals have even been investigated recently, for example, the decarboxylation of levulinic acid.30 Considering that sugar alcohols (glycerol, xylitol, and sorbitol) are listed as C3, C5, and C6 platform chemicals, respectively, erythritol can also be a C4 platform chemical because it is produced by the fermentation of glucose and (crude) glycerol.31,32 All the carbon atoms in erythritol are functionalized and this enables derivatization to a variety of C4 chemicals. While the possibility of erythritol as a platform chemical was discussed in our previous review paper,33 the chemistry of hydrodeoxygenation of erythritol and its dehydrated derivative (1,4-anhydroerythritol (1,4-AHERY)) has been growing rapidly. Here we summarize recent developments in section 5.
In addition, we mention the possibility of N-acetylglucosamine as an N-containing C6 platform chemical because its polymer, chitin, is regarded as an abundant biomass resource (section 6).
2. Levoglucosan and levoglucosenone
Levoglucosan can be produced by the pyrolysis of cellulose in good yields. The yield varies depending on the reactor type and conditions, and there are several reports with yields higher than 60%.34 High temperature, such as 773 K, has been applied. As shown by the high temperature required for synthesis, levoglucosan is thermally stable and easy to handle. The production of levoglucosan from starch is also possible;35 however, production from cellulose is preferable because the use of starch as a source of chemicals competes with food supply. Levoglucosan is also one of the main components of the pyrolysis product of raw lignocellulose (bio-oil).36,37 The relatively easy production from biomass makes levoglucosan a potential platform chemical. In the DOE report from 2004, levoglucosan was selected as one of the top 30 candidates; however, it was not selected as a final top 12 building block because it has “somewhat lower potential” in transformation to useful products.4 After publication of the DOE report, more and more research was carried out for the transformation of any biomass-derived molecules including levoglucosan and its derivatives. A comprehensive review paper on the synthesis and application of levoglucosan was published in this journal in 2020.38 Fermentation is the main transformation method of levoglucosan. There are two major pathways of levoglucosan metabolism: one is the combination of oxidation, hydrolysis and reduction to glucose, and the other is direct phosphorylation to glucose-6-phosphate (Fig. 3). Both products are further consumed in the same way as that in the fermentation of glucose.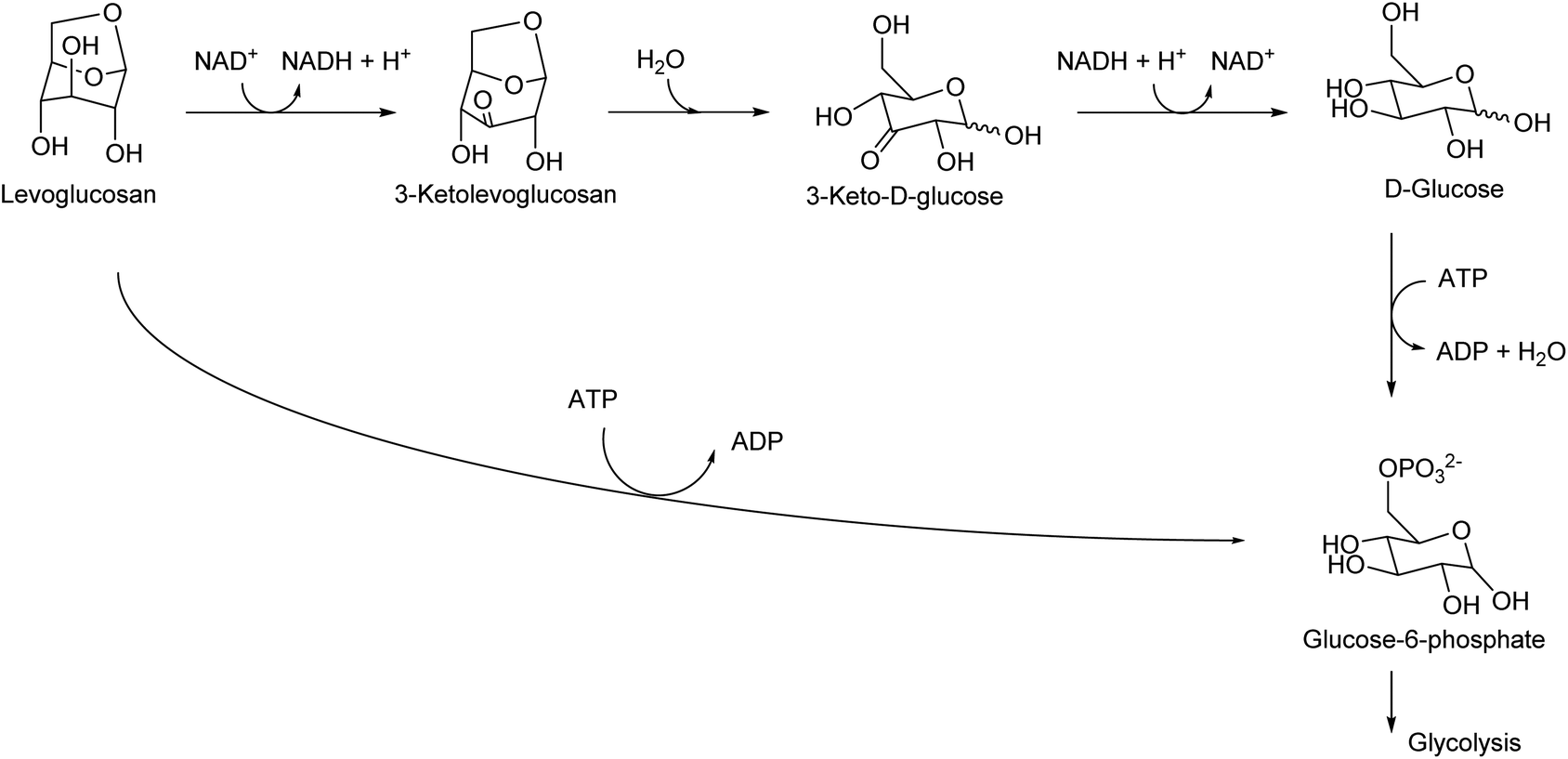 | ||
| Fig. 3 Pathways of levoglucosan metabolism. | ||
Levoglucosan can be re-hydrated to glucose over acid catalysts. Many of the products of the chemical transformation of levoglucosan are compounds that can also be produced from glucose. This is true for the reductive conversion of levoglucosan. Heeres et al. reported the conversion of levoglucosan into sorbitol over a mesoporous-carbon-supported Ru catalyst with H2 in high yield (96%) at 453 K.39 On the other hand, sorbitol can be produced by the hydrogenation of glucose or even cellulose.40,41 The production of sorbitol by the hydrogenation of glucose can proceed almost quantitatively. Although the sorbitol yield from cellulose was generally not high (typically around 60%), the performance of direct cellulose hydrogenation to sorbitol has been steadily improved in extensive recent studies.42,43 Bindwal and Vaidya reported the hydrogenolysis of levoglucosan over a Ru/C catalyst in water as a solvent.44 The main products were 1,2-propanediol and ethylene glycol with a combined yield of 35%-C (94.8% conversion) at 413 K. These glycols can also be produced by the hydrogenolysis of various sugars and sugar alcohols such as cellulose,45,46 sorbitol,47–49 xylitol,50 and glycerol.51,52 The combined yields of glycols from cellulose and sugar alcohols reached over 80%, and that from levoglucosan was still significantly lower.
Levoglucosan has three OH groups, and regioselective derivatization is not easy. The selective hydrogenolysis of levoglucosan at a specific C–OH group has not yet been reported. Limited examples of the regioselective derivatization of OH groups in levoglucosan include enzymatic acylation53 and continuous-flow benzylation over a BaO catalyst.54 However, the products of these systems are not useful as substrates for reductive conversions. Deoxydehydration (DODH) and double-hydrodeoxygenation reactions, which have been recently attracting much attention in the reductive conversion of sugar-derived compounds and are explained later, cannot be applied to levoglucosan because both pairs of vicinal OH groups (i.e. 2- and 3-positions as well as 3- and 4-positions) have the trans configuration.
On the other hand, double-dehydration of levoglucosan over acid catalysts can selectively produce the enone product, levoglucosenone (Fig. 4). The direct production of levoglucosenone from cellulose is also possible by pyrolysis in the presence of acid catalysts, and this reaction has been investigated intensively in recent years.55–57 The yield of levoglucosenone from levoglucosan and cellulose reached 48% (propylsulfonic-acid-functionalized silica catalyst, 483 K)58 and 51% (H2SO4 catalyst, 483 K),59 respectively. Levoglucosenone also has potential as a platform chemical. Levoglucosenone has only one reactive functional group in each category (OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), and therefore, selective conversions of levoglucosenone are easier than those of levoglucosan.
C), and therefore, selective conversions of levoglucosenone are easier than those of levoglucosan.
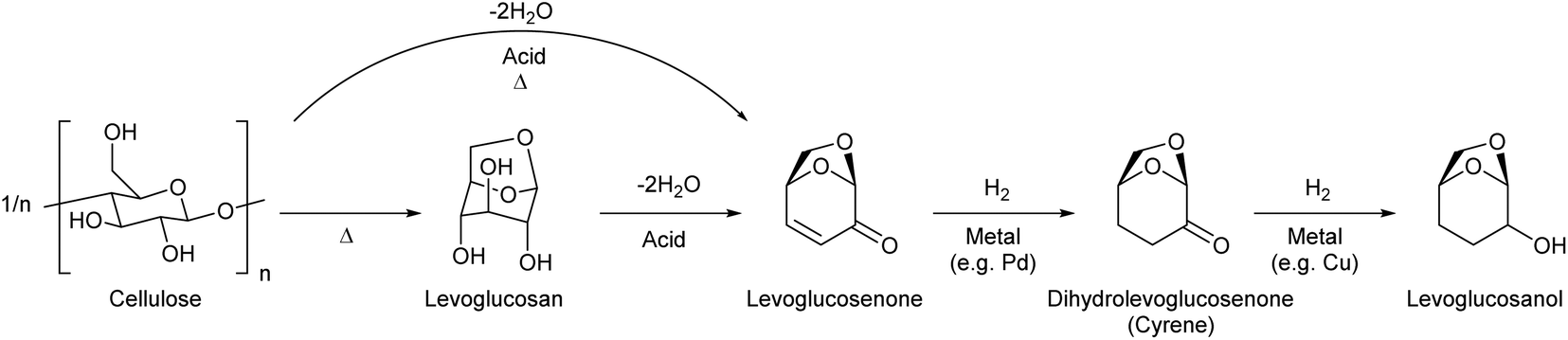 | ||
| Fig. 4 Production route for levoglucosanol from cellulose. | ||
The treatment of levoglucosenone in acidic hot water gives HMF (Fig. 5), and then HMF is hydrolyzed to levulinic acid and formic acid.60–62 The yield of HMF was about 60%. This isomerization reaction may be regarded as an indirect synthesis of HMF from cellulose, while it is a side reaction in the conversion of levoglucosenone into another product.
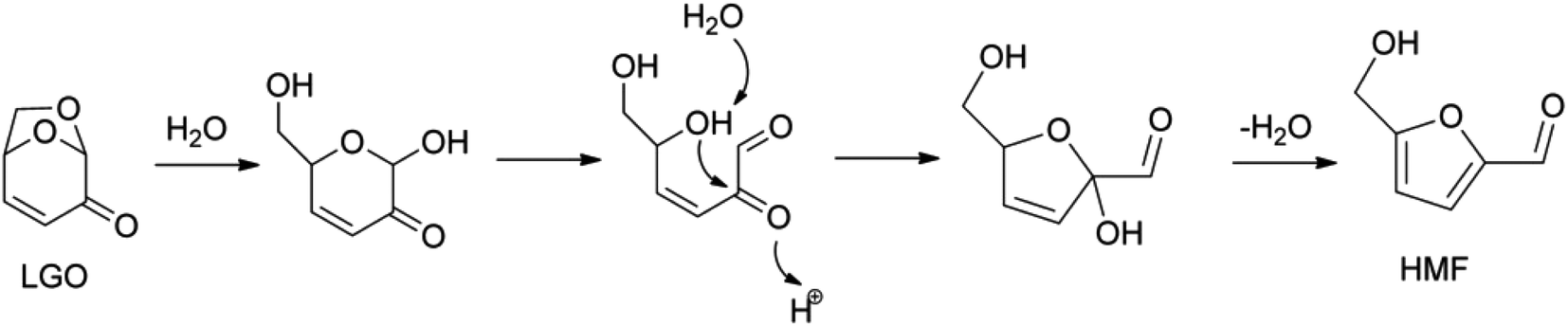 | ||
| Fig. 5 Proposed mechanism for levoglucosenone (LGO) isomerization to HMF. Reprinted from ref. 60 with permission from John Wiley and Sons, copyright 2016. | ||
The reduction of levoglucosenone first hydrogenates the CC bond to give dihydrolevoglucosenone (Cyrene),63 and further CO hydrogenation gives levoglucosanol.64–66 Cyrene has been used as a nontoxic renewable dipolar aprotic solvent. These hydrogenation reactions are not difficult and good yields (≥90%) have been reported.
Huber et al. have intensively investigated the hydrogenolysis of levoglucosanol. The hydrogenolysis of levoglucosanol in water over supported Pt catalysts combined with acidic components such as Pt/SiO2–Al2O3 gave 1,2,5,6-hexanetetraol, which is a potential polymer precursor,67 in excellent yield (maximum 94%; Fig. 6).68 A more recent paper reported a slightly lower yield (ca. 90% selectivity at 91% conversion) with a Pt–WOx/TiO2 catalyst; however, the stability of this catalyst was higher than that of Pt/SiO2–Al2O3.69 1,2,5,6-Hexanetetraol production from sorbitol by Cu-catalyzed hydrogenolysis has been reported in patents with about 40% yield.70,71 While the overall yields from cellulose with these two routes, via sorbitol and via levoglucosanol, are comparable, the high selectivity to 1,2,5,6-hexanetetraol in levoglucosanol hydrogenolysis can decrease the cost of separation from byproducts with high boiling points such as polyols. The hydrogenolysis of levoglucosanol in a non-water solvent produces the dehydrated form of 1,2,5,6-hexanetetraol, tetrahydrofuran-2,5-dimethanol.72–74 Both Pd/SiO2–Al2O3 and Pt/SiO2–Al2O3 catalysts have been tested, and slightly higher selectivity was reported with Pt/SiO2–Al2O3 (73%) (Fig. 7).75 The main byproducts were 2-hydroxymethyltetrahydropyran-5-one and 2-hydroxymethyltetrahydropyran-5-ol. Tetrahydrofuran-2,5-dimethanol can be further converted into 1,2,6-hexanetriol and 1,6-hexanediol by selective hydrogenolysis.76,77 Tetrahydrofuran-2,5-dimethanol and its hydrogenolysis products are also potential monomers. Tetrahydrofuran-2,5-dimethanol can also be synthesized through the total hydrogenation of HMF,78,79 although the cis/trans ratio was different for these two methods (mostly cis (ca. 90%) by hydrogenation of HMF; mixture of similar amounts by hydrogenolysis of levoglucosanol). When searching the literature, care must be taken to note that tetrahydrofuran-2,5-dimethanol is sometimes called by other names such as 2,5-bis(hydroxymethyl)tetrahydrofuran and 2,5-dihydroxymethyltetrahydrofuran.
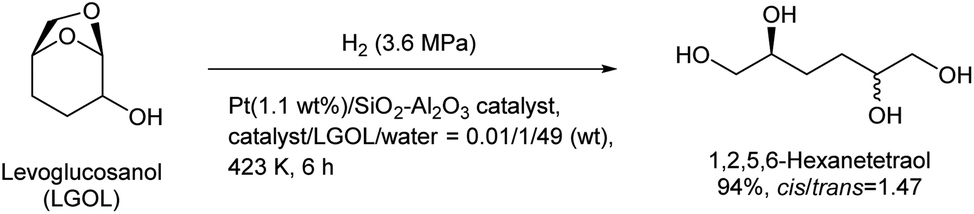 | ||
| Fig. 6 Levoglucosanol hydrogenolysis in water as a solvent.68 | ||
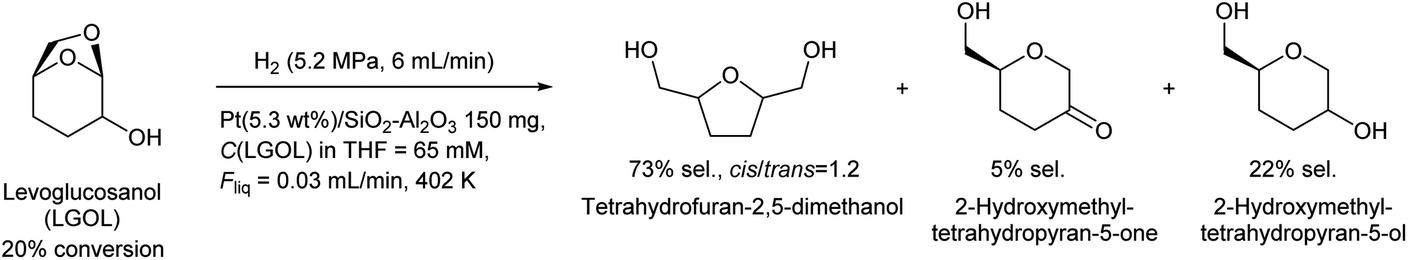 | ||
| Fig. 7 Levoglucosanol hydrogenolysis in a non-water solvent.75 | ||
The mechanisms of the hydrogenolysis reactions of levoglucosanol in both water and non-water solvents have been investigated in detail (Fig. 8). The reaction starts with protonation of the oxygen atom bridging the 1- and 6-positions, because of the large distortion of the 7-membered ring structure. The O–C bond at the 1-position is dissociated to produce a cationic intermediate. Dissociation at the 6-position does not occur because this dissociation would produce a less stable primary carbocation. In water as a solvent, the produced cationic intermediate quickly reacts with water to give 3,4-dideoxysugars (3,4-dideoxyglucose or 3,4-dideoxymannose, depending on the stereochemistry of the levoglucosanol substrate).68,80 The hydrogenation of 3,4-dideoxysugars over metal catalysts gives the target product 1,2,5,6-hexanetetraol. In non-water solvents, the cationic intermediate is rearranged.73 There are two pathways: one is hydride transfer from the 2-position to the 1-position. Subsequent deprotonation gives 2-hydroxymethyltetrahydropyran-5-one. Further hydrogenation over metal catalysts produces 2-hydroxymethyltetrahydropyran-5-ol. The other pathway is the dissociation of the C–O bond at the 5-position, which is facilitated by the oxocarbenium ion resonance state. The recombination of the carbocation at the 5-position with the OH group at the 2-position produces a tetrahydrofuran ring. Further deprotonation and hydrogenation give tetrahydrofuran-2,5-dimethanol.
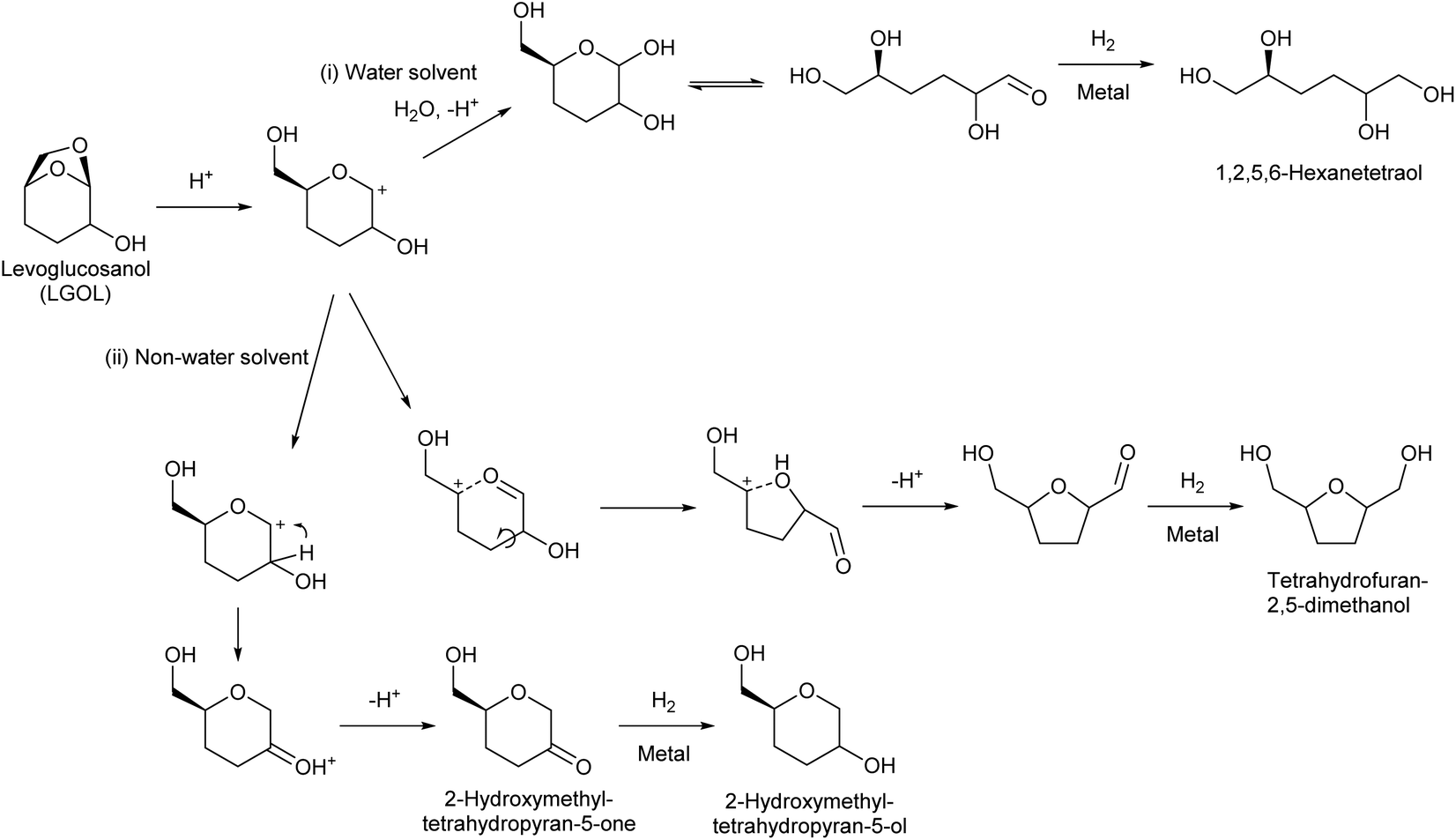 | ||
| Fig. 8 Reaction routes for levoglucosanol hydrogenolysis. | ||
The catalyst for levoglucosanol hydrogenolysis is bifunctional: it has Brønsted acidity to activate the C–O bond and a noble metal surface to hydrogenate the unsaturated bond(s). This combination, as exemplified by Pt/SiO2–Al2O3 and metal/acidic zeolite, is also the most typical class of catalysts in many C–O hydrogenolysis reactions.81,82 Although it is difficult to change largely the selectivity pattern of the main reaction paths over these catalysts, tuning the acidity appropriately as well as selecting the reaction conditions can control undesirable side reactions derived from the unsaturated intermediates.
Selective functionalization and C–C bond formation of levoglucosenone and its hydrogenation product, Cyrene, are possible. In particular, elongation of the carbon framework by C–C bond formation can enlarge the scope of the products. Useful reactions for C–C bond formation include the Michael addition83 and the Diels–Alder reaction84,85 of levoglucosenone at the electron-deficient CC moiety and the condensation of Cyrene with another Cyrene molecule or an aldehyde (Fig. 9).86 The products of these reactions can be used as intermediates in the syntheses of fine chemicals and as the substrates of total hydrodeoxygenation toward branched hydrocarbons, which can be blended into aviation fuel.87
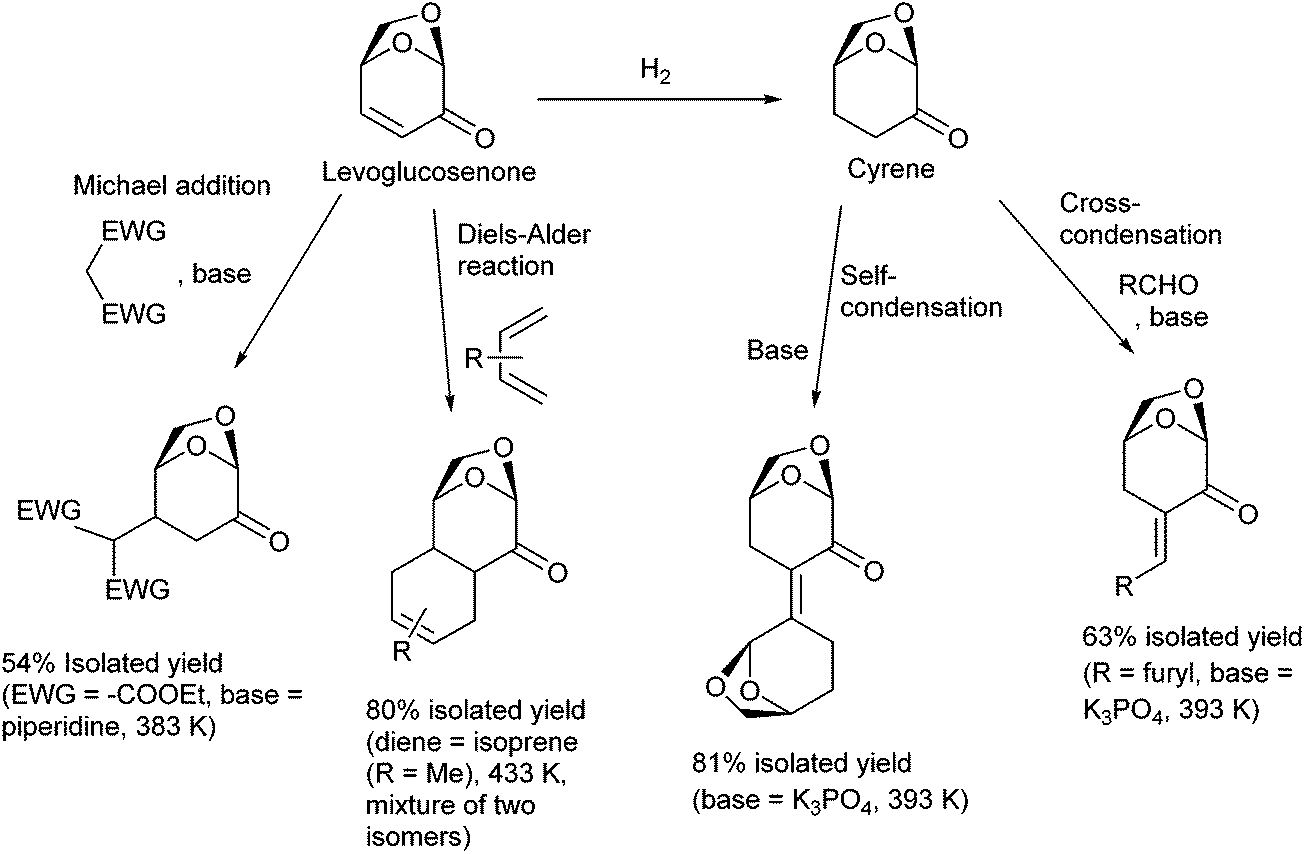 | ||
| Fig. 9 C–C bond formation reactions on levoglucosenone and Cyrene.83,84,86 | ||
As described above, the hydrogenolysis of levoglucosan/levoglucosenone and their derivatives with the same bicyclic structure can convert them into various products; however, most of them can also be synthesized from sugars via other methods starting from sorbitol and HMF. Levoglucosan and levoglucosenone are cellulose-derived molecules, and the amount of hydrogen consumed in the total reaction routes involving levoglucosan or levoglucosenone as an intermediate is the same as that required for other routes to the same target from sugars. The answer to the question of which route is more competitive depends on the yield of each step and the energy consumption, especially during separation. Generally, the levoglucosan/levoglucosenone-based routes have an advantage in view of the separation cost because of the lower polarity than sugars and sugar alcohols and the high thermal stability. Improvement of the product yield is necessary for both steps of levoglucosan/levoglucosene production and their conversion.
3. Alkyl glycosides
3.1. Production of alkyl glycosides
Alkyl glycosides are acetal compounds that are synthesized from sugars and alcohols and have been used as non-ionic, biodegradable, and environmentally benign surfactants due to their hydrophilic and hydrophobic properties resulting from sugar and alkyl moieties, respectively.88,89 The worldwide market for natural surfactants, which include alkyl glycosides, was 18.16 billion USD in 2020 and is projected to grow to 24.78 billion USD in 2028.90 The traditional approach for synthesizing alkyl glycosides is known as Fischer glycosylation (also called Fischer glycosidation, Fig. 10), where a sugar compound is reacted directly with an alcohol (used as both a solvent and reactant).91 Hydrochloric acid is a classical catalyst for Fischer glycosylation,91 and other homogeneous strong acids including trifluoromethanesulfonic acid92 and dodecylbenzenesulfonic acid93 are also effective for this type of glycosylation. Besides, to improve the reusability of catalysts and/or efficiency of the reaction, heterogeneous catalysts such as sulfuric acid immobilized on silica,94 silica-alumina,95 strongly acidic resin,96 and sulfonated mesoporous carbons97 as well as microwave-assisted reaction systems96 have been developed thus far. In all cases, a mixture of α- and β-anomers is obtained, and their molar ratio is dependent on the class of alcohols and heating methods (i.e., typical heating in an oil bath and microwave irradiation). Instead of a leaving group in the Fischer glycosylation (i.e. hydroxide), more reactive leaving groups as exemplified by bromide and chloride (Koenigs–Knorr glycosylation),98 fluoride (Mukaiyama–Suzuki glycosylation),99–101 and trichloroimidate (Schmidt glycosylation)102 have also been used in the field of organic chemistry to synthesize a variety of glycosides with precisely designed molecular structures.103 For these types of glycosylation, heavy metal salts consisting of Ag, Hg, and Sn play key roles as Lewis acidic activators toward such leaving groups.104,105 From the viewpoints of ease of operation and unnecessity of harmful heavy metals, among the glycosylation techniques described above, Fischer glycosylation is a more practically applicable means especially in the large-scale production of alkyl glycosides. The combination with epimerization of naturally abundant sugars should pave the way to using naturally rare or even inaccessible sugar-based alkyl glycosides.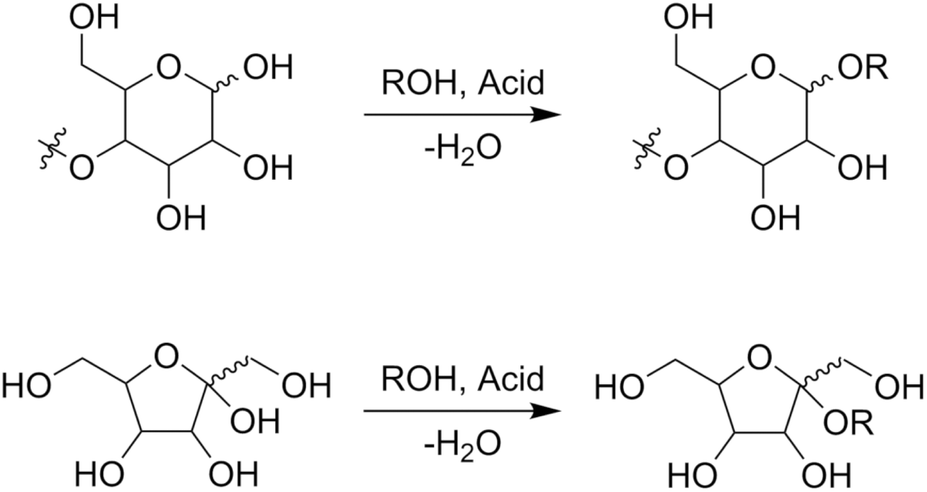 | ||
| Fig. 10 Fischer glycosylation of sugars to alkyl glycosides, as exemplified by (poly)aldohexopyranose (upper) and ketohexofuranose (lower). | ||
Another possible approach for synthesizing alkyl glycosides is the alcoholysis of oligo- and poly-saccharides. In this type of reaction, O-glycosidic bonds originally present in such saccharides are cleaved and alkoxide species then attack oxocarbenium ions, which are intermediate species typically found in a variety of glycosylation reactions,106 to form alkyl glycosides. The most attractive substance for alcoholysis is cellulose, which is a polymer of glucose linked by β-1,4-glycosidic bonds, and this polysaccharide was converted in various alcohols with 1–12 carbon atom(s) into the corresponding alkyl glucosides (Fig. 11). Similar to the hydrolysis of cellulose in water,107–110 alcoholysis is also accelerated in the presence of acid catalysts such as heteropolyacids,111 sulfonated carbon,112 and mesoporous zeolite.113 Likewise, the combination of acid catalysts and ionic liquids, the latter of which are effective for improving the reactivity of cellulose, has achieved outstanding yields of alkyl glucosides;114–116 for example, a hydrophobic and acidic bifunctional ionic liquid produced 93.1% yield of methyl glucosides including both α- and β-anomers from ball-milled cellulose (i.e., amorphous cellulose) at 413 K for 4 h.115 Jérôme et al. reported that after the sulfuric acid-assisted mechanochemical reaction of cellulose induced by ball-milling, which was the same procedure first reported for cellulose hydrolysis,117–119 the resulting cello-oligosaccharides were easily converted in 1-butanol into n-butyl glucosides in 70% yield at a relatively low temperature of 390 K for 3 h.120 Such mild alcoholysis conditions are favorable for the production of desired alkyl glucosides, since under harsh conditions in the presence of acids, alkyl glucosides undergo further transformation into 5-alkoxymethylfurfural and alkyl levulinate.121,122 The direct production of alkyl glycosides via alcoholysis has also been reported for other poly- or oligo-saccharides such as starch,123,124 which consists of glucose linked by α-1,4-glycosidic bonds and is known to undergo depolymerization more easily than cellulose,108 and mannotetraose,125 which is a tetramer of mannose connected by β-1,4-glycosidic bonds and a model compound of naturally occurring β-mannan. For the alcoholysis of starch, sub-critical alcohols, methanol124 and isooctyl alcohol,123 were applied to the synthesis of methyl glucosides and isooctyl glucosides, respectively; these reactions proceeded without any catalysts, because of the high reactivity of both starch and sub-critical alcohols. In the case of mannotetraose, β-mannase from Trichoderma reesei, which is a hydrolase for β-1,4-glycosidic bonds in β-mannan, was employed in 1-hexanol.125 This enzyme cleaved the glycosidic bonds in mannotetraose partially to produce n-hexyl mannobioside and n-hexyl mannotrioside, but the yield of the former product was at most 1.9% (the yield of the latter product was not reported in the cited reference). As described here, the variation of poly- and oligo-saccharides investigated as substrates for alcoholysis has been limited thus far, yet these examples imply the possibility of the alcoholysis of other naturally occurring poly- and oligo-saccharides to produce a variety of alkyl glycosides.
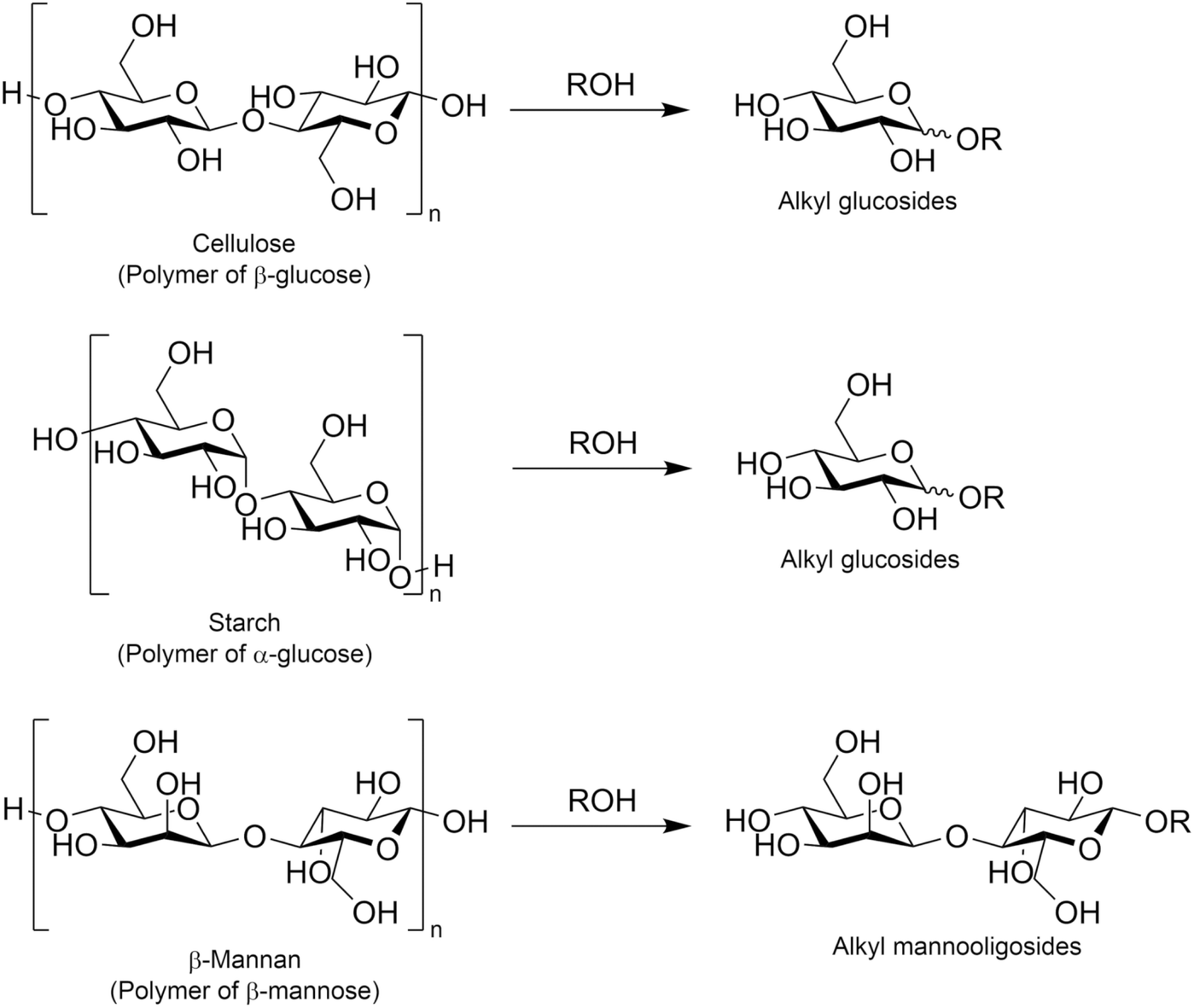 | ||
| Fig. 11 Alcoholysis of polysaccharides to alkyl glycosides. | ||
3.2. Single-hydrodeoxygenation of related substrates
There are a variety of advantages of the hydrodeoxygenation of alkyl glycosides compared to that of monosaccharides. In the case of monosaccharides, the hydrogenation of monosaccharides into the corresponding sugar alcohols readily proceeds; this means that the hydrodeoxygenation of monosaccharides provides very similar results to that of the corresponding sugar alcohols. In addition, monosaccharides have high reactivity due to the presence of formyl or carbonyl groups in their structure. These highly reactive groups are protected almost completely in alkyl glycosides, which leads to the suppression of side reactions and high selectivity to the target product in the case of alkyl glycosides. Another advantage is due to maintaining the ring structure in alkyl glycosides; the well-organized geometry (cis, trans) of OH groups and other substituents in the ring structure is beneficial for selective transformation. In contrast, once the deprotection of ring-structural alkyl glycosides (i.e., hydrolysis of alkoxy group) proceeds to form sugars and/or sugar alcohols, subsequent epimerization, hydrogenation, or dehydrogenation occurs easily over a suitable catalyst.126Monodeoxysugars and dideoxysugars are typical target products for the hydrodeoxygenation of alkyl glycosides. This is because mono- and dideoxy-sugars occupy the core structure of natural products and are important intermediates for the synthesis of rare sugars,127 medicines (e.g., antibiotics and antitumor agents),128,129 and insecticides.130 In addition, the alkyl monodeoxy/dideoxy glycosides can be further converted into the corresponding partially deoxygenated chiral acyclic polyols with high yield and high stereoselectivity; these compounds can be used for the production of fine chemicals (e.g., cosmetics, food additives, and surfactants) and polymers (e.g., alkyd resins, polyurethanes, and polyesters) due to their stereochemistry and multiple functional groups. One problem in the synthesis of monodeoxy- and dideoxy-sugars is the high E-factor of the overall process containing multi-step reactions, a variety of reagents, and protection and deprotection of functional groups.131–133 Therefore, environmentally benign synthesis methods should be developed, and one promising means is the hydrodeoxygenation of alkyl glycosides.
Generally speaking, the reaction temperature for hydrodeoxygenation is comparable to or higher than 373 K, and the stability of alkyl glycosides at this temperature enables their selective transformation into the desired products. Yet, the development of highly active hydrodeoxygenation catalysts that can work at lower reaction temperatures is always desired.
A variety of catalysts have been proposed for the hydrodeoxygenation of polyols and other substrates, as mentioned later, while the application of such catalysts to the hydrodeoxygenation of other substrates is not easy. The Ir–ReOx and Pt–WOx catalysts, which are typical hydrodeoxygenation catalysts that have been developed mainly for glycerol hydrogenolysis, exhibited a high catalytic performance in aqueous solutions.134,135 However, water is not a suitable solvent for the hydrodeoxygenation of alkyl glycosides because the alkoxy group (RO–) in alkyl glycosides is readily hydrolyzed to the hydroxy group (HO–) in water, resulting in the opening of their ring structure to cause hydrogenation of the formyl group. Yet, the electron-withdrawing nature of the −OH group at the 2-position on the ring destabilizes the carbocation transition state and results in the low reactivity of alkyl glycosides toward hydrolysis.136 These could be the reasons for the lack of reports on the hydrodeoxygenation of alkyl glycosides to the corresponding monodeoxysugars using the developed hydrodeoxygenation catalysts and H2 reductant. Therefore, in section S1 (ESI†), we mentioned the reported results on the hydrodeoxygenation of model diols with cyclic structures.
3.3. Double-hydrodeoxygenation of alkyl glycosides with cis-vicinal OH groups
Double-hydrodeoxygenation of alkyl glycosides is easier than single-hydrodeoxygenation when the alkyl glycosides possess cis-vicinal OH groups because there is an effective reaction for the removal of cis-vicinal OH groups, deoxydehydration (DODH). Various catalysts and reductants have been proposed for DODH and there are many good review papers that summarize general DODH reactions.137–143 In this review, we focus on cases using H2 as a reductant. The general scheme of DODH with H2 reductant is shown in Fig. 12, and the reported catalysts for this type of reaction are ReOx–Au/CeO2,144–146 ReOx–Ag/CeO2,147 MoOx–Au/TiO2,148 ReOx/C,149 and CH3ReO3.150 Most of these catalysts are bimetallic. In these bimetallic catalysts, the role of added metal (Au and Ag) is the activation of H2 molecules to reduce DODH-active species such as Re and Mo. In the case of Au- and Ag-added catalysts, DODH products (alkenes) become major products since the consecutive hydrogenation of CC in DODH products does not proceed over Au144–146 or Ag.147 In contrast, ReOx–Pd/CeO2 catalysts, in which Pd species have the same role as Au and Ag to reduce Re, gave the saturated products through the high hydrogenation activity of Pd.151–154 This reaction system is called “DODH + hydrogenation”, and the reaction scheme is shown in Fig. 13. The overall reaction for DODH + hydrogenation is regarded as simultaneous hydrodeoxygenation of vicinal OH groups, which is called double-hydrodeoxygenation in this review.
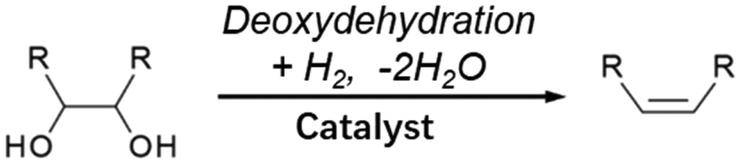 | ||
| Fig. 12 Generalized scheme for DODH with H2 reductant. | ||
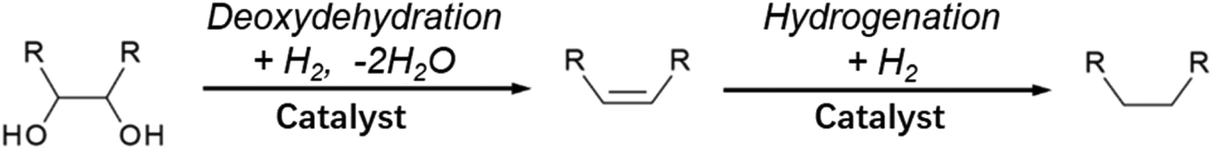 | ||
| Fig. 13 Generalized scheme for DODH + hydrogenation (double-hydrodeoxygenation) with H2 reductant. | ||
It is known that DODH is catalyzed by homogeneous Re and Mo complexes with high valence states, and the reaction proceeds by the redox cycle between the higher valent species (Re7+ and Mo6+) and lower valent ones (Re5+ and Mo4+). An important point is that typical reductants are secondary alcohols and triphenylphosphine, and H2 cannot be used with these homogeneous catalysts. It is characteristic that the reported heterogeneous DODH catalysts such as ReOx–Au/CeO2, ReOx–Ag/CeO2, MoOx–Au/TiO2, and ReOx–Pd/CeO2 enable the use of H2 reductant. Based on characterization of the catalyst and the dependence of the Re loading amount on the catalytic performance, it has been proposed that monomeric Re species on CeO2 were the active sites for DODH. The catalytic cycles for DODH on heterogeneous Re species on CeO2 and homogeneous Re species are illustrated in Fig. 14. All the steps in Fig. 14 do not require acids, and therefore, the use of strong acids, which may lead to side reactions, can be avoided in DODH (+hydrogenation). DODH systems do not need water as a solvent (rather, water generally retards DODH reactions significantly), and many organic solvents such as hydrocarbons, ethers, and alcohols can be used, enabling the conversion of a wide range of substrates.
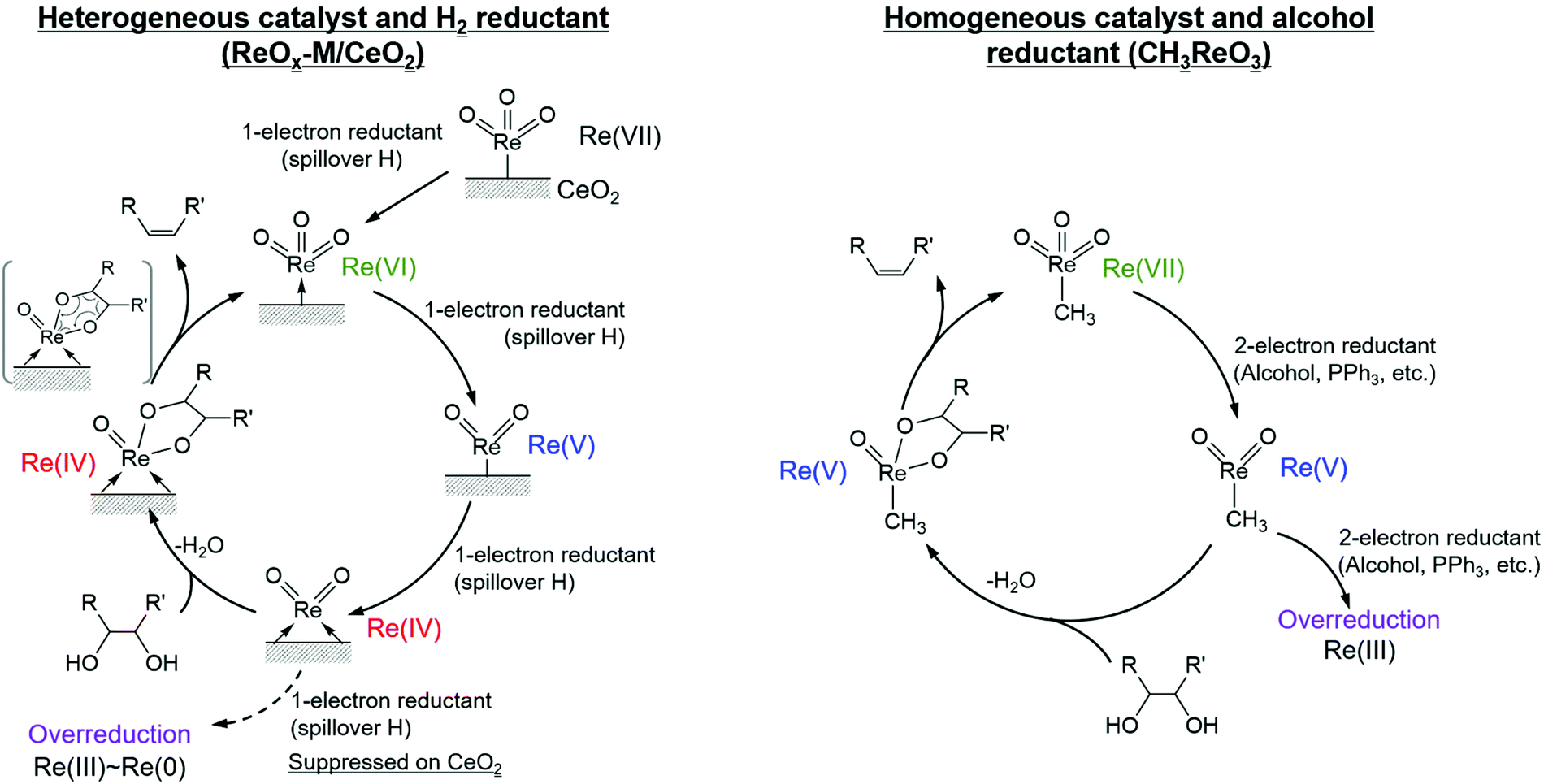 | ||
| Fig. 14 Catalytic cycles for DODH on heterogeneous Re on CeO2 and homogeneous Re species. Reproduced from ref. 155 with permission from Elsevier, copyright 2020. | ||
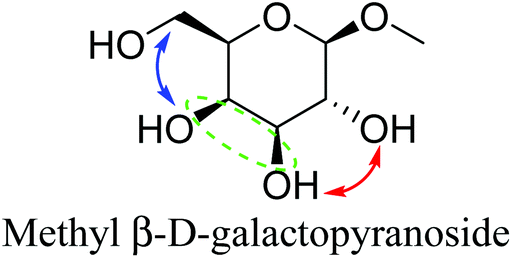
Another important feature of the DODH of cyclic vicinal diols is the much higher reactivity of cis-vicinal diols than that of trans-vicinal diols. The reactivity tendency of cis- and trans-vicinal diols can be explained by the stability of the DODH products, as illustrated in Fig. 15, where the DODH of 1,2-cyclohexanediols is used as a model reaction. The reactivity difference between cis- and trans-vicinal OH groups in DODH is very useful for the selective transformation of methyl glycosides. Alkyl glucoside, whose availability is the highest among alkyl glycosides, contains only trans-vicinal OH groups, and unfortunately, it has very low reactivity in DODH. In contrast, the DODH of alkyl glycosides with cis-vicinal OH groups is possible. In particular, the combination of ReOx–M/CeO2 (M = Pd, Ag, and Au) catalysts with H2 reductant enhances the DODH activity and decreases the reaction temperature, which allows the application of ReOx–M/CeO2 (M = Pd,156,157 Ag,147 and Au146) to the DODH of methyl glycosides with cis-vicinal OH groups.
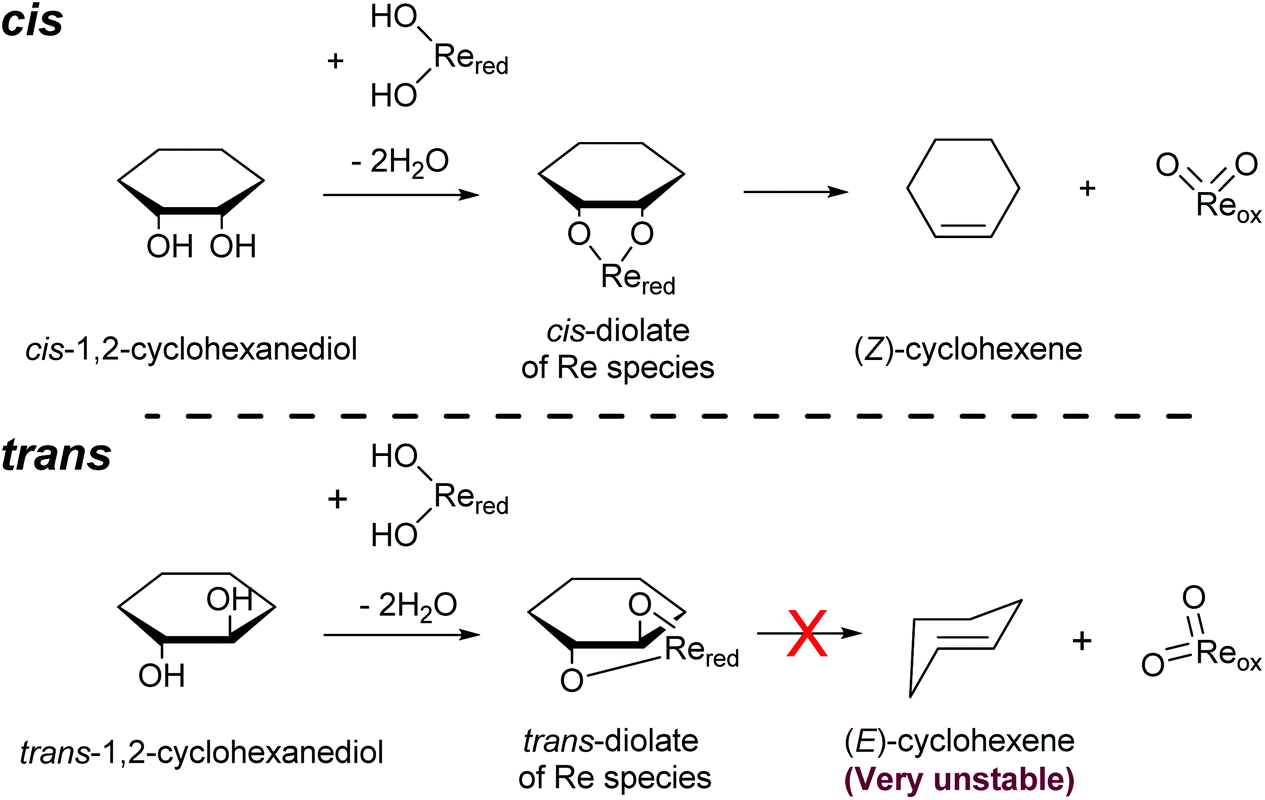 | ||
| Fig. 15 DODH of cis- and trans-1,2-cyclohexanediols. | ||
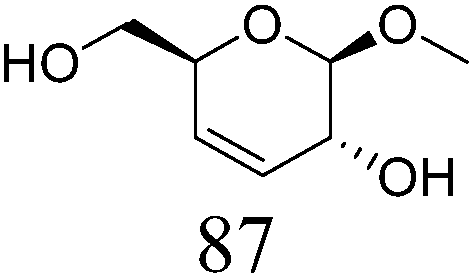
Table 2 lists the results for the DODH (+hydrogenation) of several methyl glycosides with cis-vicinal OH groups using ReOx–M/CeO2 (M = Pd, Au, and Ag) catalysts and H2 reductant. It is demonstrated that the corresponding saturated and unsaturated dideoxy-products are synthesized in high yields in one step using suitable catalysts and H2 reductant. Generally speaking, the protection and deprotection of OH groups are required for the transformation of specific OH groups of sugars. In contrast, the present method relying on DODH does not need the protection or deprotection of OH groups. The highly selective conversion of methyl glycosides with cis-vicinal OH groups in Table 2 is due to the selective recognition of cis-vicinal OH groups as illustrated in Fig. 16.
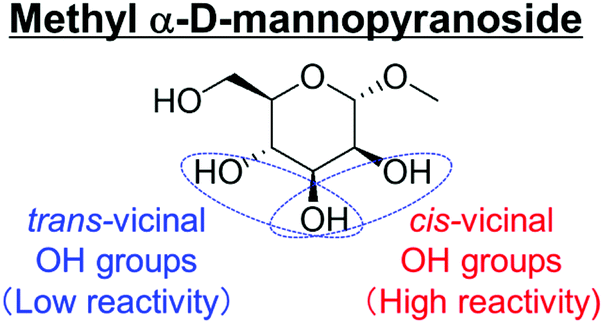 | ||
| Fig. 16 Reactive cis- and unreactive trans-vicinal OH groups of methyl α-D-mannopyranoside in DODH. | ||
| Entry | Substrate | Catalyst [amount per g] | t [h] | Conv. [%] | Main product yield [%] | |
|---|---|---|---|---|---|---|
| DODH product | Dideoxy product | |||||
| Reaction conditions: methyl glycoside (1.29 mmol), 1,4-dioxane (10 g), 413 K.a ReOx–dpAu0.3/CeO2 (1 wt% Re, Au/Re = 0.3, prepared via deposition precipitation method), H2 (1.7 MPa) (at 413 K).b ReOx–Pd/CeO2 (2 wt% Re, Pd/Re = 0.25), H2 (8.0 MPa) (at 413 K).c ReOx–Ag/CeO2 (1 wt% Re, Ag/Re = 0.3). | ||||||
| 1a |
|
ReOx–Au/CeO2 (0.50) | 8 | 99 |
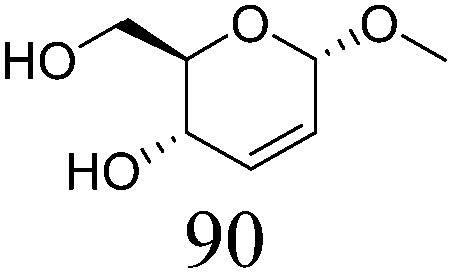
|
|
| 2b | Methyl α-D-mannopyranoside | ReOx–Pd/CeO2 (0.15) | 51 | 99 |
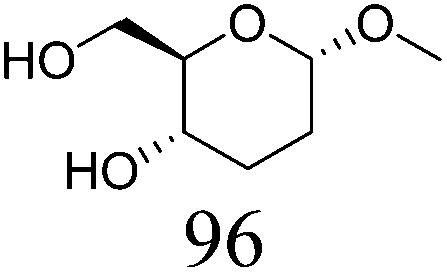
|
|
| 3a |
|
ReOx–Au/CeO2 (0.50) | 48 | >99 |
|
|
| 4b | Methyl β-D-galactopyranoside | ReOx–Pd/CeO2 (0.30) | 72 | 94 |
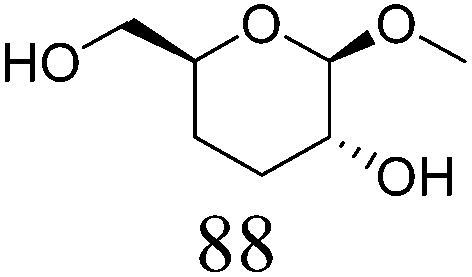
|
|
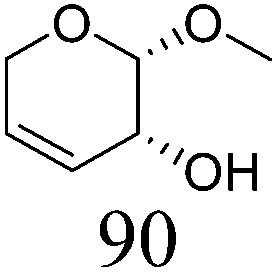
| 5a |
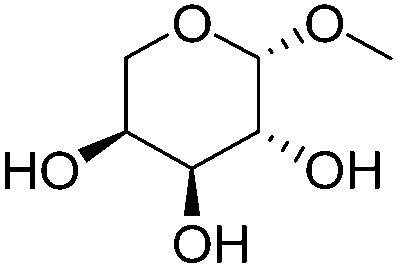
|
ReOx–Au/CeO2 (0.50) | 8 | 97 |
|
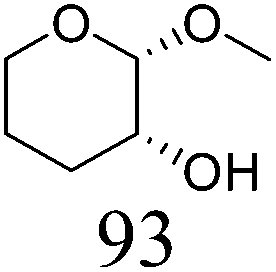
|
| 6b | Methyl β-L-arabinopyranoside | ReOx–Pd/CeO2 (0.15) | 72 | >99 |
|
|
| 7a |
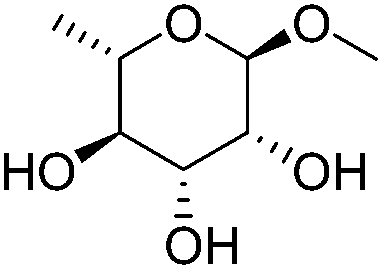
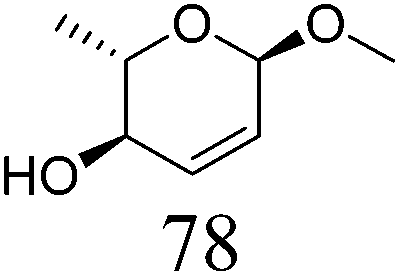
|
ReOx–Au/CeO2 (0.50) | 16 | 93 |
|
|
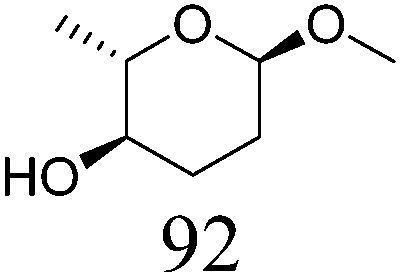
| 8b | Methyl α-L-rhamnopyranoside | ReOx–Pd/CeO2 (0.15) | 36 | >99 |
|
|
| 9c |
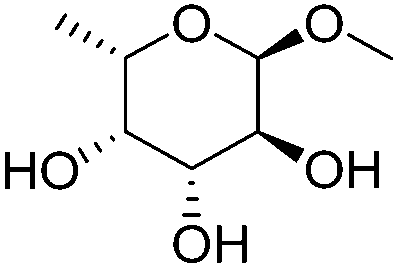
|
ReOx–Ag/CeO2 (1.00) | 20 | 94 |
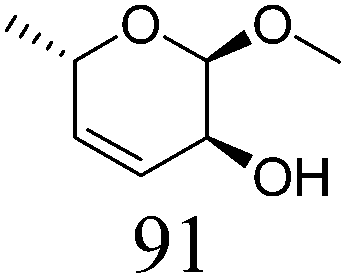
|
|
| 10b | Methyl α-L-fucopyranoside | ReOx–Pd/CeO2 (0.45) | 95 | 97 |

|
|
| 11c |
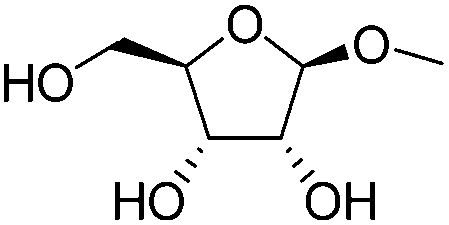
|
ReOx–Ag/CeO2 (0.25) | 10 | 99 |

|
|
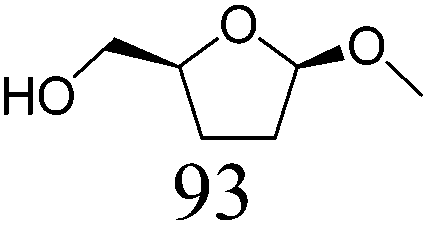
| 12b | Methyl β-D-ribofuranoside | ReOx–Pd/CeO2 (0.10) | 24 | >99 |
|
|
In addition, the obtained saturated dideoxy methyl glycosides can be further converted into valuable chiral acyclic polyols via hydrolysis of the methoxy moiety and hydrogenation steps (Fig. 17). (2S)-1,2,5-Pentanetriol was obtained from methyl β-D-ribofuranoside in a yield of 97% with 95% ee through a one-pot two-step reaction over ReOx–Pd/CeO2 as the common catalyst for both steps.156 Meanwhile, the DODH by the same ReOx–Pd/CeO2 catalyst and subsequent transformation over Rh–ReOx/SiO2 produced (2R)-1,2,5-pentanetriol from methyl β-L-arabinopyranoside in a yield of 95% with 95% ee. Moreover, the hexane-tetraols and -triols with fixed OH group positions and stereochemistry could be obtained over Pt-based catalysts (Pt/SiO2 or Pt/SiO2–Al2O3) in 77–95% yield with >92% diastereoselectivity from the synthesized saturated dideoxy methyl glycosides.136
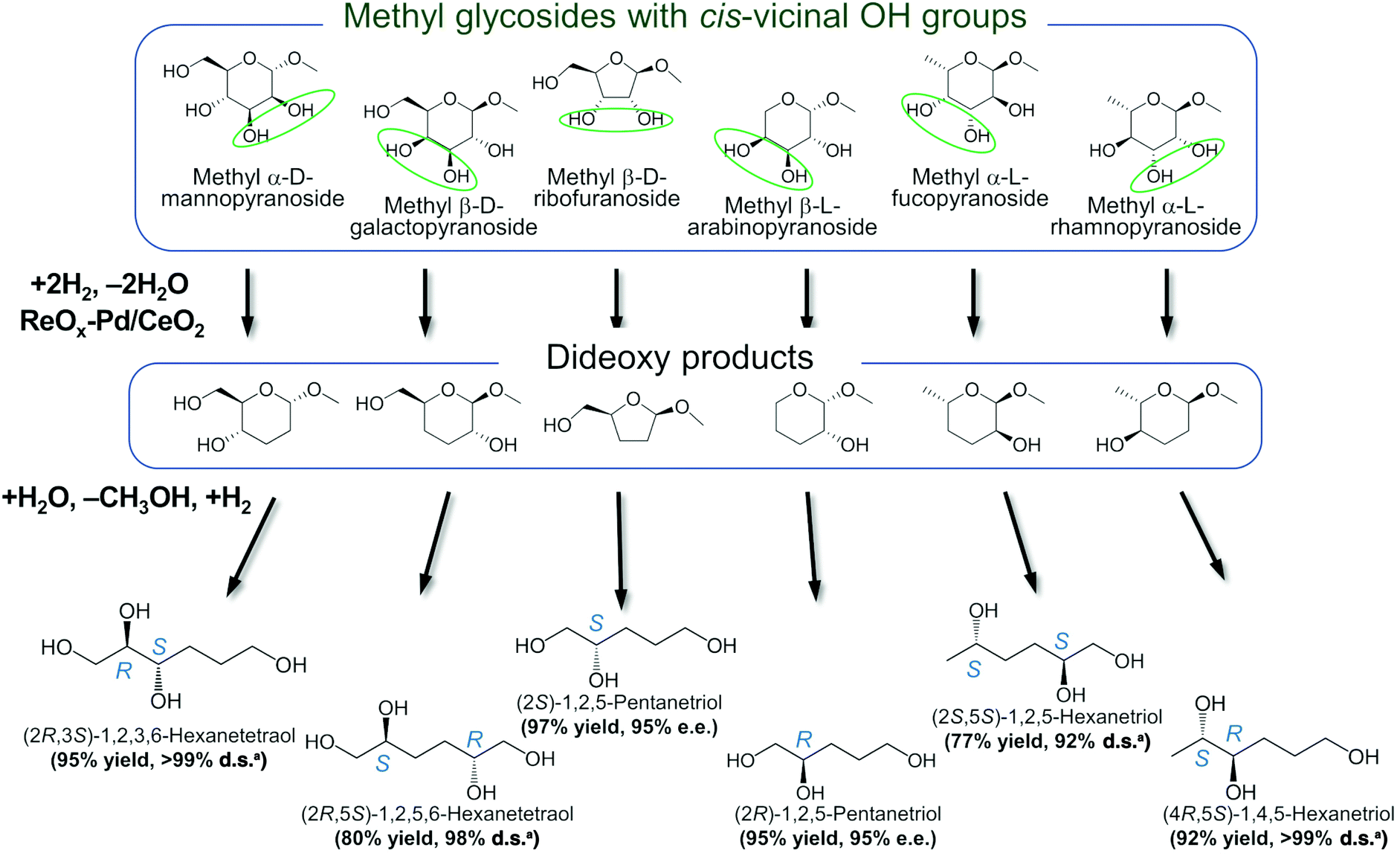 | ||
Fig. 17 Conversion of methyl glycosides into chiral pentanetriols, hexanetriols, and hexanetetraols.136,156a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Diastereoselectivity measured by 13C NMR. Diastereoselectivity measured by 13C NMR. | ||
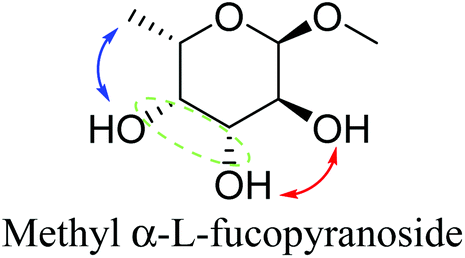
Based on data for catalyst amounts and reaction times in Table 2, it is expected that the reactivity would be different for different substrates. Therefore, the reactivity of the substrates was compared on the basis of the results of reaction rate measurements for the DODH + hydrogenation of 6-membered ring methyl glycosides with cis-vicinal OH groups on ReOx–Pd/CeO2 as listed in Table 3. The obtained reaction orders with respect to substrate concentration and H2 pressure are also listed in Table 3. Both reaction orders with respect to the substrate concentration and H2 pressure in the DODH + hydrogenation on ReOx–Pd/CeO2 were almost zero in all cases, suggesting that the reaction mechanism for the DODH + hydrogenation was the same. The DODH of the diolate formed from the substrate and ReOx species was the rate-determining step and the coverage of the catalytic active sites by diol molecules was close to the saturation level. Therefore, the reactivity difference cannot be explained by the difference in the reaction mechanism and/or the rate-determining step. More reactive methyl glycosides commonly have substituents juxtaposed to cis-vicinal OH groups on the opposite side, and less reactive methyl glycosides have substituents neighboring cis-vicinal OH groups on the same side. A detailed comparison was carried out by using more reactive methyl α-L-rhamnopyranoside and less reactive methyl α-L-fucopyranoside. The apparent activation energies of the reaction of methyl α-L-rhamnopyranoside and methyl α-L-fucopyranoside were found to be 63 and 73 kJ mol−1, respectively. The adsorption and transition states of these two methyl glycosides were also analyzed by density functional theory (DFT) calculations, and the results are shown in Fig. 18. The energy difference between these two substrates at their transition states was about 10 kJ mol−1, which agreed well with the difference in the experimentally obtained activation energies. In contrast, the energy difference at the adsorbed states was much smaller. The energy difference at the transition states can be explained by steric hindrance, as illustrated in Fig. 19. The angles between the substituent neighboring cis-vicinal OH groups and C–OH in the cis-vicinal OH groups in the Re diolate from methyl α-L-rhamnopyranoside at the transition state were calculated to be 72.4° and 155.8°, while the angles in the case of Re diolate from methyl α-L-fucopyranoside were 37.6° and 73.7°. It is clear that the angles are wider for methyl α-L-rhamnopyranoside than methyl α-L-fucopyranoside, meaning that less steric hindrance gives a lower energy at the transition state.
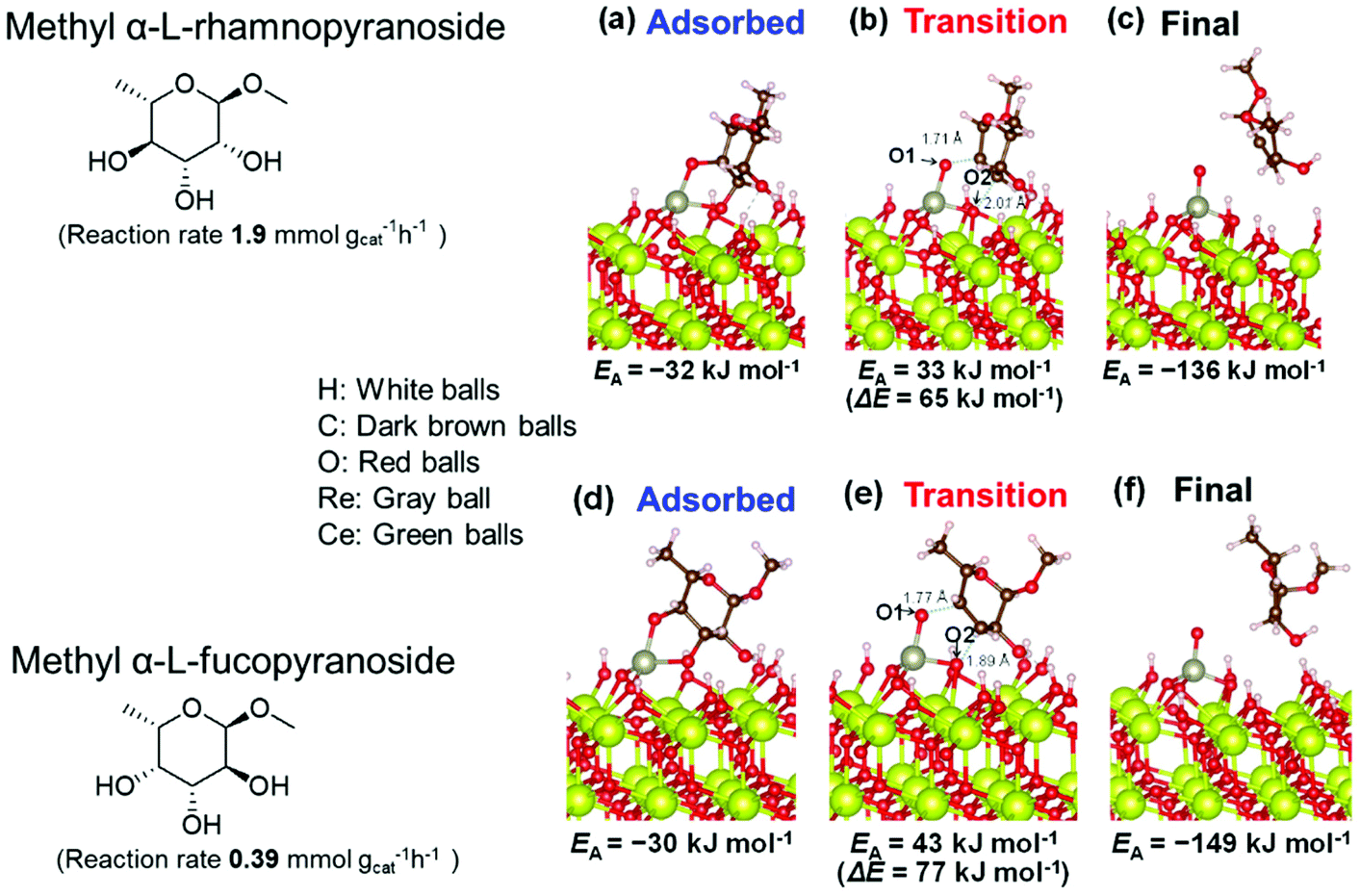 | ||
| Fig. 18 DFT calculations of the adsorption and transition states of the DODH of methyl α-L-rhamnopyranoside and methyl α-L-fucopyranoside on ReOx/CeO2(111). Reproduced from ref. 157 with permission from the American Chemical Society, copyright 2020. | ||
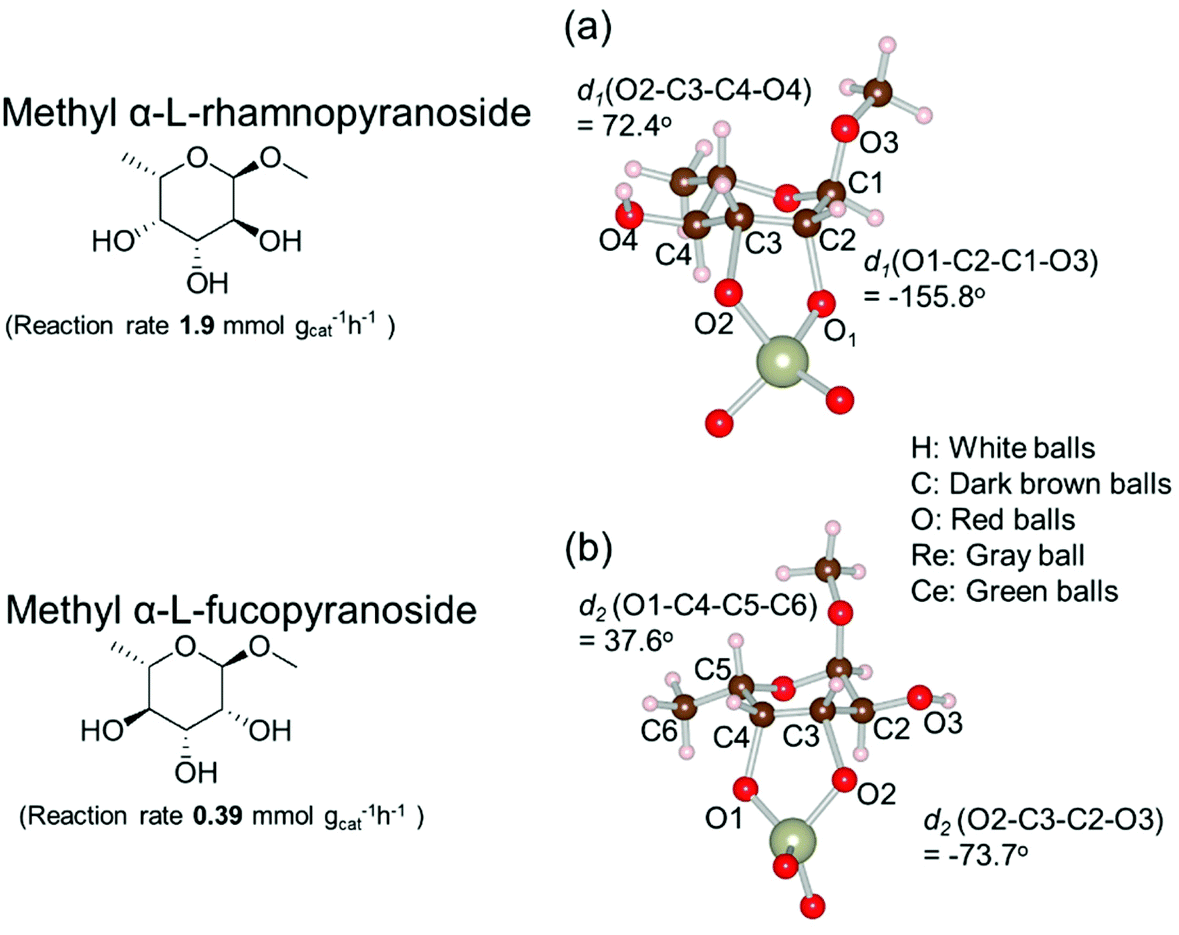 | ||
| Fig. 19 Structure of the transition states of diolates from methyl α-L-rhamnopyranoside and methyl α-L-fucopyranoside on ReOx/CeO2(111). Reproduced from ref. 157 with permission from the American Chemical Society, copyright 2020. | ||
146
| Entry | Substrate | Reaction rate [mmol gcat−1 h−1] | Reaction order | |
|---|---|---|---|---|
| Concentration | H2 pressure | |||
| Reaction conditions: ReOx–Pd/CeO2 catalyst (0.15 g) (Re = 2 wt%, Pd/Re = 0.25), substrate (1.3 mmol), 1,4-dioxane (10 g), 7.7 MPa H2 at 413 K (initial 1.0 MPa H2 at r.t.), 413 K. Red arrows: opposite direction between the functional group and its adjacent cis-vicinal OH group, blue arrows: same direction between the functional group and its adjacent cis-vicinal OH groups. | ||||
| 1 |
|
1.9 | 0.1 | 0.2 |
| 2 |
|
1.7 | –0.1 | 0.3 |
| 3 |

|
1.7 | 0.0 | 0.2 |
| 4 |
|
0.73 | –0.1 | 0.2 |
| 5 |
|
0.38 | 0.0 | 0.3 |
Compared to conventional organochemical approaches described roughly in section 3.2, one-pot DODH + hydrogenation using heterogeneous catalysts is a more useful and environmentally benign means of synthesizing dideoxysugars from alkyl glycosides. The use of hydrogen as a reducing agent in the synthesis of dideoxysugars is favorable in terms of the reagent cost and atom efficiency, which are especially important in the chemical industry operating on a large scale, while the use of high-pressure H2 is a barrier to laboratory-scale syntheses. Considering that dideoxysugars are fine chemicals, expanding the scope of the reducing agents may have merit. The use of homogeneous catalysts may also be acceptable in the synthesis of fine chemicals, although the turnover number (TON) of typical homogeneous DODH catalyst systems is not large (<102) especially when using an inexpensive reducing agent such as simple secondary alcohols and H2.138 Meanwhile, the substrate scope is an apparent issue: it is limited to sugars (and sugar derivatives) that possess cis-vicinal diols in their structure. This fact means that the most common naturally occurring monosaccharide, glucose, cannot be transformed into dideoxysugars due to the lack of a cis-vicinal diol structure. In this respect, different types of reaction that enable epimerization of glucose to access rare sugars containing a cis-vicinal diol are necessary. Mannose has been reported to be synthesized from glucose directly via epimerization by Mo-based catalysts158,159 and Sn-beta zeolites,160,161 while other rare sugars are difficult to produce from glucose except for biochemical methods using epimerase. In order to make DODH + hydrogenation more useful and attractive, highly efficient chemical techniques, which have been very limited thus far,162,163 need to be devised.
4. Glucaric acid and mucic acid
Glucaric acid is the most popular aldaric acid (aldose-derived dicarboxylic acid), an oxidized derivative of glucose. Glucaric acid has been industrially manufactured on the order of 104 t per year and used for a number of purposes such as a food additive and a corrosion inhibitor.164 Glucaric acid was selected for the “Top 12” in the DOE report in 2004 (see Table 1).4 The expected utilization of glucaric acid is as a monomer of polyamides and polyesters. However, because of “limited research activity” in the utilization of glucaric acid, it was not selected for the revised 2010 list.5Aldaric acids including glucaric acid are generally synthesized by nitric acid oxidation of the corresponding sugars. Modest yields (40–45%) of glucaric acid are obtained.165,166 Catalytic oxidation with molecular oxygen is preferable to traditional nitric acid oxidation. An excess amount of base is frequently added to the oxidation systems for sugars and sugar alcohols;167 however, neutralization is necessary to obtain the free acid, leaving a large amount of salt. Therefore, base-free systems are preferable. Up to about 70% yield of glucaric acid through the base-free catalytic oxidation of glucose with O2 was reported in the literature.168–170 It is generally energy-consuming to separate sugar acids from an aqueous reaction mixture containing various compounds with high boiling points. Biocatalytic, electrocatalytic and photocatalytic oxidation systems have also been investigated;166,171 however, these systems tend to be energy consuming. Energy consumption should be carefully assessed for the practical application of any systems.
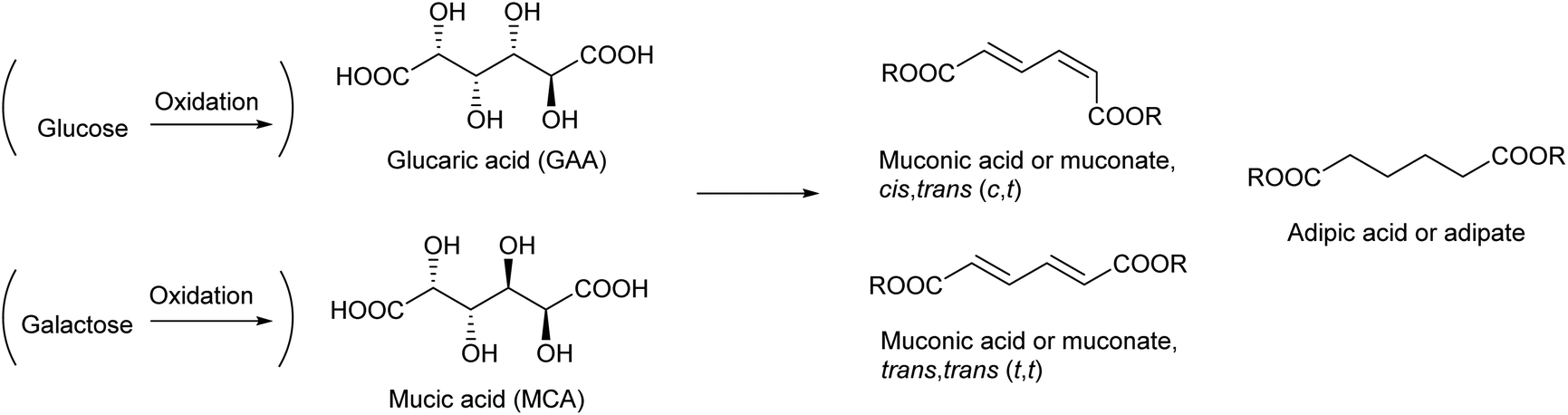
The recent development of the deoxydehydration (DODH) reaction has again shed light on the use of glucaric acid as a source of chemicals. DODH of glucaric acid produces cis,trans-muconic acid (2,4-hexadienedioic acid). The most stable isomer of muconic acid (trans,trans) is also produced.172 The hydrogenation of muconic acids produces adipic acid, which is a very important monomer for a polyamide, 6,6-nylon, in industry.173 Currently, adipic acid is manufactured by the hydrogenation of petroleum-derived benzene and subsequent stepwise oxidations. The final step of adipic acid manufacture uses nitric acid. The manufacture of adipic acid has low atom efficiency in the overall process, and an efficient synthesis method for adipic acid from renewable resources is highly desirable.
Reports on the DODH of glucaric acid and its related reactions are summarized in Table 4. Examples of the DODH of glucaric acid are rather limited, and the DODH of mucic acid (galactaric acid), which is the galactose-derived aldaric acid, is more frequently carried out. The product of the DODH of mucic acid is trans,trans-muconic acid, which is the most stable isomer. Mucic acid is more reactive in DODH than glucaric acid172,174 probably because of the higher stability of the DODH product. Typical systems used a homogeneous Re catalyst and an alcohol as both a reductant and solvent.172,175–182 Because of the excess use of alcohol, the product is usually obtained in the form of an ester. A one-pot system using the combination of a DODH catalyst (Re) + alcohol and hydrogenation catalyst + H2 to directly obtain adipic acid from aldaric acids was also reported.174,183,184 Bimetallic solid catalysts containing both Re and a noble metal for hydrogenation have been reported recently.185,186 Considering that adipic acid is a bulk chemical, it is also better to use H2 as a reducing agent in the DODH step. However, the use of H2 as a reducing agent for the DODH of aldaric acids is difficult; the ReOx–M/CeO2 system mentioned in the previous section does not work probably because of the strong adsorption of the carboxylate group on the basic CeO2 surface. Toste et al. reported the production of adipic acid from glucarodilactone, where the OH groups at the 3- and 4-positions in glucaric acid are condensed with the carboxylic groups at the 6- and 1-positions, respectively, by DODH + hydrogenation with H2 as a reducing agent.183 This system used water as a solvent, which generally retards DODH reactions significantly, although a large amount of catalyst and additives such as active carbon were necessary. The TONs of all the reported systems for the DODH of aldaric acid or its ester were still low. As another approach, the hydrodeoxygenation of glucaric acid using a large amount of halogen ions, where nucleophilic substitution of an alcohol with a halogen ion and subsequent hydrogenolysis of an alkyl halide proceed, has been reported.187 The use of a large amount of corrosive acid is a problem for this halogen-mediated hydrodeoxygenation system. Grilc et al. reported the hydrodeoxygenation of mucic acid by a commercial sulfided NiMo/Al2O3 catalyst, and a small amount of adipic acid was obtained.188
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Substrate | Catalyst | Reducing agent | Temp. [K] | Product (yield [%]) | TONa | Average TOFa [h−1] | Ref. |
| TsOH = trifluoromethanesulfonic acid; DMAP = 4-dimethylaminopyridine; [BMIM]OTf = 1-butyl-3-methylimidazolium trifluoromethanesulfonate; Cptt = 1,3-di-tert-butylcyclopentadienyl.a Based on Re amount. One turnover means two DODH reactions (e.g., mucic acid to muconate). | |||||||
| GAA | HReO4 | 1-Butanol | 433 | Muconate, c,t:t,t = 18:7 (25) |
3.1 | 0.21 | 172 |
| MCA | HReO4 | 1-Butanol | 433 | Muconate, t,t (62) | 8.9 | 0.59 | 172 |
| MCA | CH3ReO3 | 3-Pentanol | 428 | Muconate, t,t (57) | 11 | 0.76 | 172 |
| MCA | CH3ReO3 | 3-Pentanol | 393 | Muconate, t,t (99) | 20 | 0.83 | 175 |
| MCA | CH3ReO3 + TsOH + Pt/C | 3-Pentanol | 473 | Adipate (75) | 15 | 0.31 | 175 |
| MCA | NH4ReO4 | Indoline + 1-butanol | 423 | Muconate, t,t (57) | 5.7 | 0.24 | 176 |
| MCA | NH4ReO4 | 3-Pentanol | 393 | Mucic acid, t,t (82) + muconate, t,t (16) | 20 | 0.82 | 177 |
| Glucarate-6,3-lactone | KReO4 + Pd/C + H3PO4 + C | Methanol + H2 (0.5 MPa) | 423 | Adipate (88) | 88 | 4.9 | 183 |
| Glucarodilactone | KReO4 + Pd/C + C + DMAP | H2 (0.5 MPa) | 423 | Adipic acid (72) | 7.2 | 0.5 | 183 |
| Dibutyl mucate | Re2O7 + TsOH + [BMIM]OTf | 1-Butanol | Reflux | Muconate, t,t (>99) | 20 | 1.7 | 178 |
| MCA | CpttReO3 | 3-Pentanol | 393 | Muconate, t,t (75) | 15 | 1.3 | 179 |
| MCA | CH3ReO3 | Indoline + 1-butanol | 463 | Muconate, t,t (36) | 3.6 | 0.075 | 180 |
| MCA-1,4-lactone | ReOx/ZrO2, Pd/C | 1-Butanol + H2 (0.1 MPa) | 393, 393 | Adipate (82) after two DODH + hydrogenation cycles | 8.3 | 0.35 | 184 |
| MCA | L(NNO)Re(CO)3 | 3-Octanol | 453 | Muconate, t,t (46) | 23 | 3.8 | 181 |
| MCA | [L(py4)ReO2]+ | 3-Pentanol | 453 | Muconate, t,t (65) | 33 | 11 | 182 |
| MCA | Pt-ReOx/C | 2-Propanol | 443 | Adipate (85) | 24 | 0.98 | 185 |
| GAA | Re/C + Pd/C | Methanol + H2 (0.5 MPa) | 393 | Adipate (25) | 2.1 | 0.030 | 174 |
| MCA | Re/C + Pd/C | Methanol + H2 (0.5 MPa) | 393 | Adipate (58) | 6.8 | 0.094 | 174 |
| GAA | Pd–ReOx/C + Amberlyst | Methanol + H2 (2 MPa) | 383 | Adipate (95) | 18 | 0.74 | 186 |
| MCA | Pd–ReOx/C + Amberlyst | Methanol + H2 (2 MPa) | 383 | Adipate (95) | 18 | 0.74 | 186 |
| MCA | NiMoS/Al2O3 | H2 (5 MPa) | 498 | Adipic acid (4.3) | — | — | 188 |
| GAA | Rh/C + HI | H2 (2.8 MPa) | 403 | 3-Iodoadipic acid (67) + adipic acid (9) | — | — | 187 |
There are many issues for the industrialization of adipic acid production by DODH + hydrogenation of aldaric acids, considering that adipic acid is a commodity chemical with a low price, in contrast to the deoxysugars discussed in the previous section. The catalyst life (i.e., TON) should be much improved. The cost of Re catalysts is very high, and their replacement with a less expensive active metal such as Mo is desirable. The use of H2 as a reducing agent is essential. Tolerance to water content in the reaction media may be necessary. The substrate should be glucose-derived (glucaric acid), or at least the system should accept a mixture of glucaric acid and mucic acid as substrates because the main natural source of galactose is lactose, which is composed of galactose and glucose units.
Because of the importance of adipic acid, there are several other approaches to synthesizing adipic acid from renewable resources: direct production by fermentation,189 fermentative muconic acid production and subsequent hydrogenation,177,190 hydrodeoxygenation of 2,5-furandicarboxylic acid,15,191,192 carbonylation of C5 platform chemicals,193 and oxidative cleavage of lignin-derived di-functionalized cyclohexanes.194 The feasibility of each method depends on various factors: in addition to the yield and separation cost of each conversion step, the amount of H2 and other reagents (CO in carbonylation) and availability of substrates (intermediates from raw biomass) affect the feasibility. The availability of the substrate in the future will depend on the utilization scope of the same compound for other purposes; for example, future wide use of polyethylene furanoate, which is a co-polymer of 2,5-furandicarboxylic acid and ethylene glycol, will enlarge the supply of 2,5-furandicarboxylic acid and encourage the production of adipic acid from 2,5-furandicarboxylic acid. Similarly, once the polyester or polyamide of glucaric acid is commercialized, the use of glucaric acid as the source of adipic acid will be spotlighted.
5. Erythritol and 1,4-anhydroerythritol (1,4-AHERY)
Erythritol is already commercially manufactured by fermentation as a sweetener. The fermentation process is not so difficult because of various factors: (i) erythrose-4-phosphate, which is a precursor of erythritol, is involved in the pentose phosphate pathway of metabolism and is a common compound in nature; (ii) yeasts can withstand high concentrations of erythritol; and (iii) erythritol has good crystallinity.33 Furthermore, the recent development of the fermentative production of erythritol showed that glycerol can also be used as the feed.31,32,195 It is known that glycerol is the main byproduct in the large-scale production of biodiesel fuel through the transesterification of vegetable oils with methanol or ethanol, and glycerol is a waste product that is not fully utilized. Crude glycerol obtained from biodiesel fuel manufacturing is normally strongly alkaline and contains various minor impurities derived from the mother oil. Generally speaking, the purification of glycerol is energy consuming; this can be connected to the high price of glycerol and its chemically transformed products as chemicals. On the other hand, the removal of impurities from crude glycerol may not be necessary for the fermentative production of erythritol since it has been found that Moniliella megachiliensis SN-G42 assimilates nonrefined glycerol derived from palm oil and converts it efficiently into erythritol.196 For example, the carbon-based yield of erythritol was approximately 60% in a 500 mL flask batch culture after 3 days; this yield was slightly higher than that obtained with glucose. Further improvement in cell growth and erythritol production might be possible. In addition, a synthetic enzyme cascade was developed for the production of the functional C4 sugar erythrulose by utilizing formaldehyde as a C1 carbon source, where formaldehyde could be sustainably derived from other C1 feedstocks.197 The conversion of erythrulose into the corresponding sugar alcohols is easy like in the cases of xylose to xylitol and glucose to sorbitol. Based on this and related reports, we believe that erythritol will be able to be regarded as a platform chemical in the biomass refinery.33 Although erythritol itself can be used as a raw material for the production of polymers and chemicals,198–200 examples are very limited.The dehydration of erythritol gives 1,4-anhydroerythritol (1,4-AHERY), which is catalyzed by a strong Brønsted acid such as mineral acids and ion-exchange resin.201,202 A reactive distillation system for collecting the 1,4-AHERY product in high yield (∼90%) has been reported.201 The dehydration of erythritol proceeded effectively in carbonated water to give >70% yield of 1,4-AHERY at 573 K, and this could be catalyzed by protons generated from carbonic acid in the presence of high-pressure CO2.203 The relatively easy conversion of erythritol into 1,4-AHERY is thought to enable the utilization of 1,4-AHERY as a biomass-derived platform chemical. The hydrodeoxygenation of erythritol and 1,4-AHERY gives a variety of chemicals as shown in Fig. 20 and 21, where the reducing agent is hydrogen. Butanediols are important targets for erythritol hydrodeoxygenation (Fig. 20) because they have higher demands than butanetriols and also a lower amount of H2 is required for their production compared to butanols. However, the separation of one specific butanediol from a mixture of butanediols is difficult, and high selectivity is especially necessary from a practical viewpoint.
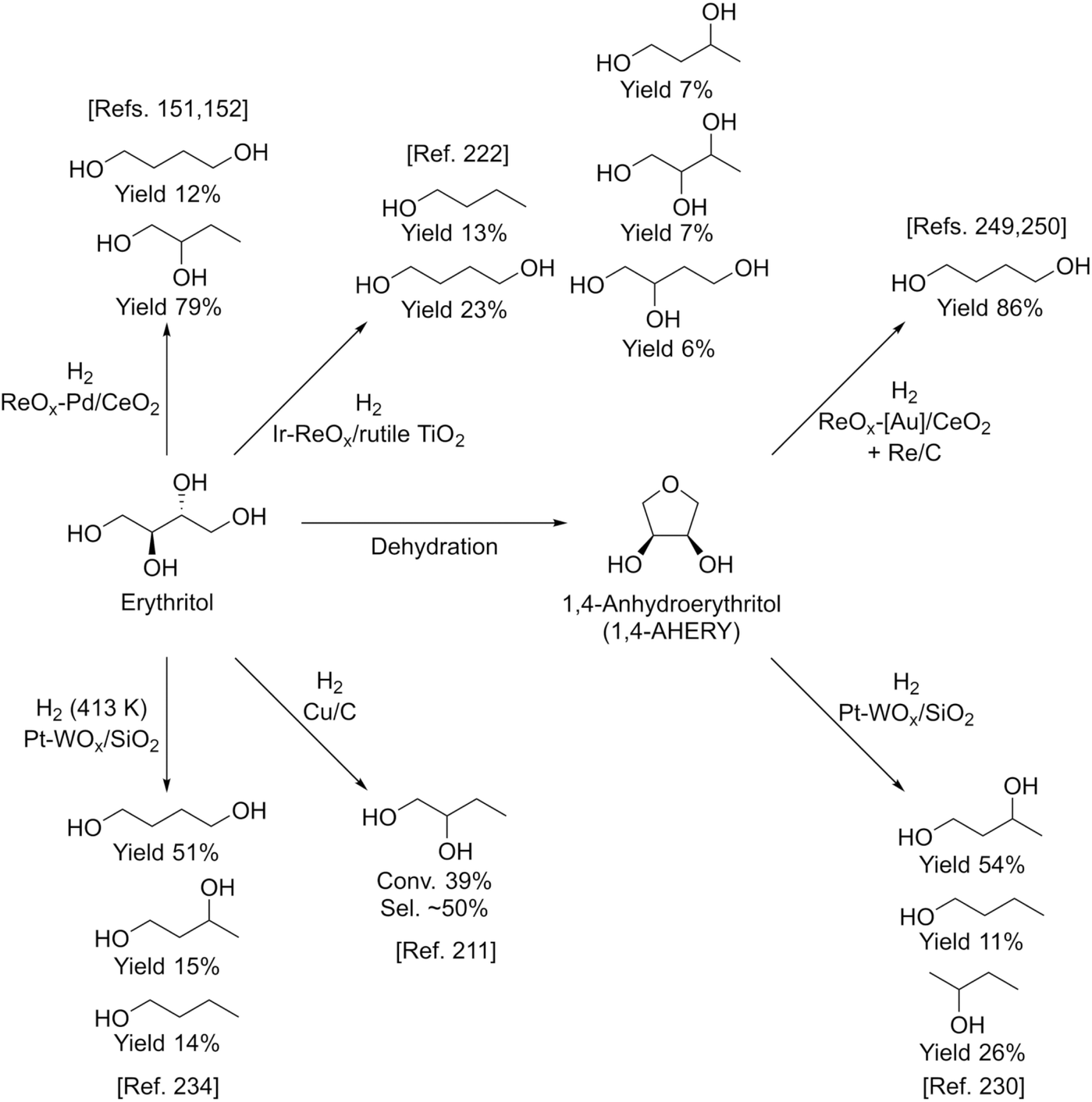 | ||
| Fig. 20 Transformation of erythritol and 1,4-AHERY into C4 diols with H2. | ||
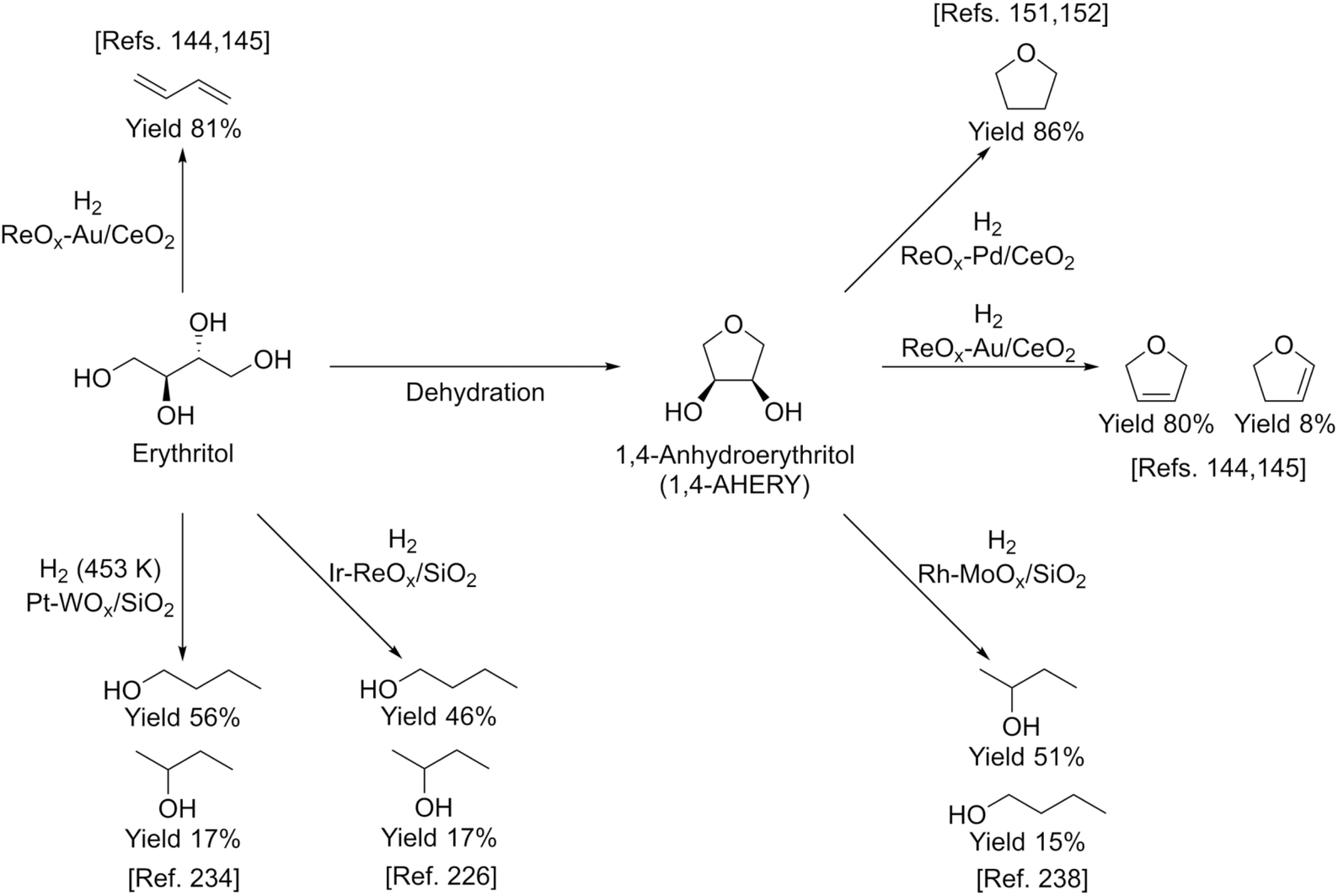 | ||
| Fig. 21 Transformation of erythritol and 1,4-AHERY into C4 monoalcohols and other products with H2. | ||
5.1. Hydrogenolysis of erythritol as a variant of glycerol
The hydrogenolysis of C–O bonds in erythritol potentially produces a variety of products; for example, two triols (1,2,3- and 1,2,4-), four diols (1,2-, 1,3-, 1,4-, and 2,3-), and two mono-alcohols are possibly produced from erythritol, even though side reactions such as C–C dissociation and cyclization are excluded. This means that the control of regio-selectivity is essential. On the other hand, the hydrogenolysis of C–O bonds in glycerol has been intensively investigated because of the large supply of glycerol as a byproduct in the biodiesel industry. Glycerol hydrogenolysis can be used as a model reaction of erythritol hydrogenolysis, and some catalysts developed for glycerol hydrogenolysis have been applied to erythritol hydrogenolysis. Although the hydrogenolysis of glycerol has also been achieved by the combination of homogeneous acid catalysts and Ru complex catalysts, examples of such systems are very limited and mainly produce 1-propanol or propane.204–206 In a related reaction of biomass-derived furanic compounds, Pd/C combined with metal triflates, which act as homogeneous Lewis acids, accelerated the complete hydrogenolysis to give alkanes.207–209 These examples indicate that the use of homogeneous catalysts has great potential for the complete removal of OH groups from biomass-derived substrates, while the regioselective transformation is rather difficult. Therefore, from the viewpoint of the regioselective hydrogenolysis of biomass-derived substrates, we focus on the reaction systems using heterogeneous catalysts in this review.There are two important mechanisms of indirect hydrogenolysis of glycerol that can work over a broad range of catalysts:51 dehydration + hydrogenation, which mainly proceeds over metal + acid catalysts, and dehydrogenation + dehydration + hydrogenation, which mainly occurs over non-acidic metal catalysts (Fig. 22). A key reaction in the latter mechanism is the second step, dehydration at C–H and C–OH at the α- and β-positions, respectively, of the carbonyl group. The acidic nature of C–H at the α-position of the carbonyl group promotes dehydration. A similar dehydration process also occurs during the dehydration + hydrogenation mechanism, which directly forms dideoxygenated products. We calculated the energy profiles of both mechanisms by DFT and details are shown in section S2 (ESI†). Here we only provide a summary: in the dehydration + hydrogenation mechanism, the main product is 1,2-propanediol via removal of the OH group at the 1-position. However, the energy difference between the removal of the OH group at the 1- and 2-positions is not so significant. In addition, when removal of the OH group at the 2-position occurs, further dehydration of 3-hydroxypropanal to acrolein easily proceeds because of the presence of C–H and C–OH at the α- and β-positions of the carbonyl group in 3-hydroxypropanal. Therefore, 1,3-propanediol and 1-propanol can also be produced. In the case of the dehydrogenation + dehydration + hydrogenation mechanism, only 1,2-propanediol can be produced. Furthermore, 1,2-propanediol is not reactive in the dehydrogenation + dehydration + hydrogenation mechanism, but it can be further hydrodeoxygenated to propanols and propane by the dehydration + hydrogenation mechanism.
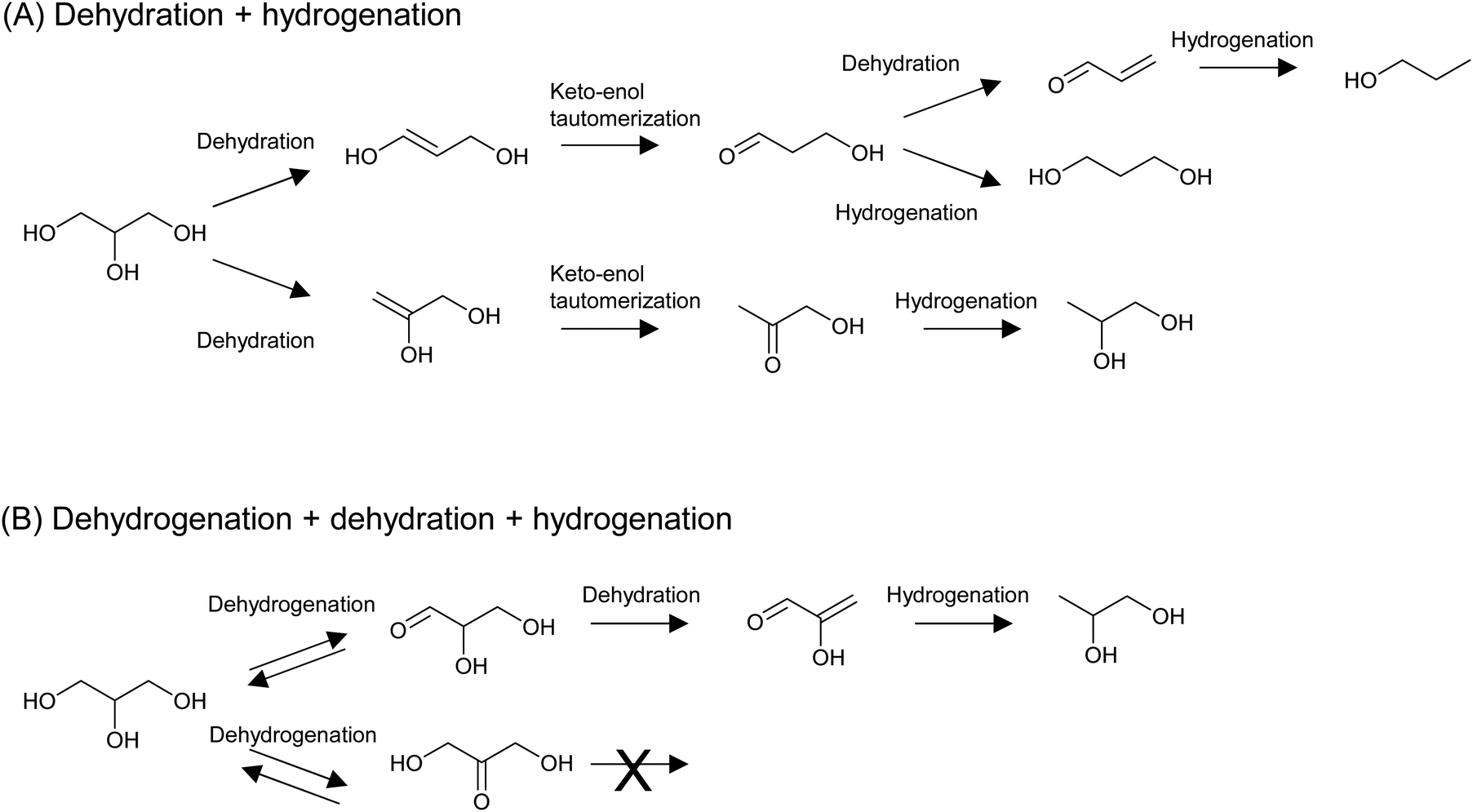 | ||
| Fig. 22 Typical mechanisms of the indirect hydrogenolysis of glycerol: dehydration + hydrogenation (A) and dehydrogenation + dehydration + hydrogenation (B). | ||
Similar analyses of the energy profiles for erythritol hydrogenolysis by the two indirect different mechanisms (dehydration + hydrogenation, dehydrogenation + dehydration + hydrogenation) were carried out and detailed results are shown in section S2 (ESI†). The reaction routes are more complicated than those for glycerol hydrogenolysis; in addition to an increase in the number of possible intermediates, the possible involvement of cyclization complicates the reaction. In summary, in the dehydration + hydrogenation mechanism, the reaction is unselective to form various products or even does not proceed by the formation of 1,4-AHERY. In fact, typical catalyst systems with the dehydration + hydrogenation mechanism such as Ru + acidic resin have not been tested for erythritol hydrogenolysis. In the dehydrogenation + dehydration + hydrogenation mechanism for erythritol hydrogenolysis, dehydrogenation at a terminus in the initial step produces 1,2,4-butanetriol + 1,2-butanediol while that at an internal position provides 1,2,3-butanetriol. The ratio of 1,2,4-butanetriol + 1,2-butanediol to 1,2,3-butanetriol may depend on the catalyst. Further conversions of both triols are possible through this mechanism because both triols have a 1,3-diol structure, while 1,2-butanediol is not reactive due to the lack of such a structure.
Erythritol hydrogenolysis was reported over Cu catalysts, which are typically utilized in glycerol hydrogenolysis to 1,2-propanediol through the dehydrogenation + dehydration + hydrogenation mechanism.210,211 The reported results for erythritol and related substrates over the Cu/C catalyst are shown in Fig. 23.211 The main products of erythritol hydrogenolysis were 1,2,4-butanetriol and 1,2-butanediol. An important point is that data were recorded at a low conversion level and it appeared that 1,2-butanediol was also a primary product. The reaction temperature was rather high (453 K) and a negative reaction order with respect to H2 pressure was mentioned in the same report. This suggests that the reaction of erythritol over Cu/C proceeds through the dehydrogenation + dehydration + hydrogenation route, which also opens the reaction of erythritol to 1,2-butanediol through the dehydrogenation + (double) dehydration + hydrogenation route as described above. The reactivity of 1,2-butanediol was much lower than that of erythritol, 1,2,3- and 1,2,4-butanetriols over Cu/C, which was also in agreement with the discussion above for the dehydrogenation + dehydration + hydrogenation route.
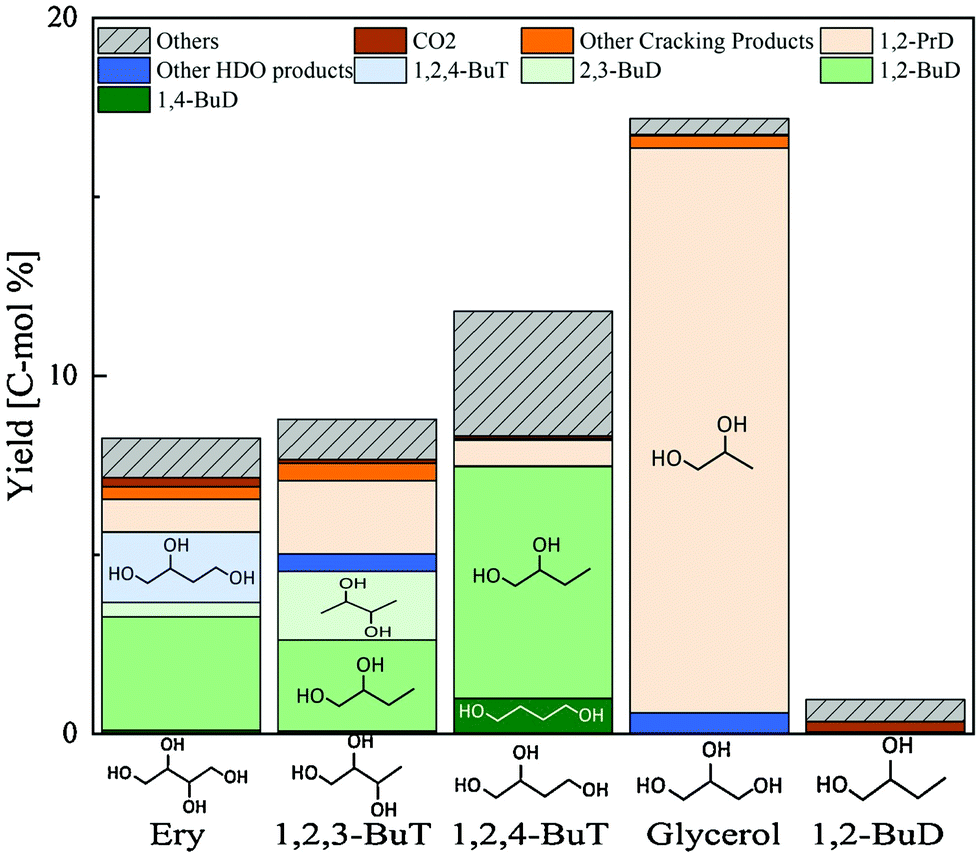 | ||
| Fig. 23 Hydrogenolysis of various substrates over Cu/C. Reaction conditions: substrate (4.5 g), water (25.5 g), catalyst (0.9 g-Cu), H2 (1.8 MPa), 453 K, 24 h. Ery = erythritol; BuT = butanetriol; BuD = butanediol; PrD = propanediol; HDO = hydrodeoxygenation. Reprinted from ref. 211 with permission from Elsevier, copyright 2021. | ||
Neither of the indirect mechanisms in Fig. 22 give 1,3-propanediol as the main product from glycerol, and another mechanism is necessary for its production. Indeed, there are systems for selective glycerol hydrogenolysis to 1,3-propanediol.212,213 Typical catalysts are supported Pt–WOx and Ir–ReOx, and selected systems with high yields are summarized in section S3 (ESI†). Generally, Ir–ReOx catalysts have an advantage in terms of catalytic activity, which permits hydrogenolysis at lower reaction temperatures, while Pt–WOx catalysts have an advantage from the viewpoint of high yield and selectivity of the target products. One proposed mechanism is the acidic activation of a secondary C–O bond combined with SN2-like hydride attack on a secondary carbon (Fig. 24) in the 1,2-diol moiety of the substrate.214 With this mechanism, erythritol will be converted into 1,2,4-butanetriol and then 1,4-butanediol, while the selective production of 1,4-butanediol from erythritol is very difficult with the two indirect mechanisms outlined in Fig. 22.
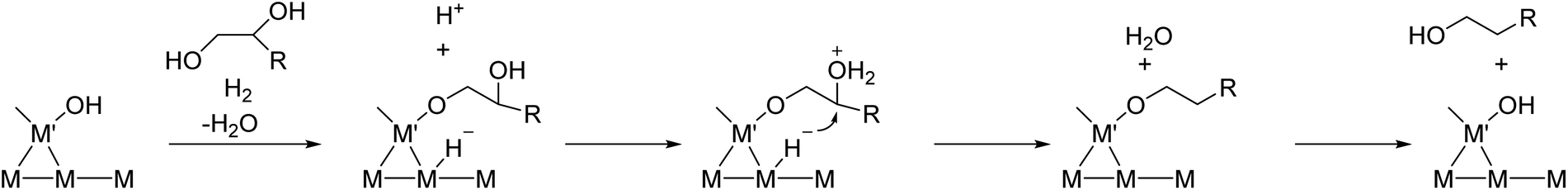 | ||
| Fig. 24 Proposed reaction mechanism for glycerol hydrogenolysis to 1,3-propanediol over M–M′Ox. R = CH2OH. | ||
The structure of the Ir–ReOx/SiO2 catalyst has been reported to be Ir metallic nanoparticles partially covered with three-dimensional ReOx clusters.215 There are direct bonds between the Ir metal surface and Re atoms in the ReOx clusters, as expected from observation of the Re–Rh bond in Rh–ReOx catalysts216 and the Re–Pt bond in Pt–ReOx catalysts217 by extended X-ray absorption fine structure (EXAFS) analysis. The direct interaction between Ir and ReOx is strongly connected to the reaction mechanism of C–O hydrogenolysis outlined in Fig. 24. The Ir–ReOx/SiO2 catalysts maintain very small Ir metal particles (ca. 2–3 nm) even when the loading amount of Ir is increased (from 4 wt% to 20 wt%).218 Interestingly, the Ir-amount-based catalytic activity for glycerol hydrogenolysis slightly increased and the optimum Re addition amount per Ir decreased with an increase of the Ir loading amount. The catalytically active site (the interface between Ir metal surfaces and ReOx clusters) effectively formed with a high loading amount of Ir and Re species. Based on this finding, the preparation of Ir–ReOx species on oxide supports with a much lower surface area than SiO2 has been attempted, and rutile TiO2 (6 m2 g−1) was found to be a suitable support.219 The transmission electron microscope (TEM) images and model structures are shown in Fig. 25 and 26, respectively. Ir nanoparticles attach to the rutile TiO2 surface strongly, maintaining a small particle size even when the density is high. The advantage of the formation of dense metallic particles is a reduction in the amount of modifier (ReOx clusters in this case) by sharing it between two metal particles as illustrated in Fig. 26. In particular, this kind of catalyst design is more efficient in the case of the expensive modifier such as Re. The TiO2 support has also been proposed to have a role in the suppression of the over-reduction of Re species, leading to the interaction between an erythritol molecule and catalytic active site.220,221
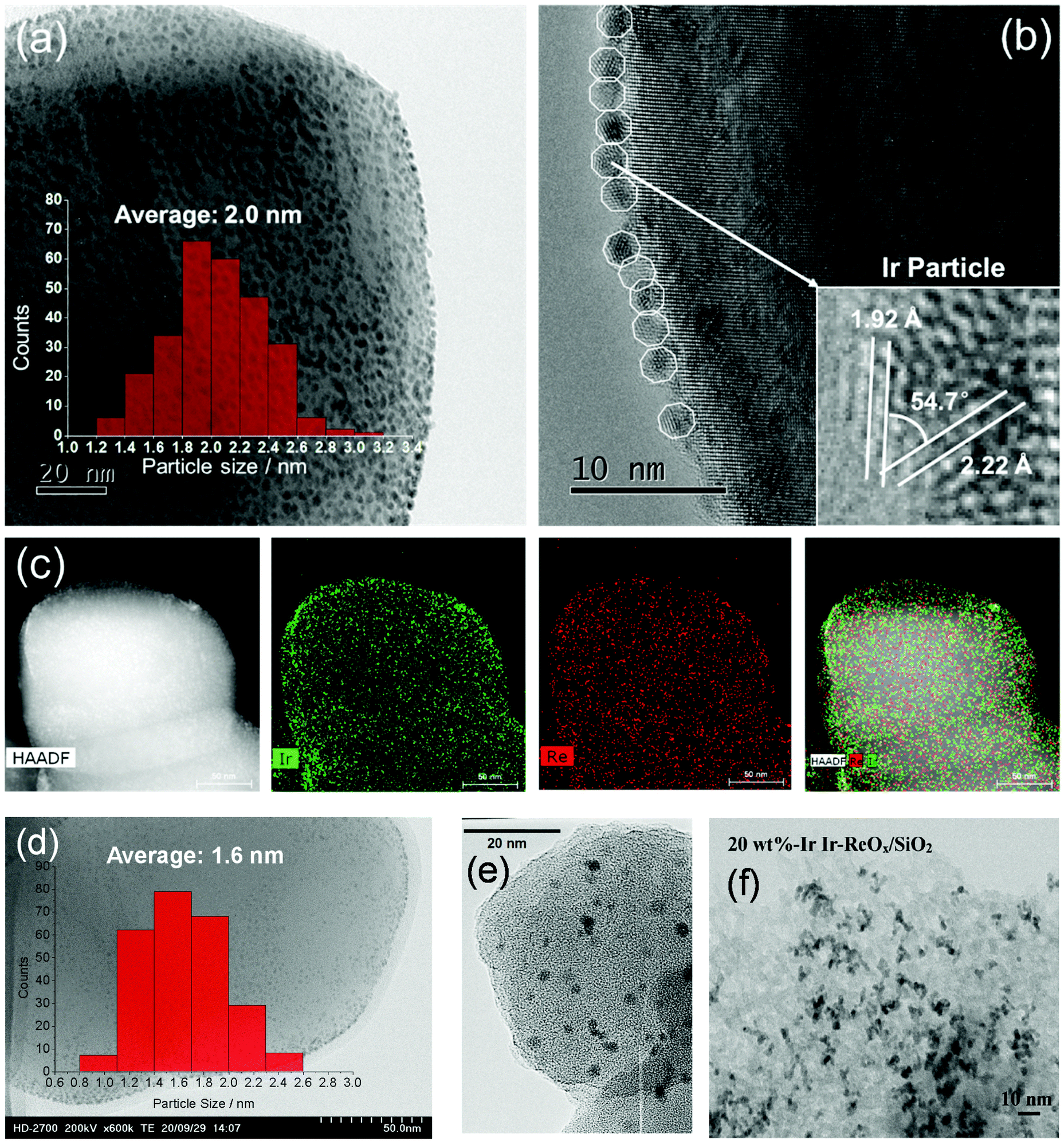 | ||
| Fig. 25 TEM images of Ir–ReOx/rutile TiO2 (4 wt% Ir, Re/Ir = 0.24) (a–c), Ir–ReOx/rutile TiO2 (1 wt% Ir, Re/Ir = 0.24) (d), Ir–ReOx/SiO2 (4 wt% Ir, Re/Ir = 0.83) (e), and Ir–ReOx/SiO2 (20 wt% Ir, Re/Ir = 0.34) (f). Reprinted from ref. 222 with permission from John Wiley and Sons, copyright 2020, for (a–d). Reproduced from ref. 225 with permission from Elsevier, copyright 2010, for (e) and from ref. 218 with permission from Elsevier, copyright 2019, for (f). | ||
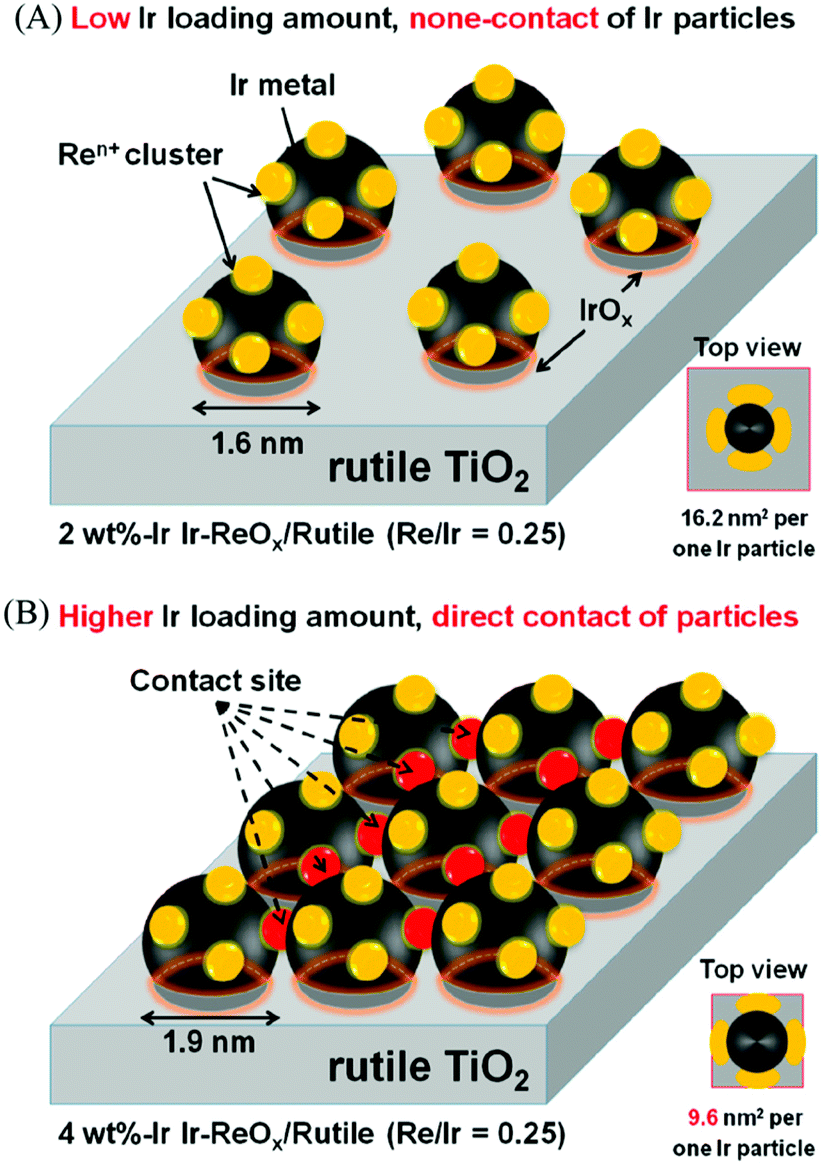 | ||
| Fig. 26 Model structure of Ir–ReOx/rutile TiO2 catalysts. Reprinted from ref. 219 with permission from the American Chemical Society, copyright 2019. | ||
Fig. 27 shows the conversion and selectivity as a function of reaction time for erythritol hydrogenolysis over Ir–ReOx/rutile TiO2 (4 wt% Ir, Re/Ir = 0.25) at 353 K.222 The reaction temperature (353 K) for the Ir–ReOx catalyst was clearly lower than that (453 K) for the Cu catalyst, which was due to the very high activity of Ir–ReOx catalysts for C–O hydrogenolysis of various polyfunctionalized compounds such as sugars,223,224 sugar alcohols,222,225–227 and cyclic ones.228 The selectivity of the primary products can be estimated from the data at a lower conversion level (i.e., shorter reaction time) (Fig. 27). The selectivities of 1,2,4-butanetriol, 1,2,3-butanetriol, 1,4-butanediol, and 1,3-butanediol in the initial stage were estimated to be 55%, 20%, 15%, and 10%, respectively, at 373 K.222 The C–O bond at the 2-position of erythritol was mainly dissociated first over Ir–ReOx/rutile TiO2. In the case of glycerol hydrogenolysis, the initial selectivities of 1,3-propanediol and 1-propanol were estimated to be 70% and 20%, respectively, at 393 K over the same Ir–ReOx/rutile TiO2 catalyst.219 The reactivity of erythritol is lower (about half) than that of glycerol under similar reaction conditions over Ir–ReOx/rutile TiO2. An important feature of the Ir–ReOx/TiO2-catalyzed hydrogenolysis of erythritol and glycerol was the relatively high selectivity of 1,4-BuD and 1-propanol, respectively, even at a low conversion level. Similar behavior was observed in the cases of erythritol and glycerol hydrogenolysis over Ir–ReOx/SiO2 catalysts.226,227 This behavior suggests that 1,4-butanediol is produced directly on the active site from erythritol without desorption of 1,2,4-butanetriol as an intermediate. The product distribution over Ir–ReOx was clearly different from the case of Cu/C; this could be related to direct C–O hydrogenolysis by SN2 attack of the hydride. The highest yield of 1,4-butanediol was 23%, which is far from satisfactory.222 From the viewpoint of the yield of 1,3-propanediol in glycerol hydrogenolysis, Pt–WOx catalysts are superior to Ir–ReOx catalysts.212,229 The applicability of Pt–WOx catalysts to erythritol hydrogenolysis should be mentioned.
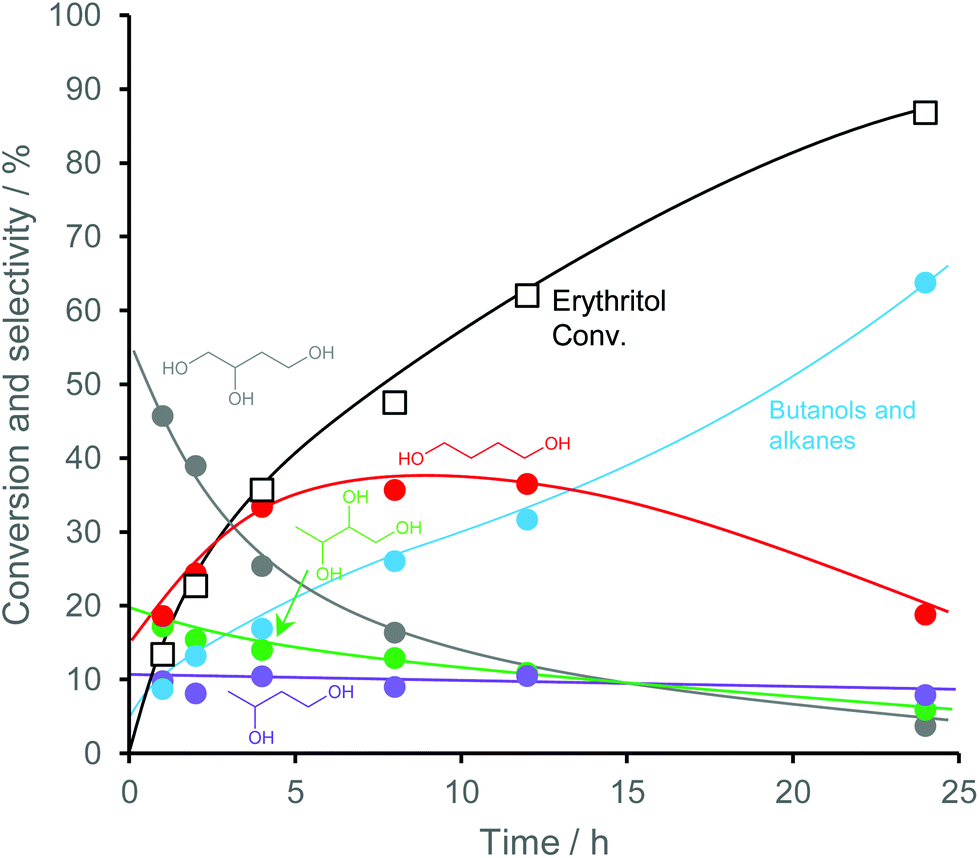 | ||
| Fig. 27 Time course of erythritol hydrogenolysis over Ir–ReOx/TiO2 (4 wt% Ir, Re/Ir = 0.25).222 Reaction conditions: 20 wt% aqueous solution of erythritol (5 g), catalyst (300 mg), 353 K, H2 (8 MPa), 373 K. | ||
For Pt–WOx catalysts, there are many reports on glycerol hydrogenolysis to 1,3-propanediol;135 however, the relationship between structure and performance is complex because the reported performance varies extensively even among catalysts with similar supports and compositions. Nonetheless, recent developments in this research area show that silica-based supports and a low W amount are effective in terms of both activity at low reaction temperature and selectivity to 1,3-propanediol.230–233 Therefore, Pt–WOx/SiO2 is one of the most suitable model catalysts and it is thought to be possible to reveal the essence of Pt–WOx catalysts. The Pt–WOx/SiO2 (4 wt% Pt, W/Pt = 0.25) catalyst after the reaction contains about 5 nm Pt metal particles, and the presence of the direct W–Pt bond is strongly suggested by EXAFS analysis.231 Elemental mapping in the TEM observation indicates that the W species are located around Pt metal particles. An interesting result is that the signal intensity ratio of W to Pt in the X-ray photoelectron spectroscopy (XPS) results is much lower than that of the catalyst composition (W/Pt = 0.25). At present, it is interpreted as W species (W4+) being located between the Pt metal and SiO2 surface selectively as illustrated in Fig. 28. The structure of Pt–WOx/SiO2 is rather different from that of Ir–ReOx catalysts; however, the reaction kinetics such as the reaction orders with respect to the concentrations of the substrates and H2 pressure is similar, suggesting that the reaction mechanisms over these two catalyst systems are also similar. The catalytically active site of Pt–WOx/SiO2 is the interface between the Pt metal surface and WOx species and the mechanism could be SN2-like hydride attack on the secondary carbon as shown in Fig. 24.231
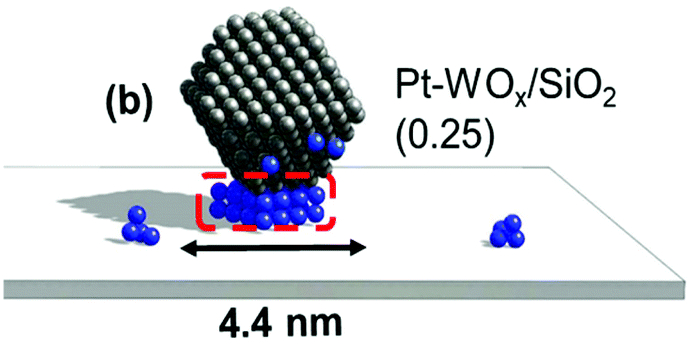 | ||
| Fig. 28 Proposed structure of Pt–WOx/SiO2. Reprinted from ref. 231 with permission from Elsevier, copyright 2021. | ||
Fig. 29 shows the time course of erythritol hydrogenolysis over Pt–WOx/SiO2 (4 wt% Pt, W/Pt = 0.25).234 The selectivities of 1,2,4- and 1,2,3-butanetriols at the initial stage were about 80% and 20%, respectively. The very high selectivity of 1,3-propanediol from glycerol (57% yield of 1,3-propanediol)230 is connected to that of 1,2,4-butanetriol from erythritol. This excellent selectivity of Pt–WOx/SiO2 in the C–O hydrogenolysis of the C–O bond neighboring –CH2OH enables a high yield (51%) of 1,4-butanediol. Another important point is that 1,4-butanediol is formed by consecutive C–O hydrogenolysis of erythritol (erythritol → 1,2,4-butanetriol → 1,4-butanediol), considering the very low initial selectivity of 1,4-butanediol formation.
 | ||
| Fig. 29 Time course of erythritol hydrodeoxygenation over Pt–WOx/SiO2 (4 wt% Pt, W/Pt = 0.25). Reaction conditions: erythritol (0.5 g), H2O (4 g), catalyst (0.2 g), H2 (8 MPa), 413 K. Reduction conditions: liquid-phase reduction at 473 K. BuT = butanetriol, BuD = butanediol, BuOH = butanol. Reprinted from ref. 234 with permission from the Royal Society of Chemistry, copyright 2021. | ||
The above results of erythritol hydrogenolysis over Ir–ReOx and Pt–WOx catalysts are directed to the synthesis of butanediols. On the other hand, Ir–ReOx and Pt–WOx catalysts are also effective for the synthesis of butanols when the catalysts are applied to more severe reaction conditions such as higher reaction temperature and longer reaction time. This is because butanols are formed by the further hydrogenolysis of butanediols. When the Ir–ReOx/SiO2 catalyst was used, 43% yield of 1-butanol and 17% yield of 2-butanol were reported.226 Meanwhile, the Pt–WOx/SiO2 catalyst was reported to produce 57% yield of 1-butanol and 18% yield of 2-butanol.234 It should be noted that Ir–ReOx/SiO2 catalysts were also effective for producing hexanols (3-hexanol 41% yield, 2-hexanol 18% yield, 1-hexanol 1% yield) from mechanocatalytically depolymerized cellulose in a biphasic reaction system (n-decane and H2O) with the aid of H2SO4.223 These high yields of mono-alcohols in the hydrogenolysis of polyols are due to the lower reactivity of mono-alcohols over Ir–ReOx/SiO2134 and Pt–WOx/SiO2.230
5.2. Deoxydehydration (DODH) and DODH + hydrogenation of erythritol
DODH is also a useful reaction in the conversion of erythritol, although examples are limited especially for those using H2 as a reducing agent. The results for the DODH and DODH + hydrogenation of erythritol with H2 as a reducing agent are summarized in Table 5. 1,2-Butanediol can be synthesized from erythritol by the DODH of erythritol to 1-butene-3,4-diol and the consecutive hydrogenation of the CC bond in 1-butene-3,4-diol to 1,2-butanediol. These two reactions proceed in one pot over ReOx–Pd/CeO2.151,152 The reaction time should be carefully controlled in order to obtain 1,2-butanediol in high yield, because 1,2-butanediol is converted into n-butane after consumption of erythritol. Still, the 1,2-butanediol yield for DODH + hydrogenation over ReOx–Pd/CeO2 was much higher than that obtained with other systems such as Cu catalysts as described in section 5.1.
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Solvent | P(H2) [MPa] | Temp. [K] | Time [h] | Conv. [%] | Selectivity [%] | TONa | Ref. | |||
| 1,3-Butadiene | 1,2-BuD | 1,4-BuD | Butenes and butane | ||||||||
| BuD: butanediol.a TON = ((mol of hydrocarbons) × 2 + (mol of butanediols) + (mol of butenediols))/(mol of Re). | |||||||||||
| ReOx–Pd/CeO2 | 1,4-Dioxane | 8 | 433 | 24 | 98 | 0 | 79 | 12 | 5 | 249 | 151 |
| ReOx–Au/CeO2 | 1,4-Dioxane | 8 | 413 | 60 | 97 | 83 | 0 | 1 | 3 | 426 | 144 |
| ReOx–Ag/CeO2 | 1,4-Dioxane | 8 | 413 | 20 | >99 | 86 | 0 | 0 | 6 | 245 | 147 |
| ReOx–Ag/CeO2 | None | 8 | 413 | 24 | >99 | 91 | 0 | 0 | 3 | 240 | 147 |
| ReOx–Cu/CeO2 | 1,4-Dioxane | 8 | 413 | 24 | >99 | 0 | 50 | 4 | 22 | 129 | 147 |
| ReOx–Ni/CeO2 | 1,4-Dioxane | 8 | 413 | 24 | 25 | 0 | 0 | 0 | 0 | 26 | 147 |
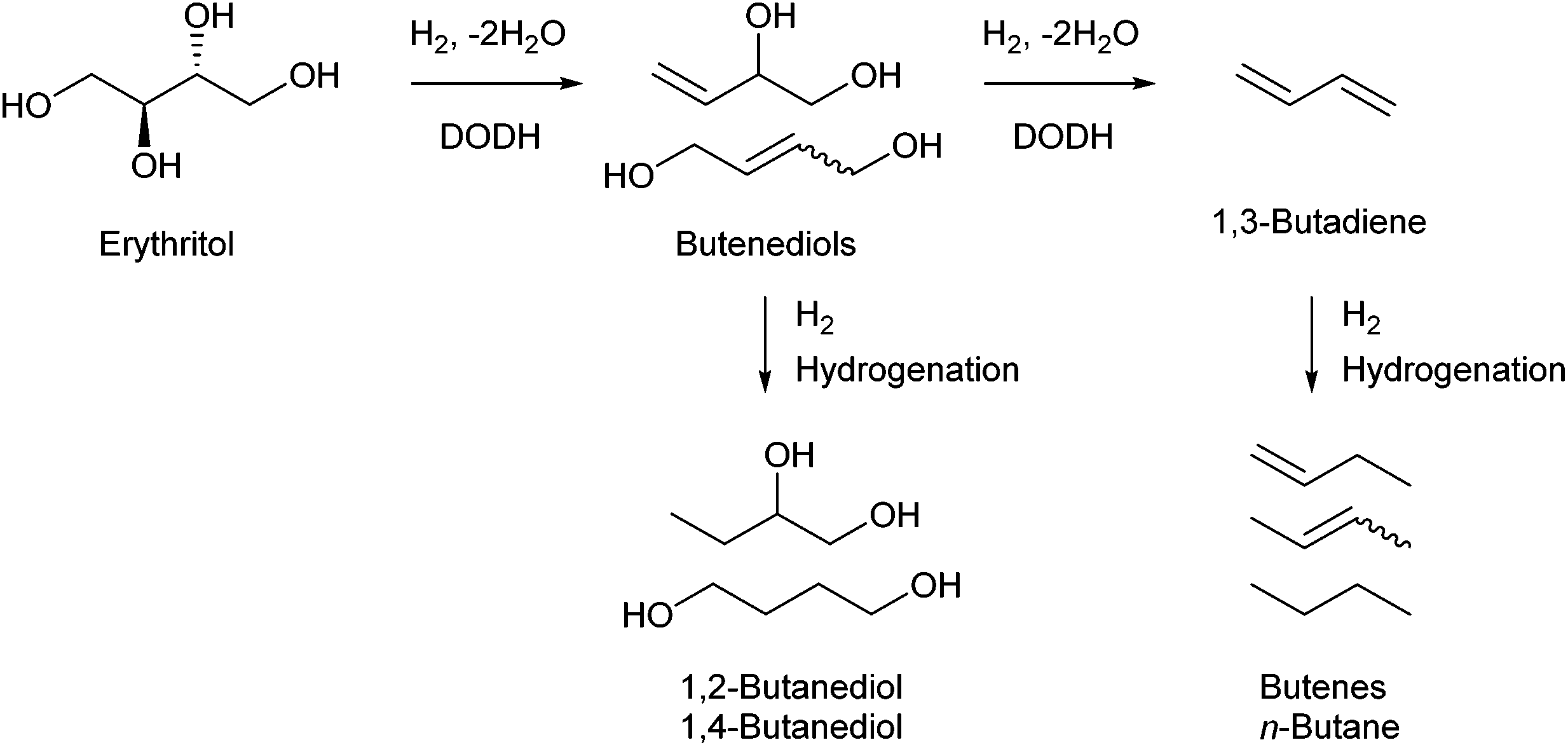
The DODH of erythritol was catalyzed by ReOx–Au/CeO2 and a high yield of 1,3-butadiene (∼80%) was achieved (see Table 5).144,145 It has been recently reported that ReOx–Ag/CeO2 exhibited a higher activity than ReOx–Au/CeO2 and a comparably high yield of 1,3-butadiene.147 Furthermore, the application of “solventless” conditions further increased the yield of 1,3-butadiene, probably due to the efficient transfer of 1,3-butadiene from the liquid phase (erythritol and butenediols for the “solventless” conditions) to the gas phase. In addition, the “solventless” conditions greatly improved the E-factor.
5.3. Production of 1,3-butanediol: hydrogenolysis of 1,4-anhydroerythritol (1,4-AHERY)
The main primary product in the hydrogenolysis of erythritol is 1,2,4-butanetriol on both Ir–ReOx and Pt–WOx catalysts as well as Cu catalysts. This means that it is difficult to obtain a high yield of 1,3-butanediol from erythritol because the hydrogenolysis of primary C–OH bonds in glycols is very challenging and suitable catalysts for this hydrogenolysis are too limited. A recent example was the hydrogenolysis of 1,2-propanediol to 2-propanol catalyzed by Ru–ReOx/SiO2; however, the obtained yield of 2-propanol was as low as 23% and not satisfactory.235 The selective hydrogenolysis of 1,2,4-butanetriol to 1,3-butanediol is regarded to be very challenging at present. 1,3-Butanediol is a possible target from 1,4-AHERY. One of the possible routes is the conversion of 1,4-AHERY to 3-hydroxytetrahydrofuran236 and the consecutive conversion of 3-hydroxytetrahydrofuran to 1,3-butanediol.237 The conversion of 1,4-AHERY into 3-hydroxytetrahydrofuran was catalyzed by the WOx–Pd/C catalyst and the yield of 3-hydroxytetrahydrofuran was 72% (Fig. 30).236 The hydrogenolysis of 3-hydroxytetrahydrofuran to 1,3-butanediol was catalyzed by Ir–ReOx/SiO2 and the selectivity of 1,3-butanediol formation was 81% at 24% conversion.237 The overall selectivity of 1,3-butanediol from 1,4-AHERY was calculated to be 58%. It has been recently reported that the hydrogenolysis of 3-hydroxytetrahydrofuran to 1,3-butanediol was also catalyzed by Pt–WOx/SiO2 and the selectivity of 1,3-butanediol formation was 90% at 40% conversion,230 and the overall selectivity of 1,3-butanediol from 1,4-AHERY increased to 65%. Actually, the solvent for the conversion of 1,4-AHERY into 3-hydroxytetrahydrofuran over WOx–Pd/C was 1,4-dioxane, and that for the hydrogenolysis of 3-hydroxytetrahydrofuran to 1,3-butanediol over Ir–ReOx/SiO2 and Pt–WOx/SiO2 was water. The difference in suitable solvents for the process including a series of two reactions decreases the feasibility of the conversion process of 1,3-butanediol from 1,4-AHERY. The one-pot conversion of 1,4-AHERY was thus desired.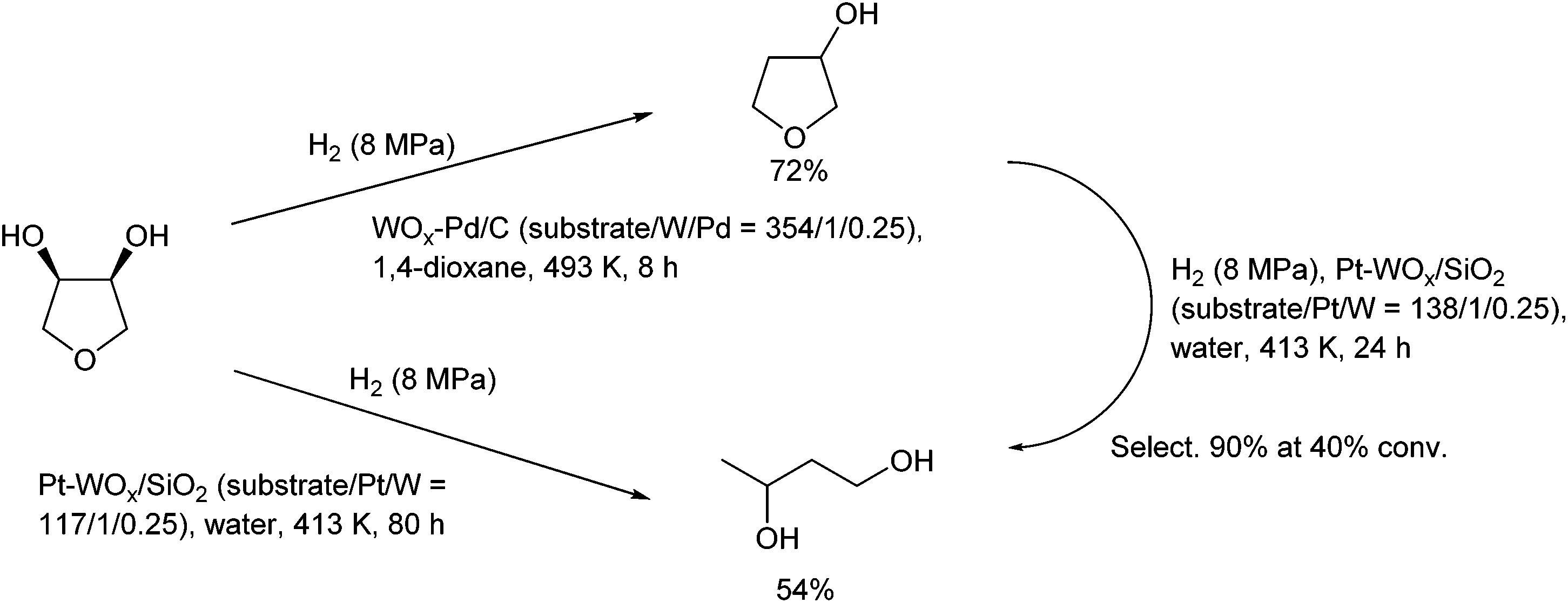 | ||
| Fig. 30 Selective hydrogenolysis systems of 1,4-AHERY.230,236 | ||
A possible one-pot route is the combination of the hydrogenolysis of 1,4-AHERY to 1,2,3-butanetriol and the successive one of 1,2,3-butanetriol to 1,3-butanediol. It has been reported that Rh–MoOx/SiO2 catalyzed the one-pot hydrogenolysis of 1,4-AHERY to 2-butanol with the highest yield of 2-butanol (51%).238 Such Rh–MoOx catalysts were also found to be effective for the C–O hydrogenolysis of related compounds.238–241 It has been proposed that the primary ring-opening C–O hydrogenolysis of 1,4-AHERY over Rh–MoOx/SiO2 gave 1,2,3-butanetriol, and the reaction mechanism was thought to be similar to that of glycerol hydrogenolysis over Ir–ReOx catalysts. From the viewpoint of the synthesis of 1,3-butanediol from 1,2,3-butanetriol, Rh–MoOx/SiO2 is not suitable on the basis of the low selectivity to 1,3-propanediol through glycerol hydrogenolysis.242 Ir–ReOx/SiO2 was also investigated for the hydrogenolysis of 1,4-AHERY,238 as listed in Table 6. The selectivity to 1,2,3-butanetriol + 1,3-butandiol was around 60%; however, the selectivity to 2-butanol was 14% even at a low conversion level, suggesting that a high yield of 1,3-butanediol is difficult on the Ir–ReOx/SiO2 catalyst. This can be explained by the not so high yield of 1,3-propanediol through glycerol hydrogenolysis on Ir–ReOx catalysts due to the consecutive hydrogenolysis of 1,3-propanediol to 1-propanol as mentioned above. Based on this discussion, it is thought that Pt–WOx is more suitable than Ir–ReOx in the one-pot hydrogenolysis of 1,4-AHERY to 1,3-butanediol.
| Entry | Catalyst | Time [h] | Conv. [%] | Selectivity [%] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1,2,3-BuT | 3-HTHF | 1,2-BuD | 2,3-BuD | 1,3-BuD | 1-BuOH | 2-BuOH | Alkanes | Others | ||||
| BuT: butanetriol; HTHF: hydroxytetrahydrofuran; BuD: butanediol; BuOH: butanol. Reaction conditions: catalyst (0.1 g) (Rh or Ir 4 wt%, Mo/Rh = 0.13 or Re/Ir = 1), 1,4-AHERY (1 g), water (4 g), H2 (8 MPa), 393 K. | ||||||||||||
| 1 | Rh–MoOx/SiO2 | 1 | 21 | 37 | 9 | 9 | 12 | 2 | 4 | 23 | 2 | 2 |
| 2 | Rh–MoOx/SiO2 | 48 | 100 | <1 | <1 | <1 | 8 | <1 | 15 | 51 | 24 | 2 |
| 3 | Ir–ReOx/SiO2 | 1 | 8 | 43 | 8 | 4 | 2 | 25 | 3 | 14 | 1 | <1 |
| 4 | Ir–ReOx/SiO2 | 24 | 63 | 35 | 4 | 4 | 2 | 23 | 11 | 19 | 3 | 1 |
It has been recently demonstrated that Pt–WOx/SiO2, which is the same catalyst for erythritol hydrogenolysis to 1,4-butanediol (Fig. 29), was effective for the one-pot synthesis of 1,3-butanediol by 1,4-AHERY hydrogenolysis230 and 54% yield of 1,3-butanediol from 1,4-AHERY was achieved (Fig. 30). Fig. 31 shows the time course of the hydrogenolysis of 1,4-AHERY over Pt–WOx/SiO2 (4 wt% Pt, W/Pt = 0.25). The selectivity of 1,2,3-butanetriol was high (about 80%) at a low conversion level. The reaction routes for 1,4-AHERY and erythritol over Pt–WOx/SiO2 indicated that the key intermediates were 1,2,3-butanetriol and 1,2,4-butanetriol, respectively, as shown in Fig. 32. Regarding the formation of 1,3-butanediol, the reaction proceeds via the ring-opening hydrogenolysis of 1,4-AHERY followed by selective removal of secondary OH groups in 1,2,3-butanetriol. In the mechanism of the ring-opening hydrogenolysis of 1,4-AHERY, the anti-conformation of C–O in the tetrahydrofuran ring with C–OH is connected to high selectivity to the formation of 1,2,3-butanetriol. It is necessary to suppress the further hydrogenolysis of 1,3-butanediol in order to increase the yield of 1,3-butanediol from 1,4-AHERY.
 | ||
| Fig. 31 Time course of the hydrogenolysis of 1,4-AHERY over 4 wt% Pt–WOx/SiO2 (W/Pt = 0.25). Reaction conditions: 1,4-AHERY (0.5 g), H2O (4 g), catalyst (0.2 g), H2 (8 MPa), 413 K. BuD: butanediol; BuT: butanetriol; BuOH: butanol; HTHF: hydroxytetrahydrofuran. Reprinted from ref. 230 with permission from the Royal Society of Chemistry, copyright 2020. | ||
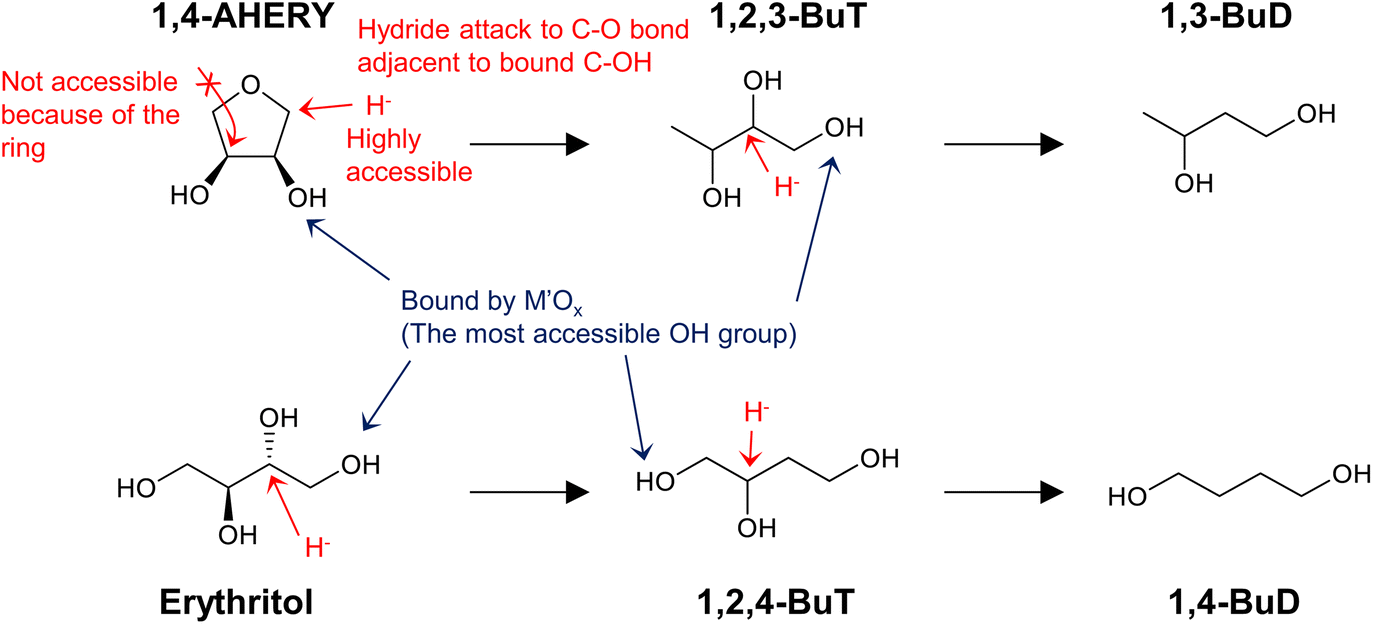 | ||
| Fig. 32 Main reaction routes of 1,4-AHERY and erythritol over 4 wt% Pt–WOx/SiO2 (W/Pt = 0.25) catalyst. M′: W; BuT: butanetriol; BuD: butanediol. | ||
5.4. Deoxydehydration (DODH) and DODH + hydrogenation of 1,4-anhydroerythritol (1,4-AHERY)
1,4-AHERY has cis-vicinal OH groups with a 5-membered ring structure. Compared to the 6-membered ring structure, the angle between the C–OH bonds is much smaller in the 5-membered ring structure, which leads to a higher DODH reactivity of 1,4-AHERY. Therefore, 1,4-AHERY is a typical model substrate in the DODH. Table 7 summarizes the DODH of 1,4-AHERY and its related reactions using H2 as a reducing agent. The DODH of 1,4-AHERY gives 2,5-dihydrofuran, and the consecutive hydrogenation of 2,5-dihydrofuran gives tetrahydrofuran. Homogeneous catalysts such as CH3ReO3 are not effective, while good 2,5-dihydrofuran yields have been reported when other reducing agents such as 3-octanol were used.243 ReOx–Pd/CeO2 catalyzed the one-pot reaction of 1,4-AHERY with H2 to tetrahydrofuran in high yield (>99%).151,152 ReOx–Au/CeO2 catalyzed the DODH of 1,4-AHERY with H2 to provide 2,5-dihydrofuran in high yield (∼80%) with 8% yield of 2,3-dihydrofuran.144,145 ReOx–Ag/CeO2 catalyzed the DODH of 1,4-AHERY with H2 to produce 2,5-dihydrofuran in high yield (∼88%) with 5% yield of 2,3-dihydrofuran.147 It was also reported recently that MoOx–Au/TiO2 and MoOx–Pd/TiO2 emerged as good catalysts for the DODH of 1,4-AHERY with H2 to form 2,5-dihydrofuran (77% yield)148 and tetrahydrofuran,148,244 respectively. Although high selectivity was reported, the activity of the MoOx–M/TiO2-catalyzed system was significantly lower than that of the ReOx–M/CeO2-catalyzed systems (higher reaction temperature or lower initial reaction rate), because of the intrinsic lower activity of Mo species in DODH than that of Re. Furthermore, the final yield was not high, suggesting that the MoOx–M/TiO2 catalysts tended to be deactivated rapidly. Further improvement of the performance of Mo-based heterogeneous DODH systems, especially at low reaction temperature, is necessary for their application to substrates with more complex structures such as erythritol and the alkyl glycosides mentioned in the previous section. Note that homogeneous catalysts combined with H2 did not work well for this reaction,245–247 demonstrating the superiority of heterogeneous catalytic systems to homogeneous ones.| Catalyst | Solvent | P(H2) [MPa] | Temp. [K] | Time [h] | Conv. [%] | Selectivity [%] | TONa | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| THF | 2,5-DHF | 2,3-DHF | 1,4-BuD | ||||||||
| DHF: dihydrofuran; BuD: butanediol; DME: dimethoxyethane; n.r.: not reported; TMSO: tetramethylene sulfoxide; Cp*: 1,2,3,4,5-pentamethylcyclopentadienyl.a TON = ((mol of THF) + (mol of dihydrofurans) + (mol of 1,4-BuD))/(mol of Re, Mo, or Ru).b Total yield of 2,5-DHF and 2,3-DHF.c Yield values. | |||||||||||
| ReOx–Pd/CeO2 | 1,4-Dioxane | 8 | 413 | 64 | >99 | >99 | 0 | 0 | 0 | 598 | 151 |
| ReOx–Au/CeO2 | 1,4-Dioxane | 8 | 413 | 24 | 93 | 3 | 86 | 9 | 0 | 269 | 144 |
| ReOx–Ag/CeO2 | 1,4-Dioxane | 8 | 413 | 2 | 98 | 4 | 90 | 5 | 0 | 149 | 147 |
| ReOx/CeO2 + ReOx/C | 1,4-Dioxane | 8 | 413 | 24 | 97 | 7 | 0 | 0 | 86 | 134 | 250 |
| ReOx–Au/CeO2 + ReOx/WO3–ZrO2 | 1,4-Dioxane | 8 | 413 | 24 | >99 | 35 | 3 | 0 | 53 | 174 | 252 |
| ReOx/C | None | 0.4 | 473 | 2 | 74 | 0.1 | 34b | – | 0.4 | 9 | 251 |
| MoOx–Au/TiO2 | 1,2-DME | 8 | 463 | 24 | 67 | 0.8 | 96 | 0.3 | 0.7 | 197 | 148 |
| MoOx–Pd/TiO2 | 1,2-DME | 8 | 463 | 24 | 65 | 98 | 0.4 | 0 | 0 | 192 | 148 |
| MoOx–Pd/TiO2 | 1,4-Dioxane | 5.2 | 413 | 8 | 29 | 98 | <1 | <1 | 0 | 44 | 244 |
| CH3ReO3 | 1,4-Dioxane | 3.4 | 423 | 16 | n.r. | 5b | 25c | n.r. | n.r. | 6 | 245 |
| Ru(TMSO)(DMSO)4 | Benzene | 6.8 | 463 | 48 | n.r. | 23b | n.r. | n.r. | n.r. | 2 | 246 |
| [Cp*Ru(CO)3]2 | Benzene | 0.4 | 443 | 72 | n.r. | 12b | n.r. | n.r. | n.r. | 0.6 | 247 |
5.5. 1,4-Anhydroerythritol (1,4-AHERY) to 1,4-butanediol via 2,5-dihydrofuran
The synthesis of 2,5-dihydrofuran in high yield enables the utilization of 2,5-dihydrofuran as an intermediate. Pinkos et al. at BASF found that 1,4-butanediol can be produced in high yield by hydration–reduction of 2,5-dihydrofuran over ReOx/TiO2 (Re = 6 wt%) catalysts with 1,4-dioxane–water or tetrahydrofuran–water as solvents and H2 in a flow reactor.248 The maximum yield of 1,4-butanediol was 85% at 427 K. Based on this previous work and reports on the selective synthesis of 2,5-dihydrofuran, the development of one-pot systems for the conversion of 1,4-AHERY to 1,4-butanediol has been attempted.It is found that a physical mixture of ReOx–Au/CeO2 + ReOx/C catalysts was effective at converting 1,4-AHERY to 1,4-butanediol in high yield (∼90%) (Fig. 33).249 Activity tests using possible intermediates as substrates suggested that the formation of 1,4-butanediol involved several steps: deoxydehydration (DODH) of 1,4-AHERY to 2,5-dihydrofuran (which is catalyzed by ReOx–Au/CeO2), isomerization of 2,5-dihydrofuran to 2,3-dihydrofuran, hydration of 2,3-dihydrofuran to 2-hydroxytetrahydrofuran, and hydrogenation of 2-hydroxytetrahydrofuran or the ring-opened form (4-hydroxybutanal) to 1,4-butanediol. The isomerization step is catalyzed by ReOx on ReOx/C. The hydration of 2,3-dihydrofuran is catalyzed by a weak acid and proceeds over the carbon support. The final hydrogenation step is catalyzed by metallic Re species on ReOx/C. During the conversion of 1,4-AHERY into 1,4-butanediol, it is necessary to suppress the hydrogenation of unsaturated intermediates like 2,5- and 2,3-dihydrofurans, and promote higher activity for the hydrogenation of 4-hydroxybutanal.
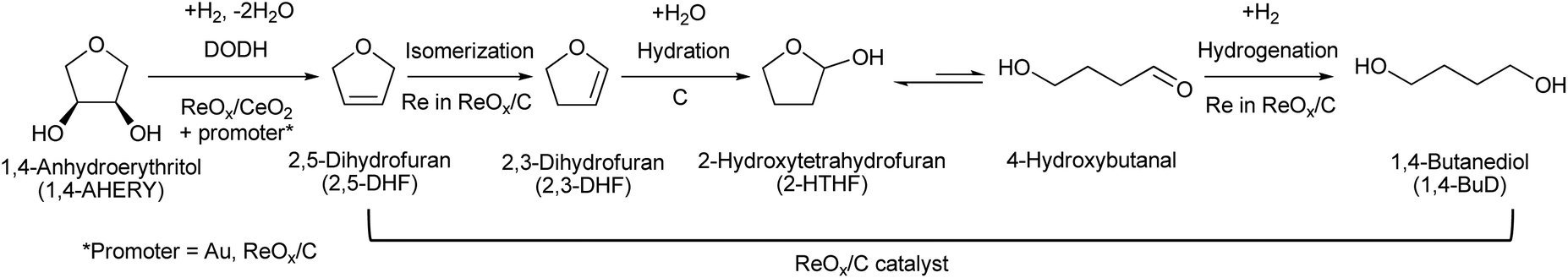 | ||
| Fig. 33 One-pot conversion of 1,4-AHERY to 1,4-butanediol over ReOx(–Au)/CeO2 + ReOx/C. Reprinted from ref. 252 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Later, it was also found that the Au promoter is not necessary in the mixed catalysts.250 A simple mixture of ReOx/CeO2 + ReOx/C showed almost the same activity and selectivity in the conversion of 1,4-AHERY into 1,4-butanediol as the case of ReOx–Au/CeO2 + ReOx/C, while ReOx/C alone did not produce 1,4-butanediol as a major product.251 It is possible that metallic Re species on the carbon support help to reduce ReOx species on CeO2via hydrogen spillover.
The bottleneck of the catalyst systems of ReOx–Au/CeO2 + ReOx/C and ReOx/CeO2 + ReOx/C is catalyst reusability because it is impossible to calcine carbon-supported catalysts for regeneration to remove deposited species from catalyst surfaces. In order to acquire catalyst reusability, the screening of oxides was conducted for the reaction of 2,5-dihydrofuran.252 As a result, the ReOx/WO3–ZrO2 catalyst showed a higher performance in the conversion of 2,5-dihydrofuran into 1,4-butanediol than other oxide-supported ReOx catalysts, although its performance was clearly lower than that of ReOx/C. Then, ReOx/WO3–ZrO2 was used as a co-catalyst with ReOx–Au/CeO2 for the one-pot conversion of 1,4-AHERY. Fig. 34 shows the time courses of the reaction of 1,4-AHERY over ReOx–Au/CeO2 + ReOx/C249 and ReOx–Au/CeO2 + ReOx/WO3–ZrO2.252 The highest yield of 1,4-butanediol over ReOx–Au/CeO2 + ReOx/WO3–ZrO2 was 55%, which was clearly lower than that over ReOx–Au/CeO2 + ReOx/C. After a longer reaction time, the main byproduct was tetrahydrofuran in both cases. There are three formation routes for tetrahydrofuran: dihydrofuran hydrogenation, dihydrofuran disproportionation (to tetrahydrofuran and furan), and dehydration of 1,4-butanediol. At a low conversion level, in the case of ReOx–Au/CeO2 + ReOx/C, the selectivity of the acetal (whose structure is shown in Fig. 34) and 1,4-butanediol was 55% and 28%, respectively, at 33% conversion.249 The low selectivity of tetrahydrofuran at this stage and the low selectivity to furan suggest that the main formation route of tetrahydrofuran over ReOx–Au/CeO2 + ReOx/C was the dehydration of 1,4-butanediol. In the case of ReOx–Au/CeO2 + ReOx/WO3–ZrO2, the selectivities of the acetal, 2,5-dihydrofuran, tetrahydrofuran, and 1,4-butanediol were 37%, 26%, 22%, and 7%, respectively, at 45% conversion.252 Furan, which is the byproduct co-produced by the disproportionation of dihydrofuran, was almost not formed. The higher levels of dihydrofuran and tetrahydrofuran selectivity at such low conversion suggested that dihydrofuran hydrogenation before hydration of dihydrofuran was the main formation route for tetrahydrofuran over ReOx–Au/CeO2 + ReOx/WO3–ZrO2. On the other hand, the reuse of ReOx–Au/CeO2 + ReOx/WO3–ZrO2 was almost possible (1,4-butanediol yield: 53% → 49% → 43%) by calcination at 573 K for 3 h, while the reuse of ReOx–Au/CeO2 + ReOx/C was difficult (1,4-butanediol yield: 86% → 76% → 44%) by N2 treatment at 773 K for 1 h.252
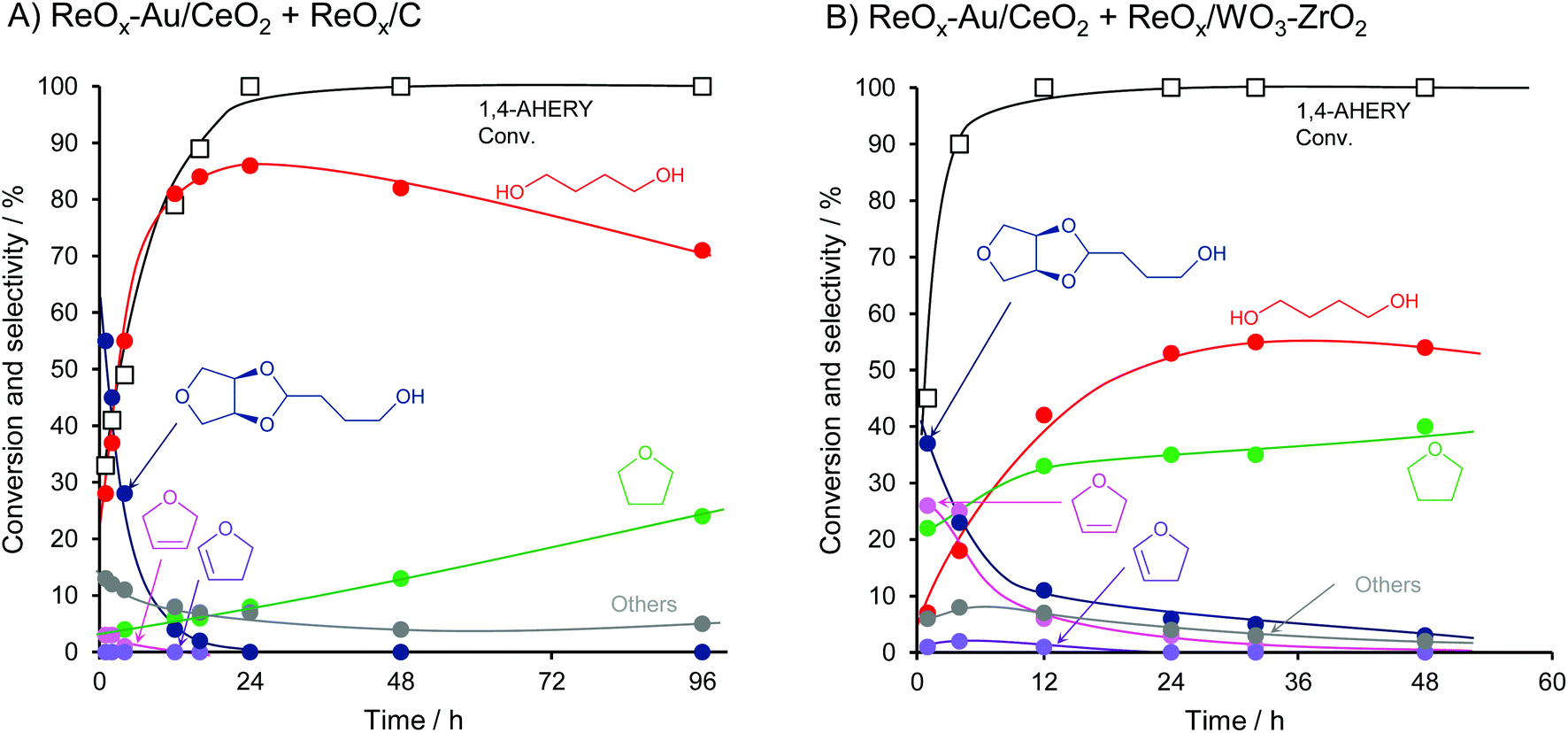 | ||
| Fig. 34 Catalytic reduction of 1,4-AHERY to 1,4-butanediol.249,252 (A) ReOx–Au/CeO2 + ReOx/C catalyst mixture. Conditions: 1,4-AHERY (0.5 g), ReOx–Au/CeO2 (Re 1 wt%, Au 0.3 wt%) (0.15 g), ReOx/C (Re 3 wt%) (0.15 g), 1,4-dioxane (4 g), H2 (8 MPa), 413 K. (B) ReOx–Au/CeO2 + ReOx/WO3–ZrO2 catalyst mixture. Conditions: 1,4-AHERY (0.3 g), ReOx–Au/CeO2 (Re 1 wt%, Au 0.3 wt%) (0.15 g), ReOx/WO3–ZrO2 (Re 1 wt%, WO3 10 wt%) (0.15 g), 1,4-dioxane (4 g), H2 (8 MPa), 413 K. Others = 1-butanol, γ-butyrolactone, furan, 2-(4-hydroxybutyloxy)tetrahydrofuran and unidentified byproducts. | ||
5.6. Summary of erythritol/1,4-anhydroerythritol hydrodeoxygenation
The number of research on the hydrodeoxygenation of erythritol and 1,4-AHERY is growing rapidly, because erythritol is now regarded as a platform chemical rather than a model substrate. Specific butanediols except 2,3-butanediol can be produced from erythritol by hydrodeoxygenation with >50% yield. A further increase of the yields and selectivities is expected, while an increase of selectivity is essential because of the difficulty in the separation of polyols. Tetrahydrofuran and 1,3-butadiene can also be produced in high yields (>80%). The hydrogen consumption amount for the production of these chemicals from erythritol or 1,4-AHERY is not high (2 equiv.) and lower than that for succinic acid reduction to 1,4-butanediol or tetrahydrofuran (4 equiv.).Most chemicals synthesized via hydrodeoxygenation of erythritol or 1,4-AHERY can also be produced from biomass through another method such as direct fermentation (for butanediols253 and 1-butanol254), ethanol conversion (for 1,3-butadiene255), succinic acid hydrogenation (for 1,4-butanediol and tetrahydrofuran256,257) and decarbonylation/decarboxylation of C5 platform molecules (for tetrahydrofuran and 2-butanol30,258). The costs can be compared for erythritol-based and other methods. There is uncertainty in the future price of erythritol because the main use of erythritol is as a low-calorie sweetener, the demand for which significantly depends on its reputation for consumers. Nevertheless, the versatile nature of erythritol as a platform chemical is attractive and we hope many researchers will join the study of erythritol/1,4-AHERY conversions.
6. N-Acetyl-D-glucosamine (NAG)
Next to cellulose, chitin is the second most abundant biomass resource found in crustacean shells and the exoskeletons of insects and has emerged as a core compound in the “shell biorefinery” for this decade due to its attractive N-containing structure.259–261 The extraction and purification of this polysaccharide from natural sources requires excess amounts of HCl and NaOH for demineralization and deproteinization, respectively, and is still under development due to its low efficiency and high environmental impact.262,263 Yet, the use of chitin as a source of N-containing chemicals has been examined in recent decades. Chitin is initially depolymerized into NAG, whose structure is identical to that of glucose (monomer of cellulose) except for an acetamide group at the 2-position, via traditional enzymatic hydrolysis264 and H2SO4-assisted mechanochemical depolymerization + subsequent thermal hydrolysis.265 Note that the undesired deacetylation to form glucosamine did not occur in these cases. In the former case, NAG could be produced in excellent yields of over 90 wt% even from untreated chitin,266,267 which typically requires pretreatment to improve its poor reactivity due to the robust crystalline structure of chitin. Although conventional hydrolysis in a concentrated HCl solution also provided a good yield of NAG (65%),268,269 a large amount of acidic waste was co-produced and needed to be neutralized. A diluted HCl solution led to the deacetylation of NAG along with the hydrolysis of β-glycosidic bonds, to yield glucosamine as a major product. Likewise, a mechanochemical technique using NaOH caused depolymerization accompanied by partial deacetylation, which produced low molecular weight chitosan.270 Such deacetylation led to the production of mixtures containing NAG-rich and glucosamine-rich polysaccharides, making their further utilization complicated.In recent years, the transformation of NAG into a variety of N-containing compounds has been explored. Similar to the glucose-to-HMF transformation, the most extensively investigated route for NAG is dehydration to 2-acetamido-2,3-dideoxy-D-erythro-hex-2-enofuranose (Chromogen I), 3-acetamido-5-(1′,2′-dihydroxyethyl)furan (Chromogen III), and 3-acetamido-5-acetylfuran (3A5AF). Due to the reaction scheme depicted in Fig. 35, the good yields of two Chromogen compounds (37.0% and 34.5% for Chromogen I and III, respectively) were achieved in subcritical water in the absence of catalysts;271 the presence of acid catalysts decreased the yield of Chromogen III because of its further dehydration to 3A5AF. Due to the presence of vicinal hydroxy groups, both Chromogen I and III can be assumed to undergo further transformation into N-containing unsaturated products via deoxydehydration (DODH) reactions. The use of the appropriate combination of solvent(s) and additive(s) in NAG transformation provided 3A5AF as a major product; for example, the system using boric acid in 1-butyl-3-methylimidazolium ([BMIM]) chloride afforded 60.0% yield of 3A5AF272 and a different system employing boric acid and sodium chloride in a mixture of [BMIM]Cl and [BMIM]HSO4 with ethyl acetate as an in situ extractant produced 3A5AF in 55.6%.273 Other routes for NAG transformation into N-containing chemicals have also been developed as exemplified by the retro-aldol reaction + hydrogenation to N-acetylmonoethanolamine,274 its further oxidation to acetylglycine,275 and hydrogenation + dehydration to 2-acetamide-2-deoxyisosorbide276,277 (see Fig. 35(ii) and (iii)).
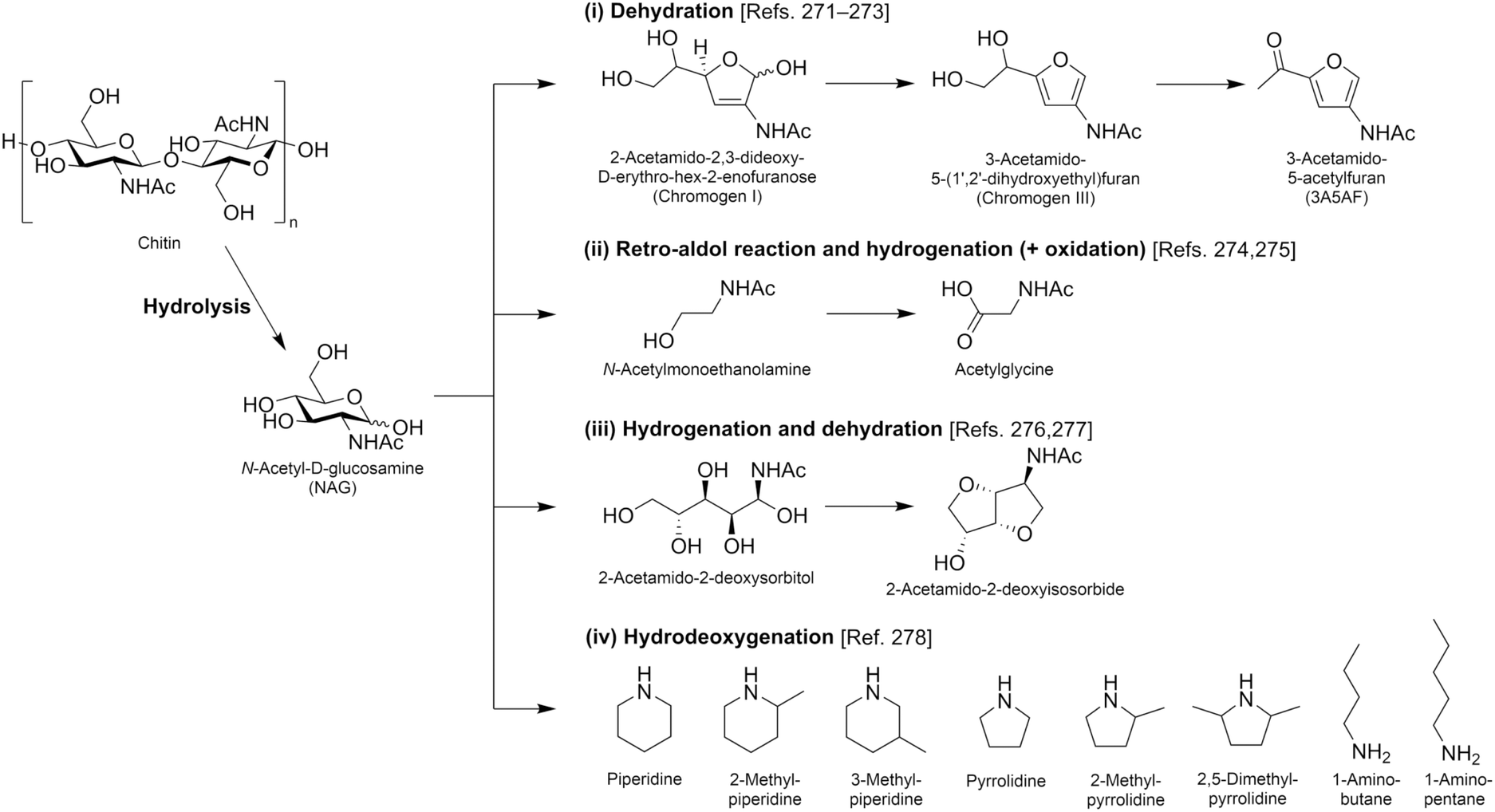 | ||
| Fig. 35 Derivatization of chitin-derived N-acetyl-D-glucosamine (NAG). | ||
The hydrodeoxygenation of NAG has recently been achieved by using a 3 mol% Ru/C catalyst in a 0.6 M phosphoric acid solution, by which a mixture of various aliphatic amines were produced in ca. 50% of their total yield under optimized conditions (Fig. 35(iv)).278 In this system, both Ru/C and phosphoric acid played important roles: the Ru/C catalyst promoted the hydrodeoxygenation of NAG to form aliphatic amines and phosphoric acid was proposed to form adducts with such amine products and stabilize them. When the system lacked either Ru/C or phosphoric acid, the total yield of aliphatic amines became less than half or even zero. Although the yield and selectivity toward desired amine(s) need to be improved, this report demonstrated the possibility of the production of useful amines from chitin-derived NAG. By contrast, the removal of all the hydroxy groups from NAG requires a lot of hydrogen and also narrows the application of NAG-derived compounds. In this context, the partial and selective removal of specific hydroxy group(s) from NAG molecules can be invoked as a promising means. For this objective, we infer that the examples of the hydrodeoxygenation of cellulose-derived compounds in the previous sections as well as the ones summarized in our previous review214 will be helpful for the precise design of catalysts and reaction systems due to the structural similarity between glucose and NAG.
The characteristics of chitin—which occurs naturally and abundantly as well as containing nitrogen atoms—make it attractive as a source of valuable N-containing chemicals. Yet, all the reactions of NAG exemplified above still suffer from low yield and/or selectivity of the desired products and necessitate further improvement by modifying and optimizing the catalytic systems. Considering that N-containing compounds are often used as fine chemicals, the well-controlled and selective (i.e., site-specific) transformation of chitin-derived compounds is required. For such an objective, homogeneous catalysts, which can be designed and synthesized to gain specific function(s) more precisely than heterogeneous catalysts, should have great potential. Yet, when there is the desire to convert chitin-derived compounds into bulk chemicals like building blocks for polymers due to the presence of both N- and O-containing functionalities, heterogeneous catalysts, which enable the large-scale production of desired products using continuous flow-type reactors, are worth investigating. We further note that in some cases, chitin and NAG have been converted into non-N-containing chemicals as summarized in previous review papers;260,264 however, without a specific reason, such transformations accompanied by denitrogenation should be avoided since non-N-containing products can also be synthesized from cellulosic biomass.
7. Conclusion and outlook
Hydrodeoxygenation is a common method for biomass valorization to fuels and chemicals, and remarkable progress has been achieved in the field for the development of heterogeneous catalysts and reaction routes. This review focuses on heterogeneous catalysts, which are generally more suitable than homogeneous catalysts because hydrogen molecules can be activated easily on a solid metal surface. For the production of bulk chemicals, heterogeneous catalysts are advantageous over homogeneous ones because of the ease of separation and applicability to continuous flow-type reactors. In most cases, a liquid-phase reaction is inevitable because of the low vapor pressure of the biomass-derived substrates. Mass transfer is sometimes problematic in the case of heterogeneous catalysts and needs to be improved by altering their structures such as morphology and pores (from micropores in crystals to macropores in pelletized catalysts). Leaching and in the worse case the involvement of leached species in the reaction are critical issues in liquid-phase heterogeneous catalysis and should be carefully checked. Meanwhile, the production of fine chemicals from biomass-derived compounds that typically possess multiple functionalities requires site-specific and/or chemoselective transformation. For such selective transformation, homogeneous catalysts, which can be designed and synthesized more precisely than heterogeneous ones, are promising candidates. Another advantage of homogeneous catalytic systems is the ease of scale up at the level of productivity for fine chemicals and a lack of mass transfer issues. In contrast, the development of highly durable homogeneous catalysts for selective hydrodeoxygenation is very challenging because of the reactivities of C–X (X = O, N, halogen, etc.) bonds in ligands and the difficulty of hydrogen activation. Alcohols are relatively inexpensive reducing agents that can be utilized in homogeneous catalysis; however, their use as reducing agents for deoxygenation reactions leads to a loss of selectivity because the biomass-derived substrates also contain alcoholic OH groups. The combination of homogeneous catalyst complexes for C–O bond activation with metallic catalysts for hydrogen activation is also a possible approach, although examples of such systems are limited thus far.As mentioned in previous sections, a variety of reaction routes such as direct C–O hydrogenolysis, dehydration + hydrogenation, dehydrogenation + dehydration + hydrogenation, and deoxydehydration ( + hydrogenation) are regarded as one-pot hydrodeoxygenation. Appropriate catalysts for each reaction process that give high yields of target products have also been developed, and progress will contribute to the wider scope of biomass-derived platform chemicals and products. Therefore, we selected and proposed potential platform chemicals for the biomass refinery in this review article, which included erythritol (+1,4-anhydroerythritol), levoglucosan (+levoglucosenone), glucaric acid, alkyl glycosides, and N-acetylglucosamine.
Practically feasible methods for the production of potential platform chemicals from biomass have not yet been established, but are under development. However, we believe that progress in efficient conversion technology for the potential platform chemicals introduced here will also promote the development of upstream processes for producing such platform chemicals. Of course, the efficiency of the current conversion technology of potential platform chemicals is not satisfactory and needs to be improved. One of the important future tasks is the substitution of noble metal catalyst components with non-noble metal ones; even if noble metal species are used, the development of catalysts with a very large TON, good reusability, and regenerability is necessary. From the viewpoint of catalytic processes, namely those using heterogeneous catalysts, the proposed catalysts are more applicable to continuous flow reactors, where leaching of the catalyst components must be completely suppressed. These tasks are very challenging and will be connected to decarbonization without using fossil resources.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (S) (KAKENHI, 18H05247) and a Grand-in-Aid for Early-Career Scientists (19K15355 and 21K14458) from the Japan Society for the Promotion of Science (JSPS).References
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS.
- A. Gulzar, A. Gulzar, M. B. Ansari, F. He, S. Gai and P. Yang, Chem. Eng. J. Adv., 2020, 3, 100013 CrossRef CAS.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, 2004, DOI:10.2172/15008859.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- G. G. Millán, S. Hellsten, J. Llorca, R. Luque, H. Sixta and A. M. Balu, ChemCatChem, 2019, 11, 2022–2042 CrossRef.
- B. Danon, G. Marcotullio and W. de Jong, Green Chem., 2014, 16, 39–54 RSC.
- T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, ACS Sustainable Chem. Eng., 2019, 7, 9601–9612 CrossRef CAS.
- T. Asano, H. Takagi, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2019, 21, 6133–6145 RSC.
- T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, Appl. Catal., A, 2020, 602, 117723 CrossRef CAS.
- A. Jaswal, P. P. Singh and T. Mondal, Green Chem., 2022, 24, 510–551 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- H. Li, S. Yang, A. Riisager, A. Pandey, R. S. Sangwan, S. Saravanamurugan and R. Luque, Green Chem., 2016, 18, 5701–5735 RSC.
- X. Kong, Y. Zhu, Z. Fang, J. A. Kozinski, I. S. Butler, L. Xu, H. Song and X. Wei, Green Chem., 2018, 20, 3657–3682 RSC.
- Y. Nakagawa, M. Yabushita and K. Tomishige, Trans. Tianjin Univ., 2021, 27, 165–179 CrossRef CAS.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 2976–2982 CrossRef CAS PubMed.
- G. R. Akien, L. Qi and I. T. Horváth, Chem. Commun., 2012, 48, 5850–5852 RSC.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC.
- S. P. Teong, G. Yi and Y. Zhang, Green Chem., 2014, 16, 2015–2026 RSC.
- M. Yabushita, P. Li, T. Islamoglu, H. Kobayashi, A. Fukuoka, O. K. Farha and A. Katz, Ind. Eng. Chem. Res., 2017, 56, 7141–7148 CrossRef CAS.
- K. I. Galkin, E. A. Krivodaeva, L. V. Romashov, S. S. Zalesskiy, V. V. Kachala, J. V. Burykina and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 8338–8342 CrossRef CAS PubMed.
- M. Kim, Y. Su, A. Fukuoka, E. J. M. Hensen and K. Nakajima, Angew. Chem., Int. Ed., 2018, 57, 8235–8239 CrossRef CAS PubMed.
- J. J. Wiesfeld, M. Kim, K. Nakajima and E. J. M. Hensen, Green Chem., 2020, 22, 1229–1238 RSC.
- M. C. Allen, A. J. Hoffman, T.-w. Liu, M. S. Webber, D. Hibbitts and T. J. Schwartz, ACS Catal., 2020, 10, 6771–6785 CrossRef CAS.
- F. J. A. G. Coumans, Z. Overchenko, J. J. Wiesfeld, N. Kosinov, K. Nakajima and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2022, 10, 3116–3130 CrossRef CAS.
- Y. Nakagawa and K. Tomishige, Catal. Commun., 2010, 12, 154–156 CrossRef CAS.
- A. Turkin, S. Eyley, G. Preegel, W. Thielemans, E. Makshina and B. F. Sels, ACS Catal., 2021, 11, 9204–9209 CrossRef CAS.
- G. J. S. Dawes, E. L. Scott, J. Le Nôtre, J. P. M. Sanders and J. H. Bitter, Green Chem., 2015, 17, 3231–3250 RSC.
- D. A. Rzechonek, A. Dobrowolski, W. Rymowicz and A. M. Mirończuk, Crit. Rev. Biotechnol., 2018, 38, 620–633 CrossRef CAS PubMed.
- M. Rakicka-Pustułka, A. M. Mirończuk, E. Celińska, W. Białas and W. Rymowicz, J. Cleaner Prod., 2020, 257, 120533 CrossRef.
- Y. Nakagawa, T. Kasumi, J. Ogihara, M. Tamura, T. Arai and K. Tomishige, ACS Omega, 2020, 5, 2520–2530 CrossRef CAS PubMed.
- S. Maduskar, V. Maliekkal, M. Neurock and P. J. Dauenhauer, ACS Sustainable Chem. Eng., 2018, 6, 7017–7025 CrossRef CAS.
- C. M. Lakshmanan, B. Gal-or and H. E. Hoelscher, Ind. Eng. Chem. Prod. Res. Dev., 1969, 8, 261–267 CrossRef CAS.
- L.-Q. Jiang, Z. Fang, Z.-L. Zhao, A.-Q. Zheng, X.-B. Wang and H.-B. Li, Renewable Sustainable Energy Rev., 2019, 105, 215–229 CrossRef CAS.
- I. G. Hakeem, P. Halder, M. H. Marzbali, S. Patel, S. Kundu, J. Paz-Ferreiro, A. Surapaneni and K. Shah, J. Environ. Chem. Eng., 2021, 9, 105614 CrossRef CAS.
- I. I. Junior, M. A. do Nascimento, R. O. M. A. de Souza, A. Dufour and R. Wojcieszak, Green Chem., 2020, 22, 5859–5880 RSC.
- W. Yin, Z. Tang, R. H. Venderbosch, Z. Zhang, C. Cannilla, G. Bonura, F. Frusteri and H. J. Heeres, ACS Catal., 2016, 6, 4411–4422 CrossRef CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- X. Liu, X. Liu, N. Li, P. Ma and Y. Zhang, Green Chem., 2021, 23, 1353–1360 RSC.
- O. V. Manaenkov, O. V. Kislitsa, V. G. Matveeva, E. M. Sulman, M. G. Sulman and L. M. Bronstein, Front. Chem., 2019, 7, 834 CrossRef CAS PubMed.
- L. S. Ribeiro, J. J. M. Órfão and M. F. R. Pereira, Mater. Today Sustainability, 2021, 11–12, 100058 CrossRef.
- A. B. Bindwal and P. D. Vaidya, Ind. Eng. Chem. Res., 2013, 52, 17781–17789 CrossRef CAS.
- K. Fabičovicová, M. Lucas and P. Claus, Green Chem., 2015, 17, 3075–3083 RSC.
- C. Li, G. Xu, K. Li, C. Wang, Y. Zhang and Y. Fu, Chem. Commun., 2019, 55, 7663–7666 RSC.
- Y. Jia and H. Liu, Catal. Sci. Technol., 2016, 6, 7042–7052 RSC.
- S. Zhu, X. Gao, Y. Zhu and Y. Li, Green Chem., 2016, 18, 782–791 RSC.
- X. Wang, A. K. Beine, P. J. C. Hausoul and R. Palkovits, ChemCatChem, 2019, 11, 4123–4129 CrossRef CAS.
- Q. Xia, X. Jin, G. Zhang, M. Liu, J. Wang, Y. Li, T. Fang, J. Ding, D. Zhang, K. Meng, X. Chen and C. Yang, Chem. Rec., 2021, 21, 133–148 CrossRef CAS PubMed.
- Y. Nakagawa, M. Tamura and K. Tomishige, Res. Chem. Intermed., 2018, 44, 3879–3903 CrossRef CAS.
- S. Kandasamy, S. P. Samudrala and S. Bhattacharya, Catal. Sci. Technol., 2019, 9, 567–577 RSC.
- P. Galletti, F. Moretti, C. Samorì and E. Tagliavini, Green Chem., 2007, 9, 987–991 RSC.
- K. C. Marion, Z. Wooke and N. L. B. Pohl, Carbohydr. Res., 2018, 468, 23–29 CrossRef CAS PubMed.
- S. Kudo, Z. Zhou, K. Norinaga and J. Hayashi, Green Chem., 2011, 13, 3306–3311 RSC.
- X.-n. Ye, Q. Lu, X. Wang, H.-q. Guo, M.-s. Cui, C.-q. Dong and Y.-p. Yang, ACS Sustainable Chem. Eng., 2017, 5, 10815–10825 CrossRef CAS.
- S. Saragai, S. Kudo, J. Sperry, U. P. M. Ashik, S. Asano and J. Hayashi, Bioresour. Technol., 2022, 344, 126323 CrossRef CAS PubMed.
- O. Oyola-Rivera, J. He, G. W. Huber, J. A. Dumesic and N. Cardona-Martínez, Green Chem., 2019, 21, 4988–4999 RSC.
- F. Cao, T. J. Schwartz, D. J. McClelland, S. H. Krishna, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
- S. H. Krishna, T. W. Walker, J. A. Dumesic and G. W. Huber, ChemSusChem, 2017, 10, 129–138 CrossRef CAS PubMed.
- X. Huang, S. Kudo, J. Sperry and J. Hayashi, ACS Sustainable Chem. Eng., 2019, 7, 5892–5899 CrossRef CAS.
- X. Huang, A. Xu, X. Bu, Y. Huang and J. Ran, Biomass Convers. Biorefin., 2021 DOI:10.1007/s13399-021-01549-z.
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS PubMed.
- J. Mazarío, M. P. Romero, P. Concepción, M. Chávez-Sifontes, R. A. Spanevello, M. B. Comba, A. G. Suárez and M. E. Domine, Green Chem., 2019, 21, 4769–4785 RSC.
- M. De bruyn, C. Sener, D. D. Petrolini, D. J. McClelland, J. He, M. R. Ball, Y. Liu, L. Martins, J. A. Dumesic, G. W. Huber and B. M. Weckhuysen, Green Chem., 2019, 21, 5000–5007 RSC.
- X. Huang, T. Liu, J. Wang, F. Wei, J. Ran and S. Kudo, J. Energy Inst., 2020, 93, 2505–2510 CrossRef CAS.
- M. De bruyn, P. Cuello-Penaloza, M. Cendejas, I. Hermans, J. He, S. H. Krishna, D. M. Lynn, J. A. Dumesic, G. W. Huber and B. M. Weckhuysen, ACS Sustainable Chem. Eng., 2019, 7, 13430–13436 CrossRef CAS PubMed.
- S. H. Krishna, M. De bruyn, Z. R. Schmidt, B. M. Weckhuysen, J. A. Dumesic and G. W. Huber, Green Chem., 2018, 20, 4557–4565 RSC.
- P. Cuello-Penaloza, S. H. Krishna, M. De bruyn, B. M. Weckhuysen, E. A. Lebrón-Rodríguez, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Sustainable Chem. Eng., 2021, 9, 16123–16132 CrossRef CAS.
- B. Urbas, US Pat, 4820880A, 1989 Search PubMed.
- K. Strensrud and C.-c. Ma, US Pat, 0044123A1, 2017 Search PubMed.
- S. H. Krishna, D. J. McClelland, Q. A. Rashke, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1278–1285 RSC.
- J. He, S. P. Burt, M. Ball, D. Zhao, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8, 1427–1439 CrossRef CAS.
- S. H. Krishna, L. Zhang, I. Hermans, G. W. Huber, T. F. Kuech and J. A. Dumesic, J. Catal., 2020, 389, 111–120 CrossRef CAS.
- S. H. Krishna, R. S. Assary, Q. A. Rashke, Z. R. Schmidt, L. A. Curtiss, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8, 3743–3753 CrossRef CAS.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS.
- J. He, S. P. Burt, M. R. Ball, I. Hermans, J. A. Dumesic and G. W. Huber, Appl. Catal., B, 2019, 258, 117945 CrossRef.
- Y. Nakagawa, M. Tamura and K. Tomishige, J. Jpn. Pet. Inst., 2017, 60, 1–9 CrossRef CAS.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS.
- M. Zhou, S. H. Krishna, M. De bruyn, B. M. Weckhuysen, L. A. Curtiss, J. A. Dumesic, G. W. Huber and R. S. Assary, ACS Sustainable Chem. Eng., 2020, 8, 16339–16349 CrossRef CAS.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
- W. Luo, W. Cao, P. C. A. Bruijnincx, L. Lin, A. Wang and T. Zhang, Green Chem., 2019, 21, 3744–3768 RSC.
- F. Shafizadeh, D. D. Ward and D. Pang, Carbohydr. Res., 1982, 102, 217–230 CrossRef CAS.
- M. S. Miftakhov, I. N. Gaisina and F. A. Valeev, Russ. Chem. Bull., 1996, 45, 1942–1944 CrossRef.
- M. B. Comba, Y.-h. Tsai, A. M. Sarotti, M. I. Mangione, A. G. Suárez and R. A. Spanevello, Eur. J. Org. Chem., 2018, 590–604 CrossRef CAS.
- L. Hughes, C. R. McElroy, A. C. Whitwood and A. J. Hunt, Green Chem., 2018, 20, 4423–4427 RSC.
- Y. Nakagawa, M. Tamura and K. Tomishige, Fuel Process. Technol., 2019, 193, 404–422 CrossRef CAS.
- W. von Rybinski, Curr. Opin. Colloid Interface Sci., 1996, 1, 587–597 CrossRef CAS.
- K. Holmberg, Curr. Opin. Colloid Interface Sci., 2001, 6, 148–159 CrossRef CAS.
- Natural Surfactants Market Size, Share & COVID-19 Impact Analysis, By Product (Anionics, Nonionics, Cationics, and Amphoterics), By Application (Detergents, Industrial & Institutional, Personal Care, Oilfield Chemicals, and Others), and Regional Forecast, 2021–2028, https://www.fortunebusinessinsights.com/natural-surfactants-market-105407, (accessed May 2022).
- E. Fischer, Ber. Dtsch. Chem. Ges., 1893, 26, 2400–2412 CrossRef.
- H. P. Wessel, J. Carbohydr. Chem., 1988, 7, 263–269 CrossRef CAS.
- N. Aoyama and S. Kobayashi, Chem. Lett., 2006, 35, 238–239 CrossRef CAS.
- B. Roy and B. Mukhopadhyay, Tetrahedron Lett., 2007, 48, 3783–3787 CrossRef CAS.
- A. T. J. W. de Goede, M. P. J. van Deurzen, I. G. van der Leij, A. M. van der Heijden, J. M. A. Baas, F. van Rantwijk and H. van Bekkum, J. Carbohydr. Chem., 1996, 15, 331–349 CrossRef CAS.
- L. F. Bornaghi and S.-A. Poulsen, Tetrahedron Lett., 2005, 46, 3485–3488 CrossRef CAS.
- W. G. Ramdani, A. Karam, K. De Oliveira Vigier, S. Rio, A. Ponchel and F. Jérôme, Mol. Catal., 2019, 468, 125–129 CrossRef CAS.
- W. Koenigs and E. Knorr, Ber. Dtsch. Chem. Ges., 1901, 34, 957–981 CrossRef.
- T. Mukaiyama, Y. Murai and S. Shoda, Chem. Lett., 1981, 10, 431–432 CrossRef.
- T. Matsumoto, H. Maeta, K. Suzuki and G. Tsuchihashi, Tetrahedron Lett., 1988, 29, 3567–3570 CrossRef CAS.
- K. Suzuki, H. Maeta, T. Matsumoto and G. Tsuchihashi, Tetrahedron Lett., 1988, 29, 3571–3574 CrossRef CAS.
- R. R. Schmidt and J. Michel, Angew. Chem., Int. Ed. Engl., 1980, 19, 731–732 CrossRef.
- M. M. Nielsen and C. M. Pedersen, Chem. Rev., 2018, 118, 8285–8358 CrossRef CAS PubMed.
- K. Toshima and K. Tatsuta, Chem. Rev., 1993, 93, 1503–1531 CrossRef CAS.
- B. K. Gorityala, J. Ma, K. K. Pasunooti, S. Cai and X.-W. Liu, Green Chem., 2011, 13, 573–577 RSC.
- D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 CrossRef CAS PubMed.
- Y.-B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC.
- M. Yabushita, H. Kobayashi and A. Fukuoka, Appl. Catal., B, 2014, 145, 1–9 CrossRef CAS.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768 CrossRef CAS PubMed.
- Z. Zhou, D. Liu and X. Zhao, Renewable Sustainable Energy Rev., 2021, 146, 111169 CrossRef CAS.
- W. Deng, M. Liu, Q. Zhang and Y. Wang, Catal. Today, 2011, 164, 461–466 CrossRef CAS.
- Z. Wen, Z. Ma, F. Mai, F. Yan, L. Yu, M. Jin, Y. Sang, Y. Bai, K. Cui, K. Wu, M. Chen, H. Chen and Y. Li, Catal. Today, 2020, 355, 272–279 CrossRef CAS.
- L. Xue, K. Cheng, H. Zhang, W. Deng, Q. Zhang and Y. Wang, Catal. Today, 2016, 274, 60–66 CrossRef CAS.
- N. Villandier and A. Corma, ChemSusChem, 2011, 4, 508–513 CrossRef CAS PubMed.
- H.-c. Zhou, B. Wang, X.-f. Guo, X.-y. Zhang, X.-h. Wei, C. Peng, D. R. MacFarlane and Y.-z. Yuan, Chem. Commun., 2018, 54, 11969–11972 RSC.
- W. Zheng, Y. Cui, Z. Xu, L. Zhao and W. Sun, Can. J. Chem. Eng., 2018, 96, 1250–1255 CrossRef CAS.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–474 RSC.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- A. Shrotri, L. K. Lambert, A. Tanksale and J. Beltramini, Green Chem., 2013, 15, 2761–2768 RSC.
- F. Boissou, N. Sayoud, K. De Oliveira Vigier, A. Barakat, S. Marinkovic, B. Estrine and F. Jérôme, ChemSusChem, 2015, 8, 3263–3269 CrossRef CAS PubMed.
- S. Zhu, J. Guo, X. Wang, J. Wang and W. Fan, ChemSusChem, 2017, 10, 2547–2559 CrossRef CAS PubMed.
- A. M. R. Galletti, C. Antonetti, S. Fulignati and D. Licursi, Catalysts, 2020, 10, 1221 CrossRef CAS.
- M. Zou, J. Chen, Y. Wang, M. Li, C. Zhang and X. Yang, J. Surfactants Deterg., 2016, 19, 879–884 CrossRef CAS.
- J. Chen, J. Li, K. Liu, M. Hong, R. You, P. Qu and M. Chen, ACS Omega, 2019, 4, 16372–16377 CrossRef CAS PubMed.
- J. Morrill, A. Månberger, A. Rosengren, P. Naidjonoka, P. von Freiesleben, K. B. R. M. Krogh, K.-E. Bergquist, T. Nylander, E. N. Karlsson, P. Adlercreutz and H. Stålbrand, Appl. Microbiol. Biotechnol., 2018, 102, 5149–5163 CrossRef CAS PubMed.
- P. J. C. Hausoul, L. Negahdar, K. Schute and R. Palkovits, ChemSusChem, 2015, 8, 3323–3330 CrossRef CAS PubMed.
- J. M. Harris, M. D. Keranen and G. A. O'Doherty, J. Org. Chem., 1999, 64, 2982–2983 CrossRef CAS PubMed.
- T. Hyodo, Y. Katayama and Y. Kobayashi, Tetrahedron Lett., 2009, 50, 3547–3549 CrossRef CAS.
- M. Saquib, I. Husain, S. Sharma, G. Yadav, V. K. Singh, S. K. Sharma, P. Shah, M. I. Siddiqi, B. Kumar, J. Lal, G. K. Jain, B. S. Srivastava, R. Srivastava and A. K. Shaw, Eur. J. Med. Chem., 2011, 46, 2217–2223 CrossRef CAS PubMed.
- S. J. Danishefsky, D. M. Armistead, F. E. Wincott, H. G. Selnick and R. Hungate, J. Am. Chem. Soc., 1989, 111, 2967–2980 CrossRef CAS.
- H.-Y. L. Wang and G. A. O'Doherty, Chem. Commun., 2011, 47, 10251–10253 RSC.
- W. Muramatsu, S. Tanigawa, Y. Takemoto, H. Yoshimatsu and O. Onomura, Chem. – Eur. J., 2012, 18, 4850–4853 CrossRef CAS PubMed.
- W. Song, S. Wang and W. Tang, Chem. – Asian J., 2017, 12, 1027–1042 CrossRef CAS PubMed.
- Y. Nakagawa, K. Mori, K. Chen, Y. Amada, M. Tamura and K. Tomishige, Appl. Catal., A, 2013, 468, 418–425 CrossRef CAS.
- J. Wang, M. Yang and A. Wang, Chin. J. Catal., 2020, 41, 1311–1319 CrossRef CAS.
- S. H. Krishna, J. Cao, M. Tamura, Y. Nakagawa, M. De Bruyn, G. S. Jacobson, B. M. Weckhuysen, J. A. Dumesic, K. Tomishige and G. W. Huber, ACS Sustainable Chem. Eng., 2020, 8, 800–805 CrossRef CAS.
- J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767–775 CrossRef CAS PubMed.
- S. Raju, M.-E. Moret and R. J. M. K. Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS.
- A. R. Petersen and P. Fristrup, Chem. – Eur. J., 2017, 23, 10235–10243 CrossRef CAS PubMed.
- L. J. Donnelly, S. P. Thomas and J. B. Love, Chem. – Asian J., 2019, 14, 3782–3790 CrossRef CAS PubMed.
- K. A. DeNike and S. M. Kilyanek, R. Soc. Open Sci., 2019, 6, 191165 CrossRef PubMed.
- N. N. Tshibalonza and J.-C. M. Monbaliu, Green Chem., 2020, 22, 4801–4848 RSC.
- M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2020, 10, 3805–3824 RSC.
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393–6397 CrossRef CAS.
- Y. Nakagawa, S. Tazawa, T. Wang, M. Tamura, N. Hiyoshi, K. Okumura and K. Tomishige, ACS Catal., 2018, 8, 584–595 CrossRef CAS.
- J. Cao, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Catal., 2019, 9, 3725–3729 CrossRef CAS.
- K. Yamaguchi, J. Cao, M. Betchaku, Y. Nakagawa, M. Tamura, A. Nakayama, M. Yabushita and K. Tomishige, ChemSusChem, 2022, 15, e202102663 CrossRef CAS PubMed.
- S. Hacatrjan, L. Liu, J. Gan, Y. Nakagawa, J. Cao, M. Yabushita, M. Tamura and K. Tomishige, Catal. Sci. Technol., 2022, 12, 2146–2161 RSC.
- A. L. Denning, H. Dang, Z. Liu, K. M. Nicholas and F. C. Jentoft, ChemCatChem, 2013, 5, 3567–3570 CrossRef CAS.
- M. Lupacchini, A. Mascitti, V. Canale, L. Tonucci, E. Colacino, M. Passacantando, A. Marrone and N. d'Alessandro, Catal. Sci. Technol., 2019, 9, 3036–3046 RSC.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 1897–1900 CrossRef CAS PubMed.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 3213–3226 CrossRef CAS.
- B. MacQueen, E. Barrow, G. R. Castro, Y. Pagan-Torres, A. Heyden and J. Lauterbach, Ind. Eng. Chem. Res., 2019, 58, 8681–8689 CrossRef CAS.
- B. MacQueen, M. Royko, B. S. Crandall, A. Heyden, Y. J. Pagán-Torres and J. Lauterbach, Catalysts, 2021, 11, 108 CrossRef CAS.
- K. Tomishige, Y. Nakagawa and M. Tamura, Chin. Chem. Lett., 2020, 31, 1071–1077 CrossRef CAS.
- M. Tamura, N. Yuasa, J. Cao, Y. Nakagawa and K. Tomishige, Angew. Chem., Int. Ed., 2018, 57, 8058–8062 CrossRef CAS PubMed.
- J. Cao, M. Tamura, R. Hosaka, A. Nakayama, J. Hasegawa, Y. Nakagawa and K. Tomishige, ACS Catal., 2020, 10, 12040–12051 CrossRef CAS.
- V. Bilik and K. Tihlárik, Chem. Zvesti, 1973, 28, 106–109 Search PubMed.
- F. Ju, D. VanderVelde and E. Nikolla, ACS Catal., 2014, 4, 1358–1364 CrossRef CAS.
- R. Bermejo-Deval, M. Orazov, R. Gounder, S.-J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288–2297 CrossRef CAS.
- S.-J. Hwang, R. Gounder, Y. Bhawe, M. Orazov, R. Bermejo-Deval and M. E. Davis, Top. Catal., 2015, 58, 435–440 CrossRef CAS.
- V. R. Jumde, N. N. H. M. Eisink, M. D. Witte and A. J. Minnaard, J. Org. Chem., 2016, 81, 11439–11443 CrossRef CAS PubMed.
- I. C. Wan, T. A. Hamlin, N. N. H. M. Eisink, N. Marinus, C. de Boer, C. A. Vis, J. D. C. Codée, M. D. Witte, A. J. Minnaard and F. M. Bickelhaupt, Eur. J. Org. Chem., 2021, 632–636 CrossRef CAS.
- K. K. Kapanji, S. Farzad and J. F. Görgens, J. Cleaner Prod., 2021, 295, 126339 CrossRef CAS.
- T. N. Smith, K. Hash, C.-L. Davey, H. Mills, H. Williams and D. E. Kiely, Carbohydr. Res., 2012, 350, 6–13 CrossRef CAS PubMed.
- Q. Zhang, Z. Wan, I. K. M. Yu and D. C. W. Tsang, J. Cleaner Prod., 2021, 312, 127745 CrossRef CAS.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- J. Lee, B. Saha and D. G. Vlachos, Green Chem., 2016, 18, 3815–3822 RSC.
- H. Shi, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, Org. Process Res. Dev., 2018, 22, 1653–1662 CrossRef CAS.
- E. Derrien, M. Ahmar, E. Martin-Sisteron, G. Raffin, Y. Queneau, P. Marion, M. Beyerle, C. Pinel and M. Besson, Ind. Eng. Chem. Res., 2018, 57, 4543–4552 CrossRef CAS.
- R. Sakuta and N. Nakamura, Int. J. Mol. Sci., 2019, 20, 3660 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 12905–12909 CrossRef CAS PubMed.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- B. Hočevar, A. Prašnikar, M. Huš, M. Grilc and B. Likozar, Angew. Chem., Int. Ed., 2021, 60, 1244–1253 CrossRef PubMed.
- X. Li, D. Wu, T. Lu, G. Yi, H. Su and Y. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4200–4204 CrossRef CAS PubMed.
- C. Boucher-Jacobs and K. M. Nicholas, Organometallics, 2015, 34, 1985–1990 CrossRef CAS.
- H. Zhang, X. Li, X. Su, E. L. Ang, Y. Zhang and H. Zhao, ChemCatChem, 2016, 8, 1500–1506 CrossRef CAS.
- N. Shin, S. Kwon, S. Moon, C. H. Hong and Y. G. Kim, Tetrahedron, 2017, 73, 4758–4765 CrossRef CAS.
- J. Li, M. Lutz, M. Otte and R. J. M. K. Gebbink, ChemCatChem, 2018, 10, 4755–4760 CrossRef CAS PubMed.
- A. Jefferson and R. S. Srivastava, Polyhedron, 2019, 160, 268–271 CrossRef CAS.
- J. Li, M. Lutz and R. J. M. K. Gebbink, Catal. Sci. Technol., 2020, 10, 3782–3788 RSC.
- J. Li, M. Lutz and R. J. M. K. Gebbink, Catalysts, 2020, 10, 754 CrossRef.
- R. T. Larson, A. Samant, J. Chen, W. Lee, M. A. Bohn, D. M. Ohlmann, S. J. Zuend and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 14001–14004 CrossRef CAS PubMed.
- J. Lin, H. Song, X. Shen, B. Wang, S. Xie, W. Deng, D. Wu, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 11017–11020 RSC.
- J. H. Jang, I. Ro, P. Christopher and M. M. Abu-Omar, ACS Catal., 2021, 11, 95–109 CrossRef CAS.
- W. Deng, L. Yan, B. Wang, Q. Zhang, H. Song, S. Wang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 4712–4719 CrossRef CAS PubMed.
- H. Shi, L. Zhang, Y. Wu, R. Yu, Y. Peng, Y. Wang, T. Li and W. Yang, Catal. Lett., 2021, 151, 338–343 CrossRef CAS.
- B. Hočevar, M. Grilc and B. Likozar, Catalysts, 2019, 9, 286 CrossRef.
- E. Skoog, J. H. Shin, V. Saez-Jimenez, V. Mapelli and L. Olsson, Biotechnol. Adv., 2018, 36, 2248–2263 CrossRef CAS PubMed.
- I. Khalil, G. Quintens, T. Junkers and M. Dusselier, Green Chem., 2020, 22, 1517–1541 RSC.
- T. Asano, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2016, 4, 6253–6257 CrossRef CAS.
- M. J. Gilkey, H. J. Cho, B. M. Murphy, J. Wu, D. G. Vlachos and B. Xu, ACS Appl. Energy Mater., 2020, 3, 99–105 CrossRef CAS.
- A. Marckwordt, F. El Ouahabi, H. Amani, S. Tin, N. V. Kalevaru, P. C. J. Kamer, S. Wohlrab and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 3486–3490 CrossRef CAS PubMed.
- K. Hatakeyama, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2020, 22, 4962–4974 RSC.
- L. Tomaszewska, A. Rywińska and W. Gładkowski, J. Ind. Microbiol. Biotechnol., 2012, 39, 1333–1343 CrossRef CAS PubMed.
- Y. Kobayashi, H. Iwata, D. Mizushima, J. Ogihara and T. Kasumi, Lett. Appl. Microbiol., 2015, 60, 475–480 CrossRef CAS PubMed.
- S. Güner, V. Wegat, A. Pick and V. Sieber, Green Chem., 2021, 23, 6583–6590 RSC.
- S. Schmidt, F. J. Gatti, M. Luitz, B. S. Ritter, B. Bruchmann and R. Mülhaupt, Macromolecules, 2017, 50, 2296–2303 CrossRef CAS.
- A. S. Amarasekara, S. R. Ali, H. Fernando, V. Sena and T. V. Timofeeva, SN Appl. Sci., 2019, 1, 212 CrossRef.
- P. Jin, J. Zhu, S. Yuan, G. Zhang, A. Volodine, M. Tian, J. Wang, P. Luis and B. Van der Bruggen, Chem. Eng. J., 2021, 406, 126796 CrossRef CAS.
- K. G. Childers, S. D. Dreher, J. Lee and J. M. Williams, Org. Process Res. Dev., 2006, 10, 934–936 CrossRef CAS.
- E. Arceo, P. Marsden, R. G. Bergman and J. A. Ellman, Chem. Commun., 2009, 3357–3359 RSC.
- K. Taniguchi, H. Nanao, O. Sato, A. Yamaguchi and M. Shirai, Mol. Catal., 2019, 477, 110519 CrossRef CAS.
- M. Schlaf, P. Ghosh, P. J. Fagan, E. Hauptman and R. M. Bullock, Adv. Synth. Catal., 2009, 351, 789–800 CrossRef CAS.
- D. Taher, M. E. Thibault, D. Di Mondo, M. Jennings and M. Schlaf, Chem. – Eur. J., 2009, 15, 10132–10143 CrossRef CAS PubMed.
- M. E. Thibault, D. V. DiMondo, M. Jennings, P. V. Abdelnur, M. N. Eberlin and M. Schlaf, Green Chem., 2011, 13, 357–366 RSC.
- A. D. Sutton, F. D. Waldie, R. Wu, M. Schlaf, L. A. P. Silks III and J. C. Gordon, Nat. Chem., 2013, 5, 428–432 CrossRef CAS PubMed.
- H.-J. Song, J. Deng, M.-S. Cui, X.-L. Li, X.-X. Liu, R. Zhu, W.-P. Wu and Y. Fu, ChemSusChem, 2015, 8, 4250–4255 CrossRef CAS PubMed.
- D.-H. Liu, T. J. Marks and Z. Li, ChemSusChem, 2019, 12, 5217–5223 CrossRef CAS PubMed.
- X. Jin, J. Shen, W. Yan, M. Zhao, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2015, 5, 6545–6558 CrossRef CAS.
- W. Wang, K. Nakagawa, T. Yoshikawa, T. Masuda, E. Fumoto, Y. Koyama, T. Tago and H. Fujitsuka, Appl. Catal., A, 2021, 619, 118152 CrossRef CAS.
- Y. Nakagawa, M. Tamura and K. Tomishige, J. Mater. Chem. A, 2014, 2, 6688–6702 RSC.
- A. D. da Silva Ruy, R. M. de Brito Alves, T. L. R. Hewer, D. de Aguiar Pontes, L. S. G. Teixeira and L. A. M. Pontes, Catal. Today, 2021, 381, 243–253 CrossRef CAS.
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC.
- Y. Amada, H. Watanabe, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, J. Phys. Chem. C, 2012, 116, 23503–23514 CrossRef CAS.
- S. Koso, H. Watanabe, K. Okumura, Y. Nakagawa and K. Tomishige, J. Phys. Chem. C, 2012, 116, 3079–3090 CrossRef CAS.
- T. Ebashi, Y. Ishida, Y. Nakagawa, S. Ito, T. Kubota and K. Tomishige, J. Phys. Chem. C, 2010, 114, 6518–6526 CrossRef CAS.
- L. Liu, S. Kawakami, Y. Nakagawa, M. Tamura and K. Tomishige, Appl. Catal., B, 2019, 256, 117775 CrossRef.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, ACS Catal., 2019, 9, 10913–10930 CrossRef CAS.
- E. M. Virgilio, C. L. Padró and M. E. Sad, ChemCatChem, 2021, 13, 3889–3906 CrossRef CAS.
- E. M. Virgilio, M. E. Sad and C. L. Padró, Appl. Catal., A, 2022, 643, 118691 CrossRef CAS.
- M. Gu, L. Liu, Y. Nakagawa, C. Li, M. Tamura, Z. Shen, X. Zhou, Y. Zhang and K. Tomishige, ChemSusChem, 2021, 14, 642–654 CrossRef CAS PubMed.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, ChemSusChem, 2015, 8, 628–635 CrossRef CAS PubMed.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Green Chem., 2016, 18, 165–175 RSC.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS.
- Y. Amada, H. Watanabe, Y. Hirai, Y. Kajikawa, Y. Nakagawa and K. Tomishige, ChemSusChem, 2012, 5, 1991–1999 CrossRef CAS PubMed.
- Y. Nakagawa, X. Ning, Y. Amada and K. Tomishige, Appl. Catal., A, 2012, 433–434, 128–134 CrossRef CAS.
- K. Tomishige, M. Tamura and Y. Nakagawa, Chem. Rec., 2014, 14, 1041–1054 CrossRef CAS PubMed.
- K. Tomishige, Y. Nakagawa and M. Tamura, Curr. Opin. Green Sustainable Chem., 2020, 22, 13–21 CrossRef.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2020, 22, 2375–2380 RSC.
- L. Liu, T. Asano, Y. Nakagawa, M. Gu, C. Li, M. Tamura and K. Tomishige, Appl. Catal., B, 2021, 292, 120164 CrossRef CAS.
- Y. Fan, S. Cheng, H. Wang, D. Ye, S. Xie, Y. Pei, H. Hu, W. Hua, Z. H. Li, M. Qiao and B. Zong, Green Chem., 2017, 19, 2174–2183 RSC.
- S. Cheng, Y. Fan, X. Zhang, Y. Zeng, S. Xie, Y. Pei, G. Zeng, M. Qiao and B. Zong, Appl. Catal., B, 2021, 297, 120428 CrossRef CAS.
- L. Liu, J. Cao, Y. Nakagawa, M. Betchaku, M. Tamura, M. Yabushita and K. Tomishige, Green Chem., 2021, 23, 5665–5679 RSC.
- B. Liu, N. Sekine, Y. Nakagawa, M. Tamura, M. Yabushita and K. Tomishige, ACS Sustainable Chem. Eng., 2022, 10, 1220–1231 CrossRef CAS.
- Y. Amada, N. Ota, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2014, 7, 2185–2192 CrossRef CAS PubMed.
- K. Chen, K. Mori, H. Watanabe, Y. Nakagawa and K. Tomishige, J. Catal., 2012, 294, 171–183 CrossRef CAS.
- T. Arai, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2016, 9, 1680–1688 CrossRef CAS PubMed.
- S. Koso, N. Ueda, Y. Shinmi, K. Okumura, T. Kizuka and K. Tomishige, J. Catal., 2009, 267, 89–92 CrossRef CAS.
- S. Koso, H. Watanabe, K. Okumura, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2012, 111–112, 27–37 CrossRef CAS.
- R. Toyoshima, J. Kawai, K. Isegawa, H. Kondoh, A. Junkaew, A. Nakayama, T. Asano, M. Tamura, Y. Nakagawa, M. Yabushita and K. Tomishige, J. Phys. Chem. C, 2021, 125, 4540–4549 CrossRef CAS.
- A. Shimao, S. Koso, N. Ueda, Y. Shinmi, I. Furikado and K. Tomishige, Chem. Lett., 2009, 38, 540–541 CrossRef CAS.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed.
- S. Albarracin-Suazo, L. F. de L. e Freitas, B. MacQueen, A. Heyden, J. A. Lauterbach, E. Nikolla and Y. J. Pagán-Torres, ACS Sustainable Chem. Eng., 2022, 10, 5719–5727 CrossRef.
- J. E. Ziegler, M. J. Zdilla, A. J. Evans and M. M. Abu-Omar, Inorg. Chem., 2009, 48, 9998–10000 CrossRef CAS PubMed.
- S. Murru, K. M. Nicholas and R. S. Srivastava, J. Mol. Catal. A: Chem., 2012, 363–364, 460–464 CrossRef CAS.
- S. Stanowski, K. M. Nicholas and R. S. Srivastava, Organometallics, 2012, 31, 515–518 CrossRef CAS.
- R. Pinkos, R. H. Fischer, B. Breitscheidel and P. Polanek, CA Pat, 2168458C, 2004 Search PubMed.
- T. Wang, S. Liu, M. Tamura, Y. Nakagawa, N. Hiyoshi and K. Tomishige, Green Chem., 2018, 20, 2547–2557 RSC.
- T. Wang, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2019, 12, 3615–3626 CrossRef CAS PubMed.
- L. E. Manzer, US Pat, 6593481B1, 2003 Search PubMed.
- T. Wang, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, React. Chem. Eng., 2020, 5, 1237–1250 RSC.
- Y. Zhang, D. Liu and Z. Chen, Biotechnol. Biofuels, 2017, 10, 299 CrossRef PubMed.
- T. C. Ezeji, N. Qureshi and H. P. Blaschek, Chem. Rec., 2004, 4, 305–314 CrossRef CAS PubMed.
- G. Pomalaza, P. A. Ponton, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2020, 10, 4860–4911 RSC.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Catal. Sci. Technol., 2016, 6, 5668–5683 RSC.
- S. D. Le and S. Nishimura, Appl. Catal., B, 2021, 282, 119619 CrossRef CAS.
- J.-P. Lange and S. H. Wadman, ChemSusChem, 2020, 13, 5329–5337 CrossRef CAS PubMed.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- X. Chen, H. Yang and N. Yan, Chem. – Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- X. Chen, S. Song, H. Li, G. Gözaydın and N. Yan, Acc. Chem. Res., 2021, 54, 1711–1722 CrossRef CAS PubMed.
- A. Percot, C. Viton and A. Domard, Biomacromolecules, 2003, 4, 12–18 CrossRef CAS PubMed.
- H. El Knidri, R. Belaabed, A. Addaou, A. Laajeb and A. Lahsini, Int. J. Biol. Macromol., 2018, 120, 1181–1189 CrossRef CAS PubMed.
- S. Cao, Y. Liu, L. Shi, W. Zhu and H. Wang, Green Chem., 2022, 24, 493–509 RSC.
- M. Yabushita, H. Kobayashi, K. Kuroki, S. Ito and A. Fukuoka, ChemSusChem, 2015, 8, 3760–3763 CrossRef CAS PubMed.
- N. Nguyen-Thi and N. Doucet, J. Biotechnol., 2016, 220, 25–32 CrossRef CAS PubMed.
- A. Zhang, C. Gao, J. Wang, K. Chen and P. Ouyang, Green Chem., 2016, 18, 2147–2154 RSC.
- J. A. Bohlmann, D. O. Schisler, K.-O. Hwang, J. P. Henning, J. R. Trinkle, T. B. Anderson, J. D. Steinke and A. Vanderhoff, US Pat, 6693188B2, 2004 Search PubMed.
- J.-K. Chen, C.-R. Shen and C.-L. Liu, Mar. Drugs, 2010, 8, 2493–2516 CrossRef CAS PubMed.
- X. Chen, H. Yang, Z. Zhong and N. Yan, Green Chem., 2017, 19, 2783–2792 RSC.
- M. Osada, S. Shoji, S. Suenaga and M. Ogata, Fuel Process. Technol., 2019, 195, 106154 CrossRef CAS.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642–4644 RSC.
- X. Chen, Y. Liu, F. M. Kerton and N. Yan, RSC Adv., 2015, 5, 20073–20080 RSC.
- F. D. Bobbink, J. Zhang, Y. Pierson, X. Chen and N. Yan, Green Chem., 2015, 17, 1024–1031 RSC.
- K. Techikawara, H. Kobayashi and A. Fukuoka, ACS Sustainable Chem. Eng., 2018, 6, 12411–12418 CrossRef CAS.
- T. Sagawa, H. Kobayashi, C. Murata, Y. Shichibu, K. Konishi and A. Fukuoka, ACS Sustainable Chem. Eng., 2019, 7, 14883–14888 CrossRef CAS.
- T. Sagawa, H. Kobayashi and A. Fukuoka, Mol. Catal., 2020, 498, 111282 CrossRef CAS.
- S. Xie, C. Jia, S. S. G. Ong, Z. Wang, M.-j. Zhu, Q. Wang, Y. Yang and H. Lin, iScience, 2020, 23, 101096 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Discussion about the model reactions of single-hydrodeoxygenation, energy profiles for the hydrogenolysis of glycerol and erythritol, and the hydrogenolysis of glycerol to 1,3-propanediol. See DOI: https://doi.org/10.1039/d2gc01289h |
| This journal is © The Royal Society of Chemistry 2022 |