
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAvoiding water reservoir effects in ALD of functional complex alkali oxides by using O3 as the oxygen source†
Henrik H.
Sønsteby
 *a,
Veronica A.-L. K.
Killi
a,
Linn M.
Rykkje
a,
Justin R.
Bickford
b,
Eric G.
Martin
b,
Robert C.
Hoffman
b and
Ola
Nilsen
*a
*a,
Veronica A.-L. K.
Killi
a,
Linn M.
Rykkje
a,
Justin R.
Bickford
b,
Eric G.
Martin
b,
Robert C.
Hoffman
b and
Ola
Nilsen
*a
aDepartment of Chemistry, University of Oslo, Blindern, 0315 Oslo, Norway. E-mail: henrik.sonsteby@kjemi.uio.no; ola.nilsen@kjemi.uio.no
bU.S. Army Research Laboratory, 2800 Powder Mill Rd., Adelphi, Maryland 20783, USA
First published on 14th December 2021
Abstract
Toxic Pb-containing piezo-, pyro- and ferroelectrics continue to dominate the market even though they were banned from use in consumer products more than a decade ago. There is a strong need for sustainable alternatives, but the lack of facile synthesis routes for thin films exhibiting suitable functional properties have limited the transition from Pb workhorse materials like Pb(Zr,Ti)O3 and Pb(Mg,Nb)O3 – PbTiO3. Atomic layer deposition has proven capable of the deposition of possible successors, such as LiNbO3, (K,Na)NbO3 and K(Ta,Nb)O3, albeit with limited control due to water reservoir effects resulting from the hygroscopicity of intermediate products. In this article, we show that replacing H2O with O3 in the deposition of complex alkali oxides provides an alternative and much more controlled process. We exemplify this by deposition of crystalline K(Ta,Nb)O3 with high compositional control and over a larger composition range than previously reported. This opens new doors to a simplified synthesis of polar functional lead-free alternatives.
Introduction
Piezo-, pyro- and ferroelectric materials are essential in modern world technology and constitute a multibillion US$ market. They find frequent use in sensors, actuators, transducers and power generators, and in more exotic technology devices such as non-volatile memory and electro-optics. Unfortunately, the major workhorse materials in current use typically contain lead (Pb), which is a potent environmental pollutant and acutely toxic to most life on earth. Although lead recycling is efficient and exposure from consumer products is kept at a minimum, lead mining continues to be a concern for workers, especially in developing countries. As a result, large economies such as the European Union and the Unites States enforce bans on use of lead in consumer products. The challenge is that synthesis of replacement materials is essentially non-existent, and thus exemptions from the bans are necessary until a new material technology is in place. This has resulted in a surge of effort to develop new, sustainable polar materials that can reduce or eliminate the use of lead.Alkali metal containing complex oxide perovskites is one family of materials that is predicted to challenge its lead-containing counterparts. Examples like (K,Na)NbO3 (KNN) and K(Ta,Nb)O3 (KTN) exhibit the desired functional properties for a range of applications within sensing, actuation and electro-optics.1–3 The challenge for both of these compounds is the lack of straightforward methods to deposit high quality crystals and/or thin films. Bulk crystal growth is slow, ineffective, and with unavoidable, large composition gradients throughout the resulting boule, limiting useful implementation in devices.3–6 Thin film techniques such as sol–gel methods, pulsed laser deposition (PLD) and metalorganic chemical vapor deposition (MOCVD) are also employed, but with severe limitations on device integration due to high-temperature processing or lack of uniformity. These films very often exhibit coarse grains, voids and/or inhomogeneous composition over applicable areas.7–15
Atomic layer deposition (ALD) is considered a promising technique for device integration of functional materials containing alkali metals, due to low deposition temperature and high conformity of growth. Based on self-limiting saturative surface chemistry, ALD has the potential to work around issues with precursor pre-reactions and non-uniformity.16 ALD processes for Na- and K-containing compounds were reported in 2014, and have since been expanded to include KNN and KTN, among others.17–21 The large majority of these processes utilize alkali metal tert-butoxides (e.g. NaOtBu or KOtBu, (Fig. 1)) and water, together with other binary metal process (e.g. Nb(OEt)5 + H2O). While this has proven to provide decent films of e.g. KNN and KTN, process control is significantly more difficult than for most ALD routes. This is likely due to a large water reservoir resulting from the hygroscopicity of the intermediary products, such as alkali metal hydroxides.22 Such effects have been shown to exhibit strong instrumental dependency, meaning that transferring the process between labs or to industry requires significant reoptimization.23
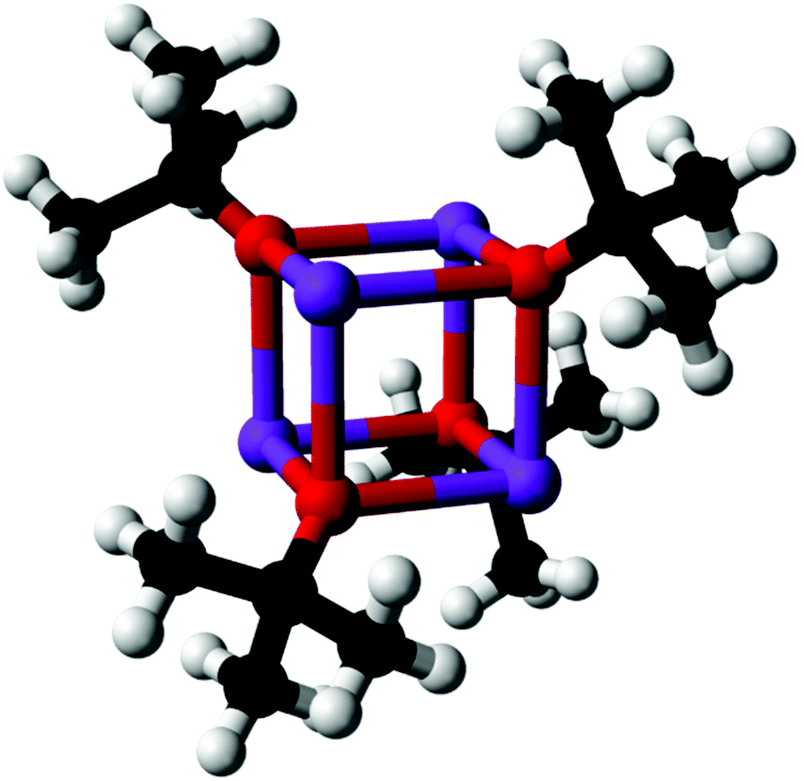 | ||
| Fig. 1 Tetramer of KOtBu, the predominant gas phase species of the precursor used. | ||
Our aim with this work is to remove the reservoir effect to provide a more elegant and reproducible route for deposition of films containing alkali metals.
In traditional metal oxide ALD use of water as a reactant is considered an advantage due to its availability, high vapor pressure and ease of use. For processes with hygroscopic intermediates, however, water as the oxygen source may constitute a silent detrimental factor. Controlling the water dose to precisely a monolayer (saturating) coverage is difficult to achieve reproducibly. Underdosing the water leads to oxidizer deficient growth yielding poor uniformity and a lean growth rate, while overdosing water leads to intermediates that persist from cycle to cycle which act as a water reservoir yielding poor uniformity and excessive growth rate. A pertinent example here is the deposition of LaAlO3, where the binary Al(CH)3 + H2O process could not be successfully employed due to formation of La(OH)x, acting as a water reservoir.24 This was avoided by using O3 as the oxygen source for both La(thd)3 and Al(CH)3, removing water outright, resulting in a process with high uniformity and reproducibility.
In this article, we use O3 as an alternative to H2O as the oxygen source for the deposition of alkali metal containing complex oxides, exemplified by KTN. We show that the group V ethoxides are compatible with O3, and that there is no water reservoir clouding the growth when including the alkali metal tert-butoxides. This provides a much more controlled and reproducible route for deposition of technologically relevant polar materials for sustainable technology.
Experimental
Thin films were deposited in an F-120 Sat ALD reactor (ASM Microchemistry). The deposition temperature was kept at 250 °C under an operating pressure of 2.3 mbar, maintained by 300 cm3 min−1 primary flow rate of N2. Nitrogen was supplied from gas cylinders (Praxair, 99.999%) and run through a Mykrolis purifier for removal of any oxygen or water contamination. KOtBu (Aldrich, 97%), Ta(OEt)5 (Sigma-Aldrich, 99%) and Nb(OEt)5 (Sigma-Aldrich, 99%) were used as cation sources, supplied from open boats inside the reactor at 150, 70 and 70 °C, respectively. Fresh KOtBu was extracted from a container kept inside an argon glovebox for each run. O3 was used as the oxygen source, supplied from an AC-2505 (In USA) generator fed with O2 gas (Praxair, >99.5%). The effective upstream gas mixture was 15 wt% O3 in O2. 3 × 3 cm2 Si (100) substrates were used for compositional analysis. Selected films were deposited on cubic perovskite La0.18Sr0.82Al0.59Ta0.41O3 (100) (LSAT, Crystal GmbH) and cubic spinel MgAl2O4 (100) (MAO, Crystal GmbH) substrates for crystallization.Post-deposition crystallization was carried out using an OTF-1200X (MTI Corp.) rapid thermal annealing furnace, heating the films to 650 °C In air at a ramp rate of ≈40 °C min−1. The films were subsequently held at 650 °C for 15 minutes, before cooling to room temperature by shutting down the furnace. The samples were extracted at 100 °C after approximately 30 minutes of cooling.
In situ quart crystal microbalance (QCM) investigation was carried out using two 6 MHz AT-cut quartz crystals (Inficon, ∅ = 1.4 cm), mounted approximately 5 cm apart in a holder made in-house. The resonance frequency was monitored by a Colnatec Eon-LT unit and logged with a device made in-house. Variations in surface area of the QCM crystals due to evolution of texture during growth were removed by using internal standards of Nb2O5 deposition at even intervals throughout the campaign. These were also used as a basis to convert the signal from Hz to mass per area.
The chemical composition was analyzed using an Axios Max Minerals X-ray fluorescence (XRF) system (Panalytical), equipped with a 4 kW Rh-tube. The system runs using Omnian and Stratos options for standardless measurements of thin films. Routine measurements of thickness on as deposited films were carried out on an alpha-SE spectroscopic ellipsometer (J. A. Woollam) in the 390–900 nm wavelength range. We successfully employed a Cauchy function to model the collected data. X-ray reflectivity (XRR) was used as a complementary method to determine thickness and densities of the Nb2O5 internal standards. XRR was carried out on a Panalytical Empyrean diffractometer equipped with a mirror providing parallel CuKα radiation. A proportional point counter was used to detect the reflected beam. X-ray diffraction measurements were carried out on a Bruker AXS D8 Discover diffractometer equipped with a LynxEye strip detector and a Ge (111) focusing monochromator, providing CuKα1 radiation.
Results and discussion
A key challenge with alkali metal processes is that the binary alkali metal oxides are not stable in air. A hypothetical KOx film deposited using KOtBu and O3 will immediately react with air upon breaking vacuum, forming hydroxides and/or carbonates. This makes the traditional way of understanding the complex process (i.e. combining known binary oxide processes) impractical. As a result, the alkali metal oxide process must be understood as an addition to a single known binary oxide process, such as those for Nb2O5 or Ta2O5. In the case of KTN, none of the binary oxide processes have been studied in depth using O3 as the oxygen source. I.e., to understand the growth of the alkali metal oxide, we must first understand the ozone-based processes for Nb2O5 and Ta2O5.Nb(OEt)5 + O3 – an alternative route to Nb2O5
Nb2O5 is a highly unreactive and passive material that finds use as a dielectric for example in solid electrolyte capacitors. Several ALD processes have been reported for Nb2O5, mostly utilizing Nb(OEt)5 or Nb(NtBu)(NEt2)3 and water. The former is a well-understood ALD process, described by the following surface reactions:| x –OH + Nb(OEt)5 → –OxNb(OEt)5−x + x EtOH | (1) |
| –OxNb(OEt)5−x + (5 − x) H2O → –OxNb(OH)5−x + (5 − x) EtOH | (2) |
Using O3 as an oxygen source for growth of Nb2O5 with Nb(OEt)5 has, however, remained unexplored until very recently, when Kukli et al. utilized O3 to deposit multilayers of SiO2 + Nb2O5 for use as a switching media.25,26 This study confirms that the O3 process provides films with very low hydrogen content, albeit very little is known regarding the mechanism of growth. As a starting point, we confirmed that the process does indeed exhibit self-limiting growth, with a saturation growth-per-cycle (GPC) of 0.40 ± 0.01 Å (ESI Fig. A†). This is very comparable with what we achieved with the water-based process in the same reactor setup, with a GPC of 0.38 ± 0.02 Å.22
To uncover the mechanism of growth, we carried out an in situ QCM investigation of deposition of pristine Nb2O5. O3 processes are a known clouding factor in QCM experiments due to the exothermic decomposition of O3. Regardless of that, and by calibrating towards the known average growth of the process and the resulting film density, a close estimate of the mass gain per pulse can be revealed (Fig. 2). Compared to the water-based process, the mass gain during the Nb(OEt)5 pulse is significantly higher.22 This suggests that the Nb(OEt)5 molecule is coordinating to the surface through activated oxygen from the previous O3 pulse without significant detachment of ethyl groups. Such detachment takes place during the subsequent O3 pulse, during which the mass gain drops to 17.6 ng cm−2 for the full cycle. This is very comparable to the 16 ng cm−2 found for a full water-based cycle, with the slight increase justified by a small increase in density (4.41 vs. 4.32 g cm−3) and GPC (0.40 vs. 0.38 Å). In conclusion for the Nb(OEt)5 + O3 process; it forms thin films with growth, control, and quality comparable to the water based process.
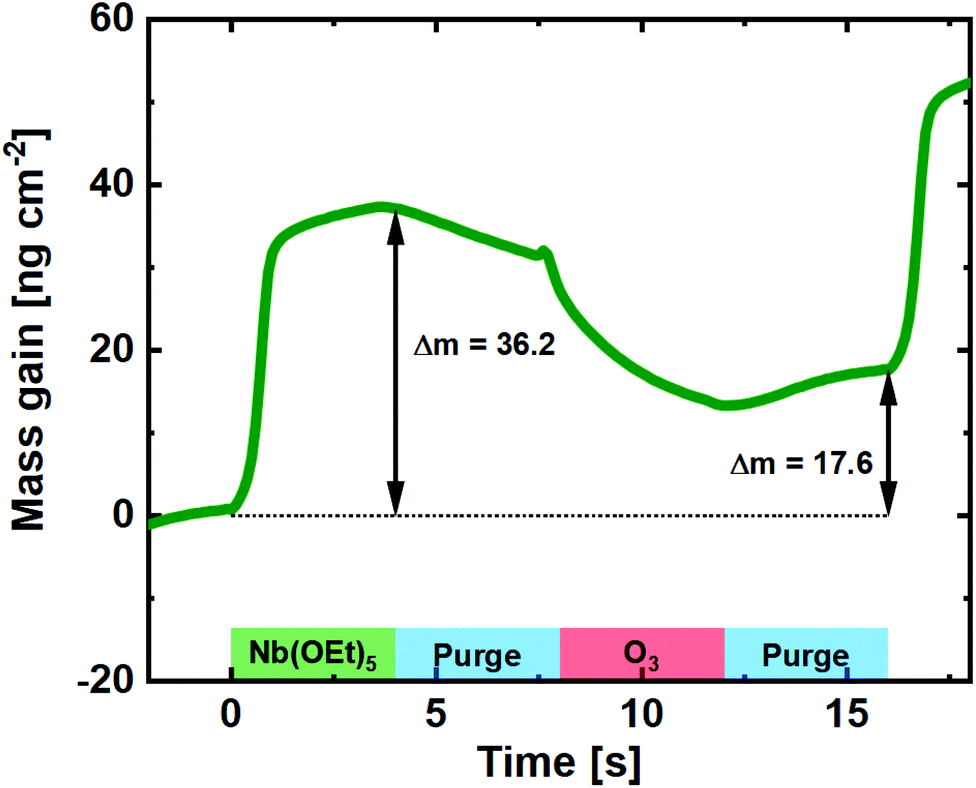 | ||
| Fig. 2 Mass gain as a function of time for one cycle of Nb(OEt)5 + O3 taken as an average of 100 cycles on a pristine QCM crystal. | ||
Ta(OEt)5 + O3 – An alternative route to Ta2O5
Ta2O5 is a high band gap high-k material, finding use in a range of electronics. In ALD it is usually deposited using a Ta(OEt)5 and water, albeit with several other processes reported. The water-based process is well understood, and the mechanism is in agreement with that for its niobium counterpart:| x –OH + Ta(OEt)5 → –OxTa(OEt)5−x + x EtOH | (3) |
| –OxTa(OEt)5−x + (5 − x) H2O → –OxTa(OH)5−x + (5 − x) EtOH(4) | (4) |
The use of O3 as the oxygen source for reaction with Ta(OEt)5, however, has not been reported in the literature. It is expected to behave very similar to its Nb counterpart, due to the similarity in chemistry and precursor structure. A QCM experiment was carried out for comparison (Fig. 3), and the basic shape of the reaction mass gain is, as expected, very similar to that of Nb(OEt)5 + O3. The major difference lies in the higher mass gain on the Ta(OEt)5 pulse, which is expected given the much heavier metal ion.
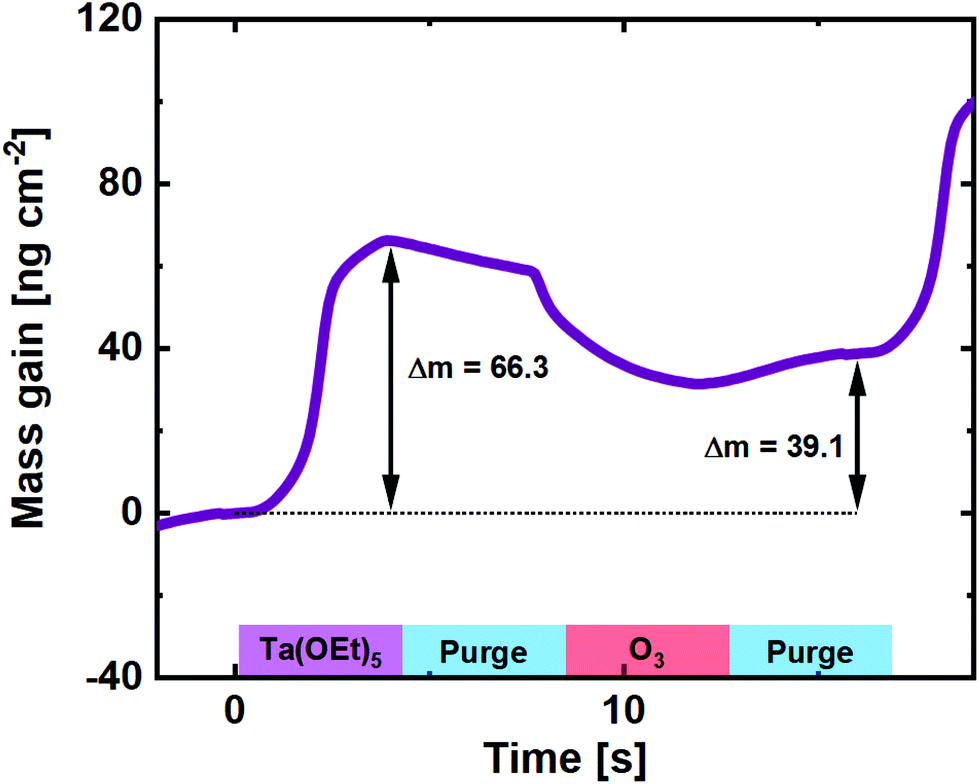 | ||
| Fig. 3 Mass gain as a function of time for one cycle of Ta(OEt)5 + O3 taken as an average of 100 cycles on a pristine QCM crystal. | ||
From the GPC of the Ta(OEt)5 + O3 process (0.50 ± 0.02 Å) and the density of the resulting Ta2O5 film (7.82 g cm−3), the average mass gain per cycle is found to be 39.1 ng cm−2.
The mass increase on Ta(OEt)5 pulses is the 66.3 ng cm−2, a factor of 1.83 higher than for the Nb process. This is slightly more than expected when taking the molar masses of the ethoxides into account (406 g mol−1 for Ta(OEt)5vs. 318 g mol−1 for Nb(OEt)5). This indicates either a higher concentration of adsorbed molecules or fewer ethyl groups detached during Ta(OEt)5 pulsing. The first is made probable by a 30% higher GPC for binary Ta2O5, but is likely accompanied also by reduced detachment of ethyl groups. This also explains the relative higher mass drop upon pulsing ozone.
Introducing potassium – higher compositional control
With confirmation that both Nb(OEt)5 and Ta(OEt)5 form compatible processes with O3, potassium can be included in the mix in the form of KOtBu. Note here that the pure KOtBu + O3 process was tested and seemed to form relatively uniform films before breaking vacuum. At that point it immediately reacts with air, forming a gel-like substance that is likely a mixture of KOH and K2CO3. These samples were very impractical to handle, but clearly proved that the KOtBu + O3 process provides growth.Although the KOtBu + O3 process is difficult to explore ex situ, we carried out an in situ QCM linearity test to see whether it is significantly different from the water-based process. As previously reported, the KOtBu + H2O process exhibits diverging growth for repeated cycles without passivation, as it acts as a water reservoir that allows KOtBu to react uncontrolled.22 The ozone-based process, however, does not exhibit this feature, and remains remarkably linear upon subsequent cycling (Fig. 4). To further compare with the water-based process, we investigated the growth of the K–Nb–O process for different ratios of the binary processes; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:3 and 1:5 (K:Nb).
:1, 1:3 and 1:5 (K:Nb).
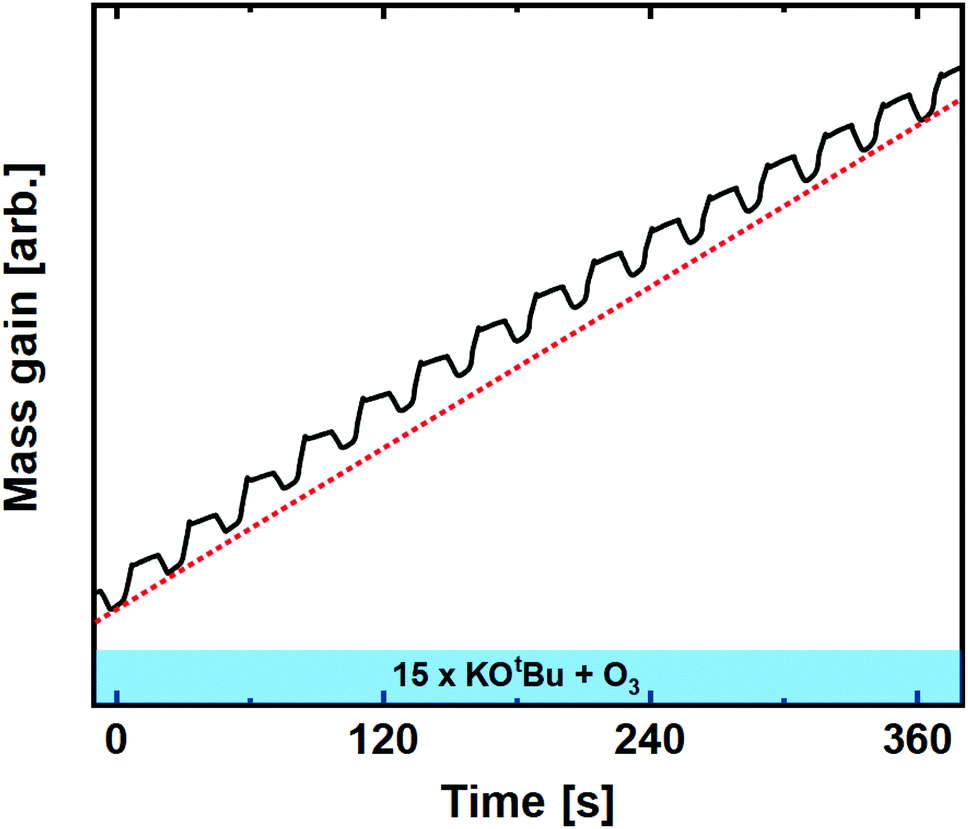 | ||
| Fig. 4 Mass gain as a function of time for 15 subsequent cycles of KOtBu + O3 compared to linear average (dashed red line). | ||
These were chosen to comply with previous reports for the water-based process in which 1:5 was the only ratio that enabled controlled growth.22 For the water-based process, we observed an extremely high mass gain upon pulsing KOtBu, which can only be explained by reaction with a water reservoir.22 The ozone-based process deviates significantly from this observation, showing no signs of uncontrolled growth at any pulsed ratio.
For the 1:5 pulsed ratio, we observe that the Nb(OEt)5 + O3 cycles are similar to what is found for growth of pure Nb2O5 (Fig. 5). The average mass gain per-cycle is found to be 17.4 ng cm−2.
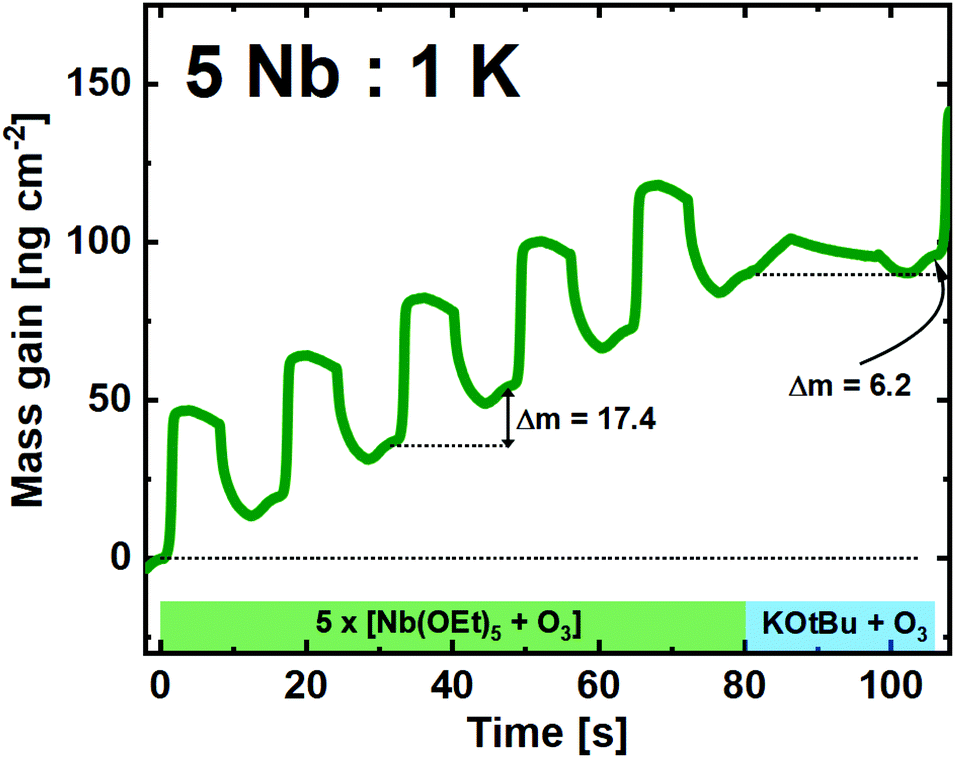 | ||
| Fig. 5 QCM campaign for a supercycle of 5:1 (Nb:K), showing the mass gain per time for the unique subcycles. | ||
We also observe that the single KOtBu + O3 cycle behaves very similar compared to that of repeated cycles, however, with a significantly lower mass gain response. Note here that the overall mass gain for a KOtBu + H2O cycle with the same K:Nb ratio was previously reported to be as high as 116 ng cm−2, a factor of 20 higher than what is observed in the ozone-based process, likely due to an overdose of water that is hard to control.22 The control that the O3 offers is important, as this will open for a much higher regulation of the overall K:Nb composition in the resulting films.
A similar trend was seen for the 1:3 pulsed ratio (ESI Fig. B†), albeit with a notably higher mass gain (25.8 ng cm−2) for the KOtBu + O3 cycle. It is probable that this is an effect of the number of preceding Nb(OEt)5 + O3 cycles, as a pristine Nb2O5 surface seems to slow down the growth of KOx. Interestingly, the 1:1 pulsed ratio continued to show remarkably linear and stable growth (Fig. 6). Although the shape of the single Nb(OEt)5 + O3 cycle is slightly altered compared to growth of pure Nb2O5, the overall mass gain is comparable. The KOtBu + O3 cycle is very similar to what was observed in the linearity test (Fig. 4), and the overall growth remains linear in nature. It is important to remember that a mass gain of 20.6 ng cm−2 for the KOtBu + O3 cycle is still quite high taking the molar mass and density of potassium oxide into account. If we hypothesize that the intermediary product of a KOtBu + O3 cycle is K2O; the molar gain of K for such a cycle will still be ≈3.3 times higher than for Nb. This is likely to be an effect of the cubane-like shape of the potassium precursor that allows a large molar concentration of K-ions to be adsorbed on the surface. The advantage here lies in the possibility of finer control compared to the water-based process that led to an extreme and uncontrolled amount of K to be incorporated in the sample.22
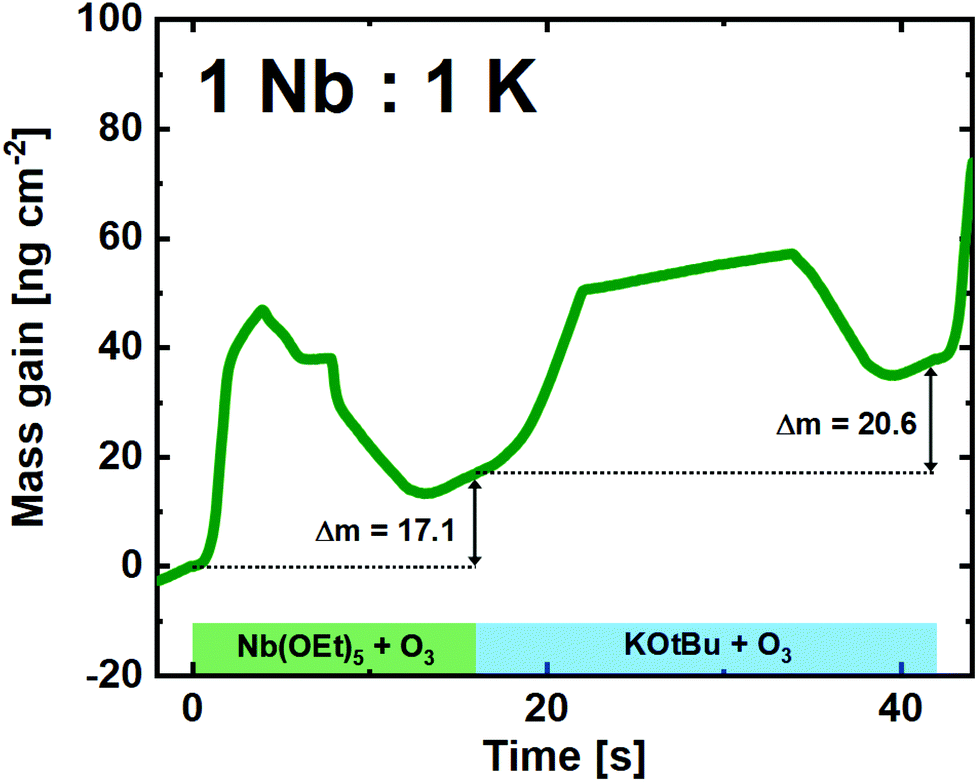 | ||
| Fig. 6 QCM campaign for a supercycle of 1:1 (Nb:K), showing the mass gain per time for the unique subcycles. | ||
Towards K(Ta,Nb)O3 – finer composition control
Lower potassium incorporation per cycle and non-existent reservoir effects for succeeding cycles should in principle lead to higher control of composition in complex systems. To explore this, we turned to one of the more challenging and functionally interesting compounds that includes potassium and group V metals: K(Ta,Nb)O3. This system adds an additional complexity factor, being a quaternary system where the relative ratio of all three cations is important. In principle, two major ratios are crucial:1. The K:(Ta + Nb) ratio, which needs to be as close to 1 as possible to achieve growth of perovskite KTN.
2. The Ta:Nb ratio, affecting the Curie temperature of the paralectric → ferroelectric phase transition.
We have previously shown that KTN can be viewed as a pseudoternary system, due to the similarity of growth for Nb(OEt)5 and Ta(OEt)5. These can be interchanged in the complex process to tune the relative Ta:Nb-ratio, without affecting the K:(Ta + Nb) ratio significantly. Hence, the goal with this study was to achieve a finer control of the K-content by using the new ozone based route. To explore this, we deposited a range of samples where the relative ratio of pulsed KOtBu with respect to the other metal precursors ranged from 16.7 to 50% (Fig. 7). Note that a 50% pulsed ratio of K in the water-based system led to films with very low uniformity and strong composition gradients, degrading immediately in air.22
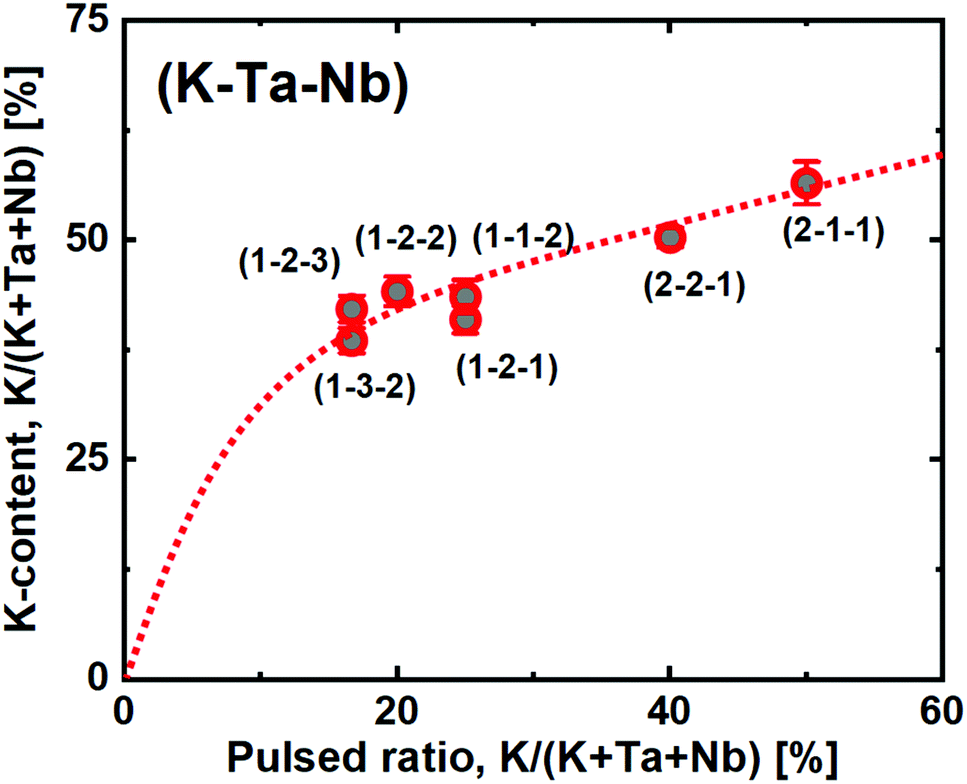 | ||
| Fig. 7 Potassium content as a function of the pulsed ratio of KOtBu, showing rapid K-incorporation at low ratios before responding linearly. Dashed red line is a guide of eye, error bars are based on reported confidence in XRF measurements. | ||
In the ozone-based system, we observe a large initial incorporation of K, which is expected due to the high molar gain of K during a KOtBu pulse. This can again be explained by the high K-density cubane like shape of the precursor.
However, after the initial increase, we observe a close to linear trend as a function of pulsed KOtBu. This allows us to directly tune the stoichiometric KTN composition, which is achieved at a 2:3 pulsed K:(Ta + Nb) ratio. The films are highly uniform, and with no observed composition gradients over our 7 × 5 cm2 substrate as mapped by ellipsometry. Also note that changing the relative Ta:Nb ratio does not drastically change the overall composition. For such a quaternary process, we use the shorthand notation (#K – #Ta – #Nb) where the #-symbols note the number of each metal cycles in the overall supercycle. This notation does not represent the individual pulsing sequence. In this work we have arranged the pulses to maximize a mixing of the elements. Hence a (2 − 2 − 1) process was arranged as [K + Ta + K + Nb + Ta] to enable an as homogeneous mixture of the cation constituents as possible.
This sample resulted in a composition of KTa0.67Nb0.35O3, which is right in the sweet spot for obtaining a Curie temperature close to room temperature; important for maximizing the electro-optical Kerr coefficient, which is strongest close to the morphotropic phase boundary between the para- and ferroelectric states.27
Another interesting observation is the close to linear relationship between the film refractive index and K incorporation (Fig. 8), as measured by spectroscopic ellipsometry. Increased potassium content decreases the refractive index, which provides a route for quick identification of approximate K-incorporation for a given Ta:Nb-ratio.
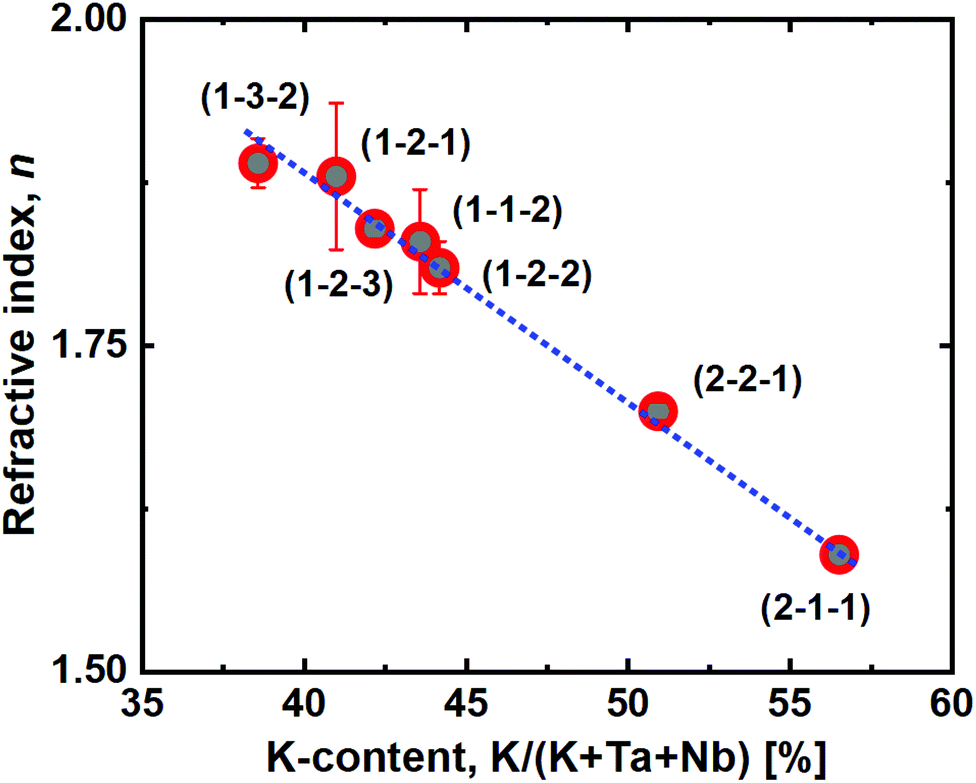 | ||
| Fig. 8 Refractive index, n, as a function of the film potassium cation content. The blue dashed line is a linear fit of the measured data. | ||
Crystal quality – major improvement
The functionality of polar materials is closely linked to the structure, hence, it is important to obtain the best possible crystallographic integrity. This is especially true for materials applicable in electro-optics, where structural defects alter optical properties.When deposited using the water based route, KTN exhibited a strong tendency to form unwanted pyrochlore, or a mixed pyrochlore-perovskite phase.20 The pyrochlore structure, K2(Ta,Nb)2O6, does not exhibit the useful electro-optic properties.
With higher control of the cation composition and less water stored in the films, we expected a better crystal quality, i.e. phase-pure KTN perovskite. To investigate this, we deposited a series of (2 − 2 − 1) samples with composition KTa0.67Nb0.35O3 on LSAT (100) and MAO (100). These substrates provide different strain scenarios and are both optimal in optical applications due to low interaction with visible light.
MAO adopts a cubic spinel structure with a = 8.089 Å, providing a surface lattice with as = 4.045 Å that is slightly larger, but close to the cubic perovskite lattice of KTN (a ≈ 3.98 Å). LSAT is a cubic mixed perovskite with a = 3.868 Å, imposing compressive strain.
All films were found to be amorphous as deposited, and required post deposition annealing to crystallize. The films were subsequently annealed at 650 °C, where major differences between the film-substrate systems were observed.
On LSAT (100), KTN adopts strong out-of-plane (100) orientation with a c-axis cell parameter of 3.987 Å (2θ(200) = 45.46°, Fig. 9). Being slightly compressed in-plane by the substrate, this c-axis elongation is expected. No secondary orientation or traces of other phases were observed, including that of the unwanted pyrochlore phase. The films are also found to be transparent to visible light upon inspection, which can be advantageous for electro-optical applications.
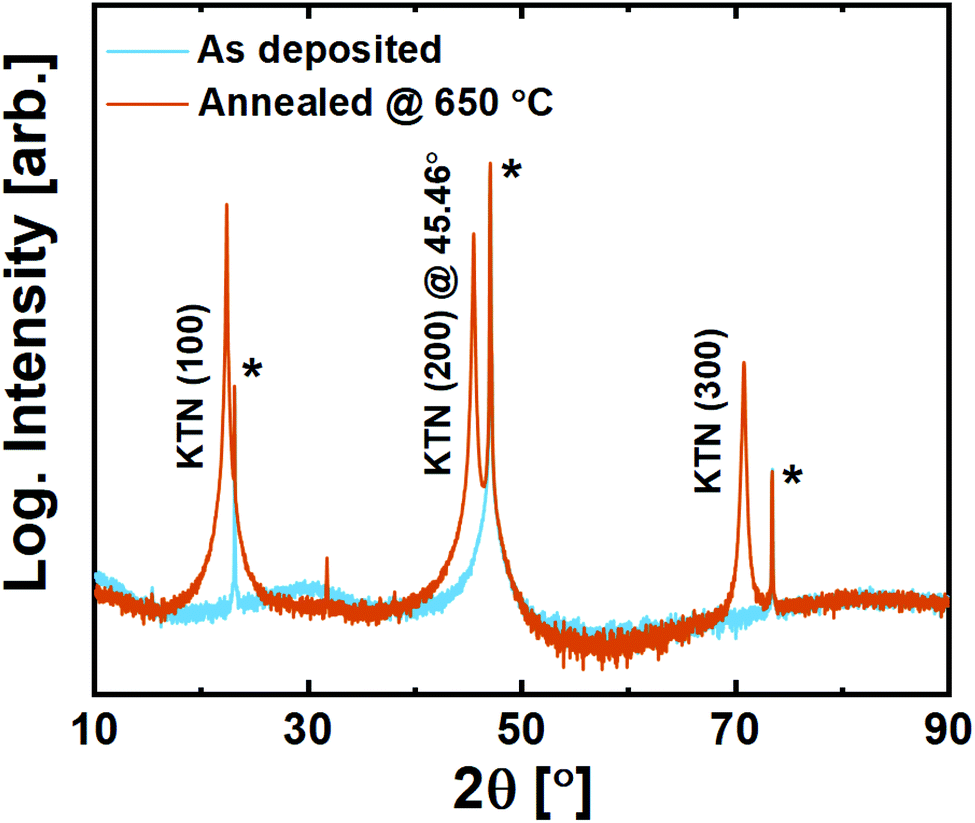 | ||
| Fig. 9 X-ray diffraction of 200 nm KTa0.67Nb0.35O3 thin films deposited on LSAT (100) substrates. Substrate reflections are marked with*. | ||
It is evident that the films deposited with the ozone process provide a far superior starting-point for solid phase epitaxy compared to what was seen for the water-based process.20 Note here that the compressive strain imposed by the LSAT substrate is ≈−3%.This becomes important when comparing it to what is observed on spinel MAO (100). In principle, the tensile strain imposed by MAO (100) on KTN is ≈1.4%, which should be more tolerable than on LSAT. The (100) surface oxygen lattice is similar for the two substrates apart from a small buckling in the O–Al–O bonds that distort the surface of MAO (100) (and the slightly larger O–O-distance in MAO).
Films on MAO are still amorphous as deposited. Upon annealing, however, we observe that while it does exhibit a minor preference for growth in the (100)-direction based on higher intensities for this family of reflections, it is effectively polycrystalline with all allowed reflections visible (Fig. 10).
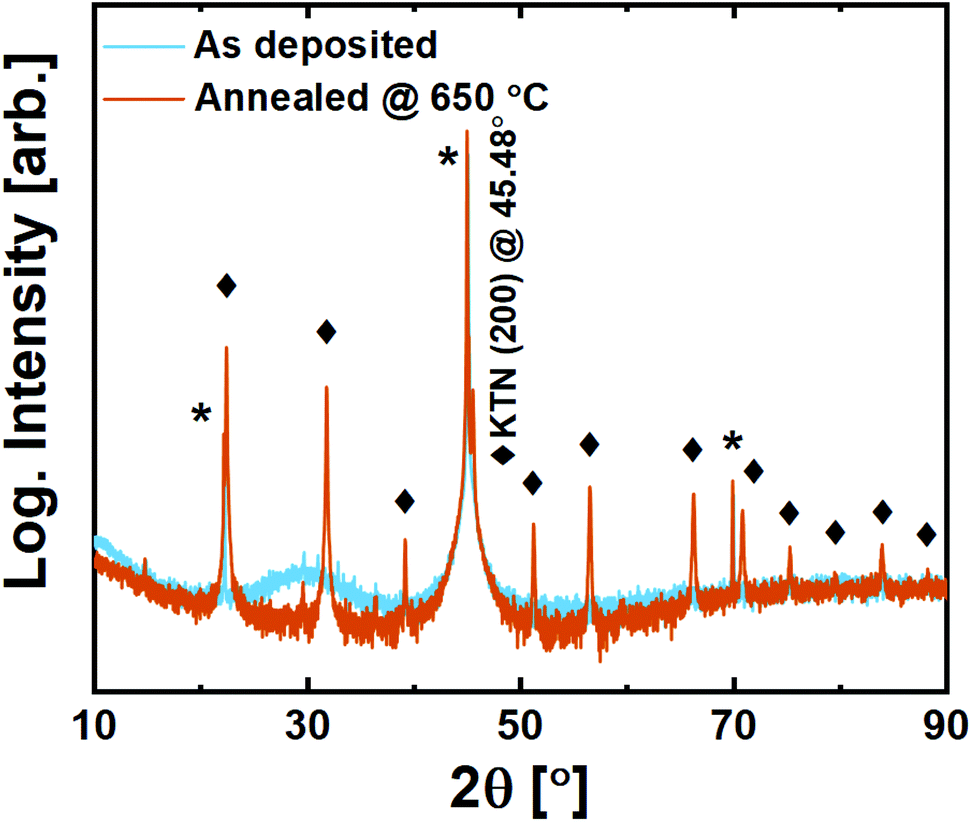 | ||
| Fig. 10 X-ray diffraction of 200 nm KTa0.67Nb0.35O3 thin films deposited on MgAl2O4 (100) substrates. Substrate reflections are marked with *, while perovskite film reflections are marked with ◆. | ||
The growth of crystallites upon annealing seems to take place without any template influencing from the substrate. While this close-to random orientation may be challenging in terms of application, it brings an opportunity to investigate the symmetry of the structure. The {110} reflection is not split, and its corresponding plane distance is found to be exactly aKTN/√2. This confirms that KTN does indeed adopt the interesting paraelectric cubic phase for the Ta:Nb ratio in question, meaning that the Curie temperature is lower than room temperature. This is important for future application and use of the new reported process.
The random polycrystallinity of KTN deposited on MAO (100) is hypothesized to be an effect of either the O–Al–O bond-buckling present on the MAO (100) surface, or that the KTN structure does not comply with tensile strain, or both. Future work on substrates like BaTiO3, imposing tensile strain without oxygen lattice buckling, could help clarify this question.
We furthermore carried out a Williamson–Hall analysis of the broadening of the reflections, showing that the broadening is predominantly due to particle size and not strain (ESI Fig. C†).
This is as expected in a polycrystalline film sample. In addition, we used the same result to estimate the average minimum crystallite size to be ≈320 nm, which is close to the 380 nm thickness of the as deposited films. The large crystallite size and visually transparent nature of the sample indicates that this still may find application within electro-optics.
Conclusions
Herein, we have shown that replacing the water-based routes for deposition of alkali metal containing complex oxides with novel ozone-based routes significantly enhances process control. A finer composition control is achieved as a result of not having to deal with water reservoir effects throughout growth. We show that the ozone-based routes can be used to deposit high quality alkali metal containing quaternary oxides, exemplified by paraelectric KTN. KTN films can be grown with high compositional control, yielding uniform films with high crystalline quality. The degree of preferred orientation can be tuned by selecting appropriate substrates, ranging from perfectly (100) oriented on LSAT (100) to random polycrystalline on MAO (100).These ozone-based provide a more elegant growth of alkali metal containing oxides, which may find applications in novel piezo- and ferroelectrics, electro-optics and sodium-/potassium battery technology.
Author contributions
HHS: Conceptualization, data curation, investigation, methodology, supervision, validation, visualization, writing – original draft, writing – review & editing, project administration, funding acquisition. VA-LKK: Data curation, investigation, validation, writing – review and editing. LMR: Data curation, investigation, validation, writing – review and editing. JRB: Conceptualization, investigation, methodology, writing – review and editing. EGM: Investigation, methodology, writing – review and editing. RCH: Conceptualization, investigation, methodology, writing – review and editing. ON: Investigation, supervision, validation, writing – review and editing, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. Nicholas Strnad for fruitful discussions in the infancy of the project.References
- J. E. Geusic, S. K. Kurtz, L. G. Van Uitert and S. H. Wemple, Appl. Phys. Lett., 1964, 4, 141 CrossRef CAS.
- F. S. Chen, J. E. Geusic, S. K. Kurtz, J. G. Skinner and S. H. Wemple, J. Appl. Phys., 1966, 37, 388 CrossRef CAS.
- T. Imai, M. Sasaura, K. Nakamura and K. Fujiura, NTT Rev., 2007, 5, 1 Search PubMed.
- P. Guenter, Ferroelectrics, 1980, 24, 35 CrossRef CAS.
- W. A. Bonner, E. F. Dearborn and L. G. Van Uitert, Ceram. Bull., 1965, 44, 23 Search PubMed.
- D. Rytz and H. J. Scheel, J. Cryst. Growth, 1982, 59, 468 CrossRef CAS.
- S. Hirano, T. Yogo, K. Kikua, T. Morisihta and Y. Ito, J. Am. Ceram. Soc., 1992, 75, 1701 CrossRef CAS.
- J. Burši'k, V. Železný and P. Vaněk, J. Eur. Ceram. Soc., 2004, 24, 455 CrossRef.
- S. Yilmaz, T. Venkatesan and R. Gerhard-Multhaupt, Appl. Phys. Lett., 1991, 58, 2479 CrossRef CAS.
- A. Rousseau, M. Guilloux-Viry, E. Doghesche, M. Bensalah and D. Remiens, J. Appl. Phys., 2007, 102, 093106 CrossRef.
- W. Yang, Z. Zhou, B. Yang, Y. Jiang, H. Tian, D. Gong, H. Sun and W. Chen, Appl. Surf. Sci., 2011, 257, 7221 CrossRef CAS.
- C. H. Jung, M. S. Choi, K. S. Lee and D. H. Yoon, Opt. Mater., 2011, 34, 269 CrossRef CAS.
- W. Yang, Z. Zhou, B. Yang, Y.- Jiang, Y. Pei, H. Sun and Y. Wang, Appl. Surf. Sci., 2012, 258, 3986 CrossRef CAS.
- A. Bouyfasi, M. Mouttalie, V. Demange, B. Gautier, A. Grandfond, S. Députier, S. Ollivier, L'H Hamedi and M. Guilloux-Viry, Appl. Surf. Sci., 2012, 258, 9297 CrossRef.
- B. M. Nichols, “Structural, dielectric, and optical properties of potassium tantalate niobate thin films prepared by metalorganic chemical vapor deposition”, dissertation, Northwestern University, IL, USA, 2000 Search PubMed.
- S. M. George, Chem. Rev., 2010, 110, 111 CrossRef CAS PubMed.
- E. Østreng, H. H. Sønsteby, S. Øien, O. Nilsen and H. Fjellvåg, Dalton Trans., 2014, 43, 16666 RSC.
- H. H. Sønsteby, O. Nilsen and H. Fjellvåg, J. Vac. Sci. Technol., A, 2016, 34, 041508 CrossRef.
- H. H. Sønsteby, O. Nilsen and H. Fjellvåg, Glob. Chall., 2019, 3, 1800114 CrossRef PubMed.
- J. R. Bickford, H. H. Sønsteby, N. A. Strnad, P. Y. Zavalij and R. C. Hoffman, J. Vac. Sci. Technol., A, 2019, 37, 020904 CrossRef.
- H. H. Sønsteby, J. E. Bratvold, V. A.-L. K. Killi, D. Choudhury, J. W. Elam, H. Fjellvåg and O. Nilsen, J. Vac. Sci. Technol., A, 2020, 38, 060804 CrossRef.
- H. H. Sønsteby, V. A.-L. K. Killi, T. A. Storaas, D. Choudhury, J. W. Elam, H. Fjellvåg and O. Nilsen, Dalton Trans., 2020, 49, 13233 RSC.
- H. H. Sønsteby, A. Yanguas-Gil and J. W. Elam, J. Vac. Sci. Technol., A, 2020, 38, 020804 CrossRef.
- H. H. Sønsteby, E. Østreng, H. Fjellvåg and O. Nilsen, Thin Solid Films, 2014, 550, 90 CrossRef.
- K. Kukli, M. Kemell, M. J. Heikkilä, H. Castán, S. Dueñas, K. Mizohata, M. Ritala and M. Leskelä, Nanotechnology, 2020, 31, 195713 CrossRef CAS PubMed.
- K. Kukli, M. Ritala and M. Leskelä, J. Electrochem. Soc., 1995, 142, 1670 CrossRef CAS.
- S. Triebwasser, Phys. Rev., 1959, 114, 63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt03960a |
| This journal is © The Royal Society of Chemistry 2022 |