
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into the NH3-selective catalytic reduction of NO over Cu-ZSM-5 as revealed by operando spectroscopy†
Xinwei
Ye
 ab,
Ramon
Oord
b,
Matteo
Monai
b,
Joel E.
Schmidt
b,
Tiehong
Chen
a,
Florian
Meirer
b and
Bert M.
Weckhuysen
*b
ab,
Ramon
Oord
b,
Matteo
Monai
b,
Joel E.
Schmidt
b,
Tiehong
Chen
a,
Florian
Meirer
b and
Bert M.
Weckhuysen
*b
aInstitute of New Catalytic Materials Science, School of Materials Science and Engineering, Key Laboratory of Advanced Energy Materials Chemistry (MOE), Nankai University, Tianjin 300350, China
bInorganic Chemistry and Catalysis Group, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl
First published on 28th February 2022
Abstract
To control diesel vehicle NOx emissions, Cu-exchanged zeolites have been applied in the selective catalytic reduction (SCR) of NO using NH3 as reductant. However, the harsh hydrothermal environment of tailpipe conditions causes irreversible catalyst deactivation. The aggregation of isolated Cu2+ brings about unselective ammonia oxidation along with the main NH3-SCR reaction. An unusual ‘dip’ shaped NO conversion curve was observed in the steamed zeolite Cu-ZSM-5, resulting from the undesired NH3 oxidation that produced NO. Here we gain further insights into the NH3-SCR reaction and its deactivation by employing operando UV-vis diffuse reflectance spectroscopy (DRS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) on fresh and steamed zeolite Cu-ZSM-5. We found that tetragonally distorted octahedral Cu2+ with associated NH3 preferentially forms during low temperature NH3-SCR (<250 °C) in fresh Cu-ZSM-5. The high coordination number of Cu2+ ensures the availability for high coverage of nitrate intermediates. Whilst in the steamed Cu-ZSM-5, [Cux(OH)2x−1]+ oligomers/clusters in pseudo-tetrahedral symmetry with coordinated NH3 accumulated during the low-temperature NH3-SCR reaction. These clusters presented a strong adsorption of surface NH3 and nitrates/nitric acid at low temperatures and therefore limited the reaction between surface species in the steamed Cu-ZSM-5. Further release of NH3 with increased reaction temperature favors NH3 oxidation that causes the drop of NO conversion at ∼275 °C. Moreover, competitive adsorption of NH3 and nitrates/nitric acid occurs on shared Lewis-acidic adsorption sites. Prompt removal of surface nitrates/nitric acid by NO avoids the surface blockage and tunes the selectivity by alternating nitrate–nitrite equilibrium. The formation of adsorbed NO2 and HNOx points to the necessity of an acid adsorbent in practical applications. The structural similarity under the NH3-SCR reaction and unselective NH3 oxidation confirmed the entanglement of these two reactions above 250 °C.
1. Introduction
Emission control of NOx (i.e., NO, N2O and NO2) has been mandated in applications such as stationary power plants and diesel engine vehicles. Vanadia-based NH3-selective catalytic reduction (NH3-SCR) catalysts are rather efficient and economical in stationary NOx abatement, but failed to adapt to diesel vehicles because of the low activity at a high air/fuel-ratio and their high SO2 oxidation activity.1,2 Considering the dominant emission of NO compared to N2O and NO2 in NOx-lean automotive exhausts, the standard NH3-SCR reaction (4NH3 + 4NO + O2 = 4N2 + 6H2O), where stoichiometrically equal amounts of NH3 as NO are employed, is the main focus in catalyst development.3 Catalysing such a redox reaction, involving electron transfer processes, requires a catalyst that can accept and donate electrons when encountering reactant molecules or bind with reaction intermediates. Transition metal-based catalysts are thus promising candidates for NH3-SCR, due to the modifiable electron configuration of their d orbitals.Since the high NO decomposition activity of zeolite Cu-ZSM-5 was discovered in 1980s, Cu-exchanged zeolites have been widely investigated for the NH3-SCR reaction.4 Although Cu-exchanged zeolites exhibit high NH3-SCR activity over a wide temperature window, the automotive industry is still facing the dilemma of choosing a suitable catalyst for commercialization – medium/large pore zeolite structures, such as MFI and BEA, are limited by their low hydrothermal stability, while the more robust small pore zeolite CHA (i.e., SSZ-13 and SAPO-34) has a higher cost. The irreversible hydrothermal aging of zeolites is a subtle yet permanent process, during which the functional moieties in Cu-exchanged zeolites undergo a dynamic transformation starting from local distortion of the structural unit regardless of the type of zeolite framework. The deactivation of catalysts should be particularly considered for the rational design of emission control systems for vehicle tailpipes.
The ideal Cu species in Cu-exchanged zeolites are isolated Cu2+ balanced by an Al pair and [CuOH]+ balance by a single Al site. When the Cu-exchanged zeolites undergo hydrothermal treatment or experience a deactivation process, the degradation of Cu increases the heterogeneity of Cu species. The CuxOy clusters/nanoparticles, spinel phase CuAl2O4, as well as Cu(OH)2 can form and are considered to be detrimental for the standard NH3-SCR reaction.5–9 Various Cu species in the zeolites provide multiple possible sites for catalytic reactions at NH3-SCR reaction conditions. Undesired byproducts, for instance NO2 and N2O, can be selectively formed during the NH3-SCR reaction.10 Additionally, with multiple evolutionary Cu species in the Cu-exchanged zeolites, the unwanted side reactions such as NO oxidation (2NO + O2 = 2NO2) and unselective NH3 oxidation to NO (4NH3 + 5O2 = 4NO + 6H2O) can also take place under standard NH3-SCR reaction conditions.11–14
In our previous study of steamed Cu-ZSM-5 zeolites we have observed an unusual NO conversion curve with a ‘dip’ shape at around 300 °C.15 A similar drop of NO conversion was reported at ∼270 °C with the hydrothermally treated zeolite Cu-SSZ-13 and was simply explained by the accelerated unselective NH3 oxidation promoted by CuxOy clusters/nanoparticles.16,17 However, the detailed structural reasons for the low NH3-SCR activity have not yet been well understood due to the interference of multiple Cu sites and side reactions.
In this study, the catalytic performance and structural properties of a series of fresh and steamed Cu-ZSM-5 zeolites were investigated for a more complete understanding of NH3-SCR catalysis utilizing Cu-exchanged zeolites by mimicking different aging severities. Operando UV-vis diffuse reflectance spectroscopy (DRS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) were conducted to gain mechanistic insight into the NH3-SCR reaction and its deactivation, and to gain a deeper understanding of the unusual catalytic behaviour of the steamed zeolite Cu-ZSM-5 material. The dynamic of structural changes of the Cu2+ site under reaction conditions were followed by operando UV-vis DRS, specifically by interpretation of the ligand-to-metal charge transfer (LMCT) as well as the d–d transition bands based on crystal field theory. The behaviour of adsorbed species, including chemisorbed NH3 and nitrates/nitric acid, were investigated utilizing their development and consumption under various reaction conditions. Finally, the ‘dip’-shaped NO conversion curve (Fig. 1) could be explained by the side reaction of unselective NH3 oxidation, which is structurally ascribed to the possible formation of [Cux(OH)2x−1]+ oligomers/clusters with a pseudo-tetrahedral Cu2+ center, coordinated with NH3 in the steamed Cu-ZSM-5 material. The slow rate of surface reaction between adsorbed NH3 and surface nitrites/nitrates or nitrous/nitric acid limits the low-temperature NH3-SCR.
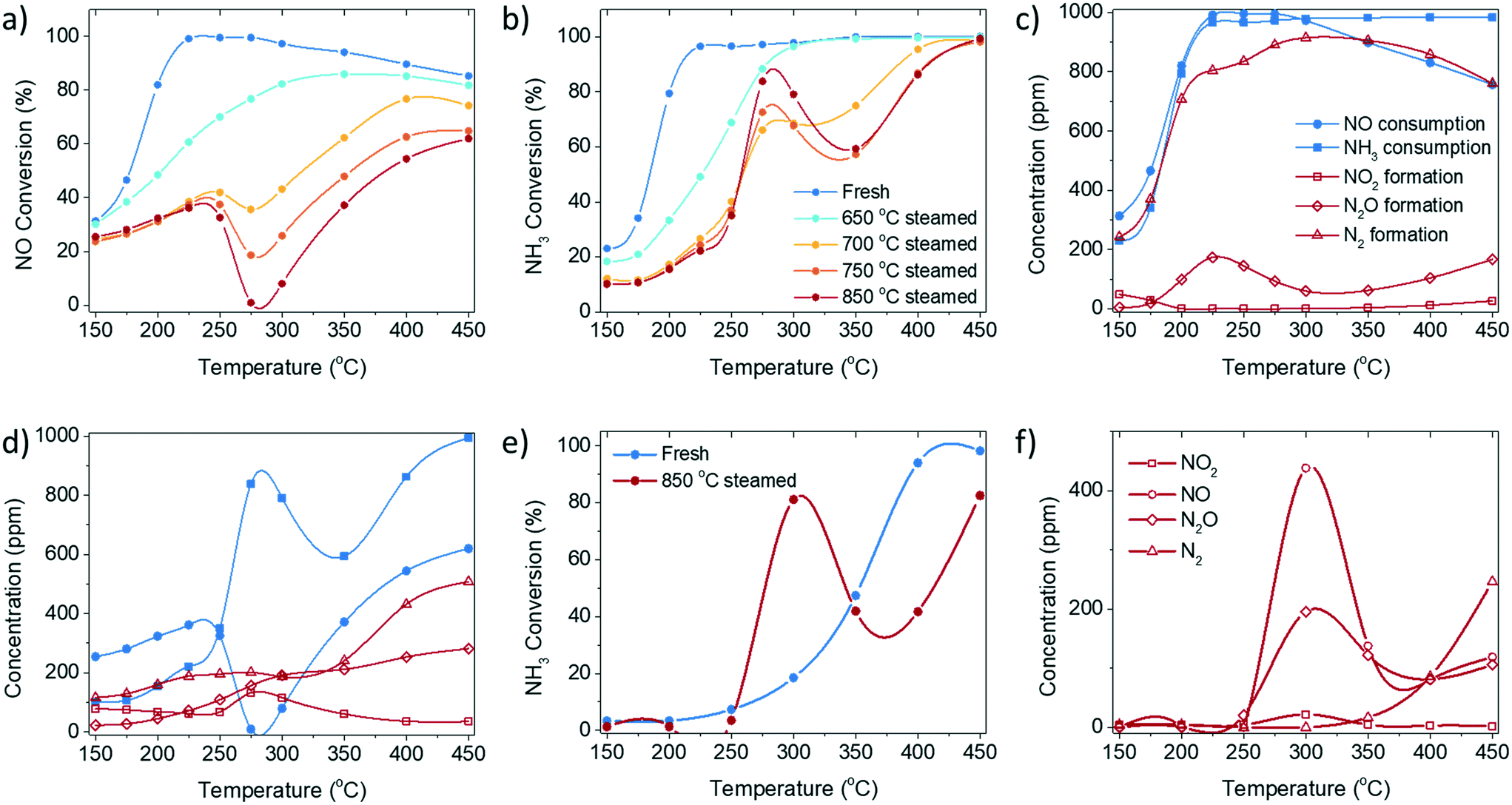 | ||
Fig. 1 a) NO conversion and b) NH3 conversion of the standard NH3-selective catalytic reduction (SCR) reaction performed over fresh and steamed Cu-ZSM-5. The reactant consumption and product formation in standard NH3-SCR on c) fresh and d) 850 °C steamed zeolite Cu-ZSM-5. e) The conversion of NH3 during the NH3 oxidation reaction over the fresh and steamed Cu-exchanged ZSM-5. f) Product concentration from the NH3 oxidation reaction over 850 °C steamed Cu-ZSM-5. The standard NH3-SCR reaction was conducted with a gas hourly space velocity (GHSV) of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 with 1000 ppm NO, 1000 ppm NH3, 5% O2 and balanced with He. The NH3 oxidation reaction was conducted with a GHSV of 100000 h−1 with 1000 ppm NH3, 5% O2 and balanced with He. 000 h−1 with 1000 ppm NO, 1000 ppm NH3, 5% O2 and balanced with He. The NH3 oxidation reaction was conducted with a GHSV of 100000 h−1 with 1000 ppm NH3, 5% O2 and balanced with He. | ||
2. Results and discussion
2.1 NH3-Selective catalytic reduction, NO oxidation and NH3 oxidation
The fresh zeolite Cu-ZSM-5 underwent a steaming pre-treatment to simulate the working catalysts after various degrees of deactivation. The standard NH3-SCR reaction was performed on fresh and steamed Cu-ZSM-5 catalysts in the temperature range from 150 to 450 °C (Fig. 1, S1 and S2†). A varying extent of deactivation of NH3-SCR activity was observed in the steamed zeolites Cu-ZSM-5. The fresh Cu-ZSM-5 showed the highest NO conversion and N2 selectivity in the whole temperature range. The steaming process mainly caused the loss of NO conversion at low reaction temperature. It is notable that unusual catalytic behaviour was observed over the zeolites that underwent the 700–850 °C steaming pre-treatment. The NO conversion had a drop starting at 250 °C followed by a continuous increase in conversion from 300 °C, exhibiting a distinct valley-shaped NO conversion curve. Meanwhile, a peak was observed in the NH3 conversion curve at the exact temperature of the observed ‘dip’ in NO conversion. This points to the non-equivalent consumption of NO and NH3 in the reaction, which contradicts the identical stoichiometric ratio of NO and NH3 in a standard NH3-SCR reaction.The conversion and formation of nitrogen-containing compounds in the NH3-SCR reaction over fresh and 850 °C steamed Cu-ZSM-5 zeolites are shown in Fig. 1c and d. From the consumption difference between NO and NH3, depicted by blue curves, the occurrence of side reactions such as NO or NH3 oxidation in standard NH3-SCR could be determined. In fresh Cu-ZSM-5, NH3-SCR was the favoured reaction, and was only slightly affected by NO oxidation below 200 °C and by NH3 oxidation above 300 °C. In contrast, side reactions had more significant impact on the steamed Cu-ZSM-5. This ‘critical temperature’ of 250 °C divides the temperature range into a low- and a high-temperature regime: when the reaction temperature was below 250 °C, the converted NO was overall higher than the converted NH3, which implied the involvement of the undesirable NO oxidation, confirmed by the additional production of NO2. NH3 oxidation hardly contributed to the low temperature regime, proven by no conversion of NH3 in the NH3 oxidation reaction (Fig. 1e). When the reaction temperature was higher than 250 °C, the consumption of NH3 overtook NO consumption, suggesting the involvement of NH3 oxidation along with the standard NH3-SCR reaction. Especially in the intermediate reaction temperature range of 250–300 °C, the apparent NO conversion dropped to near 0%, while NH3 conversion increased, because the NH3 oxidation reaction to NO facilitated over the steamed Cu-ZSM-5 (Fig. 1e and f). The produced NO from the NH3 oxidation replenished the consumed NO from NH3-SCR, and consequently led to the apparent drop in NO conversion from 250 °C in standard NH3-SCR (Fig. 1a). In return, the residue NH3 was insufficient for the reduction of the surplus NO. As for the N2O byproduct, it is formed in the NH3-SCR reaction as a partially reduced product of NO through the formation of HNO intermediate.18 N2O can also be the product of unselective oxidation of NH3 (2NH3 + 2O2 = N2O + 3H2O). At low reaction temperature, the activity of NH3-SCR reaction was high on fresh Cu-ZSM-5, resulting higher N2O yield compared to the 850 °C steamed Cu-ZSM-5. With elevated reaction temperatures, the N2O generated from both NH3-SCR and NH3 oxidation reaction kept increasing.
Although the side reaction of NH3 oxidation explained the ‘dip’ shape in the NO conversion curve during the NH3-SCR reaction, it put forward another puzzle for NH3 oxidation conducted over steamed zeolite Cu-ZSM-5, where a peak was observed in the NH3 conversion curve at around 300 °C (Fig. 1e). A possible interpretation can be found from a kinetic model of NH3 oxidation over Cu-exchanged zeolite Cu-SSZ-13. The reaction at 250–400 °C occurs on Cu-exchanged sites but the NH3 conversion decreases with the lower NH3 coverage with increasing reaction temperature, while the high temperature reaction (>400 °C) starts to take place on the over-exchanged sites, for instance the CuxOy species achieving high conversion at elevated temperature.19
2.2 Changes of the structural properties upon steaming
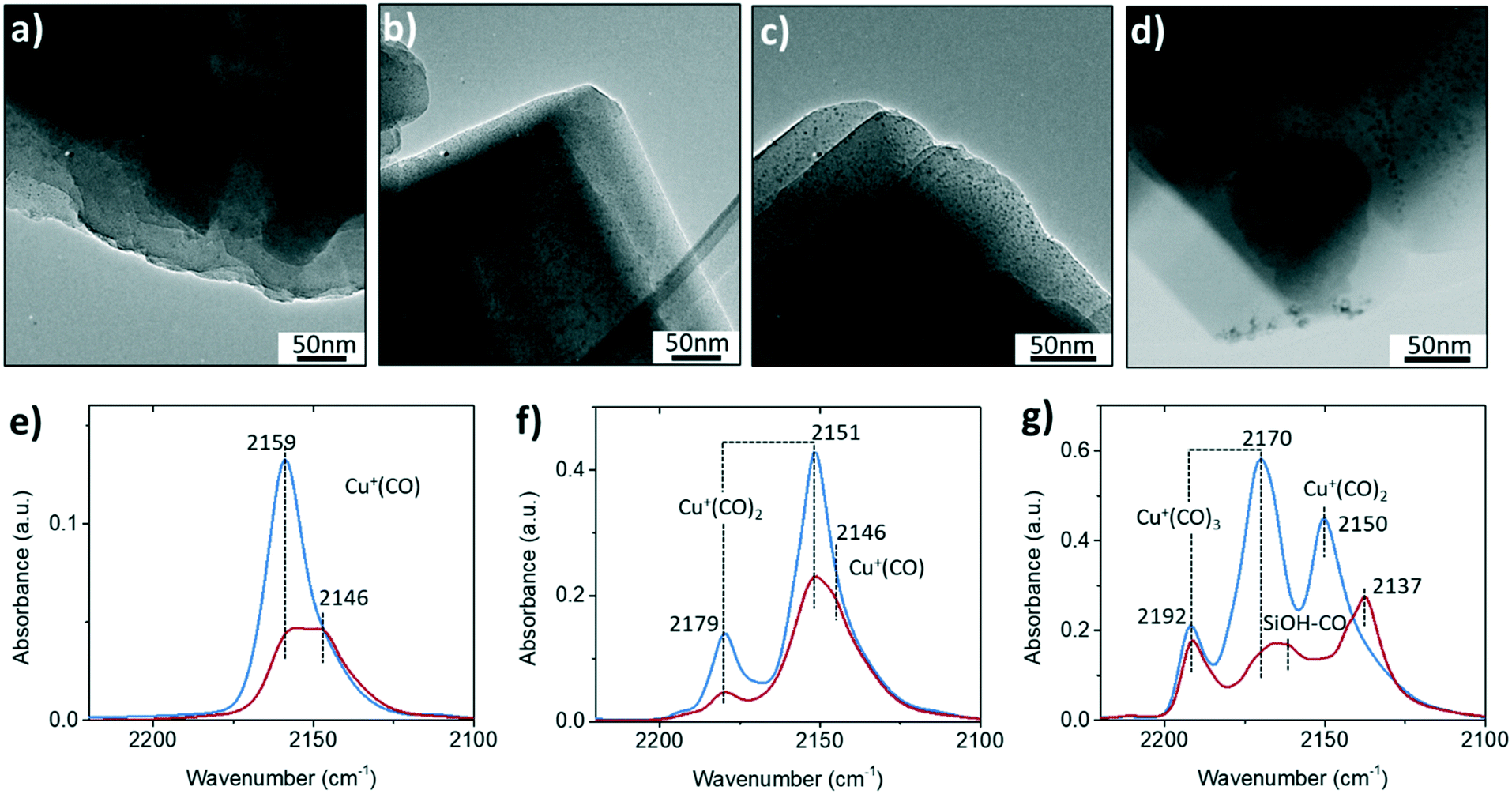 | ||
| Fig. 2 Transmission electron microscopy (TEM) images of a) fresh, b) 650 °C steamed and c–d) 850 °C steamed zeolite Cu-ZSM-5 in bright field. The CO-adsorbed Fourier transform-infrared (FTIR) spectroscopy of fresh and 850 °C steamed zeolite Cu-ZSM-5 under CO pressure of e) 0.015 mBar, f) 0.1 mBar and g) 0.5 mBar at liquid N2 temperature. Blue and red lines represent fresh and 850 °C steamed zeolite Cu-ZSM-5, respectively. | ||
CO is a universal probe molecule in FTIR spectroscopy experiments to detect the metal sites by charge donation/back-donation between metal center and CO molecule. The interaction of CO with Cu2+ is weak, and therefore only Cu+ and the hydroxyl group could be probed by CO in Cu-zeolites.24Fig. 2e–g shows the FTIR spectra with different CO coverages. One of the differences between fresh and steamed Cu-ZSM-5 in CO-adsorbed FTIR spectra is the stronger peak intensities found in the fresh Cu-ZSM-5, indicating larger numbers of available sites for CO adsorption in the fresh catalyst. The adsorption band of cuprous mono-carbonyls adducts centered at 2159 cm−1 coordinated up to three CO molecules with increasing CO pressure, which is well-documented.25 This probed Cu+ originated from [CuOH]+, which experienced auto-reduction during the dehydration pre-treatment under high vacuum.26 The loss of [CuOH]+ was confirmed in the steamed Cu-ZSM-5 in CO-adsorbed FTIR spectroscopy, and it was accompanied by the co-existence of another cuprous site coordinated with CO with a lower C–O frequency of 2146 cm−1, which was also reported in zeolite Cu-ZSM-5 with high Cu-exchanged levels.27 This cuprous site had higher coordinative saturation since only the mono-carbonyl was observed. When the CO dosage was high, the CO adsorption on the silanol became detectable only on the steamed catalyst,28 consistent with the observation of abundant internal silanol groups in the 850 °C steamed zeolite Cu-ZSM-5.
The perturbed framework T–O–T vibration is directly influenced by the interaction between the Cu ion and the framework. Fig. 3 shows the perturbed framework vibration of fresh and 850 °C steamed Cu-ZSM-5 after dehydration and subsequent NH3 treatment. The background spectrum was recorded for the hydrated form of zeolites as fully hydrated Cu2+ is mobile.29 The ammoniated Cu2+ hardly interacts with the zeolite framework, showing no perturbed T–O–T band. Upon removal of NH3, the Cu2+ is stabilized by the framework oxygen and consequently perturbs the framework T–O–T vibration. The perturbance of the framework generally depends on the charge of the interacting cation such that the higher the net charge of the interacting cation, the lower is the value of the T–O–T vibration, because a stronger interaction between opposite charges weakens the original framework vibration to a greater extent.30 The transformation from the framework stabilized Cu+ to Cu2+ causes the band shift of the asymmetric T–O–T vibration from 970 to 910 cm−1.31,32 The ∼930 cm−1 and ∼950 cm−1 bands have been assigned to bare Cu2+ and [Cu2+O−]+/[Cu2+OH−]+/O2-associated Cu+, respectively.30,33 In the fresh zeolite Cu-ZSM-5, [CuOH]+ was not shown in the perturbed framework vibration band, although its existence was clearly indicated by the CO-FTIR results and its OH stretching band at 3660 cm−1. The signal from the [CuOH]+ perturbance might be covered by the strong and broad band originating from bare Cu2+. However, in addition to the Cu2+ and Cu+ perturbed vibrational modes, the ammoniation process unveiled the 954 cm−1 band in the 850 °C steamed Cu-ZSM-5 although it lost the isolated [CuOH]+. Only the isolated or clustered Cu ions influence the perturbed framework vibration by ligand removal or addition, because interaction between large particles and zeolite framework could be hardly affected by replacement of ligands. The 954 cm−1 band is hereby supposed to be relative to the charged Cu oligomers/clusters [Cux(OH)2x−1]+ that could interfere with the framework vibrations. The adjacency of the hydroxyl group to the Cu2+ is later implied by operando DRIFTS results.
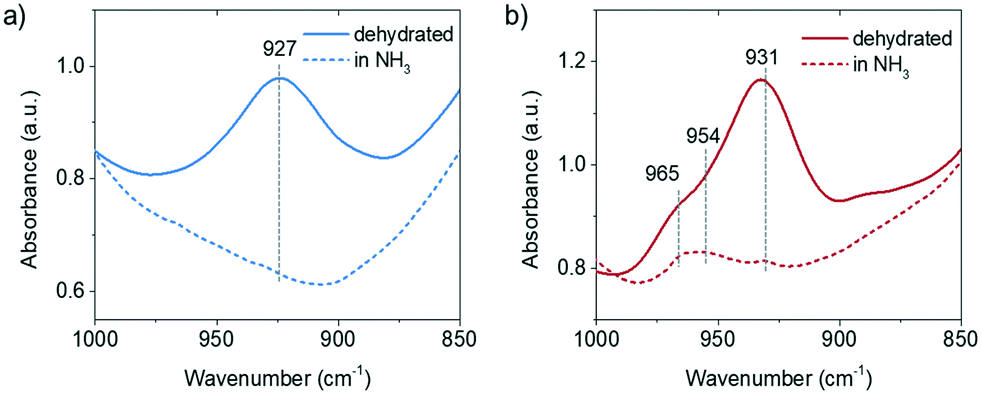 | ||
| Fig. 3 Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) data of perturbed framework T–O–T vibration of a) fresh and b) 850 °C steamed Cu-ZSM-5 after dehydration and drying under NH3/He flow. | ||
Both NO oxidation and NH3 oxidation have been under investigation in Cu-exchanged zeolites suggesting the potential contribution of isolated sites, such as Cu2+ and [CuOH]+, to the side reactions observed in the catalytic test.14,21,34 However, with the steaming-induced local damage of the zeolite framework and the formation of [Cux(OH)2x−1]+ oligomers/clusters, the NO conversion above 250 °C dropped due to the large contribution from unselective NH3 oxidation. The detrimental effect of CuxOy clusters/nanoparticles on the NH3-SCR reaction has been demonstrated to promote NH3 oxidation, and promising NH3 conversion was even observed over a physical mixture of CuO and H-SAPO-34.17,20,35
2.3 Cu2+ dynamics unravelled by operando UV-vis diffuse reflectance spectroscopy
The Cu aggregation in the steamed Cu-ZSM-5 zeolites was proposed to be responsible for the occurrence of the unselective NH3 oxidation reaction that caused the unusual NO and NH3 conversion in NH3-SCR. However, a more detailed structural correlation of the deactivated component contributions to the loss of NH3-SCR activity and the promotion of side reaction NH3 oxidation has not yet been well-understood. This requires real-time monitoring of the catalysts in a reaction to establish the structure–activity relationships for further understanding of the key structure involved in the reaction. One of the most facile means to study transition metals under working conditions is UV-vis DRS employing a high temperature UV-vis optical fibre probe.36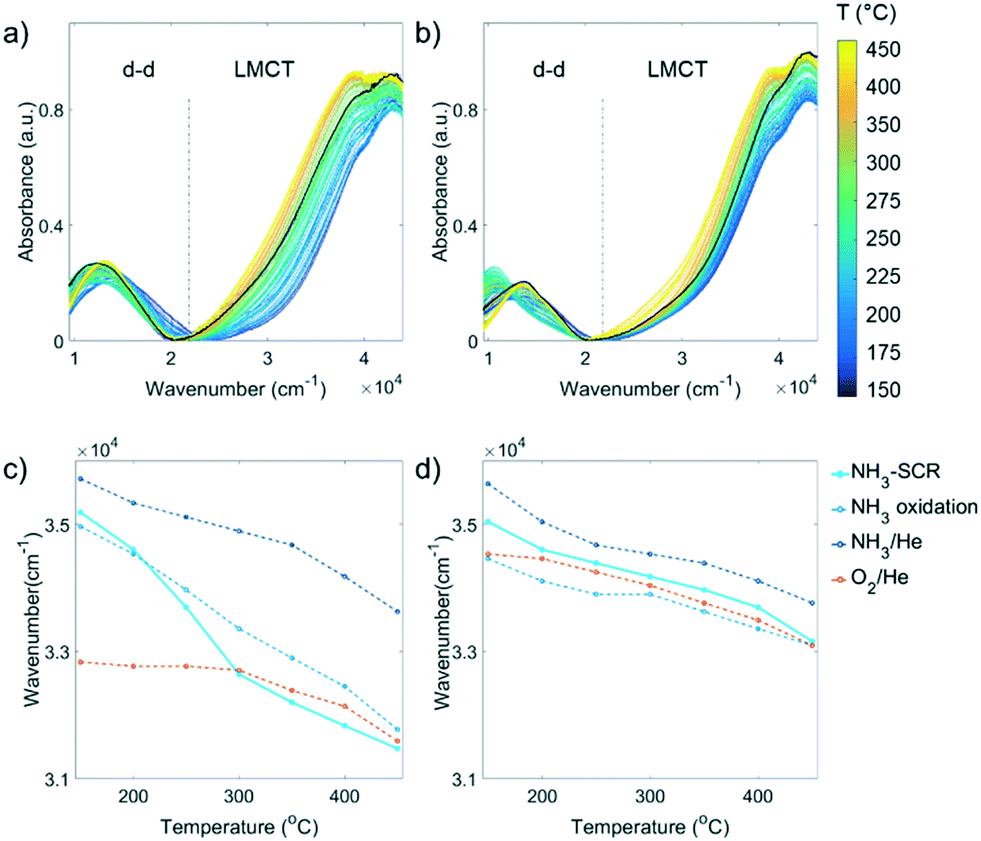 | ||
| Fig. 4
Operando UV-vis diffuse reflectance spectroscopy (DRS) data of the a) fresh and b) 850 °C steamed zeolite Cu-ZSM-5 during the NH3-selective catalytic reduction (SCR) of NO. The black solid line is the spectrum recorded at 150 °C in O2/He before the reactants were fed. The wavenumber at half height of the ligand-to-metal charge transfer (LMCT) band at ca. 39000 cm−1 in different reaction conditions was obtained at steady state for c) fresh and d) 850 °C steamed Cu-ZSM-5. | ||
To gain an intuitive look into the replacement of ligands in the reaction process, the wavenumber at half height of the LMCT maximum was followed in the NH3-SCR reaction, by comparing to that of inflow of O2/He, NH3/He and NH3 oxidation feeds (Fig. 4c and d). In the fresh zeolite Cu-ZSM-5, the reaction could again be clearly divided into two regimes including the low-temperature (150–250 °C) and high temperature (300–450 °C) NH3-SCR mechanism according to the position of the LMCT band half height. In the low temperature regime, the position of the LMCT band is shown at a high wavenumber, resembling that of the NH3 oxidation condition and being close to the LMCT band position in NH3/He, while at a temperature higher than 300 °C, the evolution of the LMCT band position is similar to that found in oxidative gases. This strongly suggests the predominant replacement of the first coordination shell atom to isolated Cu2+ from N to O during the reaction, in agreement with similar findings revealed by in situ XAS and the proposed different reaction mechanism in low- and high-temperature NH3-SCR.40 In contrast, a less significant shift of the LMCT band during the reaction was observed in the steamed zeolite Cu-ZSM-5, suggesting a less notable change of the coordinated ligands because fewer Cu2+ sites were available to take part in the reaction on the surface of [Cux(OH)2x−1]+ oligomers/clusters. No clear demarcation line between the low and high temperature regime was found in the steamed sample, though it had been in the fresh Cu-ZSM-5 zeolite material. The LMCT band positions in NH3-SCR reaction and O2/He environments are similar, indicating the dominant coordinated ligand was O2− in the steamed Cu-ZSM-5. However, the LMCT band position under NH3-SCR reaction conditions lies between its position in the NH3/O2 environment and NH3/He environment, and thereby, the coordination with NH3 cannot be ruled out.
000 cm−1, 12000–16000 cm−1 and above 16000 cm−1. Despite the band broadening and overlapping resulting from the variety of ligand combinations and heterogeneity of Cu2+ symmetry, the appearance of two spectroscopic signatures at 10350 cm−1 and 13700 cm−1 are clearly identified in the UV-vis spectra of 850 °C steamed Cu-ZSM-5. The symmetry change of Cu2+ was followed using ligand-field theory as well as previous experimental/theoretical studies on the Cu2+ UV-vis spectrum.
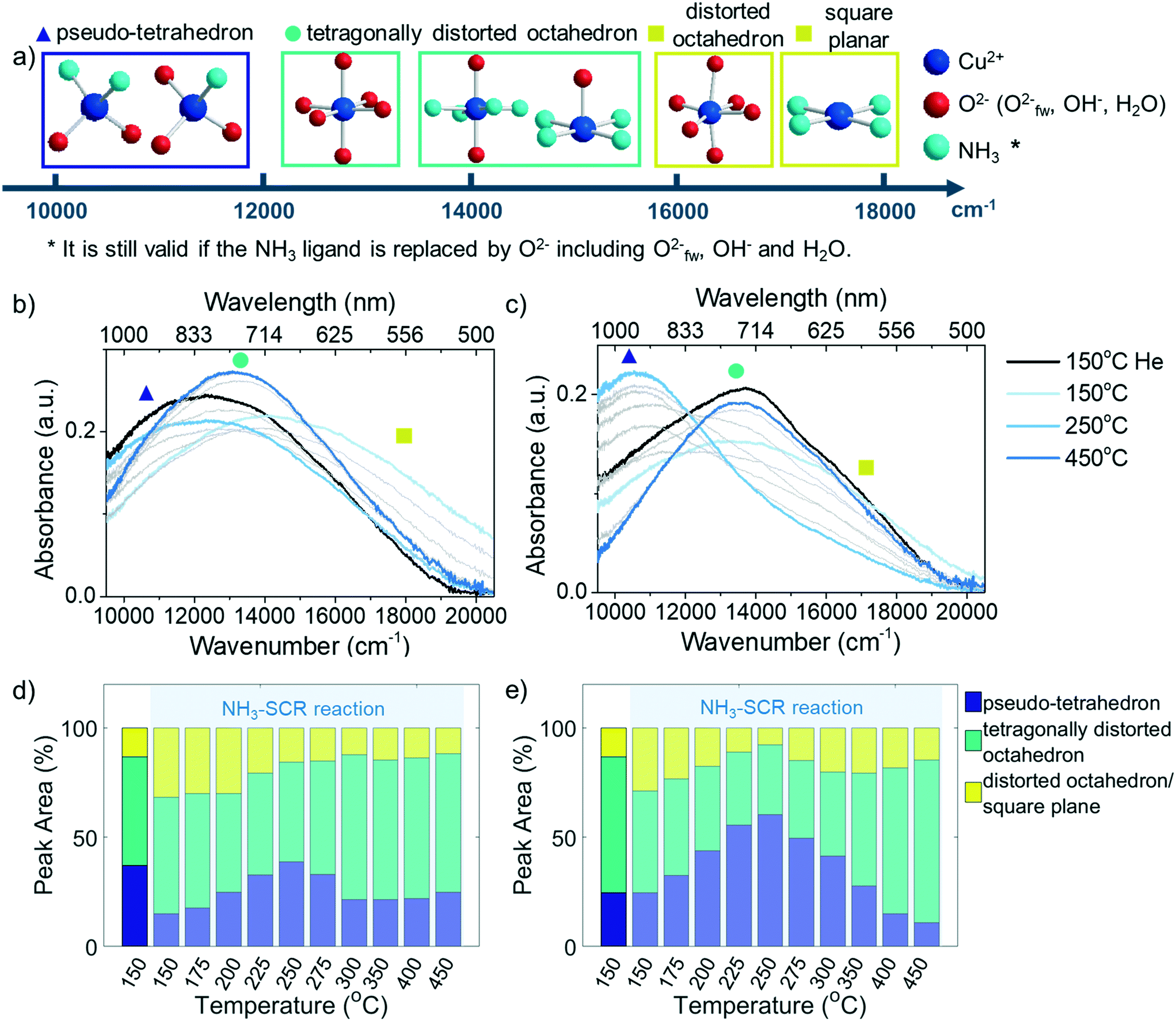 | ||
| Fig. 5 a) Schematic of the possible Cu-complexes in zeolite Cu-ZSM-5 in the NH3-SCR reaction and the approximate positions in wavenumbers of their corresponding bands, estimated based on ligand-field imposed d–d splitting of Cu2+. O2−fw represents framework oxygen. The d–d transition band and the peak fitting results of the (b and d) fresh and (c and e) 850 °C steamed Cu-ZSM-5 zeolites collected at steady-state at each temperature during the NH3-SCR reaction performance test. The bands in grey color are collected in intermediate reaction temperatures between 150–450 °C. The positions of both main peak and shoulder are indicated in (a) by a solid triangle (Prussian blue), a circle (cyan) and a square (yellow), respectively. | ||
As indicated by Fig. 5b and c, low, medium, and high wavenumber bands could be identified in the d–d transition region. The adsorption band at ∼12500 cm−1 is the typical band that arises from isolated Cu2+ in an octahedral crystal field generated by oxide ligands.41 The [Cu(H2O)6]2+ is the tetragonally distorted complex in the fully hydrated zeolite due to framework confinement. The dehydration process generates the axial elongation of Cu2+ tetragonal bipyramidal geometry upon water removal and framework attachment, which causes further splitting of octahedral ligand field and therefore the slight blue shift of the band maximum of the d–d transition.25 The Cu2+ complex in a square-planar environment has a larger splitting of the d orbital compared to octahedral, according to crystal-field theory. The simulated d–d transition band energy for the near square planar complex [Cu(NH3)4]2+ with optimized structure presented its maximum absorption at 19800 cm−1, which was in agreement with experimental observation.42,43 The high wavenumber shoulder appeared in the low temperature NH3-SCR reaction shown in Fig. 5b and c centered at ca. 17000 cm−1, suggesting the likely incorporation of a weaker field ligand O2− that might cause the redshift of the peak, i.e. the formation of [Cu(NH3)4(H2O)x]2+ (x = 1,2). The high wavenumber band could also stem from the Cu2+ complex with low symmetry that enlarges the splitting of the d orbital. At a reaction temperature between 175–300 °C, the 10350 cm−1 band became apparent especially in the steamed zeolite Cu-ZSM-5. The lower wavenumber feature of such a band implicated the smaller splitting of d orbital compared to octahedral symmetry, probably due to the tetrahedral crystal field environment (ΔT ≈ 4/9 ΔO).44 The 10350 cm−1 band was related to the coordinated NH3 since it was also present in NH3/O2 and NH3/He environments (Fig. S9†).
Partial de-ammoniation of Cu2+ caused a transition to square planar geometry, an extreme case of tetragonal distortion from octahedral symmetry, which is high in energy and is prone to relax to tetrahedral symmetry. In fact, the pseudo-tetrahedral Cu2+-complex could be expected in zeolite Cu-ZSM-5. The Cu2+ lying in a defect site or on the surface of [Cux(OH)2x−1]+ oligomers/clusters probably had then required steric hinderance for the coordinating ligand to reach octahedral symmetry; instead, the more stable tetrahedron is favoured. Indeed, the contribution of this low-wavenumber band increases with the steaming severity (Fig. S10†). A similar band maximum at 11000 cm−1 has been reported in ammoniated Cu-exchanged zeolite Y with a low coordination number.45 This low-frequency band has been hypothesized as the O3–Cu2+–NH3 structure in de-ammoniated Cu-zeolite, which was proven by multiconfigurational perturbation theory based simulation and an electron paramagnetic resonance (EPR) study.42,46 Hence, the low frequency feature is also proposed to be the pseudo-tetrahedral Cu2+ with a mix of NH3 and O2−-oriented ligands (O3–Cu2+–NH3 or O2–Cu2+–(NH3)2).
To follow the evolution of Cu-complexes throughout the NH3-SCR reaction, the d–d transition bands were fitted with three Gaussian functions by restricting fitting model parameters such as their wavenumber position based on the inspection of the eigen spectra from principal component analysis (PCA, Fig. S11†). The such established fitting model was then applied to the entire dataset, and the fitting results can be found in Fig. S12–S14.† The evolution of pseudo-tetrahedral Cu2+, tetragonally distorted Cu2+ and low symmetrical Cu2+ in fresh and 850 °C steamed zeolite Cu-ZSM-5 at steady-state during the NH3-SCR reaction are given in Fig. 5d and e. Both Cu-ZSM-5 samples behaved in a similar manner during the reaction when following the peak contributions of pseudo-tetrahedral and distorted octahedron/square planar Cu2+ during the reaction. Upon the exposure to the reactant gases, ammoniated Cu2+ developed along with the coordination with H2O. This feature gradually diminished with increasing reaction temperature (150–250 °C) because of the detachment of NH3 from Cu2+. At the same time, the contribution of a pseudo-tetrahedral Cu2+-complex with mixed NH3 and O2−-oriented ligands increased sharply. When the NH3-SCR reaction took place above 250 °C, the amount of pseudo-tetrahedral Cu2+-complex with mixed ligands started to decrease due to the continuous freeing of coordinated NH3. In this scenario, the d–d transition band of UV-vis diffuse reflectance spectra in the NH3-SCR reaction feed were eventually identical to that under O2/He flow (Fig. S15†), suggesting complete removal of coordinated NH3 and the presence of tetragonally distorted octahedral Cu2+ with an O2− ligand.
It is important to note that the change of Cu2+ geometry was due to the reaction-related dynamic but not the irreversible change of Cu2+ structure since the NO/NH3 conversion as well as the geometry were unchanged when the NH3-SCR reaction was conducted in a cyclic manner (Fig. S16†). In the low temperature regime (<250 °C), the NH3-SCR reaction was the preferential reaction according to the catalytic results. The mobile [Cu(NH3)4]2+ complex is the proposed catalytic active site that is ready to react with NO at a reaction temperature under 250 °C.40,47,48 A higher portion of NH3-solvated Cu2+ observed in the fresh zeolite Cu-ZSM-5 was attributed to the higher NO conversion at a low reaction temperature compared to the steamed Cu-ZSM-5. At a reaction temperature of 150–250 °C, the adsorbed NH3 either desorbed or reacted with intermediates, resulting in partially de-ammoniated Cu2+ with pseudo-tetrahedral symmetry, which was simultaneously coordinated with O2− or with the intermediate NOx−.49,50 The accumulation of Cu2+ in pseudo-tetrahedral symmetry with coordinated NH3, which is stable below 250 °C, limits the NO and NH3 conversion to a great extent particularly in steamed Cu-ZSM-5. As the reaction temperature increased from 250 °C, the coordinated NH3 in pseudo-tetrahedral Cu2+ started to disassociate, which could be proven by the desorption of NH3 adsorbed on Cu2+ with Lewis acidity (Fig. S7†). However, such desorption of NH3 from pseudo-tetrahedral Cu2+ provoked unselective NH3 oxidation rather than the NH3-SCR reaction, which is clear in the steamed Cu-ZSM-5 from the rapid increase of NH3 conversion and a dramatic drop of NO conversion between 250–300 °C. Finally, in high-temperature NH3-SCR above 300 °C, the adsorption of NH3 weakens, rather, the fully de-ammoniated Cu2+ tends to anchor on the framework O2− with coordination of four,42,51 resulting in the identical geometric structure as it has in O2/He flow. The Cu2+ complex with an O2−-directing ligand is the key species for the high temperature NH3-SCR reaction in fresh and steamed Cu-ZSM-5, allowing the maximum Cu2+ coordination number to be a distorted octahedron by interaction with external ligands, for example the possible reaction intermediates NO2 or NOx−.
Particularly for 850 °C steamed Cu-ZSM-5, the Cu2+ geometry is identical in the NH3-SCR reaction and unselective NH3 oxidation reactions (Fig. S9†), which is strong proof of the great impact of unselective NH3 oxidation in the NH3-SCR reaction. It also points out the similarity of the Cu2+ local structure that is responsible for NH3-SCR and unselective NH3 oxidation. In the intermediate reaction temperature of 250–300 °C, desorption of NH3 mainly took place on the surface of [Cux(OH)2x−1]+ oligomers/clusters and was followed by the rapid oxidation into NO, resulting in the sudden increase of NH3 conversion.
2.4 Adsorption competition revealed by operando diffuse reflectance infrared Fourier transform spectroscopy
The low-temperature standard NH3-SCR reaction (<250 °C) attracts extra attention because improved NO conversion is needed in this temperature range. The NH3-SCR catalysts inevitably deactivate from steam produced by fuel combustion. As indicated in the catalytic performance results, 250 °C is a critical point, from which the low-temperature NH3-SCR starts to transition to the high-temperature reaction. At this temperature, no apparent side reaction takes place in the NH3-SCR reaction even in the 850 °C steamed Cu-ZSM-5, as NH3 and NO have the same conversion (Fig. 1d), which is beneficial for investigating the behaviour in NH3-SCR without interference. Adsorbed surface species are potentially useful to gain insight into the reaction and deactivation pathways. The experimental protocol for the DRIFTS experiment is described in Fig. 6a. The experiment was conducted at 250 °C after a calcination step. The reaction started with NH3 oxidation, followed by NO addition and subsequent NH3 removal to achieve NH3-SCR and NO oxidation, respectively.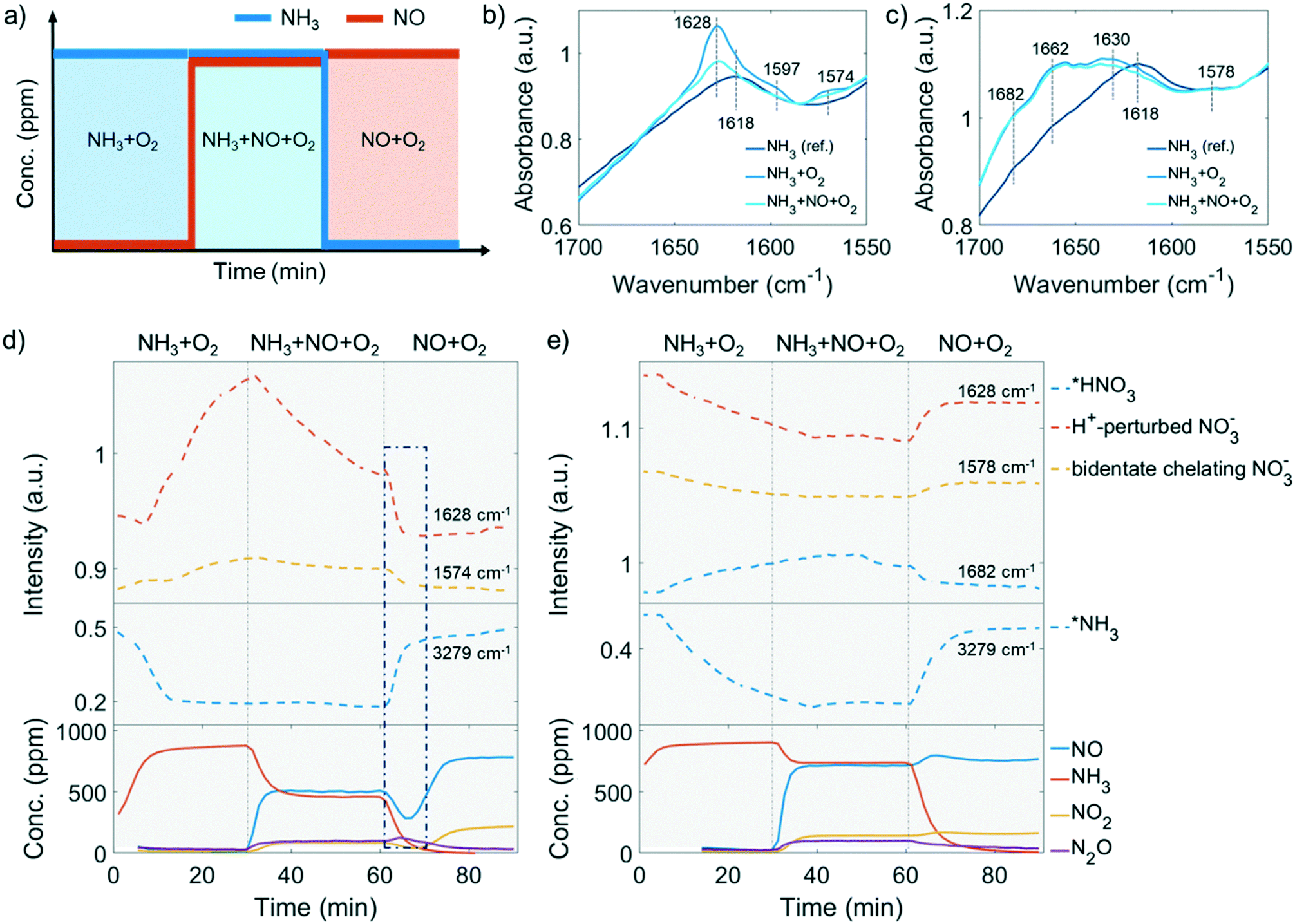 | ||
| Fig. 6 a) Procedure of the operando diffuse reflectance Fourier transform spectroscopy (DRIFTS) experiment performed. The experiment was conducted after O2 calcination at 550 °C followed by NH3 + O2, NH3 + NO + O2 and NO + O2 reaction at 250 °C with 1000 ppm of NH3 and/or 1000 ppm NO balanced by 5% O2/He. The obtained operando DRIFTS spectra of surface nitrates in b) fresh and c) 850 °C steamed zeolite ZSM-5. The evolution of selective bands representing surface nitrates (upper panel) and adsorbed NH3 (middle panel) were followed and the recorded concentration of effluent gas composition (bottom panel) in the d) fresh and e) 850 °C steamed zeolite Cu-ZSM-5. Missing datapoints at the beginning of the experiment (Fig. 6d and e, bottom panel) are due to values below the limit of detection. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of a bridging nitrate where two vicinal Cu atoms are required to anchor two nitrate O2−.55 With this postulation, a stronger 1628 cm−1 band should be found in the steamed zeolite Cu-ZSM-5 as more adjacent Cu atoms are available when Cu aggregation has happened. However, this is contradicted by the fact that the 1628 cm−1 band is a shoulder in the steamed Cu-ZSM-5, while it is an intense peak in its fresh counterpart (Fig. 6b and c). Alternatively, the 1628 cm−1 feature has been proposed to be the proton-perturbed chelating nitrate whose NO vibration is affected by the nearby Brønsted acid site; this assignment was shown by a systematic FTIR spectroscopy study of surface Cu2+(N,O) species on Cu-CHA,56 which could explain why the pronounced 1628 cm−1 band was more intense on the fresh Cu-ZSM-5 with well-defined Brønsted acid sites. The 1662 cm−1 and 1682 cm−1 band related to the adsorbed NO2 are unique in the 850 °C steamed zeolite Cu-ZSM-5, where the former one can be attributed to adsorbed NO2 while the latter one is from the protonated NO2.57,58 It is not surprising to observe NOx adsorption on the surface of steamed Cu-ZSM-5 zeolites with abundant hydroxyl groups, which leads to the formation of HNOx during the operando DRIFTS experiment where NH3-SCR, NH3 oxidation, or NO oxidation took place. The conversion between nitrous acid and nitric acid was kept in balance depending on the local concentration of NO and NO2. Finally, the 1682 cm−1 band was tentatively attributed to the adsorbed HNO3, considering that HNO2 is easily oxidized to HNO3 at reaction conditions.
O of a bridging nitrate where two vicinal Cu atoms are required to anchor two nitrate O2−.55 With this postulation, a stronger 1628 cm−1 band should be found in the steamed zeolite Cu-ZSM-5 as more adjacent Cu atoms are available when Cu aggregation has happened. However, this is contradicted by the fact that the 1628 cm−1 band is a shoulder in the steamed Cu-ZSM-5, while it is an intense peak in its fresh counterpart (Fig. 6b and c). Alternatively, the 1628 cm−1 feature has been proposed to be the proton-perturbed chelating nitrate whose NO vibration is affected by the nearby Brønsted acid site; this assignment was shown by a systematic FTIR spectroscopy study of surface Cu2+(N,O) species on Cu-CHA,56 which could explain why the pronounced 1628 cm−1 band was more intense on the fresh Cu-ZSM-5 with well-defined Brønsted acid sites. The 1662 cm−1 and 1682 cm−1 band related to the adsorbed NO2 are unique in the 850 °C steamed zeolite Cu-ZSM-5, where the former one can be attributed to adsorbed NO2 while the latter one is from the protonated NO2.57,58 It is not surprising to observe NOx adsorption on the surface of steamed Cu-ZSM-5 zeolites with abundant hydroxyl groups, which leads to the formation of HNOx during the operando DRIFTS experiment where NH3-SCR, NH3 oxidation, or NO oxidation took place. The conversion between nitrous acid and nitric acid was kept in balance depending on the local concentration of NO and NO2. Finally, the 1682 cm−1 band was tentatively attributed to the adsorbed HNO3, considering that HNO2 is easily oxidized to HNO3 at reaction conditions.
In the first step of the operando DRIFTS experiment on the fresh Cu-ZSM-5 zeolite, nitrates developed in NH3/O2 flow (Fig. 6d), suggesting the full oxidation of surface NH3, which was also observed in in situ FTIR studies on the NH3 oxidation reaction.59,60 The formed nitrates replaced the pre-adsorbed NH3 on Cu2+ and weakened the adsorbed NH3 signal (Fig. 6 and S17†). The re-appearance of the Brønsted acid site (Fig. S17a†) might be due to the reaction between the Brønsted acid adsorbed NH3 (B-NH3) and L-NO3− followed by restoration of the proton from H-cleavage of NH3.61 The NH3 oxidation reaction was followed by the NH3-SCR reaction where NO participated and reacted with surface nitrates, resulting in the formation of NO2 and NO2− (reaction 2, Scheme 1) that avoid the surface blockage by nitrates.3 Unfortunately, we cannot confirm the formation of surface nitrites as the O–N–O stretching frequency was covered by the signal from symmetric stretching of N–H as well as the intense signal from the zeolite structure.62,63 In the last step of NH3 removal, consumption of surface nitrate species accelerated. The involvement of NO in nitrate depletion is now strongly supported by the simultaneous drop of NO concentration as surface nitrates are decreasing, which is indicated by the blue rectangle in Fig. 6d. Interestingly, once the nitrates were depleted, the Cu2+ site is re-occupied by the residual NH3 (Fig. 6d, bottom panel). In fact, a similar phenomenon of NH3-nitrates competitive adsorption has been reported in Cu-exchanged zeolites, where adsorbed nitrates and NH3 on Lewis acid sites could be replaced by each other depending on reaction conditions.10,64,65 The Lewis acid, which is isolated Cu2+ in our case of fresh Cu-ZSM-5, is thus the suggested main site for NH3 adsorption and nitrate formation/adsorption according to the changes of the perturbed framework vibration with the surface species involved (Fig. S17†).
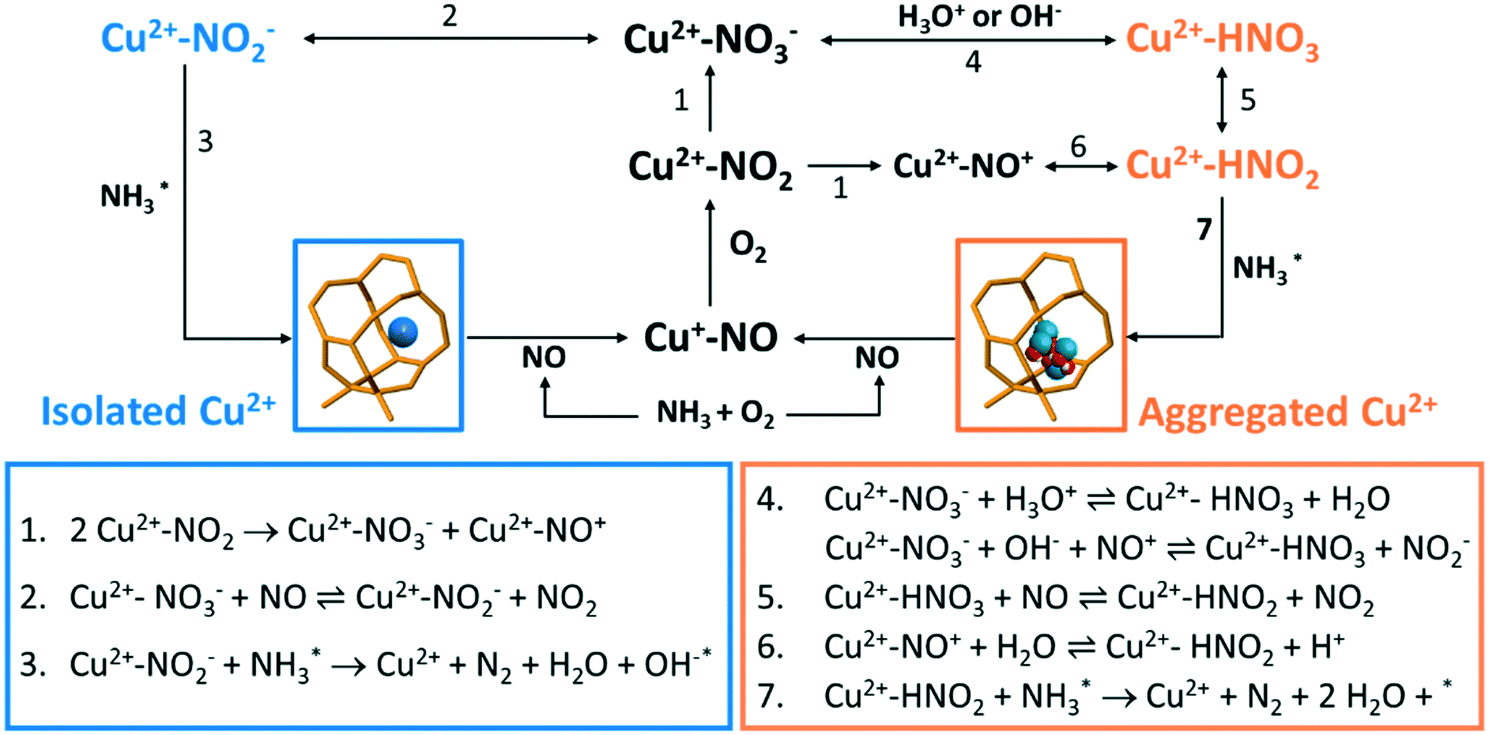 | ||
| Scheme 1 Possible reactions that take place in the low-temperature NH3-selective catalytic reduction (SCR) reaction over Cu–zeolites. The blue colour indicates dominant reactions that can take place on isolated Cu2+, while the orange colour stands for possible reactions over aggregated Cu2+ species, such as [Cux(OH)2x−1]+ oligomers/clusters. | ||
The causes of deactivation were revealed from the operando DRIFTS data collected on the 850 °C steamed Cu-ZSM-5 zeolite (Fig. 6e). In NH3/O2 flow, adsorbed NH3 and nitrates developed in the initial stage, followed by the disappearance of both surface species along with the increased adsorbed nitric acid (Fig. 6e, S18a and c†). No significant change of surface species was observed upon the subsequent addition of NO in the second step. Similarly, the NH3 desorption in NH3/O2 flow could be explained by the competitive adsorption between NH3 and nitric acid, because these two species exhibited opposite trends throughout the whole DRIFTS experiment (Fig. 6e). The production of nitric acid is related to the attenuation of surface nitrates (Fig. 6e), which can react with the adjacent proton H+/hydronium or surface hydroxyl group to from nitric acid (reaction 4, Scheme 1).53 The observed adsorbed NO2 was the precursor for nitric acid formation, showing good agreement with an in situ FTIR study conducted on hydroxyl-rich or hydrated silica,γ-Al2O3 and TiO2.58,66,67 In the final step in NO/O2, analogous to the nitrate depletion in the fresh sample, nitric acid was reduced by NO (Fig. 6e), producing NO2 that could turn into nitrate by disproportionation (reaction 5 and 1, Scheme 1).58,68 The surface coverage of nitrates and nitric acid results from the competition between their formation and consumption.
The competitive adsorption of NH3 and surface nitrates/nitric acid occurred on both fresh and 850 °C steamed Cu-ZSM-5 zeolites. With the replacement of adsorbed NH3, nitrates/nitric acid was generated with the appearance of perturbed framework vibration by Cu2+ (in the fresh Cu-ZSM-5) and [Cux(OH)2x−1]+ oligomers/clusters (in the 850 °C steamed Cu-ZSM-5) as demonstrated in Fig. S17 and S18.† Not all the [CuOH]+ sites were involved in the reaction since they were partially preserved in NH3-rich flow and were not perturbed by surface nitrates (Fig. S17a, d and g†). Nitric acid rather than nitrates was the more stable intermediate that was more ready to react with adsorbed NH3 in the 850 °C steamed Cu-ZSM-5.
The coupling of NO oxidation and NH3 oxidation with the standard NH3-SCR reaction is through surface nitrates, which are formed from adsorbed NO2. NO2 can be formed through several reaction pathways at NH3-SCR reaction conditions according to the catalytic results (Fig. 1). NO2 is one of the byproducts generated from the participation of either NO oxidation reaction below 250 °C or unselective NH3 oxidation reaction above 250 °C. The NO oxidation reaction produces NO2, which is formed via reaction between dissociated O2 and loosely adsorbed NO according to a detailed kinetic model of NO oxidation.70 For NH3 oxidation, although its reaction mechanism is still under debate, a two-step reaction pathway named ‘the internal SCR mechanism’ has been proposed where NH3 is first oxidized to NOx followed by the NH3-SCR reaction.1,61
In the NH3-SCR reaction, the NO oxidation with molecule O2 into nitrates through the formation of NO2 has been stressed as it is suggested as a rate-determined step.50 The formation of NO2 promotes the formation of surface nitrates and meanwhile boosts the reoxidation of Cu+ to Cu2+ in the oxidation half cycle.71–74 A Cu monomer in Cu-exchanged zeolites has been reported as the NO2 adsorption site, enabling NH3-SCR reaction to proceed with NO2 intermediate.75 Nevertheless, NO2 could be also detected as an undesired side product. Upon encountering the hydroxylated or hydrated surface, nitrates could be protonated to form surface nitric acid, which happened in the 850 °C steamed Cu-ZSM-5 as shown in Fig. 6c. However, the surface nitrite rather than nitrate is the key intermediate for the desired N2 production, demonstrating the importance of nitrate–nitrite equilibrium (reaction 2 and 4, Scheme 1) which greatly influences the reaction selectivity. Shifting the equilibrium towards nitrite formation was witnessed in both fresh and steamed Cu-ZSM-5 zeolites, deduced by the nitrate depletion with the introduction of NO in NH3/O2 feed in the operando DRIFTS experiments. A similar founding was also described in a combined FTIR-XAS study on zeolite Cu-SSZ-13.76 Successive reaction of nitrites/nitrous acid with L-NH3 lead to products formation (reaction 3 and 7, Scheme 1). Participation of B-NH3 was not observed based on the operando DRIFTS data because of the observation of well-preserved of Brønsted acid sites, although some previous studies suggested the surface NH4NO3/NH4NO2 is reaction intermediate that decompose to N2O/N2 at reaction temperatures.53,72,77
2.5 Structure–intermediate–performance relationship
A structure–intermediate–performance relationship can be established to elucidate the reasons behind the high activity at low temperature for the NH3-SCR reaction in fresh Cu-ZSM-5 and the undesirable side reactions in the steamed Cu-ZSM-5 by correlating the results from the operando UV-vis DRS and DRIFTS experiments. NH3 did not completely desorb from Lewis sites according to NH3-TPD (Fig. S7†) and operando UV-vis diffuse reflectance spectra at a low reaction temperature (Fig. 5).Adsorbed NH3 is prerequisite for the low temperature reaction (<250 °C), which is ready to react with the surface nitrites/nitrates intermediates. The higher coordination number of Cu2+ in tetragonally-distorted octahedral symmetry in the fresh Cu-ZSM-5 ensures that enough empty orbitals are available for stabilizing nitrites/nitrates, together with NH3 ligands judged by the evolution of the LMCT band. It is noted that the complete desorption of L-NH3 happened at ∼400 °C in NH3-TPD. Therefore, low surface coverage of L-NH3 at 250 °C during the NH3-SCR reaction observed in operando DRIFTS experiment (Fig. S17d†) implied the highly active nature of adsorbed NH3 on isolated Cu2+ for the formation of nitrites/nitrates intermediates.
As for the 850 °C steamed Cu-ZSM-5 zeolite, NO and NH3 conversion was low at 150–250 °C during which the pseudo-tetrahedral Cu2+ (O3–Cu2+–NH3 or O2–Cu2+–(NH3)2) accumulated likely due to the nitrates/nitric acid adsorbed [Cux(OH)2x−1]+ oligomers/clusters coordinated with a NH3 ligand. A slower reaction rate between adsorbed NH3 and nitrates/nitric acid was found on [Cux(OH)2x−1]+ oligomers/clusters because of the co-existence of these surface species. And the surface coverage of nitrites/nitrates intermediates was also lower in the steamed Cu-ZSM-5 zeolite compared to that of in its fresh counterpart (Fig. 6d and e). When the L-NH3 desorption started from 250 °C, the Cu2+ in pseudo-tetrahedral symmetry also began to disappear due to NH3 removal (Fig. 5c and e). In the steamed Cu-ZSM-5, the freed NH3 was expeditiously oxidized and released NO from the surface of [Cux(OH)2x−1]+ oligomers/clusters because of its weaker coordinating ability to stabilize reaction intermediates. This caused the peaked conversion of NH3 and the undesired NO production at 250–300 °C. Additionally, in the 850 °C steamed Cu-ZSM-5, the same active unit and the same intermediate resulted in the same pseudo-tetrahedral Cu2+ structure with mixed NH3 and nitrates/nitric acid in the NH3-SCR and NH3 oxidation reaction process. The [Cux(OH)2x−1]+ oligomers/clusters could be further aggregated into Cu(OH)2, which was recently proposed as the precursor of the inactive CuAl2O4 species.9
2.6 Practical implication of detected acidic products
Understanding the relationship between the NH3-SCR reaction and its side reactions including NH3 oxidation and NO oxidation is required to address the practical problems that the NH3-SCR catalysts, which are Cu-based zeolites, inevitably experience during hydrothermal aging in the exhaust pipe of a vehicle. This causes irreversible structural damage that starts from local degradation of framework Al or Cu migration/aggregation. Changes in the structural properties tune the reaction direction and consequently the reaction activity and selectivity. Protonated NO2 was identified as a significant spectator based on the operando DRIFTS experiment over the steamed zeolite Cu-ZSM-5, suggesting a hydroxylated environment around the NO2 adsorption site, for example the [Cux(OH)2x−1]+ oligomers/clusters found in this study. Moreover, the effects of abundant internal silanol groups generated from the steaming process cannot be ruled out, which is facilitated for H2O adsorption via hydrogen bonding. Hydroxyl groups and H2O should be considered in the nitrate-mediated reaction network, resulting in the formation of surface HNO3 and HNO2 according to reactions 4–6 in Scheme 1. The acid–base reaction between HNOx and NH3 occurs to form NH4NOx which is capable of decomposing into N2 or N2O.53,78,79 It should be stressed that surface HNO3/HNO2 is not exclusive to the steamed zeolite since it has also been reported to be involved in elementary steps in the NH3-SCR reaction in a microkinetic model over Cu-ZSM-5.78Although NO2 incorporates in the reaction through fast NH3-SCR or is converted into surface NOx− or HNOx, excessive NO2 was still detected from the outlet even from the fresh zeolite Cu-ZSM-5. Considering the practical reaction conditions after a vehicle engine, the limited amount of acidic NO2 byproduct can convert to nitric acid in H2O vapor (4NO2 + 2H2O + O2 ⇌ 4HNO3) produced by diesel combustion, while the nitric acid can also be reversely decomposed to NO2 at relatively high operational temperatures. Therefore, another adsorbent/catalyst to trap or further remove of possible undesired acidic components is still necessary after the NH3-SCR unit in an automotive emission control system. Alkali or alkaline earth metal oxide based catalysts are promising lean NOx trap (LNT) catalysts,80 which can be placed at the exist of the NH3-SCR unit to limit the emission of acidic byproducts.
Conclusions
Side reactions in the NH3-SCR reaction, such as NO oxidation and NH3 oxidation, should not be neglected, especially when considering the practical application of the Cu–zeolite-based catalysts in the tailpipe exhaust treatment of diesel vehicles. The contribution of NO oxidation was found at low reaction temperatures (<250 °C), which is considered an essential temperature range for Cu+ reoxidation in the redox cycle. When the reaction temperature is higher than 250 °C, the contribution from unselective NH3 oxidation accelerated and produced NO, resulting in a ‘dip’ shape of the NO conversion curve in the NH3-SCR reaction in steamed Cu-ZSM-5 zeolites. The occurrence of unselective NH3 oxidation was found to be more significant in the more severely steamed Cu-ZSM-5 samples, which was ascribed to the aggregation of Cu whose structure is postulated to be [Cux(OH)2x−1]+ oligomers/clusters.Combining the results from the operando UV-vis DRS and DRIFTS experiments, we propose that Cu2+ probably degraded into [Cux(OH)2x−1]+ oligomers/clusters, which can further grow into Cu(OH)2 nanoparticles in the steamed zeolite Cu-ZSM-5. The dynamic changes in the symmetry of the Cu2+ complex revealed via operando UV-vis DRS show the structural reason for the high NH3-SCR reaction activity of the fresh Cu-ZSM-5 and the deactivation of steamed Cu-ZSM-5. Octahedral Cu2+ with a coordination number of six can be formed during the NH3-SCR reaction in the fresh Cu-ZSM-5, facilitating the reaction with a high surface coverage of intermediates. The low temperature reaction showed the preference for NH3 coordination, which is replaced by O2−-oriented ligand coordination at elevated reaction temperature, confirming different reaction mechanisms in low- and high-temperature NH3-SCR. There is a more pronounced formation of pseudo-tetrahedral Cu2+ in the steamed Cu-ZSM-5 during the low temperature reaction. The pseudo-tetrahedral symmetry is closely related to partially de-ammoniated Cu2+ and its adsorption on the surface of [Cux(OH)2x−1]+ oligomers/clusters. The relaxation of this distorted structure by further removal of NH3 ligand brings about the undesired NH3 oxidation reaction. It should be noted that the same geometric structure of a Cu2+ center is shared with low-temperature NH3-SCR and NH3 oxidation, but higher NH3 conversion was found in the NH3-SCR reaction, stressing the important role of NO in NH3-SCR reaction.
The performed operando DRIFTS experiments suggest that isolated Cu2+ in the fresh Cu-ZSM-5 and the [Cux(OH)2x−1]+ oligomers/clusters in the steamed Cu-ZSM-5 are the main sites participating in the NH3-SCR reaction up to 250 °C. This can be concluded from the competitive adsorption between NH3 and surface nitrates/nitric acid at 250 °C, because they share the same adsorption sites on Lewis acid sites. Surface nitrates are the key surface species to bridge the NH3-SCR, NH3 oxidation and NO oxidation reactions. However, surface nitric acid was more prevalent in the steamed Cu-ZSM-5 because of the presence of high density of hydroxyl groups. The high surface coverage of nitrates/nitric acid was reconciled by the reaction with NO to avoid the surface blockage; this reaction governs the nitrate–nitrite equilibrium that determines the selectivity of the reaction. Additionally, no direct involvement of Brønsted acid sites in the NH3-SCR reaction was observed at steady state, instead, the surface nitrates were perturbed by the nearby proton, probably from the Brønsted acid sites. Finally, a structure–intermediate–performance relationship could be established to elucidate the low NH3-SCR activity and the ‘dip’ shape of NO conversion curve in the steamed Cu-ZSM-5: the pseudo-tetrahedral Cu2+ complex of [Cux(OH)2x−1]+ oligomers/clusters with associated NH3 and nitrates/nitric acid exhibited low activity below 250 °C due to the relatively strong adsorption of surface species; the further increase in temperature (above 250 °C) released the NH3 and directed the unselective NH3 oxidation. For practical implications, the formation of adsorbed NO2 and surface nitric acid should be considered for a better design of vehicle exhaust control systems to meet the requirement of future stringent regulations.
Experimental
See ESI.†Author contributions
X. Ye designed and performed the experiments, as well as processed the acquired data, and drafted the manuscript. R. Oord and M. Monai participated in the discussion of the results, while J. Schmidt provided scientific suggestions and revised the manuscript. T. Chen, F. Meirer and B. M. Weckhuysen supervised the research and the preparation and writing of the article.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work is supported by the Netherlands Organization for Scientific Research (NWO) in the frame of a Gravitation Program (MCEC, Multiscale Catalytic Energy Conversion). X. Y. acknowledges support from China Scholarship Council (CSC). Ru-Pan Wang (Department of Physics, University of Hamburg) is acknowledged for the discussion of the operando UV-vis DRS results.Notes and references
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC
.
- J. Wang, H. Zhao, G. Haller and Y. Li, Appl. Catal., B, 2017, 202, 346–354 CrossRef CAS
- F. Gao, J. H. Kwak, J. Szanyi and C. H. F. Peden, Top. Catal., 2013, 56, 1441–1459 CrossRef CAS
- M. Iwamoto, H. Furukawa, Y. Mine, F. Uemura, S. Mikuriya and S. Kagawa, J. Chem. Soc., Chem. Commun., 1986, 1272–1273 RSC
- D. Wang, Y. Jangjou, Y. Liu, M. K. Sharma, J. Luo, J. Li, K. Kamasamudram and W. S. Epling, Appl. Catal., B, 2015, 165, 438–445 CrossRef CAS
- W. Su, Z. Li, Y. Peng and J. Li, Phys. Chem. Chem. Phys., 2015, 17, 29142–29149 RSC
- A. Wang, Y. Chen, E. D. Walter, N. M. Washton, D. Mei, T. Varga, Y. Wang, J. Szanyi, Y. Wang, C. H. F. Peden and F. Gao, Nat. Commun., 2019, 10, 1137 CrossRef PubMed
- J. Song, Y. Wang, E. D. Walter, N. M. Washton, D. Mei, L. Kovarik, M. H. Engelhard, S. Prodinger, Y. Wang, C. H. F. Peden and F. Gao, ACS Catal., 2017, 7, 8214–8227 CrossRef CAS
- Y. Ma, X. Wu, L. Liu, L. Cao, R. Ran, Z. Si, F. Gao and D. Weng, Appl. Catal., B, 2020, 278, 119306 CrossRef CAS
- H. Sjövall, L. Olsson, E. Fridell and R. J. Blint, Appl. Catal., B, 2006, 64, 180–188 CrossRef
- M. P. Ruggeri, I. Nova, E. Tronconi, J. A. Pihl, T. J. Toops and W. P. Partridge, Appl. Catal., B, 2015, 166–167, 181–192 CrossRef CAS
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2014, 319, 1–14 CrossRef CAS
- T. Yu, J. Wang, Y. Huang, M. Shen, W. Li and J. Wang, ChemCatChem, 2014, 6, 2074–2083 CrossRef CAS
- P. S. Metkar, V. Balakotaiah and M. P. Harold, Catal. Today, 2012, 184, 115–128 CrossRef CAS
- X. Ye, J. E. Schmidt, R. Wang, I. K. Ravenhorst, R. Oord, T. Chen, F. Groot, F. Meirer and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15610–15617 CrossRef CAS PubMed
- F. Gao, Y. Wang, N. M. Washton, M. Kollár, J. Szanyi and C. H. F. Peden, ACS Catal., 2015, 5, 6780–6791 CrossRef CAS
- D. Wang, L. Zhang, J. Li, K. Kamasamudram and W. S. Epling, Catal. Today, 2014, 231, 64–74 CrossRef CAS
- W. B. Williamson and J. H. Lunsford, J. Phys. Chem., 1976, 80, 2664–2671 CrossRef CAS
- L. Olsson, K. Wijayanti, K. Leistner, A. Kumar, S. Y. Joshi, K. Kamasamudram, N. W. Currier and A. Yezerets, Appl. Catal., B, 2015, 174–175, 212–224 CrossRef CAS
- Y. J. Kim, P. S. Kim and C. H. Kim, Appl. Catal., A, 2019, 569, 175–180 CrossRef CAS
- F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. F. Peden, J. Catal., 2013, 300, 20–29 CrossRef CAS
- J. Holzinger, P. Beato, L. F. Lundegaard and J. Skibsted, J. Phys. Chem. C, 2018, 122, 15595–15613 CrossRef CAS
- J. A. van Bokhoven, D. C. Koningsberger, P. Kunkeler and H. van Bekkum, J. Catal., 2002, 211, 540–547 CrossRef CAS
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741–12761 RSC
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, J. Phys. Chem. C, 2019, 123, 15139–15144 CrossRef CAS
- G. J. Millar, A. Canning, G. Rose, B. Wood, L. Trewartha and I. D. R. Mackinnon, J. Catal., 1999, 183, 169–181 CrossRef CAS
- P. N. R. Vennestrøm, T. V. W. Janssens, A. Kustov, M. Grill, A. Puig-Molina, L. F. Lundegaard, R. R. Tiruvalam, P. Concepción and A. Corma, J. Catal., 2014, 309, 477–490 CrossRef
- C. W. Andersen, E. Borfecchia, M. Bremholm, M. R. V. Jørgensen, P. N. R. Vennestrøm, C. Lamberti, L. F. Lundegaard and B. B. Iversen, Angew. Chem., Int. Ed., 2017, 56, 10367–10372 CrossRef CAS PubMed
- J. Sárkány, Appl. Catal., A, 1999, 188, 369–379 CrossRef
- H. Y. Chen, L. Chen, J. Lin, K. L. Tan and J. Li, Inorg. Chem., 1997, 36, 1417–1423 CrossRef CAS PubMed
- E. Broclawik, J. Datka, B. Gil and P. Kozyra, Phys. Chem. Chem. Phys., 2000, 2, 401–405 RSC
- P. E. Fanning and M. A. Vannice, J. Catal., 2002, 207, 166–182 CrossRef CAS
- H.-Y. Chen, Z. Wei, M. Kollar, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, Catal. Today, 2016, 267, 17–27 CrossRef CAS
- F. Gao and C. Peden, Catalysts, 2018, 8, 140 CrossRef
- M. H. Groothaert, K. Lievens, H. Leeman, B. M. Weckhuysen and R. A. Schoonheydt, J. Catal., 2003, 220, 500–512 CrossRef CAS
- R. A. Schoonheydt, Chem. Soc. Rev., 2010, 39, 5051–5066 RSC
- E. Borfecchia, C. Negri, K. A. Lomachenko, C. Lamberti, T. V. W. Janssens and G. Berlier, React. Chem. Eng., 2019, 4, 1067–1080 RSC
- C. Negri, M. Signorile, N. G. Porcaro, E. Borfecchia, G. Berlier, T. V. W. Janssens and S. Bordiga, Appl. Catal., A, 2019, 578, 1–9 CrossRef CAS
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed
- S. A. Yashnik, Z. R. Ismagilov and V. F. Anufrienko, Catal. Today, 2005, 110, 310–322 CrossRef CAS
- A. Delabie, K. Pierloot, M. H. Groothaert, B. M. Weckhuysen and R. A. Schoonheydt, Microporous Mesoporous Mater., 2000, 37, 209–222 CrossRef CAS
- V. F. Anufrienko, R. A. Shutilov, G. A. Zenkovets, V. Y. Gavrilov, N. T. Vasenin, A. A. Shubin, T. V. Larina, A. V. Zhuzhgov, Z. R. Ismagilov and V. N. Parmon, Russ. J. Inorg. Chem., 2012, 57, 1285–1290 CrossRef CAS
-
P. Atkins, T. Overton, J. Rourker and M. Weller, Shriver and Atkin's Inorganic Chemistry, Oxford University Press, Oxford, 5th edn, 2010 Search PubMed
-
W. de Wilde, R. A. Schoonheydt and J. B. Uytterhoeven, in ACS Symposium Series, Washington, 1977, pp. 132–143 Search PubMed
- E. F. Vansant and J. H. Lunsford, J. Phys. Chem., 1972, 76, 2860–2865 CrossRef CAS
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Science, 2017, 357, 898–903 CrossRef CAS PubMed
- A. Marberger, A. W. Petrov, P. Steiger, M. Elsener, O. Kröcher, M. Nachtegaal and D. Ferri, Nat. Catal., 2018, 1, 221–227 CrossRef CAS
- C. Negri, E. Borfecchia, M. Cutini, K. A. Lomachenko, T. V. W. Janssens, G. Berlier and S. Bordiga, ChemCatChem, 2019, 11, 3828–3838 CrossRef CAS
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS
- D. Nachtigallová, P. Nachtigall and J. Sauer, Phys. Chem. Chem. Phys., 2001, 3, 1552–1559 RSC
- J. Szanyi, J. H. Kwak, R. J. Chimentao and C. H. F. Peden, J. Phys. Chem. C, 2007, 111, 2661–2669 CrossRef CAS
- D. Wang, L. Zhang, K. Kamasamudram and W. S. Epling, ACS Catal., 2013, 3, 871–881 CrossRef CAS
- G. M. Underwood, T. M. Miller and V. H. Grassian, J. Phys. Chem. A, 1999, 103, 6184–6190 CrossRef CAS
- K. I. Hadjiivanov, Catal. Rev.: Sci. Eng., 2000, 42, 71–144 CrossRef CAS
- C. Negri, P. S. Hammershøi, T. V. W. Janssens, P. Beato, G. Berlier and S. Bordiga, Chem. – Eur. J., 2018, 24, 12044–12053 CrossRef CAS PubMed
- A. Penkova, K. Hadjiivanov, M. Mihaylov, M. Daturi, J. Saussey and J. C. Lavalley, Langmuir, 2004, 20, 5425–5431 CrossRef CAS PubMed
-
A. Davydov, Molecular Spectroscopy of Oxide Catalyst Surfaces, Wiley, Chichester, 2003 Search PubMed
- X. Zhang, H. Wang, L. Meng, X. Nie and Z. Qu, ACS Appl. Energy Mater., 2020, 3, 3465–3476 CrossRef CAS
- E. K. Dann, E. K. Gibson, R. H. Blackmore, C. R. A. Catlow, P. Collier, A. Chutia, T. E. Erden, C. Hardacre, A. Kroner, M. Nachtegaal, A. Raj, S. M. Rogers, S. F. R. Taylor, P. Thompson, G. F. Tierney, C. D. Zeinalipour-Yazdi, A. Goguet and P. P. Wells, Nat. Catal., 2019, 2, 157–163 CrossRef CAS
- M. Jabłońska, ChemCatChem, 2020, 12, 4490–4500 CrossRef
- R. E. Weston and T. F. Brodasky, J. Chem. Phys., 1957, 27, 683–689 CrossRef CAS
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed
- H. Zhu, J. H. Kwak, C. H. F. Peden and J. Szanyi, Catal. Today, 2013, 205, 16–23 CrossRef CAS
- H. Sjövall, E. Fridell, R. J. Blint and L. Olsson, Top. Catal., 2007, 42–43, 113–117 CrossRef
- A. L. Goodman, G. M. Underwood and V. H. Grassian, J. Phys. Chem. A, 1999, 103, 7217–7223 CrossRef CAS
- J. Baltrusaitis, J. Schuttlefield, J. H. Jensen and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 4970–4980 RSC
- T. Yu, T. Hao, D. Fan, J. Wang, M. Shen and W. Li, J. Phys. Chem. C, 2014, 118, 6565–6575 CrossRef CAS
- M. Moreno-González, R. Millán, P. Concepción, T. Blasco and M. Boronat, ACS Catal., 2019, 9, 2725–2738 CrossRef
- L. Olsson, H. Sjövall and R. J. Blint, Appl. Catal., B, 2009, 87, 200–210 CrossRef CAS
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. A. Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed
- Y. Zhang, Y. Peng, K. Li, S. Liu, J. Chen, J. Li, F. Gao and C. H. F. Peden, ACS Catal., 2019, 9, 6137–6145 CrossRef CAS
- C. Paolucci, A. A. Verma, S. A. Bates, V. F. Kispersky, J. T. Miller, R. Gounder, W. N. Delgass, F. H. Ribeiro and W. F. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11828–11833 CrossRef CAS PubMed
- M. P. Ruggeri, I. Nova and E. Tronconi, Top. Catal., 2013, 56, 109–113 CrossRef CAS
- L. Chen, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, ACS Catal., 2020, 10, 5646–5656 CrossRef CAS
- C. Tyrsted, E. Borfecchia, G. Berlier, K. A. Lomachenko, C. Lamberti, S. Bordiga, P. N. R. Vennestrøm, T. V. W. Janssens, H. Falsig, P. Beato and A. Puig-Molina, Catal. Sci. Technol., 2016, 6, 8314–8324 RSC
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Appl. Catal., B, 2014, 156–157, 428–437 CrossRef CAS
- H. Sjövall, R. J. Blint and L. Olsson, Appl. Catal., B, 2009, 92, 138–153 CrossRef
- M. Bendrich, A. Scheuer, R. E. Hayes and M. Votsmeier, Appl. Catal., B, 2018, 222, 76–87 CrossRef CAS
- H. Mahzoul, J. F. Brilhac and P. Gilot, Appl. Catal., B, 1999, 20, 47–55 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and additional characterization results. See DOI: 10.1039/d1cy02348a |
| This journal is © The Royal Society of Chemistry 2022 |