
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intercalation and reactions of CO under single layer graphene/Ni(111): the role of vacancies
Rocco
Davì
ac,
Giovanni
Carraro
 c,
Marija
Stojkovska
ac,
Marco
Smerieri
c,
Letizia
Savio
c,
Jean-Jacques
Gallet
de,
Fabrice
Bournel
de,
Mario
Rocca
bc and
Luca
Vattuone
*bc
c,
Marija
Stojkovska
ac,
Marco
Smerieri
c,
Letizia
Savio
c,
Jean-Jacques
Gallet
de,
Fabrice
Bournel
de,
Mario
Rocca
bc and
Luca
Vattuone
*bc
aDCCI, Università degli Studi di Genova, Via Dodecaneso 31, 16146 Genova, Italy
bDIFI, Università degli Studi di Genova, Via Dodecaneso 33, 16146 Genova, Italy. E-mail: vattuone@fisica.unige.it
cIMEM-CNR, UOS di Genova, Via Dodecaneso 33, 16146 Genova, Italy
dSorbonne Université, CNRS, Laboratoire de Chimie Physique Matière et Rayonnement, UMR 7614, Campus Pierre et Marie Curie, 4 Place Jussieu, 75252 Paris Cedex 05, France
eSynchrotron SOLEIL, L’Orme des Merisiers, Saint-Aubin, F-91192 Gif-sur-Yvette, France
First published on 14th November 2022
Abstract
We use synchrotron radiation-induced core level photoemission spectroscopy to investigate the influence of vacancies, produced by ion bombardment, on monolayer graphene/Ni(111) exposed to CO at pressures ranging from ultra-high vacuum (10−10 mbar) up to near ambient (5.6 mbar) conditions. CO intercalates at a rate which is comparable to the one observed in absence of defects and reacts via the Boudouard reaction producing additional carbon atoms and CO2. While the former attach to the graphene layer and extend it over areas previously covered by carbide, the CO2 molecules bind to the graphene vacancies forming epoxy-like bonds across them, thus mending the defects. The so-formed complexes give rise to a peak at 533.4 eV which persists upon evacuating the vacuum chamber at room temperature and which we assign to a covalently bonded species containing C and O.
Introduction
The influence of vacancies in gas intercalation below supported graphene (G) layers and their effect on the chemical reactivity is a major issue addressed in several theoretical papers.1–6 Still, so far, this process has only been little explored experimentally. Theoretical investigations addressed, in particular, the role of single and double vacancies.1,2 They predict an increased chemical reactivity for several molecules like CO, NO, NO2, NH3 and H2,3–5 thus paving the way for using graphene in catalysis and for sensors. The experimental studies addressing this topic demonstrated, e.g., the role of vacancies for the adsorption of oxygen on graphene/Ru7 and the feasibility of a graphene sensor exploiting adsorption at vacancies.8 Intercalation of molecules and atoms can also be facilitated by structural defects,9e.g. in the presence of N dopants.10 In a previous study performed under ultra-high vacuum (UHV) conditions,11 some of us showed that a relatively strong CO adsorption occurs on single G/Ni(111) layers previously defected by ion bombardment, which generates mono- and double-vacancies. The vibrational fingerprints of adsorbed CO persist up to Room Temperature (RT), while CO desorption from a pristine G layer takes place around 200 K.12 The reported CO vibrational modes at 237 meV and 253 meV are close to those of CO chemisorbed at bridge and atop sites of bare Ni(111), leading to the conclusion that CO passes through the vacancy and binds to the metal substrate. Mild annealing to 380 K leads to CO desorption and healing of the vacancies since no CO fingerprints were detected after a few annealing/adsorption cycles. The mending of the vacancies was proposed to occur via the Boudouard reaction:| CO + CO → CO2 + C |
In order to investigate this phenomenon under more realistic reaction conditions, we performed a Near Ambient Pressure X-Ray Photoemission Spectroscopy (NAP-XPS) synchrotron radiation study of CO intercalation at pressures ranging from UHV up to P ∼ 5.6 mbar through a single G/Ni(111) layer defected following the protocol described in ref. 11.
Experimental
Experiments were performed at the Tempo Beamline of the SOLEIL Synchrotron Radiation source (Saint-Aubin, France).13 The experimental setup13 of Sorbonne Université allows for real-time NAP-XPS measurements in the pressure range from UHV up to 20 mbar. The maximum pressure reached in the present experiment is determined by the attenuation of the photoemitted electron signal caused by CO in the gas phase.Briefly, the X-ray beam enters the main chamber through a differentially pumped stage at an incidence angle of 54° with respect to the surface normal and illuminates a spot of approx. 0.1 mm diameter on the sample. The photoemitted electrons are collected along the surface normal through a nozzle leading to the electron energy analyser (Specs Phoibos 150 NAP). The distance between the sample and the nozzle is obtained by optimising the signal and fixed by the focal point of the analyser in order to minimise the attenuation of the photoemitted current under NAP conditions. It is comparable with the nozzle cone aperture, that is 300 μm.
High purity CO (purity > 99.99%) is introduced into the analysis chamber through a dedicated gas line. The pressure is read by a capacitance gauge for P > 10−2 mbar.
The Ni crystal is mounted on a sample holder heated by a ceramic heater module. The Ni(111) surface is cleaned in UHV by sputtering cycles with 3 keV Ar+ ions followed by annealing to a temperature T ∼ 1000 K and by chemical cleansing, in P = 5 × 10−7 mbar O2 pressure at 1000 K. This procedure led to a surface with some residual carbide, which could not be removed entirely. G is grown following the protocol of ref. 14, i.e. exposing the sample at T = 830 K to P = 1.7 × 10−5 mbar of C2H4 for 300 s, corresponding to an exposure of ∼3800 L. A controlled number of defects was introduced in the graphene layer by sputtering with 150 eV Ar+ ions at a background pressure of 1 × 10−5 mbar. According to literature, this treatment produces mainly isolated single-vacancies (with ∼50% probability) and double-vacancies (with ∼30% probability).15 The probability of creating a single vacancy is then close to the one expected upon Ne+ ion bombardment with the same kinetic energy (conditions used in ref. 11), while the probability of creating a double vacancy is about 1.5 times higher for Ar+ than for Ne+.
XPS spectra were calibrated towards the Fermi Level and fitted following the procedure described in ref. 16.
The CO coverage was determined by carefully fitting the O 1s region and following the procedure described in the Discussion section.
Results
CO exposure at low pressure on the sputtered surface
In Fig. 1 we show the effect of sputtering on the graphene layer.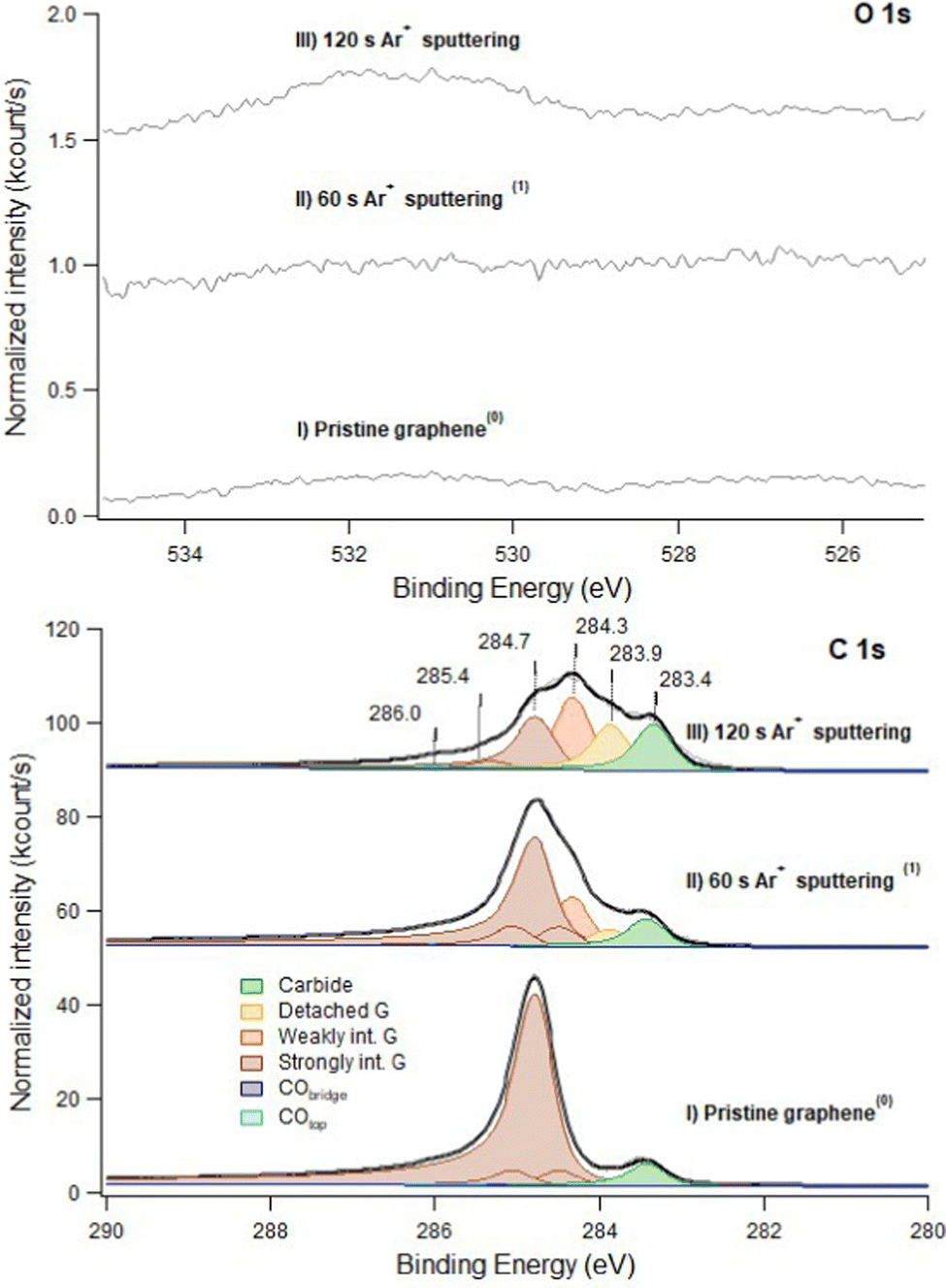 | ||
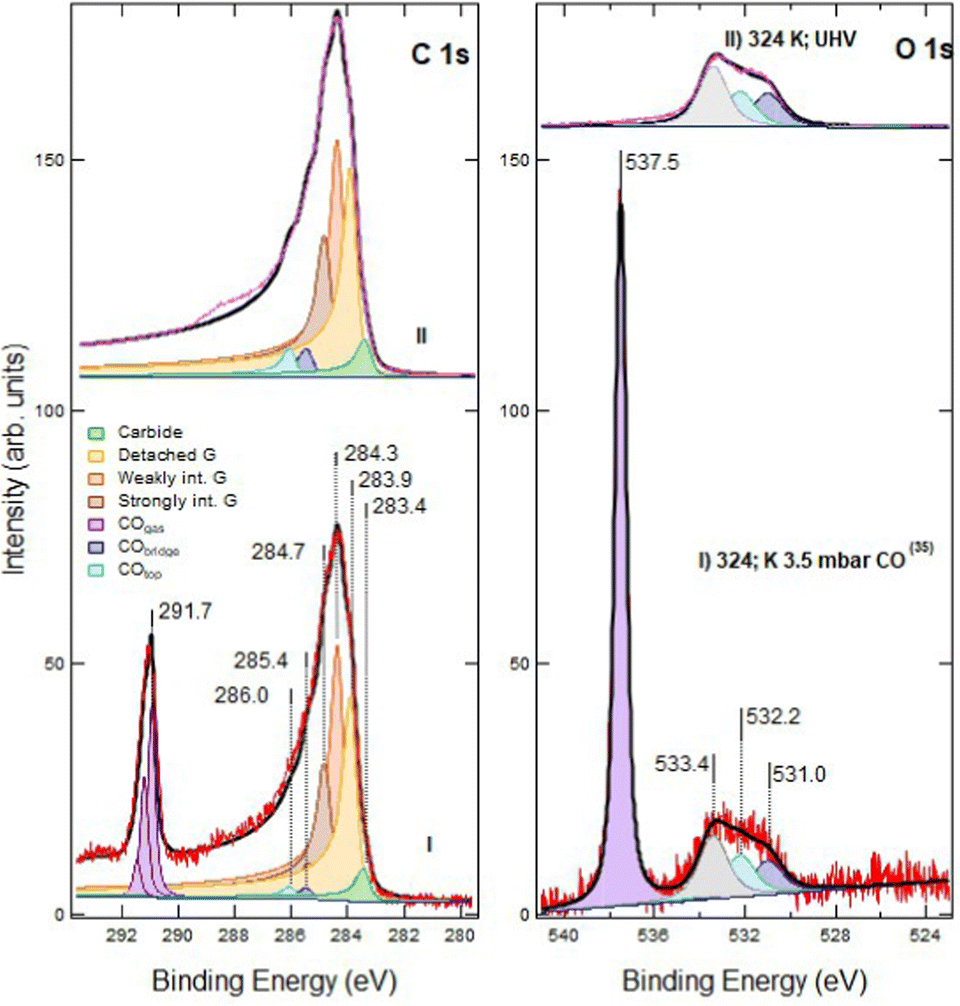
| Fig. 1 C 1s (bottom panel) and O 1s (top panel) spectra recorded at hν 650 eV. Trace I: pristine layer, recorded immediately after graphene growth. Trace II: defected layer obtained after 60 s sputtering with Ar+ ions (kinetic energy of 150 eV; Ar pressure of 1 × 10−5 mbar). Trace III: same as II after 120 s sputtering in identical conditions. Only very weak signals are present in the O 1s spectrum (note the difference in the intensity scale between the C 1s and O 1s regions). This indicates that no extended Ni areas are exposed to the residual gas since they would be promptly covered by CO. The apex numbers appearing after the description of the spectra refer to their position in the sequence reported in Fig. 6. | ||
For pristine G/Ni(111) (trace I) only a negligible contamination is evident in the O 1s spectrum. It consists mainly of CO and of an unidentified O-containing species with O 1s binding energy Eb = 526.7 eV. Inspection of the C 1s region indicates that pristine graphene interacts strongly with the Ni(111) substrate and is mainly in the bridge-top configuration (Eb = 284.7 eV – see Table 1 for the assignment of the different XPS components), with only a minor fraction of top-fcc. The residual carbide coverage (Eb = 283.4 eV) is estimated to be ∼0.13 ML by comparing the intensity of its peak with the one of graphene (see Discussion section for details).
| Adsorbed/absorbed species | E b(C 1s) (eV) | E b(O 1s) (eV) | |
|---|---|---|---|
| Nickel carbide | Ni2C | 283.4 | — |
| C atoms at defects & detached graphene | Gdefect | 283.9 | — |
| Weakly interacting graphene | Gweak | 284.3 | — |
| Strongly interacting graphene | Gstrong | — | |
| Bridge-top | 284.7 | ||
| Top-fcc | 284.5 + 285.0 | ||
| CO bridge | CObridge | 285.4 | 531.0 |
| CO top | COtop | 286.0 | 532.2 |
COC/O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (this work) O (this work) |
COC | 288.4 | 533.4 |
| COC19 | 287.1 | 533.1 | |
| O–CO20 |
289.0 | 533.4 | |
Upon 60 s Ar+ ion bombardment, the intensity of the strongly interacting component (Gstrong) decreases while the amount of carbide increases to ∼0.17 ML. Additional components appear at 284.3 eV and 283.9 eV. Following ref. 17 the former is assigned to G bubbles produced by intercalated Ar atoms and hence weakly interacting with the substrate (Gweak). The lower intensity component at 283.9 eV is ascribed to the formation of single vacancies and larger defects (Gdefect in Fig. 2). We mention that, unfortunately, in absence of defects, CO intercalation causes a G component at a very close binding energy (detached G in ref. 16 and 18) which cannot be separated.
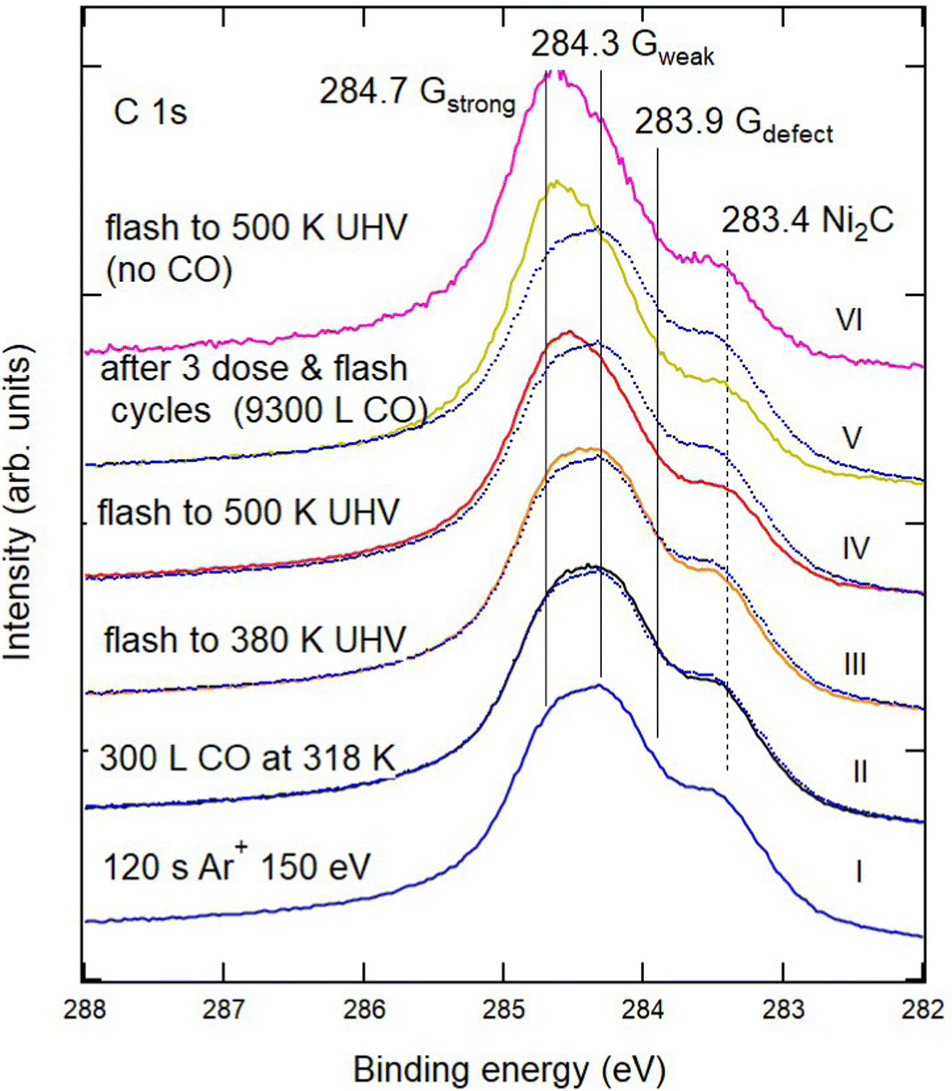 | ||
| Fig. 2 Sequence of XPS spectra of the C 1s region showing the effect of CO exposure and annealing cycles on defected G/Ni(111). From bottom to top: (I) G/Ni(111) defected by 120 s sputtering with 150 eV Ar+ ions (blue). (II) Exposure to CO (400 s, PCO = 1 × 10−6 mbar). (III and IV) Annealing to 380 K and 500 K, respectively. (V) After the third exposure annealing cycle (2nd cycle: CO dose of 400 s at PCO = 1 × 10−5 mbar and annealing at 380 K; 3rd cycle: CO dose of 400 s at PCO = 2 × 10−5 mbar and annealing to 500 K), reaching a total CO exposure of 9300 L. Spectrum I (blue dotted trace) is superimposed on the other spectra to highlight the modifications induced by the treatments. Little or no change in the spectral shape occurs when the sample is annealed to 500 K without CO, as witnessed by the comparison of spectra V and VI and in agreement with the information obtained in ref. 11 by vibrational spectroscopy. | ||
When doubling the Ar+ dose, the Gstrong peak decreases further while the components due to Gweak and Gdefect become dominant. Carbide also increases significantly, reaching 0.25 ML. Traces of CO are now more evident in the O 1s spectrum.
Unfortunately, during this beamtime we could not measure the sputtering current. However, the coverage of intercalated Ar can be estimated using overview spectra recorded with hν = 970 eV by the ratio between the Ar 2p intensity (intercalated Ar) and the total C 1s signal (data not shown). After 60 s and 120 s of sputtering, an Ar coverage of the order of ∼0.03 ML Ni (111) and of ∼0.12 ML Ni(111), respectively, can be estimated. The non-linearity in the Ar coverage indicates that its intercalation is favoured in the presence of vacancies. Notably, the overall shape of our spectrum for an estimated Ar coverage of 0.12 ML is similar to the one reported by Larciprete et al.17 for 0.27 ML of Ar. The difference can be accounted for by the larger kinetic energy of the Ar+ ions (150 eV instead of 100 eV) employed in the present study.
After 60 s and 120 s sputtering at normal incidence, the total carbon intensity has decreased by ∼6% and ∼12% respectively.
Vice versa, the total fraction of carbon atoms covering Ar bubbles and/or involved in vacancies, which was initially negligible, is now 31% and 51%, respectively.
Fig. 2 shows the evolution of the C 1s region for the most defected sample (120 s Ar+ bombardment – trace I) after exposure to CO at RT and under UHV conditions (trace II) and subsequent annealing in vacuum to increasing T (traces III and IV). The annealing temperature is lower than the one at which Larciprete et al.17 start observing Ar desorption, so that the changes in the spectral shape should not be related to the emptying of the Ar bubbles at the surface.
Upon CO exposure and annealing to 500 K, the amount of Gdefect decreases significantly, as evidenced by the reduction of the low Eb shoulder in the spectra and by the decrease of the nickel carbide component. These changes are accompanied by a recovery of the intensity of Gstrong, witnessed by the upshift of the binding energy value corresponding to the maximum of the C 1s intensity. Further CO uptake and annealing cycles (trace V) have a smaller effect on the global shape of the XPS spectrum. These results confirm our previous claim that the vacancies in the graphene film may be healed by CO exposure and annealing.11 Indeed, heating the sample obtained after three doses and annealing cycles (trace V) to 500 K without exposing it to CO (trace VI) produces no appreciable change in the overall shape of the C 1s region.
The significant changes in the C 1s spectra of Fig. 2 are not accompanied by a corresponding evolution of the O 1s region (not shown), which shows only a very weak signal as in Fig. 1. Most likely the latter is caused by the rapid saturation of the newly generated bare Ni sites by CO adsorption from the residual gas. Therefore, at low pressure, the presence of vacancies does not lead to an intercalated CO coverage large enough to be detected. Only mending of the vacancies occurs via the Boudouard reaction, which leads to the production of carbon atoms and CO2. Such CO2 is produced close to the repaired vacancy and thus promptly desorbs thus escaping detection by XPS.
On Ni(110) and in UHV conditions, both chemisorbed and physisorbed CO2 were identified by XPS upon exposure at 90 K22 but, to the best of our knowledge, no CO2 formation has ever been reported. As we demonstrated in ref. 21, this process can be observed at RT only under NAP conditions thanks to the accumulation of CO2 under the graphene cover.
In that study, the formation of physisorbed CO2 is witnessed by the appearance of an O 1s peak at Eb = 533.4 eV and of a C 1s signal at 291.3 eV. The physisorbed nature of such species is confirmed by the fact that it promptly disappears upon evacuation of the experimental chamber at RT.16 Indeed, given the low adsorption energy, CO2 must be trapped under the graphene cover since it would otherwise rapidly desorb.
In ref. 16, we also inferred the existence at RT of a weakly bonded CO species adsorbed above the graphene layer, confirming our previous finding obtained at low temperatures under UHV.12
CO exposure of the sputtered surface under NAP conditions
Exposure of defected G/Ni(111) to CO under NAP conditions was performed on the sample sputtered for 60 s only, in order to have a significant density of still isolated vacancies. In Fig. 3 we report the effect of CO exposure at different pressure, ranging from PCO = 1 × 10−5 mbar to several mbar. CO is admitted into the chamber with PCO = 1 × 10−5 mbar at RT and then the temperature is increased (see also Fig. 6 panels f and g).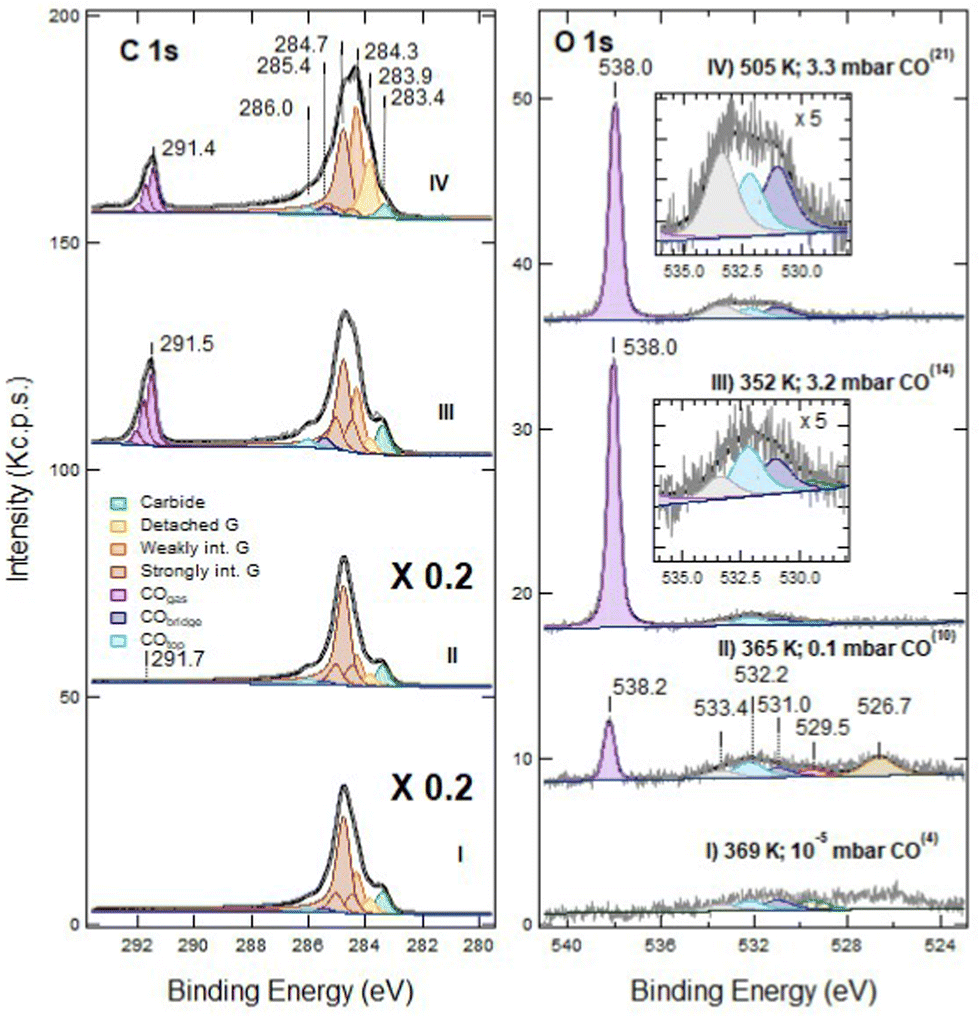 | ||
| Fig. 3 NAP-XPS spectra of the C 1s (left) and O 1s (right) regions for defected G/Ni(111) (60 s at RT; ion energy of 150 eV): (I) at T = 369 K and PCO = 10−5 mbar; (II) at 365 K and PCO = 0.1 mbar; (III) at 352 K and PCO = 3.2 mbar; (IV) at 505 K and PCO = 3.3 mbar. Spectra are shown without normalisation. The apex numbers reported in brackets indicate the position of the spectrum in the sequence of Fig. 6. Insets contain an enlargement of the O 1s region of the corresponding spectra. Note that C 1s spectra (I) and (II) are scaled by a factor 5; as a consequence, the C 1s signal due to gas phase CO at PCO = 0.1 mbar CO is only apparently small compared to the O 1s counterpart. | ||
Exposure to CO at 1 × 10−5 mbar between RT and 369 K (trace I) does not modify the XPS spectrum significantly (compare trace I of Fig. 3 with trace II of Fig. 1). In the O 1s region, weak signals due to dissociated oxygen atoms and to the unassigned oxygen-containing species are present at 529.5 eV and 526.7 eV, respectively, together with small contributions at 532.2 eV and possibly at 533.4 eV.
When increasing the CO pressure to PCO = 0.1 mbar (trace II) and above (traces III and IV), additional peaks appear at Eb = 291.7 eV (C 1s) and at 538.2 eV (O 1s). They are due to gas-phase CO and, being referred to the vacuum level and they shift with the work function change of the sample caused by surface modification. The evident asymmetric shape of this C 1s feature in spectra III and IV is due to the presence of vibrational overtones. Small signals corresponding to adsorbed CO species are now detected in the O 1s region (features at 532.2 eV and 531.0 eV, corresponding to CO at on-top and bridge Ni sites, respectively) together with those previously detected at 529.5 eV, 526.7 eV and 533.4 eV.
The XPS spectra change even more after increasing the CO pressure above 3 mbar (traces III and IV). In the O 1s region, the impurity signal at 526.7 eV and the dissociated oxygen feature at 529.5 eV disappear, possibly due to the removal of that oxygen by reaction with CO, while the component at 533.4 eV increases and eventually becomes dominant when heating the sample to 505 K (trace IV). This peak cannot be attributed to physisorbed CO2 as in the twin experiment performed on pristine graphene,16 since: (i) it is not accompanied by its C 1s companion at Eb = 291 eV, which would be apparent on the right-hand side of the gas-phase CO signal; (ii) it does not disappear upon evacuation of the chamber, while physisorbed CO2 is expected to desorb in these conditions.
In the C 1s region, we observe a strong evolution of the graphene layer. More than half of the strongly interacting graphene converts into components at 284.3 and 283.9 eV. The former corresponds to weakly interacting graphene. The latter could be due either to graphene defects17 or to detached graphene,16,18,19 species which cannot be resolved by a fitting procedure. However, since an increase of the defect concentration by NAP CO exposure is improbable, we ascribe the raise of the 283.9 eV component to detached graphene originated from further delamination induced by CO intercalation. Although a comparison among spectra is impeded by the change in pressure and to the correlated decrease in the absolute intensities, there is evidence for an increase in the adsorbed CO coverage (see also the discussion session below). Upon CO exposure at 505 K, the fractions of detached and weakly interacting graphene grow further at the expense of the strongly interacting one (trace IV).
No qualitative changes are observed in the C 1s and O 1s regions upon increasing PCO to 5.6 mbar at 500 K (Fig. 4, trace I). However, after cooling the sample (trace II), the intensity of CO at atop sites (Eb = 286.0 eV) increases. This effect, already observed on pristine G/Ni(111), is ascribed to the transient adsorption of weakly bonded CO, which has the same binding energy, above strongly interacting graphene.16 Its presence causes a change in the work function, which explains the shift of the Eb values of gas-phase CO.16
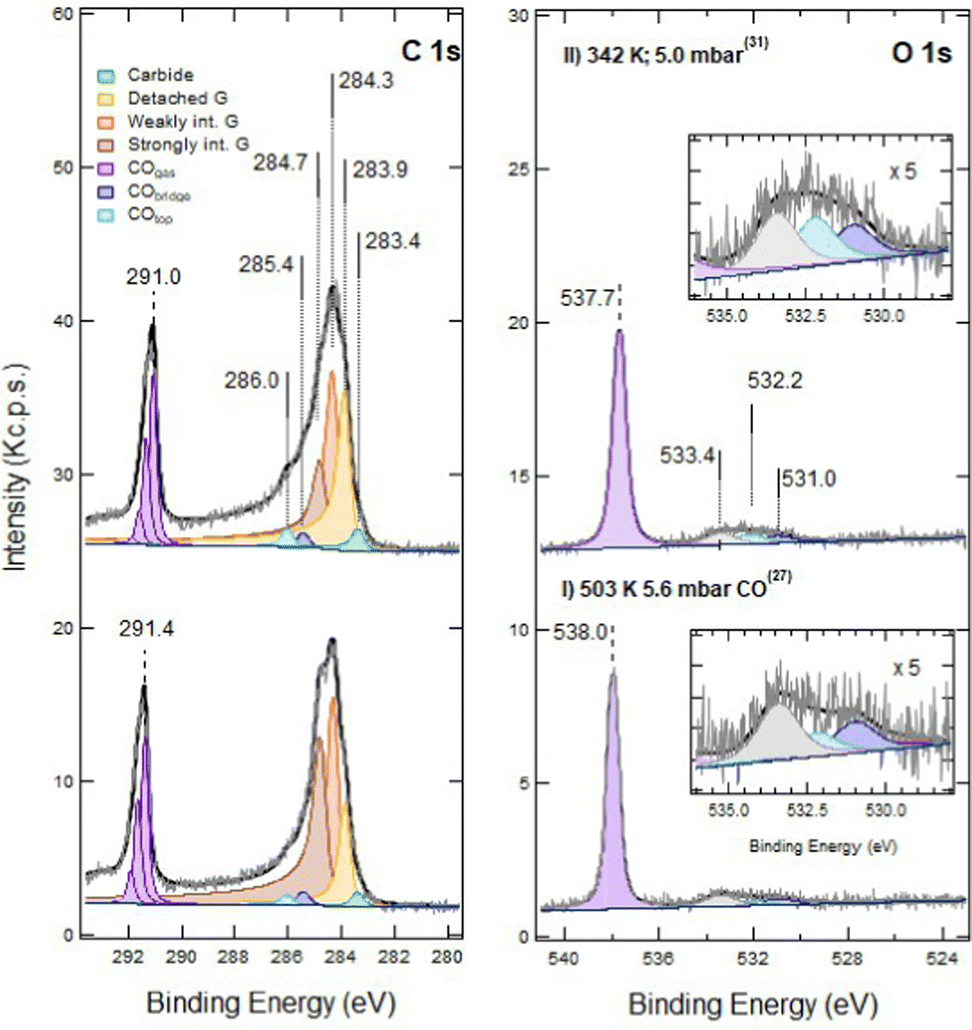 | ||
| Fig. 4 NAP-XPS spectra of the C 1s (left) and O 1s (right) regions for sputtered G/Ni(111) (60 s at RT; ion energy of 150 eV) during CO exposure: (I) at 503 K and PCO = 5.6 mbar; (II) after cooling down to 342 K and PCO = 5.0 mbar. The spectra are plotted in absolute counts since the data were recorded in similar NAP conditions and at different sample temperatures so that no evident normalisation protocol can be applied. | ||
Moreover, we notice an increased intensity in the region between 286.0 eV and 289.0 eV. This tail is also present after CO removal (as discussed later) and indicates an increased peak asymmetry.
The spectra collected before and after evacuation are compared in Fig. 5. The relative fractions of strongly interacting, weakly interacting and detached graphene are unaffected by evacuation, indicating that CO deintercalation can be neglected. The decrease of the on-top CO component can thus be safely ascribed to the desorption of metastable CO previously adsorbed above Gstrong.
 | ||
| Fig. 5 XPS spectra of the C 1s (left) and O 1s (right) regions recorded before (I) and after (II) evacuation of the experimental chamber. Since in UHV conditions, the photoemitted intensity is no longer attenuated by scattering off gas-phase CO, the signal increases significantly. In order to compare the spectra, the O 1s region is normalised to the intensity of the CO bridge component at 531.0 eV. Being intercalated and strongly bonded to the Ni substrate, such species are unlikely to be affected by the change in CO pressure. The C 1s region is normalised by imposing that the area of the graphene components is independent of CO pressure. Note that the C 1s intensity for CO is inferred from the O 1s intensity using the cross sections for photoemission under UHV and the ratio of gas phase lines under NAP (see the Discussion session): for this reason, a direct comparison of the intensities of the different species would thus be misleading. The comparison of the spectrum recorded under NAP with the spectrum recorded after evacuation indicates that the data are compatible with the presence of the 288 eV feature under NAP conditions. | ||
Since only overview spectra were recorded in the Ar 2p region, it is not possible to determine safely whether some Ar has desorbed during the NAP experiment.
Discussion
To gain further insight into the observed phenomena, a more quantitative analysis of the whole sequence of spectra is reported in Fig. 1, 3 and 4 is necessary. The outcome is summarised in Fig. 6vs. spectrum position in the recorded sequence. Panel (a) shows the work function change (Δϕ) induced by CO exposure in NAP conditions, and panel (b) the asymmetry (α) of Gstrong peak. Panels (c) and (d) report the G and Ni2C coverage and the fractional area of the different G species, respectively. Panel (e) focuses on the coverage of the adsorbed species. Finally, panels (f) and (g) provide information on the temperature and pressure conditions while recording the spectra.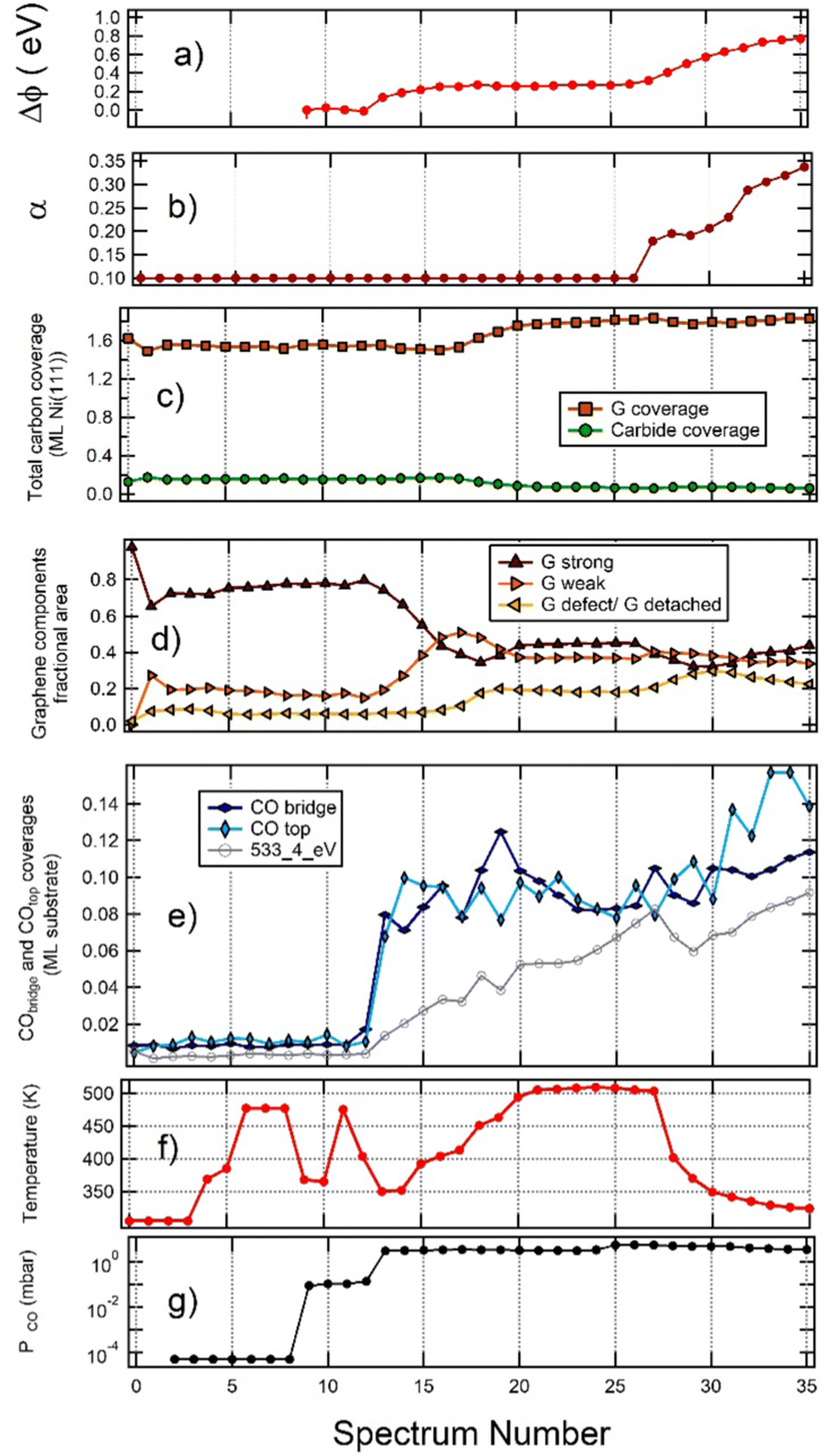 | ||
| Fig. 6 Analysis of the whole sequence of spectra reported in Fig. 1, 3 and 4, plotted vs. spectrum number. (a) Work function change Δϕ (equal to the opposite of the apparent Eb shift of gas-phase CO); (b) asymmetry (α) of the strongly interacting graphene peak; (c) carbon coverage of graphene (brown) and carbide (green); (d) fractional areas of the different graphene components. Colour code: strongly interacting G is brown; weakly interacting G is mustard; detached G is light blue; (e) coverage of CO top (sum of intercalated and weakly adsorbed on graphene – light blue), CO bridge (blue) and of the 533.4 eV component (grey); (f) sample temperature; (g) CO pressure in the chamber. The initial point (point 0) corresponds to pristine graphene and the second point (point 1) to the sample immediately after 60 s sputtering. | ||
To perform this analysis, we applied the same procedure used in ref. 16 and 21. Since the adsorbed coverage cannot be determined directly from the XPS intensities due to the temperature and pressure dependent attenuation of the photoemission signals, we evaluate them by assuming that the carbon layer is closed. Considering that a full monolayer (ML) of graphene and of Ni2C corresponds to 2 ML and 0.5 ML of C (in ML of Ni(111)), respectively, the fraction of surface area covered by graphene (fG) and by carbide (fNi2C) can be obtained from the intensities IG and INi2C of the corresponding C 1s lines from the formulas:
| fG = (IG)/(IG + 4INi2C) and fNi2C = 1 − fG. |
The O 1s and C 1s spectra were recorded at the same photon energy (hν = 650 eV) in order to monitor the evolution of the spectra in real-time without changing the gap of the photon monochromator. This procedure, however, implies a different attenuation of the photoemitted intensity due to scattering off gas phase CO because of the different kinetic energies of the photoelectrons.
To determine the O 1s and C 1s intensities due to the oxygen-containing species consistently, we firstly find the intensity of the different O 1s components and then we fit the C 1s region by assigning the corresponding C 1s intensity in two different ways, depending on the pressure range:
(1) Under UHV conditions, the intensity ratio between O 1s and C 1s components is determined by the corresponding photoemission cross-sections (б(C 1s) = 1.5 Mb; б(O 1s) = 3.5 Mb at hν = 650 eV) and by the stoichiometry of the species (the transmission function of the analyser is constant over the scanned energy interval).
(2) Under NAP conditions (PCO > 1 mbar), the photoemission intensity is corrected for the electron kinetic energy-dependent attenuation of the signal due to scattering off gas-phase molecules. IC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1sads, must then satisfy the relationship:
1sads, must then satisfy the relationship:
| IO1sads/IO1sCOgas = εIC1sads/IC1sCOgas |
1sads is the intensity of the O 1s line of the adsorbed moiety and IO1sgas, and IC1sgas are the intensities of the corresponding gas-phase CO lines. For spectra acquired in the intermediate pressure range 10−2 mbar < PCO < 10−1 mbar, we compared the expected intensities following both methods. The matching between the two procedures is acceptable, and thus our analysis can also be trusted in this pressure range.
By comparing the C 1s intensity for adsorbed species to the C 1s intensity of graphene, we obtain the coverage of oxygen-containing species.16
From inspection of the graphs in Fig. 6, we gain the following information:
(a) The relative amount of Gstrong, Gweak and defect/detached graphene remains constant when increasing PCO from UHV to 0.1 mbar, thus indicating that exposure to CO at pressure as high as 0.1 mbar does not lead to significant CO intercalation. Until intercalation starts, the peak with Eb(C 1s) = 283.9 eV corresponds thus to C at defected graphene sites.
(b) For PCO ∼ 3 mbar, the fraction of the surface covered by Gstrong decreases with increasing CO coverage. Such behaviour indicates that CO intercalation and partial delamination of graphene occurs at this pressure already around 400 K. This change is indeed correlated with an increase of the work function, which is higher for Gweak than for Gstrong.24
(c) Annealing from 400 K to 500 K determines an increase in the fractional coverage of detached G and an increase in the total amount of G, which cannot be explained by the corresponding, smaller reduction of the Ni2C signal. Hence, an additional supply of carbon atoms is required. In this regime the 283.9 eV peak is due to detached graphene.
(d) Cooling the sample under PCO = 5 mbar (spectra 30 to 35) causes an increase in the coverage of on-top CO. This result is ascribed to the adsorption of a weakly bound CO species above strongly interacting G domains and not to further intercalation since this change is not accompanied by an increase in the fractional area of detached and weakly interacting G. Indeed the fractional area of Gstrong increases slightly since CO adsorbs only on such patches and the process is energetically favourable. For these spectra, we also observe an increase of the work function. Since the latter is lower for Gstrong than for detached G, this observation is consistent with the build-up of a layer of weakly adsorbed CO.
(e) Contrary to previous experiments performed on the same system at lower pressure,18 only incomplete delamination of G occurs when the sample is exposed to CO under NAP conditions in the mbar pressure range, a phenomenon we also observed in the absence of artificially generated defects. We can rationalise this surprising finding considering that the adsorption of CO on G determines a decrease in the energy of the system.16 The energy gain due to further CO intercalation (which would give rise to full delamination) competes, therefore, with the energy gain due to CO chemisorption, which occurs at strongly interacting graphene patches and becomes important at high CO pressure. Interestingly, comparing Fig. 6 with our previous experiment on pristine G,16 it is evident that the defects created by sputtering do not allow to reach a higher CO coverage than observed for pristine graphene. This result suggests that the holes created by ion bombardment do not open important new paths for CO intercalation.
We note that the first part of the experiment yields similar results to those observed for CO exposure on pristine G;16 on the contrary, the sputtered layer behaves differently upon CO removal (see Fig. 5). Indeed, in the presence of vacancies, after removal of the CO partial pressure, a shoulder between 286.0 eV and 289 eV becomes evident (see Fig. 5). An asymmetry also remains in the C 1s peak shape. Moreover, the O 1s intensity at 533.4 eV persists after evacuation, which is at odds with its assignment to physisorbed CO2.
To rationalise these findings, we propose the following possible explanation:
(a) Ion bombardment produces mostly single and some double vacancies. The Ar atoms form nanobubbles which are expected to be stable up to 500 K17 and probably act as spectators.
(b) As reported for similar systems,25 we believe that the low coordination atoms at the vacancies bind to the Ni substrate, thus explaining why intercalated CO is comparable with the one we observed in absence of defects.
(c) While exposure to CO in UHV conditions causes healing of the vacancies, explained by the occurrence of the Boudouard reaction,11 the evolution is more complex under NAP conditions.
We speculate that two possible processes can occur:
| CO + G* → C + G*–O |
| CO + CO + G* → C + G*–CO2 |
O) bridge across the vacancy. Even if our experimental data does not allow to rule out the former mechanism, the second path seems more likely since it explains the absence of physisorbed CO2, the presence of which is evident in absence of defects.
Whatever the actual mechanism, the produced C atoms contribute to the observed increase in the graphene coverage.
This interpretation is coherent with the increase of the graphene coverage since additional carbon is made available and with the growth of a tail of the graphene peaks, particularly evident after evacuation. Indeed, such a feature, together with the persisting O 1s intensity at Eb = 533.4 eV, witnesses the presence of C–O–C19 or O–CO20 groups, which change the doping of the graphene layer and thus the intensity of the inelastic tail of the graphene peaks. Moreover, the damaged G layer is unable to convert back into strongly interacting graphene due to the presence of such covalently bonded groups. We recall that “scars” were observed by STM inspection after CO dosing of the defected graphene layer in UHV.11
An alternative explanation could relate the feature at 533.4 eV and the graphene tail to the interaction of the system with a highly reactive species produced by the photolysis of the gas molecules in the NAP regime. Indeed, the feature at 533.4 eV has been assigned in literature to OH bonded to aromatic C (phenolic oxygen)26,27 and the presence of water traces is unavoidable under NAP conditions.
Such an explanation can, however, hardly explain the present data since, under similar NAP conditions but in the absence of vacancies, no species with Eb at 533.4 eV is observed after the evacuation of the vacuum chamber16 while effects due to photolysis of water admolecules should be independent of the presence of defects.
Moreover, C–OH should contribute in the C 1s region around 285.5 eV, contrary to our experimental evidence, the extra-feature present in the C 1s spectrum after evacuation being centered around 288 eV (upper spectrum in Fig. 5).
We conclude, therefore, that the formation of such strongly bonded species is related to the defects created by ion bombardment.
Conclusions
We investigated the effect of vacancies on the interaction of graphene with CO under NAP conditions. They do not significantly affect the coverage of intercalated CO under the graphene cover, which came out to be similar to the one observed for pristine graphene when exposed to a comparable CO pressure. This result is explained by the fact that the C atoms at the vacancies saturate their dangling bond towards the Ni substrate.Exposure to CO under UHV repairs the vacancies, partly restoring strongly interacting graphene. We suggest that the vacancy mending mechanism involves the formation of CO2via the Boudouard reaction. Under UHV the so-formed CO2 promptly desorbs, and thus it is not observed while the residual C atoms repair the vacancy.
Under NAP conditions, on the contrary, most likely CO2 molecules reach the vacancies where they bind to their edges and form C–O–C/O–CO bridges. The 533.4 eV peak is most likely due to these structures.
Author contributions
R. Davì performed the measurements and the analysis, prepared the figures and wrote the manuscript. G. Carraro, M. Stojkovska, M. Smerieri performed the measurements. M. Bournel and J. Gallet provided assistance before and during the beamtime. L. Savio and M. Rocca contributed to the discussion and to the analysis of the data and wrote the manuscript. L. Vattuone performed the measurements, wrote the manuscript, and coordinated the collaboration. All authors revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from MIUR through projects PRIN2017 no. 2017NYPHN8 and 2017KFMJ8E_003 and from Fondazione Compagnia di S. Paolo through project MC-nano.Notes and references
- I. A. Pašti, A. Jovanović, A. S. Dobrota, S. v Mentus, B. Johansson and N. v Skorodumova, Atomic adsorption on graphene with a single vacancy: systematic DFT study through the periodic table of elements, Phys. Chem. Chem. Phys., 2018, 20, 858–865 RSC.
- B. Sanyal, O. Eriksson, U. Jansson and H. Grennberg, Molecular adsorption in graphene with divacancy defects, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 113409 CrossRef.
- Y.-H. Zhang, Y. Chen, K.-G. Zhou, C. Liu, J. Zeng, H. Zhang and Y. Peng, Improving gas sensing properties of graphene by introducing dopants and defects: a first-principles study, Nanotechnology, 2009, 20, 185504 CrossRef PubMed.
- G. Kim, S.-H. Jhi, S. Lim and N. Park, Effect of vacancy defects in graphene on metal anchoring and hydrogen adsorption, Appl. Phys. Lett., 2009, 94, 173102 CrossRef.
- N. Tit, K. Said, N. M. Mahmoud, S. Kouser and Z. H. Yamani, Ab-initio investigation of adsorption of CO and CO2 molecules on graphene: Role of intrinsic defects on gas sensing, Appl. Surf. Sci., 2017, 394, 219–230 CrossRef CAS.
- G. Carraro, L. Savio and L. Vattuone, Coatings, 2022, 12, 397 CrossRef CAS.
- T. Li and J. A. Yarmoff, Defect-induced oxygen adsorption on graphene films, Surf. Sci., 2018, 675, 70–77 CrossRef CAS.
- Y. Liu, H. Liu, Y. Chu, Y. Cui, T. Hayasaka, V. Dasaka, L. Nguyen and L. Lin, Defect-Induced Gas Adsorption on Graphene Transistors, Adv. Mater. Interfaces, 2018, 5, 1701640 CrossRef.
- A. Ibrahim and Y. S. Lin, Gas permeation and separation properties of large-sheet stacked graphene oxide membranes, J. Membr. Sci., 2018, 550, 238–245 CrossRef CAS.
- D. Perilli, S. Fiori, M. Panighel, H. Liu, C. Cepek, M. Peressi, G. Comelli, C. Africh and C. di Valentin, Mechanism of CO Intercalation through the Graphene/Ni(111) Interface and Effect of Doping, J. Phys. Chem. Lett., 2020, 11, 8887–8892 CrossRef CAS.
- E. Celasco, G. Carraro, A. Lusuan, M. Smerieri, J. Pal, M. Rocca, L. Savio and L. Vattuone, CO chemisorption at vacancies of supported graphene films: a candidate for a sensor?, Phys. Chem. Chem. Phys., 2016, 18, 18692–18696 RSC.
- M. Smerieri, E. Celasco, G. Carraro, A. Lusuan, J. Pal, G. Bracco, M. Rocca, L. Savio and L. Vattuone, Enhanced Chemical Reactivity of Pristine Graphene Interacting Strongly with a Substrate: Chemisorbed Carbon Monoxide on Graphene/Nickel(1 1 1), ChemCatChem, 2015, 7, 2328–2331 CrossRef.
- H. Liu, A. Zakhtser, A. Naitabdi, F. Rochet, F. Bournel, C. Salzemann, C. Petit, J. J. Gallet and W. Jie, Operando Near-Ambient Pressure X-ray Photoelectron Spectroscopy Study of the CO Oxidation Reaction on the Oxide/Metal Model Catalyst ZnO/Pt(111), ACS Catal., 2019, 9, 10212–10225 CrossRef.
- E. Celasco, G. Carraro, M. Smerieri, L. Savio, M. Rocca and L. Vattuone, Influence of growing conditions on the reactivity of Ni supported graphene towards CO, J. Chem. Phys., 2017, 146, 104704 CrossRef PubMed.
- O. Lehtinen, J. Kotakoski, A. v Krasheninnikov, A. Tolvanen, K. Nordlund and J. Keinonen, Effects of ion bombardment on a two-dimensional target: Atomistic simulations of graphene irradiation, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 153401 CrossRef.
- R. Davì, G. Carraro, M. Stojkovska, M. Smerieri, L. Savio, M. Lewandowski, J.-J. Gallet, F. Bournel, M. Rocca and L. Vattuone, Boudouard reaction under graphene cover on Ni(1 1 1), Appl. Surf. Sci., 2022, 599, 154065 CrossRef.
- R. Larciprete, S. Colonna, F. Ronci, R. Flammini, P. Lacovig, N. Apostol, A. Politano, P. Feulner, D. Menzel and S. Lizzit, Self-Assembly of Graphene Nanoblisters Sealed to a Bare Metal Surface, Nano Lett., 2016, 16, 1808–1817 CrossRef CAS PubMed.
- M. Wei, Q. Fu, Y. Yang, W. Wei, E. Crumlin, H. Bluhm and X. Bao, Modulation of Surface Chemistry of CO on Ni(111) by Surface Graphene and Carbidic Carbon, J. Phys. Chem. C, 2015, 119, 13590–13597 CrossRef CAS.
- L. Stobinski, B. Lesiak, A. Malolepszy, M. Mazurkiewicz, B. Mierzwa, J. Zemek, P. Jiricek and I. Bieloshapka, Graphene oxide and reduced graphene oxide studied by the XRD, TEM and electron spectroscopy methods, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 145–154 CrossRef CAS.
- R. Larciprete, S. Gardonio, L. Petaccia and S. Lizzit, Atomic oxygen functionalization of double walled C nanotubes, Carbon, 2009, 47, 2579–2589 CrossRef CAS.
- R. Davì, G. Carraro, M. Stojkovska, M. Smerieri, L. Savio, M. Lewandowski, J.-J. Gallet, F. Bournel, M. Rocca and L. Vattuone, Graphene growth on Ni (1 1 1) by CO exposure at near ambient pressure, Chem. Phys. Lett., 2021, 774, 138596 CrossRef.
- M. Roiaz, E. Monachino, C. Dri, M. Greiner, A. Knop-Gericke, R. Schlögl, G. Comelli and E. Vesselli, Reverse Water-Gas Shift or Sabatier Methanation on Ni(110)? Stable Surface Species at Near-Ambient Pressure, J. Am. Chem. Soc., 2016, 138, 4146–4154 CrossRef CAS PubMed.
- J. Kraus, R. Reichelt, S. Günther, L. Gregoratti, M. Amati, M. Kiskinova, A. Yulaev, I. Vlassiouk and A. Kolmakov, Photoelectron spectroscopy of wet and gaseous samples through graphene membranes, Nanoscale, 2014, 6, 14394–14403 RSC.
- G. Giovannetti, P. Khomyakov, G. Brocks, V. Karpan, J. van den Brink and P. Kelly, Doping Graphene with Metal Contacts, Phys. Rev. Lett., 2008, 101, 026803 CrossRef.
- L. Ferrighi, D. Perilli, D. Selli and C. di Valentin, Water at the Interface between Defective Graphene and Cu or Pt (111) Surfaces, ACS Appl. Mater. Interfaces, 2017, 9, 29932–29941 CrossRef.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, Probing the Thermal Deoxygenation of Graphene Oxide Using High-Resolution In Situ X-ray-Based Spectroscopies, J. Phys. Chem. C, 2011, 115, 17009–17019 CrossRef.
- S. Yamamoto, K. Takeuchi, Y. Hamamoto, R. Y. Liu, Y. Shiozawa, T. Koitaya, T. Someya, K. Tashima, H. Fukidome, K. Mukai, S. Yoshimoto, M. Suemitsu, Y. Morikawa, J. Yoshinobu and I. Matsuda, Enhancement of CO2 adsorption on oxygen-functionalized epitaxial graphene surface under near-ambient conditions, Phys. Chem. Chem. Phys., 2018, 20, 19532–19538 RSC.
| This journal is © the Owner Societies 2022 |