
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-crystallization of organic chromophore roseolumiflavin and effect on its optical characteristics†
Takin
Haj Hassani Sohi
 a,
Felix
Maass
a,
Constantin
Czekelius
b,
Markus
Suta
c and
Vera
Vasylyeva
*a
a,
Felix
Maass
a,
Constantin
Czekelius
b,
Markus
Suta
c and
Vera
Vasylyeva
*a
aLaboratory for Molecular Crystal Engineering, Department of Inorganic Chemistry and Structural Chemistry I, Heinrich-Heine-University Duesseldorf, Universitaetstr. 1, 40225 Duesseldorf, Germany. E-mail: vera.vasylyeva-shor@hhu.de
bLaboratory for Asymmetric Synthesis and Catalysis, Department of Organic Chemistry and Macromolecular Chemistry, Heinrich-Heine-University Duesseldorf, Universitaetstr. 1, 40225 Duesseldorf, Germany
cLaboratory for Inorganic Photoactive Materials, Department of Inorganic Chemistry and Structural Chemistry II, Heinrich-Heine-University Duesseldorf, Universitaetstr. 1, 40225 Duesseldorf, Germany
First published on 1st August 2022
Abstract
A structural study on the solid, microcrystalline chromophore roseolumiflavin was carried out. Based on rational design considerations three binary co-crystals were prepared with a primary focus set on utilizing the imide functional group. Therefore, robust supramolecular synthons, including a carboxylic acid–amide dimer, halogen bonding, as well as a hydrogen-bonded three-point binding motif, were successfully incorporated into the multicomponent systems. All the co-crystals showed a blue-shifted fluorescence in the red range compared to the near-infrared-emitting solid roseolumiflavin. Solvatochromic studies finally elucidated that the degree of close packing between the flavin molecules within the crystal structures was decisive for the emission colour. These results render new possibilities to investigate flavins in the aspect of crystal engineering to tune optical properties of organic chromophores in the solid state.
Introduction
Chirality, catalytic activity, drug-efficacy, charge-transfer processes, thermal behaviour, and mechanical or optical properties1,2 are some of the possible fields where the application of the crystal engineering concept is manifold. One topic of interest in this area is the modification of organic solid-state chromophores. The applicability of such solids in diverse fields, such as organic or polymeric light-emitting diodes (OLEDs/PLEDs),3,4 solid-state lasers,5,6 or fluorescent chemosensors,7 is attracting great interest in a broad range of research. Moreover, tunability, flexibility, and low-cost characterization add to the great advantages for the potential usability of organic chromophores.8 The tuneable character has already led to various studies in recent years in the aspect of crystal engineering, focussing on co-crystallization strategies to effectively modulate optical properties.9–14 Flavins, as a subset of organic solid-state chromophores, belong to the class of isoalloxazines and are especially interesting for biological processes, adapting the role of coenzymes or photoreceptors.15,16 With a diversity of spectral properties, flavins may act as natural reporters to monitor processes in vivo.17 Here, their selective autofluorescence activity can be utilized to apply these organic compounds as biomarkers in metabolic processes.18 A comprehensive study in 2004 by Sikorska et al. reported strongly red-shifted fluorescence emission spectra in solution for isoalloxazines compared to alloxazines and their constitutional isomers.19 Isoalloxazines have also exhibited longer decay times and more intense fluorescence.19–21 Overall, these properties are strongly correlated with the number and position of the substituents. Thus, a common route to affect the luminescence properties of organic chromophores, particularly flavins, is based on derivatisation, with various functional groups affecting the polarity or rigidity of the system.22 Hereby, in some cases, the fluorescence activity might be hindered. Quenching processes can have several causes, including excited-state reactions, molecular rearrangements, energy transfer, ground-state complex formation, and collisional or configurational crossover-based quenching.23 In the solid state, organic molecules might exhibit self-quenching processes that result from aggregation and consequent efficient non-radiative relaxation23 or from close packing in the crystal structures forming strong π⋯π contacts or hydrogen bonds, which in turn may cause severe fluorescence quenching.24–26 To overcome this problem, we suggest an alternative strategy to tune the luminescence of flavins upon co-crystallization. It should be noted that experimental studies on flavin derivatives in the solid state are conspicuously scarce. A Cambridge Structural Database survey27 only indicated a scant amount of available structural investigations of flavins. However, the crystal structures of several simple flavin derivatives28–30 have been reported along with some solvates31 and salts.32–34 Also several co-crystals,35–38 referred to as molecular complexes, were structurally characterised, with all of them exhibiting strong hydrogen-bonded interaction patterns. The majority of these investigations were carried out in the 1970s. However, no investigations on the structure–property relations for the reported structures have thus far been performed.The present work is concerned with a structural characterization of 8-(dimethylamino)-7,10-dimethylbenzo[g]pteridine-2,4(3H,10H)-dione, also known as roseolumiflavin (R), and the synthesis of its multicomponent crystals (Chart 1). A notable focus was further set on an investigation of the luminescence. Until now, R has only been a topic of discussion on a theoretical level by Karasulu and Thiel in 2014, in which the photophysical properties of R in different environments were investigated.39 Experimental data are yet to be published and no structural investigations have been carried out until now. An insight into the molecular self-assembly in the solid state, however, opens a great prospective towards better understanding and controlling the optical properties of flavins.
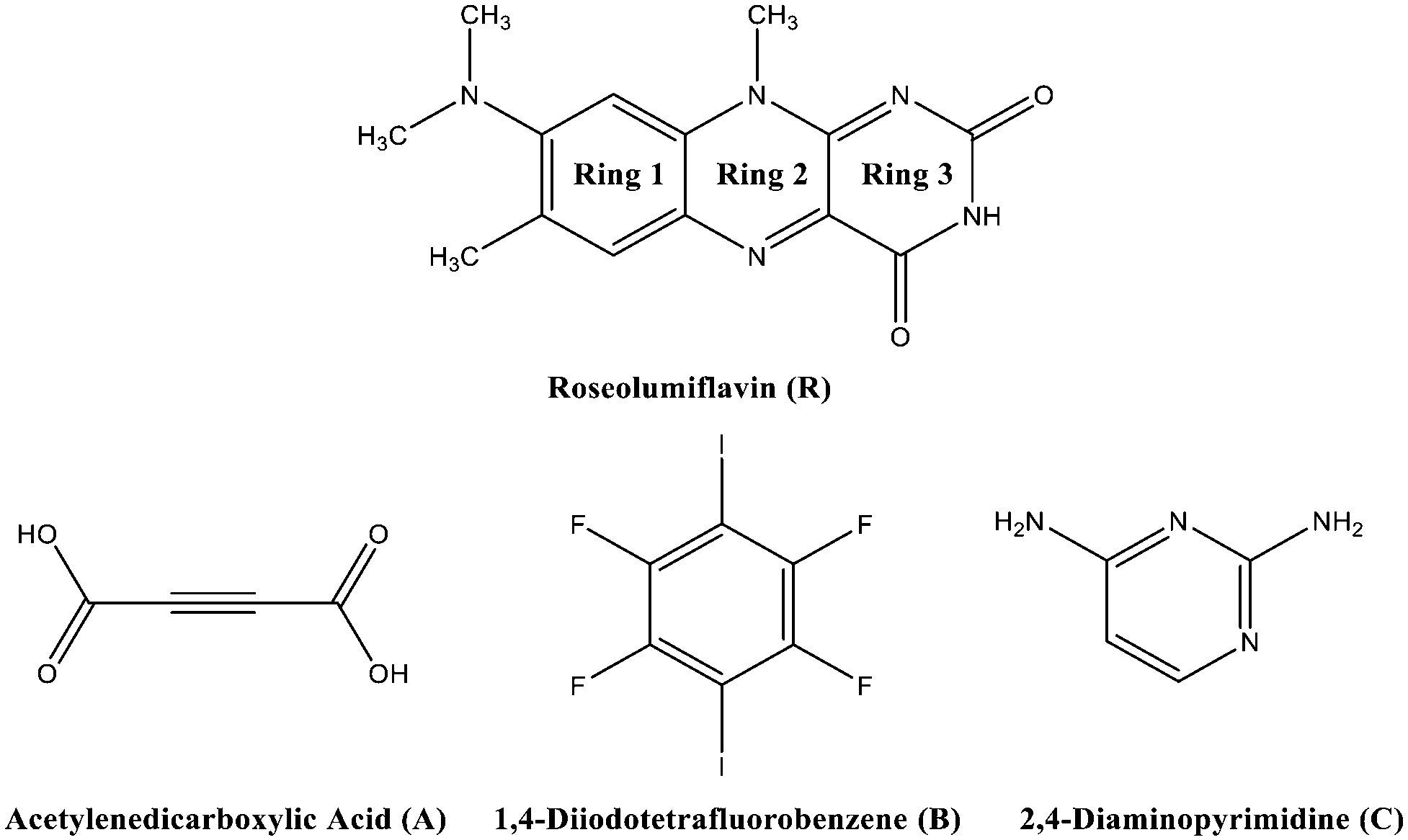 | ||
| Chart 1 Chemical structures of R and the co-formers A–C; aromatic rings of R denote Rings 1–3. | ||
In this study, we wanted to verify if the implementation of a second entity into the crystal lattice would disturb an anticipated aggregation of R and even lead to an enhancement of the luminescence. In addition, it was of interest to clarify which kind of intermolecular interactions and supramolecular synthons were accessible for the co-crystal formation and how they affect the luminescent character of R. For this purpose, three co-formers were chosen based on rational design considerations to result in binary co-crystalline phases with R.
Hereby, emphasis was placed on utilizing the imide group, which offers donor as well as acceptor functionalities to allow different kinds of intermolecular interactions and to allow it to undergo one-, two-, and three-point recognition patterns during the self-assembly. The high acidity on the N–H centre was assumed to favour hydrogen-bond interactions. Via a retrosynthetic approach, both –N–H and –O–H based supramolecular synthons are commonly represented in the literature, promising sufficient adaptability.40,41 Acetylene-dicarboxylic acid (A) with its two carboxylic sides is a promising candidate to undergo an acid–amide heterodimer formation, which has been shown to be more favourable over amide–amide or acid–acid homodimers.42 Another hydrogen-bonded motif, the DAD (donor–acceptor–donor) three-point recognition pattern, was expected to result in a co-crystallisation with 2,4-diamino-pyrimidine (C). Further, the nucleophilic character of unsaturated heteroatoms in R suggested an affinity towards halogen bonding. Accordingly, a model halogen bond co-former 1,4-diiodotetrafluorobenzene (B) was introduced as a possible building block for the one-point molecular recognition. Moreover, the aromatic rings in B and C could exhibit an additional effect on the overall crystal packing involving π-interactions.
Here, we present a structural characterization of R along with a successful synthesis and crystal structure analysis of three new roseolumiflavin co-crystals R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :A, R:B, and R:C. A careful crystal packing analysis was performed, which presented various effects of different types of intermolecular interactions on the overall topology. Spectroscopic analyses of all the obtained structures uncovered possibilities to modify the luminescent properties of the organic chromophore via co-crystallization. Based on the results, the main purpose of our research was to obtain a better understanding about a potential targeted tuning of the optical properties of roseolumiflavin in the solid state.
:A, R:B, and R:C. A careful crystal packing analysis was performed, which presented various effects of different types of intermolecular interactions on the overall topology. Spectroscopic analyses of all the obtained structures uncovered possibilities to modify the luminescent properties of the organic chromophore via co-crystallization. Based on the results, the main purpose of our research was to obtain a better understanding about a potential targeted tuning of the optical properties of roseolumiflavin in the solid state.
Experimental section
Synthesis
:1) co-crystal (R:A).
A is highly hygroscopic and tends to form a monohydrate, which does not participate in a co-crystallization, according to Tantardini et al.44 Thus, to synthesize the R:A co-crystal, an excess amount of A was used. A and R were mixed in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in a micro-centrifuge tube with 15 μL acetonitrile and six milling balls. The sample was ground for 25 min at 15 Hz frequency in a Retsch MM 400 ball mill. Clear bright red needle-shaped single crystals were obtained via seeding in acetonitrile and by slow evaporation of the solvent for 2 weeks at room temperature. IR: 3506, 3440, 3255, 1695, 1684, 1632 cm−1. PXRD (characteristic Bragg reflections): 6.29°, 11.59°, 13.55°, 25.22°, 25.97°, 27.78° 2θ.
:3) co-crystal (R:B).
R:B was prepared in a 1:1 molar ratio of R and B in a micro-centrifuge tube with 20 μL methanol and eight milling balls. The sample was ground for 25 min at 15 Hz frequency. Red block-shaped single crystals were obtained via seeding in methanol and by a slow solvent evaporation for 1 week at room temperature. All attempts to achieve a 1:1 co-crystal by variation of the crystallization parameters and/or molar ratio of the starting materials resulted in the same R:B (2:3) phase. IR: 3359, 3237, 3126, 2986, 2806, 1683, 1623 cm−1. PXRD (characteristic Bragg reflections): 12.93°, 16.63°, 16.94°, 24.28°, 24.96°, 26.65°, 26.94° 2θ.
:1) co-crystal (R:C).
R and C were dissolved in methanol in a 1:1 molar ratio and crystallized after slow evaporation of the solvent at room temperature to give orange-red needle-shaped crystals of R:C. IR: 3372, 3322, 3192, 3114, 2978, 2804, 1688, 1652 cm−1. PXRD (characteristic Bragg reflections): 6.14°, 11.47°, 12.64°, 24.84°, 27.15° 2θ.
:1 molar ratio in a micro-centrifuge tube with 15 μL acetonitrile and six milling balls. The sample was ground for 25 min at 15 Hz frequency in a Retsch MM 400 ball mill. Clear bright red needle-shaped single crystals were obtained via seeding in acetonitrile and by slow evaporation of the solvent for 2 weeks at room temperature. IR: 3506, 3440, 3255, 1695, 1684, 1632 cm−1. PXRD (characteristic Bragg reflections): 6.29°, 11.59°, 13.55°, 25.22°, 25.97°, 27.78° 2θ.
:3) co-crystal (R:B).
R:B was prepared in a 1:1 molar ratio of R and B in a micro-centrifuge tube with 20 μL methanol and eight milling balls. The sample was ground for 25 min at 15 Hz frequency. Red block-shaped single crystals were obtained via seeding in methanol and by a slow solvent evaporation for 1 week at room temperature. All attempts to achieve a 1:1 co-crystal by variation of the crystallization parameters and/or molar ratio of the starting materials resulted in the same R:B (2:3) phase. IR: 3359, 3237, 3126, 2986, 2806, 1683, 1623 cm−1. PXRD (characteristic Bragg reflections): 12.93°, 16.63°, 16.94°, 24.28°, 24.96°, 26.65°, 26.94° 2θ.
:1) co-crystal (R:C).
R and C were dissolved in methanol in a 1:1 molar ratio and crystallized after slow evaporation of the solvent at room temperature to give orange-red needle-shaped crystals of R:C. IR: 3372, 3322, 3192, 3114, 2978, 2804, 1688, 1652 cm−1. PXRD (characteristic Bragg reflections): 6.14°, 11.47°, 12.64°, 24.84°, 27.15° 2θ.
Thermogravimetric analysis
A Netzsch TG 209 was operated in the range between 30 °C and 600 °C with a 10 °C min−1 heating rate under a nitrogen atmosphere. No melting point could be obtained because R decomposed before melting.Infrared spectroscopy
The FT/IR spectra were measured on a Bruker Tensor 37 with an ATR-unit at room temperature (25 °C).UV-vis and luminescence spectroscopy
Absorption spectra in solution were measured in a UV-vis SPECORD S600 system at room temperature (25 °C). The emission spectra in solution were measured by a HORIBA FluoroMax 4 system at room temperature (25 °C). Qualitative measurements were performed upon excitation with a UV lamp emitting at 366 nm. The photoluminescence excitation spectra were acquired on an Edinburgh Instruments FLS980 spectrometer with a 450 W continuous Xe arc lamp, double grating monochromators for both excitation and emission compartments, and a Hamamatsu R928P thermoelectrically cooled photomultiplier tube for detection. Photoluminescence emission spectra were acquired on an Edinburgh FLS1000 spectrometer with similar features and upon excitation with a VPL-420 laser in continuous wave mode. All excitation spectra were corrected for the lamp intensity and grating efficiency, while emission spectra were corrected for the grating efficiency and sensitivity of the photomultiplier. Time-resolved luminescence measurements were performed using pulsed EPL diode lasers from Edinburgh Instruments with adjustable repetition rates and temporal pulse widths of around 75 ps.X-Ray diffraction
Powder X-ray diffraction (PXRD) analyses were performed by a Rigaku Miniflex diffractometer in θ/2θ geometry at ambient temperature using Cu-Kα radiation (λ = 1.54182 Å). Single-crystal X-ray diffraction (SCXRD) measurements were carried out on a Bruker APEX Duo diffractometer with a CCD detector, micro-focus X-ray tube, and Mo-Kα radiation (λ = 0.71073 Å) at 140(2) K for R and a Rigaku XtaLAB Synergy-S diffraction system with a HiPyx 6000 photon detector, and micro-focus X-ray tube with Cu-Kα radiation (λ = 154182 Å) measured at 100(2) K for the co-crystals. Cell refinement, data collection, and data reduction on the Rigaku Synergy-S system were performed with CrysAlisPro.45 On the Bruker APEX Duo, data collection and cell refinement were achieved with APEX2 (ref. 46) and data reduction was performed via SAINT.47 Structure solution was performed by SHELXT 2014/5 (ref. 48) for R and R:B and SHELXT 2018/2 (ref. 48) for R:A and R:C. R:B was refined with SHELXL-2014/7,48R was refined with SHELXL-2017/1.49R:B and R:C were refined with SHELXL-2018/3.49 The co-crystals were additionally refined with the Olex2 software package.50 All the non-hydrogen atoms were refined with anisotropic displacement parameters. All the hydrogen atoms were experimentally refined. R:A and R:C crystallized as very thin plates/needles and exhibited an extremely poor diffracting ability, which was reflected in the data quality and quality values. Numerous attempts to obtain larger single crystals failed. The crystallographic data are summarized in Table 1.
:A, R:B, and R:C
| Name | R |
R:A (1:1) |
R:B (2:3) |
R:C (1:1) |
|---|---|---|---|---|
| Empirical formula | C14H15N5O2 | C14H15N5O2, C4H2O4 | C14H15N5O2, 1.5(C6F4I2) | C14H15N5O2, C4H6N4 |
| Formula weight [g mol−1] | 285.31 | 354.35 | 888.10 | 395.44 |
| Temperature [K] | 140(2) | 100(10) | 100(2) | 100.01(13) |
| Space group (no.) | P21/c (14) | Pbca (61) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) (2) |
P (2) |
| Crystal system | Monoclinic | Orthorhombic | Triclinic | Triclinic |
| a [Å] | 12.7469(10) | 6.9603(4) | 7.7683(3) | 8.0245(5) |
| b [Å] | 7.0326(8) | 18.1475(7) | 12.2451(5) | 8.4915(5) |
| c [Å] | 14.3336(13) | 28.0356(12) | 14.6375(5) | 14.9566(7) |
| α [°] | 90 | 90 | 108.280(4) | 87.548(5) |
| β [°] | 99.135(5) | 90 | 91.090(3) | 74.813(5) |
| γ [°] | 90 | 90 | 96.577(4) | 65.130(6) |
| Volume [Å3] | 1268.6(2) | 3541.2(3) | 1311.17(9) | 889.69(10) |
| Z/Z′ | 4/1 | 8/1 | 2/1 | 2/1 |
| ρ calc [g cm−3] | 1.494 | 1.498 | 2.249 | 1.476 |
| μ [mm−1] | 0.105 | 0.976 | 28.774 | 0.854 |
| T min/Tmax | 0.6559/0.7455 | 0.844/1.000 | 0.327/0.597 | 0.698/1.000 |
| F(000) | 600 | 1664 | 834 | 416 |
| Crystal size [mm3] | 0.05 × 0.10 × 0.15 | 0.02 × 0.02 × 0.18 | 0.02 × 0.05 × 0.05 | 0.03 × 0.03 × 0.06 |
| θ range [°] | 1.987/24.998 | 3.1490/69.2990 | 3.185/78.301 | 3.071/78.995 |
| Completeness [%] | 99.6 | 99.5 | 99.7 | 99.7 |
| Recorded reflections | 8604 | 14153 |
15384 |
12348 |
| Indep. reflections | 2224 | 3325 | 5298 | 3599 |
| Goodness-of-fit F2 | 1.007 | 0.996 | 1.112 | 1.006 |
| X-ray source (wavelength [Å]) | MoKα (λ = 0.71073 Å) | CuKα (λ = 1.54184 Å) | CuKα (λ = 1.54184 Å) | CuKα (λ = 1.54184 Å) |
| R 1 [I > 2σ(I)]/wR2 [all reflns.] | 0.0493, 0.1399 | 0.0617, 0.1645 | 0.0342, 0.0941 | 0.0557, 0.1678 |
Software
Crystal structure figures were prepared by Mercury 2020.2.0.51 Verbose crystal structure information, including Z′, was obtained via PLATON for the Windows Taskbar (Version 1.19).52 All the spectra were plotted with OriginPro 2019.53Results and discussion
Characterization of R
R crystallizes in the monoclinic space group P21/n (no. 14). The asymmetric unit contains one molecule of R, four of which shape the unit cell. The crystal structure is based on dimeric supramolecular assemblies connected via N–H⋯O interactions (N2–H⋯O2: 2.813(2) Å; note: all distances were measured between donor and acceptor molecules unless noted otherwise). Both moieties are twisted with an angle of 8.65° and build up along the crystallographic a axis (Fig. 1).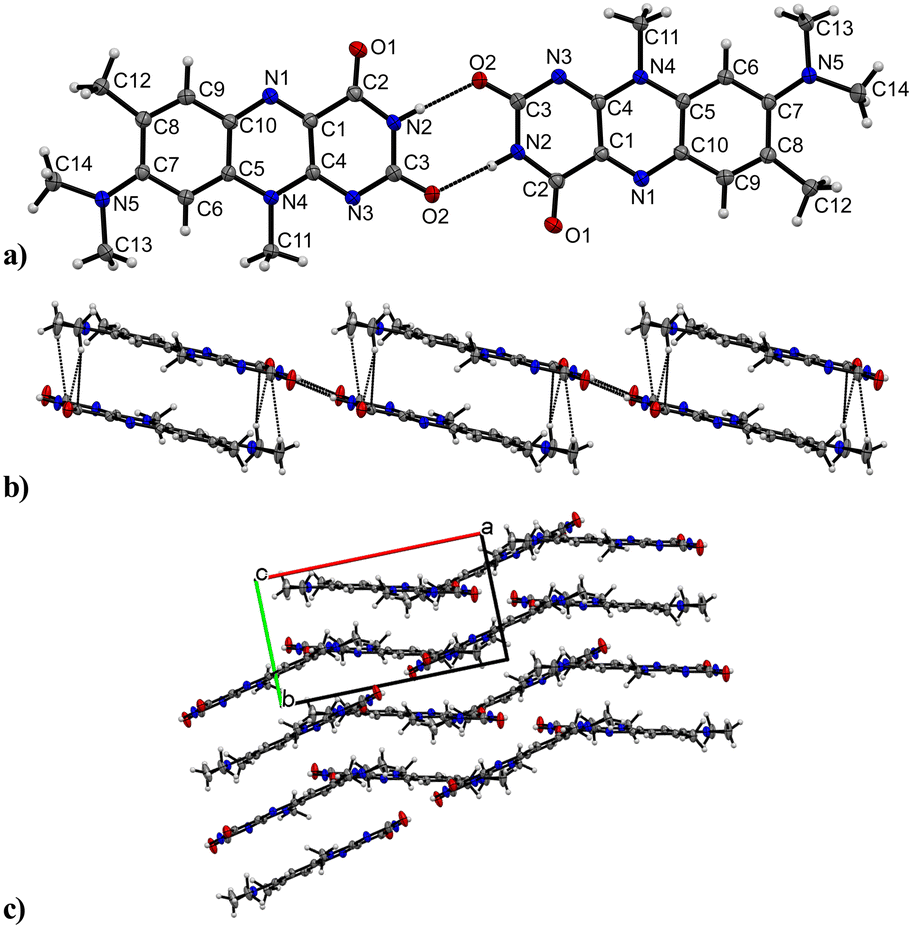 | ||
| Fig. 1 Crystal packing and intermolecular interactions of R. a) Head-to-head dimer motif formed by amide–amide synthon. Labels for H-atoms are omitted for clarity. View along the crystallographic b axis. b) π⋯π interactions stabilize the stacking patterns along the a axis. View along the c axis. c) Alternating R stacks along the c axis with each stack tilted at a 26.64° angle. View along the c axis. | ||
Short hydrogen bonds (C14–H⋯C3: 2.87(4) Å, C13–H⋯O1: 2.62(3) Å, C13–H⋯C2: 2.79(3) Å) along the b axis allow for the dimer motifs to form chain-like networks with strong displaced stacking interactions where Ring 2 is stacked above the Ring 1 (centroidRing 2⋯centroidRing 1: 3.576(4) Å and 3.6280(13) Å), expanding along the a axis. Each unit is further supported on either side via short hydrogen bonds (C11–H⋯O1: 3.147(2) Å, C13–H⋯O1: 3.177(2) Å, C6–H⋯O1: 2.66(2) Å, C12–H⋯O2: 2.51(3) Å) building up in a spiral form along the b axis, which results from the tilted nature of each stacked dimeric motif alternating along the crystallographic c axis. This allows the amide groups to be in a position in close proximity to the methyl groups, ultimately spanning the crystal packing. Additionally, 1H-NMR analysis was performed for confirmation of the purity and for characterization of the compound (for details see the ESI†). The corresponding IR spectrum in Fig. 2 illustrates strong peaks at 1690 and 1633 cm−1, which were assigned to amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes, while two bands at 3380 and 3250 cm−1 originated from the mono-substituted amide N–H stretching mode. The band at 3129 cm−1 may have resulted from combination vibrations ν(CO) + δ(N–H). The vibrational band at 2799 cm−1 likely originated from the NC–H stretching mode, while the band at 2984 cm−1 could be assigned to C–H stretching vibrations. In addition, bands of aryl C–H vibrations (3063 cm−1) were present.
O stretching modes, while two bands at 3380 and 3250 cm−1 originated from the mono-substituted amide N–H stretching mode. The band at 3129 cm−1 may have resulted from combination vibrations ν(CO) + δ(N–H). The vibrational band at 2799 cm−1 likely originated from the NC–H stretching mode, while the band at 2984 cm−1 could be assigned to C–H stretching vibrations. In addition, bands of aryl C–H vibrations (3063 cm−1) were present.
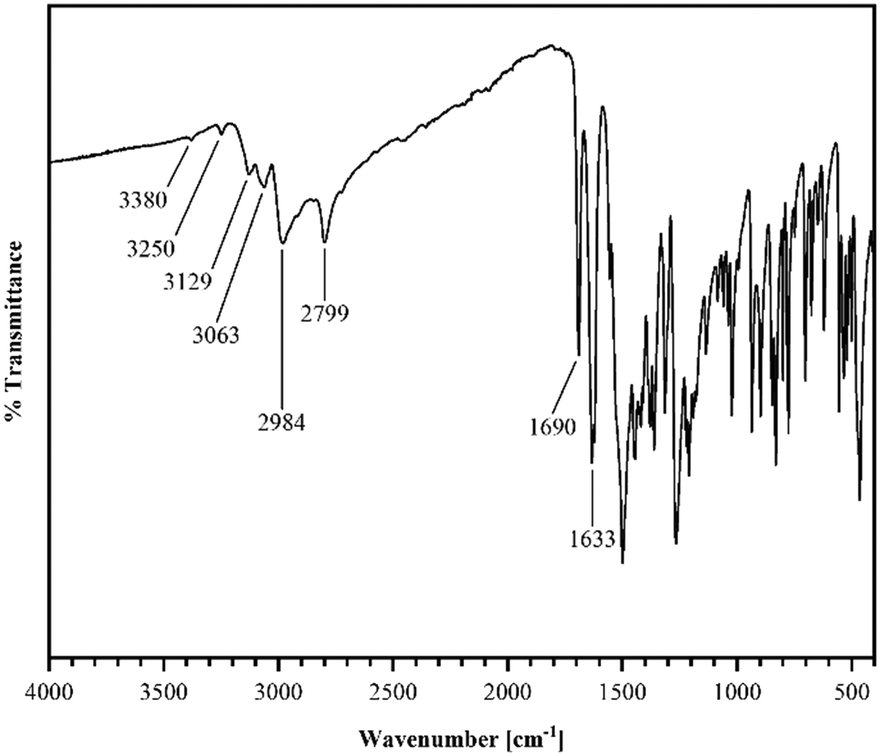 | ||
| Fig. 2 Infrared spectrum (ATR) of R recorded from 400 to 4000 cm−1. | ||
PXRD analysis
Powder diffraction patterns of the samples R:A, R:B, and R:C synthesized via liquid-assisted grinding are depicted in Fig. 3 and distinctly different in comparison to pure R, which suggested the formation of new crystal phases. The overall purity of all the co-crystals was in good agreement with the available single-crystal data simulations (Fig. S7–S9 in the ESI†).
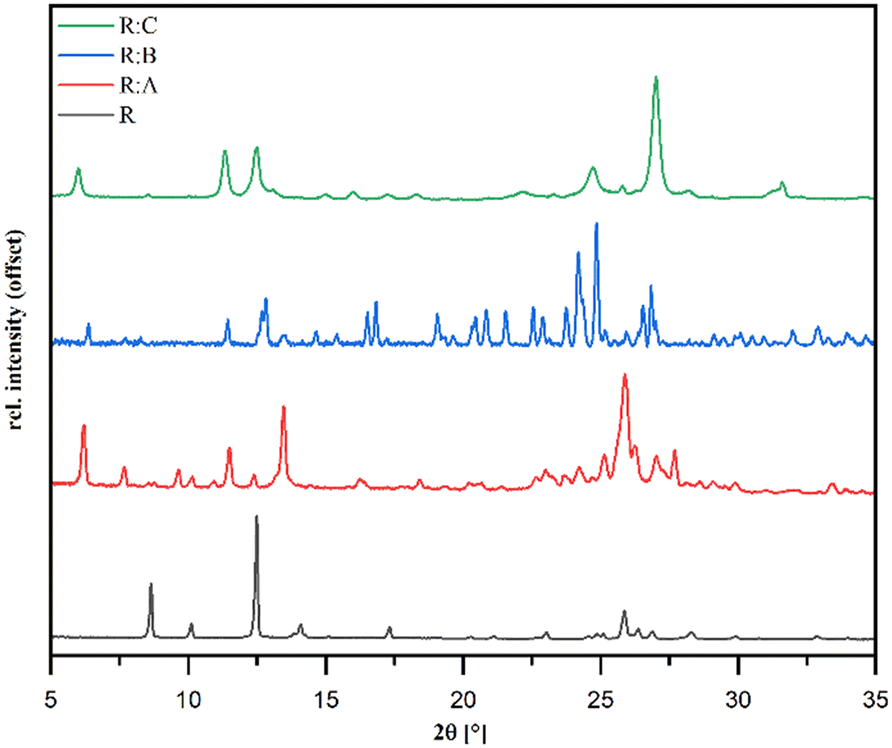 | ||
| Fig. 3 Powder X-ray diffraction patterns (Cu Kα) of R and the co-crystals R:A, R:B and R:C. | ||
SC-XRD analysis of the co-crystals
R:A crystallizes in the orthorhombic space group Pbca (no. 61) with one of each of the R and A molecules setting up the asymmetric unit (Fig. 4). The dicarboxylic functionality of the co-former potentially allows the formation of two hydrogen-bonded interactions with R mediated by two carboxylic groups. Indeed, we observed both ends took part in the intermolecular assembly however, each side was connected uniquely. A classic linear assembly between the carboxylic group and the R amide group supplanted the favoured dimeric amide–amide formation (donor–acceptor distances N2–H⋯O3: 2.922(3) Å, O4–H⋯O2: 2.538(3) Å).
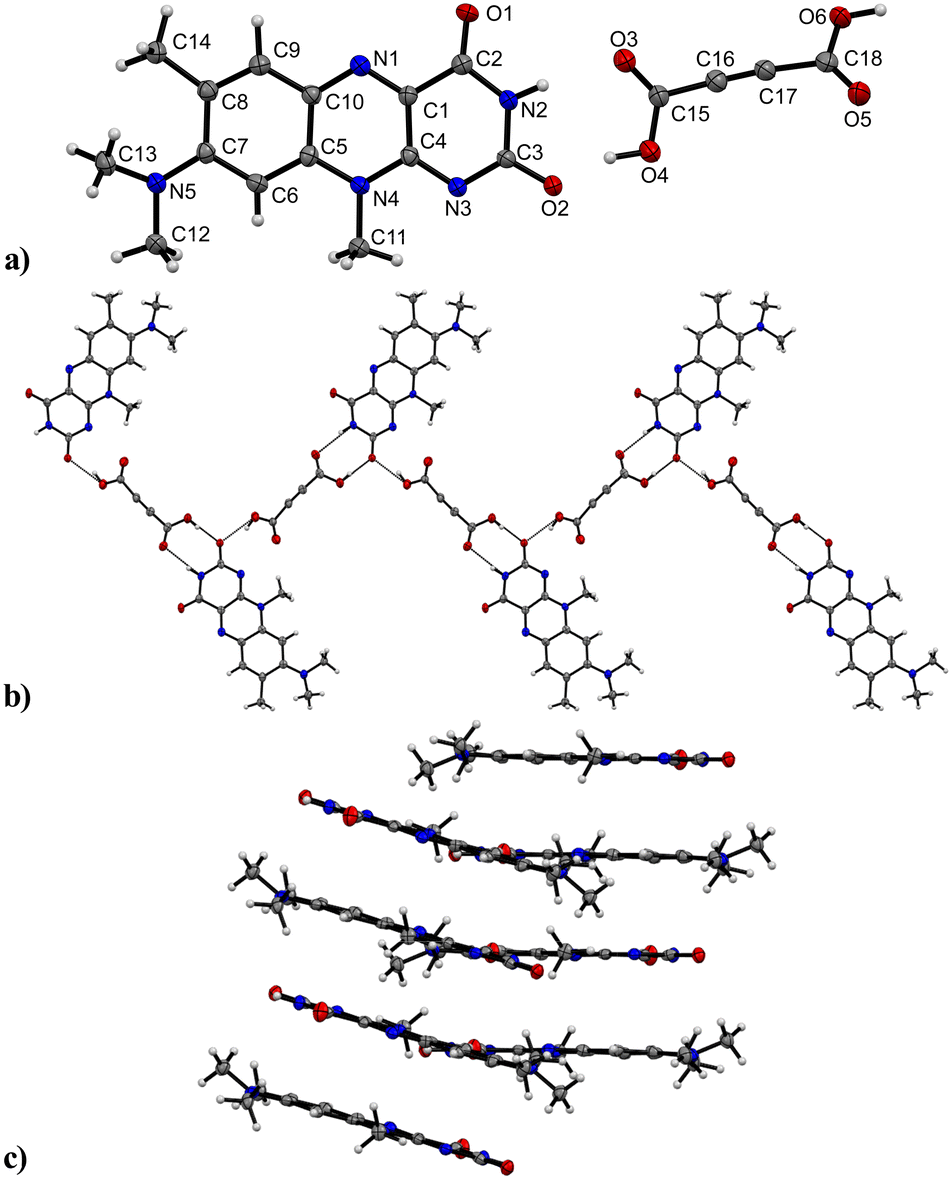 | ||
| Fig. 4 Crystal structure of R:A. a) Asymmetric unit of R:A. Labels for H-atoms are omitted for clarity. View along the crystallographic a axis. b) Zig-zag chains formed by O–H⋯O and N–H⋯O interactions between A and R along the b axis. View along the a axis. c) Tilted π⋯π stacks of R molecules. A removed for clarity. View along the b axis. | ||
Intermolecular O6–H⋯O2 interactions (2.616(3) Å) between the second carboxylic group and another unit of R ultimately allowed the formation of hydrogen-bonded zig-zag chains to propagate along the crystallographic b axis.
The zig-zag chains further intertwine to increase the close-packing efficiency in the crystal structure. This is also due to the carboxylic groups of the A units being twisted at an angle of 46.09° between the planes through each carboxylic group. Along the a axis, the flavin molecules interact via slightly disordered π⋯π stacks (centroidRing 3⋯centroidRing 1: 3.5297(12) Å and 3.5090(12) Å, centroidRing 2⋯centroidRing 2: 3.5312(15) Å and 3.5087(13) Å), each flavin molecule being inversely oriented to the stacking partner. The stacks are tilted by 17.53° relative to the neighbouring stacks.
R in combination with a model halogen bond donor 1,4-diiodotetrafluorobenzene resulted in a formation of the R:B (2:3) co-crystal with one molecule of R and 1.5 co-former molecules, assigned as B1 and B2 in Fig. 5 in the asymmetric unit cell. The compound crystallizes in the triclinic space group P (no. 2) and contains the familiar amide–amide synthon, which leads to a dimer formation (N1–H⋯O2: 2.834(6) Å) similar to that in pure roseolumiflavin. Here, moderate π⋯π stacking interactions were observed again between two flavin molecules with inverse directionality which, mediated by the dimeric motifs, propagate along the b axis (centroidout,Ring 2⋯centroidout,Ring 1: 3.684(3) Å, centroidRing 2⋯centroidRing 2: 3.727(3) Å). The stacks are displaced along the c axis surrounded by B molecules offering halogen bonds to build up linear chains.
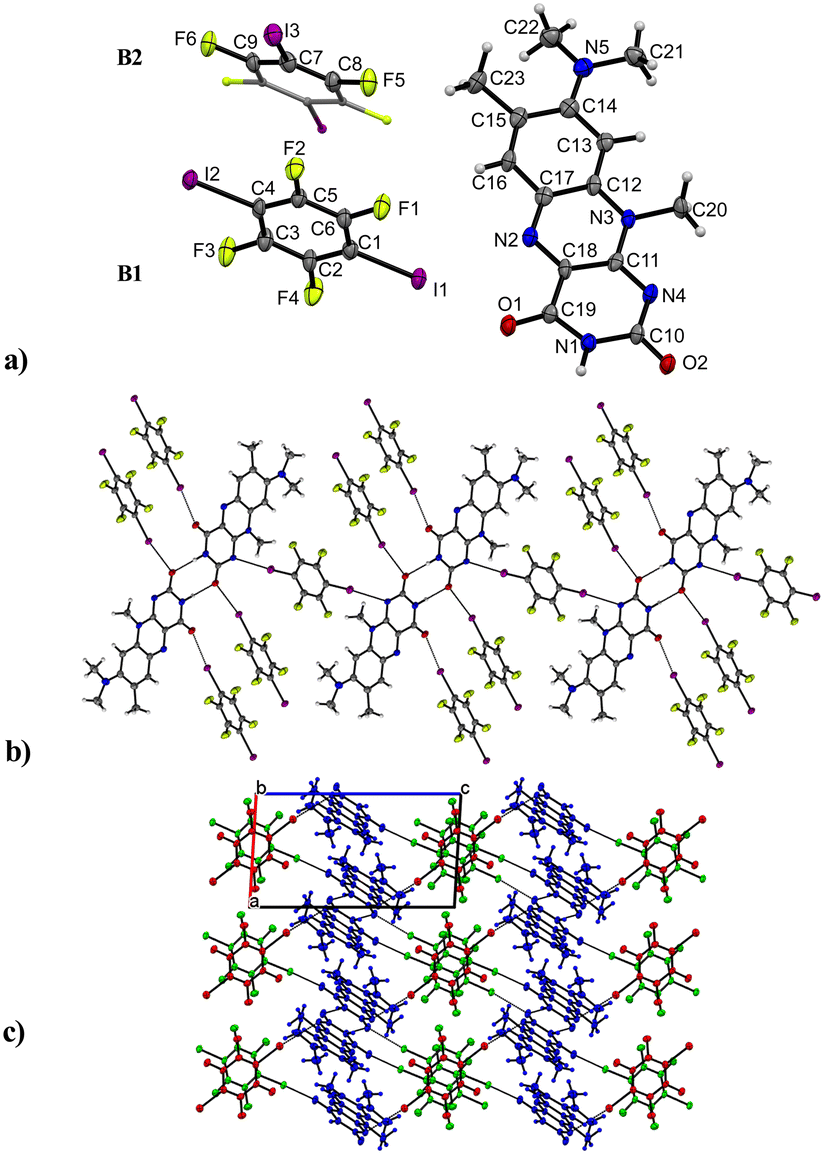 | ||
| Fig. 5 Intermolecular interactions and crystal packing of R:B. a) Asymmetric unit of R:B. The symmetry inequivalent co-formers are labelled as B1 and B2. Symmetry equivalent part of B2 molecule is shown as capped sticks. Labels for H-atoms are omitted for clarity. View perpendicular to the bc plane b) I⋯N and I⋯O interactions connect dimeric motifs to build up 2D networks in the crystal packing. View along the a axis. c) Symmetry inequivalent stacked co-formers (B1: green, B2: red) propagate along the b axis, I⋯O and I⋯N interactions stabilize R stacks. View along the b axis. | ||
Through the conjugation of the iodine substituents to the carbonyl groups, the stacked entities are stabilized on either side (I1⋯O1: 2.800(3) Å, I2⋯O2: 2.899(3) Å) establishing halogen-bonded chains between the co-former and flavin molecules along the c axis. Additional halogen bonds are generated by the symmetry inequivalent second B entity positioned above and below the flavin dimers with slight displacement when connecting with the N4 ring atom (I3⋯N4: 3.163(4) Å) and thus building up a complex 2D network. Slightly distorted stacks of the symmetry inequivalent co-former entities propagate along the b axis with a repetition pattern of two consecutive units of B1 and one unit of B2.
The third and final successfully obtained co-crystal R:C crystallizes in a triclinic P (no. 2) space group with the asymmetric unit containing one of both a R and C molecule, respectively (Fig. 6). The C and R molecules are connected via a three-point recognition pattern between the imide group and the amine groups along the crystallographic c axis (N9–H⋯O1: 2909(3) Å; N2–H⋯N6: 2937(3) Å and N8–H⋯O2: 2938(3) Å). A similar interaction pattern was found by Scarbrough et al. in 1977 between lumiflavin and 2,6-diamino-9-ethylpurine.35 Two additional hydrogen bonds connect two co-formers (N8–H⋯N7: 2977(3) Å) to build tetrameric units, which propagate along the b axis mediated by strong N–H⋯N (N9–H⋯N3: 3190(3) Å) and N–H⋯O (N9–H⋯O2: 3097(3) Å) hydrogen bonds as well as C–H⋯O interactions (C17–H⋯O2: 3381(3) Å) between C and R entities.
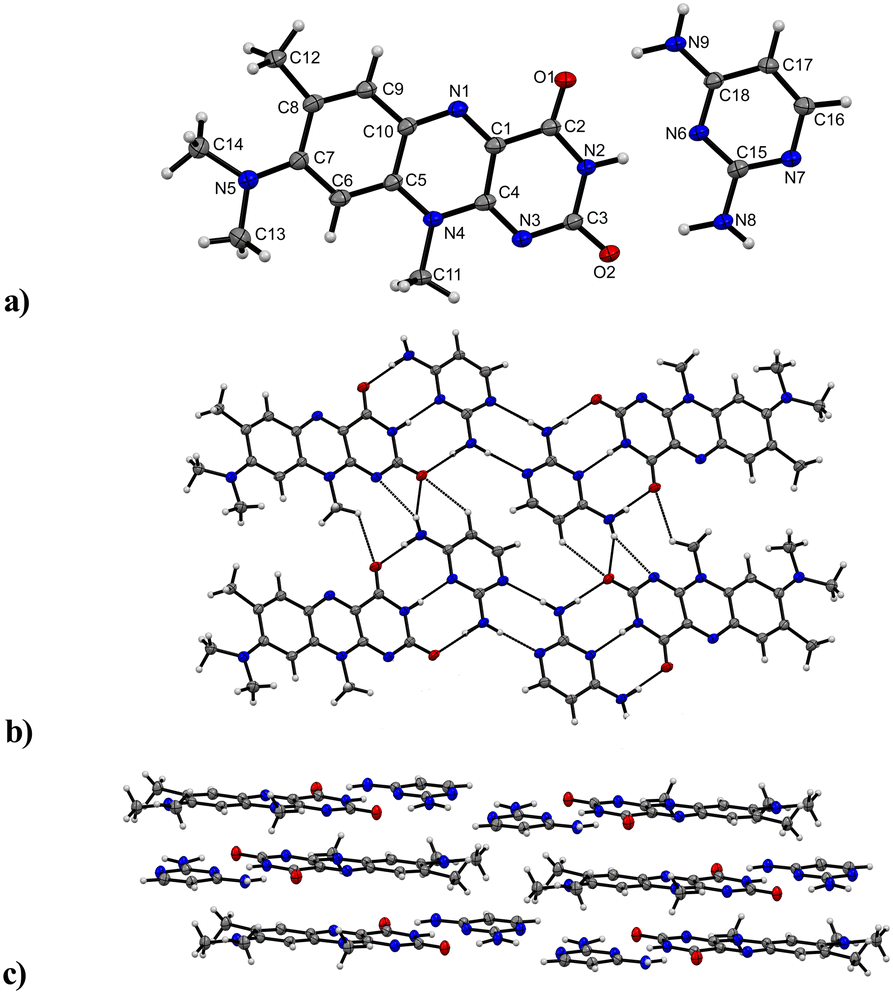 | ||
| Fig. 6 Intermolecular interactions and crystal packing of R:C a) asymmetric unit of R:C. Labels for H-atoms are omitted for clarity. View perpendicular to the bc plane. b) Tetrameric motifs connected by hydrogen bonds propagate along the b axis mediated by the C unit. View along the a axis. c) π⋯π contacts between C and R. View along the b axis. | ||
Further, weak hydrogen bonds C–H⋯O (C11–H⋯O1: 3085(3) Å) between adjacent R molecules are present as well. The resulting layers are parallel to the bc plane. π⋯π stacks between co-facially adjacent flavin molecules govern an expansion along the a axis, where the tetrameric subunits alternate with an inverse orientation of the flavin units on each layer. 2,4-Diaminopyrimidine and flavin molecules arranged in slipped stack formations are additionally stabilized by π⋯π interactions.
To conclude, R:A with its edge-to-face arrangement displays a crystal packing driven by the formation of the carboxylic acid–amide dimer, which replaces the amide–amide dimeric supramolecular synthon observed in R. In contrast, in R:B the mentioned amide–amide dimer of R molecules is present; however, the dimers are connected with I⋯O halogen bonds over B molecules to give chains of alternating co-former and flavin molecules. Additionally, two flavin molecules are further stabilized by an extra B molecule connected via short N⋯I contacts. The R:C co-crystal contains π⋯π contacts between the co-former molecules and R. These stacked arrangements are further connected by hydrogen-mediated donor–acceptor–donor three-point bonding motifs via N–H⋯O and N–H⋯N interactions between C and R entities. The dimeric flavin motifs connected via amide–amide synthons only remained in the R:B co-crystal. These were responsible for the shortest-range interactions in the pure form R. The hydrogen-bonded D⋯A distances in the R:B crystal structure were in a similar range compared to pure R, while in both other co-crystal structures the distances were significantly larger. Here, it is noteworthy that a mere focus on the N–H⋯O interaction would only describe one part of the imide group interaction. Since the dimeric R motifs are detached, we also consider the CO group separately. In the case of R:A, significantly stronger hydrogen bond interactions were observed, mediated by two separate A entities towards the flavin CO group at a 105.76° angle between both interactions. The O6–H⋯O2 (1.68(5) Å) as well as O4–H⋯O2 (1.53(5) Å) hydrogen bond distances were significantly shorter than the N–H⋯O interactions (Table 2).
| Structure | D–H⋯A | D–H [Å] | H⋯A [Å] | D⋯A [Å] |
|---|---|---|---|---|
| R | N2–H1⋯O2 | 0.84(2) | 1.98(2) | 2.813(2) |
|
R:A
|
N2–H2⋯O3 | 0.88(4) | 2.05(4) | 2.922(3) |
|
R:B
|
N1–H1⋯O2 | 0.96(5) | 1.88(5) | 2.834(6) |
|
R:C
|
N9–H9A⋯O1 | 0.98(3) | 1.94(3) | 2.909(3) |
| N2–H2⋯N6 | 0.94(4) | 2.00(4) | 2.937(3) | |
| N8–H8A⋯O2 | 0.99(3) | 1.96(3) | 2.938(3) |
The dimeric motif was replaced by a three-point recognition pattern provided by the co-former component in R:C where the overall D⋯A distances were slightly longer; however, the H⋯O1 and H⋯O2 distances were slightly shorter than the equivalent H⋯A interactions in pure R, which can be considered as a driving factor for the substitution of the dimer motif. Lastly, π⋯π stacking interactions were shown to be affected as a result of the co-crystal formations (Table 3). The aromatic rings of R are referred to as noted in Chart 1.
| Structure | π⋯π | π⋯π [Å] |
|---|---|---|
| i 1 − x, 1 − y, 1 − z; ii −x, 1 − y, 1 − z; iii 2 − x, 1 − y, 1 − z. | ||
| R | Ring 2–Ring 3i | 3.576(4) & 3.6280(13) |
|
R:A
|
Ring 2–Ring 2i | 3.5312(15) |
| Ring 2–Ring 2ii | 3.5087(13) | |
| Ring 1–Ring 3i | 3.5297(12) | |
| Ring 1–Ring 3ii | 3.5090(12) | |
|
R:B
|
Ring 2–Ring 2iii | 3.727(3) |
| Ring 1–Ring 3iii | 3.684(3) | |
|
R:C
|
Ring 1–Ring 3i | 3.3948(15) |
| Ring 2–Ring 2i | 3.3869(15) | |
| Ring 2–Ring 3iii | 4.1262(16) | |
| Ring 3–Ring 3iii | 3.9787(15) | |
| Ring 1–Ciii | 3.5814(15) | |
Thermal analysis
Thermal stability was investigated by means of thermogravimetric analysis. The thermograms for R and its co-crystals are depicted in Fig. S13–S15 in the ESI.† While a decomposition point for pure R was observed at 350 °C, the corresponding thermograms for the co-crystals displayed additional plateaus under 350 °C, indicating the decomposition of the co-crystals with the plateaus assigned to the respective components.The thermogram for R:A displayed two processes with the decomposition of A located at TR:A,1 = 130 °C causing a mass loss of 28% and the decomposition point of R is shifted to TR:A,2 = 343 °C with a mass loss of 47%. The thermogram for R:B exhibited two distinct endothermic processes starting at TR:B,1 = 117 °C and TR:B,2 = 348 °C, which could be clearly assigned to the respective components of the co-crystal. TR:B,1 = 117 °C likely originated from C–I bond breaking processes, resulting in a mass decrease of 68%. TR:B,2 = 348 °C represents the decomposition point of R, with a 24% mass loss. The thermogram for R:C contained two processes, the first of which at TR:C,1 = 244 °C could be attributed to the decomposition process of the co-former component C with a mass loss of 23%. The second point originated from the decomposition process of R starting at TR:C,2 = 322 °C with a mass loss of 37%. With regard to the thermal stability of R, we hence observed a roughly similar decomposition point in R:B compared to pure R, whereas the thermal stability was decreased significantly by −28 °C in R:C and −7 °C in R:A.
Optical properties
The co-crystallization exerted an effect on the luminescence properties of roseolumiflavin in the solid state. Fig. 7 presents the powdered, microcrystalline compounds prepared under daylight and excited with a conventional UV lamp at λexc = 366 nm. While R only showed weak visible luminescence (but near-infrared luminescence as will be discussed below), the co-crystals showed a comparably more intense orange-red luminescence. When observing the single and multicomponent crystals under daylight the latter exhibit modified colours compared to R. | ||
| Fig. 7
R, R:A, R:B, and R:C under daylight (top) and excited at λexc = 366 nm (bottom). | ||
The luminescence emission spectra confirmed a generally blue-shifted luminescence in the co-crystals compared to solid R. The selected excitation wavelength was based on the employed continuous-wave laser, which allowed recording the emission spectra with a higher signal-to-noise ratio. While R emitted light in the near-infrared area (λmaxem = 733 nm), the co-crystals R:A, R:B, and R:C showed blue-shifted emission bands with maxima at λmaxem = 638 nm for R:A, λmaxem = 643 nm for R:B, and λmaxem = 628 nm for R:C with red emission (Fig. 8) at room temperature. These results indicate that the co-crystallization and the consequent inhibition of certain interaction modes in the pure solid R could also be noted in the luminescence properties. The emission spectra and excitation spectra of the respective co-formers are available in the ESI.† All the data on the optical properties are summarized in Table 4.
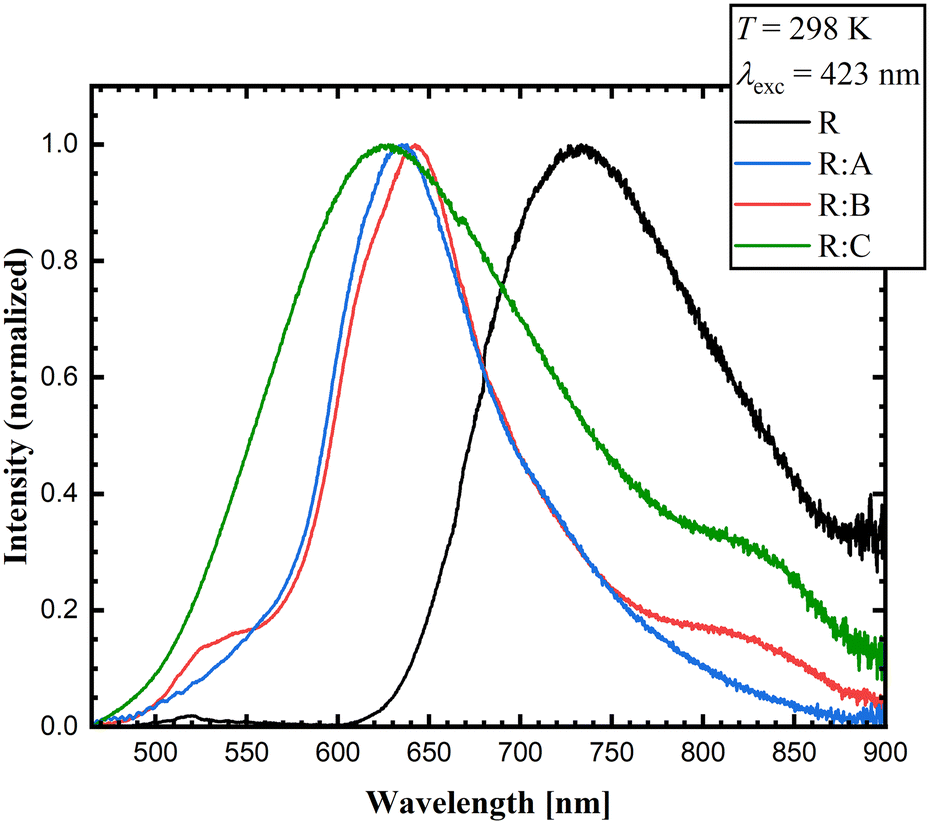 | ||
| Fig. 8 Experimental emission solid-state spectra of R, R:A, R:B, and R:C (λexc = 423 nm) at room temperature. | ||
| Solid state | Solution | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| τ [ns] | λ maxexc,ss [nm] | λ maxem,ss [nm] | λ maxabs,solv [nm] | λ maxem,solv [nm] | |||||
| CHCl3 | ACN | MeOH | CHCl3 | ACN | MeOH | ||||
| R | 2.85 | 633 | 733 | 487 | 488 | 491 | 533 | 512 | 542 |
|
R:A
|
0.99 | 592, 350 | 638 | 483 | 486 | 491 | 524 | 519 | 542 |
|
R:B
|
1.42 | 520, 402 | 643 | 490 | 486 | 490 | 546 | 527 | 546 |
|
R:C
|
0.98 | 565, 339 | 628 | 486 | 485 | 485 | 536 | 540 | 552 |
Time-resolved luminescence measurements in the solid state provide additional insights into the photophysical properties of the respective samples. The corresponding decay curves are shown in Fig. 9. The luminescence decay times for the co-crystals R:A and R:C are close to 1 ns, while for R:B a value of 1.42 ns was measured. Yet, the average decay time for pure R is around 3 ns. According to the luminescence decay data and the small Stokes shift upon comparison of the emission spectra with the corresponding excitation spectra (see ESI,† Fig. S26–32), the emission of R and the co-crystals R:A–R:C could be assigned to fluorescence. The red-shifted luminescence in the case of pure R was related to the stacking of molecular units within the crystalline phase and is typically assigned to aggregate-based emission. Usually, this is accompanied by severe quenching based on the higher probability of non-radiative relaxation for lower energetic transitions. On the other hand, if radiative decay remains the dominant mechanism, the respective decay rate should decrease with the decreasing emission energy due to the lower photon density of states at higher energies.
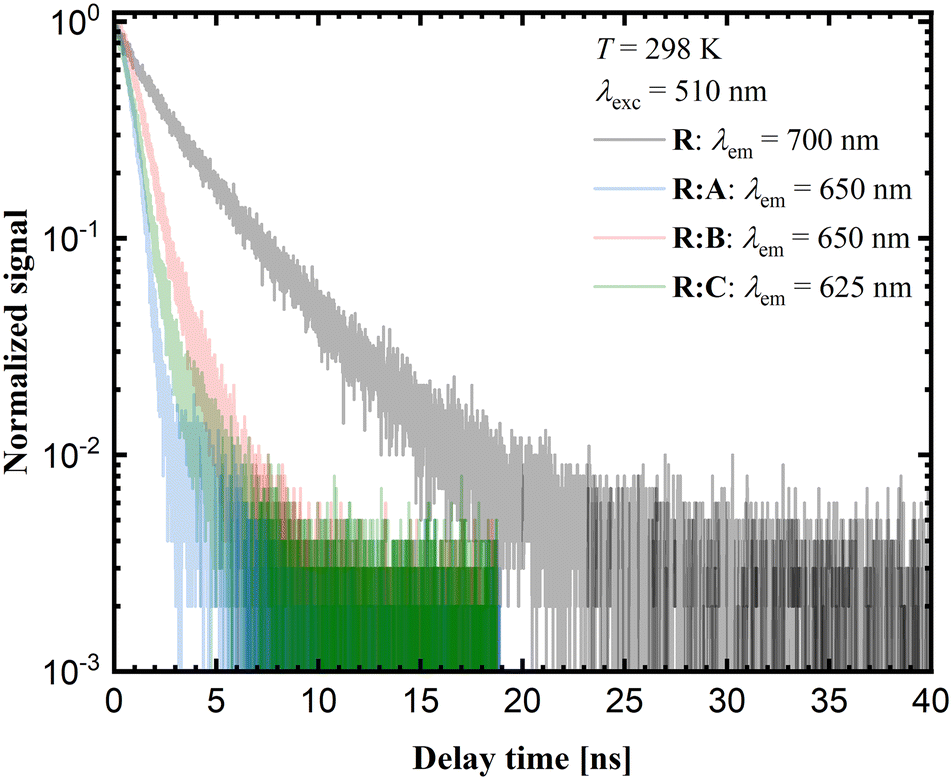 | ||
| Fig. 9 Experimental solid-state decay curves of R, R:A, R:B, and R:C, acquired at room temperature. | ||
The observed trend in the time-resolved luminescence data indicates that aggregation-induced quenching was clearly of minor relevance, suggesting that the quantum yield of solid R at room temperature cannot be too small.
In order to verify whether the correlation between the aggregation of molecules in the solid and a red-shifted luminescence is given, the absorption and emission spectra of R in dilute solutions were also measured. Since R generally exhibits a very poor solubility in a number of solvents and the respective co-crystals are not soluble in apolar solvents, the optical properties were investigated in chloroform, acetonitrile, and methanol. Indeed, the emission bands of R in solution were even additionally blue-shifted towards the green range, clearly suggesting a trend of the degree of supramolecular bonding within the crystalline solid with the red-shift in the luminescence of R. Variations in the solvent polarity did, however, only slightly affect the luminescence, excluding a significant charge-transfer-type nature of the radiatively emitting state (Fig. S20–S22 in the ESI†).
The absorption maxima are all in the range of 483–496 nm depending on the system and solvent, which indicated that also the excited state cannot involve a significant charge-transfer-type nature.
No clear fluorescence dependence from the polarity could be observed either. Within the co-crystals, incremental shifts were observed from R:A over R:B to R:C in methanol and acetonitrile. This trend was, however, not consistent for chloroform. Here, R:C exhibited the strongest Stokes shift of 56 nm (2093 cm−1). In general, the relatively small Stokes shifts in all systems suggest an excitation from the ground state into the locally exited (LE) state, with no significant relaxation indicating a limited degree of reorganization in the excited state. This is in good agreement with the theoretical calculations reported previously.39 The charge-transfer processes described by Thiel et al., however, would imply a remarkable reorganization of the charge density and cause a large Stokes shift as well as a rearrangement of the molecular geometry, which was neither observed in solution nor in the solid state.
Based on the experimental results, we propose a mechanism by which the emission bands of R and co-crystals correlate with the degree of the crystal packing alteration. In the pure solid roseolumiflavin, the hydrogen-bonded amide–amide dimeric motifs and π⋯π stacking allow close-packing, which results in strongly red-shifted emission bands based on an aggregation-induced emission. In the solvent environment, this aggregation is inhibited. In solution, both pure R and the dissolved co-crystallized compounds showed luminescence with similar wavelengths strongly blue-shifted from the solid R. Upon co-crystallization and the introduction of each respective co-former into the crystal lattice, the stacking between different flavin molecules was gradually disturbed but not completely inhibited. Common for all the co-crystals hereby is the partial disruption of the dimeric synthon compared to R. Whereas in R:A and R:C, the amide–amide dimers were resolved and replaced by two- and three-point binding motifs with the co-agents, R:B showed just the formation of an additional halogen bond. The last, however, seemed to influence the strength of the hydrogen bonds in the amide–amide dimer. Further, rearranged stacking motifs were noticed in all the multicomponent crystal structures, which resulted in stacking arrangements closer to cofacial patterns, while the pure R crystal structure showed merely π⋯π stacks between Ring 2 and Ring 3. In R:A and R:B nevertheless, the π-stacks stayed intact, resulting in emission band shifts in similar ranges, with R:B showing a smaller shift in accordance with the slightest interference in the interacting modes. R:C, on the other hand, partly modified the π-stacks by incorporating C units in the interaction pattern. Here, the offset π⋯π interactions between flavin and 2,4-diaminopyrimidine entities led to significantly shortened flavin–flavin stacks. Thus, the crystal packing of R experienced the strongest distortion in R:C, yielding the largest blue-shift of all the solid, co-crystalline systems. In solution, the distortion and inhibition of stacking interaction was then extreme, leading to the strongest blue-shift to green luminescence compared to the solid compounds.
Thus, upon comparison with the solvent-based luminescence measurements, we arrived at the conclusion that it is not the nature of the co-former or particular intermolecular interaction itself that leads to changes in the luminescence of the co-crystals compared to the pure solid roseolumiflavin, but rather their effect on the crystal packing; in particular, the distortion of the flavin–flavin hydrogen-bonded motifs and π⋯π stacking caused by the intermolecular interactions involved. The new environments provided by the respective interaction partners caused a disruption of the close packing of R molecules that was translated in their spectroscopic properties in a classic sense of a structure–property relationship.
Conclusions
In this work, we presented the structural analysis of roseolumiflavin (R). X-ray analyses on the obtained single crystals helped identify the strong hydrogen-bonded amide–amide synthons in R as well as π⋯π stacking interactions as the driving factors for the molecular self-assembly processes and allowing the construction of the crystal packing. Based on the rational design with specifically selected co-formers, three new co-crystals were synthesized driven by hydrogen and halogen bond interactions. Impressively, the co-crystals showed different interaction patterns (carboxylic acid–amide synthon, three-point binding motifs, halogen bonding) which promises great accessibility for R in the aspect of crystal engineering and may serve as grounds for further research in this area on flavins in general. It was shown that the structural versatility also influences the photophysical properties. While the solid R showed near-infrared luminescence, the new co-crystals R:A, R:B, and R:C showed blue-shifted emission in the orange-red range. In solution, the dissolved molecules showed green fluorescence. This implies an aggregation-induced red-shift of the luminescence, which, however, could not be severely quenched given the observed elongation of the decay rates with decreasing emission energy. The negligible degree of solvatochromism in both the luminescence and absorption spectra indicated that both the excited and ground state were rather molecularly localized states without a significant degree of charge transfer and formation of a strong electric dipole moment, in agreement with previous theoretical predictions. Overall, our findings indicate a way of tailoring the optical properties of flavins with the aid of crystal engineering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Roseolumiflavin synthesis and SC-XRD analyses were funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 396890929/GRK 2482 and 440366605. We thank the CeMSA@HHU (Center for Molecular and Structural Analytics @ Heinrich Heine University) for recording the NMR- and MS-spectroscopic data. We further thank Tobias Heinen for valuable discussions and the research group of Prof. Peter Gilch for the possibility to measure the emission spectra in solution. The authors are grateful to Prof. Matthias Karg (HHU Düsseldorf) for the access to the FLS980 spectrometer and to PD Klaus Schaper and Prof. Thomas Müller (HHU Düsseldorf) for the possibility to use the pulsed EPL-375, EPL-450, and EPL-510 laser diodes from Edinburgh Instruments for the time-resolved luminescence measurements of the solid compounds.Notes and references
- G. R. Desiraju, Chemistry beyond the molecule, Nature, 2001, 412, 397–400 CrossRef CAS PubMed.
- D. Braga, F. Grepioni, L. Maini and M. Polito, in Molecular Networks. Structure and Bonding, ed. M. W. Hosseini and D. Braga, Springer-Verlag, Berlin, Heidelberg, 2009, pp. 25–50 Search PubMed.
- C. W. Tang and S. A. VanSlyke, Organic electroluminescent diodes, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Electroluminescence in conjugated polymers, Nature, 1999, 397, 121–128 CrossRef CAS.
- A. J. C. Kuehne and M. C. Gather, Organic Lasers: Recent Developments on Materials, Device Geometries, and Fabrication Techniques, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- J. Gierschner, S. Varghese and S. Y. Park, Organic Single Crystal Lasers: A Materials View, Adv. Opt. Mater., 2016, 4, 348–364 CrossRef CAS.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, New sensing mechanisms for design of fluorescent chemosensors emerging in recent years, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- M. K. Bera, P. Pal and S. Malik, Solid-state emissive organic chromophores: design, strategy and building blocks, J. Mater. Chem. C, 2020, 8, 788–802 RSC.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, A cocrystal strategy to tune the luminescent properties of stilbene-type organic solid-state materials, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- S. Li, Y. Lin and D. Yan, Two-component molecular cocrystals of 9-acetylanthracene with highly tunable one-/two-photon fluorescence and aggregation induced emission, J. Mater. Chem. C, 2016, 4, 2527–2534 RSC.
- Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Piezochromic Luminescence of Donor-Acceptor Cocrystals: Distinct Responses to Anisotropic Grinding and Isotropic Compression, Angew. Chem., 2018, 130, 15896–15900 CrossRef.
- Y. Liu, A. Li, S. Xu, W. Xu, Y. Liu, W. Tian and B. Xu, Reversible Luminescent Switching in an Organic Cocrystal: Multi-Stimuli-Induced Crystal-to-Crystal Phase Transformation, Angew. Chem., Int. Ed., 2020, 59(35), 15098–15103 CrossRef CAS PubMed.
- J. Vainauskas, F. Topić, O. S. Bushuyev, C. J. Barrett and T. Friščić, Halogen bonding to the azulene π-system: cocrystal design of pleochroism, Chem. Commun., 2020, 56, 15145–15148 RSC.
- T. Feiler, B. Bhattacharya, A. A. L. Michalchuk, V. Schröder, E. List-Kratochvil and F. Emmerling, Mechanochemical Syntheses of Isostructural Luminescent Cocrystals of 9-Anthracenecarboxylic Acid with two Dipyridines Coformers, Crystals, 2020, 10, 889 CrossRef CAS.
- S. O. Mansoorabadi, C. J. Thibodeaux and H. Liu, The diverse roles of flavin coenzymes--nature's most versatile thespians, J. Org. Chem., 2007, 72, 6329–6342 CrossRef CAS PubMed.
- H. Grajek, Review - Flavins as photoreceptors of blue light and their spectroscopic properties, Curr. Top. Biophys., 2011, 34, 53–65 CAS.
- V. Massey, The Chemical and Biological Versatility of Riboflavin, Biochem. Soc. Trans., 2000, 28, 283–296 CrossRef CAS PubMed.
- J. Galbán, I. Sanz-Vicente, J. Navarro and S. de Marcos, The intrinsic fluorescence of FAD and its application in analytical chemistry: a review, Methods Appl. Fluoresc., 2016, 4, 42005 CrossRef PubMed.
- E. Sikorska, I. V. Khmelinskii, D. R. Worrall, J. Koput and M. Sikorski, Spectroscopy and photophysics of iso- and alloxazines: experimental and theoretical study, J. Fluoresc., 2004, 14, 57–64 CrossRef CAS PubMed.
- M. Sikorski, E. Sikorska, F. Wilkinson and R. P. Steer, Studies of the photophysics and spectroscopy of alloxazine and related compounds in solution and in the solid state, Can. J. Chem., 1999, 77, 472–480 CrossRef CAS.
- S. Salzmann and C. M. Marian, The photophysics of alloxazine: a quantum chemical investigation in vacuum and solution, Photochem. Photobiol. Sci., 2009, 8, 1655–1666 CrossRef CAS PubMed.
- A. Reiffers, C. Torres Ziegenbein, A. Engelhardt, R. Kühnemuth, P. Gilch and C. Czekelius, Impact of Mono-Fluorination on the Photophysics of the Flavin Chromophore, Photochem. Photobiol., 2018, 94, 667–676 CrossRef CAS PubMed.
- Principles of fluorescence spectroscopy (3rd ed), ed. J. R. Lakowicz, Springer, New York, Berlin, 3rd edn, 2006 Search PubMed.
- E. Horiguchi, S. Matsumoto, K. Funabiki and M. Matsui, Optical Properties of Novel 2,3-Dicyano-5-methyl-6 H -1,4-diazepine Dyes in the Solid State, Bull. Chem. Soc. Jpn., 2005, 78, 1167–1173 CrossRef CAS.
- C.-L. Chiang, M.-F. Wu, D.-C. Dai, Y.-S. Wen, J.-K. Wang and C.-T. Chen, Red-Emitting Fluorenes as Efficient Emitting Hosts for Non-Doped, Organic Red-Light-Emitting Diodes, Adv. Funct. Mater., 2005, 15, 231–238 CrossRef CAS.
- Y. Ooyama, T. Nakamura and K. Yoshida, Heterocyclic quinol-type fluorophores. Synthesis of novel imidazoanthraquinol derivatives and their photophysical properties in benzene and in the crystalline state, New J. Chem., 2005, 29, 447 RSC.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, New software for searching the Cambridge Structural Database and visualizing crystal structures (CCDC, ConQuest Version 2020.3.0), 2002, 58, 389–397 Search PubMed.
- M. Á. Farrán, R. M. Claramunt, C. López, E. Pinilla, M. R. Torres and J. Elguero, Structural characterization of alloxazine and substituted isoalloxazines: NMR and X-ray crystallography, ARKIVOC, 2006, 2007(4), 20–38 Search PubMed.
- M. Wang and C. J. Fritchie, Geometry of the unperturbed flavin nucleus. The crystal structure of 10-methylisoalloxazine, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 2040–2045 CrossRef CAS.
- R. Miura, K. Matsui, K. Hirotsu, A. Shimada, M. Takatsu and S. Otani, X-Ray crystallographic determination of a derivative of a new flavin compound, roseoflavin, J. Chem. Soc., Chem. Commun., 1973, 703 RSC.
- S. T. Caldwell, L. J. Farrugia, S. G. Hewage, N. Kryvokhyzha, V. M. Rotello and G. Cooke, Model systems for flavoenzyme activity: an investigation of the role functionality attached to the C(7) position of the flavin unit has on redox and molecular recognition properties, Chem. Commun., 2009, 1350–1352 RSC.
- R. B. Bates, T. C. Sneath and D. N. Stephens, Crystal structure of 10-methylisoalloxazinium bromide dihydrate, J. Org. Chem., 1970, 35, 1625–1627 CrossRef CAS.
- J. S. Sherfinski, A. J. deArmendi and C. J. Fritchie Junior, American Crystallographic Association Abstracts, Papers (Summer), 1974, vol. 238 Search PubMed.
- C. J. Fritchie Jr., G. D. Sproul and T. D. Wade, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1972, 28, S48 Search PubMed.
- F. E. Scarbrough, H.-S. Shieh and D. Voet, The X-ray crystal structure of the molecular complex bis(lumiflavin–2,6-diamino-9-ethylpurine)–ethanol–water, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 2512–2523 CrossRef.
- B. L. Trus, J. L. Wells, R. M. Johnston, C. J. Fritchie Jr. and R. E. Marsh, American Crystallographic Association Abstracts, Papers (Winter), 1970, vol. 36 Search PubMed.
- M. C. Kuo, J. B. R. Dunn and C. J. Fritchie, The crystal structure of a flavin molecular complex: 10-propylisoalloxazine–bis(naphthalene-2,3-diol), Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1766–1771 CrossRef.
- D. Voet and A. Rich, The crystal and molecular structure of an intermolecular complex between riboflavin and an adenosine derivative, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1151–1156 CrossRef CAS PubMed.
- B. Karasulu and W. Thiel, Photoinduced intramolecular charge transfer in an electronically modified flavin derivative: roseoflavin, J. Phys. Chem. B, 2015, 119, 928–943 CrossRef CAS PubMed.
- A. Nangia and G. R. Desiraju, Supramolecular Synthons and Pattern Recognition, Design of Organic Solids, Top. Curr. Chem., 1998, 198, 57–95 CrossRef CAS.
- G. R. Desiraju, J. J. Vittal and A. Ramanan, Crystal engineering, A textbook, World Scientific, Singapore, 2011 Search PubMed.
- S. Saha and G. R. Desiraju, Acid···Amide Supramolecular Synthon in Cocrystals: From Spectroscopic Detection to Property Engineering, J. Am. Chem. Soc., 2018, 140, 6361–6373 CrossRef CAS PubMed.
- S. Kasai, R. Miura and K. Matsui, Chemical Structure and Some Properties of Roseoflavin, Bull. Chem. Soc. Jpn., 1975, 48, 2877–2880 CrossRef CAS.
- C. Tantardini, S. G. Arkhipov, K. A. Cherkashina, A. S. Kil'met'ev and E. V. Boldyreva, Crystal structure of a 2:1 co-crystal of meloxicam with acetyl-endi-carb-oxy-lic acid, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 1856–1859 CrossRef CAS PubMed.
- CrysAlisPRO, Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England Search PubMed.
- APEX2, Bruker AX Inc., Madison, 2012 Search PubMed.
- SAINT, Bruker Analytical X-Ray Systems, Madison, 2018 Search PubMed.
- G. M. Sheldrick, SHELXT - integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- J. Lübben, C. M. Wandtke, C. B. Hübschle, M. Ruf, G. M. Sheldrick and B. Dittrich, Aspherical scattering factors for SHELXL - model, implementation and application, Acta Crystallogr., Sect. A: Found. Adv., 2019, 75, 50–62 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: from visualization to analysis, design and prediction, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- OriginPro, OriginLab Corporation, Northampton, MA, USA, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CIFs, preparation of R, IR, 1H-NMR, 13C-NMR and HRMS spectra, solid-state time-resolved luminescence data, solid-state emission and excitation spectra, solution emission and absorption spectra, thermal (DSC and TG) data. CCDC 2168404–2168407. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00589a |
| This journal is © The Royal Society of Chemistry 2022 |