
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the origin of chaotrope-modulated electrocatalytic activity of cytochrome c at electrified aqueous|organic interfaces†
Alonso
Gamero-Quijano
 *ab,
Pierre-André
Cazade
ac,
Shayon
Bhattacharya
ac,
Sarah
Walsh
b,
Grégoire
Herzog
d,
Damien
Thompson
*ac and
Micheál D.
Scanlon
*abe
*ab,
Pierre-André
Cazade
ac,
Shayon
Bhattacharya
ac,
Sarah
Walsh
b,
Grégoire
Herzog
d,
Damien
Thompson
*ac and
Micheál D.
Scanlon
*abe
aThe Bernal Institute, University of Limerick (UL), Limerick V94 T9PX, Ireland. E-mail: daniel.gamero@ua.es
bDepartment of Chemical Sciences, School of Natural Sciences, University of Limerick (UL), Limerick V94 T9PX, Ireland. E-mail: micheal.scanlon@ul.ie
cDepartment of Physics, School of Natural Sciences, University of Limerick (UL), Limerick V94 T9PX, Ireland. E-mail: damien.thompson@ul.ie
dLaboratoire de Chimie Physique et Microbiologie pour les Matériaux et l’Environnement, Université de Lorraine, CNRS, LCPME, F-54000 Nancy, France
eAdvanced Materials & Bioengineering Research (AMBER) Centre, Dublin, Ireland
First published on 26th January 2022
Abstract
Electrochemical, spectroscopic and computational methods are used to demonstrate that electrified aqueous|organic interfaces are a suitable bio-mimetic platform to study and contrast the accelerated electrocatalytic activity of cytochrome c towards the production of reactive oxygen species (ROS) in the presence of denaturing agents such as guanidinium chloride and urea.
The presence of high levels of ROS in the cells of patients with chronic disease is well known but poorly understood.1–3 This is due to a lack of model platforms to detect and evaluate how subtle changes in the cytoplasmic environment affect protein performance in vivo.4 Methodologies capable of diagnosing how these changes trigger or inhibiting a cascade of reactions occurring within cells will inform new strategies to counteract the proliferation of diseases such as breast cancer,5 acute myeloid leukaemia,6 and neurodegeneration.7
Recently, using cytochrome c (Cyt c) as a model protein, we demonstrated the capability of electrified aqueous|organic interfaces to replicate the molecular machinery of the inner mitochondrial membrane at the onset of apoptosis.8 Precise electrochemical control of Cyt c adsorption at an immiscible aqueous|organic interface formed between water and α,α,α-trifluorotoluene (TFT) activates Cyt c enzymatic production of ROS. Positive biasing of the water|TFT interface, by externally applying an interfacial Galvani potential difference 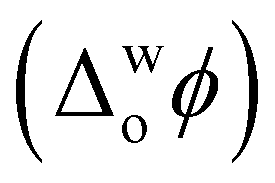 > +0.1 V, triggers ROS production in the presence of an organic electron donor, decamethylferrocene (DcMFc), whereas negative biasing (
> +0.1 V, triggers ROS production in the presence of an organic electron donor, decamethylferrocene (DcMFc), whereas negative biasing (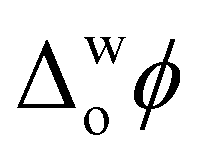 ≈ −0.2 V) does not. External polarisation regulates the electrochemical performance of the protein by modulating the binding of molecules to Cyt c at the interface, in particular, electrostatic and hydrophobic interactions between Cyt c and tetrakis(pentafluorophenyl)borate (TB−) organic electrolyte anions.8 At positive biasing, these Cyt c/TB− interactions are promoted, leading to (i) a conformational change of the adsorbed Cyt c from its native state that exposes the redox-active heme to small molecules and (ii) a favourable orientation of the heme-pocket perpendicular to the water|TFT interface. The latter facilitates interfacial electron transfer (IET) between partially exposed heme and DcMFc, thereby leading to ROS production. At negative bias, Cyt c/TB− interactions are inhibited. Thus, Cyt c does not present any preferential orientation towards the interface at negative bias and no detectable IET or ROS production occurs.8
≈ −0.2 V) does not. External polarisation regulates the electrochemical performance of the protein by modulating the binding of molecules to Cyt c at the interface, in particular, electrostatic and hydrophobic interactions between Cyt c and tetrakis(pentafluorophenyl)borate (TB−) organic electrolyte anions.8 At positive biasing, these Cyt c/TB− interactions are promoted, leading to (i) a conformational change of the adsorbed Cyt c from its native state that exposes the redox-active heme to small molecules and (ii) a favourable orientation of the heme-pocket perpendicular to the water|TFT interface. The latter facilitates interfacial electron transfer (IET) between partially exposed heme and DcMFc, thereby leading to ROS production. At negative bias, Cyt c/TB− interactions are inhibited. Thus, Cyt c does not present any preferential orientation towards the interface at negative bias and no detectable IET or ROS production occurs.8
Here, using cyclic voltammetry (CV) we demonstrate that electrocatalytic activity of Cyt c towards production of ROS at an electrified water|organic interface is significantly boosted by adding denaturing agents, such as the chaotropes guanidinium chloride (GdmCl) or urea, to the aqueous phase. Using differential capacitance measurements, UV/vis and Raman spectroscopy, and molecular dynamics (MD) simulations, we reveal the atomic-scale mechanisms by which GdmCl and urea promote electrocatalysis. All electrochemical studies were performed using a four-electrode electrochemical cell under ambient, aerobic conditions. The electrochemical cell configuration used is outlined in Scheme 1 and all electrochemical measurements were calibrated to the Galvani potential scale (for details see Fig. S1, ESI†).
 | ||
| Scheme 1 Schematic of the four-electrode electrochemical cell used for all electrochemical measurements. For blank experiments x was 0 μM, and with Cyt c in solution x was 10 μM. For experiments with the chaotropes guanidinium chloride (GdmCl) and urea, y varied from 0 to 12 M. The organic electrolyte salt was bis(triphenylphosphoranylidene)ammonium tetrakis(pentafluorophenyl)borate (BATB). All experiments were carried out under aerobic conditions. | ||
The influence of introducing the chaotropes GdmCl and urea to the aqueous phase on the electrocatalytic activity of Cyt c towards the production of ROS was revealed by CV measurements (Fig. 1a). The onset Galvani potential for IET  shifted negatively in the presence of both denaturing agents, in particular for GdmCl, with significant positive currents measured at low potentials (Fig. 1a). These positive currents are attributed to IET from organic DcMFc to aqueous O2, electrocatalysed by Cyt c.8 The negative currents at the negative end of the potential window are attributed to ion transfer of decamethylferrocenium cations (DcMFc+),9,10 generated during ROS production, from the TFT to aqueous phase. Progressive negative shifts of
shifted negatively in the presence of both denaturing agents, in particular for GdmCl, with significant positive currents measured at low potentials (Fig. 1a). These positive currents are attributed to IET from organic DcMFc to aqueous O2, electrocatalysed by Cyt c.8 The negative currents at the negative end of the potential window are attributed to ion transfer of decamethylferrocenium cations (DcMFc+),9,10 generated during ROS production, from the TFT to aqueous phase. Progressive negative shifts of 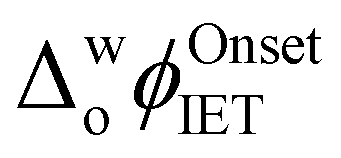 up to 250 mV were observed upon increasing the aqueous GdmCl concentration from 0 to 8 M (Fig. S2a–d and S3a, ESI†). By comparison, increasing the aqueous urea concentration from 0 to 8 M led to a small negative shift of
up to 250 mV were observed upon increasing the aqueous GdmCl concentration from 0 to 8 M (Fig. S2a–d and S3a, ESI†). By comparison, increasing the aqueous urea concentration from 0 to 8 M led to a small negative shift of 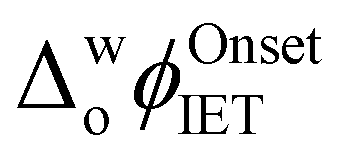 by ca. 40 mV, that was independent of the aqueous urea concentration in this range (Fig. S2e–g and S3b, ESI†). Even with a 12 M aqueous urea concentration,
by ca. 40 mV, that was independent of the aqueous urea concentration in this range (Fig. S2e–g and S3b, ESI†). Even with a 12 M aqueous urea concentration, 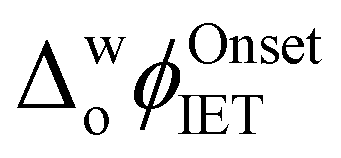 shifted only ca. 70 mV (Fig. S2h and S3b, ESI†), far less than for lower GdmCl concentrations. Comparative analysis of the charge transferred in the CVs in Fig. 1a and Fig. S3, ESI,† in the absence and presence of increasing GdmCl or urea concentrations, clearly demonstrates that ROS production dramatically increases in the presence of GdmCl compared with urea (Fig. S4, ESI†). The positive currents attributed to IET disappeared in control CVs with Cyt c in the presence of increasing concentrations of GdmCl and urea in the aqueous phase, but without DcMFc in the organic phase (Fig. S5 and S6, ESI†).
shifted only ca. 70 mV (Fig. S2h and S3b, ESI†), far less than for lower GdmCl concentrations. Comparative analysis of the charge transferred in the CVs in Fig. 1a and Fig. S3, ESI,† in the absence and presence of increasing GdmCl or urea concentrations, clearly demonstrates that ROS production dramatically increases in the presence of GdmCl compared with urea (Fig. S4, ESI†). The positive currents attributed to IET disappeared in control CVs with Cyt c in the presence of increasing concentrations of GdmCl and urea in the aqueous phase, but without DcMFc in the organic phase (Fig. S5 and S6, ESI†).
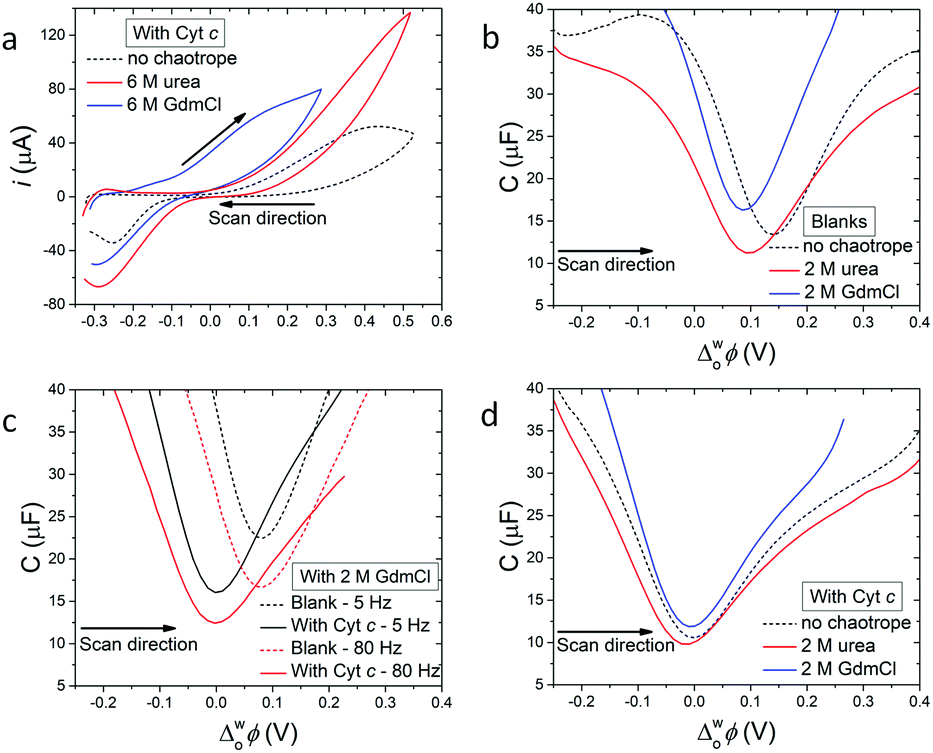 | ||
| Fig. 1 Electrochemistry of cytochrome c (Cyt c) at an electrified aqueous|α,α,α-trifluorotoluene (water|TFT) interface in the presence of the organic electron donor decamethylferrocene (DcMFc). (a) Cyclic voltammetry (CV) in the presence of Cyt c, with and without urea or GdmCl. Scan rate was 20 mV s−1. (b) Differential capacitance measurements (C/μF) in the absence of Cyt c, with and without urea or GdmCl. (c) Influence of applied AC frequency (5 vs. 80 Hz) on differential capacitance measurements in the presence of 2 M GdmCl, with and without Cyt c. (d) Differential capacitance measurements in the presence of Cyt c, with and without urea or GdmCl. All capacitances were measured at 5 Hz, unless stated otherwise. The four-electrode electrochemical cell used for all measurements is described in Scheme 1. | ||
Differential capacitance measurements probed the charge distribution at the water|TFT interface in the presence of 2 M GdmCl or urea in the aqueous phase, without (Fig. 1b and c) and with (Fig. 1d) Cyt c. For experiments without Cyt c, the differential capacitance increased in the presence of GdmCl, but decreased in the presence of urea, compared to a blank measurement without a chaotrope (Fig. 1b). The differential capacitance changed with sampling frequency (5 vs. 80 Hz) in the presence of GdmCl (Fig. 1c), but not urea (Fig. S7a, ESI†).
An increase of differential capacitance indicates that the interfacial thickness (or width of the mixed solvent region) increases in the presence of GdmCl but decreases with urea. This correlates with the dependence of the differential capacitance on sampling frequency for GdmCl (Fig. 1c), consistent with Gdm+ cations penetrating the water|TFT interface, with part of the capacitive increase attributed to faradaic processes involving Gdm+ ion transfer.11 Such ion partitioning, due to saturation of the aqueous phase with GdmCl, leads to Gdm+ acting as a co-solvent, increasing the miscibility of interfacial water and TFT. In agreement with these experimental observations, MD simulations vide infra reveal expediated interfacial water|TFT mixing in the presence of GdmCl, due to Gdm+ enhancing the hydrophobicity of the interface. Furthermore, MD simulations demonstrate that high interfacial concentrations of urea impede access of TFT to the water phase, leading to a decreased interfacial thickness.
The minimum capacitance, defined as the potential of zero charge (PZC), is sensitive to adsorption of species from both the aqueous and organic phase;12–14e.g., a negative shift implies adsorption of positive species at the interface. The PZC shifted negatively in the presence of both GdmCl and urea, compared to a measurement without a chaotrope (Fig. 1b). While such shifts were expected for adsorption of Gdm+ cations, their origin is less clear for adsorption of neutral urea molecules. One hypothesis is that urea species display a cationic behaviour, with well-defined dipole moments oriented towards the interface, when an external bias is applied. Indeed, this cationic behaviour of neutral urea molecules, leading to a negative shift of the PZC, has previously been observed at electrified water|1,2-dichloroethane interfaces.15
Upon addition of Cyt c to the aqueous phase, the PZC shifted negatively, as expected since Cyt c has a net charge of +9 in its oxidised form at pH 7 (Fig. 1d and Fig. S7b–d, ESI†).16 Also, near-identical PZC values centred at  ≈ 0 V were observed, irrespective of the presence or nature of a chaotropic agent (Fig. 1d). Increasing the concentrations of urea and GdmCl to 4 and 6 M produced the same trends in PZC observed using 2 M concentrations (Fig. S8, ESI†). Thus, when Cyt c adsorbs at the water|TFT interface in the absence or presence of a chaotrope, the resulting interfacial charge distributions are similar at
≈ 0 V were observed, irrespective of the presence or nature of a chaotropic agent (Fig. 1d). Increasing the concentrations of urea and GdmCl to 4 and 6 M produced the same trends in PZC observed using 2 M concentrations (Fig. S8, ESI†). Thus, when Cyt c adsorbs at the water|TFT interface in the absence or presence of a chaotrope, the resulting interfacial charge distributions are similar at 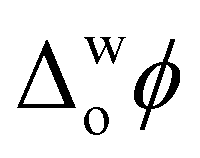 ≈ 0 V and nearby potentials (−0.1 to +0.1 V).
≈ 0 V and nearby potentials (−0.1 to +0.1 V).
A key question then is why does GdmCl promote the electrocatalytic activity of Cyt c more than urea, despite near-identical interfacial ionic environments being experienced by adsorbed Cyt c? Differential capacitance measurements probe the ionic environment at the electrified water|TFT interface as a whole, and are thus relatively insensitive to any subtle conformational changes of an adsorbed protein. In contrast, CV probes IET, dependent on local, molecular-level factors that increase the probability of simultaneous interactions of the heme active site with DcMFc and O2, especially subtle conformational changes that expose the heme active site or orientate the heme towards the interface. Hence, despite Cyt c adsorption occurring at similar potential ranges in the presence of GdmCl or urea, the electrocatalytic activity of Cyt c with GdmCl is correlated with conformational changes specific to that chaotrope. Thus, spectroscopic experiments in bulk aqueous phases and MD simulations at the water|TFT interface were employed to investigate the precise conformational changes experienced by Cyt c at the interface in the presence of GdmCl or urea and their links to the observed electrocatalytic activity.
Changes of the UV/vis absorbance spectra of aqueous Cyt c solutions in the presence of various concentrations (y M, Scheme 1) of urea or GdmCl (Fig. S9–S11, ESI†) were consistent with conformational changes leading to a moderate exposure of the heme pocket in the presence of urea, but major exposure in the presence of GdmCl. Raman spectroscopy confirmed that within the 2 to 8 M range, Cyt c passes through more conformational changes in the presence of GdmCl than urea, with larger frequency upshifts of Cyt c core size markers observed with GdmCl (Fig. S12, ESI†). The frequency shifts (e.g., ν15, ν13, ν21, ν4, ν11, etc.) are mainly seen for in-plane skeletal modes due to a combination of core-expansion and extensive Fe–porphyrin backbonding in Cyt c,17,18 with details given in Table S1, ESI.†
To further probe Cyt c conformational shifts and quantify exposure of the heme pocket in the presence of chaotropes (2 M aqueous solutions of urea and GdmCl), we used interface models (with the experimental ion distributions estimated based on the differential capacitance measurements with Cyt c present at the water|TFT interface at 0.0 V as detailed in Fig. S13, ESI†) to perform multiple, 0.5 μs long MD simulations (see Fig. 2) of the electrochemical cell (for details see Section S3, ESI†). Experimentally, in the presence of GdmCl but not urea, the bond between Met80 and the heme is broken.17 This is accounted for in the present simulations.
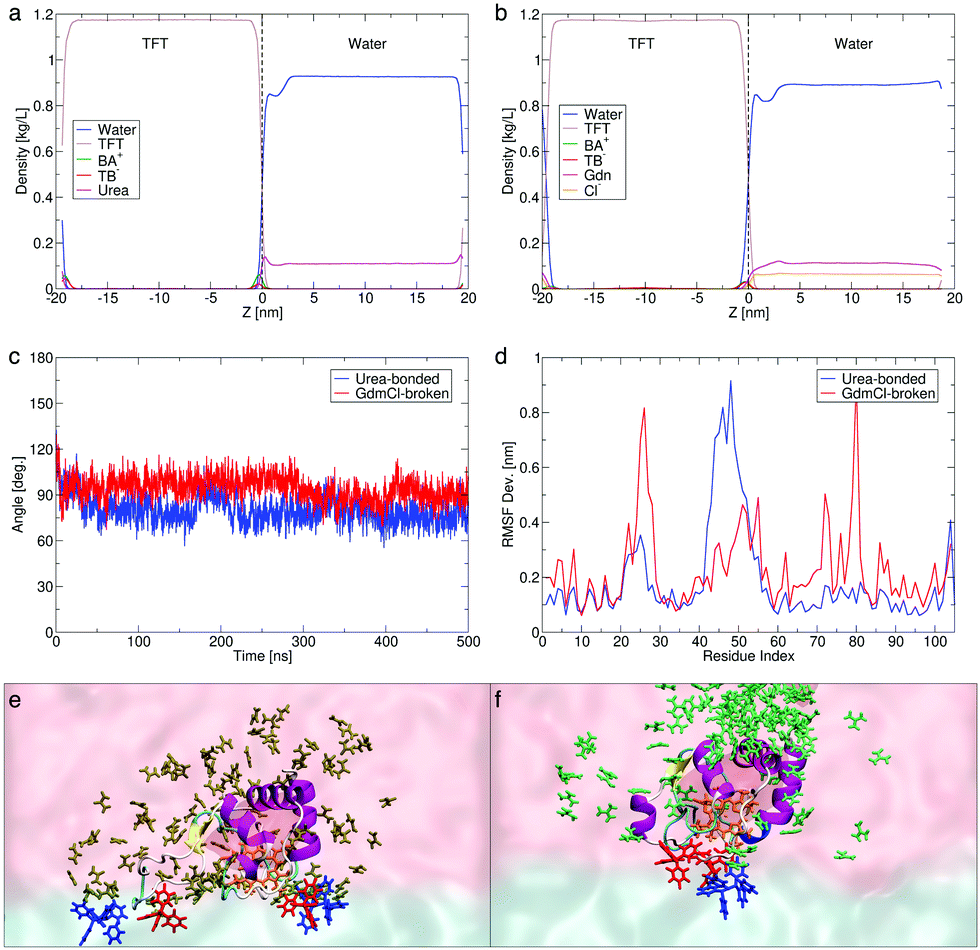 | ||
| Fig. 2 Computed density profiles across the water|TFT interface of solvents, molecular and ionic species in the presence of chaotropes (a) urea and (b) guanidinium chloride (Gdm+Cl−). (c) Simulation timelines showing near-normal orientations sampled between the heme plane and the water|TFT interface in the presence of both denaturing agents. (d) Root mean square fluctuations (RMSF) showing the flexibility of the Cyt c amino acid residues. Representative snapshots showing the orientation of the Cyt c heme active site (orange sticks, with the full protein shown in cartoon representation) in the presence of (e) urea (brown) and (f) Gdm+ (green). The BA+ and TB− ions from the organic phase are shown as blue and red sticks. Only chaotrope molecules within 1.2 nm of Cyt c, and BA+ and TB− ions within 0.3 nm of Cyt c are shown. | ||
The computed density profiles of solvents and ions reveal a dip in the water density corresponding to the position of Cyt c in the water phase, which is closer to the interface and less prominent in the presence of urea (Fig. 2a) than in the presence of GdmCl (Fig. 2b and Fig. S19a, ESI†). This is coupled with a larger urea than Gdm+ population at the interface, the latter being more distributed in the water phase with a slightly higher density near Cyt c. The observation of this dip in water density for both chaotropes, coupled with the observation of IET by CV in the presence of both chaotropes (Fig. 1a), clearly indicates that the concentration of chaotropes employed does not produce a physical screening effect between Cyt c and the organic side of the interface.
We note a predicted thicker interface in the presence of GdmCl (∼2 nm) vs. urea (∼1 nm), despite the lower local concentration of Gdm+ at the interface (Fig. 2b). Gdm+ at the interface expedites interfacial water|TFT mixing, enhancing the hydrophobicity of the interface and leading to a decrease in the computed dielectric constant (εr) from 40 in the presence of urea to 30 in the presence of GdmCl (Fig. S18a, ESI†), compared with 52 in the absence of chaotropes. By contrast, the high concentration of urea at the interface may partly limit interfacial water|TFT mixing (see Section S3.4, ESI†). These predictions are in agreement with the experimental observations that the differential capacitance is slightly increased in the presence of Gdm+ species and slightly decreased in the presence of urea (Fig. 1b). In the presence of both chaotropes, the heme active site is exposed and docked to the interface with the heme plane oriented perpendicular to the interfacial plane throughout the 0.5 μs dynamics (Fig. 2c).
The short-lived, sporadic nature of the interactions between TB− and Cyt c at the interface (Fig. S15 and S19; see details in Section S3.4, ESI†) mean that the ordering of Cyt c could not be solely attributed to the local interfacial concentration of TB− (Fig. 2a and b). To further understand the different mechanisms of pre-organisation of the Cyt c heme pocket for IET and its increased exposure to the water|TFT interface in the presence of urea and GdmCl, we calculated the root mean square fluctuations (RMSF Dev.), or flexibilities, of the protein residues (Fig. 2d). The RMSF data reveals that in the presence of urea with the Met80–heme bond intact, a mobile loop spanning residue positions 40–57 in Cyt c flips (blue line in Fig. 2d, also see Fig. S14b, ESI†) to create a more open structure that increases the accessibility of the heme pocket at the interface (see Movie S1, ESI†). The dynamicity of this so-called “Ω loop” is known to regulate unfolding of Cyt c.19,20 On the other hand, in the presence of GdmCl with the Met80–heme bond broken, residue ranges 20–30 (belonging to loop-A)21 and 76–82 (belonging to loop-D)22 undergo structural rearrangements (red line in Fig. 2d and Fig. S14b, ESI†) and open up the Cyt c structure as a first step to expose the heme pocket when the Cyt c Met80–heme bond is broken (see details in Section S3.4, ESI†), while the Ω loop remains more closed than in urea (Fig. 2d). By these two distinct chaotropic effects on different parts of Cyt c, GdmCl and urea promote exposure of the heme pocket to facilitate its near-normal orientation at the interface (see Fig. 2e and f, and also Cyt c–TB− interactions in Fig. S15, ESI†). Further analyses of control simulations are provided in Fig. S19 and S20 (ESI†).
In summary, upon adsorption at an aqueous|organic interface, Cyt c experiences equivalent ionic environments but undergoes distinct conformational shifts and rearrangements (by entirely different mechanisms) in the presence of molar concentrations of the aqueous chaotropes urea and GdmCl. We find that ROS production (by IET from DcMFc to O2 electrocatalysed by adsorbed Cyt c) is promoted more by GdmCl than urea because the presence of GdmCl leads to physicochemical changes to the water|TFT interface and conformational changes to the adsorbed Cyt c, which work in tandem to increase the probability of DcMFc and O2 reaching the heme active site. Specifically, Gdm+ cations penetrate the interface, creating a more hydrophobic environment that increases the interfacial concentration of DcMFc, leading to enhanced IET. Meanwhile, urea partially impedes access of TFT to the water phase, which may retard IET. Most importantly, the distinct Cyt c unfolding mechanism with GdmCl leads to a more open heme pocket structure than when urea is added.
Our data shows that the partial unfolding of Cyt c and its conformational changes occurring at aqueous|organic interfaces can be tracked and understood even in the presence of low concentrations of denaturing agents. The use of 2 M of chaotropes is sufficient to produce Cyt c intermediates that exhibit outstanding electrocatalytic behaviours at the interface and could lead to development of new strategies to inhibit ROS production. We have demonstrated that aqueous|organic interfaces are suitable and tuneable platforms to carry out bioelectrochemistry and direct electron transfer reactions with proteins in the presence of spectator and co-factor organic molecules, a further step toward full cytoplasmimetics.
MDS acknowledges Science Foundation Ireland (SFI) under Grant 13/SIRG/2137 and the European Research Council through a Starting Grant (716792). AG-Q acknowledges an Irish Research Council (IRC) Government of Ireland Postdoctoral Fellowship (GOIPD/2018/252). DT acknowledges support under SFI awards number 15/CDA/3491 and 12/RC/2275_P2.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Perillo, M. Di Donato, A. Pezone, E. Di Zazzo, P. Giovannelli, G. Galasso, G. Castoria and A. Migliaccio, Exp. Mol. Med., 2020, 52, 192–203 CrossRef CAS PubMed.
- Y. M. Lee, W. He and Y. C. Liou, Cell Death Dis., 2021, 12 Search PubMed.
- J. Zhang, D. Duan, Z. L. Song, T. Liu, Y. Hou and J. Fang, Med. Res. Rev., 2021, 41, 342–394 CrossRef CAS PubMed.
- P. E. Porporato, N. Filigheddu, J. M. B. S. Pedro, G. Kroemer and L. Galluzzi, Cell Res., 2018, 28, 265–280 CrossRef CAS PubMed.
- V. Raimondi, F. Ciccarese and V. Ciminale, Br. J. Cancer, 2020, 122, 168–181 CrossRef PubMed.
- S. Wu, M. Akhtari and H. Alachkar, Sci. Rep., 2018, 8, 1–9 Search PubMed.
- J. V. Leonard and A. H. V. Schapira, Lancet, 2000, 355, 389–394 CrossRef CAS.
- A. Gamero-Quijano, S. Bhattacharya, P.-A. Cazade, A. F. Molina-Osorio, C. Beecher, A. Djeghader, T. Soulimane, M. Dossot, D. Thompson, G. Herzog and M. D. Scanlon, Sci. Adv., 2021, 7, eabg4119 CrossRef CAS PubMed.
- P. Ge, M. D. Scanlon, P. Peljo, X. Bian, H. Vubrel, A. O’neill, J. N. Coleman, M. Cantoni, X. Hu, K. Kontturi, B. H. Liu and H. H. Girault, Chem. Commun., 2012, 48, 6484–6486 RSC.
- X. Bian, M. D. Scanlon, S. Wang, L. Liao, Y. Tang, B. Liu and H. H. Girault, Chem. Sci., 2013, 4, 3432–3441 RSC.
- M. F. Suárez-Herrera and M. D. Scanlon, Anal. Chem., 2020, 92, 10521–10530 CrossRef PubMed.
- C. Yufei, V. J. Cunnane, D. J. Schiffrin, L. Murtomäki and K. Kontturi, J. Chem. Soc., Faraday Trans., 1991, 87, 107–114 RSC.
- D. Momotenko, C. M. Pereira and H. H. Girault, Phys. Chem. Chem. Phys., 2012, 14, 11268–11272 RSC.
- M. F. Suárez-Herrera and M. D. Scanlon, Electrochim. Acta, 2019, 328, 135110 CrossRef.
- P. Peljo, E. Vladimirova, E. Smirnov, G. Gschwend, L. Rivier and H. H. Girault, J. Phys. Chem. C, 2018, 122, 18510–18519 CrossRef CAS.
- L. C. Petersen and R. P. Cox, Biochem. J., 1980, 192, 687–693 CrossRef CAS PubMed.
- Y. Sun, V. Karunakaran and P. M. Champion, J. Phys. Chem. B, 2013, 117, 9615–9625 CrossRef CAS PubMed.
- M. Fedurco, J. Augustynski, C. Indiani, G. Smulevich, M. Antalík, M. Bánó, E. Sedlák, M. C. Glascock and J. H. Dawson, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1703, 31–41 CrossRef CAS PubMed.
- M. M. G. Krishna, Y. Lin, J. N. Rumbley and S. W. Englander, J. Mol. Biol., 2003, 331, 29–36 CrossRef CAS PubMed.
- A. I. Karsisiotis, O. M. Deacon, M. T. Wilson, C. Macdonald, T. M. A. Blumenschein, G. R. Moore and J. A. R. Worrall, Sci. Rep., 2016, 6, 30447 CrossRef CAS PubMed.
- M. E. P. Murphy, G. D. Brayer, J. S. Fetrow and R. E. Burton, Protein Sci., 1993, 2, 1429–1440 CrossRef CAS PubMed.
- P. Mulligan-Pullyblankt, J. S. Spitzer, B. M. Gilden and J. S. Fetrow, J. Biol. Chem., 1996, 271, 8633–8645 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of methods, supporting data and analyses. See DOI: 10.1039/d1cc05293d |
| This journal is © The Royal Society of Chemistry 2022 |