
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethyltetrazine as a small live-cell compatible bioorthogonal handle for imaging enzyme activities in situ†
Diana
Torres-García
,
Merel A. T.
van de Plassche
,
Emma
van Boven
,
Tyrza
van Leeuwen
,
Mirjam G. J.
Groenewold
,
Alexi J. C.
Sarris
,
Luuk
Klein
,
Herman S.
Overkleeft
 * and
Sander I.
van Kasteren
*
* and
Sander I.
van Kasteren
*
Leiden Institute of Chemistry and The Institute for Chemical Immunology, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: s.i.van.kasteren@chem.leidenuniv.nl; h.s.overkleeft@lic.leidenuniv.nl
First published on 13th September 2022
Abstract
Bioorthogonal chemistry combines well with activity-based protein profiling, as it allows for the introduction of detection tags without significantly influencing the physiochemical and biological functions of the probe. In this work, we introduced methyltetrazinylalanine (MeTz-Ala), a close mimic of phenylalanine, into a dipeptide fluoromethylketone cysteine protease inhibitor. Following covalent and irreversible inhibition, the tetrazine allows vizualisation of the captured cathepsin activity by means of inverse electron demand Diels Alder ligation in cell lysates and live cells, demonstrating that tetrazines can be used as live cell compatible, minimal bioorthogonal tags in activity-based protein profiling.
Introduction
Activity-based protein profiling (ABPP) allows the identification and visualization of enzyme activities within complex biological systems.1 ABPP has been particularly useful in the study of proteases and other hydrolases, which are often expressed in an inactive form, also called zymogens, and have their activity tightly regulated by post-translational processes.2 Approaches geared at measuring expression levels, like Western blots, are therefore of limited use in studying their activity. ABPP employs mechanism-based inhibitors, called activity-based probes (ABPs), which are able to react with only the active form of proteases thanks to their electrophilic warhead, yielding a covalent and irreversible enzyme-inhibitor adduct. This allows the distinction between active and inactive enzyme species. Additionally, ABPs contain a chemical moiety, often a fluorophore or a biotin, that allows for detection of the probe-protease complex, often referred to as a detection tag.Fluorescent detection is by far the most frequently used technique in ABPP thanks to its rapid readout in gel screening experiments and its compatibility with localisation studies in both cell and animal studies.3,4 However, incorporating fluorophores directly into an ABP comes with some drawbacks. As fluorophores are often large and hydrophobic moieties, directly incorporating them into an ABP is likely to affect target binding and will impact the physiochemical properties of the ABP.5,6 To circumvent this issue, one can incorporate small, bioorthogonal handles into ABPs.5,7,8 Bioorthogonal tags are selected on their reactivity, chemoselectivity, bioorthogonality, and their size, which is ideally as small as possible.9 Alkynes and azides are frequently employed due to their small size, and well established protocols for Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) in cell lysates.5,10 However, due to the cytotoxicity of copper, this approach is ultimately unsuitable for experiments on live cells. To circumvent the need for a copper catalyst, strain-promoted azide-alkyne cycloadditions (SPAACs) have been developed.11 However, these strained alkynes typically suffer from high background labeling, as they are able to react with free thiols present in the cell.12,13 Alternatively, azides can also participate in the live cell compatible Staudinger–Bertozzi ligation,14 although it typically suffers from poor reaction kinetics and hydrolysis of the azide by the phosphine reagent.15–17
The inverse electron demand Diels–Alder (IEDDA) reaction has in recent years come to the fore as an attractive alternative to the aforementioned bioorthogonal reactions.18–20 IEDDA comprises a reaction between an electron-poor diene, typically a tetrazine, and a strained, electron-rich dienophile. It has attracted a lot of attention in recent years due to its excellent biocompatibility and fast reaction kinetics.20 IEDDA-compatible dienophiles like transcyclooctene (TCO)21 and vinylboronic acid22 have been successfully employed in ABPP. However, one major drawback of this approach is still the size of the participating reactive groups, and the influence they could have on the properties of the probe. For example, an expoxomicin-derived ABP bearing norbornene was shown to be less potent proteasome inhibitor than its azide counterpart.23
To overcome this problem and in line with the triazine amino acids reported by Webb and co-workers,24 we envisaged that incorporating a small, tetrazine containing amino acid into a model inhibitor could mimic the structural and spatial properties of phenylalanine. Here we explore this strategy and incorporate such an amino acid, namely L-methyltetrazinyl-alanine, into a small peptidyl inhibitor, and used a two-step labelling protocol to show that this amino acid can act as both a phenylalanine mimic and as a bioorthogonal label, allowing for a two-step labelling protocol in both cell lysates and live cells.
Results and discussion
While tetrazine-containing amino acids have been incorporated in both peptides25 and proteins,26–28 these amino acids either have a tetrazine attached to para-position of a phenyl-ring, or to the ε-amine of the lysine side chain. This makes these literature Tz-amino acids relatively bulky. Additionally, the incompatibility of tetrazines under standard Fmoc deprotection conditions limits the use of tetrazines in Fmoc solid phase peptide chemistry. However, tetrazines are stable under peptide coupling conditions, as well as acidic resin cleavage, allowing late stage introduction of the tetrazine on resin.25,27 Our aim was to generate an ABP containing a tetrazine amino acid as closely isosteric to naturally occurring aromatic amino acids as possible. We therefore chose to substitute the phenyl ring of phenylalanine analogue for a methyltetrazine.29 Although a monosubstituted tetrazine would be preferred sterically, these are known to be relatively unstable in biological systems.30For the synthesis of MeTz-Ala (2, Scheme 1A), we started from commercially available Cbz-protected asparagine. The side chain-amide was dehydrated to nitrile 1 using N,N′-dicyclohexylcarbodiimide (DCC). This was followed by tetrazine formation using a 3-mercaptopropionic acid-catalyzed one-step reaction with hydrazine hydrate and acetonitrile, followed by oxidation with sodium nitrite under acidic conditions.31 This yielded Cbz-protected MeTz-Ala (2) (Scheme 1A) in 13% yield.
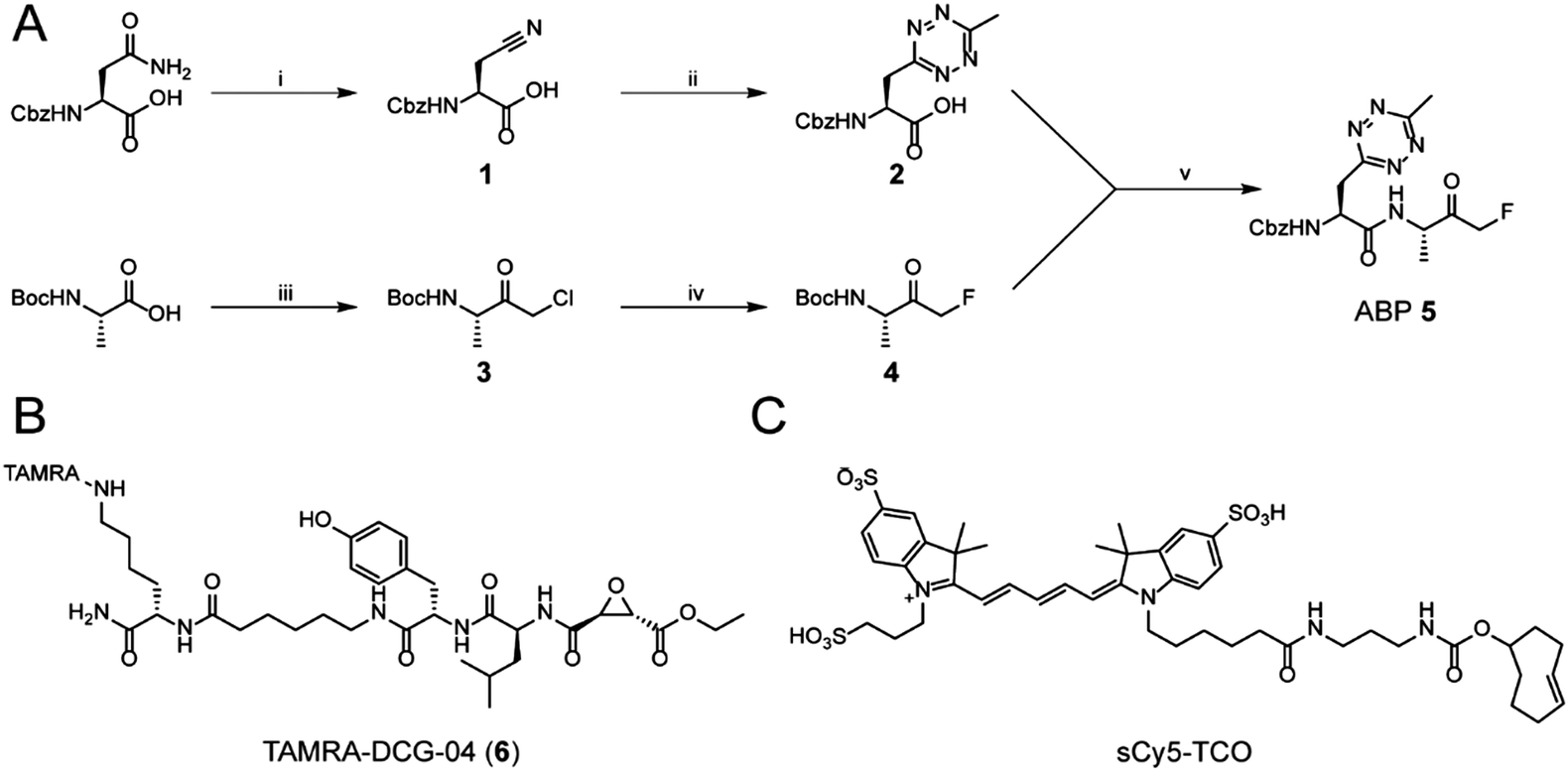 | ||
| Scheme 1 The synthesis of the methyltetrazine amino acid and its incorporation in an ABP. (i) DCC, pyridine/acetone (1/1). (ii) (1) 3-Mercaptopropionic acid, hydrazine hydrate, acetonitrile. (2) NaNO2 in AcOH/DCM (1/1). (iii) (1) Isobutylchloroformate, N-methyl morpholine. (2) Etheral diazomethane. (3) HBr/AcOH. (iv) TBAF, p-toluenesulfonic acid, THF. (v) (1) 2 M HCl/dioxane. (2) PyBoP, DIPEA, DMF. (B) The chemical structure of TAMRA-DCG-04. (C) The chemical structure of sCy5-TCO. | ||
To assess whether the methyltetrazine amino acid could function as both a bioorthogonal label and a phenylalanine isostere, we selected Z-FA-FMK, a well-characterized pan-cathepsin inhibitor, as our basis for ABP development.32 Since the phenylalanine, the alanine and the N-terminal Cbz group all play an important role in protease binding, this small dipeptidyl inhibitor has limited space to attach a handle for detection without significantly influencing its labeling affinities.33 This made it an ideal model to test the biological scope of the methyltetrazine as a bioorthogonal phenylalanine isostere. To this end, we designed probe 5, where the phenyl ring of phenylalanine in Z-FA-FMK is replaced with a methyltetrazine.
To synthesize probe 5, we started from Boc-protected alanine. The free carboxylic acid was transformed into the bromomethylketone using a two-step protocol, in which the starting material was first reacted with diazomethane to form the diazoketone, and subsequently treated with HBr/AcOH to form a bromomethylketone. The resulting intermediate 3 was then converted into the fluoromethylketone using TBAF and p-toluenesulfonic acid.34 To form final probe 5, we deprotected the N-terminal Boc group of 4, and coupled it to 2 using PyBOP/DIPEA as coupling reagents (Scheme 1B).
Z-FA-FMK was originally designed as a cathepsin B inhibitor.33 However, it was found to be a potent inhibitor of multiple cysteine proteases, including cathepsins B, L and S,33 as well as caspases 2, 3, 6 and 7.35 To compare probe 5 and Z-FA-FMK in terms of cathepsin inhibition potencies, we performed a competition experiment with the known pan-cathepsin ABP TAMRA-DCG-04 (6) (Scheme 1B).36 We selected Jurkat T-cells as a model cell line, as the activity of Z-FA-FMK towards cathepsin B has been well-characterized in this cell line.37,38 As cathepsins require a mildly acidic pH to remain active, Jurkat T-cells were lysed in a sodium acetate buffer (pH 5.5). These lysates were incubated with different concentrations of either probe 5 or Z-FA-FMK. After the first incubation, the lysates were subsequently incubated with 6 to fluorescently label the remaining cathepsin activities (Fig. 1). Differences in the potency of probe 5 compared with Z-FA-FMK were quantified by the decrease in labeling intensity yielded the apparent half-maximal inhibitory concentrations (IC50s) (Fig. S2, ESI†), showing that Z-FA-FMK is the more potent inhibitor and, both probes appear to target the same cathepsins (Fig. 1A).
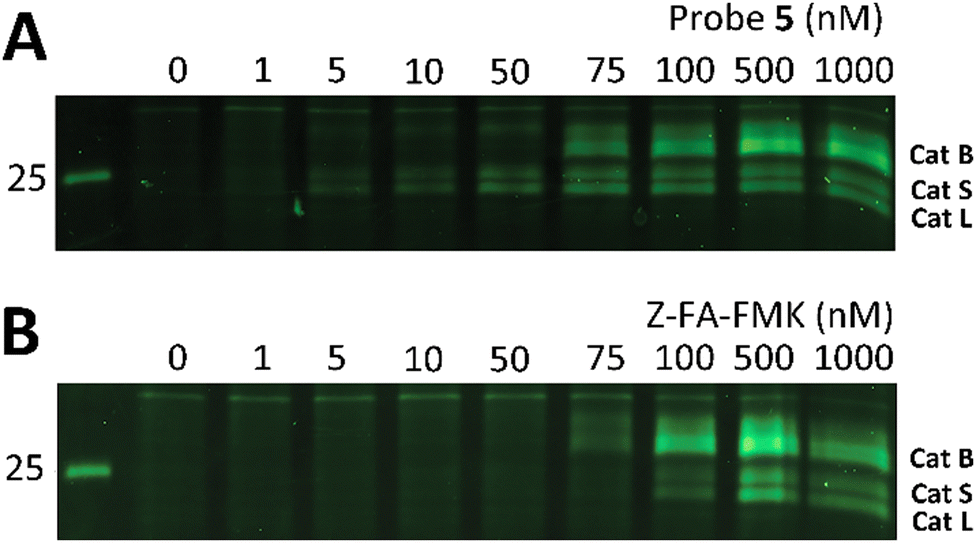 | ||
| Fig. 1 Labeling of cysteine cathepsins by either probe 5 (A) or Z-FA-FMK (B) in Jurkat T-cell lysates. The residual cathepsin activity was labeled using TAMRA-DCG-04 (green, Cy3 settings). The unprocessed gels are shown in Fig. S1 (ESI†). | ||
The competition experiment underscores the reported38 finding that, although Z-FA-FMK was originally designed as a cathepsins B inhibitor, it is also a potent inhibitor of other cysteine cathepsins and caspases. Based on molecular weight, the competed bands by probe 5 correspond to cysteine cathepsins B, S, and L (top to bottom),39 which was verified by the individual inhibition of those cathepsins in RAW 264.7 cells, and the later residual activity labeling with probe 5 and s-Cy5-TCO (Fig. 2).
 | ||
| Fig. 2 Competitive labeling of Cathepsin B (A), S (B), and L (C). Live RAW 264.7 cells were incubated for 2.5 h with the indicated concentration of specific inhibitors for each cathepsin. Afterward, the cells were lysed and residual cathepsin activity was labeled with probe 5 (10 μM) and s-Cy5-TCO (2 μM). The gels were imaged in two channels, green (Cy3) and red (Cy5). The unprocessed gels are shown in Fig. S3 (ESI†). | ||
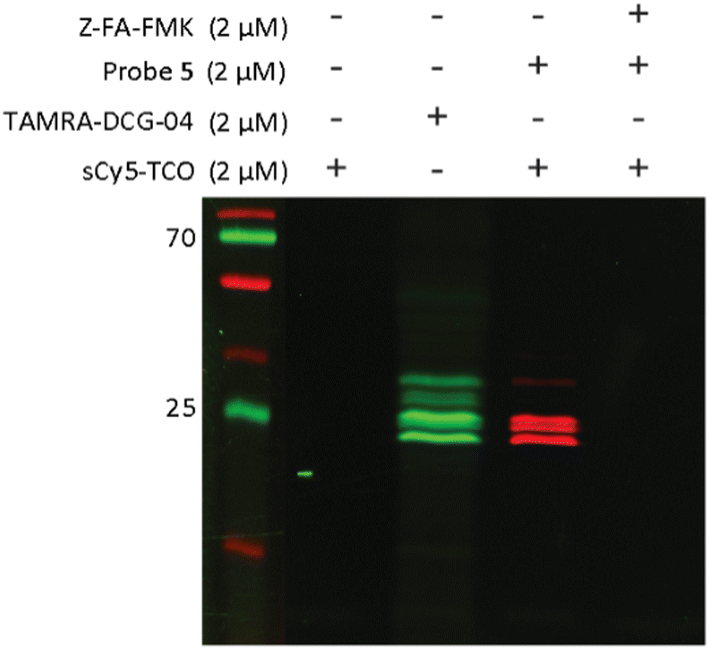
After confirming that probe 5 is able to inhibit cathepsins with a specificity similar to that of its parent compound, we set out to establish the efficiency of IEDDA bioorthogonal labeling of its targets. To this end, we incubated the Jurkat cell lysate with different concentrations of probe 5, and subsequently with the fluorophore sCy5-TCO (Scheme 1C), which can covalently react with the methyltetrazine to form a fluorescent probe–protease complex. Fluorescence imaging confirmed that probe 5 is a bona fide two-step ABPP and that treatment with probe 5-inhibited cathepsins in cell extracts with sCy5-TCO yields a labeling pattern similar to that obtained with 6 as the positive control (Fig. 3). Competition with Z-FA-FMK showed that both our two-step ABP and the one-step ABP 6, in all likelihood have the same targets.
 | ||
| Fig. 3 Competition labeling of cysteine cathepsins by probe 5 in Jurkat lysates. The gel was imaged using Cy3 (green)/Cy5 (red) settings. The unprocessed gels are shown in Fig. S4 (ESI†). | ||
Encouraged by these results, we decided to establish whether probe 5 could be used to label cathepsins in live cells. To assess potential toxicity of the probe, we compared the cytocompatibility of probe 5 to that of the parent inhibitor Z-FA-FMK using an tetrazolium (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-reduction assay.40 Both probe 5 and Z-FA-FMK showed some toxicity after 24 hours, (86 ± 0.15% and 67 ± 0.13% respectively, Fig. S5, ESI†).
To explore whether probe 5 is able to label cathepsins in live cells, we treated live Jurkat cells with probe 5 for 2 hours. As sCy5-TCO is unlikely to cross the cell membrane due to its negatively charged sulfate groups,41 we first lysed the cells and incubated the lysates with sCy5-TCO. Fluorescence imaging again showed clear bands at the same molecular weight as the DCG-04 bands, confirming that probe 5 is indeed able to cross the cell membrane and label multiple cysteine cathepsins in live cells (Fig. 4).
 | ||
| Fig. 4 Labeling of cysteine cathepsins by probe 5 in live Jurkat T-cells. Live cells were incubated with 2 μM of probe 5 for 2 hours. After incubation with probe 5, the cells were lysed and lysates were incubated with 2 μM of sCy5-TCO for 30 minutes. Lanes were rearranged for clarity, the unprocessed gel is shown Fig. S6 (ESI†). | ||
One of the largest advantages of the tetrazine-TCO ligation over the classical copper(I) promoted click reaction is that this ligation can be performed in live cells. We therefore next assessed whether both the protease labeling as well as the IEDDA could be performed in live cells, and could potentially be used to not only visualize, but also localize cathepsins activity in live cells using microscopy. As sCy5-TCO is not cell permeable, we switched to the membrane-permeant TCO fluorophore, CF©500-TCO for these experiments.42 To confirm that this TCO fluorophore is able to react with probe 5 in live cells, Jurkat cells were incubated with probe 5 and subsequently incubated with CF©500-TCO. In-gel detection showed clear bands at the correct molecular weights, proving that both the cathepsin labeling and IEDDA can take place inside live cells (Fig. S7, ESI†). However, we anticipated that the small cytosol of Jurkat cells would complicate the localization of cathepsin activity. We therefore switched to bone-marrow derived dendritic cells (BMDCs) for microscopy, as these cells have a significantly larger cytosol and well-described cathepsin activity.43,44
To assess whether probe 5 could be used to localize cathepsin activity, we incubated mature BMDCs with probe 5 for two hours, while co-stimulating with lipopolysaccharide (LPS) to increase cathepsin activity.43,45 After incubation with probe 5 and extensive washing, we incubated the BMDCs with CF©500-TCO for an additional 2 hours. Afterward, cells were fixed and washed to reduce the background signal of unreacted CF©500-TCO. In order to localize lysosomal compartments within the imaged cells, we co-stained with an anti-lysosome-associated membrane protein-1 (LAMP-1, CD107a) antibody, which did require fixation of the cells post-IEDDA. Fig. 5 shows a clear difference in the CF©500-TCO signal between the control (bottom panels) and the signal detected when adding the probe 5 (upper panels). In addition, CF©500-TCO localized partly to LAMP-1 positive vesicles (0.5 Mander's coefficient, Fig. S8, ESI†). The partial colocalization of LAMP-1 and CF©500-TCO observed in Fig. 5 is in correspondence with previous colocalization studies of cathepsin activity and LAMP-1.46,47 This confirms that probe 5 can indeed be used to visualize cathepsin activity in live cells. In addition, we confirm the colocalization with cathepsin B, which has 0.476 Mander's coefficient (Fig. S9, ESI†).
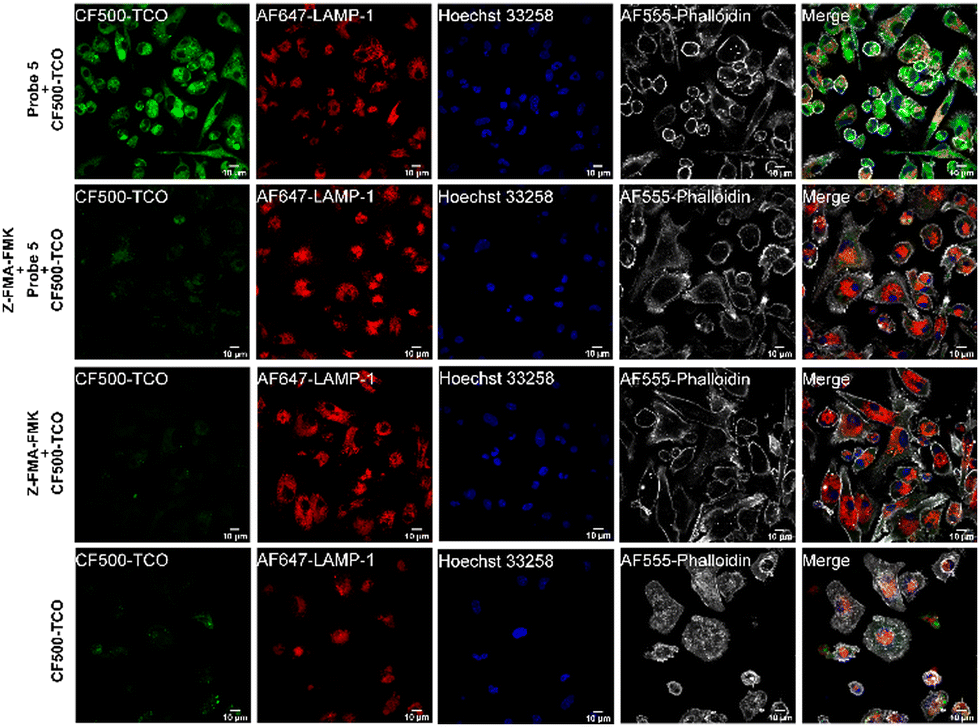 | ||
| Fig. 5 Imaging of probe 5 and CF©500-TCO ligation by confocal microscopy. BMDCs were incubated for 2 h with probe 5 and CF©500-TCO or CF©500-TCO. After uptake, BMDCs were fixed with 2% PFA and processed for immunoflurescence staining with LAMP-1 as a lysosomal marker (red). The nucleus was stained with Hoechst 33258 (blue) and actin was stained using Phalloidin AF555 (gray). Scale bar is 10 μm (white bar, right corner). | ||
As seen in Fig. 5, the fluorescence intensity of CF©500-TCO (upper panel) is higher compared with the experimental groups. To confirm this, we evaluated the cellular uptake of CF©500-TCO by cytometry (Fig. 6). In this regard, the mean fluorescence intensity (MFI) of cells incubated with Probe 5 and CF©500-TCO (Fig. 6B, purple) is higher compared with other experimental groups (p < 0.05). This result suggest that the ligation between the probe 5 and CF©500-TCO is taking place inside the cells and the difference in the fluorescence intensity relies upon this ligation and it is not a mere effect of the uptake of CF©500-TCO.
 | ||
| Fig. 6 Cellular uptake of probe 5 and ligation with CF©500-TCO. RAW 264.7 cells were incubated for 2 h with FA-FMK and/or probe 5. After uptake, RAW 264.7 cells were incubated with CF©500-TCO. (A) Flow cytometry histograms showing intensity of CF©500-TCO uptake. (B) Comparison of uptake efficiency by mean fluorescence intensity (MFI) of CF©500-TCO fluorescence. * p-value < 0.05, MFI in cells incubated with Probe 5 and CF©500-TCO (purple) compared with the rest of the experimental groups. Bars represent the mean and whiskers the SEM. Two independent experiments were performed. | ||
Conclusions
In conclusion, we successfully synthesized MeTz-Ala which was incorporated into a peptidyl inhibitor. It was confirmed that the resultant tetrazine containing probe has a similar cathepsin inhibition pattern as the parent inhibitor, and enabled visualization of the probe-protease conjugates in live cells by IEDDA ligation with TCO-modified fluorophores.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Research Council (CoG 865175) and the Institute of Chemical Immunology.Notes and references
- S. Chakrabarty, J. P. Kahler, M. A. T. van de Plassche, R. Vanhoutte and S. H. L. Verhelst, in Activity-Based Protein Profiling, ed. B. F. Cravatt, K.-L. Hsu and E. Weerapana, Springer International Publishing, Cham, 2019, pp. 253–281 Search PubMed.
- H. Ryšlavá, V. Doubnerová, D. Kavan and O. Vaněk, J. Proteomics, 2013, 92, 80–109 CrossRef PubMed.
- Y. Ben-nun, E. Merquiol, A. Brandis, B. Turk, A. Scherz and G. Blum, Theranostics, 2015, 5, 847–862 CrossRef CAS PubMed.
- I. Abd-Elrahman, H. Kosuge, T. W. Sadan, Y. Ben-Nun, K. Meir, C. Rubinstein, M. Bogyo, M. V. McConnell and G. Blum, PLoS One, 2016, 11, 1–8 CrossRef PubMed.
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS.
- B.-T. Xin, C. Espinal, G. de Bruin, D. V. Filippov, G. A. van der Marel, B. I. Florea and H. S. Overkleeft, ChemBioChem, 2020, 21, 248–255 CrossRef CAS PubMed.
- H. Ovaa, P. F. van Swieten, B. M. Kessler, M. A. Leeuwenburgh, E. Fiebiger, A. M. C. H. van den Nieuwendijk, P. J. Galardy, G. A. van der Marel, H. L. Ploegh and H. S. Overkleeft, Angew. Chem., Int. Ed., 2003, 42, 3626–3629 CrossRef CAS PubMed.
- S. H. L. Verhelst, K. M. Bonger and L. I. Willems, Molecules, 2020, 25(24), 1–21 CrossRef.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. Martell and E. Weerapana, Molecules, 2014, 19, 1378–1393 CrossRef PubMed.
- W. A. van der Linden, N. Li, S. Hoogendoorn, M. Ruben, M. Verdoes, J. Guo, G. J. Boons, G. A. van der Marel, B. I. Florea and H. S. Overkleeft, Bioorg. Med. Chem., 2012, 20, 662–666 CrossRef CAS.
- Y. Yang, X. Yang and S. H. L. Verhelst, Molecules, 2013, 18, 12599–12608 CrossRef CAS PubMed.
- R. van Geel, G. J. Pruijn, F. L. van Delft and W. C. Boelens, Bioconjugate Chem., 2012, 23, 392–398 CrossRef CAS.
- M. Verdoes, B. I. Florea, U. Hillaert, L. I. Willems, W. A. van der Linden, M. Sae-Heng, D. V. Filippov, A. F. Kisselev, G. A. van der Marel and H. S. Overkleeft, ChemBioChem, 2008, 9, 1735–1738 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- M. L. W. J. Smeenk, J. Agramunt and K. M. Bonger, Curr. Opin. Chem. Biol., 2021, 60, 79–88 CrossRef CAS PubMed.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS.
- H. Wu and N. K. Devaraj, Top. Curr. Chem., 2015, 374, 3 CrossRef.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- H. Lebraud, D. J. Wright, C. E. East, F. P. Holding, M. O’Reilly and T. D. Heightman, Mol. BioSyst., 2016, 12, 2867–2874 RSC.
- S. Eising, B.-T. Xin, F. Kleinpenning, J. J. A. Heming, B. I. Florea, H. S. Overkleeft and K. M. Bonger, ChemBioChem, 2018, 19, 1648–1652 CrossRef CAS PubMed.
- L. I. Willems, N. Li, B. I. Florea, M. Ruben, G. A. van der Marel and H. S. Overkleeft, Angew. Chem., Int. Ed., 2012, 51, 4431–4434 CrossRef CAS PubMed.
- K. A. Horner, N. M. Valette and M. E. Webb, Chem. – Eur. J., 2015, 21, 14376–14381 CrossRef CAS.
- B. M. Zeglis, F. Emmetiere, N. Pillarsetty, R. Weissleder, J. S. Lewis and T. Reiner, ChemistryOpen, 2014, 3, 48–53 CrossRef CAS PubMed.
- R. J. Blizzard, T. E. Gibson and R. A. Mehl, Methods in Molecular Biology, in Noncanonical Amino Acids, ed. E. Lemke, Humana Press, New York, 2018, vol. 13, pp. 201–217 Search PubMed.
- Z. Ni, L. Zhou, X. Li, J. Zhang and S. Dong, PLoS One, 2015, 10, 1–9 Search PubMed.
- J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898–2901 CrossRef CAS PubMed.
- J. C. T. Carlson, H. Mikula and R. Weissleder, J. Am. Chem. Soc., 2018, 140, 3603–3612 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS.
- W. Mao, W. Shi, J. Li, D. Su, X. Wang, L. Zhang, L. Pan, X. Wu and H. Wu, Angew. Chem., Int. Ed., 2019, 58, 1106–1109 CrossRef CAS PubMed.
- D. Rasnick, Anal. Biochem., 1985, 149, 461–465 CrossRef CAS.
- N. K. Ahmed, L. A. martin, L. M. Watts, J. Palmer, L. Thornburg, J. Prior and R. E. Esser, Biochem. Pharmacol., 1992, 44, 1201–1207 CrossRef CAS.
- G.-D. Roiban, M. Matache, N. D. Hădade and D. P. Funeriu, Org. Biomol. Chem., 2012, 10, 4516–4523 RSC.
- F. J. Lopez-Hernandez, M. A. Ortiz, Y. Bayon and F. J. Piedrafita, Mol. Cancer Ther., 2003, 2, 255–263 CAS.
- D. Greenbaum, K. F. Medzihradszky, A. Burlingame and M. Bogyo, Chem. Biol., 2000, 7, 569–581 CrossRef CAS PubMed.
- C. P. Lawrence, A. Kadioglu, A.-L. Yang, W. R. Coward and S. C. Chow, J. Immunol., 2006, 177, 3827–3836 CrossRef CAS PubMed.
- T. Rajah and S. C. Chow, PLoS One, 2015, 10, 1–16 CrossRef PubMed.
- L. Ben-Aderet, E. Merquiol, D. Fahham, A. Kumar, E. Reich, Y. Ben-Nun, L. Kandel, A. Haze, M. Liebergall, M. K. Kosińska, J. Steinmeyer, B. Turk, G. Blum and M. Dvir-Ginzberg, Arthritis Res. Ther., 2015, 17, 69 CrossRef PubMed.
- M. B. Hansen, S. E. Nielsen and K. Berg, J. Immunol. Methods, 1989, 119, 203–210 CrossRef CAS.
- M. S. Y. Tan, D. Davison, M. I. Sanchez, B. M. Anderson, S. Howell, A. Snijders, L. E. Edgington-Mitchell and E. Deu, PLoS One, 2020, 15, 1–15 Search PubMed.
- J. Jiang, X. Li, F. Mao, X. Wu and Y. Chen, Anal. Biochem., 2021, 614, 114063 CrossRef CAS PubMed.
- A.-M. Lennon-Duménil, A. H. Bakker, R. Maehr, E. Fiebiger, H. S. Overkleeft, M. Rosemblatt, H. L. Ploegh and C. Lagaudrière-Gesbert, J. Exp. Med., 2002, 196, 529–540 CrossRef PubMed.
- R. Maehr, H. C. Hang, J. D. Mintern, Y.-M. Kim, A. Cuvillier, M. Nishimura, K. Yamada, K. Shirahama-Noda, I. Hara-Nishimura and H. L. Ploegh, J. Immunol., 2005, 174, 7066–7074 CrossRef CAS.
- A. Saric, V. E. B. Hipolito, J. G. Kay, J. Canton, C. N. Antonescu and R. J. Botelho, Mol. Biol. Cell, 2016, 27, 321–333 CrossRef CAS PubMed.
- D. M. van Elsland, E. Bos, W. de Boer, H. S. Overkleeft, A. J. Koster and S. I. van Kasteren, Chem. Sci., 2016, 7, 752–758 RSC.
- X.-T. Cheng, Y.-X. Xie, B. Zhou, N. Huang, T. Farfel-Becker and Z.-H. Sheng, J. Cell Biol., 2018, 217, 3127–3139 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00120a |
| This journal is © The Royal Society of Chemistry 2022 |