
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive assembly of bilingual peptide nucleic acids†
Hector S.
Argueta-Gonzalez
 ,
Colin S.
Swenson
,
George
Song
and
Jennifer M.
Heemstra
*
,
Colin S.
Swenson
,
George
Song
and
Jennifer M.
Heemstra
*
Department of Chemistry, Emory University, 1515 Dickey Dr, Atlanta, Georgia, USA. E-mail: jen.heemstra@emory.edu
First published on 17th June 2022
Abstract
Peptide nucleic acids (PNAs) are high-affinity synthetic nucleic acid analogs capable of hybridization with native nucleic acids. PNAs synthesized having amino acid sidechains installed at the γ-position along the backbone provide a template for a single biopolymer to simultaneously encode nucleic acid and amino acid sequences. Previously, we reported the development of “bilingual” PNAs through the synthesis of an amphiphilic sequence featuring separate blocks of hydrophobic and hydrophilic amino acid functional groups. These PNAs combined the sequence-specific binding activity of nucleic acids with the structural organization properties of peptides. Like other amphiphilic compounds, these γ-PNAs were observed to assemble spontaneously into micelle-like nanostructures in aqueous solutions and disassembly was induced through hybridization to a complementary sequence. Here, we explore whether assembly of these bilingual PNAs is possible by harnessing the nucleic acid code. Specifically, we designed an amphiphile-masking duplex system in which spontaneous amphiphile assembly is prevented through hybridization to a nucleic acid masking sequence. We show that the amphiphile is displaced upon introduction of a releasing sequence complementary to the masking sequence through toehold mediated displacement. Upon release, we observe that the amphiphile proceeds to assemble in a fashion consistent with our previously reported structures. Our approach represents a novel method for controlled stimuli-responsive assembly of PNA-based nanostructures.
Introduction
Nature has evolved two universal “languages” to encode information, function, and structure in biopolymers. The information stored in and communicated through nucleic acid and protein sequences is essential to functional biological systems. Nucleic acid sequences feature the high specificity necessary for accurate transmission of information but are limited by their relatively low diversity of chemical functionality. Conversely, amino acids contain high chemical diversity for structure and function but lack the ability to pass along genetic information.1–3 We hypothesized that integration of these codes into a single biopolymer could overcome the inherent limitations they possess individually, as the resulting “bilingual” polymers would be able to encode information in both amino acid and nucleobase languages, thus generating materials with novel properties for biomedical and nanotechnology applications.Our lab recognized that peptide nucleic acid (PNA) was uniquely suited to generate these bilingual biopolymers. Unlike DNA–peptide conjugates and other hetero-copolymers that contain separated domains, PNA allows for both nucleobases and amino acids to be arrayed along a pseudopeptide backbone, thus integrating nucleic acid and peptide functionality together.3–5 This unnatural structure also provides key functional properties such as strong resistance to hydrolase degradation while retaining Watson–Crick–Franklin base pairing.6, 7 Additionally, solid-phase synthesis allows precise placement of amino acid side chains along the backbone, and modifications at the γ-position can be conveniently incorporated using amino acid starting materials.5, 8 Taking these advantages into account, previous efforts in our lab led to the development of a bilingual γ-PNA system that combined the structure and sequence specific activities of both classes of biomolecules into a single biopolymer. Encoding amphiphilicity through the amino acid sequence led to spontaneous assembly into micellar-like nanostructures, while the nucleic acid sequence directed stimuli-responsive disassembly through sequence-specific hybridization to a complementary DNA or RNA strand.9 While controlling disassembly offers utility for delivery applications, we recognized that potential applications could be broadened by introducing control for assembly as well.10
We recognized that this could be possible by masking the hydrophobic portion of the amphiphile with a nucleic acid strand to prevent assembly, and that if this masking strand was designed to have an unpaired toehold region, then addition of a fully complementary DNA or RNA sequence would lead to release of the amphiphilic PNA and subsequent assembly (Fig. 1).11 Using fluorophore labeled PNA and quencher labeled masking sequence, we were able to monitor hybridization using fluorescence spectroscopy and demonstrate near quantitative release of masking sequences. We also demonstrate the specificity of this system using a scrambled release sequence, which results in minimal displacement. Visualization by transmission electron microscopy (TEM) validated the stimuli-responsive assembly of micelle-sized structures, consistent with our previously reported spontaneously assembling system. This novel system demonstrates the use of toehold mediated displacement for the development of a stimuli-responsive bilingual biopolymer assembly, greatly expanding the potential for use in biomedical and nanotechnology applications.
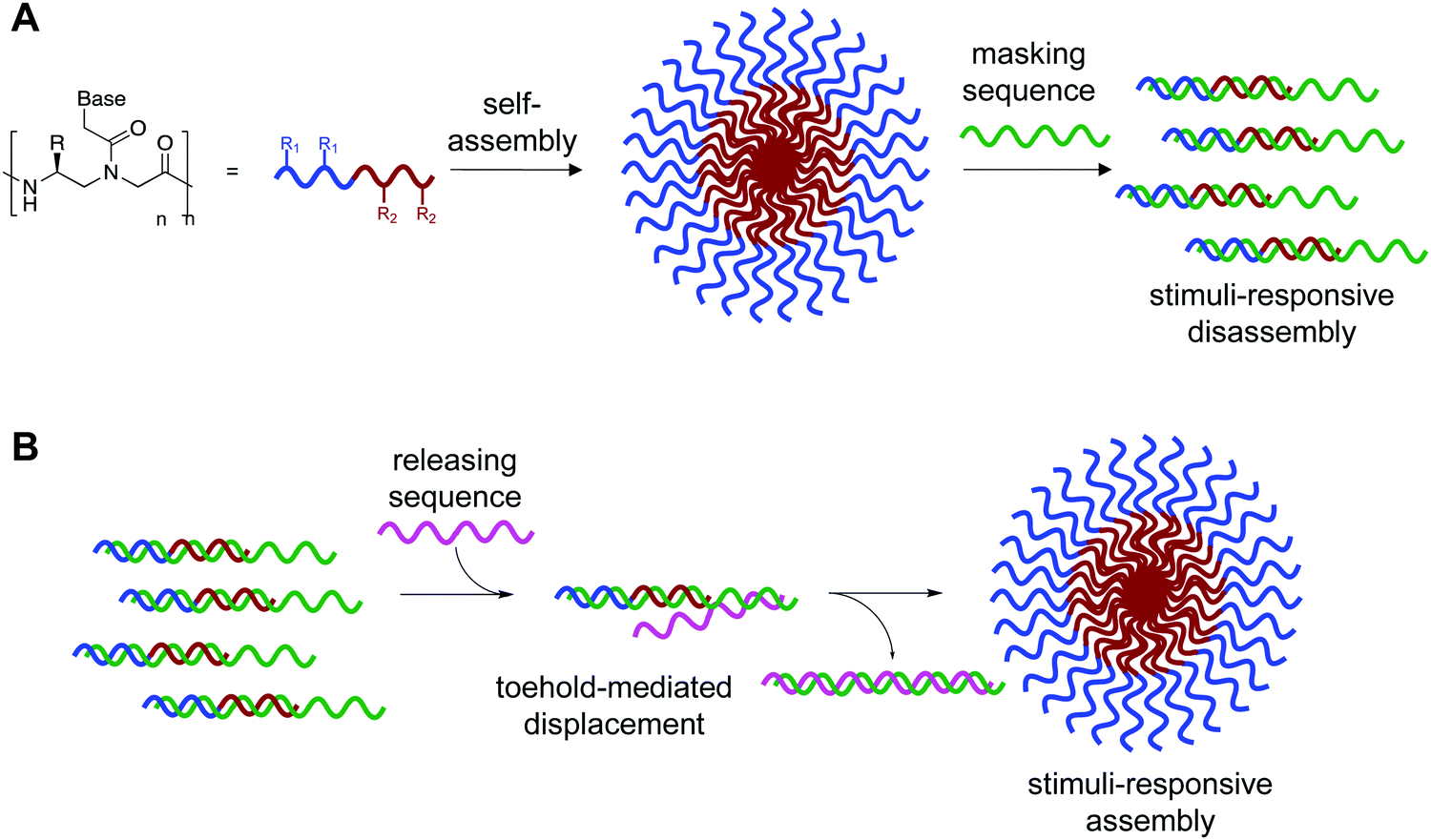 | ||
| Fig. 1 (A) Bilingual PNAs having amphiphilic side chains at the γ-position to direct self-assembly and a nucleotide sequence to direct disassembly through recognition of a complementary masking sequence. The hydrophobic group (R1) drives self-assembly by inducing aggregation, while the hydrophilic group (R2) increases solubility. (B) Stimuli-responsive assembly through toehold-mediated displacement of the masking strand. | ||
Results and discussion
Design and synthesis of amphiphilic PNA
We previously designed and characterized a dodecameric PNA amphiphile (PNA-A1) complementary to a portion of the oncogenic miRNA-21.12 The sequence was modified at the C-terminus to include 4-dimethylamino-naphthalimide (4-DMN), a fluorophore which displays a solvatochromic shift when in a hydrophobic environment, such as the hydrophobic region generated upon micellar assembly.9,13 PNA-A1 displayed spontaneous self-assembly, and the introduction of a complementary DNA or RNA was associated with a disappearance of assemblies attributable to sequence specific hybridization. Based on these results, we recognized the potential to modify the complementary DNA or RNA sequence to include a toehold for subsequent displacement and stimuli responsive PNA assembly. Toehold-mediated displacement systems function by hybridizing an oligonucleotide to a masking sequence having an unpaired toehold region. A fully complementary releasing sequence can then bind to the toehold and displace the target oligonucleotide.14,15 In this context, we recognized the potential to temporarily mask the amphiphilicity of the PNA sequence, and restore this upon displacement, thus driving assembly.In this design, we envisioned a fluorophore-labeled sequence hybridized to a quencher-labeled complementary masking sequence to maximize signal gain upon displacement. Introduction of the releasing sequence would then cause a change in hybridization state, separating the fluorophore-quencher pair and increasing fluorescence signal.15,16 This change in fluorescence signal provides a convenient output to monitor the fraction of PNA amphiphile remaining hybridized to the masking sequence.
In our previous PNA-A1 sequence, the fluorophore was located on the C-terminus such that it would be embedded in the hydrophobic micelle core upon assembly.9 However, for the current system, moving the fluorophore to the N-terminus of the sequence would enable more efficient quenching in the toehold construct. Moreover, because the solvatochromic behavior of the dye was no longer needed, the 4-DMN was replaced with 5-carboxyfluoroscein (FAM), which is commonly used in fluorophore–quencher systems.16, 17 In addition to the desired PNA-A1-FAM sequence, we also synthesized an unmodified control sequence (PNA-C1-FAM) in order to perform initial optimization of the toehold displacement construct (Table 1). Both PNA sequences were synthesized and characterized as previously reported.
| Strand | Sequence |
|---|---|
| “FAM” denotes 5-carboxyfluorescein. “BHQ-1” denotes Black Hole Quencher 1. Subscripts denote the amino acid residues incorporated at the γ-position. | |
| MS-R | 5′UAGCUUAUCAGACUGAUGUUGA/BHQ13′ |
| MS-D | 5′TAGCTTATCAGACTGATGTTGA/BHQ13′ |
| MS-D-m1 | 5′TAGCTTATCAGACTGATGTTGT/BHQ13′ |
| PNA-C1-FAM | CCTGACTACAACT/FAMN |
| PNA-A1-FAM | CCTAGACATACAKACTK/FAMN |
| RS-R | 3′AUCAAAUAGUCUGACUACAACU5′ |
| RS-D | 3′ATCAAATAGTCTGACTACAACT5′ |
| RS-D-m1 | 3′ATCAAATAGTCTGACTACAACA5′ |
| RS-D-sc | 3′AATGAATTCTGAACTAAGCTGGTATGCG5′ |
Effect of masking sequence on quenching efficiency
We were interested to explore both DNA and RNA as potential masking sequences to disrupt PNA micelle assembly. To this end, we designed complementary sequences having a 10 nt toehold region and modified each with Black Hole Quencher 1 (BHQ) on the 3′ terminus. We investigated the quenching efficiency of DNA and RNA masking sequences MS-D and MS-R, respectively, on the fluorescent PNA-C1-FAM to determine an effective concentration for hybridization and quenching.16, 18 We also aimed to explore the effect of hybridization region on toehold-mediated displacement through incorporation of a single mismatch at the 3′ end of the DNA masking sequence (MS-D-m1).15 By introducing a mismatch in the hybridization region, we anticipated that the thermodynamic preference for displacement by a fully complementary releasing strand would be increased, leading to more efficient triggering of assembly.19 We found that the quenching efficiency of all three sequences was high and reached greater than 90% at concentrations above 500 nM (Fig. 2). Moving forward, we chose 3 μM as the working concentration for our PNA amphiphile and masking strands to remain an order of magnitude above the previously reported critical micelle concentration (CMC) of 317 nM.9, 20, 21 Having optimized the concentration for duplex formation using PNA-C1-FAM, we then moved forward and replicated the hybridization of the amphiphilic sequence PNA-A1-FAM with MS-D and observed similar concentration dependence (Fig. S12, ESI†).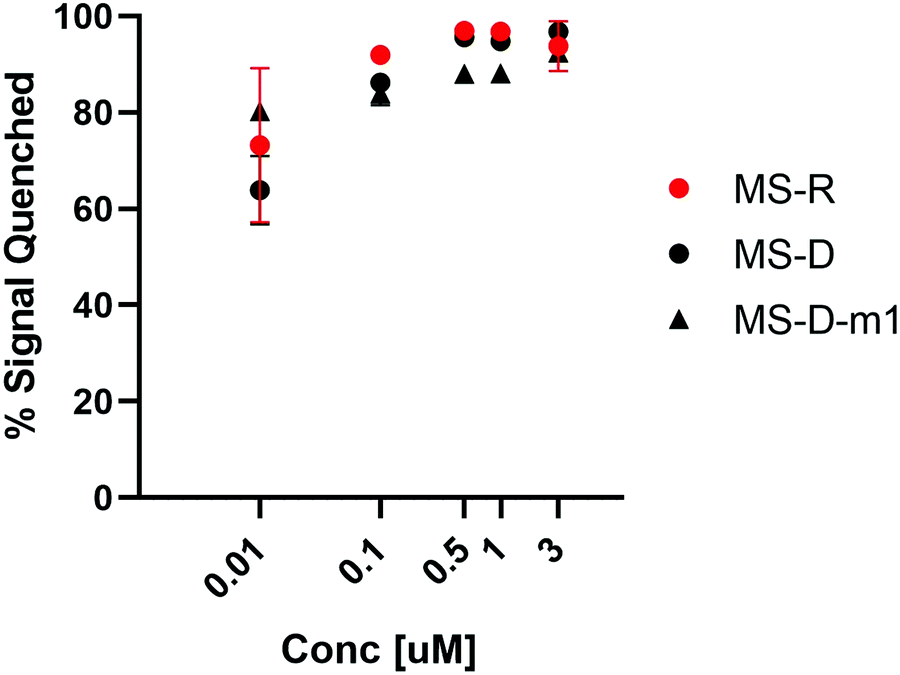 | ||
Fig. 2 Hybridization of MS-D, MS-R, and MS-D-1m to PNA-C1-FAM was monitored using fluorescence quenching. PNA and DNA sequences were at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Error bars represent standard error. (n = 3). :1. Error bars represent standard error. (n = 3). | ||
Effect of releasing sequence on displacement efficiency
We envisioned that the release of the PNA strand from the masking sequence would be largely influenced by the strength of the duplex formed between the masking and releasing sequences.15,19 Thus, we tested displacement using both DNA and RNA releasing strands, RS-D and RS-R, respectfully, at several ratios relative to the PNA-C1-FAM:MS-D/R duplex system (Fig. 3A). Displacement was quantified by restoration of fluorescence upon addition of releasing sequence and calculated using eqn (2). In the PNA-C1-FAM:MS-D system, the addition of RS-D up to a 1:1 stoichiometric equivalence resulted in linear displacement, immediately restoring up to 85% of the fluorescence signal (Fig. 3B). We continued to monitor fluorescence for 1 hour after addition of the releasing sequence, and observed no significant increase in fluorescence, indicating that equilibrium for displacement is reached very rapidly. While the response to RS-R was also observed to be linear, we only observed 20% displacement with one equivalent of releasing sequence, and additional incubation did not lead to an increase in fluorescence signal.
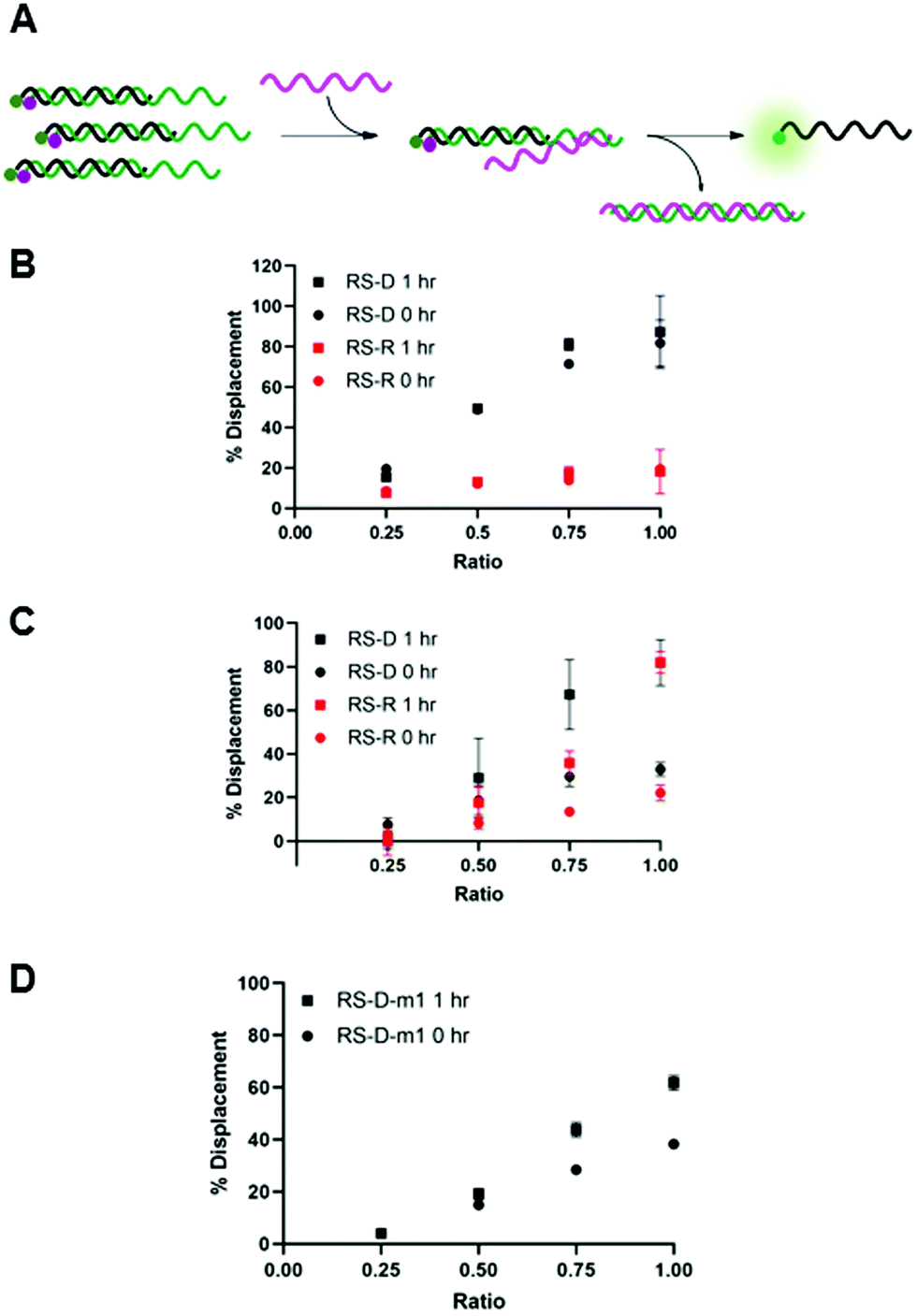 | ||
| Fig. 3 (A) Toehold mediated displacement system using PNA-C1-FAM. When hybridized to the masking sequence, fluorescence is quenched. When a releasing sequence is introduced, the PNA is released, and fluorescence intensity increases. (B–D) % Displacement from PNA-C1-FAM:MS using RS-D, RS-R, and RS-D-m1, respectively, was evaluated in relation to the equivalents of RS added. (B) MS-D. (C) MS-R. (D) MS-D-m1. PNA:MS duplex was used at 3 μM in 1× PBS. Error bars represent standard error (n = 3). | ||
Changing the masking strand to RNA to give a PNA-C1-FAM:MS-R duplex resulted in similar thermodynamic response upon addition of the releasing sequences, but with slower kinetics (Fig. 3C). Specifically, upon the introduction of RS-D and RS-R, similar levels of fluorescence recovery to the PNA-C1-FAM:MS-D system were achieved, however, an incubation period of 1 hour was required to obtain maximum displacement in both systems. Previous reports using fluorophore-quenching and melting temperature studies show the thermodynamics of duplex stability are: RNA:RNA > DNA:DNA > hybrid duplexes, which explains the superior displacement capability of RS-R with MS-R compared to MS-D.22,23 Additionally, predictive models and other kinetics studies show a difference in the rate of displacement depending on nucleic acid backbone, especially in heterochiral strand displacement and duplex formation, with a report demonstrating that RNA releasing strands provide faster rates of displacement.24,25 Interestingly, our studies show the opposite trend for RNA releasing sequences, but this may be due to differences in experimental parameters such as concentration, stoichiometry, and buffer conditions.
We hypothesized that the thermodynamics for displacement of the PNA-C1-FAM from the masking sequence would be driven by the relative stability of the PNA-MS and RS-MS duplexes. Seeking to harness this principle to obtain higher levels of displacement, we introduced a single-base mismatch at the 3′ terminus of the masking sequence (MS-D-m1) and the complementary releasing sequence (RS-D-m1) such that they retain full complementarity with each other, but hybridization between the PNA and the masking sequence is reduced (Fig. 3D). We chose to locate the mismatch at a terminal site rather than an internal site for two reasons. First, internal mismatches in PNA can have a significant impact on duplex stability and we recognized that this would likely prevent efficient hybridization at room temperature. Second, the primary role of the masking sequence is to shield the hydrophobic block of the PNA sequence, and thus we sought to locate the mismatch as far away as possible from this region of the duplex. Unexpectedly, we observed a decrease in displacement yield and kinetics for the mismatched sequences compared to the fully complementary sequences, indicating that the mechanism for PNA displacement may not be completely driven by relative duplex stability.15 While somewhat unexpected, similar results have been observed by others when investigating toehold systems.19,25 Moreover, our melting temperature studies showed that the presence of the mismatch surprisingly increased the duplex stability with PNA-C1-FAM (Table S5). Given these results, we decided to focus the following work with PNA-A1-FAM on the MS-D system because it yielded the fastest and most complete displacement of the systems tested.
After confirming and optimizing displacement using the PNA-C1-FAM duplex systems, we next tested the response of PNA-A1-FAM:MS-D using RS-R and RS-D (Fig. 4A). Ratiometric addition of RS-D resulted in a similar concentration-dependent response to that observed with the control PNA sequence, with approximately 60% immediate displacement of the amphiphile at a 1:1 ratio, which increased to 90% following a one-hour incubation (Fig. 4B). Addition of RS-R to the amphiphile masking complex resulted in much lower displacements of 30% at a 1:1 ratio immediately after addition, and this increased to 60% following incubation. While lower than the displacement observed using MS-D, this did compare favorably with the activity of the PNA-C1-FAM control with MS-R. Encouraged by the increased displacement, we tested higher ratios of RS-R up to 2:1 and observed 45% displacement immediately after addition of releasing sequence, and an increase to 90% after 1 hour of incubation (Fig. 4C). These results demonstrate that the presence of γ-modifications or the amphiphilic nature of the PNA can alter both the thermodynamics and kinetics of toehold-mediated displacement. Moreover, our results demonstrate that both DNA and RNA can serve as effective stimuli to trigger displacement and induce assembly.
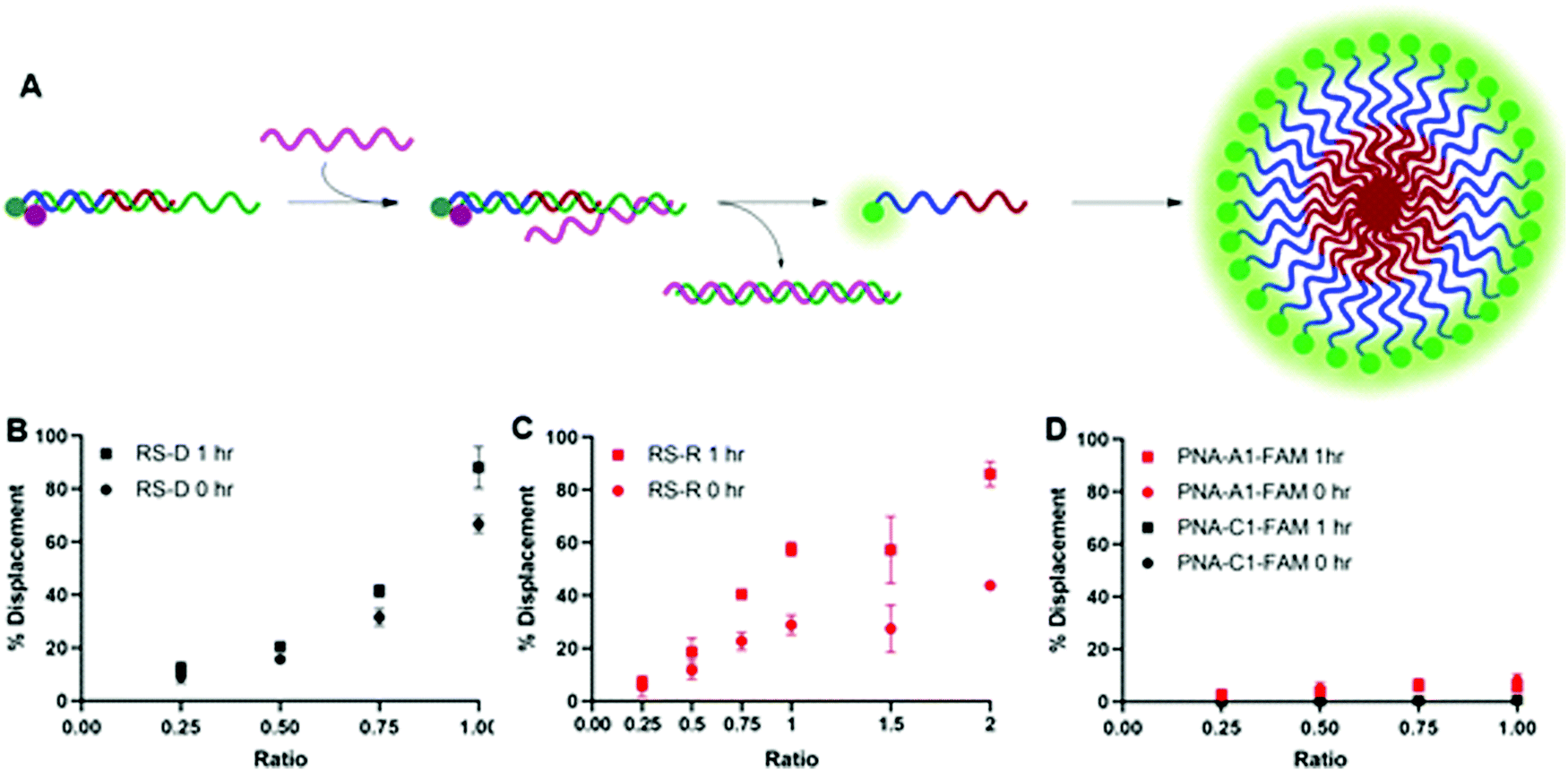 | ||
| Fig. 4 (A) Toehold mediated displacement system using PNA-A1-FAM. When hybridized to the masking sequence, fluorescence is quenched. When a releasing sequence is introduced, the PNA is released, and fluorescence intensity increases. Upon release, the amphiphile can then self-assemble. (B) % Displacement from PNA-A1-FAM:MS-D using RS-D was evaluated in relation to the equivalents of RS-D added. (C) % Displacement from PNA-A1-FAM:MS-D using RS-R was evaluated in relation to the equivalents of RS-R added. (D) % Displacement from PNA-C1-FAM:MS-D and PNA-A1-FAM:MS-D using RS-D-sc was evaluated in relation to the equivalents of RS-D-sc added. PNA:MS-D duplex was used at 3 μM in 1× PBS. Error bars represent standard error (n = 3). | ||
Finally, both control and amphiphilic systems were tested for sequence specificity through the introduction of a scrambled DNA releasing sequence, RS-D-sc. This sequence retains partial sequence complementarity but introduces several mismatches. Introduction of RS-D-sc under various conditions resulted in minimal (<10%) displacement even at a 1:1 ratio, indicating that the masked PNA is specific for its target sequence (Fig. 4D).
Characterization of PNA assembly
Upon successful toehold-mediated displacement of both control and amphiphilic PNA sequences, we sought to evaluate their assembly and hybridization properties using dynamic light scattering (DLS), circular dichroism (CD), and transmission electron microscopy (TEM). DLS analysis showed that PNA-C1-FAM and PNA-A1-FAM assemblies were similar in size to the previously published PNA-C and PNA-A structures.9 The control sequence showed a size that would be expected for an unassembled monomer and the amphiphilic sequence formed structures of 119.1 ± 34.0 nm, comparable to the size of 110.2 ± 31.9 nm of PNA-A (Fig. 5).26, 27 This indicates that neither the fluorophore nor its location affected the formation and size of assemblies.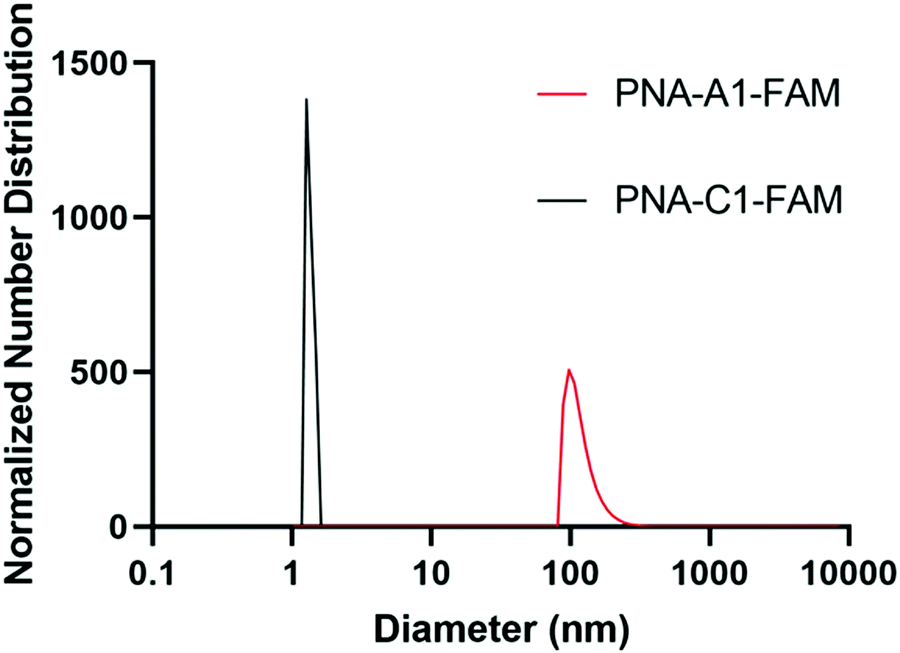 | ||
| Fig. 5 Normalized size distribution of PNA assemblies. Samples tested at 200 μM in 1× PBS. Average diameter of particles of PNA-C1-FAM = 1.5 ± 0.3 nm; PNA-A1-FAM = 119.1 ± 34.0 nm. Peaks having greater than 1% are reported. | ||
In an attempt to validate hybridization of the masking sequence and subsequent disassembly, PNA-A1-FAM was annealed to MS-D at 200 μM under previously described conditions and the complex analyzed by DLS. While we theoretically anticipated observing a shift in size back to that of PNA-C1-FAM, we instead observed the formation of larger aggregates (Fig. S13). This was not entirely unexpected, however, as our previous studies as well as the TEM data shown below for this work demonstrate that large amorphous aggregates tend to form when DNA or RNA are added to PNA in the presence of buffer salts.9 As anticipated, these aggregates persisted upon addition of RS-D to the PNA-A1-FAM:MS-D complex (Fig. S13). In DLS, the presence of large aggregates can overwhelm the signal and prevent detection of smaller assemblies, and thus we viewed the DLS data as being inconclusive as to whether we were achieving our goal of hybridization and stimuli-responsive assembly.
We next turned to CD to provide validation for the changes in hybridization upon addition of the masking and releasing sequences. Despite being chiral, PNA-A1-FAM displayed only a weak CD signal, in line with previous reports (Fig. 6).9 Upon hybridization with MS-D, we observed a strong signal with maxima at 219 nm and 262 nm and with minima at 244 nm and 201 nm, in similar agreement with previously reported right-handed PNA:DNA duplex structure.5,9,28,29 Importantly, this signal is distinct from MS-D alone (Fig. S11, ESI†). After introduction of the RS-D strand to the PNA-A1-FAM:MS-D duplex, we observed a shift in signal, indicating that the PNA amphiphile was released. Additionally, the signal observed after addition of the releasing sequence is very similar to a combination of signals from PNA alone and the MS-D:RS-D duplex, providing additional evidence for successful toehold-mediated displacement.
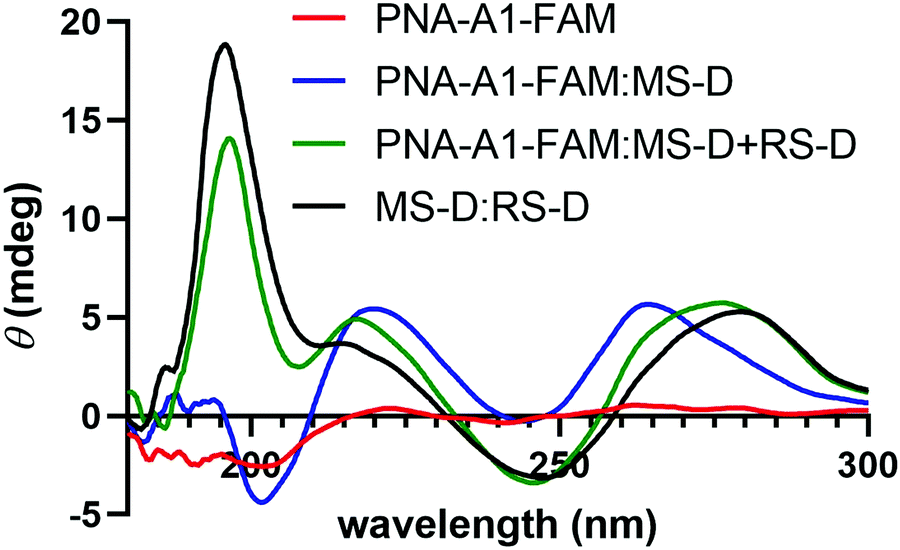 | ||
| Fig. 6 CD spectroscopy demonstrating the change in maxima and minima upon the addition of RS-D. All samples were prepared using 100 μM PNA and DNA in 1× PBS. | ||
In order to demonstrate DNA-mediated disassembly and stimuli-responsive reassembly, we turned to transmission electron microscopy (TEM). In comparison to DLS, where non-specific aggregates can block detection of smaller assemblies, TEM enables us to visualize all of these objects, and thus observe PNA assembly even in the presence of other aggregates. Assembly of PNA-A1-FAM was tested by preparing a 10 μM or 100 μM solution, spotting onto the grid, and staining with uranyl acetate for imaging. We observed relatively uniform, micelle-like structures having similar size and morphology compared to previously reported PNA-A (Fig. 7A and Fig. S8, ESI†). This was to be expected, given that both of these concentrations are above the CMC value for the PNA amphiphile. Moreover, the ability of PNA-A1-FAM to self-assemble similarly to PNA-A demonstrates that the amino acid sequence directs assembly into micelle-like structures regardless of the fluorophore and its location.
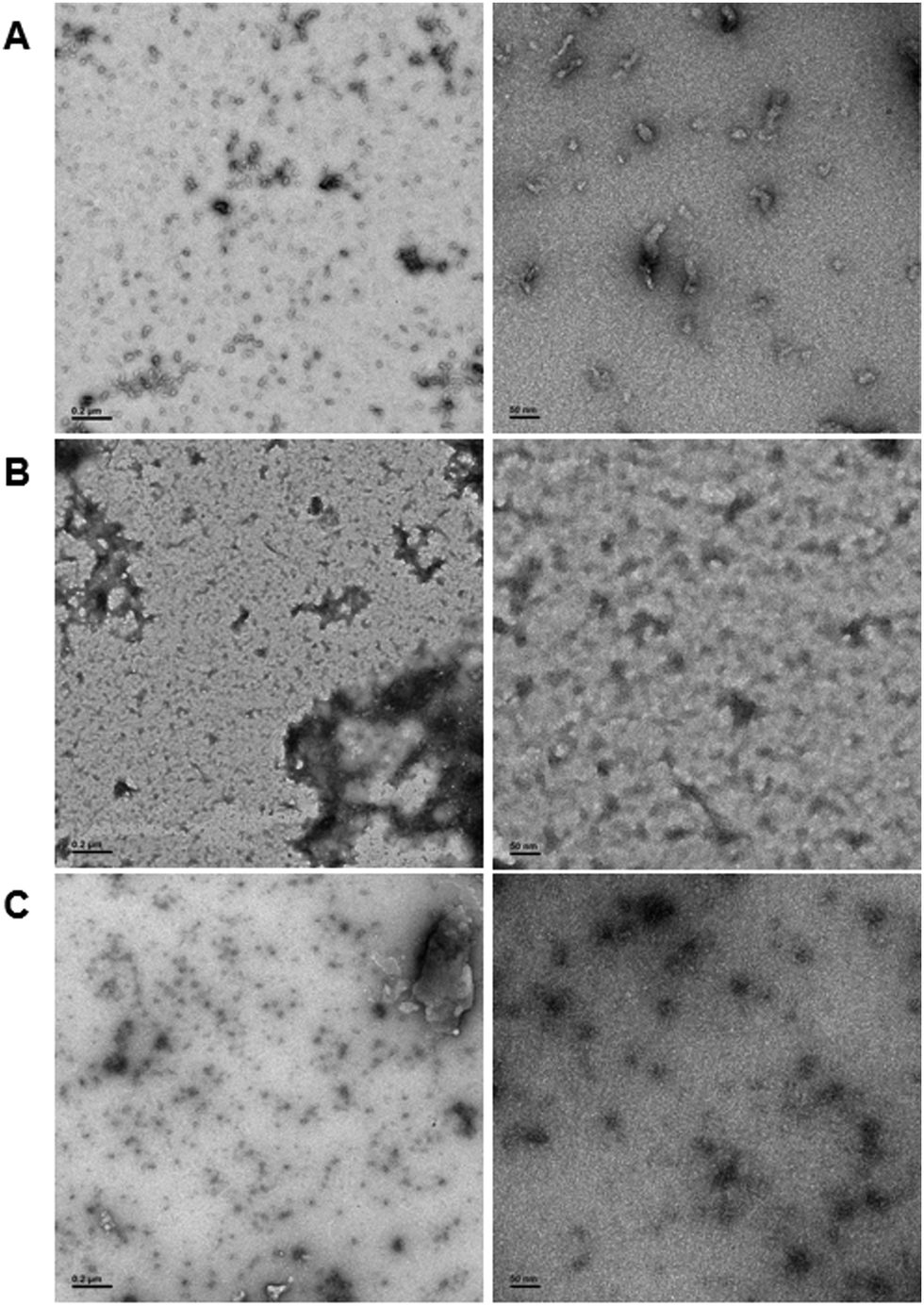 | ||
| Fig. 7 TEM images of (A) PNA-A1-FAM assembled at 10 μM, (B) PNA-A1-FAM disassembled via MS-D hybridization at 10 μM, and (C) PNA-A1-FAM reassembled via RS-D toehold release at 10 μM. Left column scale bar = 2000 nm. Right column scale bar = 50 nm. | ||
We then tested disassembly through nucleic acid recognition of the masking sequence. The PNA-A1-FAM:MS-D duplex system was initially prepared at 100 μM, annealed as described, and spotted for imaging (Fig. S9, ESI†). However, in line with our previous experience and our DLS data, we observed a very high level of non-specific aggregation, with the dark nature of these aggregates attributable to interactions between the DNA phosphate backbone and the uranyl acetate stain. Given our goal of eventually observing micelle re-assembly, we sought to minimize the presence of these non-specific aggregates and diluted the solutions to 10 μM. This reduced the quantity and size of dark aggregates, likely due to the decrease in DNA concentration. Importantly, both the high and low concentration samples show the disappearance of the ordered PNA assemblies, matching our previous report that sequence-specific recognition can induce disassembly (Fig. 7B).9
Finally, to validate stimuli-responsive assembly, we combined PNA-A1-FAM:MS-D and RS-D, incubated for 1 hour, spotted the samples onto a TEM grid, stained, and imaged. Using a concentration of 100 μM again resulted in dark amorphous aggregates which we attribute to precipitation (Fig. S10, ESI†). However, this was reduced by lowering the concentrations to 10 μM, similar to the experiments described above. While we do still observe some non-specific aggregates, we were excited to also observe a large number of ordered assemblies having an average size of 16.8 ± 5.7 nm (Fig. 7C). We do note that the structures formed upon reassembly are slightly smaller than those formed in the initial PNA assembly. This may be due to the presence of DNA in the solutions or as a result of a different assembly mechanism when the PNA is being released from a DNA complement over time. However, we were excited to observe that our bilingual PNA is able to undergo stimuli-responsive assembly as anticipated.
Conclusion
Biological information codes are extremely powerful for information storage and function, and we have shown that both peptide and nucleic acid codes can be combined into a single bilingual biopolymer using a PNA scaffold. Our initial work focused on harnessing the peptide code for assembly and the nucleic acid code for stimuli-responsive disassembly. Here we explore the extension of the nucleic acid code to also enable stimuli-responsive assembly. This is achieved through the design of a toehold-mediated displacement system in which a masking sequence hybridizes to the PNA to induce disassembly while providing a single-stranded toehold. Subsequent addition of a complementary nucleic acid results in displacement to release the PNA and trigger reassembly. Using fluorescence, we were able to monitor hybridization and subsequent displacement. We observed varying thermodynamics and kinetics for displacement when using different combinations of DNA and RNA sequences, and observed that using DNA as both the masking and releasing sequence provided highly efficient and rapid displacement. The assembled, disassembled, and reassembled structures were characterized by DLS and TEM. The presence of non-specific aggregates induced by the presence of DNA or RNA prevented direct observation of the reassembly process using DLS, however, this challenge was overcome using TEM and we were excited to observe the stimuli-responsive formation of micelle-sized assemblies. Together, this work provides a new approach to harnessing biological information codes to control assembly, providing stimuli-responsive materials that can advance applications in biotechnology and biomedicine.Experimental procedures
Abbreviations: Fmoc, fluorenylmethyloxycarbonyl; FAM, fluorescein; acetonitrile, ACN; water, H2O.PNA monomer synthesis
Unmodified PNA monomers were purchased from PolyOrg, Inc. Thymine acetic acid was purchased from Sigma-Aldrich. The protected nucleobases adenine and cytosine and Fmoc protected γ-methyl and γ-lysyl modified PNA backbones were synthesized following previously published procedures.9, 30 The Fmoc-protected L-amino acids and nucleobase starting materials were purchased from Chem-Impex Inc. All other reagents were purchased from Fisher Scientific, Chem-Impex, and Sigma-Aldrich and used without additional purification unless otherwise stated. Merck silica gel 60 F254 was used to monitor small molecule synthesis through thin layer chromatography (TLC) with UV light for visualization. Synthesized compounds were purified using flash chromatography with SiliaFlash F60 grade silica purchased from SiliCycle Inc. Characterization of compounds was completed using proton (1H) and carbon (13C) NMR using a Varian Inova 400 MHz spectrometer. The spectra were analyzed using MestReNova Software. The mass of the compounds was confirmed using an Agilent 6230 electrospray ionization time-of-flight (ESI-TOF) mass spectrometer. PNA monomer synthesis was completed as described previously.9, 31PNA oligomer synthesis
PNA oligomers were synthesized as previously described using a Biotage SP Wave semiautomatic peptide synthesizer.9 Monomer coupling efficiencies were monitored using a Nanodrop 2000 spectrophotometer to measure dibenzofulvene-piperidine adduct absorbance at 301 nm. Upon cleavage, sequences were purified through reverse-phase HPLC using an Agilent Eclipse XDB-C18 5 μm, 9.4 × 250 mm column at 60 °C with a flow rate of 2 mL min−1, monitoring 260 and 495 nm using a linear gradient (10–40%) of 0.1% TFA/CAN in 0.1% TFA/H2O. The sequences were confirmed by ESI-TOF mass spectrometry.Melting temperature analysis of PNA strands
All DNA and RNA sequences were purchased from the University of Utah DNA/Peptide Synthesis Core Facility or Integrated DNA Technologies. Samples containing 5 μM of complementary nucleic acids were prepared using stock solutions in 1× PBS. Samples were then added to an 8-cell quartz microcuvette with a 1 cm path length. A Shimadzu UV-1800 spectrophotometer equipped with a temperature controller and Julabo Corio CD water circulator were used for measurement of absorbance at 260 nm while heating from 25 to 95 °C at a rate of 0.5 °C min−1. The melting temperatures were calculated using a first derivative method and experiments were carried out in triplicate.Fluorescence monitoring of hybridization and displacement
The fluorescence quenching of the FAM-labeled PNA sequences in the PNA:MS-R or MS-D duplexes was measured by fluorescence emission of the FAM fluorophore. Samples of 100 μL containing PNA alone, as a fluorescent control sample, and PNA:MS-R or MS-D over a range of concentrations (0.01 μM to 3 μM), at a 1:1 ratio were prepared using stock solutions in 1× PBS. The samples were annealed by heating to 95 °C for 5 min and cooling 1 °C every 30 seconds until 25 °C was achieved. The samples were then transferred into a 384-well flat-bottom plate for analysis on a Biotek Cytation5 plate reader. Fluorescence was then measured using an excitation at 495 nm and emission at 520 nm. The fluorescence measurement was used to plot the % Quenched using eqn (1) using GraphPad Prism software.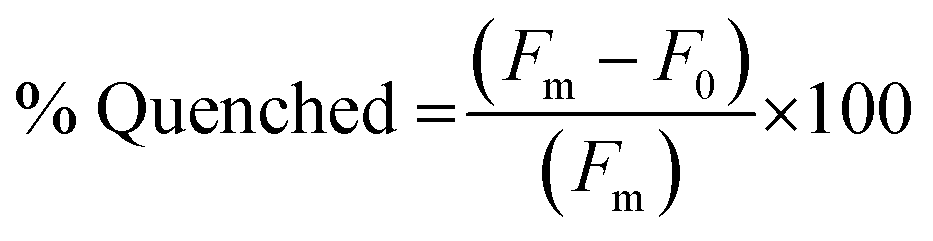 | (1) |
To measure the % Displacement via fluorescence intensity, 50 μL of samples containing 6 μM of PNA and MS-D or MS-R each in 1× PBS and 50 μL of RS-R or RS-D samples at 2× final reported quantities (0.25 to 2 equivalents) in 1× PBS were annealed as previously described. The 50 μL RS-R or RS-D samples were then transferred into the PNA:MS-D or MS-R samples to achieve the final reported concentrations. Samples containing PNA alone and PNA:MS-D or MS-R were used and had 1× PBS blank solutions added as controls. The samples were then transferred to a 384-well flat-bottom plate for analysis using the previously described protocol. The samples were measured at reported incubation intervals. The fluorescence was then used to calculate the % Displacement using eqn (2) as described and plotted using GraphPad Prism software.
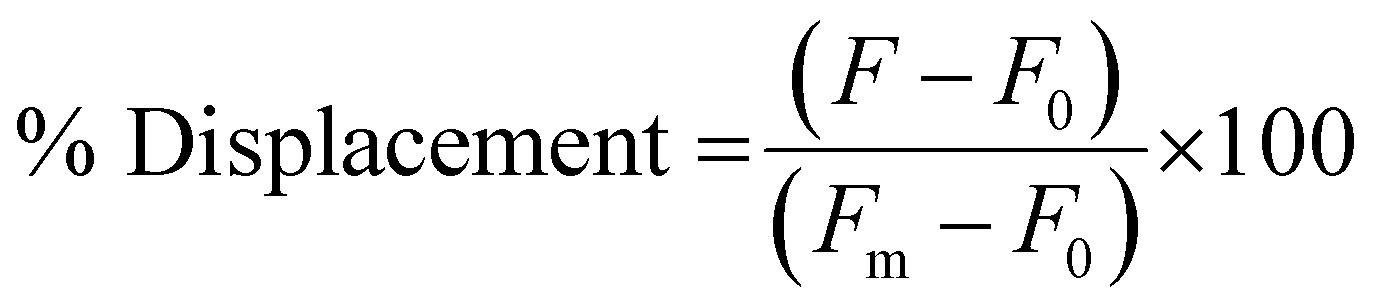 | (2) |
Preparation of PNA amphiphile assemblies
To prepare amphiphile assembly, samples of PNA, PNA:MS-D, and RS-D were dissolved in water or 1× PBS and were annealed as previously described. All samples were allowed to incubate at room temperature for 1 hour. Samples containing RS-D were then transferred to PNA:MS-D samples and allowed to incubate at room temperature for an additional hour.Determination of size using dynamic light scattering
Samples of PNA, PNA:MS-D, and PNA:MS-D were prepared in 1× PBS at 200 μM from stock solutions and were annealed through heating and slow cooling as described above. Samples were then transferred to a quartz cuvette and analyzed using a Particular Systems NanoPlus DLS nano particle analyzer and data analyzed using NanoPlus Software.Confirmation of MS-R hybridizing to PNA-A by CD
Circular Dichroism was used to confirm the hybridization of PNA-A:MS-R. Individual samples of PNA-A and MS-R were prepared at 200 μM in 1× PBS. 30 μL of PNA-A was combined with 30 μL of MS-R to form the final 100 μM in 1× PBS solutions. Samples were incubated for 1 hour, after which, they were analyzed using a JASCO J-1500 circular dichroism spectrometer. Data points from 190 to 300 nm were collected at 1 nm intervals using a continuous scanning mode of 200 nm min−1 at 23.5 °C. A sample with PNA-A alone was used as a control and had the signal overlapped using the JASCO software.Confirmation of RS-D hybridizing to MS-D in the PNA-A:MS-D duplex state by CD
Samples containing PNA-A:MS-R and MS-D were prepared at 200 μM in 1× PBS and were annealed through heating and slow cooling as described above. 30 μL of PNA-A:MS-R was combined with 30 μL of RS-D to form the final 100 μM in 1× PBS solutions and incubated for 1 hour at room temperature before being analyzed. Samples containing MS-R:RS-D and RS-D alone were used as controls.Characterization of PNA-A assemblies by TEM
Samples of PNA-A1-FAM were prepared at 100 μM in water from stock solutions. Using a 200-mesh Formvar/carbon-coated copper grid, 3.5 μL of sample was spotted for 2 minutes and was wicked away with filter paper. Then, 3.5 μL of 1% uranyl acetate stain solution was spotted for 30 seconds before wicking. Grids were dried at room temperature for 30 minutes prior to imaging using a JEOL JEM-1400 transmission electron microscope.Characterization of PNA-A disassembled using masking sequence DNA by TEM
Samples of PNA-A1-FAM:MS-D at 100 μM were prepared in 1× PBS and annealed through heating and slow cooling as described above. The duplex sample was then spotted and prepared for TEM imaging as described above.Characterization of PNA-A re-assembled using release sequence DNA by TEM
Samples containing PNA-A1-FAM:MS-D and RS-D at 200 μM were prepared in 1× PBS and annealed through heating and slow cooling as described above. After, 20 μL of RS-D was transferred to 20 μL of PNA-A1-FAM:MS-D and allowed to incubate at room temperature for 1 hour. The sample was then prepared for TEM imaging as described above.Conflicts of interest
The authors report no conflicting interests.Acknowledgements
This work was supported by the National Science Foundation (DMR 2003987 to J. M. H.). The authors also acknowledge the Robert P. Apkarian Integrated Electron Microscopy Core and NMR Research Center at Emory University for instrument access and technical assistance. They would also like to thank Mike Hanson and the oligonucleotide and peptide synthesis facility and the University of Utah for oligonucleotide materials.References
- X. Tan, et al., Nucleic acid-based drug delivery strategies, J. Controlled Release, 2020, 323, 240–252 CrossRef CAS PubMed.
- L. Wang, et al., Controlling the self-assembly of biomolecules into functional nanomaterials through internal interactions and external stimulations: A review., Nanomaterials, 2019, 9(2), 285 CrossRef CAS.
- A. M. Peterson and J. M. Heemstra, Controlling self-assembly of DNA-polymer conjugates for applications in imaging and drug delivery, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7(3), 282–297 CAS.
- M. Egholm, et al., Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral peptide backbone, J. Am. Chem. Soc., 1992, 114(5), 1895–1897 CrossRef CAS.
- M. Moccia, M. F. Adamo and M. Saviano, Insights on chiral, backbone modified peptide nucleic acids: properties and biological activity, Artif. DNA, 2014, 5(3), e1107176 CrossRef.
- S. Shakeel, S. Karim and A. Ali, Peptide nucleic acid (PNA)—a review, J. Chem. Technol. Biotechnol., 2006, 81(6), 892–899 CrossRef CAS.
- M. Egholm, et al., PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules., Nature, 1993, 365(6446), 566–568 CrossRef CAS PubMed.
- B. Sahu, et al., Synthesis of conformationally preorganized and cell-permeable guanidine-based γ-peptide nucleic acids (γGPNAs), J. Org. Chem., 2009, 74(4), 1509–1516 CrossRef CAS PubMed.
- C. S. Swenson, et al., Bilingual peptide nucleic acids: encoding the languages of nucleic acids and proteins in a single self-assembling biopolymer, J. Am. Chem. Soc., 2019, 141(48), 19038–19047 CrossRef CAS PubMed.
- L. Peng, et al., Engineering and applications of DNA-grafted polymer materials, Chem. Sci., 2013, 4(5), 1928–1938 RSC.
- H. Xu, et al., Hydrophobic-region-induced transitions in self-assembled peptide nanostructures, Langmuir, 2009, 25(7), 4115–4123 CrossRef CAS PubMed.
- R. Kumarswamy, I. Volkmann and T. Thum, Regulation and function of miRNA-21 in health and disease, RNA Biol., 2011, 8(5), 706–713 CrossRef CAS PubMed.
- H. Ikeda, Y. Nakamura and I. Saito, Synthesis and characterization of naphthalimide-containing peptide nucleic acid, Tetrahedron Lett., 2002, 43(32), 5525–5528 CrossRef CAS.
- Y. Guo, et al., Recent advances in molecular machines based on toehold-mediated strand displacement reaction, Quant. Biol., 2017, 5(1), 25–41 CrossRef.
- R. R. Machinek, et al., Programmable energy landscapes for kinetic control of DNA strand displacement, Nat. Commun., 2014, 5(1), 1–9 Search PubMed.
- Z. Tan, T. A. Feagin and J. M. Heemstra, Temporal control of aptamer biosensors using covalent self-caging to shift equilibrium, J. Am. Chem. Soc., 2016, 138(20), 6328–6331 CrossRef CAS.
- J. Chen, et al., A simple and rapid biosensor for ochratoxin A based on a structure-switching signaling aptamer, Food Control, 2012, 25(2), 555–560 CrossRef CAS.
- A. A. Sanford, et al., RE-SELEX: Restriction Enzyme-Based Evolution of Structure-Switching Aptamer, Biosensors, 2021, 12, 11692–11702 CAS.
- N. E. Haley, et al., Design of hidden thermodynamic driving for non-equilibrium systems via mismatch elimination during DNA strand displacement, Nat. Commun., 2020, 11(1), 1–11 CrossRef.
- J. P. Vernille, L. C. Kovell and J. W. Schneider, Peptide nucleic acid (PNA) amphiphiles: synthesis, self-assembly, and duplex stability, Bioconjugate Chem., 2004, 15(6), 1314–1321 CrossRef CAS PubMed.
- C. Lau, et al., Morphological characterization of self-assembled peptide nucleic acid amphiphiles., J. Phys. Chem. B, 2006, 110(18), 9027–9033 CrossRef CAS PubMed.
- M.-X. Li, et al., Exploration of the kinetics of toehold-mediated strand displacement via plasmon rulers., ACS Nano, 2018, 12(4), 3341–3350 CrossRef CAS PubMed.
- E. A. Lesnik and S. M. Freier, Relative thermodynamic stability of DNA, RNA, and DNA: RNA hybrid duplexes: relationship with base composition and structure, Biochemistry, 1995, 34(34), 10807–10815 CrossRef CAS PubMed.
- N. Srinivas, et al., On the biophysics and kinetics of toehold-mediated DNA strand displacement, Nucleic Acids Res., 2013, 41(22), 10641–10658 CrossRef CAS PubMed.
- N. Kundu, B. E. Young and J. T. Sczepanski, Kinetics of heterochiral strand displacement from PNA–DNA heteroduplexes., Nucleic Acids Res., 2021, 49(11), 6114–6127 CrossRef CAS PubMed.
- T. G. Souza, V. S. Ciminelli and N. D. S. Mohallem, A comparison of TEM and DLS methods to characterize size distribution of ceramic nanoparticles, J. Phys.: Conf. Ser., 2016, 733, 012039 CrossRef.
- R. F. Domingos, et al., Characterizing manufactured nanoparticles in the environment: multimethod determination of particle sizes, Environ. Sci. Technol., 2009, 43(19), 7277–7284 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, et al., A simple γ-backbone modification preorganizes peptide nucleic acid into a helical structure., J. Am. Chem. Soc., 2006, 128(31), 10258–10267 CrossRef CAS PubMed.
- A. Faccini, et al., Circular dichroism study of DNA binding by a potential anticancer peptide nucleic acid targeted against the MYCN oncogene, Chirality, 2008, 20(3–4), 494–500 CrossRef CAS PubMed.
- N. T. S. De Costa and J. M. Heemstra, Evaluating the effect of ionic strength on duplex stability for PNA having negatively or positively charged side chains, PLoS One, 2013, 8(3), e58670 CrossRef CAS PubMed.
- A. Porcheddu, et al., A practical and efficient approach to PNA monomers compatible with Fmoc-mediated solid-phase synthesis protocols, Wiley-VCH Verlag Weinheim, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of PNA sequences, Tm data, TEM images, and additional displacement studies. See DOI: https://doi.org/10.1039/d2cb00020b |
| This journal is © The Royal Society of Chemistry 2022 |