
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerspectives on conformationally constrained peptide nucleic acid (PNA): insights into the structural design, properties and applications
Chaturong
Suparpprom
 ab and
Tirayut
Vilaivan
*ab
ab and
Tirayut
Vilaivan
*ab
aDepartment of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Naresuan University, Tah-Poe District, Muang, Phitsanulok 65000, Thailand. E-mail: vtirayut@chula.ac.th
bOrganic Synthesis Research Unit, Department of Chemistry, Faculty of Science, Chulalongkorn University, Phayathai Road, Pathumwan, Bangkok 10330, Thailand
First published on 18th March 2022
Abstract
Peptide nucleic acid or PNA is a synthetic DNA mimic that contains a sequence of nucleobases attached to a peptide-like backbone derived from N-2-aminoethylglycine. The semi-rigid PNA backbone acts as a scaffold that arranges the nucleobases in a proper orientation and spacing so that they can pair with their complementary bases on another DNA, RNA, or even PNA strand perfectly well through the standard Watson–Crick base-pairing. The electrostatically neutral backbone of PNA contributes to its many unique properties that make PNA an outstanding member of the xeno-nucleic acid family. Not only PNA can recognize its complementary nucleic acid strand with high affinity, but it does so with excellent specificity that surpasses the specificity of natural nucleic acids and their analogs. Nevertheless, there is still room for further improvements of the original PNA in terms of stability and specificity of base-pairing, direction of binding, and selectivity for different types of nucleic acids, among others. This review focuses on attempts towards the rational design of new generation PNAs with superior performance by introducing conformational constraints such as a ring or a chiral substituent in the PNA backbone. A large collection of conformationally rigid PNAs developed during the past three decades are analyzed and compared in terms of molecular design and properties in relation to structural data if available. Applications of selected modified PNA in various areas such as targeting of structured nucleic acid targets, supramolecular scaffold, biosensing and bioimaging, and gene regulation will be highlighted to demonstrate how the conformation constraint can improve the performance of the PNA. Challenges and future of the research in the area of constrained PNA will also be discussed.
 Chaturong Suparpprom | Chaturong Suparpprom received his PhD degree in Chemistry from Chulalongkorn University (Thailand) in 2006. Under the supervision of Prof. Tirayut Vilaivan, he developed interest in the fields of peptide nucleic acid (PNA) and organic synthesis. His PhD work contributes to the development of a new family of conformationally constrained pyrrolidinyl PNA. In 2006, he started his independent career at Naresuan University, where he is currently holding an assistant professorship. He continued to develop and expand the applications of pyrrolidinyl PNA in diagnostics and biomolecular fields. His other research interests apart from PNA encompass natural fatty acid and bioactive compounds from Thai herbs. |
 Tirayut Vilaivan | Tirayut Vilaivan received his DPhil from Oxford in 1996 under the supervision of the late Professor Gordon Lowe, FRS. He started his independent career at the Department of Chemistry, Chulalongkorn University in Bangkok in 1996. He is currently a professor of chemistry at Chulalongkorn University. His research interests include (1) design, synthesis, properties, and applications of conformationally constrained peptide nucleic acid (PNA), (2) development of novel synthetic methodologies (focusing on catalysis and green processes) and (3) development of novel antimalarials based on dihydrofolate reductase inhibitors. |
Introduction
In all organisms, nucleic acids play important roles in the storage, transmission, and expression of genetic information in a precisely controlled fashion. Central to this is the unique pairing ability of the two complementary strands of nucleic acids via the highly specific Watson–Crick base-pairing scheme. The high fidelity of the base-pairing scheme suggests potential uses of nucleic acids for regulating gene expression which is indeed one of the mechanisms used by nature.1 This can have tremendous implications in the areas of therapeutics. One example that highlights the importance of nucleic acid-based therapeutics is the recent success of the mRNA vaccine accelerated by the emergence of COVID-19.2 However, due to their susceptibility to degradation by the ubiquitous endogenous nucleases, natural nucleic acids are not suitable for therapeutic purposes. Modifications are generally required to improve the stability of nucleic acids so that they can last long enough to bind to the target nucleic acids and exert the biological effects that they were designed for.3,4 Thus, the research in the area of modified nucleic acids or xeno-nucleic acids is an important field that supports the fast-growing trends of nucleic acid-based therapy. Moreover, they can be used in other areas of applications including diagnostics, biosensors, and materials science. In addition, these xeno-nucleic acids are interesting in their own right as a model for prebiotic systems chemistry.5,6Peptide nucleic acid (PNA) is one of the most fascinating members of the xeno-nucleic acid family. While modified nucleic acids generally retain the original negatively charged sugar-phosphate backbone of natural nucleic acids (or at least part of it),7 PNA carries a drastically different electrostatically neutral peptide-like backbone which makes it a unique kind of nucleic acid mimic. The first PNA reported in 1991 consisting of an N-2-aminoethylglycyl backbone (now being referred to as aegPNA) was originally designed to be a triplex-forming agent.8 However, homothymine aegPNA showed an unexpectedly strong affinity towards the DNA target and bound to it sequence-specifically in a novel triplex invasion mode. Subsequently, the ability of PNA to recognize single-stranded DNA/RNA targets following the standard Watson–Crick base-pairing rules was demonstrated.9 In addition, PNA can pair with itself to form highly stable PNA·PNA duplexes. Thus, PNA is among the first testimonies to the fact that the (deoxy)ribose-phosphate was not a requirement for base–base recognition. The electrostatically neutral backbone of PNA contributes to its exceptionally strong binding without compromising specificity. In fact, PNAs show even better mismatch discrimination than natural nucleic acids. These properties together with the complete stability towards nucleases make PNA a potential tool for nucleic-acid-based therapeutics and other applications that rely on specific base-pairing.10,11
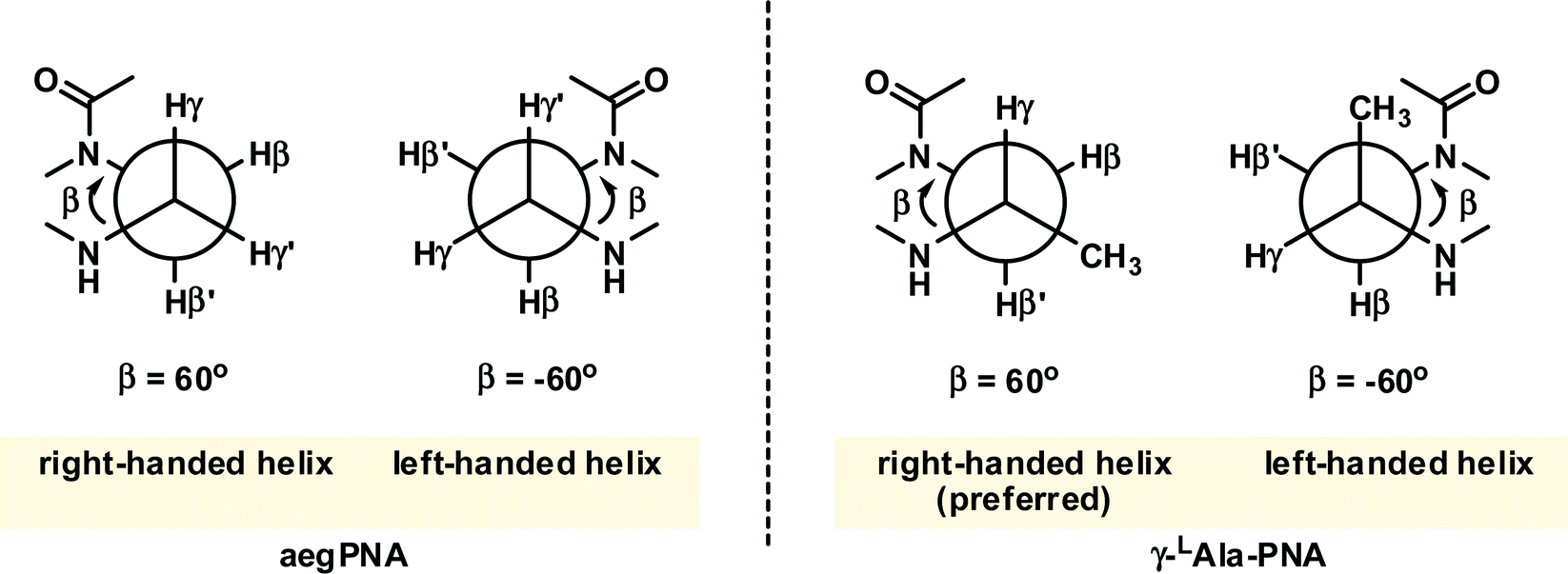
Even though the original aegPNA system already possesses many desirable characteristics and has been widely employed in various applications, there are still great demands for further improvement. During the three decades after the discovery of PNA, numerous modifications of the original aegPNA structure have been explored with the aim of improving the base-pairing efficiency and solubility, and introducing other functions in the PNA molecules such as cellular uptake or reporter groups. Chemistry-wise, the modifications of PNA can be divided into three categories: acyclic backbone modification, cyclic backbone modification, and modification that does not follow the aegPNA template. Although some earlier reviews on modified PNA exist,12–16 this review is complementary to the existing ones as it will focus mainly on the backbone modification to introduce conformational constraint in the PNA molecules. Such modification, even a minor one, will result in altering and restriction of one or more torsional angle values that will ultimately affect the global structure and binding properties of the modified PNA oligomers (Fig. 1). A broad landscape will be provided in addition to a more detailed comparison and evaluation of the impact of different structural modifications of the original aegPNA backbone on the stability and specificity of the duplex formation in a systematic way. The properties and applications of selected modified PNA are discussed in more detail. Thus, this review should be useful for specialists in the PNA area seeking a comprehensive source of information on this topic. It should also help those who are new to the PNA field to learn what modified PNA can offer and may inspire the adoption of PNA that might well suit their required applications or thinking for novel and imaginative uses of PNA.
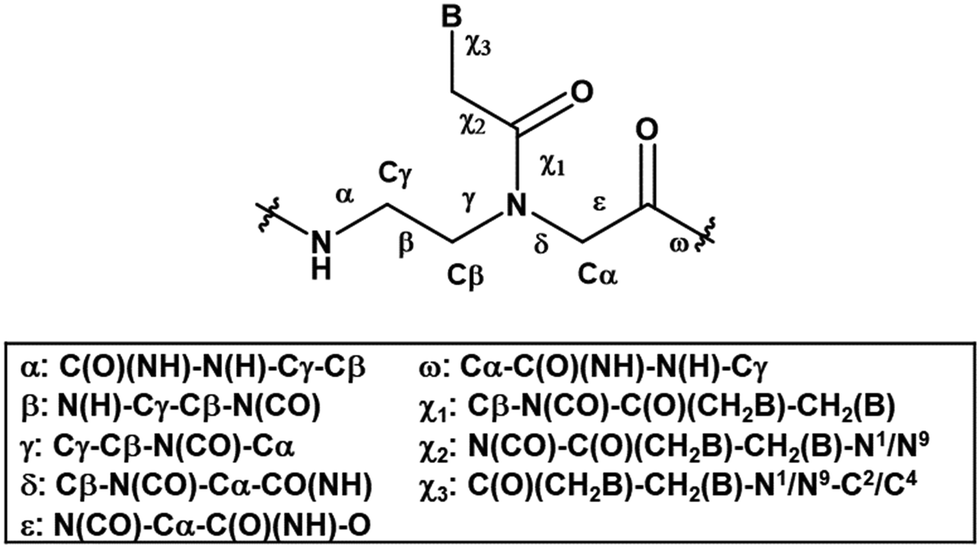 | ||
| Fig. 1 The structure of aegPNA with definition of backbone atom positions and torsional angles. | ||
Modified PNA with acyclic backbones
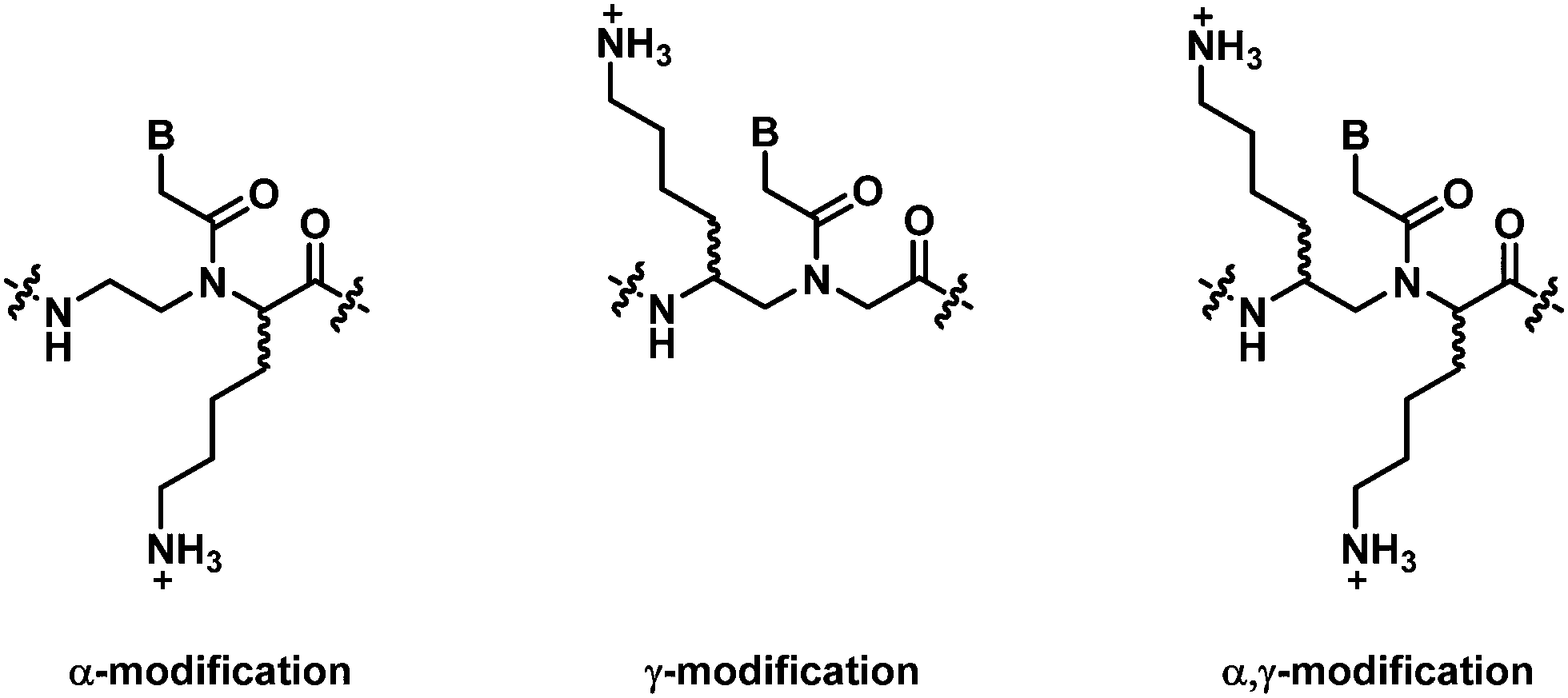
This section will cover modified PNA with an acyclic backbone that follows the generic aegPNA template (as well as its homolog) with one or more substituents on the N-aminoethylglycine backbone. There are four possible sites for substitution including α-, β-, γ-, and the secondary amide nitrogen atom (N) (Fig. 2). More than one substituent may be present on the same or different backbone carbon or nitrogen atoms, but this section will cover only the cases where the substituents are not joined to form a cyclic structure.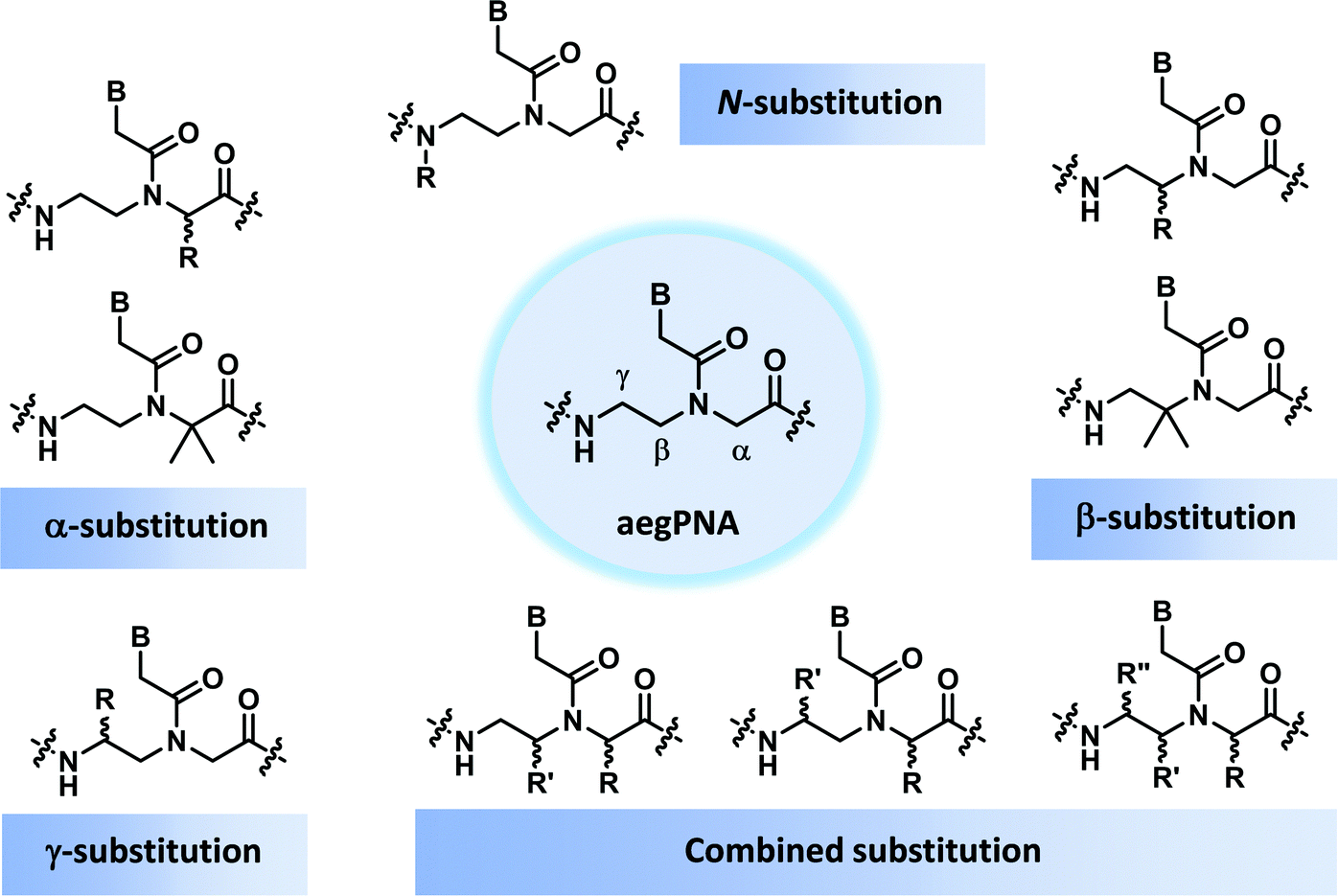 | ||
| Fig. 2 Possible sites for modification of aegPNA without forming a cyclic structure as part of the backbone. | ||
Modified PNA with α-substituents (αPNA)
Due to its synthetic accessibility from standard amino acids, αPNA is one of the earliest PNA modifications that has been introduced as early as 1994 by the same group that developed aegPNA.17 According to the thermal denaturation study on a chimeric α/aegPNA system, the introduction of a small methyl substituent at the α-position of the aegPNA backbone slightly destabilizes the PNA·DNA duplex in a stereochemically dependent fashion. The DAla αPNA modification was less destabilizing than the LAla modification. Bulky or negatively charged substituents are more destabilizing than smaller ones.18,19 The D-Lys modification showed the most promising DNA binding characteristics whereby the duplex stability was slightly enhanced due to the combination of correct stereochemistry and positively charged side chain.18,20 CD studies indicated a preferential right-handedness pre-organization of the single-stranded PNA carrying even only one D-Lys modification in the middle of the strand.20 The matching chirality of the D-Lys modified αPNA and native DNA explained the higher stability of the αPNA·DNA duplexes of the D- over the L-series. A molecular dynamics (MD) simulation of Leu-modified αPNA·DNA duplexes suggested that the amino acid side chain in the D-series is directed away from the minor groove.21 The blockage of the minor groove in the L-series was responsible for the decrease in the binding of groove binding dyes compared to the D-series or unmodified PNA. Importantly, the mismatch specificity and antiparallel selectivity of DLys αPNA were enhanced when compared to unmodified aegPNA. A stretch of three consecutive D-Lys “chiral box” modifications was sufficient to drive the binding mode to exclusively antiparallel.22Tm data of αPNA hybrids are summarized in Table 1.|
|
||||
|---|---|---|---|---|
| Sequencea (N–H/C–NH2) | Substituent (R) derived from | T m DNA (°C) | ΔTmb (°C) | Ref. |
| a The modification site is denoted by a small letter. b ΔTm refers to Tm difference between the modified PNA and aegPNA duplexes. | ||||
| GTAGATCACT | None (aegPNA) | 49 | — | 20 |
| GTAGAtCACT | L-Leu | 47 | −2 | 20 |
| GTAGAtCACT | L-Lys | 47 | −2 | 20 |
| GTAGAtCACT | D-Lys | 51 | +2 | 20 |
| GtAGAtCACt | D-Lys | 53 | +4 | 20 |
| GTAGatcACT | D-Lys | 43 | −6 | 22 |
The crystal structure of such chiral box αPNA was successfully determined at 1.66 Å resolution (Fig. 3).23 It revealed an antiparallel right-handed double helix that is grossly similar to the P-helix with a diameter of 22 Å, a helical twist of 23.2°, a rise of 3.5 Å, an x-displacement of 3.8 Å, and 15.5 base pairs per turn. The DNA sugar rings adopt both C3′-endo and C2′-endo conformations (6 vs. 4). This is in sharp contrast to the PNA2·DNA triplexes24 and PNA·DNA duplexes25, in which the DNA sugar rings adopted the C3′-endo and predominantly C2′-endo, respectively. Thus, the P-helix can accommodate both types of sugar ring puckering well. All amide bonds in the PNA backbone are in the trans-conformation without the intramolecular hydrogen bonding earlier proposed based on a molecular mechanics calculation.26 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the methylenecarbonyl linker pointed towards the C-terminus of the PNA strand to maximize the stabilizing n → π* interaction with the backbone carbonyl of the glycine unit. The lysine side chain did not electrostatically interact with the phosphate backbone of the DNA counter strand. However, it formed an ion pair with the DNA phosphate backbone of the adjacent PNA·DNA duplex in the crystal lattice to provide additional stabilization, but this interaction would likely be absent in the solution phase. The right-handed helicity was determined primarily by the steric interaction between the D-Lys side chain and the N-terminal aminoethyl group. The improved directional and mismatch specificity was probably benefitted from the conformational restriction imparted by the D-Lys substituent.
O group of the methylenecarbonyl linker pointed towards the C-terminus of the PNA strand to maximize the stabilizing n → π* interaction with the backbone carbonyl of the glycine unit. The lysine side chain did not electrostatically interact with the phosphate backbone of the DNA counter strand. However, it formed an ion pair with the DNA phosphate backbone of the adjacent PNA·DNA duplex in the crystal lattice to provide additional stabilization, but this interaction would likely be absent in the solution phase. The right-handed helicity was determined primarily by the steric interaction between the D-Lys side chain and the N-terminal aminoethyl group. The improved directional and mismatch specificity was probably benefitted from the conformational restriction imparted by the D-Lys substituent.
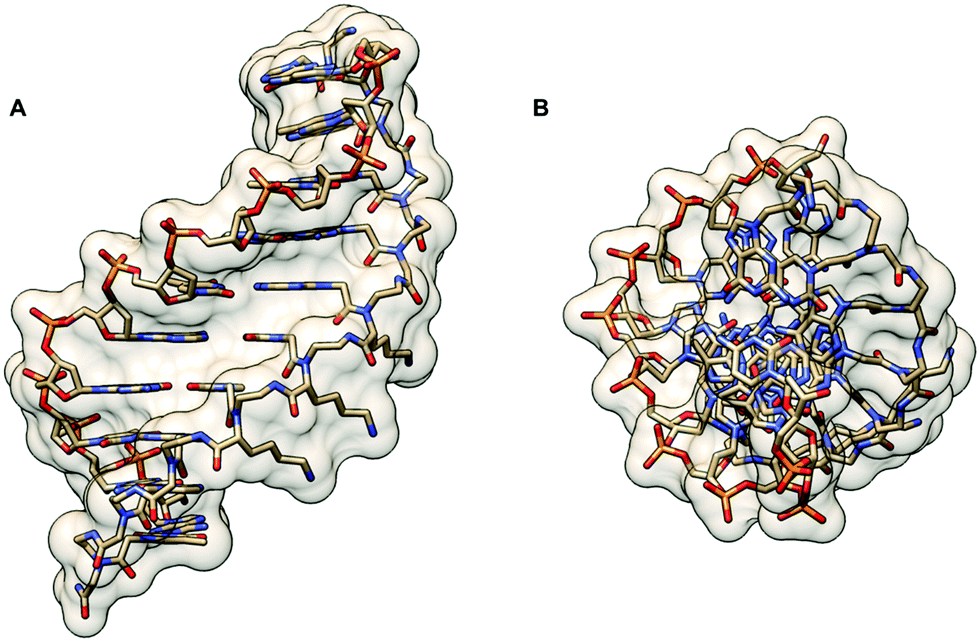 | ||
| Fig. 3 X-Ray structure of the αPNA·DNA duplex (1NR8): (A) side view from the minor groove side and (B) top view from the 3′/N-side along the helix axis.23 | ||
Modified PNA with two α-substituents
In contrast to the marginal change in the DNA/RNA duplex stability of mono-substituted αPNA, the introduction of a gem-dimethyl substituent at the α-position of the aegPNA backbone (aedmgPNA) was reported to substantially increase the stability of both DNA and RNA duplexes.27 This is true for both PNAs with chimeric and homogeneous backbones, although the data for the latter were available only for the homothymine sequence. The stability of the PNA·DNA hybrids was increased much more than that of the corresponding RNA hybrids, resulting in the reversal of DNA/RNA binding selectivity when compared to aegPNA. In addition, the antiparallel duplexes were also selectively stabilized over the parallel duplexes, making the antiparallel selectivity much better than in the case of aegPNA. The backbone-extended homolog of aegPNA with similar α-gem-dimethyl substituents (apdmgPNA) also exhibited improved stability albeit to a lesser extent. In the absence of the gem-dimethyl substituent, the backbone-extended PNA exhibited very poor binding affinity towards the DNA target. The stability against mismatched DNA was evaluated only with the homothymine sequence and was found to be more discriminating than the corresponding aegPNA hybrids (Table 2).|
|
|||
|---|---|---|---|
| Sequencea (N–H/C–NH2) | Modification | T m DNAb (°C) | T m RNAb (°C) |
| a The modification site is denoted by a small letter. b Antiparallel duplexes; Tm values for parallel duplexes are shown in parentheses. | |||
| TTTTTTT-Lys | None (aegPNA) | 43 | 54 |
| TtTtTtTt-Lys | aedmgPNA | 80 | 73 |
| ttttttttt-Lys | aedmgPNA | >85 | >85 |
| TtTtTtTt-Lys | apdmgPNA | 59 | 57 |
| tttttttt-Lys | apdmgPNA | 77 | 61 |
| GTAGATCACT | None (aegPNA) | 50 (38) | 50 (40) |
| GtAGAtCACt | aedmgPNA | 81 (56) | 72 (32) |
| GtAGAtCACt | apdmgPNA | 73 (50) | 65 (33) |
A recent structural study of the α-gem-dimethylated PNA monomer was performed by NMR and computational calculation which revealed the exclusive Z-rotamer preference of the tertiary amide bond in the PNA backbone (Fig. 4).28 On the other hand, previous studies indicate that a mixture of Z/E rotamers are formed in the case of aegPNA monomers and dimers,29–31 while only the Z-rotamers were observed in the NMR and crystal structures of PNA duplexes.32 The steric repulsion between the α-gem-dimethyl substituent and the CH2B on the tertiary amide group favored the formation of the Z-rotamer, which was also further stabilized by the n → π* interaction. This suggests that the tertiary amide bond in the α-gem-dimethylated PNA monomer is pre-organized in the correct Z-rotamer required for the duplex formation. The smaller entropy loss in the hybridization process nicely explains the higher stability of the aedmgPNA over aegPNA hybrids. At present, only the T-monomer is available; thus it is not yet known whether the conclusion will be general for mixed-sequence PNA with a fully α-gem-dimethylated backbone. In addition, the mismatch specificity has not yet been reported. Nevertheless, the preliminary results look promising. Other α-gem-disubstituted PNA monomers have been synthesized,33–35 although the incorporation into PNA oligomers and their DNA binding properties have not been reported.
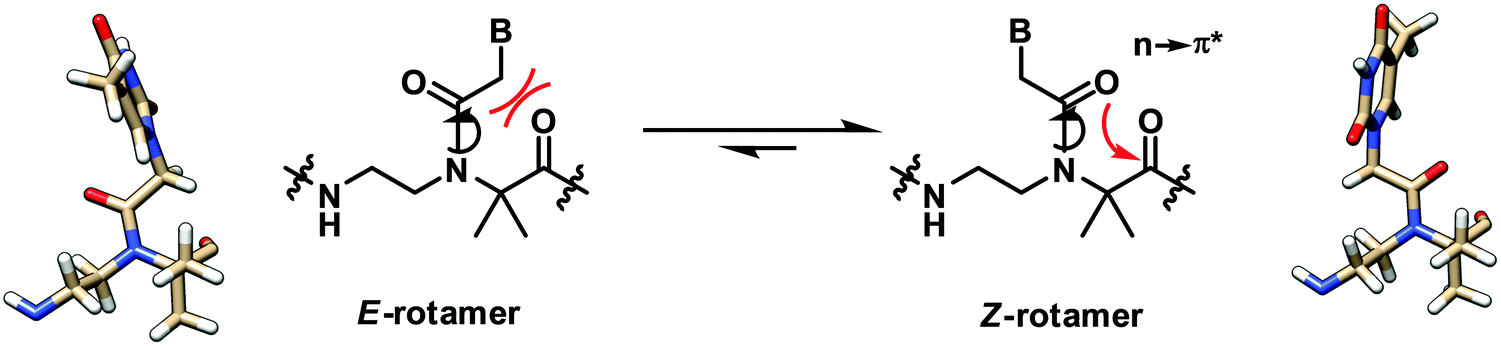 | ||
| Fig. 4 Interconversion of the E- and Z-rotamers in the α-gem-dimethylated PNA monomer.28 | ||
Modified PNA with β-substituents (βPNA)
While PNA systems with the β-carbon atom as part of a cyclic structure have been extensively studied (see Modified PNA with cyclic structure as part of the backbones), only one β-substituted PNA system with simple methyl substituents has been reported so far.36 This could be attributed to the less availability of the chiral diamine building blocks required for the synthesis of βPNA compared to the amino acid-derived αPNA and γPNA. The chimeric mixed-sequence PNA carrying three β-methyl substituents in the (S)-configuration when hybridized to DNA provided a duplex with comparable stability to unmodified aegPNA (Table 3). Similar to the case of αPNA, the stereochemistry of the substituent significantly affects the PNA·DNA duplex stability. However, the effect is more pronounced as shown by the complete absence of DNA binding of the corresponding β-methyl-substituted PNA with the (R)-configuration. Molecular modeling of the PNA·DNA duplexes indicated a steric clash between the (R)-β-methyl substituent and the CH2 group of the BCH2CO substituent on the PNA backbone (Fig. 5). In the case of αPNA, the steric clash between the less hindered CO group and the α-methyl substituent is expected to be smaller, which is consistent with the observed experimental results. In addition, CD spectra of the single-stranded βPNA suggested pre-organization into a helical conformation with opposite helical senses for the two different configurations. The (S)-β-methyl-substituted PNA adopted a right-handed helical conformation that matched the natural handedness of native DNA duplexes (Fig. 6). However, after hybridization, the CD signal changed significantly indicating a substantial conformational change which might counterbalance the effect of conformational pre-organization, resulting in the observed lack of stabilization relative to the unmodified aegPNA.
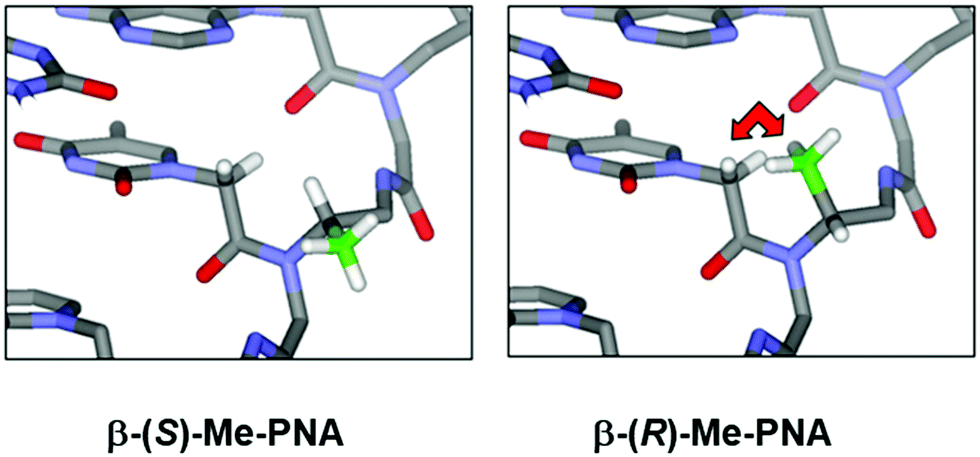 | ||
| Fig. 5 A model of the βPNA structure showing a different degree of steric clash between the β-(S)- or β-(R)-methyl groups and the nucleobase side arm (reprinted from ref. 36, Copyright (2011), with permission from Elsevier). | ||
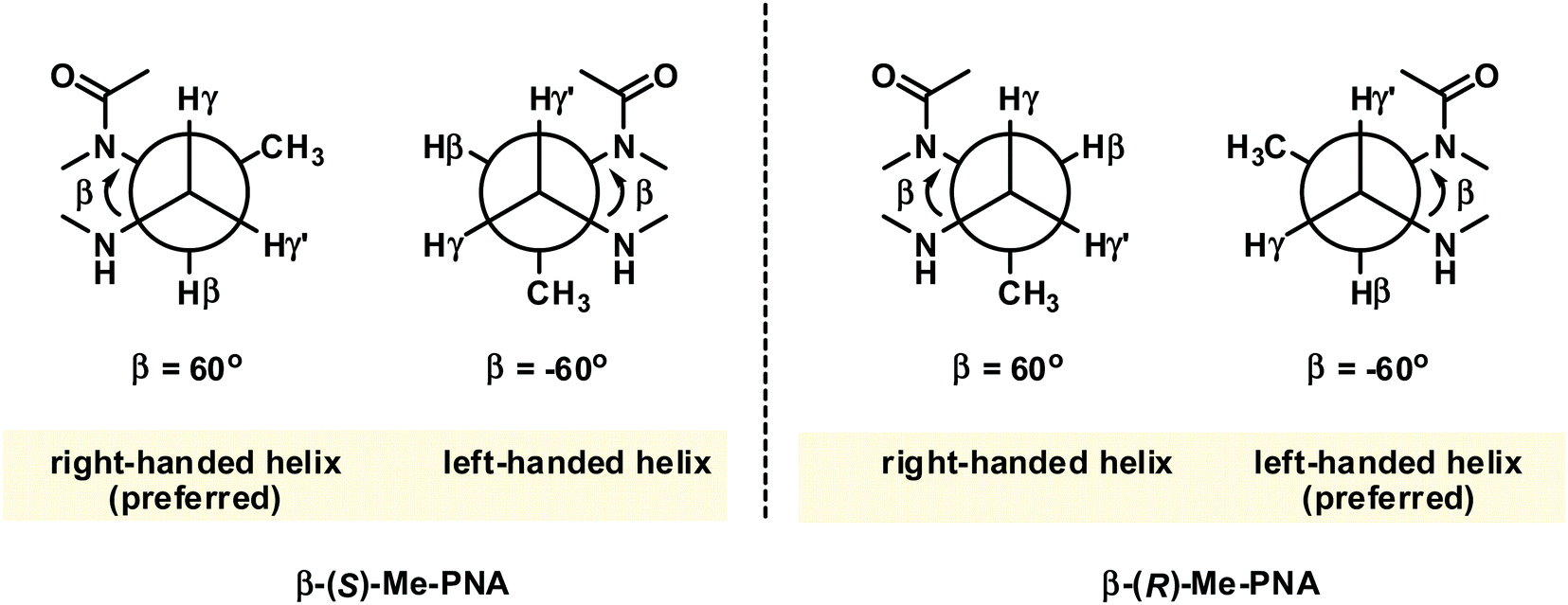 | ||
| Fig. 6 The gauche conformation of the N(H)–Cγ–Cβ–N(CO) groups necessitates pre-organization into the right-handed helix in β-(S)-methyl PNA.36 The structure is viewed along the Cγ–Cβ axis. | ||
Modified PNA with two β-substituents
In contrast to the α-gem-dimethylated PNA, the analogous NMR and theoretical study on the β-gem-dimethylated PNA monomer indicated that the E-rotamer was preferred over the Z-rotamer.28 In fact, only the E-rotamer was observed by NMR in the monomer. According to the simulation of the mono-β-methyl-substituted PNA above, the Z-rotamer would result in a severe steric clash with the pro-R methyl group, thus necessitating the adoption of the E-rotamer despite the loss of the stabilizing n → π* interaction (Fig. 7). Since the Z-rotamer was exclusively observed in the structures of all PNA duplexes and triplexes reported in the literature,32 it is unlikely that the β-gem-dimethylated PNA will interact strongly with the DNA counterpart. However, this remains to be experimentally verified.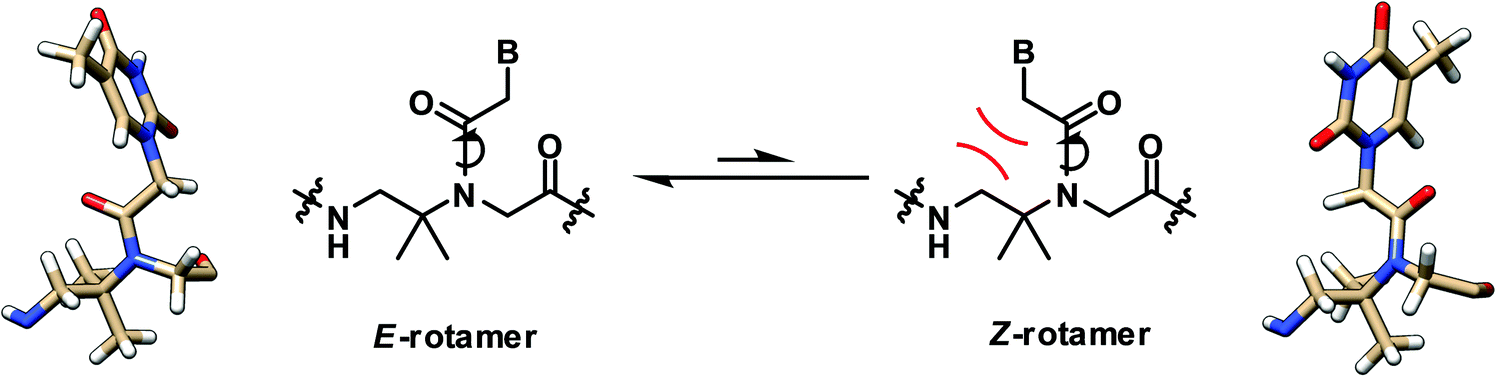 | ||
| Fig. 7 Interconversion of the E- and Z-rotamers in the β-gem-dimethylated PNA monomer.28 | ||
Modified PNA with γ-substituents (γPNA)
The γ-modified PNA monomers are conveniently obtained from standard amino acids via the corresponding N-protected α-amino aldehydes. They were first reported as early as 1994,37 but it was not until more than a decade later that the full potential of γPNA was recognized. Early studies focused on the use of γ-substituents as a handle for subsequent modification with dyes or other functional entities to create functional PNA probes38,39 or for creating long PNA strands by native chemical ligation.40,41 These reports suggest that the partial γ-substitution by the lysine or cysteine side chain does not much affect the PNA·DNA duplex stability provided that the chirality of the γ-carbon was derived from the natural L-amino acids. The remarkable stabilizing effect of the γ-substituent was revealed for the partially and fully modified γPNA derived from L-serine in the seminal paper by Ly's group in 2006.42 A single γ-hydroxymethyl modification (LSer γPNA) in a 10mer aegPNA increased the Tm of the PNA·DNA and PNA·RNA duplexes by +4 and +3 °C, respectively. In the fully γ-modified PNA, the Tm values for the PNA·DNA and PNA·RNA duplexes were increased by +19 and +10 °C with improved mismatch specificity (ΔTm = 16–19 and 12–18 °C) (Table 4). The CD spectra of single-stranded γPNA suggested the formation of helical structures – the helicity of which was determined by the configuration of the Cγ. The signal became more intense and changed from PNA·PNA- to PNA·DNA-like helices when more γPNA units were incorporated into the aegPNA strand. The observed CD signal in the single-stranded γPNA was much more pronounced than the corresponding αPNA. The degree of pre-organization is temperature-dependent – being diminished at higher temperatures – but is not sensitive to the concentration, thus indicating the intramolecular nature of the pre-organization. By varying the position of the γPNA unit in the PNA strand, it was revealed that the helical induction was unidirectional from the C-to-N termini. Even the small methyl substituent could effectively induce the pre-organization.|
|
|||||
|---|---|---|---|---|---|
| Sequence (N–H/C–LysNH2)a | Substituent (R-) | T m DNA (°C) | T m RNA (°C) | T m mmDNAb (°C) | T m mmRNAb (°C) |
| a The modification site is denoted by a small letter. b T m values for mismatched targets, expressed as a range for different mismatched bases. | |||||
| GCATGTTTGA | None (aegPNA) | 44 | 54 | 30–34 | 36–43 |
| GCATGtTTGA | (R)-HOCH2- | 48 | 57 | — | — |
| GCAtGtTtGA | (R)-HOCH2- | 53 | 60 | — | — |
| GCATGtttGA | (R)-HOCH2- | 53 | 59 | — | — |
| gcatgtttga | (R)-HOCH2- | 63 | 64 | 44–47 | 46–52 |
Conformational studies of a γPNA dimer C(LAla)T in solution by NMR indicated that it exists as a mixture of tertiary amide rotamers similar to aegPNA. However, the γPNA backbone adopted a well-defined conformation while the aegPNA dimer with the same sequence adopted a random coil structure. Detailed analysis of coupling constants revealed that the backbone of all four rotamers of γPNA adopted a right-handed helical structure with a torsional angle β [N(H)–Cγ–Cβ–N(CO)] in the range of +50 to +60° to avoid the steric clash between the γ-substituent and the C-terminal backbone tertiary amide nitrogen atom (Fig. 8). Theoretical calculation suggested that the right-handed conformation was more stable than the left-handed conformation by 3.5 kcal mol−1, i.e. >99% population excess. A solution NMR structure of a γPNA·γPNA duplex43 and an X-ray structure of a γPNA·DNA duplex are available.44 The conformation of the single-stranded γPNA dimer42 closely resembles the conformation of the γPNA·γPNA duplex,43 the γPNA·DNA duplex,44 as well as the aeg/αPNA unit from an X-ray structure of a PNA·DNA duplex (Fig. 9).23 This indicates that the γ-substituent forces the single-stranded PNA to adopt the appropriate helical conformation that is similar to those found in the duplexes thus explaining its strong binding properties. The three-dimensional structures also revealed that the γ-substituent resided at the edge of the grooves of the γPNA·γPNA and γPNA·DNA duplexes and should not significantly interfere with the duplex formation. Indeed, several other substituents in addition to CH3 including HOCH2-,42 HOCH2CH2OCH2-,45 guanidinoethyl,46 guanidinobutyl-,47 H2NCH2CH2-,46 H2NCH2CH2CH2-,48 H2NCH2CH2CH2CH2-,38,39 and HO2CCH2CH2-49 have been placed at the γ-position without significantly altering the binding properties as long as the L-configuration is preserved. Charged groups showed relatively little effects on the binding affinity and specificity. However, the electrostatic effect may be significant if the linker was short as in the case of H2NCH2-γ-PNA whereby the duplex stability was higher than in the case of the amino-modified γPNA with longer linkers (Table 5).48,50,51 Interestingly, γPNA with the negatively charged –CH2SO3− substituent adopted a more extended conformation in the single-stranded form as shown by NMR and CD analyses which could be due to the intra-strand electrostatic and steric interactions among the sulfate groups. Accordingly, the PNA2·DNA triplexes formed from γ-sulfated PNA were less stable than the corresponding unmodified PNA.52 On the other hand, the corresponding carboxyethyl-modified γ-PNA formed a highly stable triplex with oligo(dA).49 Under physiological ionic strength, γPNA with negatively charged side chain bound more strongly to RNA than DNA, and the reverse was true for γPNA with positively charged side chain.53 However, the selectivity was rather small.
 | ||
| Fig. 8 The gauche conformation of the N(H)–Cγγ–Cβ–N(CO) groups necessitates pre-organization into the right-handed helix in γ-(S)-methyl PNA. The structure is viewed along the Cγ–Cβ axis (adapted with permission from ref. 42. Copyright 2006 American Chemical Society). | ||
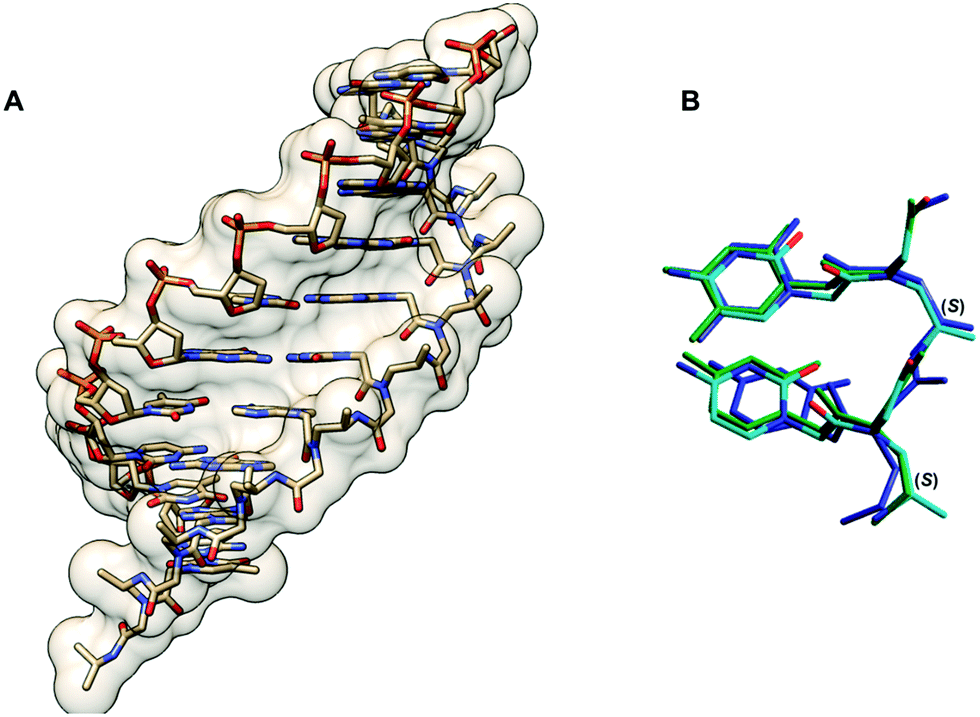 | ||
| Fig. 9 (A) X-Ray structure of the γPNA·DNA duplex (3PA0) viewed from the minor groove side.44 (B) Superposition of the PNA strand from the γPNA·DNA duplex (cyan), aegPNA·DNA duplex (green), and single-stranded γPNA dimer (blue) (reprinted with permission from ref. 44. Copyright 2010 American Chemical Society). | ||
| Sequence (N–H/C–LysNH2)a | Substituent | T m DNA (°C) | T m RNA (°C) | Ref. |
|---|---|---|---|---|
| a The modification site is denoted by a small letter. | ||||
| TTACCTCAGT | None (aegPNA) | 49 | 62 | 48 |
| TtACCtCAGT | H2NCH2-, α-(S) | 54 | — | 50 |
| TtACCtCAGT | H2NCH2-, α-(R) | 58 | — | 50 |
| TtACCtCAGT | H2NCH2-, γ-(S) | 62 | — | 50 |
| TtACCtCAGT | H2N(CH2)2-, γ-(S) | 50 | 59 | 46 |
| TtACCtCAGT | H2N(CH2)3-, γ-(S) | 53 | 63 | 48 |
Modified PNA with two γ-substituents
The solution structure of a γ-gem-dimethylated PNA monomer was also studied by NMR, which indicated that both Z- and E-rotamers exist in almost equal amounts (Fig. 10).28 Theoretical calculation confirmed the similar stability and indicated the absence of significant stabilization interactions including the n → π* interaction in both rotamers. In contrast to mono-γ-substituted PNA, the helicity induction by the chirality of the γ-substituent will be absent. This, together with the significant steric clash of the CH2B with one of the γ-methyl groups in the Z-rotamer which could not be relieved by adopting the alternative helicity, suggests that the γ-gem-dimethylated PNA may not interact strongly with the DNA counterpart. This, however, remains to be verified experimentally.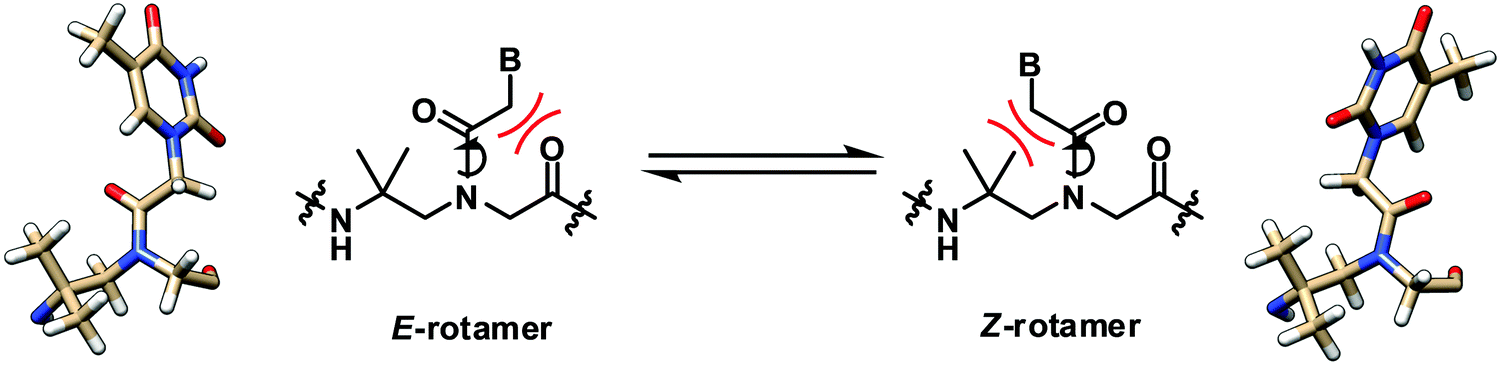 | ||
| Fig. 10 Interconversion of the E- and Z-rotamers in the γ-gem-dimethylated PNA monomer.28 | ||
N-Methylated PNA: modified PNA with N-substituents
Only one example of N-substituted PNA has been reported whereby the N-substituent was the methyl group.54 The Tm values of the duplexes of the fully N-methylated PNA decamer with both DNA and RNA were decreased compared to those of the unmethylated PNA by 12–18 °C, although partial substitution was more tolerated (Table 6). CD spectra indicated that the incorporation of up to 30% of N-methylation does not change the overall conformation of the PNA·DNA duplexes, but a distinct conformation was observed in the fully methylated PNA·DNA duplexes. The crystal structure of a 50% methylated PNA·PNA hexamer duplex was determined at 2.2 Å resolution (Fig. 11). Since the PNA·PNA duplex is achiral, both left-handed and right-handed antiparallel duplexes were observed in the asymmetric unit. The helical parameters include a diameter of 28 Å, a helical twist of 19–20°, a rise of 3.5/3.8 Å, an x-displacement of 4.8/7.2 Å, and 18 base pairs per turn which are consistent with the P-helix. Although the overall structures of the N-methylated and unmethylated PNA·PNA duplexes are similar, the major difference is the orientation of the backbone carbonyl groups. In the case of the N-methylated monomer, the amide carbonyl group turned inward to form a water-mediated hydrogen bonding with the nucleobase. For the unmethylated monomer, this amide carbonyl pointed away towards the solution and the NH group formed a water-mediated hydrogen bonding with the nucleobase instead. No other examples or applications of N-substituted PNAs have been reported so far. In view of the minimal impact on the overall conformation and the ease of modification of the nitrogen atom without the problem of introduction of additional chirality, this system offers a potential platform for the introduction of additional functions to the PNA molecules.
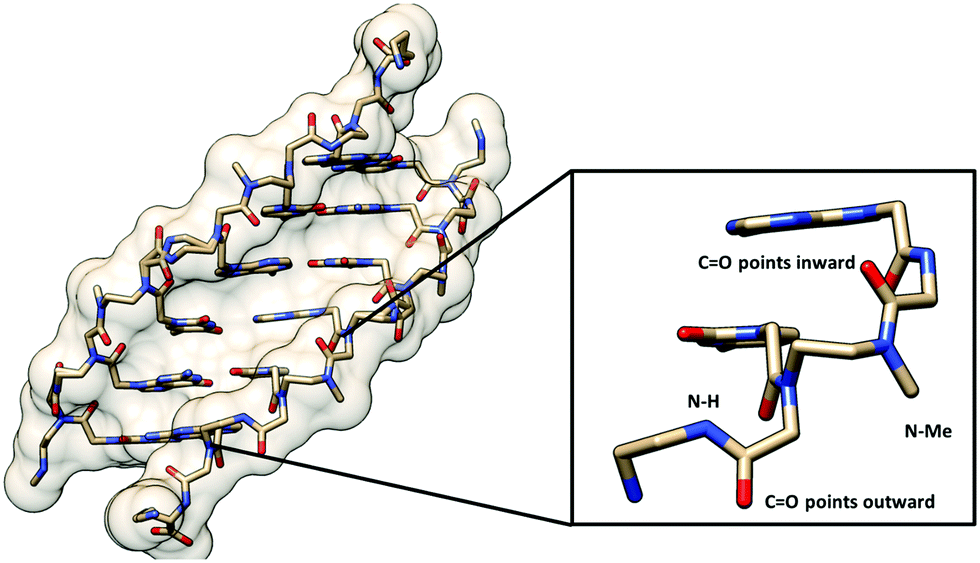 | ||
| Fig. 11 X-Ray structure of the N-Me PNA·PNA duplex (1QPY) viewed from the minor groove side with the different orientation of the backbone carbonyl groups of the N-Me and normal aegPNA monomers shown.54 | ||
Backbone-modified PNA with multiple modifications
In most PNAs with multiple modifications, the substituents are generally tied to form a ring. Such PNAs with a cyclic structure will be covered under the topic “Modified PNA with cyclic structure as part of the backbones”. This section will cover only when the substituents were not connected to form a ring. In 2005, Marchelli et al. reported a modified PNA system bearing two substituents at the α- and γ-positions – one at each position.55,56 The substituent at both positions was the same aminobutyl group derived from lysine with different stereochemistries. All four combinations (αD/γD, αD/γL, αL/γD, αL/γL), as well as the individually modified (αD, αL, γD, γL) PNAs, were synthesized and the stability of the corresponding complementary PNA·DNA hybrids was compared (Table 7). According to thermal stability measurements, the L-configuration at the γ-position is strongly preferred over the D-configuration. The chirality at the α-position showed much smaller effects on the PNA·DNA duplex stability, with the D-configuration being more preferred than the L-configuration. The results are consistent with the PNA individually modified at either the α- or γ-positions, suggesting that the two sites are independent, and the stabilization effects are additive/synergistic. Thus, the αD/γL showed the highest thermal stability, and no DNA binding was observed in the αL/γD isomer. According to CD spectroscopic studies, the helicity in α/γPNA·aegPNA and α/γPNA·DNA duplexes is primarily controlled by the configuration of the γ-position, and when the helicity induced by both positions matched that of the DNA, strong binding was observed. The preference for right- or left-handed helicities arose from the attempt to minimize the intrastrand steric clashes due to the amino acid side chains. Steric disturbances are less tolerated in the γ-position than in the α-position, and consequently, the helical induction is dominated by the stereogenic center at the γ-position. Thermodynamic parameters extracted from the melting curve data suggested that the contribution of the α-position is dominated by the enthalpic term (i.e., more negative enthalpy change), while the contribution of the γ-position is dominated by the entropic term (i.e., less negative entropy change).|
|
||||
|---|---|---|---|---|
| System | Helical induction induced by Cα | Helical induction induced by Cγ | Overall helical preference | T m DNAa (°C) |
| a T m was determined from CD melting curves at 260 nm; PNA sequence = H-GTAGAtCACT-NH2; DNA sequence = dAGTGATCTAC. The modification sites are denoted by a small letter. | ||||
| αD,γL | Right-handed | Right-handed | Right-handed | 57 |
| γL | — | Right-handed | Right-handed | 56 |
| αL,γL | Left-handed | Right-handed | Right-handed | 52 |
| αD | Right-handed | — | Right-handed | 52 |
| Unmodified | — | — | — | 50 |
| αL | Left-handed | — | Left-handed | 47 |
| αD,γD | Right-handed | Left-handed | Left-handed | 33 |
| γD | — | Left-handed | Left-handed | 32 |
| αL,γD | Left-handed | Left-handed | Left-handed | <20 |
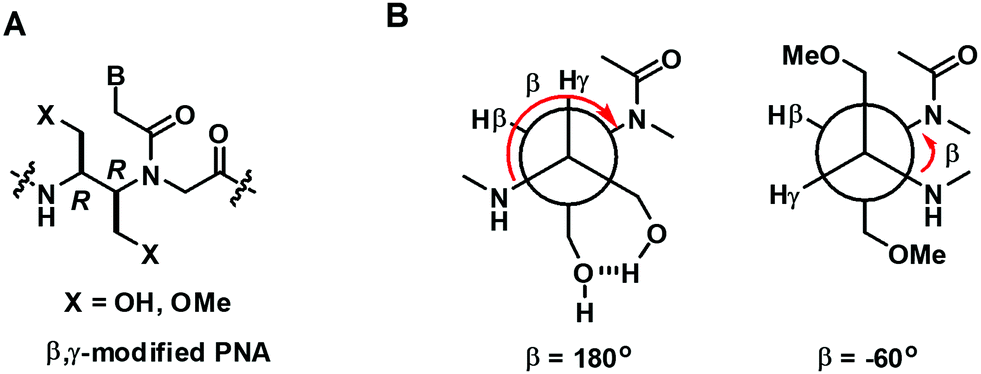
In a more recent example, Kumar et al. reported the only example of acyclic β,γ-disubstituted PNA.57 The substituents studied were methoxymethyl and hydroxymethyl in the (R,R) and (S,S)-configurations (Fig. 12A). Thermal denaturation studies on chimeric aeg/β,γ-bis(methoxymethyl)-substituted PNA with DNA/RNA showed comparable or slightly improved stability over aegPNA for the (R,R) isomer (Table 8). The duplex with parallel DNA was more destabilized than the antiparallel DNA, leading to a higher directional specificity when compared to aegPNA. Much lower stability was observed for the (S,S)-isomer, with the ΔTm in the range of −20 °C for a single modification, and the introduction of another modification completely diminished the binding. The preferred stereochemistry is in accordance with other β- or γ-substituted PNA as well as β,γ-linked PNA with cyclic structures, but the configuration notations were different due to the change in priority of the groups involved. Interestingly, the presence of two hydroxymethyl substituents was detrimental to both DNA and RNA binding as the introduction of only one modification as either (R,R) or (S,S)-isomers resulted in a complete inability to form stable hybrids with both DNA and RNA targets. It was proposed that the intramolecular hydrogen bonding between the two adjacent hydroxymethyl groups changes the torsional angle β from 60° to 180° making the conformation unsuitable for DNA/RNA binding (Fig. 12B).
 | ||
| Fig. 12 (A) Structure of β,γ-modified PNA. (B) The proposed different conformation of the OH and OMe substituted PNA (reproduced from ref. 57 with permission from the Royal Society of Chemistry). | ||
| Sequence (N–H/C–LysNH2)a | Modification | T m apDNAbcd (°C) | T m pDNAbcd (°C) | T m apRNAbcd (°C) |
|---|---|---|---|---|
| a The modification site is denoted by a small letter. b ap = antiparallel; p = parallel. c (R,R)-isomer; values for the (S,S)-isomer are shown in parentheses. d No melting transition is denoted by n.t. | ||||
| AACCGATTTCAG | None (aegPNA) | 58 | 46 | 63 |
| AACCGAtTTCAG | X = HO- | n.t. | n.t. | n.t. |
| AACCGAtTTCAG | X = MeO- | 59 (44) | 38 | 65 (52) |
| AACCGaTTTCAG | X = HO- | n.t. | n.t. | n.t. |
| AACCGaTTTCAG | X = MeO- | 58 (38) | 39 | 62 (49) |
| AACCGAtTtCAG | X = MeO- | 60 (n.t.) | n.t. | 66 (n.t.) |
Modified PNA with cyclic structure as part of the backbones
This section will discuss how conformation constraint can be introduced in the aegPNA structure by introducing a bond, an atom or a group of atoms (denoted by X) to form a bridge between different positions in the core backbone. Such bridging would create a carbocyclic or heterocyclic ring within the PNA backbone leading to new conformationally constrained PNA analogs. Possible sites for the introduction of bridging include α, β, γ, N and α′ as illustrated in Fig. 13, although not all possibilities of such bridging may have been realized as these modified PNAs are more synthetically challenging when compared to the simple backbone-substituted αPNA or γPNA. The carbonyl group on the nucleobase side arm may optionally be removed. In addition, the backbone may be extended, or some atoms swapped. The modified PNA systems will be discussed according to the bridging positions.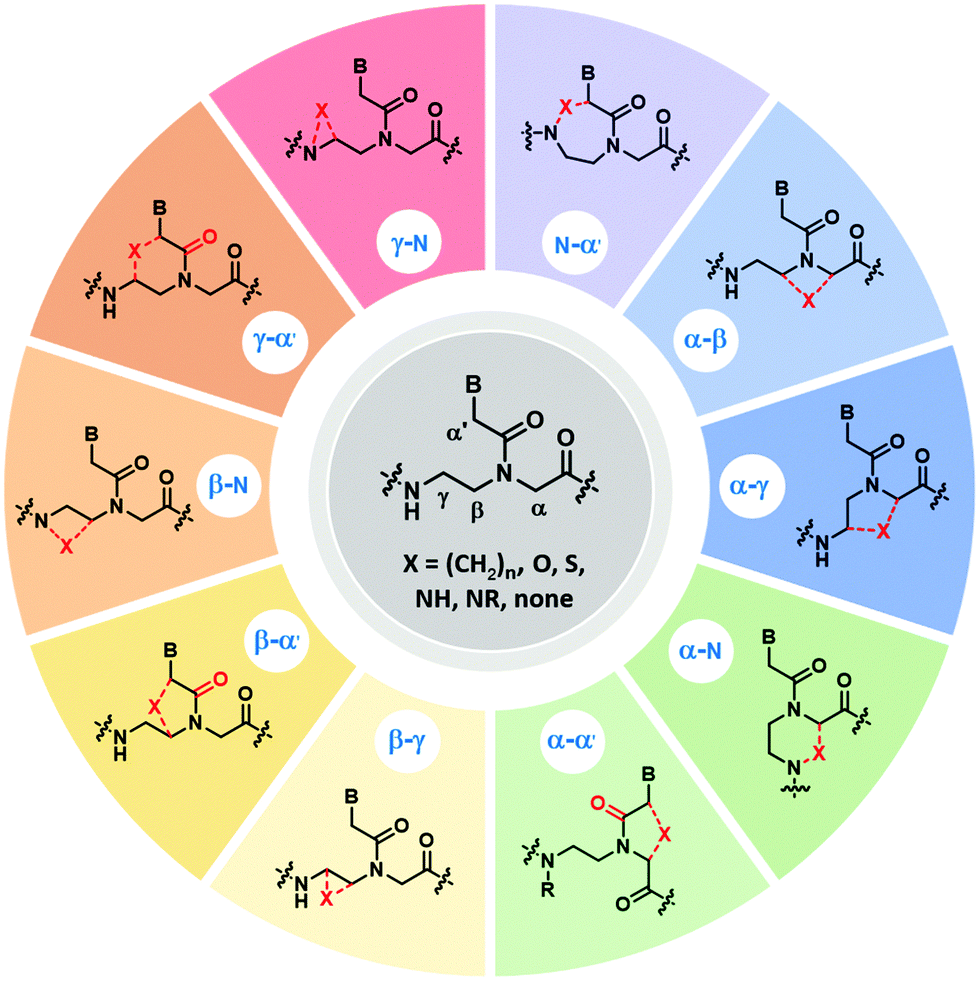 | ||
| Fig. 13 Possible sites for modification of aegPNA by forming a cyclic structure as part of the backbone. | ||
Modified PNA with (β-γ)-linkage
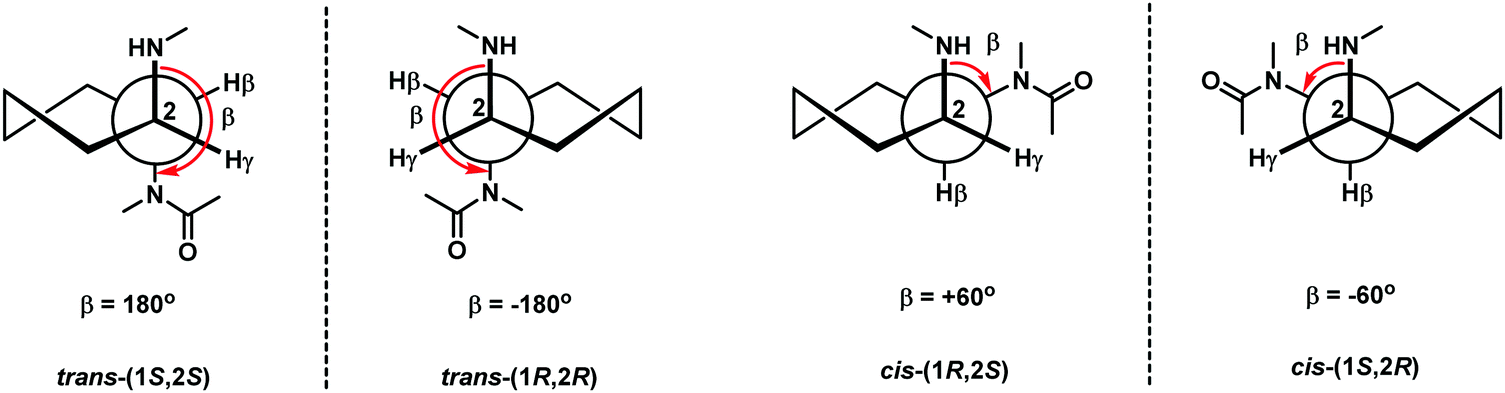
The bridging between the β- and γ- positions within the aegPNA backbone by a –(CH2)n– linkage introduces a carbocyclic ring within the aegPNA structure. Such carbocyclic bridging will increase rigidity in the structure by the limited conformations that the ring can adopt. The introduction of the ring also generates chirality that can affect pre-organization of the PNA chain, although not all possible combinations may have been investigated. Only the 3-, 5- and 6-membered carbocyclic ring (n = 1, 3 and 4) modifications have been reported in the literature (Fig. 14). The trans-cyclohexyl PNA (chPNA) was the first example of such carbocyclic PNA systems that were reported since early dates (Fig. 15).58 The DNA binding of chPNA is stereochemically dependent. The partial introduction of the (S,S)-cyclohexyl PNA monomer into the aegPNA strand resulted in a small effect on the stability of the PNA·DNA and PNA·RNA hybrids, with ΔTm = ±1 °C per (S,S)-cyclohexyl PNA unit. On the other hand, the (R,R)-cyclohexyl PNA significantly reduced the stability of the PNA·DNA and PNA·RNA hybrids. Molecular dynamics simulations based on the NMR structure of an aegPNA·DNA duplex25 suggested that the two amino groups in the diaminocyclohexane unit assumed an anti-periplanar relationship with a torsional angle β close to 180° and only minimal conformation adjustment was required for the (S,S)-chPNA binding to DNA, whereas more pronounced structural perturbation was observed in the (R,R)-chPNA. In the former case, the cyclohexane bridge was located outside the helix, while in the latter case the cyclohexane bridge resided within the major groove. However, the fully modified mixed-sequence (S,S)-chPNA decamer formed less stable duplexes with DNA and RNA when compared to aegPNA (Tm = 41 °C and 37 °C for PNA·DNA and PNA·RNA, respectively). Thermodynamic data indicated that the introduction of the (S,S)-chPNA indeed reduces the entropy loss as proposed, but it also lowers enthalpic gain resulting in lowering of the overall stability than aegPNA.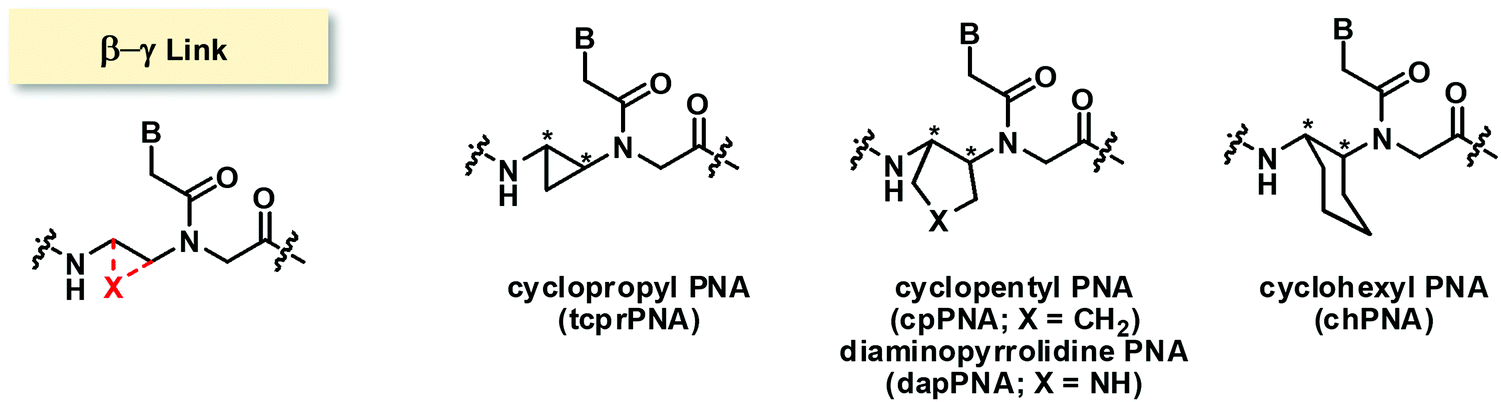 | ||
| Fig. 14 Structures of modified PNA with (β–γ)-linkage. | ||
 | ||
| Fig. 15 Structures of cyclohexane-modified PNA with (β–γ)-linkage. | ||
In a series of subsequent studies by Kumar and Ganesh, the cis-chPNA systems with the (1S,2R) and (1R,2S) configurations on the diaminocyclohexane ring were reported.59–61 The major difference between the trans- and cis-diaminocyclohexane system is the torsional angle (β; NH–CH–CH–NHCO) which should be in the range of 60°–90° for the cis-isomer (+60° in the case of (1R,2S)-chPNA and −60° in the case of (1S,2R)-chPNA62) and 180° for the trans-isomer if the two amino substituents adopt the diaxial conformation or close to 60° if they assume the diequatorial conformation (Fig. 15). This indicates that cis-(1S,2R/1R,2S)-chPNA has a torsion angle more closely similar to that of the PNA·RNA duplex than the PNA·DNA duplex based on the torsional angle β of 60–70° and 140° obtained from NMR studies of PNA·RNA63 and PNA·DNA duplexes,25 respectively.
Thermal stability data of aegPNA and chimeric ch/aegPNA with DNA and RNA are summarized in Table 9. The results suggest the importance of the stereochemistry of the cyclohexane ring that leads to the proper arrangement and specific torsion angle which could provide a basis for the selective binding of RNA over DNA targets. Given the substantial destabilization of the (1R,2R)-chPNA when compared to the (1S,2S)-chPNA systems, the relatively high stability for the PNA·RNA duplexes observed for both cis-(1S,2R)- and (1R,2S)- chPNA is quite remarkable. Nevertheless, these results are only from chimeric systems and care should be taken in interpreting the results as already noted in the case of trans-chPNA.58 At present, no Tm data for the fully modified cis-chPNA systems are available.
| PNA sequencesa (N–H/C–LysNH2) | Modification | β | T m (°C) DNA | T m (°C) RNA | Ref. |
|---|---|---|---|---|---|
| a The modification site is denoted by a small letter. b Torsional angle value of aegPNA was taken from ref. 25, and others were based on X-ray structures of monomers. | |||||
| TTTTTTTTTT | None (aegPNA) | 141 | 72 | 81 | 58 |
| TTTTtTTTTT | (1S,2S)-cHex | 70 | 76 | 58 | |
| TTTTtTTTTT | (1R,2R)-cHex | 52 | 56 | 58 | |
| TTTTtTTTTT | (1S,2R)-cHex | −63 | 62 | 77 | 59 |
| TTTTtTTTTT | (1R,2S)-cHex | 66 | 64 | 71 | 59 |
| GTAGATCACT | None (aegPNA) | 141 | 55 | 55 | 64 |
| GtAGAtCACt | (1S,2S)-cHex | 51 | 54 | 58 | |
| GtAGAtCACt | (1R,2R)-cHex | 34 | 33 | 58 | |
| GtAGAtCACt | (1S,2R)-cHex | −63 | 25 | 58 | 64 |
| GtAGAtCACt | (1R,2S)-cHex | 66 | 35 | >85 | 64 |
| gtagatcact | (1S,2S)-cHex | 41 | 37 | 58 | |
When the (β–γ)-linkage in the cyclohexyl PNA backbone was changed from –(CH2)4– to –(CH2)3–, a cyclopentane ring was formed in cyclopentyl PNA (cpPNA). The cyclopentane ring exhibits characteristic pseudoaxial/pseudoequatorial puckering properties which relaxed the torsional strain compared to the cyclohexane ring system. In addition, the five-membered ring should provide more flexibility compared to the rigid cyclohexane ring system. Such cyclopentyl PNA was introduced almost simultaneously by Kumar and Ganesh's group60,68 for the cis-(1S,2R/1R,2S)-cpPNA and Appella's group for the trans-(1S,2S/1R,2R)-cyclopentyl PNA (cpPNA).65,69–71 According to the X-ray data, the cis-diaminocyclopentane rings show β = −24° for (1S,2R)-cpPNA and +25° for (1R,2S)-cpPNA monomers.60 Hybridization studies with complementary DNA/RNA sequences by UV-Tm measurements indicate that the cp/aegPNA chimera forms thermally stable hybrids with both DNA and RNA (Table 10). While stereochemistry-dependent selective binding to RNA over DNA was observed in the chimeric homothymine sequences,60 subsequent studies in the mixed-sequence context did not show the same effect.64 Thus, both stereoisomers of cis-cpPNA appeared to strongly stabilize both DNA and RNA hybrids which is in sharp contrast to the chPNA. Again, these results are only from chimeric systems and care should be taken in generalizing the results.
| PNA sequencesa (N–H/C–LysNH2) | Modification | β | T m (°C) DNA | T m (°C) RNA | Ref. |
|---|---|---|---|---|---|
| a The modification site is denoted by a small letter. b Torsional angle value of aegPNA was taken from ref. 25, and others were based on X-ray structures of monomers. c Two different values are obtained from different sources. d n.t. = no melting transition. | |||||
| TTTTTTTT | None (aegPNA) | 141 | 45 | 62 | 60 |
| TTTtTTTT | (1S,2R)-cPen | −24 | 22 | 76 | 60 |
| TTTtTTTT | (1R,2S)-cPen | 25 | 62 | 61 | 60 |
| ttttttttt | (1S,2R)-cPen | −24 | 67 | >85 | 60 |
| ttttttttt | (1R,2S)-cPen | 25 | 72 | >85 | 60 |
| GTAGATCACT | None (aegPNA) | 141 | 55 (49)c | 55 | 64 and 65 |
| GTAGAtCACT | (1S,2S)-cPen | 86 | 55 | — | 65 |
| GTAGAtCACT | (1R,2R)-cPen | 70–90 | n.t.d | — | 65 |
| GTAGAtCACT | (1R,2R)-Pyrrolidine | 103 | 58 | — | 66 |
| GtAGAtCACt | (1S,2S)-cPen | 86 | 60 | — | 67 |
| GtAGAtCACt | (1S,2R)-cPen | −24 | 77 | 84 | 64 |
| GtAGAtCACt | (1R,2S)-cPen | 25 | 79 | >85 | 64 |
The cyclopentyl PNAs with the trans-(1S,2S) and (1R,2R)-configurations were investigated by Appella et al. The studies were more focused on DNA binding, and thus no data for RNA were available. The effect of stereochemistry on the binding properties of trans-cpPNA was remarkable as an increase in Tm of around +5 °C per (1S,2S)-cpPNA unit added to the aegPNA structure whereas no melting was observed when a single unit of (1R,2R)-cpPNA was introduced (Table 10). Thermodynamic parameters show that replacing the cyclopentyl ring in aegPNA reduces conformational flexibility and entropic cost of the hybridization, resulting in the higher thermal stability of the cpPNA·DNA hybrids.65 The Tm was increased progressively and CD spectra indicated an increasing level of pre-organization upon the introduction of more (1S,2S)-cpPNA units. Subsequent studies in a fully modified cpPNA nonamer revealed a very high Tm of 94 °C which is 52 °C higher compared to the corresponding unmodified aegPNA.71 The crystal structure of the (1S,2S)-cpPNA·DNA hybrid at 1.3 Å resolution revealed an antiparallel right-handed helix with a helical twist of 27.1 Å, a rise of 3.4 Å, and a pitch of 13.2 base-pairs per turn as shown in Fig. 16. The average torsional angle β of the trans-diaminocyclopentane unit was 86 ± 8°. This value was quite different from the aegPNA·DNA structure obtained by an earlier NMR study.25 However, it is compatible with the torsional angle β obtained from other X-ray structures of PNA·DNA duplexes.23,44 This indicates that the PNA structure in the PNA·DNA duplex may be able to adopt a broad range of conformations. Thus, from the hindsight, the apparent correlation of the torsional angle of the PNA monomer and the DNA/RNA selectivity in earlier studies of cpPNA and chPNA might be considered as over-simplification.12,64
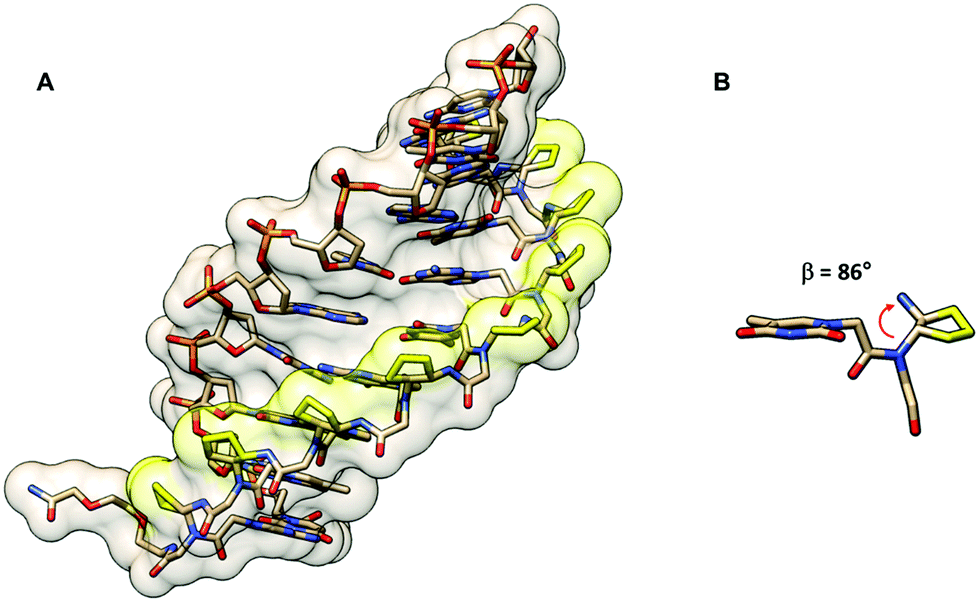 | ||
| Fig. 16 (A) X-Ray structure of the trans-(S,S)-cpPNA·DNA duplex (7KZL)71 viewed from the minor groove side. (B) Torsional angle β of the diaminocyclopentane unit from the X-ray structure of the cpPNA·DNA duplex. The cyclopentyl modification of the PNA backbone is shown in yellow. | ||
An aza analog of cpPNA in which the –(CH2)3– bridge that was part of the cyclopentane ring was replaced by a –CH2NHCH2– bridge to create a 5-membered heterocyclic ring called dapPNA was also recently reported.66 Despite carrying the wrong (R,R)-configuration, the chimeric dap/aegPNA with a single modification showed decent DNA binding (Tm = 57.2 °C, which is +6.3 °C higher than that in the case of the corresponding aegPNA). This is in sharp contrast to the corresponding carbocyclic analog reported in the literature.65 The stabilization effect was explained by the favorable electrostatic interaction due to the presence of the protonatable nitrogen atom in the dapPNA backbone. Despite the non-specific nature of such electrostatic interaction, the mismatch specificity was reported to be better than in the case of aegPNA (ΔTm for mismatched PNA·DNA duplexes: aegPNA = −6.6 °C; dapPNA = −9.2 °C).66
Only one example of PNA with trans-(S,S)-cyclopropane (tcprPNA) bridging the β–γ positions was reported in the literature.72 The extreme rigidity of the 3-membered ring was expected to make an impact on the pre-organization of the PNA. Mono-substituting the tcprPNA unit in the aegPNA results in a large decrease in thermal stability in both PNA·DNA and PNA·RNA. The torsional angle β of trans-diaminocyclopropane was not reported but it can be presumed to be more open than the trans-diaminocyclopentane analogs. The PNA2·DNA triplex showed a two-step melting indicative of different structural compatibility of tcprPNA as a Hoogsteen and Watson–Crick strand. Significant hysteresis was observed especially in the case of PNA2·RNA triplex melting, which suggested the slow kinetics of PNA·RNA binding.
Modified PNA with (α–γ)-linkage
In early attempts to constrain the conformation of aegPNA, the α- and γ-positions were linked to form 4-aminoproline isomers which generated four possible configurations (L-trans, L-cis, D-trans, D-cis) (Fig. 17).73,74 The chimeric PNA with alternating L-trans-4-aminoproline and aegPNA monomers showed marginally higher DNA·PNA duplex stability than the unmodified aegPNA in the context of the homothymine sequence. Other stereoisomers formed DNA hybrids with lower stability. Reports from another group on a similar system focused on the synthesis of the monomers and small homothymine oligomers derived from these monomers.75,76 CD studies of the single-stranded oligomers indicated well-ordered structures that are stereochemically dependent. The study on chimeric mixed-sequence PNA incorporating a single L-trans or D-trans diastereomer indicated that both isomers slightly stabilized the PNA·DNA duplexes. The parallel/antiparallel selectivity was also improved in a stereochemically dependent fashion (L-trans: antiparallel; D-trans: parallel). | ||
| Fig. 17 Structures of modified PNA with (α–γ)-linkage. | ||
Modified PNA with (β–α′)-linkage
Another common bridging strategy involves the linkage of the β- and α′-positions to form a 5-membered ring (Fig. 18). The tertiary amide carbonyl group of the nucleobase side-arm may be present or reduced to a CH2 group. In the latter situation, the backbone nitrogen atom became positively charged due to protonation under physiological pH which could provide additional electrostatic interaction with the negatively charged phosphate backbone of DNA. | ||
| Fig. 18 Structures of modified PNA with (β–α′)-linkage. | ||
A conformationally constrained pyrrolidinone PNA with (β–α′)-linkage was reported in 2001.77 All possible stereoisomers [(3S,5S), (3R,5S), (3S,5R), (3S,5S)] of the adenine pyrrolidinone PNA monomer were synthesized and incorporated in the middle of a mixed-sequence aegPNA strand to form chimeric pyrrolidinone/aegPNA. None of the four stereoisomers of pyrrolidinone PNA showed improvement in DNA and RNA binding when compared to unmodified aegPNA. However, the chimeric PNA carrying the (3S,5R) pyrrolidinone PNA monomer showed the highest preference for binding to RNA over DNA. A homoadenine sequence with the fully modified (3S,5R) pyrrolidinone PNA monomer was also synthesized, but its RNA hybrid was less stable than the corresponding aegPNA·RNA hybrid. This indicates that the conformational lock may not be in the optimal conformation to mimic the aegPNA structures in the DNA/RNA-bound forms. The six-membered ring homolog piperidinone PNA (pipPNA) was also reported.78 The incorporation of either the (3R,6R) or (3S,6R)-pipPNA monomer into a mixed-sequence chimeric pip/aegPNA resulted in significant destabilization of DNA, RNA, and aegPNA hybrids. Furthermore, two transitions were observed in most cases suggesting that the six-membered structure is less compatible with the aegPNA duplexes.
The same group also reported a similar constrained PNA design, but with the carbonyl group reduced to the CH2 group.79 Only the (2R,4S) diastereomer of the pyrrolidine PNA with the same stereochemistry as the (3S,5R) pyrrolidinone PNA was reported. The single incorporation of the pyrrolidine PNA monomer in a mixed-sequence aegPNA resulted in dramatic destabilization for both DNA (ΔTm −21 °C) and RNA hybrids (ΔTm −15 °C) which is much more than the destabilization induced by the corresponding pyrrolidinone PNA monomer under the same circumstance (ΔTm −8.5 °C for DNA and −5.5 °C for RNA). This indicates that the conformation of the pyrrolidine PNA may not be compatible with the aegPNA, and the presence of the protonatable nitrogen atom did not increase the binding affinity. On the other hand, the fully modified pyrrolidine PNA with a homoadenine sequence formed very stable DNA2·PNA triplexes with DNA (ΔTm = +23.5 °C) and RNA (ΔTm = +25.5 °C) whereas the homoadenine pyrrolidinone PNA did not show appreciable binding to both DNA and RNA (Table 11). In addition, significant hysteresis was observed in the case of pyrrolidine PNA which suggested a slow rate of hybridization. Such discrepancy between Tm data from the chimeric and fully modified PNAs emphasizes the danger of drawing general conclusions from the chimeric sequences.
| DNA or PNA sequences (N–H/C–NH2)a | Modification | T m (°C) | Ref. | |||
|---|---|---|---|---|---|---|
| apDNA | pDNA | apRNA | pRNA | |||
| a The modification site is denoted by a small letter. b Significant hysteresis was observed. Tm values were taken from the heating curves. c T m with either poly(dA), poly(rA), poly(dT), or poly(rU) as the complementary strand. | ||||||
| AAAAAAAAAA | None (aegPNA) | 56 | — | 35 | — | 79 |
| aaaaaaaaaa-Lys | Pyrrolidinone PNA (3S,5R) | n.t. | — | 26 | — | 79 |
| aaaaaaaaaa-Lys | Pyrrolidine PNA (2R,4S) | 79b | — | 60b | — | 79 |
| TACTCATACTCT-Lys | None (aegPNA) | 50 | — | 60 | — | 79 |
| TACTCaTACTCT-Lys | Pyrrolidinone PNA (3S,5R) | 41 | — | 54 | — | 79 |
| TACTCaTACTCT-Lys | Pyrrolidine PNA (2R,4S) | 28 | — | 44 | — | 79 |
| Lys-TTTTT-Lys | aegPNA | 49c | — | 54c | — | 85 |
| Lys-ttttt-Lys | POM (2R,4R) | 54bc | — | 45c | — | 85 |
| Lys-ttttt-Lys | POM (2R,4R) (pH 6) | — | — | 49c | — | 85 |
| Lys-ttttt-Lys | POM (2R,4R) (pH 8) | — | — | 40c | — | 85 |
| Lys-AAAAA | None (aegPNA) | 48c | — | 31c | — | 85 |
| Lys-aaaaa | POM (2R,4R) | 50bc | — | 70bc | — | 85 |
| TCACAACTT | DNA | 32 | n.t. | 25 | n.t. | 82 |
| Lys-TCACAACTT | None (aegPNA) | 35 | n.t. | 38 | 24 | 82 |
| Lys-tcacaactt | POM (2R,4R) | 43b | 45b | 46b | 44b | 82 |
Similar pyrrolidine PNA (POM, pyrrolidine-amide oligonucleotide mimics) with the (2R,4R)-configuration that was supposed to mimic the configuration of natural nucleosides was reported by Micklefield.80,81 From a semi-empirical quantum mechanics calculation and the X-ray crystal structure,82 the conformation of the protonated pyrrolidine ring was proposed to be similar to that of the C3′ endo-conformation of the ribose ring in native RNA (Fig. 19). Therefore, these PNA systems were proposed to preferentially bind to RNA over DNA. However, it should be noted that the nature of the substituent on the pyrrolidine ring can affect the ring puckering significantly.83,84 The non-chimeric homothymine POM binds to DNA and RNA with slightly less affinity than the corresponding aegPNA (Table 11). It appeared that the selectivity was due to kinetics rather than thermodynamics at least for the context.80 Less pronounced kinetics selectivity was observed in the homoadenine and mixed-sequence POM, and in this case, they formed more stable hybrids than aegPNA.81,85 However, the rates of hybrid formation and dissociation are significantly slower than in the case of the corresponding aegPNA. The dependency of the hybrid stability and kinetics of its formation – being more stable and forming faster at lower pH – supports the role of electrostatic interaction due to the pyrrolidine ring protonation.
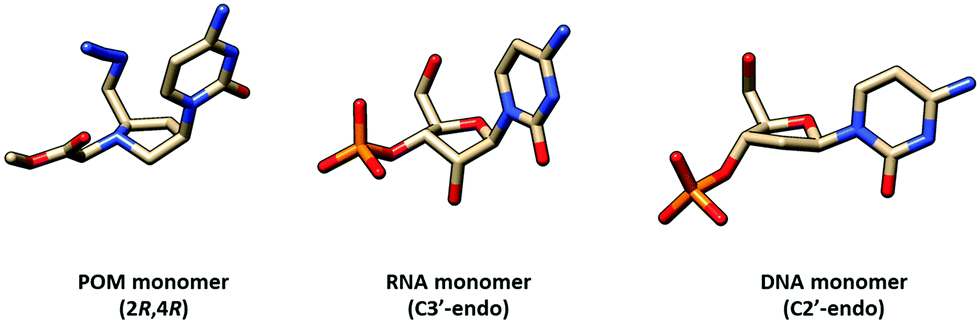 | ||
| Fig. 19 Comparison of conformation of the pyrrolidine ring in the POM monomer82 and sugar ring puckering in generic RNA and DNA structures. | ||
Modified PNA with (α–α′)-linkage
Modified PNAs with an (α–α′)-linkage were reported by several research groups under various names such as chiral PNA86 and aepPNA87–90 which are essentially the same systems but with different stereochemistry. In these PNA systems, the α and α′ positions were joined by a methylene bridge to form a five-membered pyrrolidine ring, and the tertiary amide CO group linking the nucleobase to the backbone was removed. A six-membered ring homolog with a piperidine ring instead of the pyrrolidine ring was also reported.91 In addition, a five-membered ring pyrrolidinone PNA bearing the tertiary amide CO group was also reported under the name of aeponePNA (Fig. 20).92
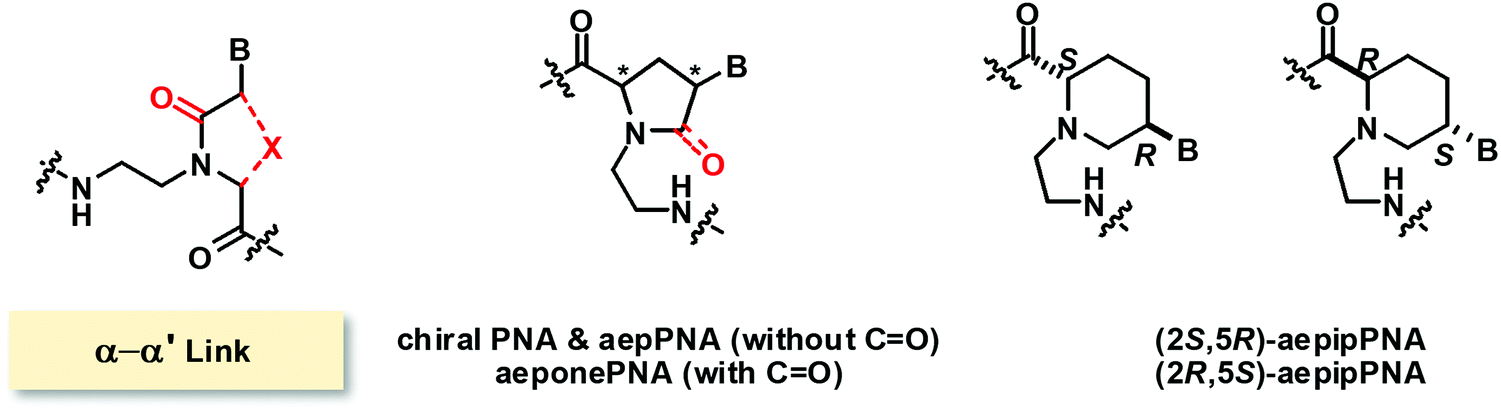 | ||
| Fig. 20 Structures of modified PNA with (α–α′)-linkage. | ||
The chiral PNA reported by Lui with a fully modified backbone with (2S,4S)-configuration showed a Tm of 46 °C for the pA12-dT10 hybrid while the pT10-dA10 and other mix-AT sequences showed no observable binding.86 Other stereoisomers of aepPNA were studied by Kumar and Ganesh87,88 and Vilaivan89,90 and the Tm data are summarized in Table 12. Selective binding to RNA was observed in the non-chimeric aepPNA with a homothymine sequence when the stereochemistry was (2R,4R) and (2S,4S). The hybrids showed similar Tm but distinctive CD features that indicated opposite handedness. The other two stereoisomers showed no binding to both RNA and DNA. The hysteresis observed in the melting curves of the aepPNA·RNA hybrids and kinetics experiments suggested a slow association rate similar to that of pyrrolidine PNA/POM.89,90 However, no binding was observed with both DNA and RNA targets in a mixed-sequence aepPNA with (2R,4R) stereochemistry in both parallel and antiparallel directions.
| PNA sequencesa | Modification | T m (°C) DNA | T m (°C) RNA | Ref. |
|---|---|---|---|---|
| a The modification site is denoted by a small letter. b With dT10. c With dA10. d With poly(dA). e With poly(rA). f With d(GCA8CG). | ||||
| H-a12-LysNH2 | aep-(2S,4S) | 46b | — | 86 |
| H-t10-LysNH2 | aep-(2S,4S) | n.t.c | — | 86 |
| H-tataaatt-LysNH2 | aep-(2S,4S) | n.t.d | — | 86 |
| Ac-t10-LysNH2 | aep-(2S,4S) | n.t.d | 42e | 90 |
| Ac-t10-LysNH2 | aep-(2R,4R) | n.t.d | 43e | 90 |
| Ac-t10-LysNH2 | aep-(2S,4R) | n.t.d | n.t.e | 90 |
| Ac-t10-LysNH2 | aep-(2R,4S) | 24d | n.t.e | 90 |
| Ac-gtagatcact-LysNH2 | aep-(2R,4R) | n.t. | n.t. | 90 |
| H-t8-β-AlaOH | aep-(2R,4S) | >80f | — | 88 |
| H-t8-β-AlaOH | aep-(2S,4S) | >80f | 35e | 92 |
| H-t8-β-AlaOH | aepone-(2S,4S) | 53f | 46e | 92 |
| H-T8-β-AlaOH | None (aegPNA) | 35f | 58e | 92 |
While Vilaivan and Liu did not observe DNA binding in the case of non-chimeric homothymine aepPNA, Kumar and Ganesh reported that the aepPNA homooligomer in both (2S,4S)- and (2R,4S)-configuration bound strongly to DNA with Tm values over 85 °C (Table 12).88 They also reported a chimeric aep/aegPNA monomer with a mixed-sequence which showed varying degrees of stabilization/destabilization of the duplex that depend on the stereochemistry and type of nucleobase. The same group also reported that when the tertiary amide CO group was present as in aeponePNA,92 the stability of the DNA hybrid was decreased while that of the RNA hybrid was increased. However, the study was only performed on the homothymine sequence and no data for the mixed-sequence are available to allow a general conclusion to be drawn.
The reported Tm data in aepPNA from three different research groups showed some discrepancies. The comparison is complicated by the non-uniform base-pairing behavior of different aepPNA monomers. The data were interpreted, with the support from NMR and theoretical calculations, as aepPNA monomers with different nucleobases might adopt different conformations that could affect the pre-organization of the PNA (Fig. 21).93 This principle could apply to other pyrrolidine-based PNAs as well, and thus the data derived from only homo-oligomers or chimeric systems with only one type of nucleobase modification should be interpreted with care.
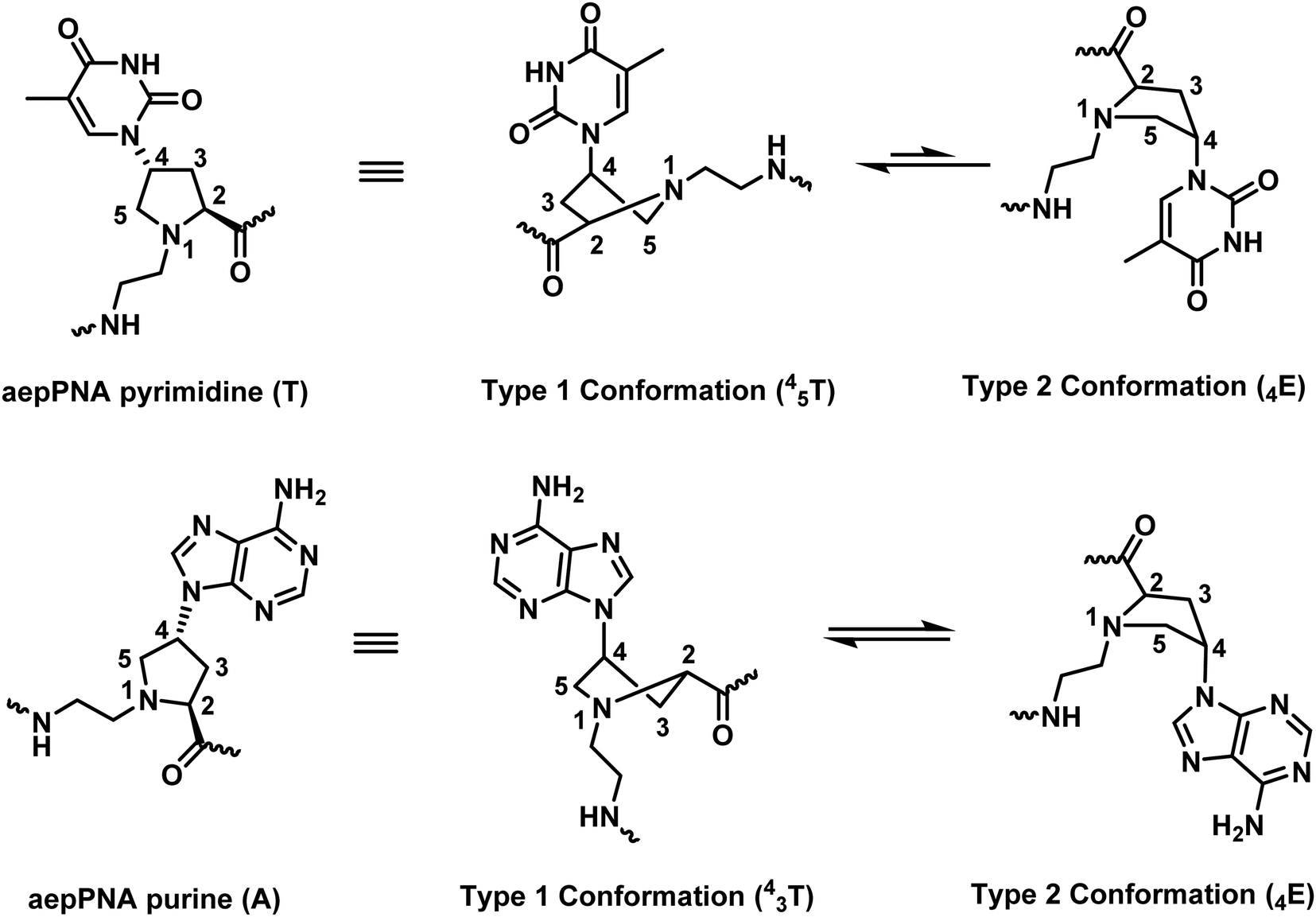 | ||
| Fig. 21 The base-dependency of the conformation of the pyrrolidine ring in aepPNA (reprinted from ref. 93, Copyright (2010), with permission from Elsevier). | ||
The six-membered ring homolog aepipPNA was studied only for the (2S,5R)- and (2R,5S)-isomers.91,94 Stabilization of the PNA·DNA duplexes and triplexes with some improvement in antiparallel selectivity over aegPNA was reported. However, the study was limited to chimeric aepip/aegPNA homopyrimidine sequences with terminal substitution, and thus general conclusion cannot be made.
Modified PNA with (γ–α′)-linkage
The only example of constrained PNA with (γ–α′)-linkage was linked by a methylene bridge to form a chiral piperidine ring. Two diastereomers [(3S,5S) and (3R,5R)] of the thymine monomer were synthesized (Fig. 22).95 The molecule was designed to have the nucleobase oriented in the axial position to mimic the structure of hexitol nucleic acids. Stabilization of the PNA·DNA triplexes was reported, but again the study was limited to chimeric homothymine sequences, and thus general conclusion cannot be made.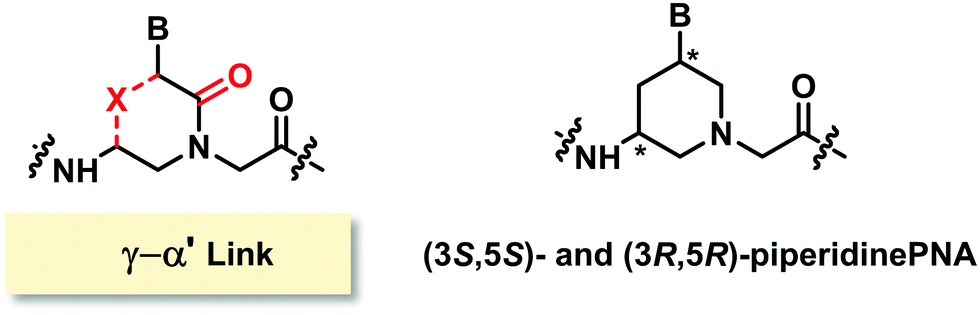 | ||
| Fig. 22 Structures of modified PNA with (γ–α′)-linkage. | ||
Modified PNA with (α−β)-linkage
The only reported constrained PNA system with (α–β)-linkage consists of sulfur-containing linkages resulting in the incorporation of a thiazolidine ring96 or a thiazane ring in the PNA structure (Fig. 23).97 The two isomers of thiazolidine PNA [(2S,4R)-anti and (2R,4R)-syn] simultaneously obtained during the cyclization could be separated and the structures were confirmed by X-ray crystallography. The presence of the thiazolidine ring constrained the torsional angles γ and δ, and also limited β and χ2 to the ranges that are compatible with the structures of aegPNA hybrids. However, it was found that the chimeric homothymine thiazolidine/aegPNA with a single thiazolidine monomer inserted in the middle showed a substantial decrease in the hybrid stability for both DNA and RNA when compared to unmodified aegPNA, although the specificity of base-pairing was still retained.96 Subsequent studies on the dimethylthiazolidine (with opposite stereochemistry at Cα) and thiazane (as a racemate) constraints in the same sequence context also showed substantial destabilization of the PNA·DNA hybrids in all cases.97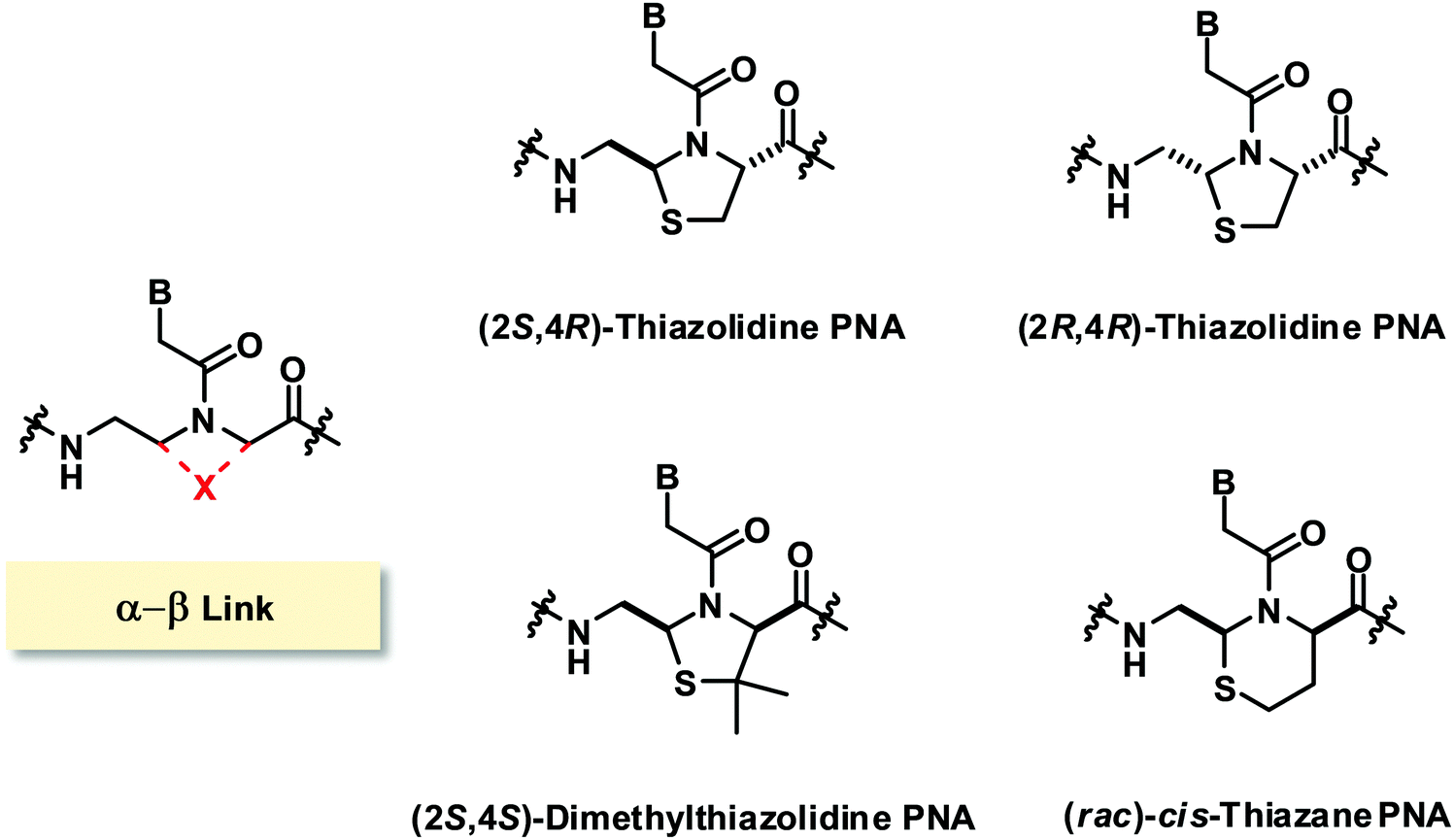 | ||
| Fig. 23 Structures of modified PNA with (α−β)-linkage. | ||
Modified PNA with (γ-N)- and (β-N)-linkages
Only one example of constrained PNA with (γ-N)-linkage has been reported.98 In the N-(pyrrolidinyl-2-methyl)glycine PNA or pmgPNA, the presence of the pyrrolidine ring generates a new chiral center, and thus both (S)-pmgPNA and (R)-pmgPNA monomers as well as their chimeric and homooligomeric PNA have been prepared. Studies in chimeric PNA suggested that the insertion of a single pmgPNA monomer into a mixed-sequence aegPNA destabilized the PNA·DNA duplexes much more than the PNA·RNA duplexes. The (R)-pmgPNA monomer was less destabilizing than the (S)-pmgPNA monomer. Unfortunately, the fully modified mixed-sequence (R)-pmgPNA decamer showed no binding to both complementary DNA and RNA. The backbone-extended version of pmgPNA was also reported but the binding properties were not better than in the case of aegPNA.99In an example of constrained PNA with (β-N)-linkage, 3-aminopyrrolidine ring was used to constrain the PNA backbone in ethano-PNA (Fig. 24).100 The structure was designed based on the proposal that the amino substituent and the ring nitrogen should adopt the gauge conformation that would restrict the torsional angle β to 60–80° similar to the conformation of aegPNA·RNA duplexes. The use of 3-aminoproline as the starting material allowed a facile placement of various ring substituents such as aminomethyl and guanidinomethyl at the 5-position through the appropriate transformation of the carboxyl group. Incorporation of the ethano-PNA monomer into the aegPNA backbone destabilized both DNA and RNA duplexes, but the effect was smaller with RNA leading to an increase in RNA binding selectivity in a sequence-specific fashion (Table 13). The related secdapPNA with the same basic scaffold as ethano-PNA, but with an additional amino substituent showed substantial destabilization of PNA·DNA hybrids when incorporated into homothymine aegPNA, and no melting was observed in the case of the mixed-sequence.66
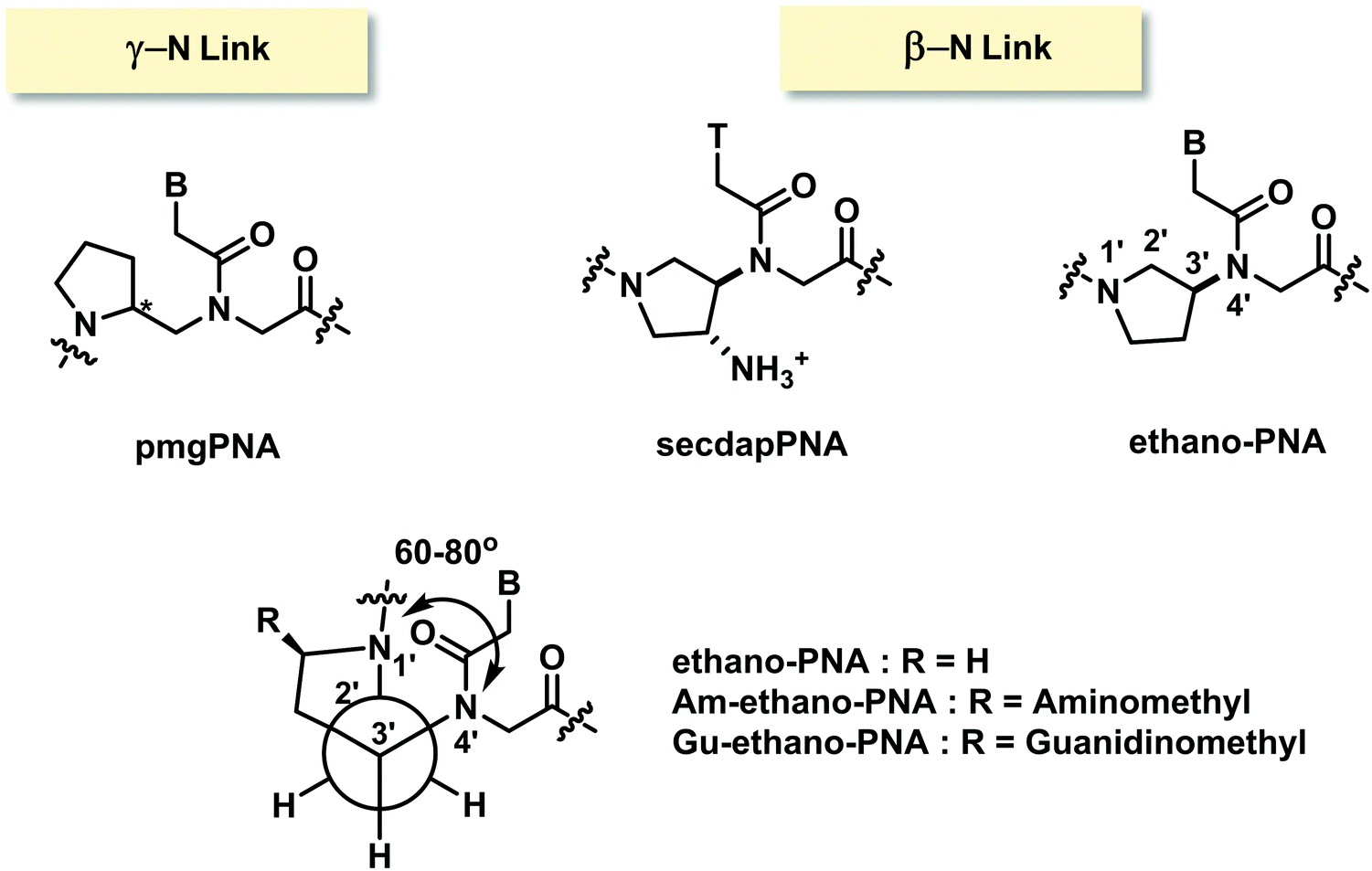 | ||
| Fig. 24 Structures of modified PNA with (γ-N)- and (β-N)-linkages. | ||
| PNA sequencea (N–H/C–LysNH2) | Modification | T m DNA (°C) | T m RNA (°C) | T m mmRNA (°C) |
|---|---|---|---|---|
| a The modification site is denoted by a small letter. | ||||
| CATTGTCACACT | None (aegPNA) | 65 | 66 | 55 |
| CATTGtCACACT | Ethano- (R = H) | 50 | 59 | 45 |
| CATTGtCACACT | Am-ethano- (R = H2NCH2–) | 52 | 60 | 47 |
| CATTGtCACACT | Gu-ethano (R = guanidinomethyl–) | 54 | 63 | 47 |
Constrained PNA designs with extended aegPNA backbones
In this section, the constrained PNA structure does not follow the generic aegPNA templates, but relates to it by backbone extension. The structures of such PNAs are included in Fig. 25. The backbone extended versions of POM reported by Mickelfield showed poorer binding when compared to the original POM. However, selective binding to RNA over DNA was observed when the extension was inserted in the right place, but the data were from the homothymine sequence only.101 The same RNA selectivity was also reported by Kumar on the bepPNA system which is essentially the same as bePOM II but with opposite stereochemistry.102 The six-membered ring constrained aminopipecolyl PNAs with the (2S,4S)-substitution and (2S,5R)-substitution patterns have been reported to show some stereochemistry- and sequence-dependent binding behavior. The chimeric aminopipecolyl/aegPNA with (2S,4S)-stereoisomer decreased the stability of the PNA2·DNA triplex but increased the stability of the PNA·DNA duplex and the opposite is true for the (2S,5R)-stereoisomer.103,104 Although technically the (2S,5R)-aminopipecolyl PNA should be regarded as an (α–γ)-linked system, it is more convenient to compare it with the (2S,4R)-aminopipecolyl PNA here.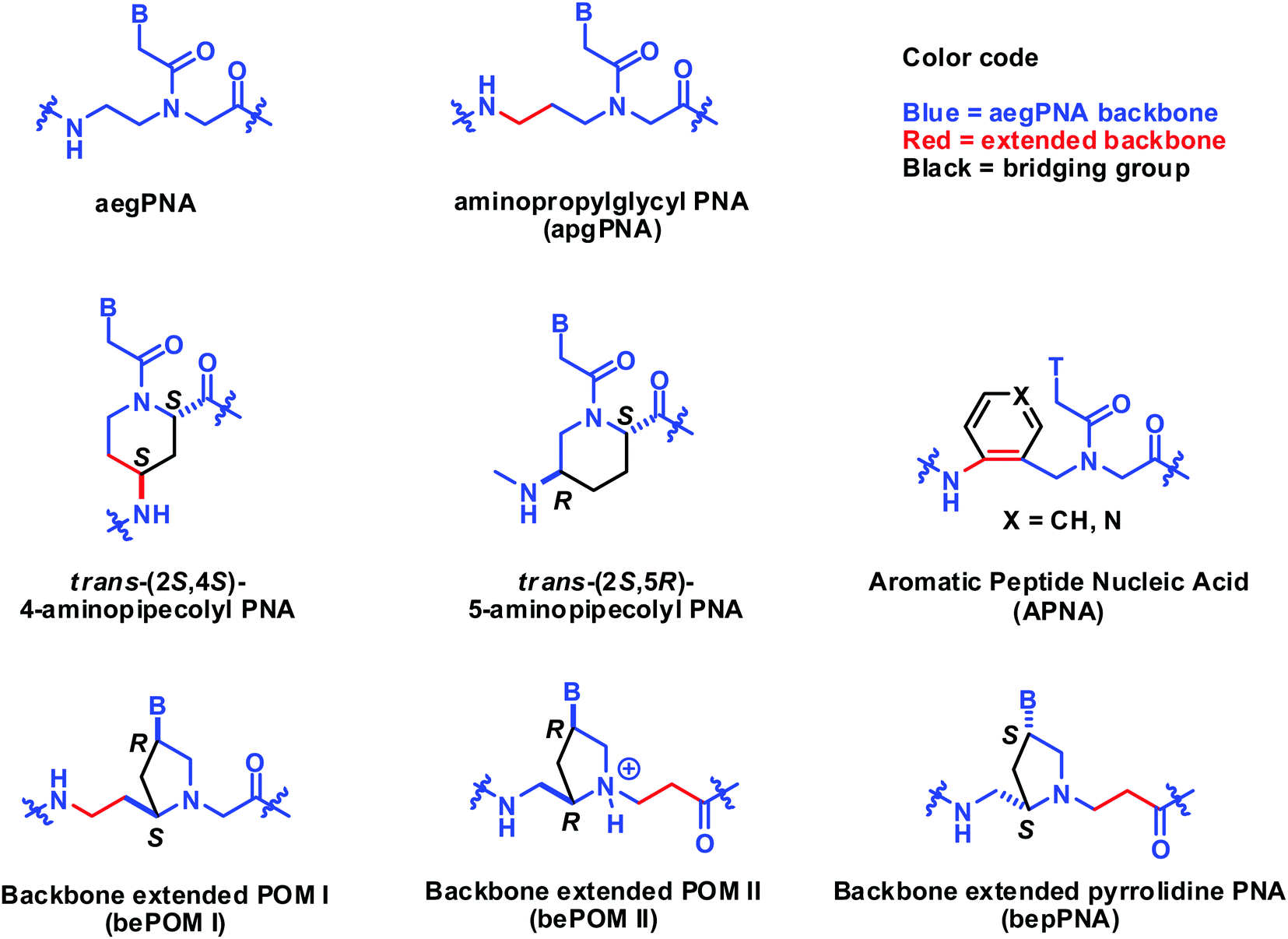 | ||
| Fig. 25 Structures of constrained aegPNA with extended backbones. | ||
Tsantrizos et al. reported another modified PNA with an aromatic ring incorporated in the extended backbone of aegPNA. This is perhaps the only constrained PNA with an achiral backbone. Incorporation of one aromatic PNA (APNA) monomer into the aegPNA backbone resulted in a small decrease in Tm for both DNA and RNA for both homothymine105 and mixed-sequence.106 However, the stability was not decreased further upon the incorporation of more APNA monomers up to 4 consecutive residues. Attempts to prepare the fully modified APNA failed due to insolubility. Replacement of the benzene ring with a more hydrophilic pyridine ring also resulted in further destabilization (Table 14).107
| Sequencea (N–H/C–NH2) | System | T m (°C) | Ref. | |||
|---|---|---|---|---|---|---|
| apDNA | pDNAb | apRNAb | pRNAb | |||
| a The modification site is denoted by a small letter. b Significant hysteresis was observed. | ||||||
| TTTTTTTT-Lys | aegPNA | 52 | — | 66 | — | 102 |
| Lys-tttttttt | POM (2R,4R) | 53b | — | 52b | — | 101 |
| Lys-tttttttt | bePOM I | n.t. | — | n.t. | — | 101 |
| Lys-tttttttt | bePOM II | n.t | — | 44b | — | 101 |
| tttttttt-Lys | bepPNA | n.t. | — | 59 | — | 102 |
| GTAGATCACT-Lys | aegPNA | 55 | — | 55 | — | 102 |
| GtAGAtCACt-Lys | bepPNA | n.t. | — | 81 | — | 102 |
| TTTTTT-Lys | aegPNA | 64 | — | 56 | — | 105 |
| TTTtTT-Lys | APNA (X = CH) | 44 | — | 50 | — | 105 |
| TTTtTT-Lys | APNA (X = N) | 39 | — | 47 | — | 107 |
| TTTcTT-Lys | APNA (X = CH) | <5 | — | 17 | — | 105 |
| GTAGATCACT-Lys | APNA (X = CH) | 54 | 39 | 58 | 42 | 106 |
| GTAGAtCACT-Lys | APNA (X = CH) | 40 | 34 | 48 | 31 | 106 |
| gtagATCACT-Lys | APNA (X = CH) | 43 | 28 | 45 | 28 | 106 |
Alternative PNA designs that do not follow the generic aegPNA template
There are relatively few PNA systems that do not fit the aegPNA template that still show decent DNA/RNA binding (Fig. 26). In 1999, Sisido proposed a new PNA system called oxy-peptide nucleic acid or OPNA.108,109 The glycine nitrogen atom in the aegPNA backbone was replaced by an oxygen atom in the OPNA and the nucleobase was attached to the Cγ via an ethylene linker. The system with the (S)-configuration derived from L-homoserine showed stronger binding than the (R)-configuration.110 OPNA hybridizes with DNA to form duplexes with lower stability than the corresponding aegPNA·DNA duplex but showing a remarkably sharp melting transition. However, the system was quite flexible, and a read-through can happen, resulting in lowering of the specificity. A marginal preference for DNA over RNA and parallel selectivity were reported under a limited sequence context. The analysis of thermodynamic parameters indicated that the OPNA system showed a large entropy loss as expected from its rather flexible structure.108 This entropy term decreases when the backbone of the OPNA was constrained by the incorporation of a pyrrolidine ring as in POPNA, at the expense of the decreased enthalpy term. Overall, the effect of the backbone constraint in terms of increasing binding affinity and specificity was relatively small.110,111 Partial replacement of the backbone oxygen atom in OPNA by a protonatable N-Me group (PAPNA) did not affect the stability of the PNA·DNA duplex while improving the cell permeability.112 The Tm data for the OPNA family are summarized in Table 15.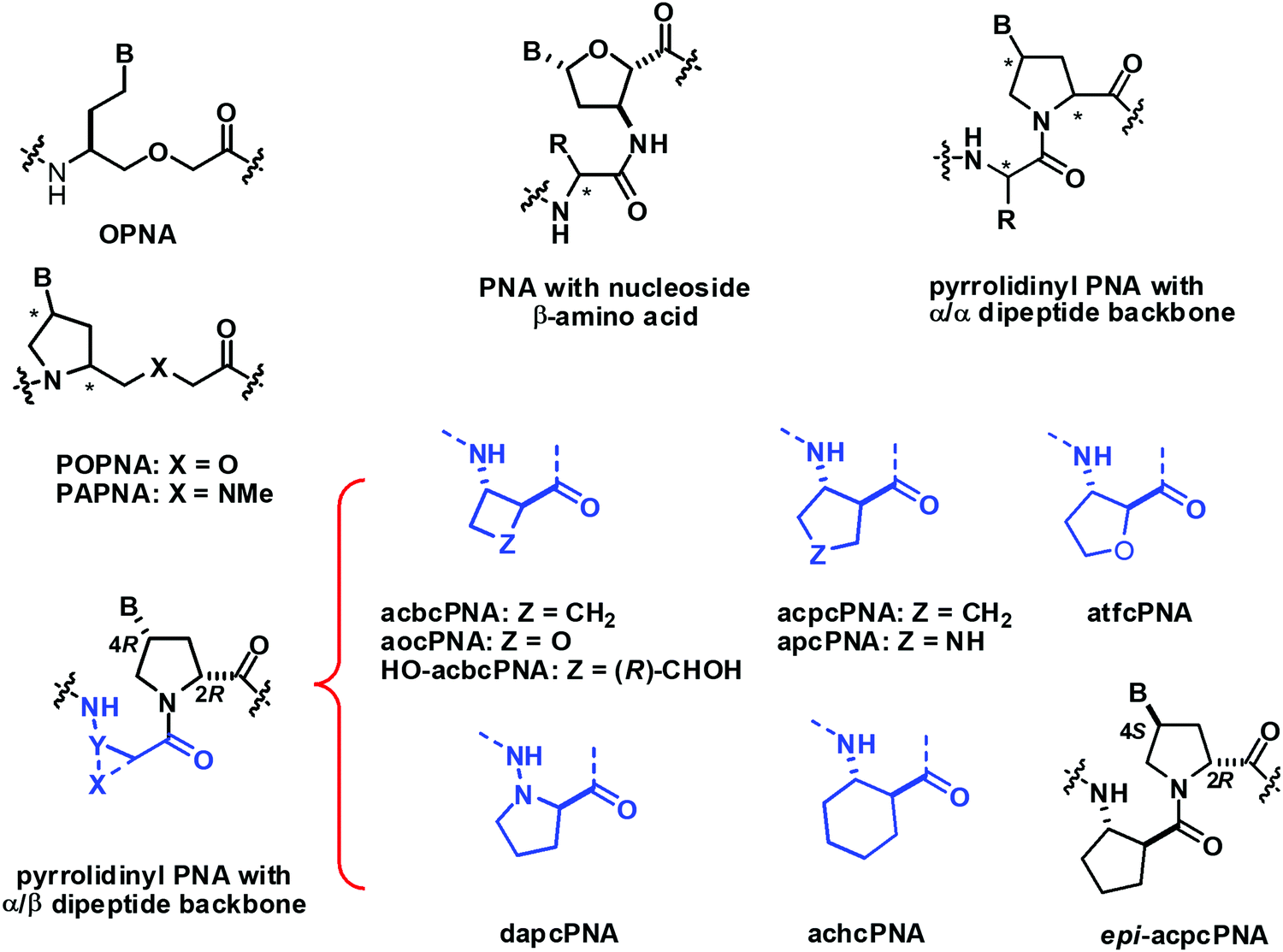 | ||
| Fig. 26 Structures of conformationally constrained PNA that does not follow the generic aegPNA template. | ||
| System (A9) | dT9 | dT4CT4 | dT4AT4 | dT4GT4 | rU9 | Ref. |
|---|---|---|---|---|---|---|
| DNA | 15 | — | — | — | — | 110 |
| aegPNA | 50 | — | — | — | — | 109 |
| OPNA (S) | 35 | 17 | 17 | 15 | 9 | 109 and 110 |
| OPNA (R) | 23 | 13 | 9 | 9 | 11 | 110 |
| POPNA (cis-L) | 34 | 19 | 13 | 15 | 12 | 110 and 111 |
| POPNA (cis-D) | 30 | 16 | 28 | 13 | 14 | 110 and 111 |
| POPNA (trans-L) | 23 | 20 | 20 | 21 | 22 | 110 and 111 |
| POPNA (trans-D) | 30 | 17 | 16 | 17 | 19 | 110 and 111 |
Kumar has reported yet another interesting PNA design derived from an alternating sequence of nucleoside β-amino acids and α-amino acid.113,114 The nucleoside β-amino acids were obtained from the corresponding deoxyribosides. The homo-oligomer of a similar nucleoside β-amino acid derived from AZT has been independently shown to form well-defined folded helical structures but no DNA/RNA binding studies have been reported.115 Various α-amino acids have been introduced as a spacer including glycine, proline, sarcosine, lysine, and methionine. Strong binding to both DNA and RNA with a preference for RNA over DNA binding was observed in a preliminary study with the homothymine PNA sequence for all α-amino acid spacers studied.113 Subsequent report with mixed-sequence PNA with glycine spacer also confirmed the generality of this preference and allowed the determination of the directional selectivity as exclusively antiparallel.114 Nevertheless, the stability difference between RNA and DNA was only marginally better than in the case of aegPNA (Table 16). In addition, the thermal stability was generally lower than that of the corresponding aegPNA duplexes with a similar level of mismatch discrimination. A single-stranded PNA dimer with a D-proline spacer showed a more pronounced CD signal than the same dimer with L-proline and glycine, which is indicative of stronger pre-organization due to the matching stereochemistry of the nucleoside β-amino acid and the spacer.116
| Sequence/system | apDNA | pDNAa | mmDNA | apRNA | pRNAa | mmRNA |
|---|---|---|---|---|---|---|
| a n.t. = no transition. b T m of DNA was measured in the presence of 100 mM NaCl. c PNA with nucleoside β-amino acids and glycine linker. | ||||||
| CTTCTTCCTT | ||||||
| DNAb | 28 | n.t. | 19 | 36 | n.t. | 29 |
| aegPNA | 47 | 33 | 37 | 51 | 38 | 41 |
| PNAc | 40 | n.t. | 31 | 48 | n.t. | 38 |
| CACTGATTTCAA | ||||||
| DNAb | 36 | n.t. | 29 | 45 | n.t. | 33 |
| aegPNA | 54 | 40 | 46 | 61 | 49 | 49 |
| PNAc | 45 | n.t. | 38 | 54 | n.t. | 43 |
Vilaivan et al. introduced an alternative design of conformationally constrained PNA featuring a dipeptide backbone derived from nucleobase-modified proline and an amino acid spacer.117 The first generation of these pyrrolidinyl PNAs carried α-amino acid spacers including glycine and L-serine. They bound weakly to DNA in a stereochemically dependent fashion118 Inspired by the ability of short oligomers of β-amino acids to form well-defined secondary structures,119 the second generation pyrrolidinyl PNA was designed to incorporate a β-amino acid spacer instead of α-amino acid. The very first member of the second generation pyrrolidinyl PNAs that show strong DNA binding properties consists of thymine-modified proline in the (2R,4R) configuration and a cyclic hydrazino acid D-aminopyrrolidine carboxylic acid (DAPC) as the spacer.120 This dapcPNA system also showed a strong preference for binding to DNA over RNA, at least in the polyT/U-polyA sequence context.121 The same PNA sequence bearing acyclic spacers including β-alanine and N-amino-N-methylglycine,122 as well as cyclic spacers with different stereochemistry including L-aminopyrrolidine carboxylic acid and (1R,2S)-2-aminocyclopentanecarboxylic acid,120 failed to form stable hybrids with the complementary DNA target. This clearly indicated the crucial role of the spacer in determining the DNA binding ability of the pyrrolidinyl PNA. Subsequent studies on diastereomeric PNAs with a homogeneous backbone derived from (2R,4R)-thymine-modified proline and all possible stereoisomers of 2-aminocyclopentanecarboxylic acid (ACPC) spacers (1R,2R; 1S,2S; 1R,2S; and 1S,2R) led to the identification of acpcPNA with (2R,4R)-proline/(1S,2S)-ACPC backbone that showed exceptionally strong DNA binding ability and very high sequence specificity (Table 17).123,124 Other unique features of acpcPNA include the exclusive antiparallel selectivity, the preference for binding to DNA over RNA, and the inability to form PNA·PNA self-duplexes.124,125 Although experimental three-dimensional structures of acpcPNA and related pyrrolidinyl PNAs are not yet available, the structural bases of their unusual behaviors have been studied in detail by MD simulations.126–129 When the stereochemistry of the ACPC spacer was fixed as (1S,2S) and the stereochemistry at the proline part was varied (2R,4R; 2S,4S; 2R,4S; and 2S,4R), only the acpcPNA stereoisomers with 2R configuration could bind to DNA whereby the configuration of the nucleobase can be either 4R or 4S. This led to the discovery of epi-acpcPNA with (2R,4S)-proline/(1S,2S)-ACPC backbone which exhibits very strong DNA binding affinity and specificity comparable to that of acpcPNA.125,130 Heteroatoms such as N131 and O132 can be incorporated in the five-membered ring of the ACPC spacer in acpcPNA to improve water solubility without significantly interfering with the DNA binding ability. The nitrogen atom can be used as a convenient handle for subsequent modification to create functional PNA probes133,134 or cell-permeable PNA.135
| Systema | apDNA | pDNA | mmDNA | apRNA | pRNA | mmRNA | Ref. |
|---|---|---|---|---|---|---|---|
a Sequences: PNA: Ac-GTAGATCACT-LysNH2; apDNA: dAGTG![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) TCTAC; pDNA: dCATCTAGATG; apRNA: rAGUGUCUAC; pRNA: dCAUCUAGAUG.
b Without C-terminal Lys.
c Without the N-terminal Ac group. TCTAC; pDNA: dCATCTAGATG; apRNA: rAGUGUCUAC; pRNA: dCAUCUAGAUG.
b Without C-terminal Lys.
c Without the N-terminal Ac group.
|
|||||||
| aegPNAb | 43 | 32 | 33–36 | 47 | 31 | 29–37 | 53 |
| acpcPNA | 53 | <20 | 24–29 | 42 | <20 | <20–24 | 125 and 132 |
| epi-acpcPNA | 51 | <20 | 27–28 | 41 | <20 | — | 130 |
| atfcPNA | 53 | <20 | 23–25 | 36 | <20 | <20 | 132 |
| aocPNAc | 46 | — | <20–28 | 30 | — | — | 136 |
| acbcPNA | 66 | <20 | 36–47 | 58 | 32 | — | 137 |
| achcPNA | n.t. | <20 | <20 | <20 | <20 | — | 137 |
The effect of ring size of the cyclic β-amino acid in the (1S,2S)-configuration was explored in a series of pyrrolidinyl PNAs with (2R,4R)-proline stereochemistry (Fig. 27). This led to the discovery of acbcPNA with a four-membered cyclic β-amino acid spacer that exhibits even stronger DNA binding affinity than acpcPNA. On the other hand, the achcPNA with a six-membered cyclic β-amino acid spacer failed to bind to DNA/RNA. The difference in the DNA binding ability was explained in terms of the degree of similarity of the torsional angles N(H)-Cα-Cβ-C(O) of the cyclic β-amino acid oligomers from the literature (ACHC,138 ACPC,139 ACBC140) and of the PNA·DNA duplexes obtained from MD simulation – the closer the values the more stable the duplexes.137 Attempts to increase the hydrophilicity of acbcPNA by incorporating an oxygen atom into the ring136 or as a side chain in the form of a hydroxyl group141 resulted in suboptimal DNA binding.
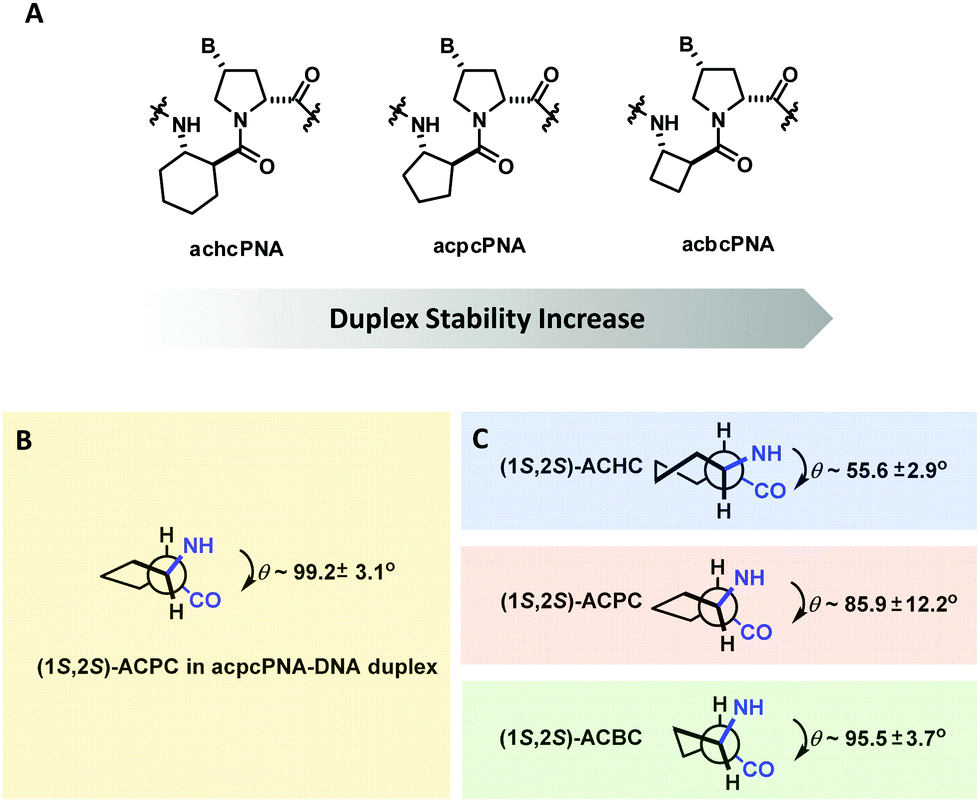 | ||
| Fig. 27 (A) Structures of pyrrolidinyl PNA homologs. (B) Torsional angle N(H)–Cα–Cβ–C(O) of (1S,2S)-ACPC in ACPC PNA. C) Native torsional angles of cyclic β-amino acid oligomers are obtained from the literature (adapted with permission from ref. 137. Copyright 2012 American Chemical Society). | ||
With regard to the backbone-modified PNA, it is worth mentioning the XNA systems developed by Asanuma et al., namely serinol nucleic acid (SNA)142 and acyclic threoninol nucleic acid (aTNA)143 (Fig. 28). Their backbones consist of phosphodiester-linked 2-amino-1,3-propanediols, and the nucleobases are attached to the backbone via the amide bond similar to the case of PNA. Despite the relatively flexible backbone, SNA and aTNA can form antiparallel self-pairing duplexes with comparable or even higher stability than that of PNA·PNA self-duplexes.144 On the other hand, the SNA and aTNA duplexes with DNA/RNA are less stable than the corresponding PNA·DNA or PNA·RNA duplexes. The stereochemistry of the methyl group in aTNA showed pronounced effects on the hybrid stability, with L-aTNA forming more stable hybrids with both DNA and RNA than D-aTNA. Interestingly, the pseudosymmetric SNA paired to DNA only in the antiparallel direction and the L-aTNA must adopt parallel orientation to pair in the same way which was indeed observed experimentally.145 Although the CD signal of single-stranded aTNA is weak, the self-duplex of L-aTNA is right-handed, suggesting the crucial role of the chiral methyl group in inducing the helicity by selectively stabilizing/destabilizing the helices. Overall, this suggests that the backbone rigidity is not a prerequisite for duplex formation but rather the delicate balance between flexibility and rigidity that should be taken into account for the design of new PNA systems.
 | ||
| Fig. 28 Structures of SNA, D-aTNA and L-aTNA. | ||
Structural aspects of the design of conformationally constrained PNA
In order to minimize the entropy loss as a result of conformational re-organization upon duplex (or higher-order structures) formation, the structure of the single-stranded PNA should closely mimic the DNA/RNA-target bound conformation. The information from three-dimensional structures of PNA·DNA, PNA·RNA, and PNA·PNA hybrids will be useful for the design of new PNA systems that can not only bind strongly but can also exhibit selectivity among different types of nucleic acid targets. The key helix parameters and torsional angle data are summarized in Tables 18 and 19, respectively. The three-dimensional structures of standard DNA and RNA duplexes as well as those of selected unmodified and modified aegPNA duplexes are shown in Fig. 29–31.| System | Method | Disp (Å) | Rise (Å) | Tilt (°) | Twist (°) | bp per turn | Ref. |
|---|---|---|---|---|---|---|---|
| a Left-handed helix. b Values for two different duplexes in the same asymmetric unit. | |||||||
| PNA·PNA (1PUP) | X-ray | 8.3 | 3.2 | 1.0 | 19.8 | 18 | 146 |
| PNA·PNAa (2K4G) | NMR | 7.9 | 3.7 | −0.2 | −17.3 | 21 | 32 |
| γPNA·γPNA (2KVJ) | NMR | −9.0 | 2.9 | 2.1 | 16.3 | 22 | 43 |
| PNA2·DNA (1PNN) | X-ray | 6.8 | 3.4 | 5.1 | 22.9 | 16 | 24 |
| PNA·DNA (1PDT) | NMR | −2.9 | 3.5 | −1.3 | 27.5 | 13 | 25 and 71 |
| αPNA·DNA (1NR8) | X-ray | −3.8 | 3.5 | 0.2 | 23.2 | 16 | 23 |
| γPNA·DNAb (3PA0) | X-ray | −5.5 | 3.2 | 0.3 | 23.9 | 16 | 44 |
| −6.3 | 2.9 | 2.3 | 23.3 | 16 | |||
| cpPNA·DNA (7KZL) | X-ray | −3.4 | 3.4 | 0.4 | 27.1 | 13 | 71 |
| PNA·RNA (5EME/5EMF) | X-ray | −6 | 2.4 | — | 26 | 14 | 147 |
| ADNA | −4.5 | 2.6 | −4.5 | 32.7 | 11 | 44 | |
| BDNA | 0 | 3.4 | −0.1 | 36.0 | 10 | 44 | |
| System | Method | α | β | γ | δ | ε | χ 1 | χ 2 | χ 3 | ω | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Average values for all residues with SD in parentheses. Terminal residues were excluded. b W: The PNA strand that forms the Watson–Crick base-pairing strand; H: the PNA strand that forms Hoogsteen base-pairing. c Values for two different duplexes in the same asymmetric unit. | |||||||||||
| PNA·PNA (1PUP) | X-ray | −120(6) | 66(5) | 74(9) | 91(11) | 174(11) | 6(6) | −175(4) | 83(3) | −170(14) | 146 |
| 83(11) | −19(3) | ||||||||||
| PNA·PNA (2K4G) | NMR | −93(24) | −66(5) | −75(4) | −88(10) | −155(17) | 2(4) | 169(10) | −82(8) | 175(10) | 32 |
| 123(16) | −3(12) | ||||||||||
| γPNA·γPNA (2KVJ) | NMR | −110 to −130 | 60 | 75 | 90 | 175/−175 | 10 to −10 | 175/−175 | 90 to 110 | 43 | |
| PNA2·DNAb (1PNN) | X-ray | −103(W) | 73(W) | 70(W) | 93(W) | 165(W) | 1(W) | −170(W) | 89(W) | −178(W) | 24 |
| −108(H) | 69(H) | 69(H) | 87(H) | 175(H) | 1(H) | −175(H) | 102(H) | −178(H) | |||
| PNA·DNA (1PDT) | NMR | 105(55) | −141(13) | 78(16) | 139(13) | 35(42) | −3(3) | 151(9) | −103(19) | 25 | |
| αPNA·DNA (1NR8) | X-ray | −114(15) | 73(9) | 67(4) | 90(5) | 179(10) | 4(6) | −178(3) | −91(6) | −178(2) | 23 |
| 72(7) | |||||||||||
| γPNA·DNAc (3PA0) | X-ray | −106(14) | 70(12) | 60(6) | 117(20) | −150(7) | 12(11) | −177(6) | 87(10) | −116(32) | 44 |
| −100(13) | 68(10) | 59(9) | 123(27) | −110(24) | 13(9) | −174(9) | 82(6) | −155(16) | 44 | ||
| cpPNA·DNA (7KZL) | X-ray | −123(15) | 86(8) | 67(4) | 109(7) | 175(10) | 0(3) | −179(3) | 89(6) | −170(4) | 71 |
| PNA·RNA (176D) | NMR | 150 to 172 | 67 to 72 | 79 to 86 | 72 to 85 | 64 to 105 | 3 to 9 | −146 to −171 | 58 to 84 | 63 | |
| PNA·RNA (5EME) | X-ray | 114(7) | 71(9) | 70(8) | 96(5) | −6 (9) | 4(4) | −172(6) | 83(8) | 147 | |
| 79(3) | 162(7) | ||||||||||
| PNA·RNA (5EMF) | X-ray | 112(8) | 66(7) | 73(7) | 94(6) | −6(10) | 6(6) | −174(4) | 81(5) | 147 | |
| 76(2) | 159(8) | ||||||||||
 | ||
| Fig. 29 Three-dimensional structures of standard DNA·DNA,151 DNA·RNA152 and RNA·RNA153 duplexes, with details of the monomer conformation and base-pairing. | ||
 | ||
| Fig. 30 Three-dimensional structures of PNA·PNA,23,146 PNA·DNA25 and PNA·RNA63,147 duplexes, with details of the monomer conformation and base-pairing. | ||
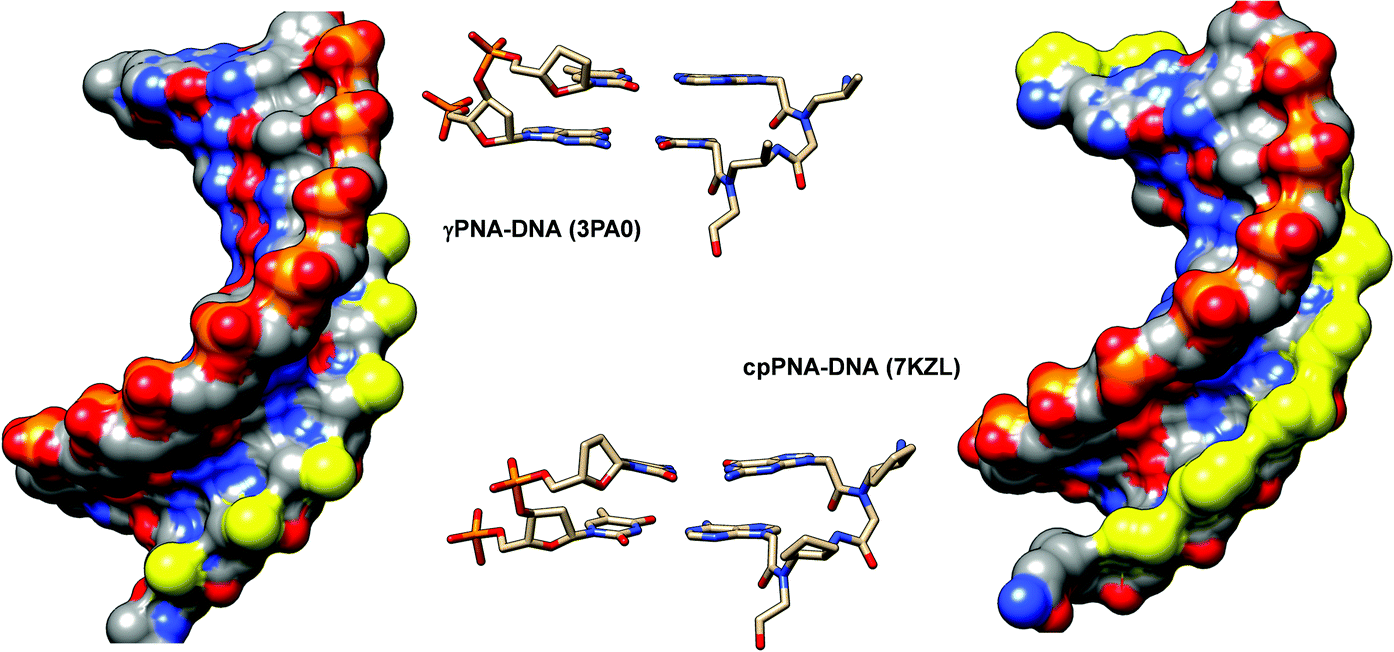 | ||
| Fig. 31 Three-dimensional structures of γPNA·PNA44 and cpPNA·DNA71 duplexes, with details of the monomer conformation and base-pairing. The backbone substituents are highlighted in yellow. | ||
X-Ray structures of native aegPNA duplexes suggested that the PNA itself exhibits an inherent propensity to form helical structures. Since the aegPNA molecule is achiral, both right-handed and left-handed are observed in equal amounts. The PNA·PNA duplex showed distinct structural features from conventional A- or B-type DNA helices. It is characterized by a more open and elongated helix (18–21 bp per turn) with a narrow and shallow minor groove and a wide, deep major groove which was described as a novel “P” type helix.146 The base pairs are more or less perpendicular to the helix axis. NMR structures of other PNA·PNA duplexes revealed the same general helix feature at the global level.32,43 In the case of γPNA·γPNA, the base pair rise was smaller, resulting in a helix that resembles the A-type helix more,43 but with the elongated character of the P-type helix (22 bp per turn). The γ-substituent was shown to align along the edge of the groove and it did not interfere with the duplex formation.
Three-dimensional structures of PNA·DNA25 and PNA·RNA duplexes63 have been studied by NMR since early dates. The first X-ray structure of the PNA·DNA duplex was published in 200323 and those of PNA·RNA duplexes were not available until 2016.147 The helical parameters of PNA·DNA duplexes obtained from both NMR (1PDT)25 and X-ray (1NR8)23 studies are grossly similar and reveal distinctive features that are different from both typical DNA·DNA helices and P-type PNA·PNA helices. This suggests that both partners (PNA and DNA) are adjusting themselves to a mutually compromised structure that is an intermediate between the two. The displacement in PNA·DNA duplexes was in the range of −2.9 to −3.8 Å which is smaller than that of the corresponding PNA·PNA duplexes (8 Å). The number of base pairs per turn was in the range of 13–16 which was larger than that of typical DNA·DNA duplexes (10) but smaller than that of PNA·PNA duplexes (18). At the individual PNA monomer level, the X-ray and NMR structures showed considerable discrepancy especially in the torsional angle value β N(H)–Cγ–Cβ–N(CO). The NMR structure25 reported a much larger β value of 141° (almost corresponded to the extended antiperiplanar conformation, with respect to the two backbone nitrogen atoms) compared to the β value of 73° (gauche conformation, with respect to the two backbone nitrogen atoms) in the X-ray structure.23 The discrepancy could in part be attributed to the difference in the PNA structures (purely aegPNA in the NMR and chimeric aeg/D-Lys αPNA in the X-ray structures). However, it should be noted that the values are quite uniform (59–83°) in the latter regardless of the type of PNA monomer (aeg/α) within the X-ray structure. Due to the somewhat limited availability of three-dimensional structures of native PNA·DNA duplex in the literature, it is not clear about the origin of such discrepancy and more structural information would be helpful in guiding the designing of new PNA systems. Nevertheless, experimental results available so far indicate that the system that permanently locks the β value in the range of 70°–83° resulted in substantial stabilization of the PNA·DNA duplexes (see below). On the other hand, the duplex stability data for the system that has the β value in the larger range which corresponded to the more extended conformation are not yet available. The cyclopropyl PNA system with a native β value of 145° is the closest candidate but only Tm data of the PNA2·DNA triplex were available, which failed to show the expected stabilization.72
The β value of 73° from the X-ray structure of the αPNA·DNA duplex23 was also in good agreement with the values obtained from other PNA structures including PNA2·DNA triplex (73°), γPNA·DNA duplex (68°), and cpPNA·DNA duplex (86°) (Table 19). Somewhat smaller torsional angle β values were found in the corresponding PNA duplexes including PNA·PNA duplex (66°) and γPNA·γPNA duplex (60°) but they were still in a similar range. Accordingly, the conformation constraint at the β and γ positions should be designed so that the β values are fixed in such range as long as the substituent does not sterically or electrostatically interfere with the binding. The remarkable stabilization of PNA·DNA duplexes derived from γPNA42 and cpPNA71 emphasizes the importance of this point. In both cases, the backbone substituents were located near the rim of the groove and pointed to the solvent, and thus their role was to guide the PNA to fold into the right conformation rather than providing additional stabilization. The values of the torsional angle χ1 are close to 0° in all PNA·DNA and PNA·PNA duplexes indicating the preferred Z-rotamers of the amide bond connecting the nucleobase to the PNA backbone, resulting in the side-chain CO being pointed in the C-terminus direction and interacting with the backbone CO by the n → π* interaction. Thus, any conformation constraint should observe this requirement, and an example in the case of α-gem-dimethyl PNA27,28 has illustrated the point very well. On the other hand, locking χ1 by replacement of the tertiary amide bond with an alkene resulted in substantial destabilization regardless of the configuration of the double bond.148 Attempts to incorporate a fluorine atom to mimic the amide carbonyl group also failed to improve the binding.149,150 This indicates the indispensable role of the amide group in addition to the appropriate χ1 value. The torsional angle values χ2 are generally close to 180° to avoid clashing with the PNA backbone. The torsional angle values ω are always fixed at more or less 180° due to the trans-amide backbone. Other torsional angle values are also quite conserved among different structures of PNA·DNA duplexes as well as PNA·PNA duplexes as can be clearly seen in Fig. 29–31. These torsional angles could be potential points for introducing constraint in the PNA structures in addition to β and χ1 that have already been extensively utilized. Overall, the experimental structural data indicate that the PNA requires little conformational adjustment to adopt the proper DNA-binding conformation, and the conformational constraint can help reduce the entropic penalty upon the conformational change further. However, it should be noted that it is difficult to modify just one torsional angle without affecting others, and unless an aromatic ring or a rigid bicyclic molecule was employed, the conformation lock will allow a certain degree of flexibility. In fact, such flexibility should be desirable, as in the case of the cpPNA bearing the trans-(S,S)-cyclopentyl PNA that formed much more stable duplexes with DNA when compared to its homolog trans-(S,S)-cyclohexyl PNA. This could be explained by the rigidity of the cyclohexane ring that probably locked the PNA molecule into the wrong conformation that could not be easily adjusted to accommodate the requirement for duplex formation.58
In most PNA·DNA structures, the sugar ring DNA can exist in both C3′-endo conformation, as found in A-DNA duplexes, and C2′-endo conformation, as found in conventional B-DNA duplexes. The two conformations can co-exist in the same structure. In the PNA·RNA structures,63,147 the sugar ring always adopted the C3′-endo conformation as expected for RNA duplexes. The same is true for the sugar ring of DNA in the DNA·RNA hybrids. Early NMR structures of a PNA·DNA duplex25 and a PNA·RNA duplex63 might suggest two distinctive values of the torsional angle β (141 vs 73°) which could provide a potential basis for the design of a PNA that can differentiate between DNA and RNA. However, as mentioned earlier, subsequent studies indicated that the PNA monomers in several PNA·DNA duplexes23,44 could also adopt torsional angles β that are more similar to those in the PNA·RNA duplexes. In view of conformational similarity of PNA in PNA·DNA and PNA·RNA duplexes, the rational design of PNA systems that can bind selectively to either RNA or DNA may not be as straightforward as designing a PNA that can bind strongly to both. In the case of γPNA, the PNA·DNA duplex was more substantially stabilized than the corresponding PNA·RNA duplex upon the introduction of the constraint. It has been suggested that the differential stabilization was the result of the less conformational flexibility of RNA to adjust itself to bind to the rigid PNA scaffold than DNA.45 Promising DNA/RNA selectivity has been achieved in some PNA systems based on trial and error. However, mostly the data are not yet sufficient to allow a general conclusion, and the structural information is still limited. Such information will be highly valuable in guiding the design of new PNA systems with enhanced DNA binding properties.
Comparison of modified PNA·DNA and PNA·RNA hybrids’ stability and specificity
For those who are interested in finding potential applications of new PNA systems, the key question is perhaps how much better they are when compared to the standard aegPNA and when compared among each other in terms of binding affinity and specificity. While a large data set for Tm values between PNA·DNA and PNA·RNA hybrids are available in the literature, a direct comparison among different types of PNA is not straightforward for many reasons. First, Tm values are sensitive to several parameters including the length, base sequence, salt concentration, buffer, and concentration of the PNA. Second, while PNA molecules are considered electrostatically neutral, they still carry the positively charged amino group at the N-terminus. Furthermore, PNA is often modified by end-capping or incorporation of one or more hydrophilic amino acid residues (typically lysine) to improve water solubility. These charged modifiers could affect the nucleic acid binding via the electrostatic interaction.154 Third, the data for many modified PNA systems are available only for the homothymine sequences or chimeric mixed-sequence that carries up to a few modified monomers – typically T – along the standard aegPNA backbone. While in some instances the stabilization/destabilization effects observed in the chimeric PNA translated well to the fully modified PNA,65,71 it is difficult to draw a general conclusion from a limited set of such data. Thus, only the Tm data that are available for fully modified, non-chimeric PNA with “average” mix base sequence with good distribution between purine and pyrimidine bases will be compared in this section (Fig. 32).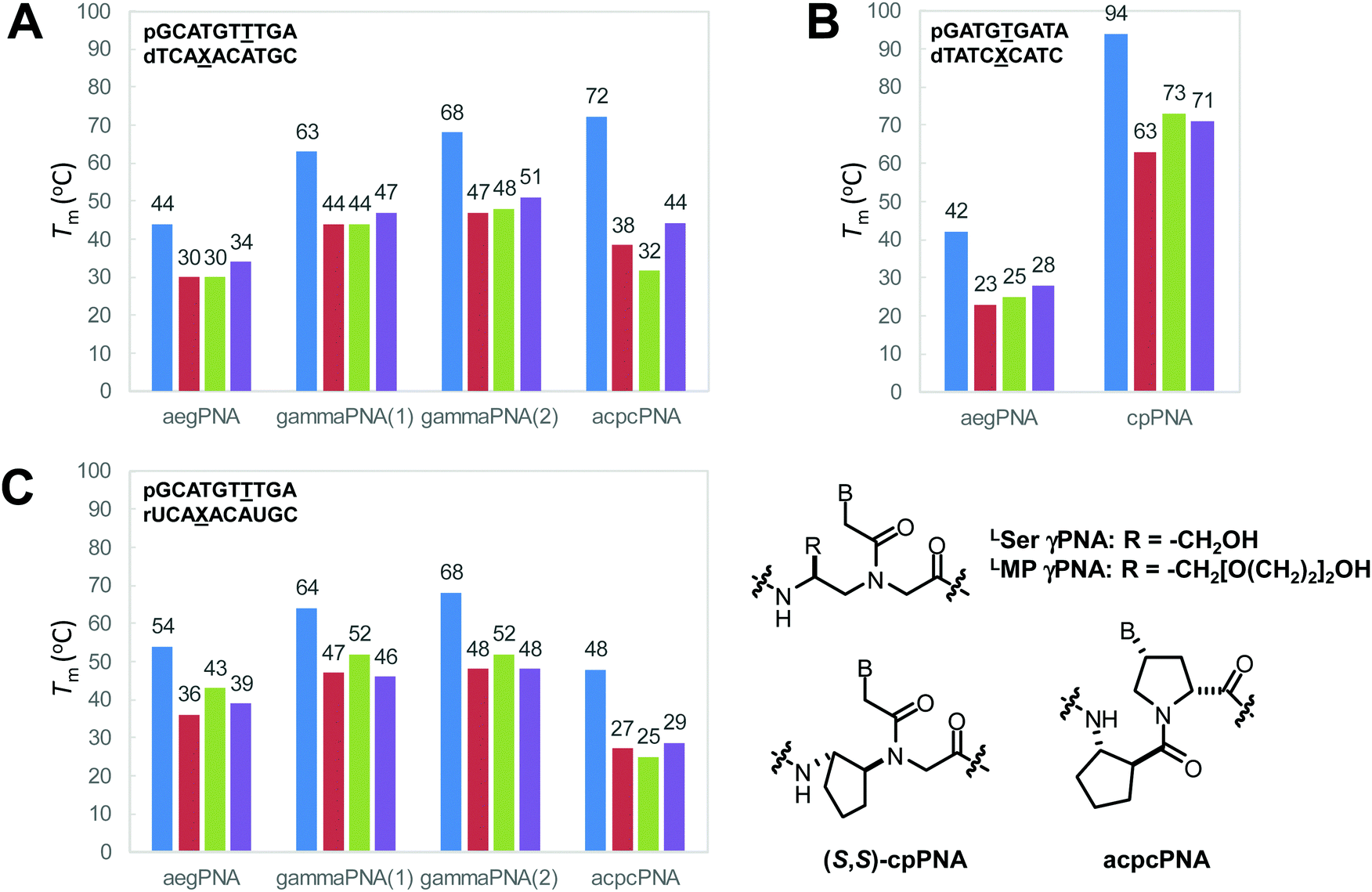 | ||
| Fig. 32 Comparison of Tm values of selected PNA·DNA duplexes (A and B) and PNA·RNA duplexes (C). Color code for X in the DNA/RNA strand: A (blue), C (red), G (green), T/U (purple). γPNA(1) = LSer γPNA; γPNA(2) = LminiPEG (LMP) γPNA. Tm data were taken from ref. 42, 45, 53, 71 and 117 (adapted with permission from ref. 117. Copyright 2015 American Chemical Society). | ||
The aegPNA duplexes in Fig. 32 showed Tm values in the range of 42–44 °C for PNA·DNA hybrids and 54 °C for PNA·RNA hybrids.53 The Tm values were decreased by 10–14 °C and 11–18 °C for the mismatched PNA·DNA and PNA·RNA hybrids, respectively. The DNA hybrids with γPNA showed considerably higher Tm values when compared to aegPNA with the same sequence (ΔTm for the complementary γPNA·DNA duplex and the aegPNA·DNA duplex = +19 to +24 °C), and the mismatch discrimination was slightly better (ΔTm for the mismatched γPNA·DNA duplex and the matched γPNA·DNA duplex = −16 to −21 °C).42,45 The pyrrolidinyl acpcPNA showed an even higher Tm for the complementary DNA hybrid (ΔTm for the complementary acpcPNA·DNA duplex and the aegPNA·DNA duplex = +28 °C) and better mismatch discrimination than both aegPNA and γPNA (ΔTm for the mismatched acpcPNA·DNA duplex and the matched acpcPNA·DNA duplex = −28 to −41 °C).117 The cpPNA showed the highest PNA·DNA duplex stability with a Tm value of 94 °C for a 9 nt mixed-sequence PNA·DNA duplex whereas the corresponding aegPNA·DNA duplex showed a much lower Tm at 42 °C (ΔTm for the complementary γPNA·DNA duplex and the aegPNA·DNA duplex = +52 °C).71 The downside of this cpPNA system is that although the Tm values were drastically decreased by 21–31 °C, the mismatched duplexes were still quite stable, with Tm values in the range of 63–73 °C. However, in practice, this may not be as bad as it sounds since only partial modifications of aegPNA can yield substantial improvement in the thermal stability of the duplex without compromising the mismatch specificity.
The aegPNA·RNA duplex was more stable than the corresponding aegPNA·DNA duplex, as shown by the increase in Tm by 10 °C for the same decameric sequence.53 The γPNA·RNA duplexes, on the other hand, showed about the same thermal stability as the corresponding γPNA·DNA duplexes. Nevertheless, the γPNA·RNA duplexes are still more stable than the aegPNA·RNA duplexes.42,45 In contrast, acpcPNA showed a distinctive preference for DNA over RNA, and thus the Tm of the acpcPNA·RNA hybrid was lower than that of the corresponding acpcPNA·DNA hybrid by 24 °C, making it the least stable RNA duplex of all PNAs. However, the mismatch specificity of acpcPNA was better than that of both γPNA and aegPNA (ΔTm for mismatched and complementary PNA·RNA duplexes acpcPNA: −19 to −23 °C; γPNA: −12 to −20 °C; aegPNA: −11 to −18 °C).117 No RNA binding data for cpPNA are available.
Applications of backbone-modified PNA in chemical biology
PNA has found widespread uses in many areas of applications that rely on the specific pairing between nucleobases.155 The characteristic advantages of PNA over conventional oligonucleotides include the following: (1) the high binding affinity of PNA allows the design of shorter probes, which result in better discrimination; (2) the binding strength of PNA to nucleic acids is less sensitive to ionic strength; (3) PNA can interact with structured nucleic acid targets in various modes – the most common being the triplex invasion which can be reliably used for site-specific targeting of dsDNA; and (4) PNA is completely stable towards nucleases and proteases. The inability of PNA as a nuclease and polymerase substrate makes it an effective blocker of enzyme activities such as in the PCR clamping technique.156 On the other hand, this makes PNA not compatible with several enzyme-mediated strategies for signal amplification. This also includes the inability of PNA to induce the RNAse-H mediated RNA degradation, and thus antisense PNA could only inhibit translation by steric blocking. In addition, the more hydrophobic nature of PNA can result in non-specific effects including self-aggregation, off-target binding, and adsorption to various hydrophobic surfaces such as plastic tubes that may complicate the interpretation of the results. In this section, applications of backbone-modified PNA related to the field of chemical biology and advantages over unmodified PNA, if comparison data are available, are discussed.Targeting of DNA/RNA duplexes
PNA can interact with DNA and RNA duplexes in many ways depending on the sequence context (Fig. 33).157 In the case of unmodified aegPNA, binding to dsDNA can occur efficiently via the triplex invasion mode with the formation of a highly stable PNA2·DNA triplex. In this binding mode, two pyrimidine-rich PNA strands bind cooperatively to the purine-rich DNA strand via the Watson–Crick and Hoogsteen base-pairings, leaving the pyrimidine-rich DNA strand unpaired. This is in contrast to other triplex-forming oligonucleotides (TFOs) such as the locked nucleic acid (LNA) whereby the TFO strand resides in the major groove of the original dsDNA and forms only the Hoogsteen base pairs.158,159 The formation of triplex invasion complexes was reported since the very early date of PNA discovery and their X-ray structure has been reported at atomic resolution.24 The triplex invasion relies on the formation of T·A·T and/or C·G·C+ base triplets (Fig. 34A); thus the DNA target sites require an extended stretch of the oligopurine tract. For the recognition of G, the protonation of C is required; otherwise an unnatural nucleobase that mimics C+ such as pseudoisocytosine (J) or N7-G should be used to allow efficient invasion at physiological pH. By appending to the PNA strand a tail-clamp consisting of two oligopyrimidine PNA blocks joined together by a flexible linker, duplex invasion by the remaining sequence on the PNA strand can be induced close to the triplex invasion site.160,161 Nevertheless, this approach still requires the presence of a stretch of oligopurine sequence in the target. The double duplex invasion strategy (Fig. 33) whereby two PNA strands bind to each DNA strand in the same region does not suffer the same limitation. However, since the two PNA strands required to invade at the same site are necessarily complementary to each other, it was necessary to modify both PNA strands to destabilize the (usually more stable) PNA·PNA hybrid and to promote PNA·DNA hybrid formation. For aegPNA, the only option was to modify the nucleobases so that they are pseudocomplementary, i.e., cannot bind to each other, but can still bind with the respective canonical complementary nucleobases. Pseudocomplementary bases are only available for the A·T pair by the replacement of T and A in the PNA strand with 2-thiouridine (US) and 2,6-diaminopurine (D), respectively (Fig. 34B).162 The development of an equivalent pseudocomplementary G·C pair has yet to be achieved, but it was recently discovered that the replacement of G in the PNA strand with N7-methylated G (G+) significantly stabilized the PNA·DNA duplexes and destabilized the PNA·PNA duplexes provided that the G+ are present on both PNA strands.163 This strategy has been employed to improve the invasion efficiency of GC-rich dsDNA targets.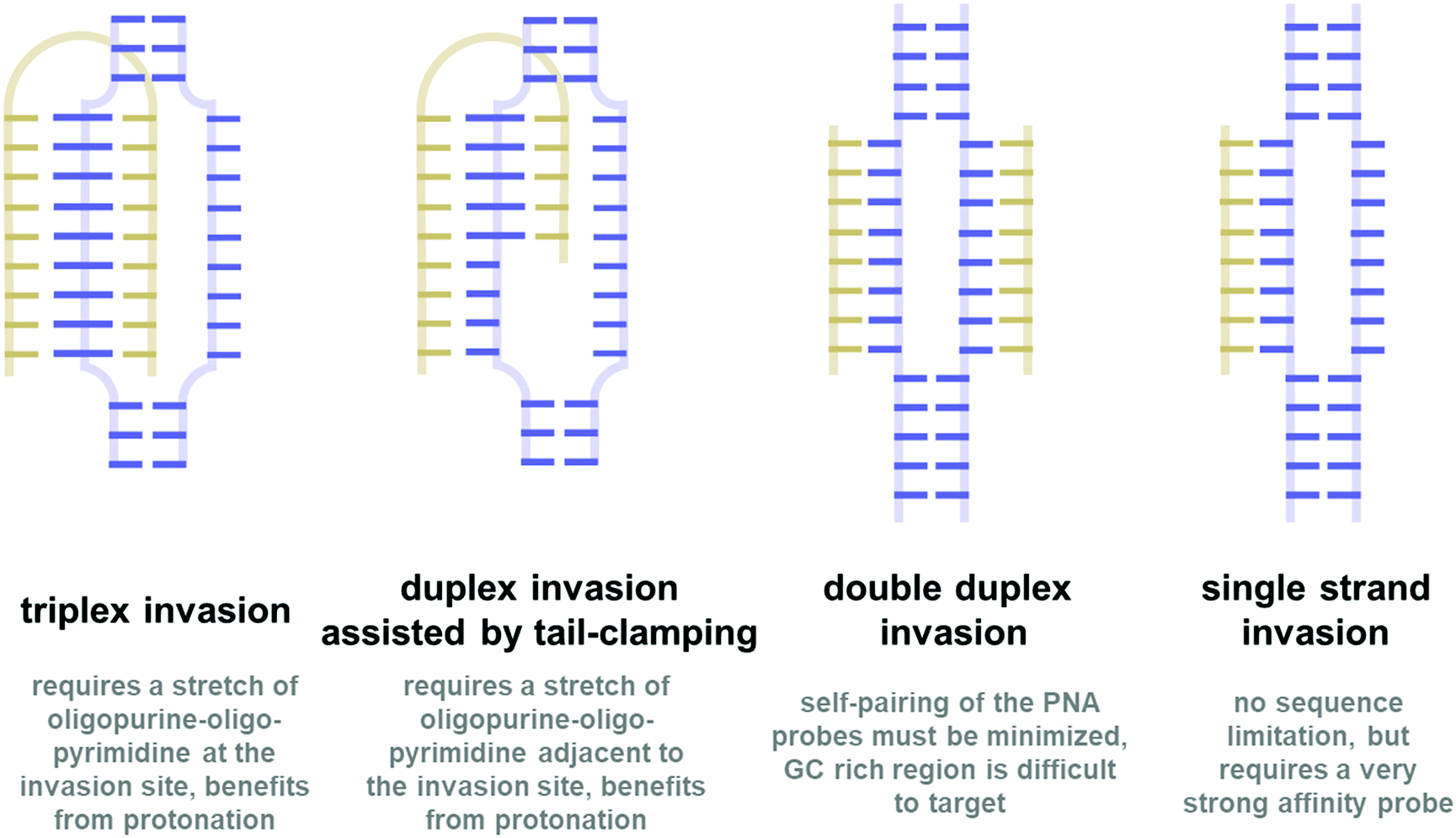 | ||
| Fig. 33 Various modes of targeting DNA duplexes (blue) by PNA (gold). | ||
 | ||
| Fig. 34 (A) Watson–Crick/Hoogsteen base triplets involved in the triplex formation between PNA (gold) and DNA (blue). (B) A pseudocomplementary base-pairing scheme for A·T pairs. | ||
Backbone modification opens new avenues for enhancing the DNA/RNA duplex recognition. In one example, the pseudocomplementary αPNA with multiple positive charges allowed more effective double duplex invasion in GC rich dsDNA targets. Normally invasion at the GC rich target is difficult since the G·C pairs in PNA·PNA duplexes tend to be more stable than the corresponding PNA·DNA duplexes and there was no effective pseudocomplementary G·C pair available. In this case, the electrostatic repulsion between the two positively charged modified PNA strands destabilized the pseudocomplementary PNA·PNA duplex and stabilized the PNA·DNA duplex further. It was also demonstrated that incorporation of multiple positive charges along the PNA strands is more effective than placing the positive charges at both ends of the PNA strands.154
The cyclic acpcPNA is unique among other PNA systems in the sense that the two strands of acpcPNA with complementary sequences could not efficiently form a self-duplex. Although the stability and directionality of the self-duplex are stereochemistry dependent,130,164 the self-duplexes are much less stable than the corresponding PNA·DNA duplexes in general. Thus, these pyrrolidinyl PNAs are inherently pseudocomplementary. Quantitative double duplex invasion of DNA duplexes by a pair of short acpcPNA sequences under low salt conditions was demonstrated by fluorescence energy transfer165,166 and gel electrophoresis studies.166 Invasion of a DNA duplex by a single-stranded acpcPNA was also observed, albeit with less efficiency.165
The strong affinity of γPNA enables efficient DNA duplex invasion by single-stranded γPNA without sequence restriction (up to 100% GC content) but longer γPNA sequences (15–20 bases) were required to invade efficiently.167,168 The specificity was demonstrated by the absence of invasion in single mismatched DNA targets. The invasion at the designated site was further confirmed by chemical probing using diethyl pyrocarbonate (DEPC)/piperidine. However, it was still necessary to perform the invasion under low salt conditions that destabilize the DNA·DNA duplexes. When a pre-formed invasion complex was incubated in a buffer at physiological ionic strength, the DNA duplex was reconstituted with the dissociation of the PNA strand.168 This suggests that the failure of the γPNA to invade under physiological conditions is due to thermodynamic rather than kinetic factors.168 For shorter γPNA sequences (10 bases), additional modifications are necessary to increase the affinity to allow the invasion. These include the incorporation of a terminal acridine group169 or a modified base (G-clamp).170,171 The latter strategy allowed duplex invasion under physiological conditions.172 The combination of a single-stranded γPNA invasion sequence and a DNAzyme designed to cleave a specific region of ssDNA formed by the DNA duplex invasion allows the introduction of nicks or double-strand breaks into dsDNA with a higher sequence fidelity than the CRISPR/Cas system.173 Recently, an efficient sequence-specific invasion of the DNA duplex by single-stranded cyclopentyl PNA (cpPNA) as short as 9 nt was reported, but the invasion failed at high salt concentrations.71
In the single strand invasion of the DNA duplex, the looped-out DNA strand is not stabilized by base-pairing as in the case of double duplex invasion. At high salt concentrations, the DNA duplex can thus easily re-form. To circumvent this problem, the chimeric duplex formed between γPNA and pyrene-modified DNA (“invader” probe) was recently proposed as an alternative pseudocomplementary PNA·DNA pair that can invade the DNA duplex in a double duplex invasion mode without the complication due to the self-complementarity of the γPNA self-duplex formation. The driving force is the strong base-pairing between DNA and γPNA as well as invader DNA, as opposed to the weak binding of γPNA and invader DNA due to the inability of the pyrene to intercalate into the γPNA·DNA duplex.175 In a different approach, γPNAs bearing a set of bifacial or Janus nucleobases were designed to recognize canonical Watson–Crick base pairs (AT, TA, CG, and GC) by inserting themselves between the base pair with a perfect shape and hydrogen bonding complementarity (Fig. 35).174 Such bifacial PNA would bind to the DNA duplex as a central strand instead of fitting into the major groove as in the case of triplex recognition. The process is expected to be enthalpically favorable since two strong PNA·DNA base pairs are simultaneously formed at the expense of one weaker DNA·DNA base pair. In addition, there is no sequence restriction as in the triplex recognition or triplex invasion that requires an oligopurine/oligopyrimidine tract. Despite the possibilities of self-pairing by the formation of E·E and E·F base pairs and the reported high stability of γPNA·γPNA duplexes,176 no self-pairing of the bifacial γPNA was observed both experimentally and in an MD simulation. A bifacial γPNA as short as 6 nt sequence can efficiently invade CG rich hairpin structures and longer DNA duplexes under physiological conditions with much faster kinetics than the usual duplex invasion (30 min rather than hours). A bifacial γPNA as short as 3 nt was shown to target dsRNA with r(CAG) repeats. An RNA-templated native chemical ligation of the short PNA chains was employed to enhance the binding efficiency further.177,178
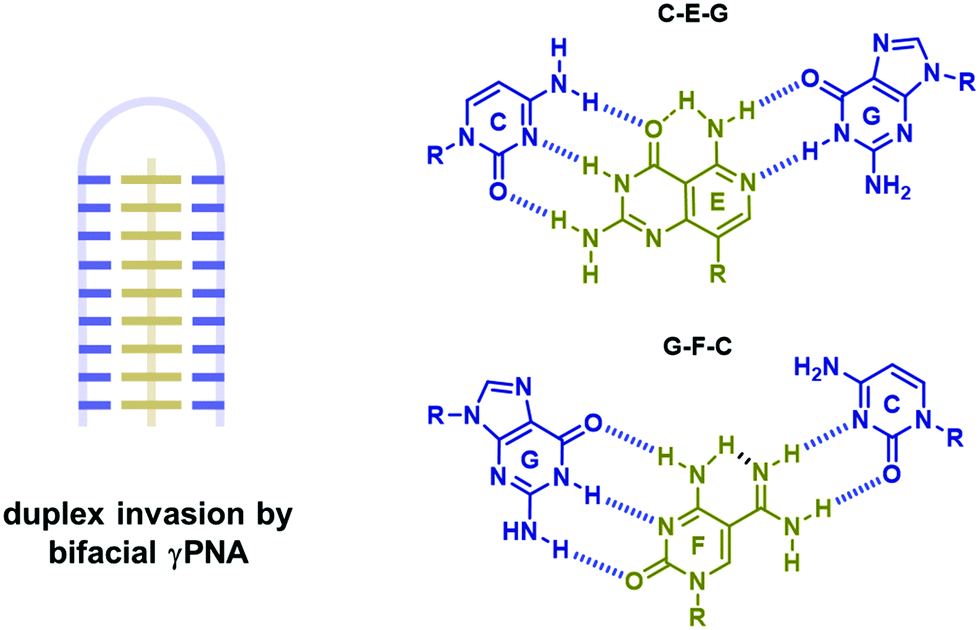 | ||
| Fig. 35 Invasion of DNA duplexes by bifacial PNA.174 | ||
PNA can also bind sequence specifically to double-stranded RNA (dsRNA) targets by triplex formation181 or triplex invasion.182 Targeting dsRNA by PNA is important since many biologically relevant RNA targets exist in structured forms.183 For example, a short PNA probe has been used to inhibit the replication of the influenza virus by targeting the viral RNA in the region that is not accessible by conventional antisense agents.184 In the triplex binding mode, aegPNA as short as 6 nt binds to an oligopurine/pyrimidine stretch region in dsRNA with a low nanomolar affinity. The affinity of PNA to dsRNA was better than that towards the corresponding dsDNA duplex by an order of magnitude, and attempts to adjust the PNA structure to favor the triplex formation with dsDNA over dsRNA binding by extending the PNA backbone to accommodate the B-DNA structure have been so far unsuccessful.185 Since the triplex formation requires a Hoogsteen base-pairing, the process is benefited from the protonation of C in the PNA strand at low pH (Fig. 36).181 Substitution of the base C with the more easily protonated 2-aminopyridine (M) promotes the triplex formation at physiological pH.179 The requirement of the oligopurine/pyrimidine stretch was relaxed by the use of 2-pyrimidinone (P) and 3-oxo-2,3-dihydropyridazine (E) bases (Fig. 36).180 Relatively few studies on the binding of PNA with backbone modifications to dsRNA targets have been published. When a few guanidine-modified αPNA monomers (GPNA) were incorporated into the aegPNA backbone, the binding to dsRNA occurred but with reduced affinity and specificity. In addition, depending on the sequence, the binding may occur in the triplex or duplex invasion modes instead of the expected triplex formation.186 Only invasion by a γPNA was observed in dsRNA with r(CUG) repeats.187 Fluorine NMR studies on the binding of PNA partially modified with LSer γPNA monomers to the miR-215 dsRNA model indicated the ability of γPNA to form a triplex invasion complex in addition to triplex formation. The preference of either of the binding modes could be fine-tuned by the appropriate placement of the γPNA monomer.188 Modified PNA with three consecutive γ-(S)-guanidinylmethyl PNA subunits showed improved affinity and selectivity for dsRNA binding via the triplex formation and enhanced cell internalization.189 The dependence of the binding mode on the precise positions of the γPNA backbone modification might explain the poorer performance of γPNA when compared to unmodified aegPNA in a templated fluorogenic reaction between two PNA probes mediated by a dsRNA template.190 Incorporation of modified bases that could not form stable Watson–Crick base pairs should offer a viable strategy to improve the selectivity of triplex binding over the triplex invasion of these modified PNAs.191
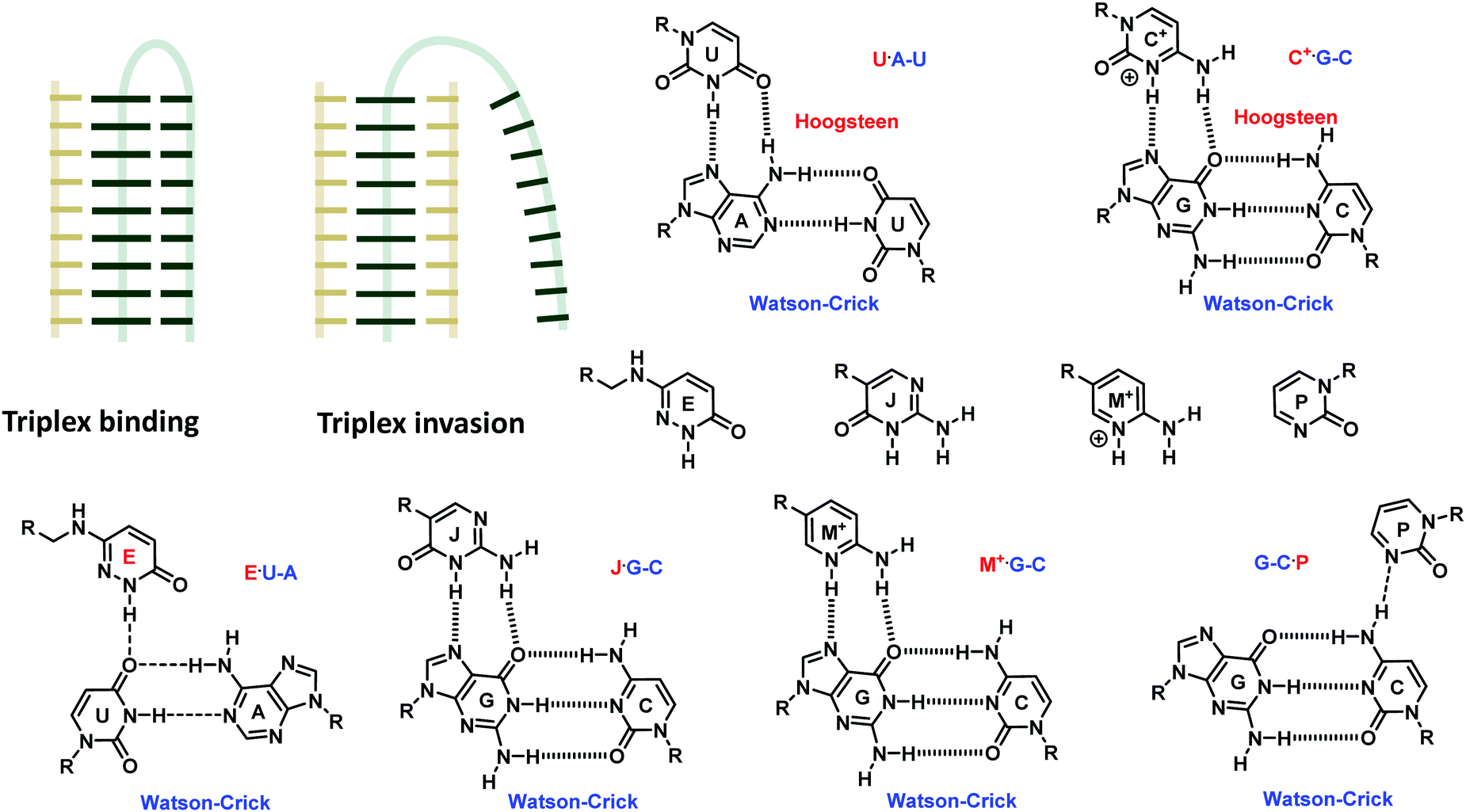 | ||
| Fig. 36 Targeting RNA duplexes (gray) by PNA (gold) via triplex formation and invasion, and the structures of the modified base that facilitate the triplex binding mode.179,180 | ||
Targeting G-quadruplexes
Targeting G-quadruplexes in a sequence-specific fashion by forming PNA·DNA heteroquadruplexes (Fig. 37A) is less straightforward than targeting duplex DNA as it is subjected to competition with the duplex formation between the G-rich PNA and the C-rich complementary DNA strand as well as self-quadruplex formation and aggregation of G-rich PNA sequences.192 Armitage et al. proposed two strategies to promote G-quadruplex formation over duplex formation193 which involves the elimination of nucleobases in the loop region and PNA backbone modification to discourage the duplex formation without affecting the (hetero)G-quadruplex formation. The use of DAla γPNA which pre-organizes into left-handed helical structures substantially destabilized the duplexes while the stability of the (hetero)-G-quadruplex was much less affected as a result of the underwound nature of the G-quadruplex structures. The combination of the two strategies allowed targeting of Myc19 DNA with low nanomolar affinities. Although the binding was weaker than that of the unmodified aegPNA or LAla γPNA, the selectivity for hetero G-quadruplex formation over the complementary DNA strand was much improved. The G-quadruplex invasion by LLys γPNA was employed for the multivalent display of the RGD peptide on a DNA G-quadruplex scaffold.194 Cyclopentyl PNA with a G4T4G4 sequence can also form self-(cpPNA·cpPNA) and hetero-(cpPNA·DNA) G-quadruplex structures. When compared to aegPNA, the self-quadruplex was destabilized and the hetero-G-quadruplex was stabilized. Thus, the use of cpPNA offers another viable strategy to modulate the preference for self- versus hetero-quadruplex formation.70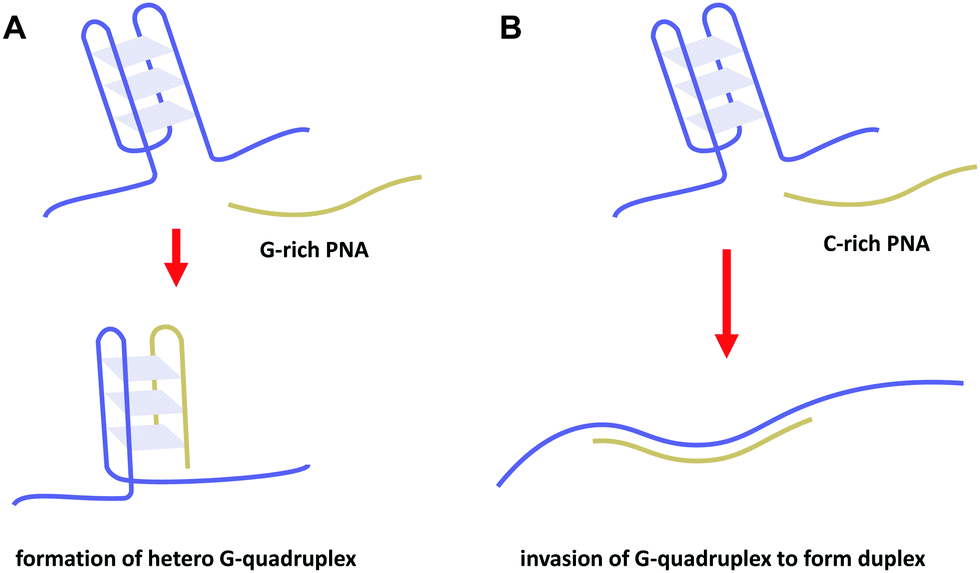 | ||
| Fig. 37 Targeting DNA G-quadruplexes (blue) by PNA (gold) by hetero G-quadruplex formation (A) or duplex formation (B). | ||
The high stability of γPNA·RNA duplexes allowed an efficient invasion of RNA G-quadruplexes by C-rich γPNA which resulted in the disruption of the G-quadruplex and concomitant formation of the γPNA·RNA duplex (Fig. 37B). The inhibition of in vitro translation of an artificial reporter mRNA transcript bearing the G-quadruplex-forming sequence inserted in the 5'-untranslated (UTR) region by the γPNA designed to target the G-quadruplex was reported to be more potent than 2′-OMe RNA and more specific than aegPNA.195
Complex structures involving backbone-modified PNA
The concept of bimodal PNA (bmPNA) that can simultaneously and independently recognize two strands of DNA has been proposed by the Ganesh group very recently. A sequence of nucleobases appended to the aegPNA backbone at the α or γ position via an appropriate linker can recognize an additional DNA strand via another set of Watson–Crick base-pairing that is independent of the first one (Fig. 38A).196–198 In the absence of the nucleobase attached to the nitrogen atom, the α or γ nucleobase-modified PNA can also recognize its DNA complementary target, albeit with somewhat weaker affinity than the original aegPNA. When both sets of nucleobase sequences are present on the same PNA strand, each sequence binds to its specific complementary DNA target independent of one another to form a ternary complex between one PNA and two DNA strands. Thermal stability measurements indicated that the melting occurred in two steps and that the two base-pairings mutually stabilized each other as shown by the increase in Tm for both steps. This led to the proposal that the melting occurred through the re-organization of the ternary complex followed by single step dissociation of the three strands rather than sequential dissociation of each strand.197 High-order complexes can also form when the PNA carries a homothymine sequence that can form a stable triplex structure with homoadenine DNA either at the α-face198 or the γ-face.199 An example is a quintuplex structure formed from two bimodal PNA, two G-rich, and one A-rich DNA strands (Fig. 38B). The central A-rich DNA strand formed a triplex with the T-rich face of the bimodal PNA strands, leaving the C-rich face available to bind to another G-rich DNA strand. The order of the hybrid formation does not affect the final composition of the ultimate complex. Such design might find new applications in the areas of nucleic acid nanotechnology or in biologically relevant systems by targeting two nucleic acid strands simultaneously.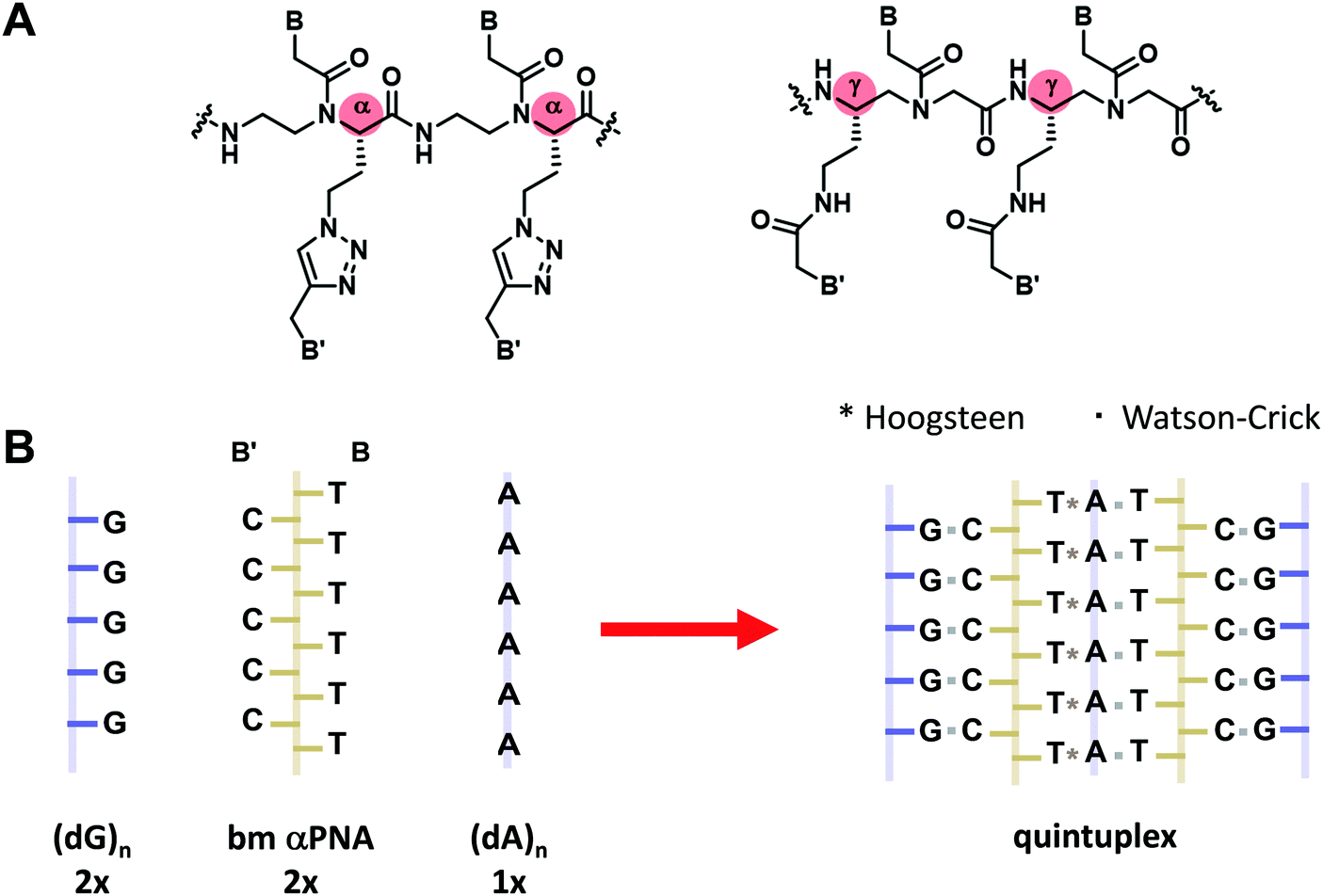 | ||
| Fig. 38 (A) Structures of bimodal αPNA and γPNA. (B) Spontaneous assembly of bimodal PNA (gold) with DNA (blue) to form complex structures.198 | ||
Another bifacial PNA (bPNA) system was recently proposed by Bong et al. Instead of using two sets of base sequences, melamine was chosen as a Janus nucleobase that allows simultaneous recognition of two thymine/uracil bases on two DNA or RNA strands (Fig. 39).200,201 In this work, the dipeptide derived from glutamyllysine202 was chosen as a scaffold rather than the aegPNA for synthetic convenience. The melamine was attached to the lysine amino group. A well-defined triplex with Kd in the low nanomolar range was formed in the presence of homothymine DNA, either as two separated strands or as a hairpin. These triplexes show a sharp cooperative melting and their formation can be detected spectrophotometrically and by gel electrophoresis. The PNA with the fully or partially methylated melamine base showed no triplex formation. Remarkably, the corresponding diaminotriazine base without the extra NH group on a similar peptide scaffold only formed a duplex with poly(dT) and poly(rU).202 The triplex formation on a single-stranded DNA/RNA target with a (T/U)n(A/C)m(T/U)n recognition site (n, m = 6–10) completely inhibited exonuclease activities and transcription/reverse transcription.203 An enhanced version with two melamine groups attached to the same peptide unit allows the design of a very short peptide that selectively targets two (U/T)6 domains of any DNA/RNA that can fold into the required triplex.204 In addition, invasion of the RNA triplex carrying a U·A·U stretch – a common motif that occurs in long non-coding RNA involved in several biologically relevant processes – by bPNA occurred with concomitant displacement of the central oligo(rA) strand. Such displacement of the oligo(rA) tail accelerates the degradation of the RNA target by exonucleases, thus allowing the possibilities of using bPNA to regulate RNA functions.205 It is conceivable that other bifacial nucleobases capable of forming similar base-triplet schemes with other combinations of nucleobases could be incorporated in the bPNA so that the recognition motif can be expanded to more diverse biologically important targets.177
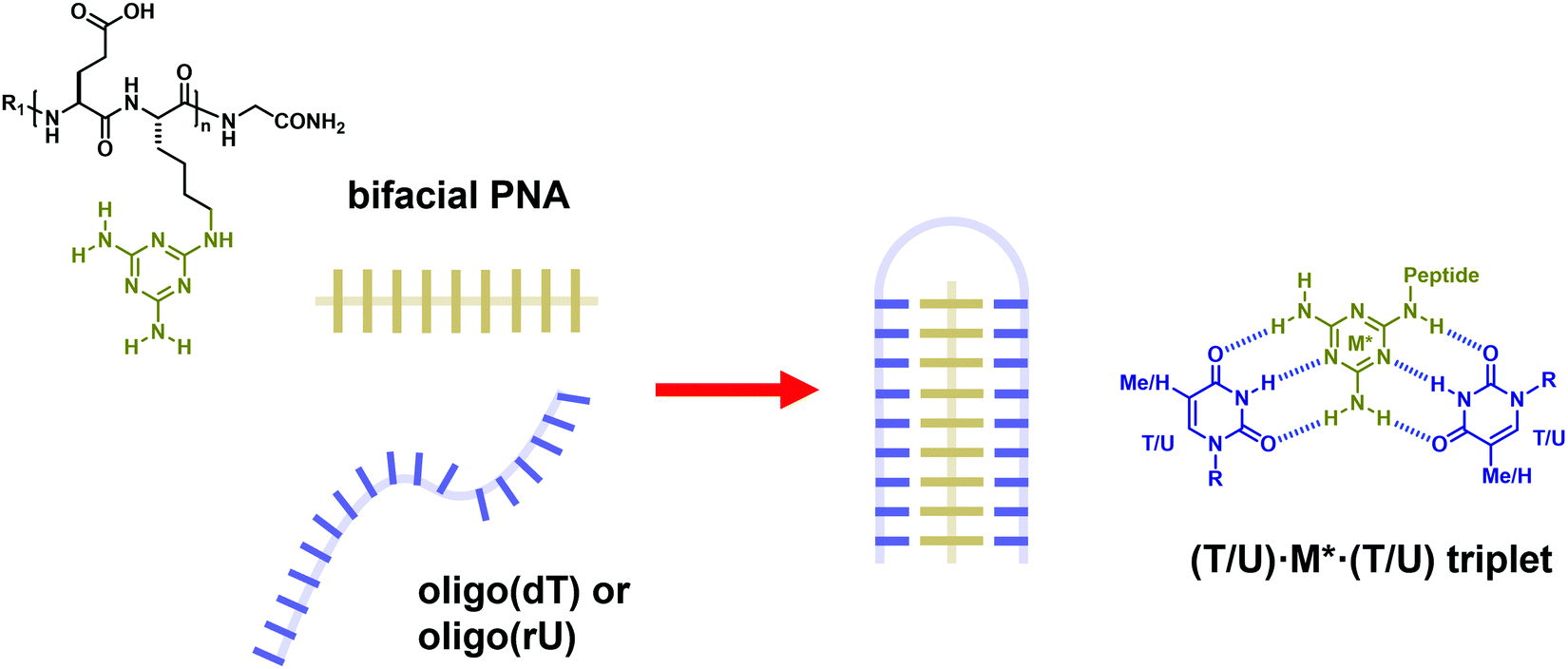 | ||
| Fig. 39 Structures of bifacial PNA containing the melamine base (gold) and its interaction with thymine in DNA (blue) or uracil in RNA strands to form a stable triplex structure (adapted with permission from ref. 200. Copyright 2012 American Chemical Society). | ||
PNA·PNA duplex formation
Due to the absence of unfavorable electrostatic repulsion, unmodified aegPNA forms highly stable PNA·PNA self-duplexes. Since aegPNA is inherently achiral, the left-handed and right-handed duplexes are formed equally well. Upon binding to DNA, the right-handed helical conformation was determined by the chirality of the DNA strand. The situation is different for modified PNA with a chiral backbone. Although most backbone-modified PNAs are chiral, they are usually not easily accessible in both enantiomers. However, the simple structure of γPNA renders both enantiomers easily accessible. It has been known since the first discovery of γPNA that single-stranded PNA with a γ-modified backbone pre-organized into a right-handed helix when the γ-substituent was in the L-configuration. It could be predicted that the opposite is true for the D-configuration and this has been experimentally verified.176,206 Two complementary γPNA sequences with the same configuration can pair to form a highly stable duplex, and such γPNA duplex formation could be used for the regulation of gene expression in a reversible manner.207 Only the right-handed γPNA can pair with natural DNA, and the two enantiomeric γPNAs do not cross-pair with each other. Accordingly, the two enantiomeric γPNAs are stereochemically orthogonal. Yet, both enantiomers of γPNA pair indiscriminately with the achiral aegPNA (Fig. 40). Such property has been utilized to create a hairpin structured probe from the chimeric γ/aegPNA sequence to improve its mismatch discrimination.208 Thus, the information between the two orthogonal γPNAs and between the left-handed γPNA and natural DNA/RNA can be transmitted through the achiral aegPNA mediator. Another level of orthogonality can be introduced by including the left-handed enantiomer of natural DNA/RNA.209,210 Several interesting applications of such orthogonal PNA systems are emerging such as in the control of nanostructure self-assemblies,176 the improvement of signal amplification by hybridization chain reaction,211 the design of a PNA-based beacon that incorporates a short DSer γPNA sequence as the signaling unit to facilitate the catalytic turnover without compromising the specificity,212 and in the supramolecular assembly of synthetic transcription factors on dsDNA without competing DNA binding.213 Recently, the concept of orthogonal PNA pairs has been extended to cpPNA probes, in which the left-handed (R,R)-cpPNA could only pair with itself, but not with natural DNA/RNA and the right-handed (S,S)-cpPNA. Importantly, only a few modifications by the chiral cpPNA monomers were sufficient to determine the helicity and the ultimate base-pairing behavior of the resulting chimeric PNA. This orthogonal strategy was employed for the development of a microfluidic-based assay of HIV-1 RNA.214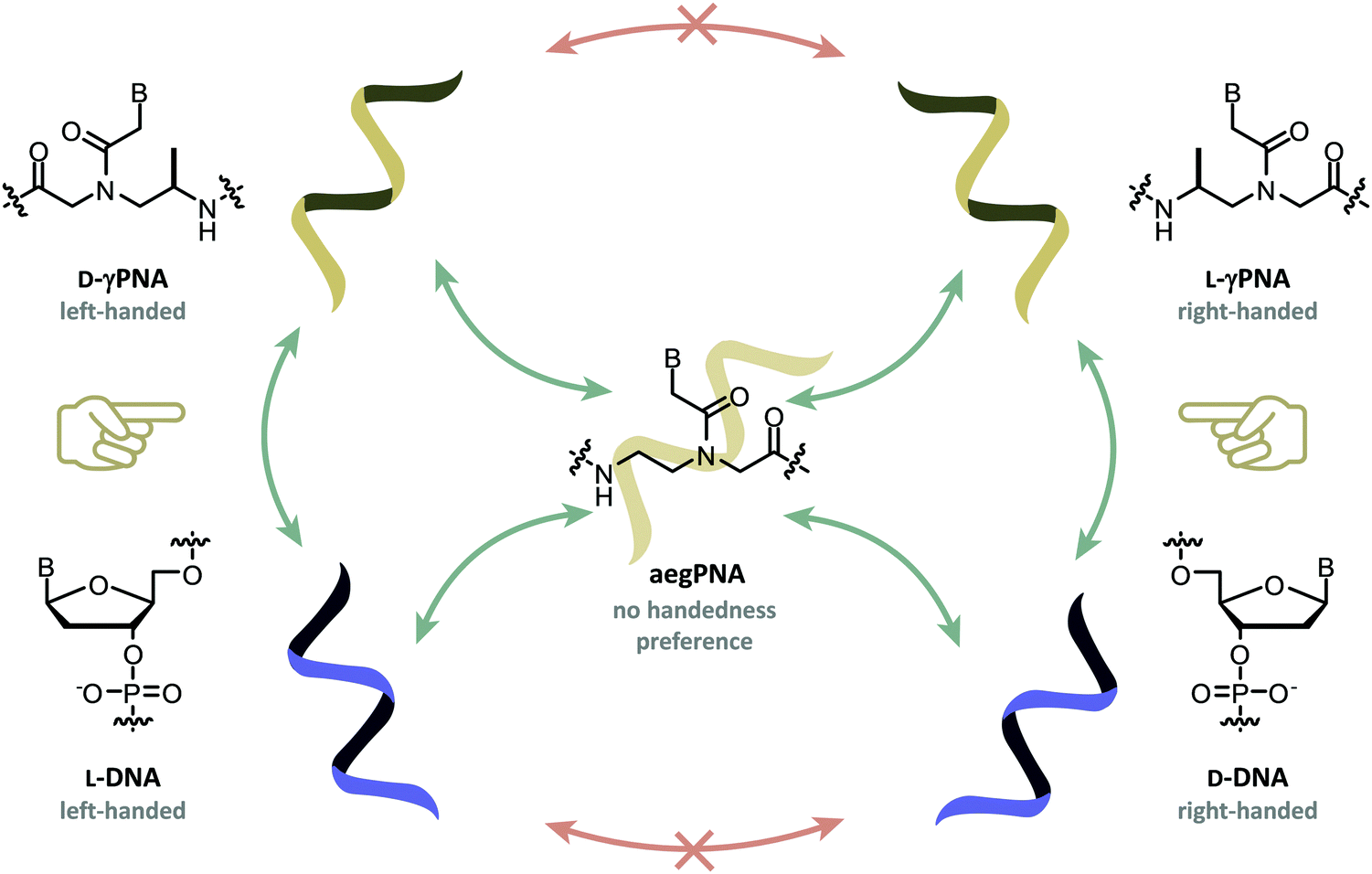 | ||
| Fig. 40 Orthogonality pairing in γPNA (gold) and DNA (blue). Only γPNA and DNA with the same handedness can self-pair and cross-pair. The achiral aegPNA pairs indiscriminately with all; thus it can act as a mediator to transmit the information between these orthogonal nucleic acids.176 | ||
Modified PNA as a supramolecular scaffold
The self-assembly of PNA, either on its own or in combination with DNA, has been exploited in creating well-defined supramolecular architectures for the construction of PNA-based nanomaterials215 or for biological applications.216 Short aegPNA can self-assemble into well-defined structures via the Watson–Crick base-pairing and base stacking to provide materials with interesting optical properties.217,218 A programmable self-assembly of short γPNA strands that are partially complementary to each other to form well-defined 3-helix nanotubes consisting of purely γPNA219 or γPNA and DNA220 was demonstrated. The use of PNA instead of DNA allows the self-assembly process to occur in organic solvents and the γPNA modification carrying a hydrophilic side chain was essential as aegPNA alone tends to form aggregates.The ability of short PNA to form a self-complementary duplex was utilized in constraining the conformation of peptides which allows enhancing and regulating their biological activities through PNA/PNA and PNA/DNA hybridization.221,222 Three-consecutive intramolecular base-pairing of γPNA has been utilized to stabilize the cyclic peptide RTD-1 in place of disulfide bonds. The retention of potent antibacterial activities of the peptide indicated that the base-pairing perfectly mimicked the disulfide bond.223 Armitage et al. also reported the use of γPNA with a hairpin construct as a staple to constrain the HIV-1 transactivator of transcription (TAT) peptide into a loop conformation which enhanced its cell penetration ability.224 The advantage of γPNA is two-fold. First, the high stability of γPNA·γPNA duplexes enables the formation of a hairpin structure with a very short stem (only 4 nt). Second, the γPNA hairpin can be designed to have an overhang sequence that can be further conjugated to other biomolecules via γPNA·DNA hybridization that occurs efficiently even with a short (6 nt) overhang sequence.
Multivalent interactions play crucial roles in several biological processes. The self-assembly of PNA or its hybridization on a DNA (or PNA) scaffold enables a multivalent display of the ligand in a programmable and precisely controlled fashion (Fig. 41A). This approach offers advantages in terms of both synthetic efficiency and control of spatial arrangement as well as density of the ligand over conventional approaches whereby the ligands are connected by a flexible linker or attached to other multivalent scaffolds.225 Winssinger pioneered the construction of multivalent oligosaccharide and peptide libraries by self-assembling two or more ligand-modified PNA on a DNA template.226–228 This provides a powerful way to generate a large number of well-defined multivalent constructs that can be subsequently screened against the respective biological receptors in a combinatorial fashion. It should be noted that strong base pairing is not required as a result of cooperativity between the base pairing and ligand–receptor binding. Thus, dynamic assemblies of L-fucose-modified short self-complementary PNA probes that were barely stable at room temperature could effectively block the binding of bacterial lectins to epithelial cells.229 Nonetheless, the use of aegPNA in these early approaches offers limited control of the ligand arrangement since only terminal modification was possible. In a subsequent work by Seitz, the incorporation of a Cys γPNA monomer in the PNA molecule allowed the introduction of the carbohydrate ligand (LacNAc) in any desired position via the thiol group. The results from the screening of ligands with different spatial arrangements agree with the expected structure of the receptor binding sites (Erythrina cristagalli lectin). In addition, the rigidity of the construct could be further fine-tuned by incorporating nick-sites or single-stranded regions (Fig. 41B).230 Appella reported a similar strategy to construct libraries of multivalent displays of various ligands including c(RGDfK) peptide,231 dopamine D2 receptor (D2R) agonist,232 and A2A adenosine receptor antagonist233 on DNA or PNA scaffolds employing Lys γPNA as the adapter.
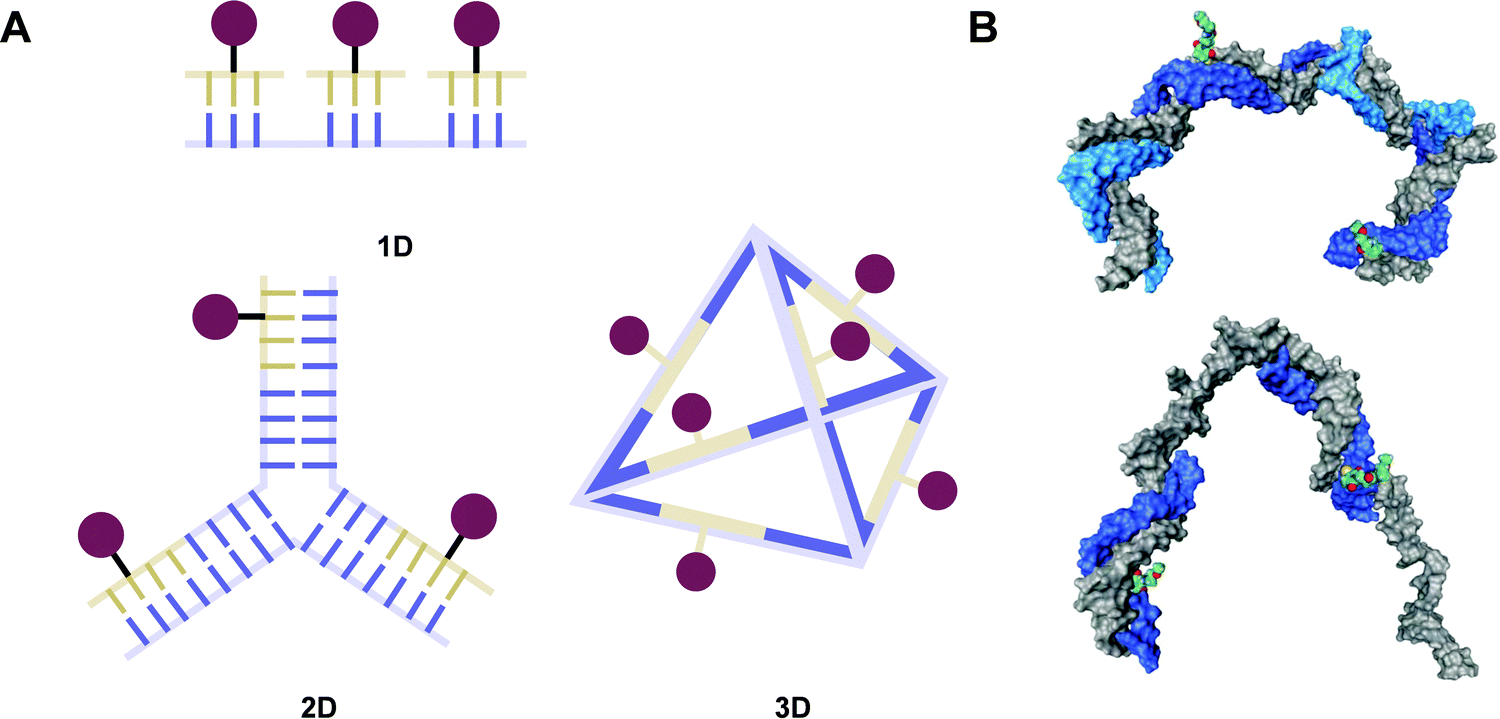 | ||
| Fig. 41 (A) Schematic representation of using PNA for multivalent display of ligands on various DNA scaffolds. (B) A model showing the spatial arrangement of two N-acetyllactosamine (LacNAc) ligands in two different multivalent constructs derived from γPNA (blue) and DNA (gray) (reproduced from ref. 230 with permission from the Royal Society of Chemistry). | ||
In a more complex architecture design, short PNA strands were used as an adapter for the rapid assembly of protein molecules on three-dimensional DNA scaffolds.234,235 The use of the γPNA backbone allows versatile attachment of the ligand at any required position along the PNA strand. Short bifunctional γPNA probes with a thiol group on the backbone could be site-specifically and stably attached to the DNA tetrahedron which could be used for programmed assembly of ligands or biomolecules.236
In the last example, the control of the supramolecular architecture of amphiphilic PNA was described by Heemstra et al. (Fig. 42).237,238 The amphiphilic PNA was created by strategic placement of hydrophobic and hydrophilic side chains on a γPNA backbone. Such amphiphilic PNA spontaneously assembled to form nanoaggregates with an average diameter around 35 nm in an aqueous environment. When the target nucleic acid (miRNA-21) was added, the hybridization between the γPNA and the nucleic acid target disrupted the aggregate structures. Thus, the aggregation state of the amphiphilic PNA was modulated by the presence or absence of its nucleic acid target. In contrast to earlier designs of amphiphilic PNA whereby the hydrophobic and hydrophilic domains are conjugated to the PNA termini,239,240 the integration of peptide side-chains directly into the PNA backbone should allow designing of more complex functional structures such as helices, sheets and their supramolecular assemblies that incorporate the base-pairing ability of PNA.
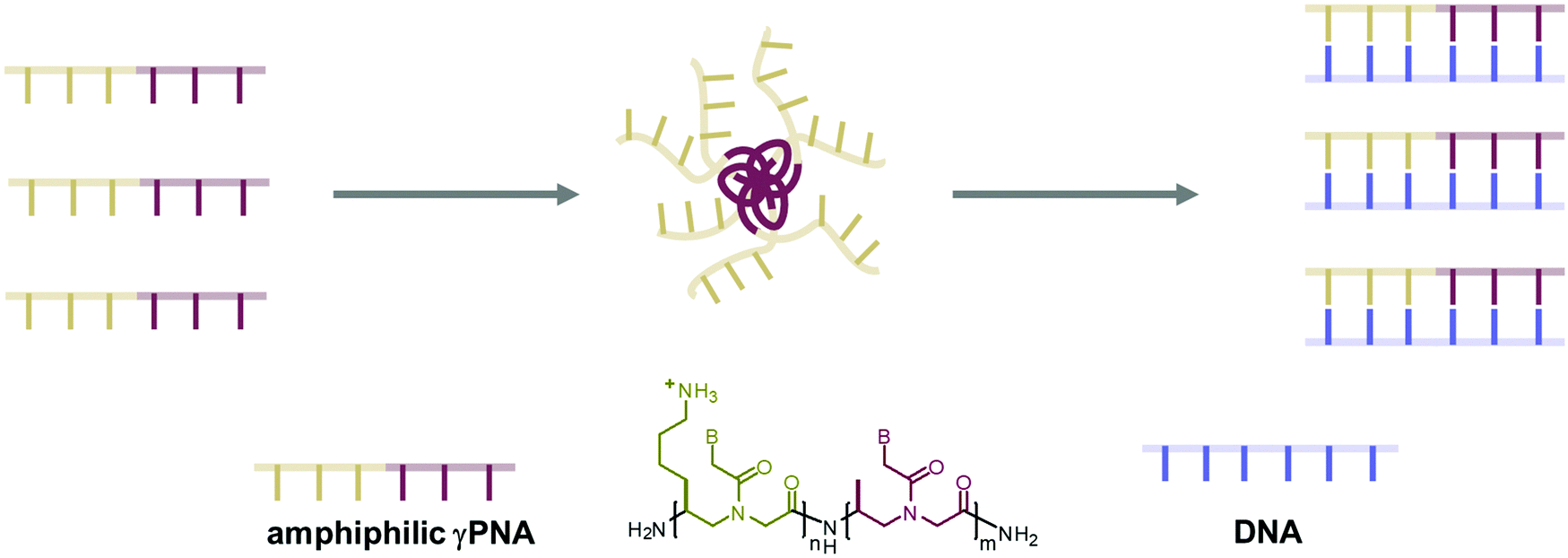 | ||
| Fig. 42 The self-assembly of amphiphilic γPNA probes is modulated by γPNA·DNA/RNA duplex formation (adapted with permission from ref. 237. Copyright 2019 American Chemical Society). | ||
Template-directed PNA oligomerization and sequence adaptive PNA
In the previous topic, the PNA was non-covalently assembled on the DNA template via stable base pairings. If the PNA strand is very short, the binding will be very weak. Despite the weak interaction of individual PNA strands, the adjacent PNA strands can be chemically joined to give a ligated product with increased stability. This principle is related to the templated ligation of PNA,241,242 but it has a more dynamic nature. Several ligation strategies have been used to join small PNA fragments on DNA or RNA templates such as native chemical ligation,178 azide–alkyne click reaction,243 and reductive alkylation (Fig. 43A).244 The last strategy allows new PNA strands to be synthesized by the assembly of short PNA fragments (4–5 nt each) on a specific DNA template whereby the sequence of the product was determined by the sequence of the template similar to the natural translation process. By incorporation of γPNA or αPNA building blocks into the tetrameric/pentameric PNA aldehyde fragments, Liu has demonstrated the use of such DNA-templated oligomerization for the synthesis of PNA oligomers bearing various functional groups in a sequence-defined fashion (Fig. 43B).244 The yield and rate of the templated reaction are related to the thermal stability of the PNA·DNA duplexes which was primarily determined by the stereochemistry and position of the side-chain attachment rather than the nature of the functional group. While similar results were observed for D-αPNA, L-αPNA, and L-γPNA building blocks, the D-γPNA showed much lower efficiency which is in good agreement with the relative stability of each modified PNA system. Such dynamic assembly of functionalized PNA on DNA templates may find application in the design and evolution of functional biopolymers by using the DNA template not only for directing the synthesis but also for facilitating identification and enrichment as well as diversification of the products.244 In the case of triazole linkage, it was reported that the 1,4-disubstituted triazole linker with appropriate spacing can mimic the trans-amide bond reasonably well as demonstrated by the similar DNA binding affinity and specificity of the unmodified and modified PNAs.243 This system should potentially be useful in DNA templating polymerization similar to the reductive alkylation described above.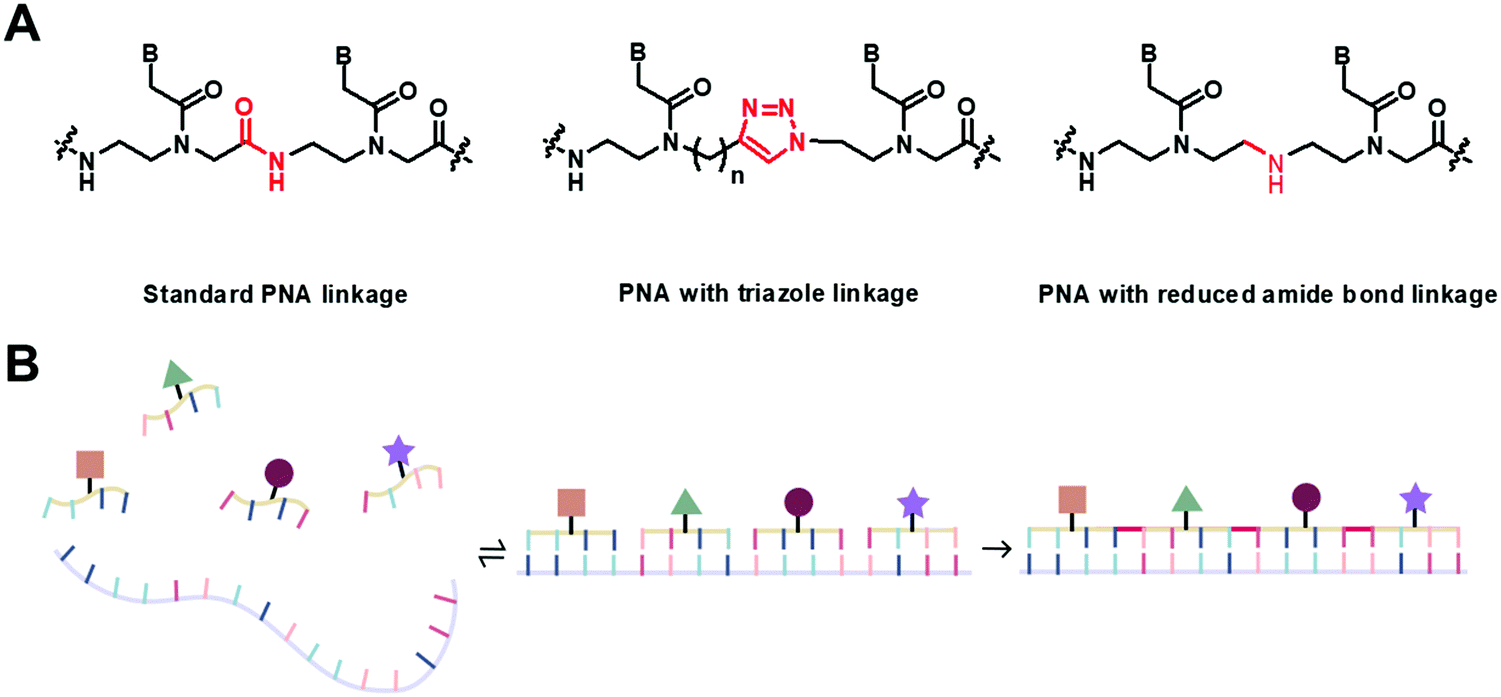 | ||
| Fig. 43 (A) Some chemistry useful for template-directed PNA oligomerization. (B) The self-assembly and oligomerization of functionalized PNA probes on a DNA template. | ||
Relating to the abovementioned template-assisted oligomerization, another kind of PNA called thioester PNA (tPNA) was designed by attaching the nucleobases, via the thiol-thioester exchange reaction, to an oligopeptide backbone bearing alternating cysteine and another amino acid (Fig. 44).245 The tPNA can form reasonably stable hybrids with DNA/RNA as well as self-hybrids. A broad range of amino acids can be accepted, with positively charged side chains forming more stable hybrids than negatively charged ones as would be expected from the electrostatic effects with the DNA backbone. Importantly, the thiol exchange reaction is readily reversible, thus allowing a dynamic exchange of the nucleobase in the presence of a template. A proof-of-concept experiment using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of adenine and deazaguanine thioester demonstrated a shift in the base ratio at equilibrium from 1:1 to 3:1 in the presence of an oligo(dT) template. The opposite results were obtained in the presence of an oligo(dC) template. Thus, this tPNA system represents an adaptive system whose sequence information can dynamically change according to the environment.
:1 mixture of adenine and deazaguanine thioester demonstrated a shift in the base ratio at equilibrium from 1:1 to 3:1 in the presence of an oligo(dT) template. The opposite results were obtained in the presence of an oligo(dC) template. Thus, this tPNA system represents an adaptive system whose sequence information can dynamically change according to the environment.
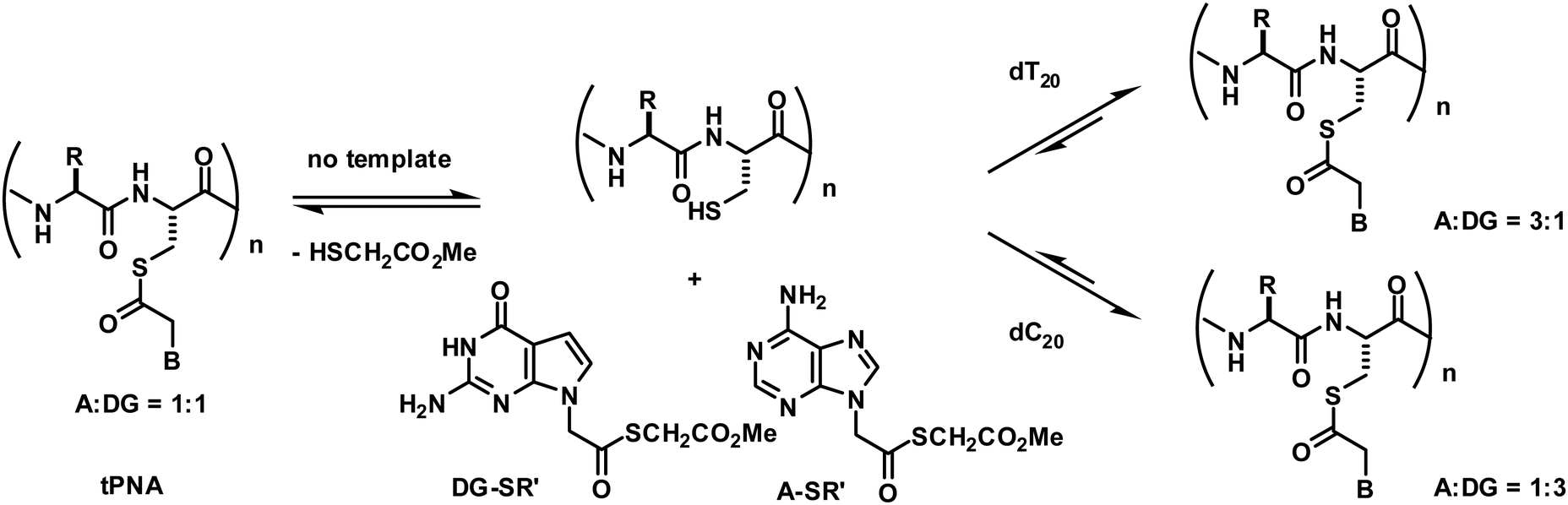 | ||
| Fig. 44 Dynamic sequence adaptive behavior of thioester peptide nucleic acids (tPNA).245 | ||
Modified PNA for nucleic acid biosensing and bioimaging
The high affinity, specificity, and biological stability of PNA suggest its obvious utility in biosensing and bioimaging applications. Accelerated by the recent discovery of many regulatory roles of RNA,246 a particularly fast-growing area that received widespread attention and could benefit from the high performance of PNA probes is the imaging of RNA in live cells.247 Biosensing and bioimaging applications of PNA are major topics that may be worth several separate monographs. Although many reviews on such topics exist,248–251 they primarily focus on conventional PNA probes with only a few examples of modified PNA probes. This section will primarily focus on the advantages of conformationally constrained PNA which has the potential to offer improved nucleic acid binding affinity and sequence specificity over conventional aegPNA probes in biosensing and bioimaging applications.The cyclopentyl-modified PNA (cpPNA) showed a substantial improvement in DNA binding affinities and mismatched specificity over aegPNA. The incorporation of only a few residues of the cpPNA monomer was sufficient to increase the stability by two orders of magnitude giving the dissociation constants in the sub-nanomolar range.71 Hence, cpPNA offers great potential to enhance the performance of PNA-based sensors. In one example, a PNA probe with single cpPNA modification was immobilized on a glass slide for the scanometric detection of anthrax lethal factor DNA in a microarray format.252 The DNA target simultaneously hybridized with the immobilized PNA probe and another DNA-functionalized gold nanoparticles, resulting in the deposition of the gold nanoparticles on the sensor chip surface. Subsequent electroless-silver deposition to enhance the signal enabled a simple colorimetric readout by a scanner. The DNA target was detectable down to 25 aM, which was much better than the 50 fM detection limit obtained by the corresponding DNA capture probe. Interestingly, no signal was obtained when unmodified aegPNA was employed as the capture probe. Although the basis of such remarkable performance improvement was not obvious, the advantage of cpPNA over aegPNA probes was demonstrated.
In another study, the cpPNA probe was employed for the detection of anthrax protective antigen DNA in a PNA-based sandwich hybridization. The cpPNA was immobilized on a 96-well plate as a capture probe, and aegPNA modified with biotin at the C-terminus was used as the reporter probe. Subsequent binding to the avidin-HRP conjugate followed by the addition of the HRP substrate resulted in color generation that was measured spectrophotometrically. As low as 10 zmol of DNA was successfully detected while the corresponding unmodified aegPNA probe gave measurable signals down to only 10 fmol levels under the same conditions.253 Quantitative detection of anthrax DNA was possible over the range of 10 to 107 copies of DNA.254 The cpPNA version of the forced intercalation PNA (FIT) probe was also recently reported.255 The FIT probe originally developed by Seitz consists of thiazole orange or similar dyes linked to the aegPNA backbone as an artificial nucleobase that lights up upon binding to the correct DNA/RNA target sequence that restricted the dyes’ motion.256 The introduction of two flanking cpPNA monomers significantly improved the performance of the FIT PNA probes in terms of both brightness and mismatch discrimination for the in vitro detection of RNA. This resulted in lowering of the limit of detection to 1.8 nM which was almost an order of magnitude better than that of conventional FIT probes based on aegPNA. However, the comparative performance in cellular RNA imaging is yet to be demonstrated.
Applications of pyrrolidinyl PNA probes with the α/β-peptide backbone (acpcPNA) for DNA sensing have already been reviewed elsewhere.117,257 The superior mismatch discrimination ability of immobilized acpcPNA over DNA and aegPNA probes was experimentally demonstrated in a surface plasmon resonance (SPR) experiment whereby the three probes were critically compared side-by-side in capturing the same ssDNA target.258 The experiments also revealed the faster kinetics of hybridization and confirmed the exclusive binding in the antiparallel direction of acpcPNA probes. On the other hand, the same comparative SPR experiments revealed that the binding of immobilized acpcPNA probes to dsDNA targets was less efficient than aegPNA likely due to its steric bulkiness.259 Thus, acpcPNA probes have been extensively used as the key recognition element in paper- or chip-based electrochemical and optical nucleic acid sensing devices for point-of-care applications.260–267 In many cases, the PNA·DNA hybridization resulted in the change of electrical properties such as capacitance and thus no labeling or other signal transduction mechanism was required.268,269 While acpcPNA probes have been primarily used for DNA detection, satisfactory performance in the electrochemical detection of RNA was successfully demonstrated.270 The differential electrostatic properties of PNA and DNA have been utilized for the detection of the DNA sequence. According to this principle, the negatively charged DNA was selectively captured via electrostatic interactions on various surfaces such as polymer beads,271,272 magnetic beads,273 cellulose paper,274 or carbon electrode275 that were coated with a positively charged polymer such as chitosan or poly(2-(dimethylamino)ethyl methacrylate) (PQDMAEMA). The neutral PNA molecule could not interact with such a surface unless it was hybridized with the captured DNA target, and the presence of the PNA probe hybridized to DNA could be detected by optical, electrochemical, or mass spectrometric detection.
The acpcPNA probes have also been used in the development of self-reporting fluorescence probes for DNA hybridization.257 These probes are designed to carry an environment-sensitive dye such as pyrene or thiazole orange attached to the terminal, backbone, or nucleobase. The fluorescence change was induced by the hybridization between the PNA probe and the DNA target that alters the environment of the dye.134,276,277 Alternatively, a simple fluorophore-labeled PNA probe was used in combination with a quencher which can be present on the same PNA strand166 or as a separate entity such as another quencher labeled PNA or DNA strand,278 oligo(dG),279 graphene oxide (GO),280 and metal nanoparticles.281 In all cases, the fluorescence of the free probe could be restored after hybridization with the correct DNA/RNA target which resulted in the separation of the probe and the quencher. Faster kinetics, superior single mismatch discrimination as well as improved sensitivity in the fluorescence restoration of GO-quenched acpcPNA over aegPNA and DNA probes by DNA targets have been demonstrated and the principle had been successfully applied for both in vitro DNA/RNA detection and cellular RNA imaging.281
The high affinity of short γPNA probes towards DNA targets enabled a sandwich hybridization assay to be performed on very short DNA and RNA targets which is typically not possible with conventional DNA or even aegPNA probes. γPNA amphiphiles have been used in combination with fluorescent DNA nanotags in micelle-tagging electrophoresis.284,285 The two short probes bound to adjacent positions on the same DNA target cooperatively stabilize each other by base stacking without compromising the specificity as generally observed when only one longer probe sequence was used. This strategy allows multiplex detection of various members of the let-7 miRNA family at a single mismatch resolution with detection limits of 10–100 pM.285 Likewise, the use of γPNA allows an efficient hybridization chain reaction (HCR) via γPNA·γPNA duplex formation to be performed using very short hairpin probes with only 5 nt stem, 5 nt loop, and 5 nt toehold making the total length of the hairpin constructs only 20 nt, far shorter than the DNA-based constructs (typically 30–40 nt). The use of miniPEG γPNA solved the problem of the limited solubility of the PNA-derived cHCR products.286 A microarray prepared from miniPEG γPNA probes was reported to distinguish between complementary and mismatched DNA targets better than aegPNA arrays,287 although the results may not be directly comparable due to the difference in the probe sequence and length as well as other factors. The DNA duplex invasion ability of single-stranded γPNA was utilized in the chemiluminescence microbeads array for the rapid and simultaneous identification of 21 blood pathogens without requiring denaturation (Fig. 45A).282 It was also used in the nanopore-based sequencing and barcoding of long DNA strands without suffering the sequence limitation as in the case of triplex-forming bis-aegPNA.288 The sequence-specific invasion of DNA duplexes by γPNA probes resulted in the change of electronic signals when the DNA strand is passing through the nanopore (Fig. 45B). This strategy allows the detection of CFTRΔF508 gene mutation289 and classification of HIV-1 sub-types283 that could not be directly performed by the nanopore alone due to the similarity between the viral subtype genes.
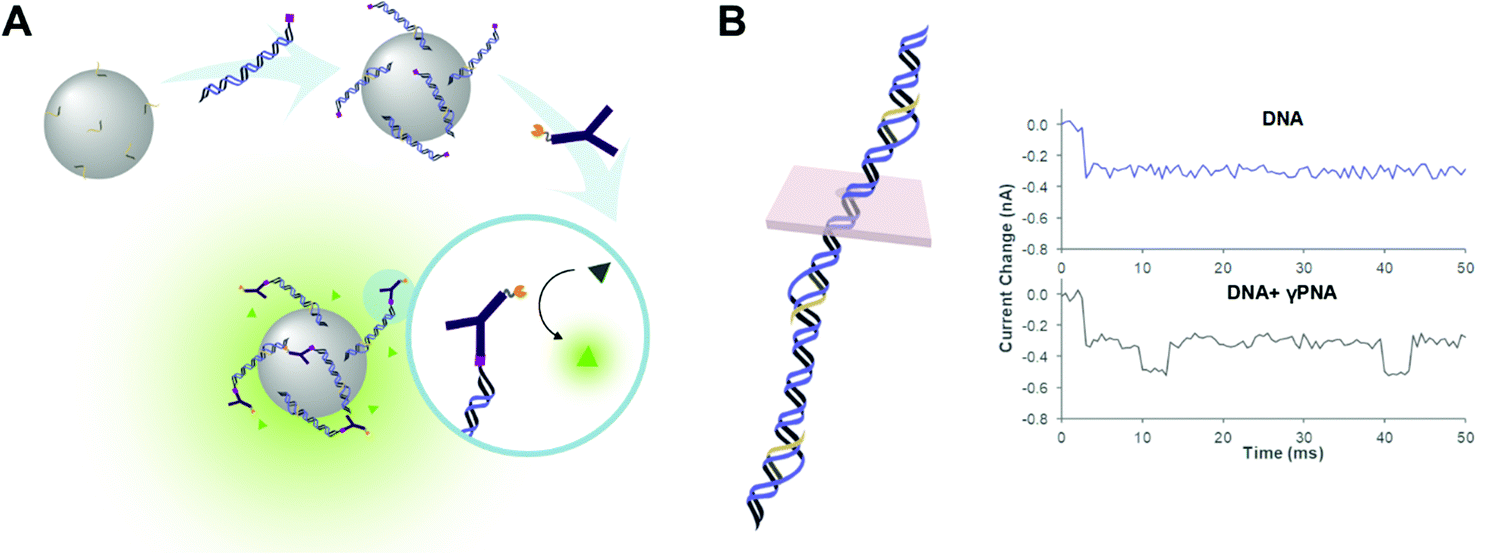 | ||
| Fig. 45 Examples of biosensing strategies that took the advantages of the duplex invasion ability of modified PNA probes. (A) Chemiluminescence microbeads γPNA array for the rapid and simultaneous identification of blood pathogens.282 (B) Nanopore-based sequencing and barcoding of long dsDNA strands by γPNA probes (adapted with permission from ref. 283. Copyright 2012 American Chemical Society). | ||
The γ-position of PNA is an ideal place for internal modification of PNA via amide bond formation38,39 or click chemistry.293 A 5-azidomethyluracil Lys γPNA monomer allows facile double functionalization at the internal positions of the PNA strand.294 The modifying group on the γ-Lys side chain pointed in the minor groove direction while the modifying group on the C5 of uracil pointed in the major groove direction and thus the strategy allows placement of the label in a controllable fashion. Chimeric γGPNA-aegPNA probes have been used for quantitative imaging of cellular miRNA-21 and miRNA-31 in different cancer cell lines via a fluorogenic RNA-templated Staudinger reaction which unmasked the fluorophore on one of the PNA strands (Fig. 46A).290 The GPNA modification facilitates the cell permeability of the probes. The short Cy5-labeled γPNA miniprobe bound cooperatively to the TTAGGG repeats in the human telomere. The binding was enhanced by the incorporation of the G-clamp into the γPNA and thus the performance of a short (6 nt) probe could approach that of longer probes (12–18 nt) without the G-clamp. The shorter probe stained faster and showed improved sensitivity due to the increasing number of labeled-probes bound to the same length of the telomere target. Its effectiveness in the staining of short telomeres in Jurkat cells that do not stain well with conventional aegPNA probes was demonstrated.295 By a strategic design of the probe sequence, two 9 nt γPNA miniprobes with a pair of FRET dyes could bind alternately on the same DNA telomere. This allows the visualization of the telomere in the cells and tissues under wash-free conditions with minimal interference from background autofluorescence (Fig. 46B).291 The application of miniPEG γPNA in combination with HCR for visualizing non-nucleic acid targets was demonstrated for the membrane protein carbonic anhydrase IX (CA IX, a cancer biomarker) expressed on the cell surface. It should be noted that in this example, the non-nucleic acid target was indirectly detected with the assistance of a CA IX ligand conjugated to an initiator γPNA strand (Fig. 46C).292
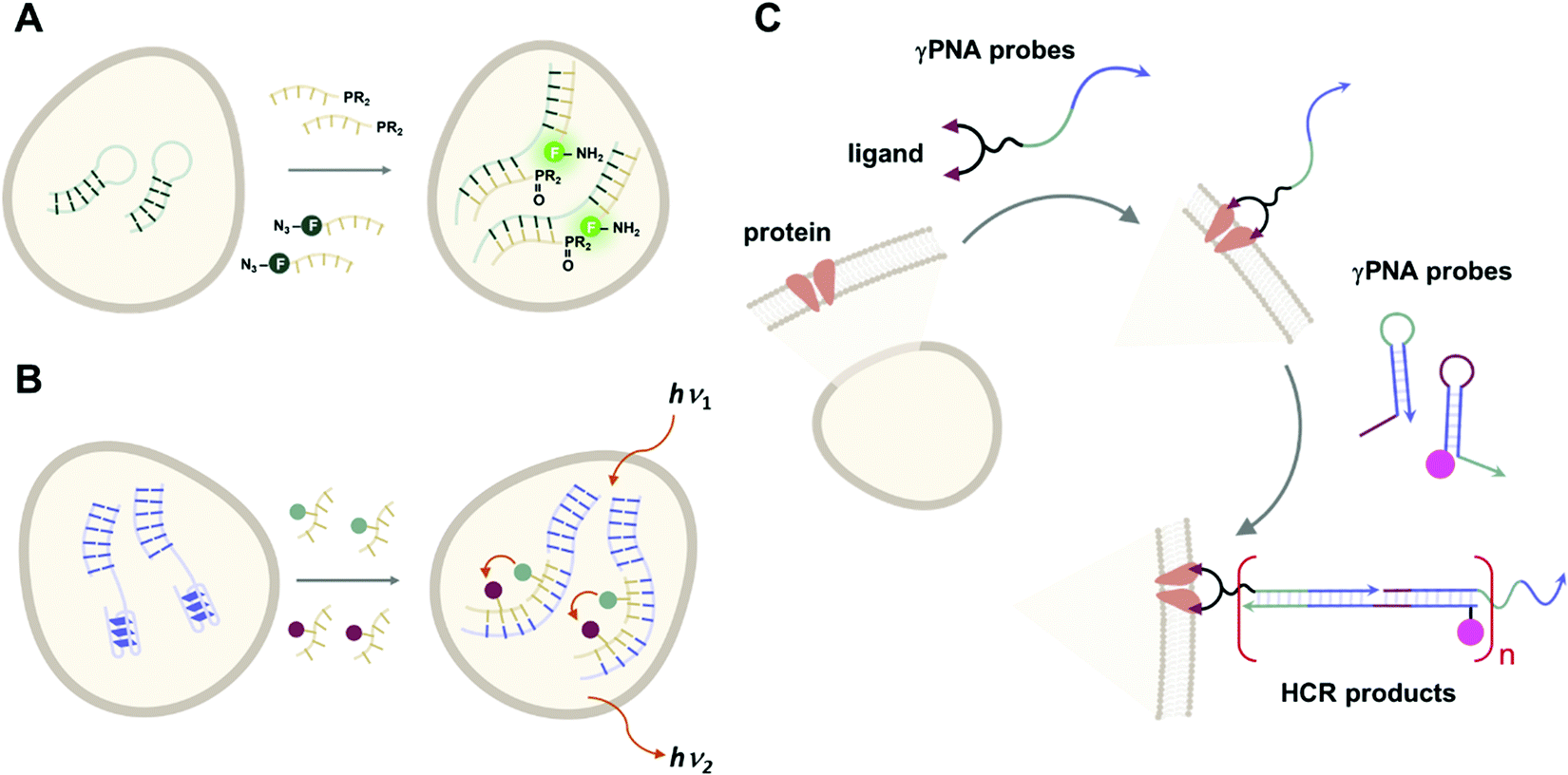 | ||
| Fig. 46 Examples of strategies for cellular imaging that took the advantages of high affinity modified PNA probes. (A) RNA imaging by templated Staudinger reaction between two PNA probes.290 (B) Visualization of telomeric DNA by PNA probes.291 (C) Visualization of carbonic anhydrase IX with the aid of γPNA probes and HCR.292 | ||
Applications of the chiral box DLys αPNA for in vitro detection of DNA single point mutations in clinical diagnostic were demonstrated by capillary electrophoresis296 and SPR.297 The improved mismatch discrimination over DNA and aegPNA probes was explained by the strong destabilizing effects of the mismatch over the complementary hybrids as a result of limited conformational flexibility of the modified PNA.
Modified PNA for enhancement of cellular uptake and gene regulation
The conventional aegPNAs have already found tremendous applications in the areas of gene regulation and editing.298–300 Backbone modification offers potential improvement of the performance of aegPNA not only by increasing binding affinities and specificity but also by providing opportunities for introducing various functional moieties to improve the cellular uptake and pharmacodynamic and pharmacokinetic properties, as well as reduce non-specific binding.Various functions can be incorporated into the PNA molecule through the modification of the α-substituents. In the simplest form, fluorophores can be incorporated at the internal positions of the PNA molecule via the lysine side-chain amino group.301 More complex functional groups such as sugars have been incorporated at the α-position of PNA to modulate its pharmacokinetic properties without deteriorating the DNA binding ability.302,303 Some glycosylated αPNAs showed improved hepatic uptake by 7–40 fold relative to unmodified PNA.303 Another related study in N-acetylgalactosamine (GalNAc)-modified γPNA suggested that exact placement of the GalNAc group produced a strong effect on the uptake efficiency whereby the introduction of three-consecutive backbone GalNAc-modification was preferred over a single terminal modification with three GalNAc groups.304 Inspired by the HIV-1 TAT transduction domain which is rich in amino acids with basic side chains, the αPNA with Arg modification (“GPNA”) was proposed to improve cellular uptake by Ly et al.305 The GPNA could indeed enter human cells and localize in the nucleus. GPNA with D-Arg modification was shown to offer superior binding affinity than L-Arg towards RNA targets,306 although the duplex stability was lower than that of the unmodified PNA·RNA hybrids, especially when consecutive modified residues were present in the PNA strand. The PNA sequences with an alternating unmodified and D-Arg modified backbone showed smaller effects on the affinity towards both DNA and RNA targets and still allowed effective cell penetration.307 At micromolar concentrations, the GPNA designed to target the 5'-UTR region of the E-cadherin mRNA transcript exerted the antisense effect by downregulating the E-cadherin expression in A549 cells as shown by immunofluorescence staining.308 A GPNA designed to target the epidermal growth factor receptor (EGFR) mRNA was shown to inhibit EGFR expression and cancer cell growth without the aid of transfection agents. Antitumor effects at the same level as that of standard EGFR inhibitors including erlotinib and cetuximab were demonstrated in xenograft mouse models.309 Another study by Mitra and Ganesh on the positively charged (R)-aminomethylene αPNA revealed similar trends in terms of DNA binding and cell permeability.51 The latter work also revealed the superiority of γ-modifications over α-modifications.
The γPNA has been extensively used for biological applications because of the excellent binding properties and ready availability. The γ-side chain modification for biological applications usually incorporates positively charged38,39,46–48,50,51 or hydrophilic groups42,45,49 to improve the water solubility and cell penetration, and reduce non-specific interactions. The miniPEG γPNA with a hydrophilic diethyleneglycol side chain45 has found widespread use in several biological applications including the control of gene expression and gene editing.
The cell-penetrable guanidine modified αGPNA and γGPNA were evaluated for anti-miRNA-210 activities in K562 chronic myelogenous leukaemic cells by Corradina et al. in 2012.310 The incorporation of the arginine carrier within the GPNA backbone was shown to improve the cellular uptake and stability towards enzymatic digestion when compared to aegPNA with oligoarginine appended at the N-terminus, although the observed anti-miRNA-210 effects were similar in cell-based assays. While the enzymatic stability issue could be solved by using oligo(D-arginine), PNA-oligoarginine conjugates have been reported to be more cytotoxic than GPNA as a result of their amphipathic nature.308 Monga et al. also reported an intrinsically cell-permeable antisense γ-guanidine-modified PNA (γGPNA)-aegPNA chimera designed to target either the transcription or translation start sites of the β-catenin gene in human hepatocellular cancer cells (HCC).311 Complementary γGPNA decreased the expression level of β-catenin mRNA and protein in HepG cells while the mismatched sequence did not. Moreover, the treatment also affects the liver tumor cell biology such as decreasing cell proliferation and survival, and expression of angiogenic factors.
Glazer et al.312 reported the cellular delivery of miniPEG γPNA and γGPNA employing poly(lactide-co-glycolide) (PLGA) nanoparticles as a carrier. The γPNA was designed to target the CCR5 chemokine receptor mRNA transcript at the middle of the mRNA transcript. The CCR5 expression level was suppressed by a half while essentially no change in the CCR5 expression was observed with the control aegPNA and a scrambled γPNA. This suggests that the high-affinity γPNA could better inhibit the translation machinery than conventional antisense aegPNA that usually requires targeting at the 5'-UTR site. A tetra-arginine-modified tail-clamp LSer γPNA designed to target miRNA-155 was shown to exhibit superior anticancer activities in lymphoma cell lines and in mouse models over anti-miR-155 aegPNA and a control γPNA sequence.313 Likewise, a 22nt miniPEG γPNA designed to target miRNA-210 can suppress tumor growth in mice much better than aegPNA with the same sequence and another miniPEG γPNA with a scrambled sequence when co-delivered with PLGA nanoparticles.314 In a more elaborated control, an antisense miniPEG γPNA conjugated with pH-low insertion peptides (pHLIP) was shown to selectively target the DNA double-strand break repair factor KU80 in tumor cells thereby making them susceptible to irradiation.315 In most cases, it was proposed that the better biological activities of the conformationally constrained γ-modified PNA might be related to the pre-organization ability that leads to higher binding affinity. However, the increased hydrophilicity of the miniPEG γPNA could also play an important role in reducing off-target interactions that could limit the availability of the PNA to exhibit the desired biological effects.
Recently, Glazer et al. have reported a series of interesting studies on site-specific genome editing induced by PNA in the presence of donor DNA.299,300,316 The genome editing in mouse bone marrow to correct the β-globin gene IVS2-654 (C → T) mutation by a miniPEG γPNA and PLGA nanoparticle platform was demonstrated ex vivo and in vivo.317 The γPNA binds at a mixed-sequence site on the gene via the DNA duplex invasion, allowing the gene edit to be performed without the need of the triplex-forming sequence typically required for the aegPNA-based gene-editing.318 Although the efficiency still requires improvement to compete with the well-developed CRISPR-Cas9 and related technologies,319 such PNA-based gene editing appears to offer promising potential for therapeutics and beyond. However, despite the seemingly solid evidence of gene-editing by γPNA including in vivo320 and in utero321 studies in mouse models, a question has been raised whether the observed effects are real or just artifacts resulting from the possible aggregation of the (miniPEG) PNA onto the donor DNA.322
The pyrrolidinyl acpcPNA has not yet been extensively studied for biological applications. In one report, the acpcPNA designed to target the NF-κB binding site in the promoter region of the Il6 gene was introduced to macrophage cells with the help of oxidized carbon nanoparticles and was shown to inhibit the stimulated production of Il6 mRNA as well as IL-6 protein levels in vitro.323 Enhanced cell membrane permeation of the acpcPNA was also realized by backbone modification with positively charged groups.135
Conclusions and outlook
This review covers the modification of PNA by introducing constraint in the aegPNA backbone. A big landscape has been provided to show what has been done and what remains to be explored. We have attempted to provide sufficient details to allow comparison of the performance and evaluation of the success of each PNA design in a systematic fashion. However, this may not be possible in all circumstances due to the availability and completeness of data issues. In many cases only data for chimeric PNA resulting from only one or two substitutions of the constrained PNA monomer along the aegPNA backbone, many of which are the homothymine or homoadenine sequences that are not typical structures, were reported. Although in some instances, the stabilization translates well into the homogeneous mixed-sequence backbone-modified PNA, the data from chimeric systems should be used with care. It is most likely that the exact stabilization/destabilization will depend on the type of nucleobase, position of modification, the extent of modification, and the nature of the nucleic acid target, among many other parameters. Destabilization upon introduction of up to a few residues may reflect the incompatibility of the modified monomer with the aegPNA duplex structure resulting in local or global distortion of the PNA structure and the modified PNA with a homogeneous backbone may show different binding profiles. Likewise, the observed stabilization should be interpreted with equal care.There are many factors to consider in the design of conformationally constrained PNA. The purpose of the introduction of conformation constraint was to reduce the entropy loss. In order to make this strategy effective, the conformation must be locked into the right one. Many examples in this review repeatedly demonstrated that conformational constraints are more often than not have detrimental effects on the binding. The aegPNA should be regarded as a fortunate success because a balance between rigidity and flexibility (“constrained flexibility”) provides a benchmark system that already outperformed natural DNA or RNA in terms of binding affinity and specificity. The availability of several NMR and X-ray structures of PNA hybrids provides a basis for the rational design of conformationally constrained PNA. However, in reality this may not be as straightforward as it sounds. Although all the torsional angle parameters are known for each type of PNA structure, there are variations among different data sets, and even in the same structures some variations exist. In attempts to control one torsional angle, one inevitably disturbs the others. This is especially true when more than one bond in the PNA backbone is part of cyclic structures. There is an inevitable trade-off between flexibility and rigidity to find a good balance. In addition, the introduction of a cyclic structure introduces stereochemical complication which grows exponentially with the number of chiral centers incorporated thus contributing to the synthetic challenges. Stereochemistry is very important in controlling the topology of the molecule such as being in extended or helical (left-handed vs right-handed) conformations. Unfortunately, the effect of stereochemistry has not been fully evaluated in many PNA systems. Thus, it is likely that several promising modified PNA systems are waiting to be discovered and show their full potential.
Despite such complications, impressive progress has been made since the first discovery of aegPNA some 30 years ago. It has become clear that the introduction of conformational constraint is a viable strategy for the improvement of PNA binding affinity, base-pairing specificity, and selectivity in terms of both directional selectivity and type of target (DNA vs RNA). One of the best-performed strategies that also happens to be the least demanding in terms of synthetic efforts is the introduction of the γ-substitution, which pre-organizes the PNA into helical conformation to avoid an unfavorable steric clash with the PNA backbone.42 Another successful strategy was the introduction of a cyclic structure to primarily control the β values, and the trans-five membered ring appeared to provide a good balance between flexibility and rigidity.70 Alternatively, one may move beyond the aegPNA template and find an alternative scaffold as demonstrated by the acpcPNA with an α/β-peptide backbone.117 This is akin to moving from the natural nucleic acid sugar-phosphate backbone to PNA which has already proven to be an exciting and rewarding discovery.8 Most chiral PNAs perform well in terms of improving the directional selectivity of the achiral aegPNA, but selectivity for either DNA or RNA has not yet been fully realized. Ironically, decent selectivity for DNA over RNA and other unusual properties such as the inability to self-pair have been realized by serendipity rather than by rational design.117
This review also highlights the practical advantages of these modified PNAs in various applications ranging from materials science, drug discovery, cellular imaging, gene regulation, and gene editing. Although the emphasis was made on the advantages of these modified PNAs in terms of higher binding affinity and specificity, this should be regarded as the minimum requirement and not the only factor that will determine the ultimate success of the modified PNA for particular applications. Of course, many of such applications indeed rely on the high-affinity binding and take full advantage of the modified PNA that can form more stable hybrids with nucleic acid targets than aegPNA. Several other applications also clearly benefited from the higher specificity of modified PNA probes as well as the ability to incorporate functional entities directly into the PNA backbone in a precisely controlled fashion. Nevertheless, for applications in complex biological systems, other properties that should be taken into consideration include solubility, cell permeability, non-specific aggregation, off-target binding, pharmacokinetics, and pharmacodynamics, among others. In many cases, it might be difficult to judge whether the apparently better performance of modified PNA over aegPNA is due to the stronger affinity to the target as a result of the backbone modification or there are complications from indirect effects that might modulate other properties such as permeability, non-specific binding, or biological stability. Currently, little is known about such indirect effects, and more studies are still required to clarify this.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the Thailand Research Fund through the International Research Network Grant (IRN62W0002) (to T. V.) is acknowledged. The authors also thank Miss Kotchakorn Supabowornsathit and Mr Kriangsak Faikhruea for preparing some illustrations. Molecular graphics and analyses were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.References
- C. Buccitelli and M. Selbach, mRNAs, proteins and the emerging principles of gene expression control, Nat. Rev. Genet., 2020, 21, 630–644 CrossRef CAS PubMed.
- N. Chaudhary, D. Weissman and K. A. Whitehead, mRNA Vaccines for infectious diseases: Principles, delivery and clinical translation, Nat. Rev. Drug Discovery, 2021, 20, 817–838 CrossRef CAS PubMed.
- K. Morihiro, Y. Kasahara and S. Obika, Biological applications of xeno nucleic acids, Mol. Biosyst., 2017, 13, 235–245 RSC.
- K. Duffy, S. Arangundy-Franklin and P. Holliger, Modified nucleic acids: Replication, evolution, and next-generation therapeutics, BMC Biolol., 2020, 18, 112 CrossRef PubMed.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Prebiotic systems chemistry: New perspectives for the origins of life, Chem. Rev., 2014, 114, 285–366 CrossRef CAS PubMed.
- A. Eschenmoser, Searching for nucleic acids alternatives, Chimia, 2005, 59, 836–850 CrossRef CAS.
- S. Ochoa and V. T. Milam, Modified nucleic acids: Expanding the capabilities of functional oligonucleotides, Molecules, 2020, 25, 4659 CrossRef CAS PubMed.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide, Science, 1991, 254, 1497–1500 CrossRef CAS PubMed.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freler, D. A. Driver, R. H. Berg, S. K. Ki, B. Norden and P. E. Nielsen, PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules, Nature, 1993, 365, 566–568 CrossRef CAS PubMed.
- P. Muangkaew and T. Vilaivan, Modulation of DNA and RNA by PNA, Bioorg. Med. Chem. Lett., 2020, 30, 127064 CrossRef CAS PubMed.
- J. D. R. Perera, K. E. W. Carufe and P. M. Glazer, Peptide nucleic acids and their role in gene regulation and editing, Biopolymers, 2021, 112, e23460 CrossRef CAS PubMed.
- V. A. Kumar and K. N. Ganesh, Conformationally constrained PNA analogs: Structural evolution toward DNA/RNA binding selectivity, Acc. Chem. Res., 2005, 38, 404–412 CrossRef CAS PubMed.
- T. Sugiyama and A. Kittaka, Chiral peptide nucleic acids with a substituent in the N-(2-aminoethyl)glycine backbone, Molecules, 2013, 18, 287–310 CrossRef CAS PubMed.
- M. Moccia, M. F. A. Adamo and M. Saviano, Insights on chiral, backbone modified peptide nucleic acids: Properties and biological activity, Artif. DNA: PNA & XNA, 2014, 5, e1107176 Search PubMed.
- A. Das and B. Pradhan, Evolution of peptide nucleic acid with modifications of its backbone and application in biotechnology, Chem. Biol. Drug Des., 2021, 97, 865–892 CrossRef CAS PubMed.
- N. Brodyagin, M. Katkevics, V. Kotikam, C. A. Ryan and E. Rozners, Chemical approaches to discover the full potential of peptide nucleic acids in biomedical applications, Beilstein J. Org. Chem., 2021, 17, 1641–1688 CrossRef CAS PubMed.
- K. L. Dueholm, K. H. Petersen, D. K. Jensen, M. Egholm, P. E. Nielsen and O. Buchardt, Peptide nucleic acid (PNA) with a chiral backbone based on alanine, Bioorg. Med. Chem. Lett., 1994, 4, 1077–1080 CrossRef CAS.
- G. Haaima, A. Lohse, O. Buchardt and P. E. Nielsen, Peptide nucleic acids (PNAs) containing thymine monomers derived from chiral amino acids: Hybridization and solubility properties of D-lysine pNA, Angew. Chem., Int. Ed. Engl., 1996, 35, 1939–1942 CrossRef CAS.
- A. Puschl, S. Sforza, G. Haaima, O. Dahl and P. E. Nielsen, Peptide nucleic acids (PNAs) with a functional backbone, Tetrahedron Lett., 1998, 39, 4707–4710 CrossRef CAS.
- S. Sforza, G. Haaima, R. Marchelli and P. E. Nielsen, Chiral peptide nucleic acids (PNAs): Helix handedness and DNA recognition, Eur. J. Org. Chem., 1999, 197–204 CrossRef CAS.
- I. Dilek, M. Madrid, R. Singh, C. P. Urrea and B. A. Armitage, Effect of PNA backbone modifications on cyanine dye binding to PNA–DNA duplexes investigated by optical spectroscopy and molecular dynamics simulations, J. Am. Chem. Soc., 2005, 127, 3339–3345 CrossRef CAS PubMed.
- S. Sforza, R. Corradini, S. Ghirardi, A. Dossena and R. Marchelli, DNA binding of a D-lysine-based chiral PNA: Direction control and mismatch recognition, Eur. J. Org. Chem., 2000, 2905–2913 CrossRef CAS.
- V. Menchise, G. De Simone, T. Tedeschi, R. Corradini, S. Sforza, R. Marchelli, D. Capasso, M. Saviano and C. Pedone, Insights into peptide nucleic acid (PNA) structural features: The crystal structure of a D-lysine-based chiral PNA–DNA duplex, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12021–12026 CrossRef CAS PubMed.
- L. Betts, J. A. Josey, J. M. Veal and S. R. Jordan, A nucleic acid triple helix formed by a peptide nucleic acid–DNA complex, Science, 1995, 270, 1838–1841 CrossRef CAS PubMed.
- M. Eriksson and P. E. Nielsen, Solution structure of a peptide nucleic acid–DNA duplex, Nat. Struct. Biol., 1996, 3, 410–413 CrossRef CAS PubMed.
- O. Almarsson and T. C. Bruice, Peptide nucleic acid (PNA) conformation and polymorphism in PNA–DNA and PNA–RNA hybrids, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9542–9546 CrossRef CAS PubMed.
- A. Gourishankar and K. N. Ganesh, (α,α-Dimethyl)glycyl (dmg) PNAs achiral PNA analogs that form stronger hybrids with CDNA relative to isosequential RNA, Artif. DNA: PNA & XNA, 2012, 3, 5–13 Search PubMed.
- P. Kulkarni, D. Datta, R. O. Ramabhadran and K. Ganesh, Gem-dimethyl peptide nucleic acid (α/β/γ-gdm-PNA) monomers: Synthesis and the role of gdm-substituents in preferential stabilisation of Z/E-rotamers, Org. Biomol. Chem., 2021, 19, 6534–6545 RSC.
- K. L. Dueholm, M. Egholm, C. Behrens, L. Christensen, H. F. Hansen, T. Vulpius, K. H. Petersen, R. H. Berg, P. E. Nielsen and O. Buchardt, Synthesis of peptide nucleic acid monomers containing the four natural nucleobases: Thymine, cytosine, adenine, and guanine and their oligomerizationsynthesis of peptide nucleic acid monomers containing the four natural nucleobases: Thymine, cytosine, adenine, and guanine and their oligomerization, J. Org. Chem., 1994, 59, 5767–5773 CrossRef CAS.
- S.-M. Chen, V. Mohan, J. S. Kiely, M. C. Griffith and R. H. Griffey, Molecular dynamics and NMR studies of single-stranded PNAs, Tetrahedron Lett, 1994, 35, 5105–5108 CrossRef CAS.
- M. Oleszczuk, S. Rodziewicz-Motowidlo and B. Falkiewicz, Restricted rotation in chiral peptide nucleic acid (PNA) monomers-influence of substituents studied by means of 1H NMR, Nucleos. Nucleot. Nucl., 2001, 20, 1399–1402 CrossRef CAS PubMed.
- W. He, E. Hatcher, A. Balaeff, D. N. Beratan, R. R. Gil, M. Madrid and C. Achim, Solution structure of a peptide nucleic acid duplex from NMR data: Features and limitationssolution structure of a peptide nucleic acid duplex from NMR data: Features and limitations, J. Am. Chem. Soc., 2008, 130, 13264–13273 CrossRef CAS PubMed.
- W. Maison, I. Schlemminger, O. Westerhoff and J. Martens, Modified PNAs: A simple method for the synthesis of monomeric building blocks, Bioorg. Med. Chem. Lett., 1999, 9, 581–584 CrossRef CAS PubMed.
- W. Maison, I. Schlemminger, O. Westerhoff and J. Martens, Multicomponent synthesis of novel amino acid–nucleobase chimeras: A versatile approach to PNA-monomers, Bioorg. Med. Chem., 2000, 8, 1343–1360 CrossRef CAS PubMed.
- J. Martens, W. Maison, I. Schlemminger, O. Westerhoff and H. Gröger, Precursors for PNA-monomers, International Patent Application, WO 00/02864, 2000 Search PubMed.
- T. Sugiyama, Y. Imamura, Y. Demizu, M. Kurihara, M. Takano and A. Kittaka, β-PNA: Peptide nucleic acid (PNA) with a chiral center at the β-position of the PNA backbone, Bioorg. Med. Chem. Lett., 2011, 21, 7317–7320 CrossRef CAS PubMed.
- L. Kosynkina, W. Wang and T. C. Liang, A convenient synthesis of chiral peptide nucleic acid (PNA) monomers, Tetrahedron Lett., 1994, 35, 5173–5176 CrossRef CAS.
- E. A. Englund and D. H. Appella, Synthesis of γ-substituted peptide nucleic acids: A new place to attach fluorophores without affecting DNA binding, Org. Lett., 2005, 7, 3465–3467 CrossRef CAS PubMed.
- E. A. Englund and D. H. Appella, γ-Substituted peptide nucleic acids constructed from L-lysine are a versatile scaffold for multifunctional display, Angew. Chem., Int. Ed., 2007, 46, 1414–1418 CrossRef CAS PubMed.
- C. Dose and O. Seitz, Convergent synthesis of peptide nucleic acids by native chemical ligation, Org. Lett., 2005, 7, 4365–4368 CrossRef CAS PubMed.
- M. C. de Koning, L. Petersen, J. J. Weterings, M. Overhand, G. A. van der Marel and D. V. Filippov, Synthesis of thiol-modified peptide nucleic acids designed for post-assembly conjugation reactions, Tetrahedron, 2006, 62, 3248–3258 CrossRef CAS.
- A. Dragulescu-Andrasi, S. Rapireddy, B. M. Frezza, C. Gayathri, R. R. Gil and D. H. Ly, A simple γ-backbone modification pre-organizes peptide nucleic acid into a helical structure, J. Am. Chem. Soc., 2006, 128, 10258–10267 CrossRef CAS PubMed.
- W. He, M. J. Crawford, M. Madrid, R. R. Gil, D. H. Ly and C. Achim, The structure of a γ-modified peptide nucleic acid duplexthe structure of a γ-modified peptide nucleic acid duplex, Mol. Biosyst., 2010, 6, 1619–1629 RSC.
- J. I. Yeh, B. Shivachev, S. Rapireddy, M. J. Crawford, R. R. Gil, S. Du, M. Madrid and D. H. Ly, Crystal structure of chiral γPNA with complementary DNA strand: Insights into the stability and specificity of recognition and conformational pre-organization, J. Am. Chem. Soc., 2010, 132, 10717–10727 CrossRef CAS PubMed.
- B. Sahu, I. Sacui, S. Rapireddy, K. J. Zanotti, R. Bahal, B. A. Armitage and D. H. Ly, Synthesis and characterization of conformationally pre-organized, (R)-diethylene glycol-containing γ-peptide nucleic acids with superior hybridization properties and water solubility, J. Org. Chem., 2011, 76, 5614–5627 CrossRef CAS PubMed.
- D. R. Jain, L. Anandi, V. M. Lahiri and K. N. Ganesh, Influence of pendant chiral Cγ-(alkylideneamino/guanidino) cationic side-chains of PNA backbone on hybridization with complementary DNA/RNA and cell permeability, J. Org. Chem., 2014, 79, 9567–9577 CrossRef CAS PubMed.
- B. Sahu, V. Chenna, K. L. Lathrop, S. M. Thomas, G. Zon, K. J. Livak and D. H. Ly, Synthesis of conformationally pre-organized and cell-permeable guanidine-based γ-peptide nucleic acids (γGPNAs), J. Org. Chem., 2009, 74, 1509–1516 CrossRef CAS PubMed.
- P. Kumar and D. R. Jain, Cγ-Aminopropylene peptide nucleic acid (amp-PNA): Chiral cationic PNAs with superior PNA:DNA/RNA duplex stability and cellular uptake, Tetrahedron, 2015, 71, 3378–3384 CrossRef CAS.
- Y. Kirillova, N. Boyarskaya, A. Dezhenkov, M. Tankevich, I. Prokhorov, A. Varizhuk, S. Eremin, D. Esipov, I. Smirnov and G. Pozmogova, Polyanionic carboxyethyl peptide nucleic acids (ce-PNAs): Synthesis and DNA binding, PLoS One, 2015, 10, e0140468 CrossRef PubMed.
- R. Mitra and K. N. Ganesh, PNAs grafted with (α/γ, R/S)-aminomethylene pendants: Regio and stereo specific effects on DNA binding and improved cell uptake, Chem. Commun., 2011, 47, 1198–1200 RSC.
- R. Mitra and K. N. Ganesh, Aminomethylene peptide nucleic acid (am-PNA): Synthesis, regio-/stereospecific DNA binding, and differential cell uptake of (α/γ,R/S)am-PNA analogs, J. Org. Chem., 2012, 77, 5696–5704 CrossRef CAS PubMed.
- C. Avitabile, L. Moggio, G. Malgieri, D. Capasso, S. Di Gaetano, M. Saviano, C. Pedone and A. Romanelli, γ-Sulphate PNA (PNA S): Highly selective DNA binding molecule showing promising antigene activity, PLoS One, 2012, 7, e35774 CrossRef CAS PubMed.
- N. T. S. De Costa and J. M. Heemstra, Differential DNA and RNA sequence discrimination by PNA having charged side chains, Bioorg. Med. Chem. Lett., 2014, 24, 2360–2363 CrossRef CAS PubMed.
- G. Haaima, H. Rasmussen, G. Schmidt, D. K. Jensen, J. S. Kastrup, P. W. Stafshede, B. Norden, O. Buchardt and P. E. Nielsen, Peptide nucleic acids (PNA) derived from N-(N-methylaminoethyl)glycine. Synthesis, hybridization and structural properties, New J. Chem., 1999, 23, 833–840 RSC.
- T. Tedeschi, S. Sforza, R. Corradini and R. Marchelli, Synthesis of new chiral PNAs bearing a dipeptide-mimic monomer with two lysine-derived stereogenic centres, Tetrahedron Lett., 2005, 46, 8395–8399 CrossRef CAS.
- S. Sforza, T. Tedeschi, R. Corradini and R. Marchelli, Induction of helical handedness and DNA binding properties of peptide nucleic acids (PNAs) with two stereogenic centres, Eur. J. Org. Chem., 2007, 5879–5885 CrossRef CAS.
- T. Bose, A. Banerjee, S. Nahar, S. Maiti and V. A. Kumar, β,γ-Bis-substituted PNA with configurational and conformational switch: Preferred binding to CDNA/RNA and cell-uptake studies, Chem. Commun., 2015, 51, 7693–7696 RSC.
- P. Lagriffoule, P. Wittung, M. Eriksson, K. K. Jensen, B. Norden, O. Buchardt and P. E. Nielsen, Peptide nucleic acids with a conformationally constrained chiral cyclohexyl-derived backbone, Chem. – Eur. J., 1997, 3, 912–919 CrossRef CAS.
- T. Govindaraju, R. G. Gonnade, M. M. Bhadbhade, V. A. Kumar and K. N. Ganesh, (1S,2R/1R,2S)-Aminocyclohexyl glycyl thymine PNA: Synthesis, monomer crystal structures, and DNA/RNA hybridization studies, Org. Lett., 2003, 5, 3013–3016 CrossRef CAS PubMed.
- T. Govindaraju, V. A. Kumar and K. N. Ganesh, (1S,2R/1R,2S)-cis-Cyclopentyl PNAs (cpPNAs) as constrained PNA analogs: Synthesis and evaluation of aeg-cpPNA chimera and stereopreferences in hybridization with DNA/RNA, J. Org. Chem., 2004, 69, 5725–5734 CrossRef CAS PubMed.
- T. Govindaraju, V. Madhuri, V. A. Kumar and K. N. Ganesh, Cyclohexanyl peptide nucleic acids (chPNAs) for preferential RNA binding: Effective tuning of dihedral angle β in PNAs for DNA/RNA discrimination, J. Org. Chem., 2006, 71, 14–21 CrossRef CAS PubMed.
- S. Sharma, U. B. Sonavane and R. R. Joshi, Molecular dynamics simulations of cyclohexyl modified peptide nucleic acids (PNA), J. Biomol. Struct., 2010, 27, 663–676 CrossRef CAS PubMed.
- S. C. Brown, S. A. Thomson, J. M. Veal and D. G. Davis, NMR solution structure of a peptide nucleic acid complexed with RNA, Science, 1994, 265, 777–780 CrossRef CAS PubMed.
- T. Govindaraju, V. A. Kumar and K. N. Ganesh, (SR/RS)-Cyclohexanyl PNAs: Conformationally pre-organized PNA analogs with unprecedented preference for duplex formation with RNA, J. Am. Chem. Soc., 2005, 127, 4144–4145 CrossRef CAS PubMed.
- J. K. Pokorski, M. A. Witschi, B. L. Purnell and D. H. Appella, (S,S)-trans-Cyclopentane-constrained peptide nucleic acids. A general backbone modification that improves binding affinity and sequence specificity, J. Am. Chem. Soc., 2004, 126, 15067–15073 CrossRef CAS PubMed.
- A. Sharma, S. H. More and K. N. Ganesh, Electrostatics Favor PNA: DNA stability over stereochemistry in pyrrolidine-based cationic dual-backbone PNA analogs, Eur. J. Org. Chem., 2021, 1146–1155 CrossRef CAS.
- H. Zheng, M. Saha and D. H. Appella, Synthesis of Fmoc-protected (S,S)-trans-cyclopentane diamine monomers enables the preparation and study of conformationally restricted peptide nucleic acids, Org. Lett., 2018, 20, 7637–7640 CrossRef CAS PubMed.
- T. Govindaraju, V. A. Kumar and K. N. Ganesh, cis-Cyclopentyl PNA (cpPNA) as constrained chiral PNA analogs: Stereochemical dependence of DNA/RNA hybridization, Chem. Commun., 2004, 860–861 RSC.
- M. C. Myers, M. A. Witschi, N. V. Larionova, J. M. Franck, R. D. Haynes, T. Hara, A. Grajkowski and D. H. Appella, A cyclopentane conformational restraint for a peptide nucleic acid: Design, asymmetric synthesis, and improved binding affinity to DNA and RNA, Org. Lett., 2003, 5, 2695–2698 CrossRef CAS PubMed.
- E. A. Englund, Q. Xu, M. A. Witschi and D. H. Appella, PNA–DNA duplexes, triplexes, and quadruplexes are stabilized with trans-cyclopentane units, J. Am. Chem. Soc., 2006, 128, 16456–16457 CrossRef CAS PubMed.
- H. Zheng, I. Botos, V. Clausse, H. Nikolayevskiy, E. E. Rastede, M. F. Fouz, S. J. Mazur and D. H. Appella, Conformational constraints of cyclopentane peptide nucleic acids facilitate tunable binding to DNA, Nucleic Acids Res., 2021, 49, 713–725 CrossRef CAS PubMed.
- J. K. Pokorski, M. C. Myers and D. H. Appella, Cyclopropane PNA: observable triplex melting in A PNA constrained with A 3-membered ring, Tetrahedron Lett., 2005, 46, 915–917 CrossRef CAS.
- S. Jordan, C. Schwemler, W. Kosch, A. Kretschmer, E. Schwenner, U. Stropp and B. Mielke, Synthesis of new building blocks for peptide nucleic acids containing monomers with variations in the backbone, Bioorg. Med. Chem. Lett., 1997, 7, 681–686 CrossRef CAS.
- S. Jordan, C. Schwemler, W. Kosch, A. Kretschmer, U. Stropp, E. Schwenner and B. Mielke, New hetero-oligomeric peptide nucleic acids with improved binding properties to complementary DNA, Bioorg. Med. Chem. Lett., 1997, 7, 687–690 CrossRef CAS.
- B. P. Gangamani, V. A. Kumar and K. N. Ganesh, Synthesis of Nα-(purinyl/pyrimidinyl acetyl)-4-aminoproline diastereomers with potential use in PNA synthesis, Tetrahedron, 1996, 52, 15017–15030 CrossRef.
- B. P. Gangamani, M. D’costa, V. A. Kumar and K. N. Ganesh, Conformationally restrained chiral PNA conjugates: Synthesis and DNA complementation studies, Nucleos. Nucleot., 1999, 18, 1409–1411 CrossRef CAS.
- A. Puschl, T. Boesen, G. Zuccarello, O. Dahl, S. Pitsch and P. E. Nielsen, Synthesis of pyrrolidinone PNA: A novel conformationally restricted PNA analog, J. Org. Chem., 2001, 66, 707–712 CrossRef CAS PubMed.
- A. Püschl, T. Boesen, T. Tedeschi, O. Dahl and P. E. Nielsen, Synthesis of (3R,6R)- and (3S,6R)-piperidinone PNA, J. Chem. Soc., Perkin Trans. 1, 2001, 2757–2763 RSC.
- A. Puschl, T. Tedeschi and P. E. Nielsen, Pyrrolidine PNA: A novel conformationally restricted PNA analog, Org. Lett., 2000, 2, 4161–4163 CrossRef CAS PubMed.
- D. T. Hickman, P. M. King, M. A. Cooper, J. M. Slater and J. Micklefield, Unusual RNA and DNA binding properties of a novel pyrrolidine–amide oligonucleotide mimic (POM), Chem. Commun., 2000, 2251–2252 RSC.
- T. H. Samuel Tan, D. T. Hickman, J. Morral, I. G. Beadham and J. Micklefield, Nucleic acid binding properties of thyminyl and adeninyl pyrrolidine-amide oligonucleotide mimics (POM), Chem. Commun., 2004, 516–517 Search PubMed.
- R. J. Worthington, A. P. O’Rourke, J. Morral, T. H. Samuel Tan and J. Micklefield, Mixed-sequence pyrrolidine-amide oligonucleotide mimics: Boc(Z) synthesis and DNA/RNA binding properties, Org. Biomol. Chem., 2007, 5, 249–259 RSC.
- J. A. Hodges and R. T. Raines, Stereoelectronic effects on collagen stability: The dichotomy of 4-fluoroproline diastereomers, J. Am. Chem. Soc., 2003, 125, 9262–9263 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, 4-Fluoroprolines: Conformational analysis and effects on the stability and folding of peptides and proteins, Top. Heterocycl. Chem., 2017, 48, 1–25 CAS.
- T. H. Samuel Tan, R. J. Worthington, R. G. Pritchard, J. Morral and J. Micklefield, Homopolymeric pyrrolidine-amide oligonucleotide mimics: Fmoc-synthesis and DNA/RNA binding properties, Org. Biomol. Chem., 2007, 5, 239–248 RSC.
- Y. Li, T. Jin and K. Liu, Synthesis and binding affinity of a chiral PNA analog, Nucleos. Nucleot. Nucl., 2001, 20, 1705–1721 CrossRef CAS PubMed.
- M. D’Costa, V. A. Kumar and K. N. Ganesh, Aminoethylprolyl (aep) PNA: Mixed purine/pyrimidine oligomers and binding orientation preferences for PNA:DNA duplex formation, Org. Lett., 2001, 3, 1281–1284 CrossRef PubMed.
- M. D’Costa, V. A. Kumar and K. N. Ganesh, Aminoethylprolyl peptide nucleic acids (aepPNA): Chiral PNA analogs that form highly stable DNA:aepPNA2 triplexes, Org. Lett., 1999, 1, 1513–1516 CrossRef PubMed.
- T. Vilaivan, C. Khongdeesameor, P. Harnyuttanakorn, M. S. Westwell and G. Lowe, Synthesis and properties of chiral peptide nucleic acids with a N-aminoethyl-D-proline backbone, Bioorg. Med. Chem. Lett., 2000, 10, 2541–2545 CrossRef CAS PubMed.
- P. Ngamwiriyawong and T. Vilaivan, Synthesis and nucleic acids binding properties of diastereomeric aminoethylprolyl peptide nucleic acids (aepPNA), Nucleos. Nucleot. Nucl., 2011, 30, 97–112 CrossRef CAS PubMed.
- P. S. Shirude, V. A. Kumar and K. N. Ganesh, (2S,5R/2R,5S)-Aminoethylpipecolyl aepip-aegPNA Chimera: Synthesis and Duplex/Triplex Stability, Tetrahedron, 2004, 60, 9485–9491 CrossRef CAS.
- N. K. Sharma and K. N. Ganesh, Expanding the repertoire of pyrrolidyl PNA analogs for DNA/RNA hybridization selectivity: Aminoethylpyrrolidinone PNA (aepone-PNA), Chem. Commun., 2003, 2484–2485 RSC.
- N. K. Sharma and K. N. Ganesh, Base dependent pyrrolidine ring pucker in aep-PNA monomers: NMR and PSEUROT analysis, Tetrahedron, 2010, 66, 9165–9170 CrossRef CAS.
- P. S. Shirude, V. A. Kumar and K. N. Ganesh, Chimeric peptide nucleic acids incorporating (2S,5R)-aminoethyl pipecolyl units: Synthesis and DNA binding studies, Tetrahedron Lett., 2004, 45, 3085–3088 CrossRef CAS.
- P. S. Lonkar and V. A. Kumar, Design and synthesis of conformationally frozen peptide nucleic acid backbone: Chiral piperidine PNA as a hexitol nucleic acid surrogate, Bioorg. Med. Chem. Lett., 2004, 14, 2147–2149 CrossRef CAS PubMed.
- S. Bregant, F. Burlina, J. Vaissermann and G. Chassaing, Synthesis and hybridization properties of thiazolidine PNAs, Eur. J. Org. Chem., 2001, 3285–3294 CrossRef CAS.
- S. Bregant, F. Burlina and G. Chassaing, New thiazane and thiazolidine PNA Monomers: Synthesis, incorporation into PNAs and hybridization studies, Bioorg. Med. Chem. Lett., 2002, 12, 1047–1050 CrossRef CAS PubMed.
- A. Slaitas and E. Yeheskiely, A novel N-(pyrrolidinyl-2-methyl)glycine-based PNA with a strong preference for RNA over DNA, Eur. J. Org. Chem., 2002, 2391–2399 CrossRef CAS.
- S. S. Gokhale and V. A. Kumar, Amino/guanidino-functionalized N-(pyrrolidin-2-ethyl) glycine-based pet-PNA: Design, synthesis and binding with DNA/RNA, Org. Biomol. Chem., 2010, 8, 3742–3750 RSC.
- A. Banerjee and V. A. Kumar, C3'-Endo-puckered pyrrolidine containing PNA has favorable geometry for RNA binding: Novel ethano locked PNA (ethano-PNA), Bioorg. Med. Chem., 2013, 21, 4092–4101 CrossRef CAS PubMed.
- R. J. Worthington, N. M. Bell, R. Wong and J. Micklefield, RNA-selective cross-pairing of backbone-extended pyrrolidine-amide oligonucleotide mimics (bePOMs), Org. Biomol. Chem., 2008, 6, 92–103 RSC.
- T. Govindaraju and V. A. Kumar, Backbone extended pyrrolidine PNA (bepPNA): A chiral PNA for selective RNA recognition, Tetrahedron, 2006, 62, 2321–2330 CrossRef CAS.
- P. S. Lonkar, V. A. Kumar and K. N. Ganesh, Constrained flexibility in PNA:DNA binding studies with bridged aminopropylglycyl PNA, Nucleos. Nucleot. Nucl., 2001, 20, 1197–1200 CrossRef CAS PubMed.
- P. S. Lonkar and V. A. Kumar, trans-5-Aminopipecolyl-aegPNA chimera: Design, synthesis, and study of binding preferences with DNA/RNA in duplex/triplex mode, J. Org. Chem., 2005, 70, 6956–6959 CrossRef CAS PubMed.
- L. D. Fader, M. Boyd and Y. S. Tsantrizos, Backbone modifications of aromatic peptide nucleic acid (APNA) monomers and their hybridization properties with DNA and RNA, J. Org. Chem., 2001, 66, 3372–3379 CrossRef CAS PubMed.
- L. D. Fader and Y. S. Tsantrizos, Hybridization properties of aromatic peptide nucleic acids: A novel class of oligonucleotide analogs, Org. Lett., 2002, 4, 63–66 CrossRef CAS PubMed.
- L. D. Fader, E. L. Myersa and Y. S. Tsantrizos, Synthesis of novel analogs of aromatic peptide nucleic acids (APNAs) with modified conformational and electrostatic properties, Tetrahedron, 2004, 60, 2235–2246 CrossRef CAS.
- M. Kuwahara, M. Arimitsu and M. Sisido, Novel peptide nucleic acid that shows high sequence specificity and all-or-none-type hybridization with the complementary DNA, J. Am. Chem. Soc., 1999, 121, 256–257 CrossRef CAS.
- M. Kuwahara, M. Arimitsu, M. Shigeyasu, N. Saeki and M. Sisido, Hybridization between oxy-peptide nucleic acids and DNAs: Dependence of hybrid stabilities on the chain-lengths, types of base pairs, and the chain directions, J. Am. Chem. Soc., 2001, 123, 4653–4658 CrossRef CAS PubMed.
- M. Kitamatsu, M. Shigeyasu, M. Saitoh and M. Sisido, Configurational preference of pyrrolidine-based oxy-peptide nucleic acids as hybridization counterparts with DNA and RNA, Biopolymers, 2006, 84, 267–273 CrossRef CAS PubMed.
- M. Kitamatsu, M. Shigeyasu, T. Okada and M. Sisido, Oxy-peptide nucleic acid with a pyrrolidine ring that is configurationally optimized for hybridization with DNA, Chem. Commun., 2004, 1208–1209 RSC.
- M. Kitamatsu, T. Kashiwagi, R. Matsuzaki and M. Sisido, Synthesis of a novel pyrrolidine-based peptide nucleic acid that contains tertiary amines in the main chain and its internalization into cells, Chem. Lett., 2006, 35, 300–301 CrossRef CAS.
- K. Gogoi and V. A. Kumar, Chimeric (α-amino acid + nucleoside-β-amino acid)n peptide oligomers show sequence specific DNA/RNA recognition, Chem. Commun., 2008, 706–708 RSC.
- A. Banerjee, S. Bagmare, M. Varada and V. A. Kumar, Glycine-linked nucleoside-β-amino acids: Polyamide analogs of nucleic acids, Bioconjugate Chem., 2015, 26, 1737–1742 CrossRef CAS PubMed.
- R. Threlfall, A. Davies, N. M. Howarth, J. Fisher and R. Cosstick, Peptides derived from nucleoside β-amino acids form an unusual 8-helix, Chem. Commun., 2008, 585–587 RSC.
- S. Bagmare, M. D’Costa and V. A. Kumar, Effect of chirality of L/D-proline and prochiral glycine as the linker amino acid in five-atom linked thymidinyl-(α-amino acid)-thymidine dimers, Chem. Commun., 2009, 6646–6648 RSC.
- T. Vilaivan, Pyrrolidinyl PNA with α/β-dipeptide backbone: From development to applications, Acc. Chem. Res., 2015, 48, 1645–1656 CrossRef CAS PubMed.
- G. Lowe, T. Vilaivan and M. S. Westwell, Hybridization studies with chiral peptide nucleic acids, Bioorg. Chem., 1997, 25, 321–329 CrossRef CAS.
- D. H. Appella, L. A. Christianson, D. A. Klein, M. R. Richards, D. R. Powell and S. H. Gellman, Synthesis and structural characterization of helix-forming β-peptides: trans-2-Aminocyclopentanecarboxylic acid oligomers, J. Am. Chem. Soc., 1999, 121, 6206–6212 CrossRef CAS.
- T. Vilaivan, C. Suparpprom, P. Harnyuttanakorn and G. Lowe, Synthesis and properties of novel pyrrolidinyl PNA carrying β-amino acid spacers, Tetrahedron Lett., 2001, 42, 5533–5536 CrossRef CAS.
- T. Vilaivan and G. Lowe, A novel pyrrolidinyl PNA showing high sequence specificity and preferential binding to DNA over RNA, J. Am. Chem. Soc., 2002, 124, 9326–9327 CrossRef CAS PubMed.
- T. Vilaivan, C. Suparpprom, P. Duanglaor, P. Harnyuttanakorn and G. Lowe, Synthesis and nucleic acid binding studies of novel pyrrolidinyl PNA carrying an N-amino-N-methylglycine spacer, Tetrahedron Lett., 2003, 44, 1663–1666 CrossRef CAS.
- C. Suparpprom, C. Srisuwannaket, P. Sangvanich and T. Vilaivan, Synthesis and oligo- deoxynucleotide binding properties of pyrrolidinyl peptide nucleic acids bearing prolyl-2-aminocyclopentanecarboxylic acid (ACPC) backbones, Tetrahedron Lett., 2005, 46, 2833–2837 CrossRef CAS.
- T. Vilaivan and C. Srisuwannaket, Hybridization of pyrrolidinyl peptide nucleic acids and DNA: Selectivity, base-pairing specificity, and direction of binding, Org. Lett., 2006, 8, 1897–1900 CrossRef CAS PubMed.
- C. Vilaivan, C. Srisuwannaket, C. Ananthanawat, C. Suparpprom, J. Kawakami, Y. Yamaguchi, Y. Tanaka and T. Vilaivan, Pyrrolidinyl peptide nucleic acid with α/β-peptide backbone a conformationally constrained PNA with unusual hybridization properties, Artif. DNA: PNA & XNA, 2011, 2, 50–59 Search PubMed.
- K. Siriwong, P. Chuichay, S. Saen-oon, C. Suparpprom, T. Vilaivan and S. Hannongbua, Insight into why pyrrolidinyl peptide nucleic acid binding to DNA is more stable than the DNA·DNA duplex, Biochem. Biophys. Res. Commun., 2008, 372, 765–771 CrossRef CAS PubMed.
- N. Poomsuk, T. Remsungnen, T. Vilaivan, A. J. Hunt and K. Siriwong, Conformational and energetic properties of pyrrolidinyl PNA–DNA duplexes: A molecular dynamics simulation, Comput. Theor. Chem., 2017, 1122, 27–33 CrossRef CAS.
- N. Poomsook, T. Vilaivan and K. Siriwong, Insights into the structural features and stability of peptide nucleic acid with a D-prolyl-2-aminocyclopentane carboxylic acid backbone that binds to DNA and RNA, J. Mol. Graph. Modell., 2018, 84, 36–42 CrossRef PubMed.
- A. Maitarad, N. Poomsuk, C. Vilaivan, T. Vilaivan and K. Siriwong, Insight into a conformation of the PNA–PNA duplex with (2'R,4'R)- and (2'R,4'S)-prolyl-(1S,2S)-2-aminocyclopentanecarboxylic acid backbones, Chem. Phys. Lett., 2018, 698, 132–137 CrossRef CAS.
- J. Taechalertpaisarn, P. Sriwarom, C. Boonlua, N. Yotapan, C. Vilaivan and T. Vilaivan, DNA-, RNA- and self-pairing properties of a pyrrolidinyl peptide nucleic acid with a (2’R,4’S)-Prolyl-(1S,2S)-2-aminocyclopentanecarboxylic acid backbone, Tetrahedron Lett., 2010, 51, 5822–5826 CrossRef CAS.
- N. Reenabthue, C. Boonlua, C. Vilaivan, T. Vilaivan and C. Suparpprom, 3-Aminopyrrolidine-4-carboxylic acid as versatile handle for internal labeling of pyrrolidinyl PNA, Bioorg. Med. Chem. Lett., 2011, 21, 6465–6469 CrossRef CAS PubMed.
- P. Sriwarom, P. Padungros and T. Vilaivan, Synthesis and DNA/RNA binding properties of conformationally constrained pyrrolidinyl PNA with a tetrahydrofuran backbone deriving from deoxyribose, J. Org. Chem., 2015, 80, 7058–7065 CrossRef CAS PubMed.
- C. Boonlua, B. Ditmangklo, N. Reenabthue, C. Suparpprom, N. Poomsuk, K. Siriwong and T. Vilaivan, Pyrene-labeled pyrrolidinyl peptide nucleic acid as a hybridization-responsive DNA Probe: Comparison between internal and terminal labeling, RSC Adv., 2014, 4, 8817–8827 RSC.
- B. Ditmangklo, C. Boonlua, C. Suparpprom and T. Vilaivan, Reductive alkylation and sequential reductive alkylation-click chemistry for on-solid-support modification of pyrrolidinyl peptide nucleic acid, Bioconjugate Chem., 2013, 24, 614–625 CrossRef CAS PubMed.
- H. PansuwanH. Pansuwan, B. Ditmangklo, C. Vilaivan, B. Jiangchareon, P. Pan-In, S. Wanichwecha, T. Palaga, T. Nuanyai, C. Suparpprom and T. Vilaivan, Hydrophilic and cell-penetrable pyrrolidinyl peptide nucleic acid via post-synthetic modification with hydrophilic side chains, Bioconjugate Chem., 2017, 28, 2284–2292 CrossRef PubMed.
- P. Seankongsuk, V. Vchirawongkwin, R. W. Bates, P. Padungros and T. Vilaivan, Enantioselective synthesis of (2S,3S)-Epi-oxetin and its incorporation into conformationally constrained pyrrolidinyl PNA with an oxetane backbone, Asian J. Org. Chem., 2017, 6, 551–560 CrossRef CAS.
- W. Mansawat, C. Vilaivan, A. Balazs, D. J. Aitken and T. Vilaivan, Pyrrolidinyl peptide nucleic acid homologues: Effect of ring size on hybridization properties, Org. Lett., 2012, 14, 1440–1443 CrossRef CAS PubMed.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, Synthesis and characterization of trans-2-aminocyclohexanecarboxylic acid oligomers: An unnatural helical secondary structure and implications for β-peptide tertiary structure, J. Am. Chem. Soc., 1999, 121, 6206–6212 CrossRef CAS.
- D. H. Appella, L. A. Christianson, D. A. Klein, M. R. Richards, D. R. Powell and S. H. Gellman, Synthesis and structural characterization of helix-forming β-peptides: trans-2-Aminocyclopentanecarboxylic acid oligomerssynthesis and structural characterization of helix-forming β-peptides: trans-2-Aminocyclopentanecarboxylic acid oligomers, J. Am. Chem. Soc., 1999, 121, 7574–7581 CrossRef CAS.
- C. Fernandes, S. Faure, E. Pereira, V. Théry, V. Declerck, R. Guillot and D. J. Aitken, 12-Helix folding of cyclobutane β-amino acid oligomers, Org. Lett., 2010, 12, 3606–3609 CrossRef CAS PubMed.
- B. Ditmangklo, W. Sittiwong, T. Boddaert, T. Vilaivan and D. J. Aitken, Pyrrolidinyl peptide nucleic acids bearing hydroxy-modified cyclobutane building blocks: Synthesis and binding properties, Biopolymers, 2021, 112, e23459 CrossRef CAS PubMed.
- H. Kashida, K. Murayama, T. Toda and H. Asanuma, Control of the chirality and helicity of oligomers of serinol nucleic acid (SNA) by sequence design, Angew. Chem., Int. Ed., 2011, 50, 1285–1288 CrossRef CAS PubMed.
- H. Asanuma, T. Toda, K. Murayama, X. Liang and H. Kashida, Unexpectedly stable artificial duplex from flexible acyclic threoninol, J. Am. Chem. Soc., 2010, 132, 14702–14703 CrossRef CAS PubMed.
- K. Murayama, Y. Tanaka, T. Toda, H. Kashida and H. Asanuma, Highly stable duplex formation by artificial nucleic acids acyclic threoninol nucleic acid (aTNA) and serinol nucleic acid (SNA) with acyclic scaffolds, Chem. – Eur. J., 2013, 19, 14151–14158 CrossRef CAS PubMed.
- K. Murayama, H. Kashida and H. Asanuma, Acyclic L-threoninol nucleic acid (L-aTNA) with suitable structural rigidity cross-pairs with DNA and RNA, Chem. Commun., 2015, 51, 6500–6503 RSC.
- H. Rasmussen, J. S. Kastrup, J. N. Nielsen, J. M. Nielsen and P. E. Nielsen, Crystal structure of a peptide nucleic acid (PNA) duplex at 1.7 Å resolution, Nat. Struct. Biol, 1997, 4, 98–101 CrossRef CAS PubMed.
- A. Kiliszek, K. Banaszak, Z. Dauter and W. Rypniewski, The First crystal structures of RNA–PNA duplexes and a PNA–PNA duplex containing mismatches–toward anti-sense therapy against TREDs, Nucleic Acids Res., 2016, 44, 1937–1943 CrossRef PubMed.
- R. Schütz, M. Cantin, C. Roberts, B. Greiner, E. Uhlmann and C. Leumann, Olefinic peptide nucleic acids (OPAs): New aspects of the molecular recognition of DNA by PNA, Angew. Chem., Int. Ed., 2000, 39, 1250–1253 CrossRef.
- M. Hollenstein and C. J. Leumann, Synthesis and incorporation into PNA of fluorinated olefinic PNA (F-OPA) monomers, Org. Lett., 2003, 5, 1987–1990 CrossRef CAS PubMed.
- M. Hollenstein and C. J. Leumann, Fluorinated olefinic peptide nucleic acid: Synthesis and pairing properties with complementary DNA, J. Org. Chem., 2005, 70, 3205–3217 CrossRef CAS PubMed.
- D. Wei, W. D. Wilson and S. Neidle, Small-molecule binding to the DNA minor groove is mediated by a conserved water cluster, J. Am. Chem. Soc., 2013, 135, 1369–1377 CrossRef CAS PubMed.
- N. C. Horton and B. C. Finzel, The structure of an RNA/DNA hybrid: A substrate of the ribonuclease activity of HIV-1 reverse transcriptase, J. Mol. Biol., 1996, 264, 521–533 CrossRef CAS PubMed.
- B. B. Chevrier, A. Podjarny, J. Johnson, J. S. de Bear, G. R. Gough, P. T. Gilham and D. Moras, Crystallographic structure of an RNA Helix: [U(UA)6A]2, J. Mol. Biol., 1989, 209, 459–474 CrossRef.
- T. Ishizuka, J. Yoshida, Y. Yamamoto, J. Sumaoka, T. Tedeschi, R. Corradini, S. Sforza and M. Komiyama, Chiral introduction of positive charges to PNA for double-duplex invasion to versatile sequences, Nucleic Acids Res., 2008, 36, 1464–1471 CrossRef CAS PubMed.
- J. Saarbach, P. M. Sabale and N. Winssinger, Peptide nucleic acid (PNA) and its applications in chemical biology, diagnostics, and therapeutics, Curr. Opin. Chem. Biol., 2019, 52, 112–124 CrossRef CAS PubMed.
- H. Ørum, P. E. Nielsen, M. Egholm, R. H. Berg, O. Buchardt and C. Stanley, Single base pair mutation analysis by PNA directed PCR clamping, Nucleic Acids Res., 1993, 21, 5332–5336 CrossRef PubMed.
- X. Liang, M. Liu and M. Komiyama, Recognition of target site in various forms of DNA and RNA by peptide nucleic acid (PNA): From fundamentals to practical applications, Bull. Chem. Soc. Jpn., 2021, 94, 1737–1756 CrossRef CAS.
- S. Buchini and C. Leumann, Recent improvements in antigene technology, Curr. Opin. Chem. Biol., 2003, 3, 717–746 CrossRef PubMed.
- B. Vester and J. Wengel, LNA (locked nucleic acid): High-affinity targeting of complementary RNA and DNA, Biochemistry, 2004, 43, 13233–13241 CrossRef CAS PubMed.
- T. Bentin, H. J. Larsen and P. E. Nielsen, Combined triplex/duplex invasion of double-stranded DNA by “Tail-Clamp” peptide nucleic acid, Biochemistry, 2003, 42, 13987–13995 CrossRef CAS PubMed.
- K. Kaihatsu, R. H. Shah, X. Zhao and D. R. Corey, Extending recognition by peptide nucleic acids (PNAs): Binding to duplex DNA and inhibition of transcription by tail-clamp PNA-peptide conjugates, Biochemistry, 2003, 42, 13996–14003 CrossRef CAS PubMed.
- J. Lohse, O. Dahl and P. E. Nielsen, Double duplex invasion by peptide nucleic acid: A general principle for sequence-specific targeting of double-stranded DNA, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11804–11808 CrossRef CAS PubMed.
- M. Hibino, Y. Aiba and O. Shoji, Cationic guanine: Positively charged nucleobase with improved DNA affinity inhibits self-duplex formation, Chem. Commun., 2020, 56, 2546–2549 RSC.
- A. Maitarad, N. Poomsuk, C. Vilaivan, T. Vilaivan and K. Siriwong, Insight into a conformation of the PNA–PNA duplex with (2’R,4’R)- and (2’R,4’S)-prolyl-(1S,2S)-2-aminocyclopentanecarboxylic acid backbones, Chem. Phys. Lett., 2018, 698, 132–137 CrossRef CAS.
- P. R. Bohländer, T. Vilaivan and H.-A. Wagenknecht, Strand displacement and duplex invasion into double-stranded DNA by pyrrolidinyl peptide nucleic acids, Org. Biomol. Chem., 2015, 13, 9223–9230 RSC.
- N. Yotapan, D. Nim-anussornkul and T. Vilaivan, Pyrrolidinyl peptide nucleic acid terminally labeled with fluorophore and end-stacking quencher as a probe for highly specific DNA sequence discrimination, Tetrahedron, 2016, 72, 7992–7999 CrossRef CAS.
- G. He, S. Rapireddy, R. Bahal, B. Sahu and D. H. Ly, Strand invasion of extended, mixed-sequence B-DNA by γPNAs, J. Am. Chem. Soc., 2009, 131, 12088–12090 CrossRef CAS PubMed.
- R. Bahal, B. Sahu, S. Rapireddy, C.-M. Lee and D. H. Ly, Sequence-unrestricted, Watson–Crick recognition of double helical B-DNA by (R)-MiniPEG-γPNAs, ChemBioChem, 2012, 13, 56–60 CrossRef CAS PubMed.
- S. Rapireddy, G. He, S. Roy, B. A. Armitage and D. H. Ly, Strand invasion of mixed-sequence B-DNA by acridine-linked, γ-peptide nucleic acid (γ-PNA), J. Am. Chem. Soc., 2007, 129, 15596–15600 CrossRef CAS PubMed.
- V. Chenna, S. Rapireddy, B. Sahu, C. Ausin, E. Pedroso and D. H. Ly, A simple cytosine to G-clamp nucleobase substitution enables chiral γ-PNAs to invade mixed-sequence double-helical B-form DNA, ChemBioChem, 2008, 9, 2388–2391 CrossRef CAS PubMed.
- H. Kuhn, B. Sahu, S. Rapireddy, D. H. Ly and M. D. Frank-Kamenetskii, Sequence specificity at targeting double-stranded DNA with A γ-PNA oligomer modified with guanidinium G-clamp nucleobases, Artif. DNA: PNA & XNA, 2010, 1, 45–53 Search PubMed.
- S. Rapireddy, R. Bahal and D. H. Ly, Strand invasion of mixed-sequence, double-helical B-DNA by γ-peptide nucleic acids containing G-clamp nucleobases under physiological conditions, Biochemistry, 2011, 50, 3913–3918 CrossRef CAS PubMed.
- M. Lyu, L. Kong, Z. Yang, Y. Wu, C. E. McGhee and Y. Lu, PNA-assisted DNAzymes to cleave double-stranded DNA for genetic engineering with high sequence fidelity, J. Am. Chem. Soc., 2021, 143, 9724–9728 CrossRef CAS PubMed.
- S. A. Thadke, V. M. Hridya, J. D. R. Perera, R. R. Gil, A. Mukherjee and D. H. Ly, Shape selective bifacial recognition of double helical DNA, Commun. Chem., 2018, 1, 79 CrossRef.
- R. G. Emehiser and P. J. Hrdlicka, Chimeric γPNA–invader probes: Using intercalator-functionalized oligonucleotides to enhance the DNA-targeting properties of γPNA, Org. Biomol. Chem., 2020, 18, 1359–1368 RSC.
- I. Sacui, W.-C. Hsieh, A. Manna, B. Sahu and D. H. Ly, Gamma peptide nucleic acids: As orthogonal nucleic acid recognition codes for organizing molecular self-assembly, J. Am. Chem. Soc., 2015, 137, 8603–8610 CrossRef CAS PubMed.
- S. A. Thadke, J. D. R. Perera, V. M. Hridya, K. Bhatt, A. Y. Shaikh, W.-C. Hsieh, M. Chen, C. Gayathri, R. R. Gil, G. S. Rule, A. Mukherjee, C. A. Thornton and D. H. Ly, Design of bivalent nucleic acid ligands for recognition of RNA repeated expansion associated with Huntington's disease, Biochemistry, 2018, 57, 2094–2108 CrossRef CAS PubMed.
- R. Bahal, A. Manna, W.-C. Hsieh, S. A. Thadke, G. Sureshkumar and D. H. Ly, RNA-Templated concatenation of triplet nucleic-acid probe, ChemBioChem, 2018, 19, 674–678 CrossRef CAS PubMed.
- T. Zengeya, P. Gupta and E. Rozners, Triple-helical recognition of RNA using 2-aminopyridine-modified PNA at physiologically relevant conditions, Angew. Chem., Int. Ed., 2012, 51, 12593–12596 CrossRef CAS PubMed.
- P. Gupta, T. Zengeyaz and E. Rozners, Triple helical recognition of pyrimidine inversions in polypurine tracts of RNA By nucleobase-modified PNA, Chem. Commun., 2011, 47, 11125–11127 RSC.
- M. Li, T. Zengeya and E. Rozners, Short peptide nucleic acids bind strongly to homopurine tract of double helical RNA at pH 5.5, J. Am. Chem. Soc., 2010, 132, 8676–8681 CrossRef CAS PubMed.
- N. Dias, C. Senamaud-Beaufort, E. le Forestier, C. Auvin, C. Helene and T. E. Saison-Behmoaras, RNA hairpin invasion and ribosome elongation arrest by mixed base PNA oligomer, J. Mol. Biol., 2002, 320, 489–501 CrossRef CAS.
- X. Zhan, L. Deng and G. Chen, Mechanisms and applications of peptide nucleic acids selectively binding to double-stranded RNA, Biopolymers, 2021, e23476 Search PubMed.
- J. Kesy, K. M. Patil, S. R. Kumar, Z. Shu, H. Y. Yong, L. Zimmermann, A. A. L. Ong, D.-F. K. Toh, M. S. Krishna, L. Yang, J.-L. Decout, D. Luo, M. Prabakaran, G. Chen and E. Kierzek, A short chemically modified dsRNA-binding PNA (dbPNA) inhibits influenza viral replication by targeting viral RNA panhandle structure, Bioconjugate Chem., 2019, 30, 931–943 CrossRef CAS PubMed.
- V. Kumar, N. Brodyagin and E. Rozners, Triplex-forming peptide nucleic acids with extended backbones, ChemBioChem, 2020, 21, 3410–3416 CrossRef CAS PubMed.
- P. Gupta, O. Muse and E. Rozners, Recognition of double-stranded RNA by guanidine-modified peptide nucleic acids, Biochemistry, 2012, 51, 63–73 CrossRef CAS PubMed.
- W.-C. Hsieh, R. Bahal, S. A. Thadke, K. Bhatt, K. Sobczak, C. Thornton and D. H. Ly, Design of a “Mini” nucleic acid probe for cooperative binding of an RNA-repeated transcript associated with myotonic dystrophy type 1, Biochemistry, 2018, 57, 907–911 CrossRef CAS PubMed.
- V. Tahtinen, L. Granqvist, M. Murtola, R. Stromberg and P. Virta, 19F NMR spectroscopic analysis of the binding modes in triple-helical peptide nucleic acid (PNA)/MicroRNA complexes, Chem. – Eur. J., 2017, 23, 7113–7124 CrossRef PubMed.
- V. Tahtinen, A. Verhassel, J. Tuomela and P. Virta, γ-(S)-Guanidinylmethyl-modified triplex-forming peptide nucleic acids increase Hoogsteen-face affinity for a MicroRNA and enhance cellular uptake, ChemBioChem, 2019, 20, 3041–3051 CrossRef PubMed.
- K. T. Kim, D. Chang and N. Winssinger, Double-stranded RNA-specific templated reaction with triplex forming PNA, Helv. Chim. Acta, 2018, 101, e1700295 CrossRef.
- G. Devi, Z. Yuan, Y. Lu, Y. Zhao and G. Chen, Incorporation of thio-pseudoisocytosine into triplex-forming peptide nucleic acids for enhanced recognition of RNA duplexes, Nucleic Acids Res., 2014, 42, 4008–4018 CrossRef CAS PubMed.
- I. G. Panyutin, M. I. Onyshchenko, E. A. Englund, D. H. Appella and R. D. Neumann, Targeting DNA G-quadruplex structures with peptide nucleic acids, Curr. Pharm. Des., 2012, 18, 1984–1991 CrossRef CAS PubMed.
- S. Lusvarghi, C. T. Murphy, S. Roy, F. A. Tanious, I. Sacui, W. D. Wilson, D. H. Ly and B. A. Armitage, Loop and backbone modifications of PNA improve G-quadruplex binding selectivity, J. Am. Chem. Soc., 2009, 131, 18415–18424 CrossRef CAS PubMed.
- P. Gupta, E. E. Rastede and D. H. Appella, Multivalent LKγ-PNA oligomers bind to a human telomere DNA G-rich sequence to form quadruplexes, Bioorg. Med. Chem. Lett., 2015, 25, 4757–4760 CrossRef CAS PubMed.
- S. N. Oyaghire, C. J. Cherubim, C. A. Telmer, J. A. Martinez, M. P. Bruchez and B. A. Armitage, RNA G-quadruplex invasion and translation inhibition by antisense γ-peptide nucleic acid oligomers, Biochemistry, 2016, 55, 1977–1988 CrossRef CAS PubMed.
- M. K. Gupta, B. R. Madhanagopal, D. Datta and K. N. Ganesh, Structural design and synthesis of bimodal PNA that simultaneously binds two complementary DNAs to form fused double duplexes, Org. Lett., 2020, 22, 5255–5260 CrossRef CAS PubMed.
- P. Bhingardeve, B. R. Madhanagopal and K. N. Ganesh, Cγ(S/R)-Bimodal peptide nucleic acids (Cγ-bm-PNA) form coupled double duplexes by synchronous binding to two complementary DNA strands, J. Org. Chem., 2020, 85, 13680–13693 CrossRef CAS PubMed.
- M. K. Gupta, B. R. Madhanagopal and K. N. Ganesh, Peptide nucleic acid with double face: Homothymine–homocytosine bimodal Cα-PNA (bm-Cα-PNA) forms a double duplex of the bm-PNA2:DNA triplex, J. Org. Chem, 2021, 86, 414–428 CrossRef CAS PubMed.
- P. Bhingardeve, P. Jain and K. N. Ganesh, Molecular assembly of triplex of duplexes from homothyminyl-homocytosinyl Cγ(S/R)-bimodal peptide nucleic acids with dA8/dG6 and the cell permeability of bimodal peptide nucleic acids, ACS Omega, 2021, 6, 19757–19770 CrossRef CAS PubMed.
- Y. Zeng, Y. Pratumyot, X. Piao and D. Bong, Discrete assembly of synthetic peptide-DNA triplex structures from polyvalent melamine-thymine bifacial recognition, J. Am. Chem. Soc., 2012, 134, 832–835 CrossRef CAS PubMed.
- X. Piao, X. Xia and D. Bong, Bifacial peptide nucleic acid directs cooperative folding and assembly of binary, ternary, and quaternary DNA complexes, Biochemistry, 2013, 52, 6313–6323 CrossRef CAS PubMed.
- G. K. Mittapalli, K. R. Reddy, H. Xong, O. Munoz, B. Han, F. De Riccardis, R. Krishnamurthy and A. Eschenmoser, Mapping the landscape of potentially primordial informational oligomers: Oligodipeptides and oligopetoids tagged with triazines as recognition elements, Angew. Chem., Int. Ed., 2007, 46, 2470–2477 CrossRef CAS PubMed.
- X. Xia, X. Piao and D. Bong, Bifacial PNA complexation inhibits enzymatic access to DNA and RNA, ChemBioChem, 2014, 15, 31–36 CrossRef CAS PubMed.
- S. Miao, Y. Liang, I. Marathe, J. Mao, C. DeSantis and D. Bong, Duplex stem replacement with bPNA+ triplex hybrid stems enables reporting on tertiary interactions of internal RNA domains, J. Am. Chem. Soc., 2019, 141, 9365–9372 CrossRef PubMed.
- S. Miao, D. Bhunia, S. Devari, Y. Liang, O. Munyaradzi, S. Rundell and D. Bong, Bifacial PNAs destabilize MALAT1 by 3′ A-tail displacement from the U-rich internal loop, ACS Chem. Biol., 2021, 16, 1600–1609 CrossRef CAS PubMed.
- A. Manicardi and R. Corradini, Effect of chirality in gamma-PNA: PNA interaction, another piece in the picture, Artif. DNA: PNA & XNA, 2014, 5, e1131801 Search PubMed.
- T. D. Canady, C. A. Telmer, S. N. Oyaghire, B. A. Armitage and M. P. Bruchez, In vitro reversible translation control using γPNA probes, J. Am. Chem. Soc., 2015, 137, 10268–10275 CrossRef CAS PubMed.
- T. D. Canady, A. S. Berlyoung, J. A. Martinez, C. Emanuelson, C. A. Telmer, M. P. Bruchez and B. A. Armitage, Enhanced hybridization selectivity using structured GammaPNA probes, Molecules, 2020, 25, 970 CrossRef CAS PubMed.
- B. E. Young, N. Kundu and J. T. Sczepanski, Mirror-image oligonucleotides: History and emerging applications, Chem. – Eur. J., 2019, 25, 7981–7990 CrossRef CAS PubMed.
- B. E. Young and J. T. Sczepanski, Heterochiral DNA strand-displacement based on chimeric D/L-oligonucleotides, ACS Synth. Biol., 2019, 8, 2756–2759 CrossRef CAS PubMed.
- W.-C. Hsieh, G. R. Martinez, A. Wang, S. F. Wu, R. Chamdia and D. H. Ly, Stereochemical conversion of nucleic acid circuits via strand displacement, Commun. Chem., 2018, 1, 89 CrossRef.
- D. Chang, E. Lindberg and N. Winssinger, Critical analysis of rate constants and turnover frequency in nucleic acid-templated reactions: Reaching terminal velocity, J. Am. Chem. Soc., 2017, 139, 1444–1447 CrossRef CAS PubMed.
- Z. Yu, W.-C. Hsieh, S. Asamitsu, K. Hashiya, T. Bando, D. H. Ly and H. Sugiyama, Orthogonal γPNA dimerization domains empower DNA binders with cooperativity and versatility mimicking that of transcription factor pairs, Chem. – Eur. J., 2018, 24, 14183–14188 CrossRef CAS PubMed.
- H. Amarasekara, K. M. Oshaben, K. B. Jeans, P. R. Sangsari, N. Y. Morgan, B. O'Farrell and D. H. Appella, Cyclopentane peptide nucleic acid: Gold nanoparticle conjugates for the detection of nucleic acids in a microfluidic format, Biopolymers, 2021, e23481 Search PubMed.
- D. Bonifazi, L.-E. Carloni, V. Corvaglia and A. Delforge, Peptide nucleic acids in materials science, Artif. DNA: PNA & XNA, 2012, 3, 112–122 Search PubMed.
- S. Barluenga and N. Winssinger, PNA as a biosupramolecular tag for programmable assemblies and reactions, Acc. Chem. Res., 2015, 48, 1319–1331 CrossRef CAS PubMed.
- O. Berger, L. Adler-Abramovich, M. Levy-Sakin, A. Grunwald, Y. Liebes-Peer, M. Bachar, L. Buzhansky, E. Mossou, V. T. Forsyth, T. Schwartz, Y. Ebenstein, F. Frolow, L. J. W. Shimon, F. Patolsky and E. Gazit, Light-emitting self-assembled peptide nucleic acids exhibit both stacking interactions and Watson–Crick base-pairing, Nat. Nanotechnol., 2015, 10, 353–360 CrossRef CAS PubMed.
- C. Avitabile, C. Diaferia, B. D. Ventura, F. A. Mercurio, M. Leone, V. Roviello, M. Saviano, R. Velotta, G. Morelli, A. Accardo and A. Romanelli, Self-assembling of Fmoc-GC peptide nucleic acid dimers into highly fluorescent aggregates, Chem. – Eur. J., 2018, 24, 4729–4735 CrossRef CAS PubMed.
- S. Kumar, A. Pearse, Y. Liu and R. E. Taylor, Modular self-assembly of gamma-modified peptide nucleic acids in organic solvent mixtures, Nat. Commun., 2020, 11, 2960 CrossRef CAS PubMed.
- S. Kumar, I. Dhami, S. A. Thadke, D. H. Ly and R. E. Taylor, Rapid self-assembly of γPNA nanofibers at constant temperature, Biopolymers, 2021, 112, e23463 CrossRef CAS PubMed.
- T. Machida, S. Dutt and N. Winssinger, Allosterically regulated phosphatase activity from peptide–PNA conjugates folded through hybridization, Angew. Chem., Int. Ed., 2016, 55, 8595–8598 CrossRef CAS PubMed.
- L. Röglin, M. R. Ahmadian and O. Seitz, DNA-Controlled reversible switching of peptide conformation and bioactivity, Angew. Chem., Int. Ed., 2007, 46, 2704–2707 CrossRef PubMed.
- S. Rapireddy, L. Nhon, R. E. Meehan, J. Franks, D. B. Stolz, D. Tran, M. E. Selsted and D. H. Ly, RTD-1 mimic containing γPNA scaffold exhibits broad-spectrum antibacterial activities, J. Am. Chem. Soc., 2012, 134, 4041–4044 CrossRef CAS PubMed.
- X. Tan, M. P. Bruchez and B. A. Armitage, Closing the loop: constraining TAT peptide by γPNA hairpin for enhanced cellular delivery of biomolecules, Bioconjugate Chem., 2018, 29, 2892–2898 CrossRef CAS PubMed.
- N. Jayaraman, Multivalent ligand presentation as a central concept to study intricate carbohydrate–protein interactions, Chem. Soc. Rev., 2009, 38, 3463–3483 RSC.
- K. Gorska, K.-T. Huang, O. Chaloin and N. Winssinger, DNA-templated homo- and heterodimerization of peptide nucleic acid encoded oligosaccharides that mimick the carbohydrate epitope of HIV, Angew. Chem., Int. Ed., 2009, 48, 7695–7700 CrossRef CAS PubMed.
- M. Ciobanu, K.-T. Huang, J.-P. Daguer, S. Barluenga, O. Chaloin, E. Schaeffer, C. G. Mueller, D. A. Mitchell and N. Winssinger, Selection of a synthetic glycan oligomer from a library of DNA-templated fragments against DC-SIGN and inhibition of HIV gp120 binding to dendritic cells, Chem. Commun., 2011, 47, 9321–9323 RSC.
- K. Gorska, J. Beyrath, S. Fournel, G. Guichard and N. Winssinger, Ligand dimerization programmed by hybridization to study multimeric ligand–receptor interactions, Chem. Commun., 2010, 46, 7742–7744 RSC.
- T. Machida, A. Novoa, P. Gillon, S. Zheng, J. Claudinon, T. Eierhoff, A. Imberty, W. Rçmer and N. Winssinger, Dynamic cooperative glycan assembly blocks the binding of bacterial lectins to epithelial cells, Angew. Chem., Int. Ed., 2017, 56, 6762–6766 CrossRef CAS PubMed.
- C. Scheibe, A. Bujotzek, J. Dernedde, M. Weberb and O. Seitz, DNA-Programmed spatial screening of carbohydrate–lectin interactions, Chem. Sci., 2011, 2, 770–775 RSC.
- E. A. Englund, D. Wang, H. Fujigaki, H. Sakai, C. M. Micklitsch, R. Ghirlando, G. Martin-Manso, M. L. Pendrak, D. D. Roberts, S. R. Durell and D. H. Appella, Programmable multivalent display of receptor ligands using peptide nucleic acid nanoscaffolds, Nat. Commun., 2012, 3, 614 CrossRef PubMed.
- A. V. Dix, J. L. Conroy, K. M. G. Rosenker, D. R. Sibley and D. H. Appella, PNA-Based multivalent scaffolds activate the dopamine D2 receptor, ACS Med. Chem. Lett., 2015, 6, 425–429 CrossRef CAS PubMed.
- A. V. Dix, S. M. Moss, K. Phan, T. Hoppe, S. Paoletta, E. Kozma, Z.-G. Gao, S. R. Durell, K. A. Jacobson and D. H. Appella, Programmable nanoscaffolds that control ligand display to a G-protein-coupled receptor in membranes to allow dissection of multivalent effects, J. Am. Chem. Soc., 2014, 136, 12296–12303 CrossRef CAS PubMed.
- J. D. Flory, C. R. Simmons, S. Lin, T. Johnson, A. Andreoni, J. Zook, G. Ghirlanda, Y. Liu, H. Yan and P. Fromme, Low temperature assembly of functional 3D DNA-PNA-protein complexes, J. Am. Chem. Soc., 2014, 136, 8283–8295 CrossRef CAS PubMed.
- J. D. Flory, S. Shinde, S. Lin, Y. Liu, H. Yan, G. Ghirlanda and P. Fromme, PNA-peptide assembly in a 3D DNA nanocage at room temperature, J. Am. Chem. Soc., 2013, 135, 6985–6993 CrossRef CAS PubMed.
- J. D. Flory, T. Johnson, C. D. Simmons, S. Lin, G. Ghirlanda and P. Fromme, Purification and assembly of thermostable Cy5 labeled γ-PNAs into a 3D DNA nanocage, Artif. DNA: PNA & XNA, 2014, 5, e992181 Search PubMed.
- C. S. Swenson, A. Velusamy, H. S. Argueta-Gonzalez and J. M. Heemstra, Bilingual peptide nucleic acids: Encoding the languages of nucleic acids and proteins in a single self-assembling biopolymer, J. Am. Chem. Soc., 2019, 141, 19038–19047 CrossRef CAS PubMed.
- C. S. Swenson and J. M. Heemstra, Peptide nucleic acids harness dual information codes in a single molecule, Chem. Commun., 2020, 56, 1926–1935 RSC.
- J. P. Vernille, L. C. Kovell and J. W. Schneider, Peptide nucleic acid (PNA) amphiphiles: Synthesis, self-assembly, and duplex stability, Bioconjugate Chem., 2004, 15, 1314–1321 CrossRef CAS PubMed.
- L.-H. Liu, Z.-Y. Li, L. Rong, S.-Y. Qin, Q. Lei, H. Cheng, X. Zhou, R.-X. Zhuo and X.-Z. Zhang, ACS Macro Lett., 2014, 3, 467–471 CrossRef CAS.
- S. Ficht, A. Mattes and O. Seitz, Single-nucleotide-specific PNA-peptide ligation on synthetic and PCR DNA templates, J. Am. Chem. Soc., 2004, 126, 9970–9981 CrossRef CAS PubMed.
- X. Peng, H. Li and M. Seidman, A template-mediated click–click reaction: PNA–DNA, PNA–PNA (or peptide) ligation, and single nucleotide discrimination, Eur. J. Org. Chem., 2010, 4194–4197 CrossRef CAS PubMed.
- D. Chouikhi, S. Barluenga and N. Winssinger, Clickable peptide nucleic acids (cPNA) with tunable affinity, Chem. Commun., 2010, 46, 5476–5478 RSC.
- R. E. Kleiner, Y. Brudno, M. E. Birnbaum and D. R. Liu, DNA-templated polymerization of side-chain-functionalized peptide nucleic acid aldehydes, J. Am. Chem. Soc., 2008, 130, 4646–4659 CrossRef CAS PubMed.
- Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Self-assembling sequence-adaptive peptide nucleic acids, Science, 2009, 325, 73–77 CrossRef CAS PubMed.
- K. V. Morris and J. S. Mattick, The rise of regulatory RNA, Nat. Rev. Genet., 2014, 15, 423–437 CrossRef CAS PubMed.
- B. A. Armitage, Imaging of RNA in Live Cells, Curr. Opin. Chem. Biol., 2011, 15, 806–812 CrossRef CAS PubMed.
- C. Briones and M. Moreno, Applications of peptide nucleic acids (PNAs) and locked nucleic acids (LNAs) in biosensor development, Anal. Bioanal. Chem., 2012, 402, 3071–3089 CrossRef CAS PubMed.
- A. Saadati, S. Hassanpour, M. de la Guardia, J. Mosafer, M. Hashemzaei, A. Mokhtarzadeh and B. Baradaran, Recent advances on application of peptide nucleic acids as a bioreceptor in biosensors development, Trends Anal. Chem., 2019, 114, 56–68 CrossRef CAS.
- M. Moccia, A. Antonacci, M. Saviano, V. Caratelli, F. Arduini and V. Scognamiglio, Emerging technologies in the design of peptide nucleic acids (PNAs) based biosensors, Trends Anal. Chem., 2020, 132, 116062 CrossRef CAS.
- Q. Lai, W. Chen, Y. Zhang and Z. Liu, Application strategies of peptide nucleic acids toward electrochemical nucleic acid sensors, Analyst, 2021, 146, 5822–5835 RSC.
- J. K. Pokorski, J.-M. Nam, R. A. Vega, C. A. Mirkin and D. H. Appella, Cyclopentane-modified PNA improves the sensitivity of nanoparticle-based scanometric DNA detection, Chem. Commun., 2005, 2101–2103 RSC.
- N. Zhang and D. H. Appella, Colorimetric detection of anthrax DNA with a peptide nucleic acid sandwich-hybridization assay, J. Am. Chem. Soc., 2007, 129, 8424–8425 CrossRef CAS PubMed.
- C. M. Micklitsch, B. Y. Oquare, C. Zhao and D. H. Appella, Cyclopentane-peptide nucleic acids for qualitative, quantitative, and repetitive detection of nucleic acids, Anal. Chem., 2013, 85, 251–257 CrossRef CAS PubMed.
- O. Tepper, H. Zheng, D. H. Appella and E. Yavin, Cyclopentane FIT-PNAs: Bright RNA sensors, Chem. Commun., 2021, 57, 540–543 RSC.
- O. Kohler, D. V. Jarikote and O. Seitz, Forced intercalation probes (FIT Probes): Thiazole orange as a fluorescent base in peptide nucleic acids for homogeneous single-nucleotide-polymorphism detection, ChemBioChem, 2005, 6, 69–77 CrossRef PubMed.
- T. Vilaivan, Fluoreogenic PNA probes, Beilstein J. Org. Chem., 2018, 14, 253–281 CrossRef CAS PubMed.
- C. Ananthanawat, T. Vilaivan, V. P. Hoven and X. Su, Comparison of DNA, aminoethylglycyl PNA and pyrrolidinyl PNA as probes for detection of DNA hybridization using surface plasmon resonance technique, Biosens. Bioelectron., 2010, 25, 1064–1069 CrossRef CAS PubMed.
- C. Ananthanawat, V. P. Hoven, T. Vilaivan and X. Su, Surface Plasmon resonance study of PNA interactions with double-stranded DNA, Biosens. Bioelectron., 2011, 26, 1918–1923 CrossRef CAS PubMed.
- P. Teengam, W. Siangproh, A. Tuantranont, T. Vilaivan, O. Chailapakul and C. S. Henry, Multiplex paper-based colorimetric DNA sensor using pyrrolidinyl peptide nucleic acid-induced AgNPs aggregation for detecting MERS-Cov, MTB, and HPV oligonucleotides, Anal. Chem., 2017, 89, 5428–5435 CrossRef CAS PubMed.
- M. Leekrajang, P. Sae-ung, T. Vilaivan and V. P. Hoven, Filter paper grafted with epoxide-based copolymer brushes for activation-free peptide nucleic acid conjugation and its application for colorimetric DNA detection, Colloids Surf., B., 2019, 173, 851–859 CrossRef CAS PubMed.
- N. Jirakittiwut, T. Munkongdee, K. Wongravee, O. Sripichai, S. Fucharoen, T. Praneenararat and T. Vilaivan, Visual genotyping of thalassemia by using pyrrolidinyl peptide nucleic acid probes immobilized on carboxymethylcellulose-modified paper and enzyme-induced pigmentation, Microchim. Acta, 2020, 187, 238 CrossRef CAS PubMed.
- C. Srisomwat, A. Yakoh, N. Chuaypen, P. Tangkijvanich, T. Vilaivan and O. Chailapakul, Amplification-free DNA sensor for the one-step detection of the hepatitis b virus using an automated paper-based lateral flow electrochemical device, Anal. Chem., 2021, 93, 2879–2887 CrossRef CAS PubMed.
- S. Naorungroj, P. Teengam, T. Vilaivan and O. Chailapakul, Paper-Based DNA sensor enabling colorimetric assay integrated with smartphone for human papillomavirus detection, New J. Chem., 2021, 45, 6960–6967 RSC.
- P. Teengam, N. Nisab, N. Chuaypen, P. Tangkijvanich, T. Vilaivan and O. Chailapakul, Fluorescent paper-based DNA sensor using pyrrolidinyl peptide nucleic acids for hepatitis C virus detection, Biosens. Bioelectron., 2021, 189, 113381 CrossRef CAS PubMed.
- N. Jirakittiwut, T. Patipong, T. Cheiwchanchamnangij, R. Waditee-Sirisattha, T. Vilaivan and T. Praneenararat, Paper-based sensor from pyrrolidinyl peptide nucleic acid for the efficient detection of Bacillus cereus, Anal. Bioanal. Chem., 2021, 413, 6661–6669 CrossRef CAS PubMed.
- C. Srisomwat, A. Yakoh, A. Avihingsanon, N. Chuaypen, P. Tangkijvanich, T. Vilaivan and O. Chailapakul, An alternative label-free DNA sensor based on the alternating-current electroluminescent device for simultaneous detection of human immunodeficiency virus and hepatitis C Co-infection, Biosens. Bioelectron., 2022, 196, 113719 CrossRef CAS PubMed.
- O. Thipmanee, A. Numnuam, W. Limbut, C. Buranachai, P. Kanatharana, T. Vilaivan, N. Hirankarn and P. Thavarungkul, Enhancing capacitive DNA biosensor performance by target overhang with application on screening test of HLA-B*58:01 and HLA-B*57:01 genes, Biosens. Bioelectron., 2016, 82, 99–104 CrossRef CAS PubMed.
- O. Thipmanee, S. Samanman, S. Sankoh, A. Numnuam, W. Limbut, P. Kanatharana, T. Vilaivan and P. Thavarungkul, Label-free capacitive DNA sensor using immobilized pyrrolidinyl PNA Probe: Effect of the length and terminating head group of the blocking thiols, Biosens. Bioelectron., 2012, 38, 430–435 CrossRef CAS PubMed.
- T. Kangkamano, A. Numnuam, W. Limbut, P. Kanatharana, T. Vilaivan and P. Thavarungkul, Pyrrolidinyl PNA polypyrrole/silver nanofoam electrode as a novel label-free electrochemical miRNA-21 biosensor, Biosens. Bioelectron., 2018, 102, 217–225 CrossRef CAS PubMed.
- B. Boontha, J. Nakkuntod, N. Hirankarn, P. Chaumpluk and T. Vilaivan, Multiplex mass spectrometric genotyping of single nucleotide polymorphisms employing pyrrolidinyl peptide nucleic acid in combination with ion-exchange capture, Anal. Chem., 2008, 80, 8178–8186 CrossRef CAS PubMed.
- J. Meebungprawa, O. Wiarachai, T. Vilaivan, S. Kiatkamjornwonge and V. P. Hoven, Quaternized chitosan particles as ion exchange supports for label-freedna detection using PNA probe and MALDI-TOF mass spectrometry, Carbohydr. Polym., 2015, 131, 80–89 CrossRef PubMed.
- B. Rutnakornpituk, T. Theppaleak, M. Rutnakornpituk and T. Vilaivan, Recyclable magnetite nanoparticle coated with cationic polymers for adsorption of DNA, J. Biomater. Sci. Polym. Ed., 2016, 27, 1200–1210 CrossRef CAS PubMed.
- P. S. Laopa, T. Vilaivan and V. P. Hoven, Positively charged polymer brushes-functionalized filter paper for DNA Sequence determination following dot blot hybridization employing pyrrolidinyl peptide nucleic acid probe, Analyst, 2013, 138, 269–277 RSC.
- J. Kongpeth, S. Jampasa, P. Chaumpluk, O. Chailapakul and T. Vilaivan, Immobilization-free electrochemical DNA detection with anthraquinone-labeled pyrrolidinyl peptide nucleic acid probe, Talanta, 2016, 146, 318–325 CrossRef CAS PubMed.
- C. Boonlua, C. Vilaivan, H.-A. Wagenknecht and T. Vilaivan, 5-(Pyren-1-yl)uracil as a base-discriminating fluorescent nucleobase in pyrrolidinyl peptide nucleic acids, Chem. Asian J., 2011, 6, 3251–3259 CrossRef CAS PubMed.
- B. Ditmangklo, J. Taechalertpaisarn, K. Siriwong and T. Vilaivan, Clickable styryl dyes for fluorescence labeling of pyrrolidinyl PNA probes for the detection of base mutations in DNA, Org. Biomol. Chem., 2019, 17, 9712–9725 RSC.
- C. Boonlua, C. Charoenpakdee, T. Vilaivan and T. Praneenararat, Preparation and performance evaluation of a pyrrolidinyl peptide nucleic-acid-based displacement probe as a DNA sensor, ChemistrySelect, 2016, 1, 5691–5697 CrossRef CAS.
- C. Charoenpakdee and T. Vilaivan, Quenching of fluorescently labeled pyrrolidinyl peptide nucleic acid by oligodeoxyguanosine and its application in DNA sensing, Org. Biomol. Chem., 2020, 18, 5951–5962 RSC.
- P. Yukhet, K. Buddhachat, T. Vilaivan and C. Suparpprom, Isothermal detection of canine blood parasite (Ehrlichia canis) utilizing recombinase polymerase amplification coupled with graphene oxide quenching-based pyrrolidinyl peptide nucleic acid, Bioconjugate Chem., 2021, 32, 523–532 CrossRef CAS PubMed.
- K. Faikhruea, I. Choopara, N. Somboonna, W. Assavalapsakul, B. H. Kim and T. Vilaivan, Enhancing peptide nucleic acid–nanomaterial interaction and performance improvement of peptide nucleic acid-based nucleic acid detection by using electrostatic effects, ACS Appl. Bio Mater., 2022, 5, 789–800 CrossRef CAS PubMed.
- J. Nölling, S. Rapireddy, J. I. Amburg, E. M. Crawford, R. A. Prakash, A. R. Rabson, Y.-W. Tang and A. Singer, Duplex DNA-invading γ-modified peptide nucleic acids enable rapid identification of bloodstream infections in whole blood, mBio, 2016, 7, e00345 CrossRef PubMed.
- A. Singer, S. Rapireddy, D. H. Ly and A. Meller, Electronic barcoding of a viral gene at the single-molecule level, Nano Lett., 2012, 12, 1722–1728 CrossRef CAS PubMed.
- J. M. Goldman, L. A. Zhang, A. Manna, B. A. Armitage, D. H. Ly and J. W. Schneider, High affinity γPNA sandwich hybridization assay for rapid detection of short nucleic acid targets with single mismatch discrimination, Biomacromolecules, 2013, 14, 2253–2261 CrossRef CAS PubMed.
- J. M. Goldman, S. Kim, S. Narburgh, B. A. Armitage and J. W. Schneider, Rapid, multiplexed detection of the let-7 miRNA family using γPNA amphiphiles in micelle-tagging electrophoresis, Biopolymers, 2021, e23479 Search PubMed.
- K. T. Kim, S. Angerani and N. Winssinger, A minimal hybridization chain reaction (HCR) system using peptide nucleic acids, Chem. Sci., 2021, 12, 8218–8223 RSC.
- B. Dong, K. Nie, H. Shi, L. Chao, M. Ma, F. Gao, B. Liang, W. Chen, M. Longa and Z. Liu, Film-spotting chiral miniPEG-γPNA array for BRCA1 gene mutation detection, Biosens. Bioelectron., 2019, 136, 1–7 CrossRef CAS PubMed.
- A. Singer, M. Wanunu, W. Morrison, H. Kuhn, M. Frank-Kamenetskii and A. Meller, Nanopore based sequence specific detection of duplex DNA for genomic profiling, Nano Lett., 2010, 2, 738–742 CrossRef PubMed.
- T. J. Morin, T. Shropshire, X. Liu, K. Briggs, C. Huynh, V. Tabard-Cossa, H. Wang and W. B. Dunbar, Nanopore-based target sequence detection, PLoS One, 2016, 11, e0154426 CrossRef PubMed.
- K. Gorska, I. Keklikoglou, U. Tschulenab and N. Winssinger, Rapid fluorescence imaging of miRNAs in human cells using templated staudinger reaction, Chem. Sci., 2011, 2, 1969–1975 RSC.
- A. Orenstein, A. S. Berlyoung, E. E. Rastede, H. H. Pham, E. Fouquerel, C. T. Murphy, B. J. Leibowitz, J. Yu, T. Srivastava, B. A. Armitage and P. L. Opresko, γPNA FRET pair miniprobes for quantitative fluorescent in situ hybridization to telomeric DNA in cells and tissue, Molecules, 2017, 22, 2117 CrossRef PubMed.
- K. T. Kim, S. Angerani and N. Winssinger, A minimal hybridization chain reaction (HCR) system using peptide nucleic acids, Chem. Sci., 2021, 12, 8218–8223 RSC.
- D. R. Jain and K. N. Ganesh, Clickable Cγ-azido(methylene/butylene) peptide nucleic acids and their clicked fluorescent derivatives: Synthesis, DNA hybridization properties, and cell penetration studies, J. Org. Chem., 2014, 79, 6708–6714 CrossRef CAS PubMed.
- A. Manicardi, A. Bertucci, A. Rozzi and R. Corradini, A bifunctional monomer for on-resin synthesis of polyfunctional PNAs and tailored induced-fit switching probes, Org. Lett., 2016, 18, 5452–5455 CrossRef CAS PubMed.
- H. H. Pham, C. T. Murphy, G. Sureshkumar, D. H. Ly, P. L. Opreskob and B. A. Armitage, Cooperative hybridization of γPNA miniprobes to a repeating sequence motif and application to telomere analysis, Org. Biomol. Chem., 2014, 12, 7345–7354 RSC.
- T. Tedeschi, M. Chiari, G. Galaverna, S. Sforza, M. Cretich, R. Corradini and R. Marchelli, Detection of the R553X DNA single point mutation related to cystic fibrosis by a “chiral box” d-lysine-peptide nucleic acid probe by capillary electrophoresis, Electrophoresis, 2005, 26, 4310–4316 CrossRef CAS PubMed.
- R. Corradini, G. Feriotto, S. Sforza, R. Marchelli and R. Gambari, Enhanced recognition of cystic fibrosis W1282X DNA point mutation by chiral peptide nucleic acid probes by a surface plasmon resonance biosensor, J. Mol. Recognit., 2004, 17, 76–84 CrossRef CAS PubMed.
- J. D. R. Perera, K. E. W. Carufe and P. M. Glazer, Peptide nucleic acids and their role in gene regulation and editing, Biopolymers, 2021, 112, e23460 CrossRef CAS PubMed.
- E. Quijano, R. Bahal, A. Ricciardi, W. M. Saltzman and P. M. Glazer, Therapeutic peptide nucleic acids: Principles, limitations, and opportunities, Yale J. Biol. Med., 2017, 90, 583–598 CAS.
- N. G. Economos, S. Oyaghire, E. Quijano, A. S. Ricciardi, W. M. Saltzman and P. M. Glazer, Peptide nucleic acids and gene editing: Perspectives on structure and repair, Molecules, 2020, 25, 735 CrossRef CAS PubMed.
- C. Dose and O. Seitz, Single nucleotide specific detection of DNA by native chemical ligation of fluorescence labeled PNA-probes, Bioorg. Med. Chem., 2008, 16, 65–77 CrossRef CAS PubMed.
- R. Hamzavi, C. Meyer and N. Metzler-Nolte, Synthesis of a C-linked glycosylated thymine-based PNA monomer and its incorporation into a PNA oligomer, Org. Biomol. Chem., 2005, 4, 3648–3651 RSC.
- R. Hamzavi, F. Dolle, B. Tavitian, O. Dahl and P. E. Nielsen, Modulation of the pharmacokinetic properties of PNA: Preparation of galactosyl, mannosyl, fucosyl, N-acetylgalactosaminyl, and N-acetylglucosaminyl derivatives of aminoethylglycine peptide nucleic acid monomers and their incorporation into PNA oligomers, Bioconjugate Chem., 2003, 14, 941–954 CrossRef CAS PubMed.
- P. Bhingardeve, B. R. Madhanagopal, H. Naick, P. Jain, M. Manoharan and K. Ganesh, Receptor-specific delivery of peptide nucleic acids conjugated to three sequentially linked N-acetyl galactosamine moieties into hepatocytes, J. Org. Chem., 2020, 85, 8812–8824 CrossRef CAS PubMed.
- P. Zhou, M. Wang, L. Du, G. W. Fisher, A. Waggoner and D. H. Ly, Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA), J. Am. Chem. Soc., 2003, 125, 6878–6879 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, P. Zhou, G. He and D. H. Ly, Cell-permeable GPNA with appropriate backbone stereochemistry and spacing binds sequence-specifically to RNA, Chem. Commun., 2005, 244–246 RSC.
- P. Zhou, A. Dragulescu-Andrasi, B. Bhattacharya, H. O’Keefe, P. Vatta, J. J. Hyldig-Nielsen and D. H. Ly, Synthesis of cell-permeable peptide nucleic acids and characterization of their hybridization and uptake properties, Bioorg. Med. Chem. Lett., 2006, 16, 4931–4935 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, S. Rapireddy, G. He, B. Bhattacharya, J. J. Hyldig-Nielsen, G. Zon and D. H. Ly, Cell-permeable peptide nucleic acid designed to bind to the 5′-untranslated region of E-cadherin transcript induces potent and sequence-specific antisense effects, J. Am. Chem. Soc., 2006, 128, 16104–16112 CrossRef CAS PubMed.
- S. M. Thomas, B. Sahu, S. Rapireddy, R. Bahal, S. E. Wheeler, E. M. Procopio, J. Kim, S. C. Joyce, S. Contrucci, Y. Wang, S. I. Chiosea, K. L. Lathrop, S. Watkins, J. R. Grandis, B. A. Armitage and D. H. Ly, Antitumor effects of EGFR antisense guanidine-based peptide nucleic acids in cancer models, ACS Chem Biol., 2013, 8, 345–352 CrossRef CAS PubMed.
- A. Manicardi, E. Fabbri, T. Tedeschi, S. Sforza, N. Bianchi, E. Brognara, R. Gambari, R. Marchelli and R. Corradini, Cellular uptakes, biostabilities and anti-miR-210 activities of chiral arginine-PNAs in leukaemic K562 Cells, ChemBioChem, 2012, 13, 1327–1337 CrossRef CAS PubMed.
- E. Delgado, R. Bahal, J. Yang, J. M. Lee, D. H. Ly and S. P. S. Monga, β-Catenin knockdown in liver tumor cells by a cell permeable gamma guanidine-based peptide nucleic acid, Curr. Cancer Drug Targets, 2013, 13, 867–878 CrossRef CAS PubMed.
- R. Bahal, N. A. McNeer, D. H. Ly, W. M. Saltzman and P. M. Glazer, Nanoparticle for delivery of antisense γPNA oligomers targeting CCR5, Artif. DNA: PNA & XNA, 2013, 4, 49–57 Search PubMed.
- K. Dhuri, R. R. Gaddam, A. Vikram, F. J. Slack and R. Bahal, Therapeutic potential of chemically modified, synthetic, triplex peptide nucleic acid–based oncomir inhibitors for cancer therapy, Cancer Res., 2021, 81, 5613–5624 CrossRef CAS PubMed.
- A. Gupta, E. Quijano, Y. Liu, R. Bahal, S. E. Scanlon, E. Song, W.-C. Hsieh, D. E. Braddock, D. H. Ly, W. M. Saltzman and P. M. Glazer, Anti-tumor activity of miniPEG-γ-modified PNAs to inhibit microRNA-210 for cancer therapy, Mol. Ther. Nucleic Acids, 2017, 9, 111–119 CrossRef CAS PubMed.
- A. R. Kaplan, H. Pham, Y. Liu, S. Oyaghire, R. Bahal, D. M. Engelman and P. M. Glazer, Ku80-Targeted pH-sensitive peptide–PNA conjugates are tumor selective and sensitize cancer cells to ionizing radiation, Mol. Cancer Res., 2020, 18, 873–882 CrossRef CAS PubMed.
- A. S. Ricciardi, E. Quijano, R. Putman, W. M. Saltzman and P. M. Glazer, Peptide nucleic acids as a tool for site-specific gene editing, Molecules, 2018, 23, 632 CrossRef PubMed.
- R. Bahal, E. Quijano, N. A. McNeer, Y. Liu, D. C. Bhunia, F. López-Giráldez, R. J. Fields, W. M. Saltzman, D. H. Ly and P. M. Glazer, Single-stranded γPNAs for in vivo site-specific genome editing via Watson–Crick recognition, Curr. Gene Ther., 2014, 14, 331–342 CrossRef CAS PubMed.
- J. Y. Chin, J. Y. Kuan, P. S. Lonkar, D. S. Krause, M. M. Seidman, K. R. Peterson, P. E. Nielsen, R. Kole and P. M. Glazer, Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13514–13519 CrossRef CAS PubMed.
- M. Adli, The CRISPR tool kit for genome editing and beyond, Nat. Commun., 2018, 9, 1911 CrossRef PubMed.
- R. Bahal, N. A. McNeer, E. Quijano, Y. Liu, P. Sulkowski, A. Turchick, Y.-C. Lu, D. C. Bhunia, A. Manna, D. L. Greiner, M. A. Brehm, C. J. Cheng, F. López-Giráldez, A. Ricciardi, J. Beloor, D. S. Krause, P. Kumar, P. G. Gallagher, D. T. Braddock, W. M. Saltzman, D. H. Ly and P. M. Glazer, In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery, Nat. Commun., 2016, 7, 13304 CrossRef CAS PubMed.
- A. S. Ricciardi, R. Bahal, J. S. Farrelly, E. Quijano, A. H. Bianchi, V. L. Luks, R. Putman, F. López-Giráldez, S. Coşkun, E. Song, Y. Liu, W.-C. Hsieh, D. H. Ly, D. H. Stitelman, P. M. Glazer and W. M. Saltzman, In utero nanoparticle delivery for site-specific genome editing, Nat. Commun., 2018, 9, 2481 CrossRef PubMed.
- P. Y. Ho, Z. Zhang, M. E. Hayes, A. Curd, C. Dib, M. Rayburna, S. N. Tam, T. Srivastava, B. Hriniak, X.-J. Li, S. Leonard, L. Wang, S. Tarighat, D. S. Sim, M. Fiandaca, J. M. Coull, A. Ebens, M. Fordyce and A. Czechowicz, Peptide nucleic acid–dependent artifact can lead to false-positive triplex gene editing signals, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109175118 CrossRef CAS PubMed.
- S. Arayachukiat, J. Seemork, P. Pan-In, K. Amornwachirabodee, N. Sangphech, T. Sansureerungsikul, K. Sathornsantikun, C. Vilaivan, K. Shigyou, P. Pienpinijtham, T. Vilaivan, T. Palaga, W. Banlunara, T. Hamada and S. Wanichwecharungruang, Bringing macromolecules into cells and evading endosomes by oxidized carbon nanoparticles, Nano Lett., 2015, 15, 3370–3376 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |