
Design of pre-catalysts for heterogeneous CO2 electrochemical reduction
Jingfu
He
 *,
Chenghui
Wu
,
Yanming
Li
and
Changli
Li
*
*,
Chenghui
Wu
,
Yanming
Li
and
Changli
Li
*
School of Materials, Sun Yat-sen University, Guangzhou 510275, Guangdong, P. R. China. E-mail: hejf27@mail.sysu.edu.cn; lichli5@mail.sysu.edu.cn
First published on 9th August 2021
Abstract
CO2 reduction reaction (CO2RR) is one of the most promising methods to alleviate the global challenge of climate change and energy supply. Metal catalysts such as copper, gold, silver, tin, bismuth, etc., attract great attention for their high efficiency to convert CO2 into valuable chemicals such as CO, HCOOH, C2H4 and C2H5OH. It is of interest that the unstable metal-based compounds can be considered as pre-catalysts due to the in situ formation of a metallic state during CO2RR, resulting in a reconstruction of surface catalytic structures that mediate much higher CO2RR activity than their metal foil counterparts. However, the precise evolution process of the catalysts during CO2RR and ultimate active sites of the catalyst have not been fully tracked and determined. In this review, we summarize the recent development of metal oxides, metal dichalcogenides, metal halides, metal nitrides and metal–organic compound pre-catalysts, and focus on the structural evolution during the activation process. The key structural factors that impact the performance of catalysts are analyzed in-depth in terms of the remnant of anion elements, enhanced surface areas, facet orientation, and grain boundaries. A perspective is also provided to tackle the critical challenges in designing highly efficient CO2RR pre-catalysts, including the manipulation and identification of surface roughness, structure disorder and partial oxidation of metal center, the monitoring and regeneration of catalysts under a long-time operation, the elimination of impurities during operation and fine control of multicomponent for structural regulation.
 Jingfu He | Jingfu He is currently an associate professor in School of Materials, Sun Yat-sen University. He received his Ph.D. in 2012 from University of Science and Technology of China. He joined the Berlinguette group at the University of British Columbia as a postdoctoral associate in 2015 and conducted research on film synthesis and CO2 electrocatalysis. His current research interests include photoelectrochemistry and electrochemistry for CO2 reduction, water splitting and in situ synchrotron radiation spectrum. |
 Changli Li | Changli Li is an Associate Professor at the School of Materials, Sun Yat-sen University, China. He received his PhD degree in Mechanical Engineering from The University of Tokyo, Japan, in 2015. After Post-Doctoral studies at Tsinghua University and The University of British Columbia, he began his independent career as a faculty member at Sun Yat-sen University. His research activities focus on the synthesis of nanostructured materials and semiconductor heterojunctions for electrochemical and photoelectrochemical energy conversion. |
1. Introduction
The development of (photo)electrochemical methods to harvest renewable energy and produce valuable chemical fuels is a key step to build a sustainable society.1–5 Electrochemical CO2 reduction (CO2RR) that utilizes clean electricity to complete carbon fixation and synthesize fuel is an effective way to alleviate the energy and environmental crisis.6–9 To realize industrialized electrochemical CO2 reduction, it is necessary to optimize the construction of an electrochemical system so that it can catalytically reduce CO2 into a single product with high energy conversion efficiency and high selectivity. CO2 reduction is a very complex reaction with a series of products with very close electrode potentials.10,11 In addition, the side reaction of hydrogen evolution is difficult to avoid, because the electrode potential of CO2 reduction is very close to that of hydrogen evolution. These difficulties have made the research of catalysts one of the most restrictive factors for CO2RR industrialization.It is generally believed that the initial reduction of CO2 can be roughly divided into two pathways, one is to generate formic acid, and the other is to generate CO.12 Formic acid cannot be further reduced in this reducing environment. On the other hand, CO may continue to be adsorbed on the surface of the catalyst and be deeply reduced to a series of products, such as methane, ethylene, ethanol, and acetic acid. The reaction pathway and termination of CO2RR is determined by the surface electron and atomic structure of the catalyst, which determines the ability of the catalyst to combine with a series of intermediate products.
Previous research on single metal has classified metals into several groups with different catalytic selectivity.10,12,13 The most inert metals that difficult to form M–C bond, such as lead, mercury, tin and indium, mainly produce formic acid. Metals with weak CO adsorption strength, such as gold and silver, produce CO as the final reduction product. Metals with very strong CO adsorption strength, such as iron, cobalt, nickel, platinum, etc., are vulnerable to the surface poisoning, resulting in the production of hydrogen. The interaction between Cu and CO is moderate, thus it is the only catalyst that can further reduce CO. However, the Cu catalytic selectivity is very sensitive to the subtle changes in the electronic structure of the surface atoms. For example, the catalytic selectivity of the (111) facet and the (110) facet of copper are significantly different.14,15 Nevertheless, for the study based on planar metal catalysts, the overall reaction rate is low, and the side reaction of hydrogen production is very strong.
Kanan research group first proposed an oxidation–reduction strategy and found that the CuOx, AuOx and other metals oxides can be utilized as pre-catalysts, resulting in the formation of nanostructured morphologies with a large number of grain boundaries and a large specific surface area.16–19 The overall overpotential for CO2RR is reduced by 400 mV compared to the planar electrode, and hydrogen production is greatly suppressed. Smith's group studied OD-Ag for CO2RR and also observed a substantial increase in energy conversion and selectivity.20,21 After that, the OD strategy has been widely used in various CO2RR metal catalysts.22–25
In recent years, other metal–nonmetal compound pre-catalysts, such as metal dichalcogenide, metal nitrite, metal halogens and MOF, were developed as promising pre-catalysts for CO2RR.26–38 Compared with metal oxides pre-catalysts, the residual nonmetal concentration of S, B, N and their influence on the metal oxidation state were different. Moreover, the different compositions of pre-catalysts can regulate the structure evolution and result in unique surface morphology and defects. Therefore, various CO2RR selectivity and activity of pre-catalysts that superior to their pristine metal foil opponent have been realized, as listed in Table 1. For instance, Sargent group found that the nanostructure catalyst derived from Cu–F pre-catalysts can realize C2+ FEs of 85.8% at 1600 mA cm−2.31 Cu2S catalysts with abundant vacancies on the surface were also created and exhibited faradaic efficiency for C3H7OH and C2H5OH of 8% and 15%.26 Although there are numerous research works on the development of pre-catalysts, a practical rule or guideline for the design of pre-catalysts still lacks due to the difference in preparation methods, test methods, and sample parameters in each research work. There is an urgent need to sort out the relationship between the structure and performance of various pre-catalysts.
| Catalysts | Identified reason (besides high surface area) | Potential vs. RHE | j total (mA cm−2) | Selectivity | Ref. | |
|---|---|---|---|---|---|---|
| Key products | Faradaic efficiency (%) | |||||
| AuOx | Grain boundaries | −0.4 | 6–10 | CO | 98% | 18 |
| AuOH | Grain boundaries | −0.59 | 2.6 | CO | 98% | 62 |
| AuOx | Surface disorder | −0.8 | 10–16 | CO | >90% | 61 |
| Au-complex | S anions | −0.68 | 4.4 | CO | 94.2% | 214 |
| AgOx | Nanostructured surface | −0.8 | 1.15 | CO | 89% | 20 |
| AgOx | Undercoordinated sites | −0.6 | 2.5 | CO | 90% | 108 |
| AgOx | Surface orientation | −0.6 | 3.7 | CO | 92.8% | 64 |
| AgClx | Cl anions | −0.5 | 2 | CO | 95% | 32 |
| AgIx | I anions | −0.7 | 16.7 | CO | 94.5% | 107 |
| AgP2 | Partially oxidized Ag | −0.8 | 9 | CO | 82% | 35 |
| Ag-complex | High surface area | −1.03 | 6 | CO | 96% | 182 |
| Ag2CO3 | Nanostructured surface | −0.55 | 1 | CO | 90% | 67 |
| Ag3PO4 | Nanostructured surface | −0.9 | 8 | CO | 93% | 156 |
| GaOx | Small metal particle | −0.71 | 5 | CO | 77% | 51 |
| CdS | Partial S coordination | −1.2 | 12 | CO | 95% | 81 |
| ZnO | Nanostructure | −0.85 | 10 | CO | 95% | 98 |
| ZnO | Zn2+ rich surface | −0.95 | 7 | CO | 95.3% | 116 |
| SnO2 | Grain boundaries | −0.8 | 6 | HCOOH | 80% | 109 |
| SnO2 | Grain boundaries | −0.99 | 7 | HCOOH | 63% | 167 |
| SnO2 | Small metal particles | −1.1 | 16 | HCOOH | 84% | 90 |
| SnS2 | Residual S anions | −0.8 | 13.9 | HCOOH | 84.5% | 84 |
| Bi2O3 | Surface defects | −0.82 | 36 | HCOOH | >98% | 23 |
| Bi2O3 | Residual O anions | −0.9 | 8 | HCOOH | 91% | 25 |
| Bi2S3 | Surface defects | −0.75 | 5 | HCOOH | 84% | 25 |
| BiOCl | Shortened interlayer bond | −1.16 | 60 | HCOOH | 95% | 33 |
| BiOBr | Surface orientation | −0.9 | 55 | HCOOH | 95% | 152 |
| BiOI | Partial oxidized Bi | −1.0 | 40 | HCOOH | 90% | 222 |
| Bi-MOF | 2D nanostructure | −0.9 | 26.5 | HCOOH | 98.6% | 223 |
| Bi-MOF | Small metal particles | −0.97 | 5.4 | HCOOH | 95% | 155 |
| Bi2O2CO3 | 2D nanostructure/subcarbonate | −0.7 | 11 | HCOOH | 85% | 68 |
| Bi2O2CO3 | 2D nanostructure | −0.8 | 5 | HCOOH | 94% | 82 |
| Cu2O | Grain boundaries | −0.5 | 2.7 | CO | 40% | 16 |
| Cu2O | Higher Cu+ coverage | −1.6 | 6 | C2+ | 59% | 140 |
| C3H7OH | 8.7% | |||||
| Cu2O | Higher Cu+ coverage | −0.9 | 20 | C2H4 | 60% | 110 |
| Regenerated CuOx | Higher Cu+ coverage | −1.0 | 5 | C2+ | 76% | 24 |
| CH3CH2OH | 32% | |||||
| CuOx | Nanostructure | −0.7 | 5 | C2H6 | 37% | 165 |
| CuOx | Residual O anions | −1.0 | 35 | C2H4 | 45% | 157 |
| CH3CH2OH | 22% | |||||
| CuSx | S anions | −0.8 | 10.7 | HCOOH | 74% | 104 |
| CuSx | S anions | −0.95 | 32 | C2+ | 51% | 26 |
| C3H7OH | 8% | |||||
| Cu(OH)Cl | Higher Cu+ coverage | −1.2 | 22.2 | C2+ | 68% | 30 |
| C2H4 | 38% | |||||
| Cu(OH)F | F anions | −0.54 | 800(flow cell) | C2+ | 84% | 31 |
| C2H4 | 60% | |||||
| CuIx | Nanostructure | −0.735 | 20 | C2+ | 57.2% | 106 |
| C2H6 | 30% | |||||
| Cu3N | Higher Cu+ coverage | −0.95 | 22 | C2+ | 64% | 224 |
| Cu3N | Nanostructure | −1.0 | 18.5 | C2+ | 68% | 55 |
| Cu(B) | Higher Cu+ coverage | −1.1 | 70 | C2H4 | 52% | 38 |
| C2H5OH | 27% | |||||
| Cu(B) | Nanostructure | −1.1 | 18.2 | C2H4 | 58.4% | 95 |
| Cu-MOF | Undercoordinated sites | −1.07 | 263(flow cell) | C2H4 | 45% | 36 |
| Cu-MOF | Higher Cu+ coverage | −0.4 | 14 | CH3COO- | 48% | 153 |
| CH3CH2OH | 32% | |||||
In this review, we systematically analyze the performance and structural evolution of pre-catalysts of metal oxides, metal dichalcogenide, metal nitrite, metal halogens and other metal salts. The composition or structural factors that strongly influence the CO2RR performance are categorized into element residue, specific surface area increase, grain boundary, surface orientation, etc., and the mechanism of their influence on performance is explained separately. The stability of the sample, especially the evolution of the element residue and the special surface structure under long-term operation is also analyzed based on the existing data. This article provides guidance on the relationship between structure and performance for future research in the field of pre-catalysts.
2. Pre-catalyst for CO2RR: synthesis and characterization technique
With the in-depth characterization of catalysts based on various advanced techniques, it is widely accepted that most of the catalysts experienced a dynamic structural change during the reaction.4,39,40 Thus, the real active sites and the finial composition/structure of the catalysts should be carefully determined. One example is the development of “bi-functional catalysts” for electrochemical water splitting. The as-synthesized metal borides, phosphides, nitrides, sulfides and selenides have been found to exhibit superior activity both for OER and HER.4,39 However, a detailed investigation on these catalysts revealed that a structural/composition change is almost inevitable after a long-term operation. For OER, the as-prepared compounds tend to change to metal oxide/(oxy)hydroxide through the oxidation of the parent catalysts, which is driven by the thermodynamically potential according to the E–pH diagram. Similarly, metal phase is proposed to be the main composition of the derived catalysts after HER. The structural, morphological and chemical state changes during other catalytic processes such as CO2RR, NO oxidation, CO oxidation, oxygen reduction etc. are also significant due to the applied potential and complex reaction environment.40 The development of various advanced characterization techniques allows researchers to gain more in-depth insight into the catalyst change under electrochemical conditions.40–42 Another issue should be considered is the unintentional introduction of impurities in derived catalysts. For example, Fe impurities are easy to be incorporated in the OER electrocatalyst during the synthesis and/or OER process, leading to the controversial conclusions and incorrect performance merits collected by different groups.4 By carefully eliminate the Fe interference during synthesis and measurement, the role of Fe is elucidated and the real OER activity trends for certain catalysts can be re-constructed.43–45 These results implied that the important role of determining the real active site of CO2RR catalysts by monitoring the structural evolution and the unintentional impurities. Thus, this section summarized the synthesis method for CO2RR pre-catalyst and highlighted the specific function of various advanced characterization techniques, which is quite important for tackling the key challenges of the catalysts.There are many effective ways to control and create the pre-catalysts from metal or metal salts precursors. Thermal treatment is one of the easiest ways to create metal oxides by placing the precursor in a muffle or tube furnace in an air atmosphere at the specified temperature and time period.16,25,46–53 By using this method, the adventive element contaminations can be largely avoided as long as high-purity metal foil or precursor is used. Generally, several μm of copper oxide layer can be formed on copper foil after annealing at 500 °C for 12 h.16,48 SnO with thickness ranging from 5.4 to 16 nm can be realized from Sn nanoparticle by changing annealing temperature between 100–180 °C and annealing period between 6–18 h.52 PbO, Bi2O3, In2O3 and Ga2O3 can also be synthesized directly from Pb and Bi metal or In(acac)3 and gallium nitrate precursor.25,49–51 The thermal annealing method can also be applied to the synthesis of copper nitride pre-catalysts by the nitridation process of copper-based precursor under NH3 flow with heating. Besides planar copper nitride, nanostructured copper nitride could also be realized by using CuOx with desired nanostructure as nitridation precursor.54,55 Worth noting that thermal annealing method is not suitable to create thick oxide layer on Au and Ag metal. Au metal is difficult to be oxidized due to the high work function of 5.1 eV. Although Au2O3 has been observed as the oxidized state of Au, it is merely a metastable state at room temperature and will quickly decompose at T > 160 °C.56,57 The thermal oxidation of Ag foils is also not effective because the instability of silver oxide at temperatures above 200 °C.58 A mixture of metallic Ag (Ag(0)) and oxidized Ag was obtained after air annealing due to thermal instability of AgOx to Ag.59,60
Electrochemical oxidation at a highly positive voltage, on the other hand, can effectively oxidize Au(0) to Au(OH)3 or Au2O3, and oxidize Ag(0) to AgO or Ag2O, depending on the electrolyte identity and applied potential.18,20,61–63 For instance, Kanan and coworkers applied a periodic symmetric square-wave potential routine at 1 kHz in 0.5 M H2SO4 for 60 min on a piece of Au foil, resulting in the formation of thick (>1 μm) and amorphous Au oxide layers.18 Changing the electrolyte to neutral would result in Au(OH)3 as products of anodization.62 The potential anodization of Ag to AgOx was usually performed under a more alkaline solution, as Ma et al. reported the fabrication of Ag2O layers by applying symmetric 50 Hz square-wave pulsed potential on polycrystalline Ag foil in 0.2 M NaOH solutions.20 Zhou et al. reported that Ag started to get oxidized to form Ag2O in neutral electrolyte when the potential was increased to 0.6 V, and was further oxidized to AgO at higher potential,64 however, in an electrolyte with pH of 3, the anodization of Ag cannot form Ag oxides. Electrochemical oxidation is also a facile and common way to create nanostructured metal oxides. Cu2O layer with nanowire morphology can be formed by anodization of a Cu layer in 3 M KOH.65 Electrochemical oxidation by linear sweep voltammetry with high positive potential limits is also feasible to synthesize cubic Bi2O3 phase.66
The electrochemical oxidation method can also introduce other nonmetal element into metal lattice by anodic treatment in electrolyte with corresponding anions. For instance, an anodic potential of 2.6 V was reported to synthesize 4.9 μm Ag2CO3 layer on Ag foil in 3 min (ref. 67) and a direct current potential difference of 10 V was applied to synthesize white solid powder of Bi2O2CO3.68 Ag halide and CuS can be fabricated by electrochemical anodic treatment in corresponding halide and thiourea contained electrolyte, respectively,32,69–71 while copper halides compounds can even be formed on the surface of Cu during CO2RR in halide salts electrolytes.72–74 Solvothermal synthesis is a very common and facile method to synthesize various of metal compounds and one of its prominent features is to create nanostructure morphologies.23,26,30,31,34,75–88 Liu et al. reported the synthesis of ultrathin sub-2 nm SnO2 quantum wires from SnCl4 and oleylamine precursor.89 A two-step strategy was developed to first synthesize Sn compound with controllable morphology, such as SnS2 nanosheet, and then converted the Sn compound to SnOx by annealing.90,91 In the work of Sun and coworkers, by raising the reaction temperature from 230 to 260 °C, the morphology of the final copper nitride products from solvothermal synthesis changed from nanoparticle to nanocube and then from 25 nm nanocube to 10 nm nanocube.79 Hollow nanotubes composed of SnS (stannous sulfide) nanosheets was synthesized by the employment of MoO3 template in solvothermal process.27 Solvothermal methods were also developed to synthesize ZnO, B–CuO, bismuth sulphide, AgBiS2, Ag2S, InS and CdS, and nanostructure such as nanoneedles, nanorods, and nanoparticles can be realized.28,29,80,81,92–98
There are also other methods that can create metal compound pre-catalysts. For instance, since Cu is vulnerable to oxidation, chemical oxidation can be applied to form Cu(OH)2 nanotube/nanowire/microflower by oxidant of (Na2S2O8/K2S2O8),99–103 and even immersing Cu foil in ammonium polysulfide solution for 5 min can result in the formation of copper sulfide.104 The direct reaction between metal foil and solution are further employed to fabricate Bi2O2CO3, Cu-halide and Ag halide.105–107 Plasma treatments can create metal oxides layer on metal and even control the nanostructure morphology and surface roughness of Cu, Ag and Sn pre-catalysts.108–111 Electrochemical deposition is another common method to achieve metal oxides and was reported to fabricate Cu2O,75,112–114 SnOx,22 Cd(OH)2 nanosheets115 and ZnO98,116 with structure parameters can be controlled by pH value, precursor concentration and deposition temperature. Compared with the high-cost vacuum deposition methods (e.g., ion-beam sputtering, magnetron sputtering, thermal evaporation), solution-based synthesis methods are the most popular means to fabricate CO2RR catalysts with controlled morphology and nanostructures. However, it should be noted that the solution method is prone to introduce impurities in catalysts during synthesis and an intentionally purified process for the electrolyte is suggested to avoid contamination.
For the characterization techniques, normal X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), Scanning electron microscope (SEM), Transmission electron microscope (TEM), Atomic force microscope (AFM), have all been applied to measure the atomic and electronic structure of pre-catalysts and derived catalysts, however, to fully understand the real structure of catalysts during CO2RR, numerous advanced operando methods have been adopted to the CO2RR system. To examine the structural and morphological change of catalysts under catalytic process, in situ and operando SEM, AFM and TEM operated under a liquid or gas-phase condition was developed to monitor the dynamic transformations.40–42 Ambient pressure X-ray photoelectron spectrum (APXPS) can provide the electric structure information of catalysts surface for electrochemical CO2RR and the system is typically performed with electrochemical treatment and XPS in the same measurement chamber with ∼10 Torr of water vapor.117–121 After the electrochemical treatment, working electrodes were pulled out of the electrolyte into the XPS measurement position that was approximately several hundreds μm away from the analyzer entrance nozzle (Fig. 1d and e).
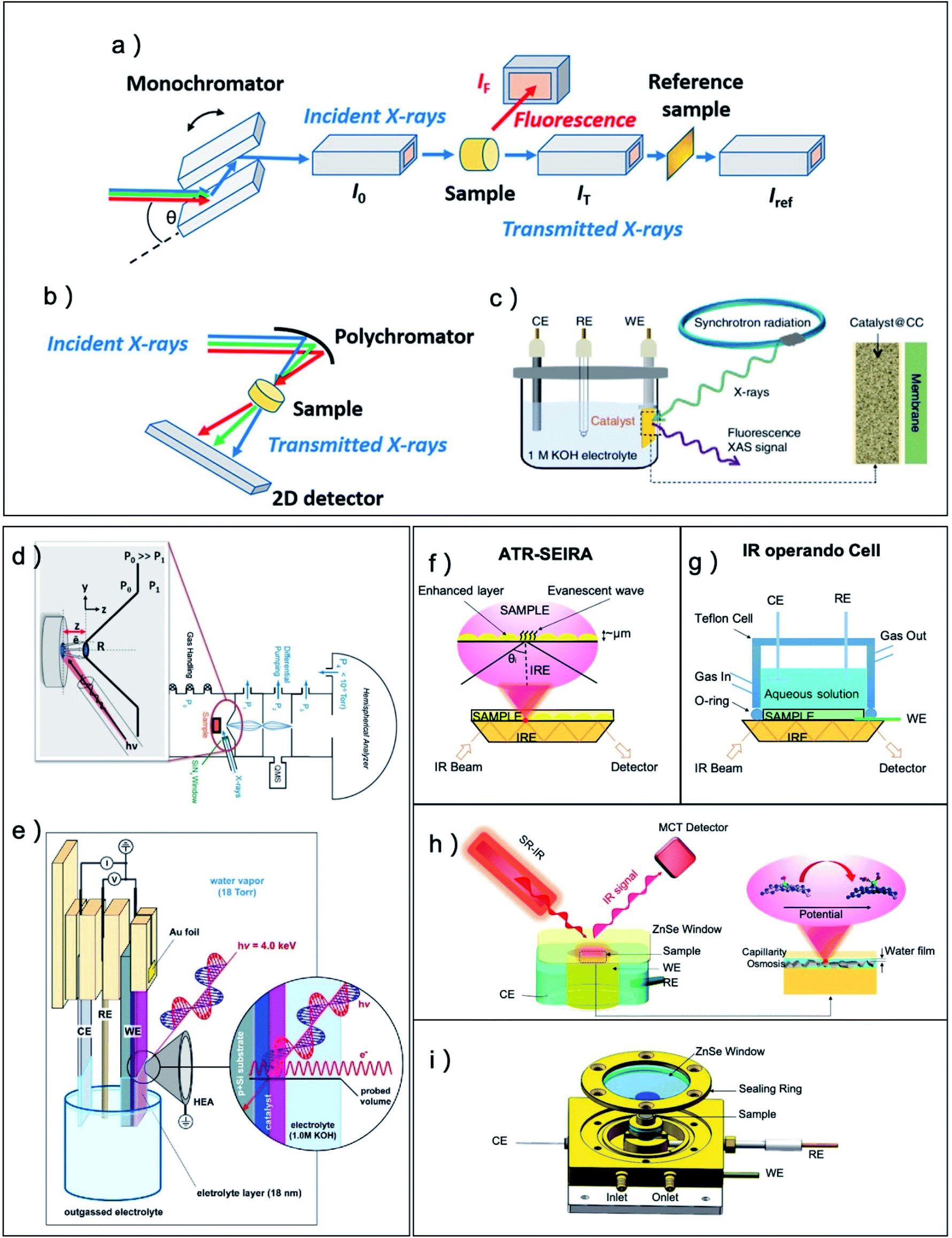 | ||
| Fig. 1 (a–c) Typical configurations for electrochemical operando XAFS. (a) Schematics of a common setup XAS measurements with monochromator front end. (b) Schematics of XAS measurement with energy-dispersive mode. (c) A typical operando electrochemical cell set-up for electrocatalysis. CE, counterelectrode; WE, working electrode; RE, reference electrode. (d and e) Typical configurations for electrochemical operando APXPS. (a and b) Adapted with permission from ref. 42 Copyright © 2021, American Chemical Society. (c) Adapted with permission from ref. 122 Copyright © 2019, Springer Nature. (d) A typical APXPS setup used in ALS endstation. (e) Schematic illustration of the reaction cell configuration used for operando electrochemical APXPS. The electrode is pulled out of the electrolyte and covered by micron meters of aqueous solution. (d) Adapted with permission from ref. 120 Copyright © 2013, Elsevier. (e) Adapted with permission from ref. 121 Copyright © 2017, American Chemical Society. (f) Schematic representations of attenuated-total-reflection surface-enhanced infrared absorption (ATR-SEIRA). (g) Schematic illustrations of operando ATR-IR cells for electrocatalytic reactions. Schematic illustrations of (h) operando SRIR methodology and (i) operando SRIR cell. (f–i) Adapted with permission from ref. 5 Copyright © 2020, Elsevier. | ||
X-ray absorption fine structure (XAFS) is a very powerful method for the operando measurement of electrochemical reaction because both incident light and outgoing light are X-ray and have a high transmission depth in water (Fig. 1a–c).42,122 XAFS are very sensitive to the local atomic and electric structure of element species in the catalysts from extended X-ray absorption fine structure and X-ray absorption near edge structure, and it can provide valuable information of catalytic metal centers for even amorphous sample. However, the XAFS method also has a high transmission depth in the sample and usually obtains the structure information of the whole bulk material. For CO2RR research, the incident mode is usually adopted to limit the detection at the surface layer.21,110Operando Raman is another method to probe the crystal structure evolution of pre-catalysts and is robust against the absorption of water.123 The operando Raman configuration is based on reflective mode which is similar to that of the normal IR that measures sample in an aqueous solution. To amplify the signal of sample, plasmonic substrate such as nanostructured Ag, Au, and Cu are usually utilized to enhance the electromagnetic field at electrode surface and multiply the resultant Raman signal by several orders of magnitude. Fortunately, Ag, Au, and Cu are the most common metal catalysts studied for CO2RR, which greatly simplify the design of surface-enhanced Raman spectroscopy in this area. Using surface-enhanced Raman spectroscopy, Han and coworkers successfully probed the difference of intermediate species on four commonly used Cu catalysts,124 and they also discovered that the CO adsorption configuration on Cu is different when changing the pH value of the electrolyte.125 For other metal catalysts that are non-plasmonic or with flat surface, a newly emerging technology of shell-isolated nanoparticle enhanced Raman spectroscopy (SHINERS) exhibit great potential to enable the amplification of Raman signal.124,126 In this method, plasmonic nanoparticles, which are enclosed within a shell composed of an insulating material, are placed in close proximity with a non-plasmonic catalyst to impart a similarly increased electromagnetic field to the material of interest.
The operando IR, on the other hand, can provide strong signal for polar groups such as C–O, O–H and C–H on the surface of the catalyst and helps to understand the reaction pathway of CO2RR on catalysts with different selectivity.5,71,127,128 The operando IR is very vulnerable to the absorption of water and usually adopts total reflection mode and the pre-catalyst sample is deposited on the surface of internal reflective element (Fig. 1f and g). The electromagnetic field of this reflected IR beam at the boundary can still extend into the sample medium and obtain the information of functional groups on the surface of the sample.5 Notably, when utilizing the synchrotron radiation (SR) light source with high brightness at a microzone, even external reflection mode IR technology can obtain sufficient signals of surface bonding groups, which can greatly simplify the experimental design for dilute catalytic site systems (Fig. 1h and i). The rational design of pre-catalysts for enhanced activity, selectivity and stability demands precise controlling of the initial composition, chemical state and nanostructure in the synthesis process. Meanwhile, the structural/chemical parameters of the catalysts should be carefully monitored under realistic operation conditions due to the pre-catalysts actively transform and sensitively respond to the electrochemical reaction environment. Thus, tremendous efforts have been conducted to correlate dynamic parameters change of the pre-catalysts to the activity trend that aims to reveal the nature of active sites in the derived catalysts.
3. The role of residual anion ions in metal–nonmetal compound derived catalysts during CO2RR
3.1. The existence of anion ions in derived catalysts
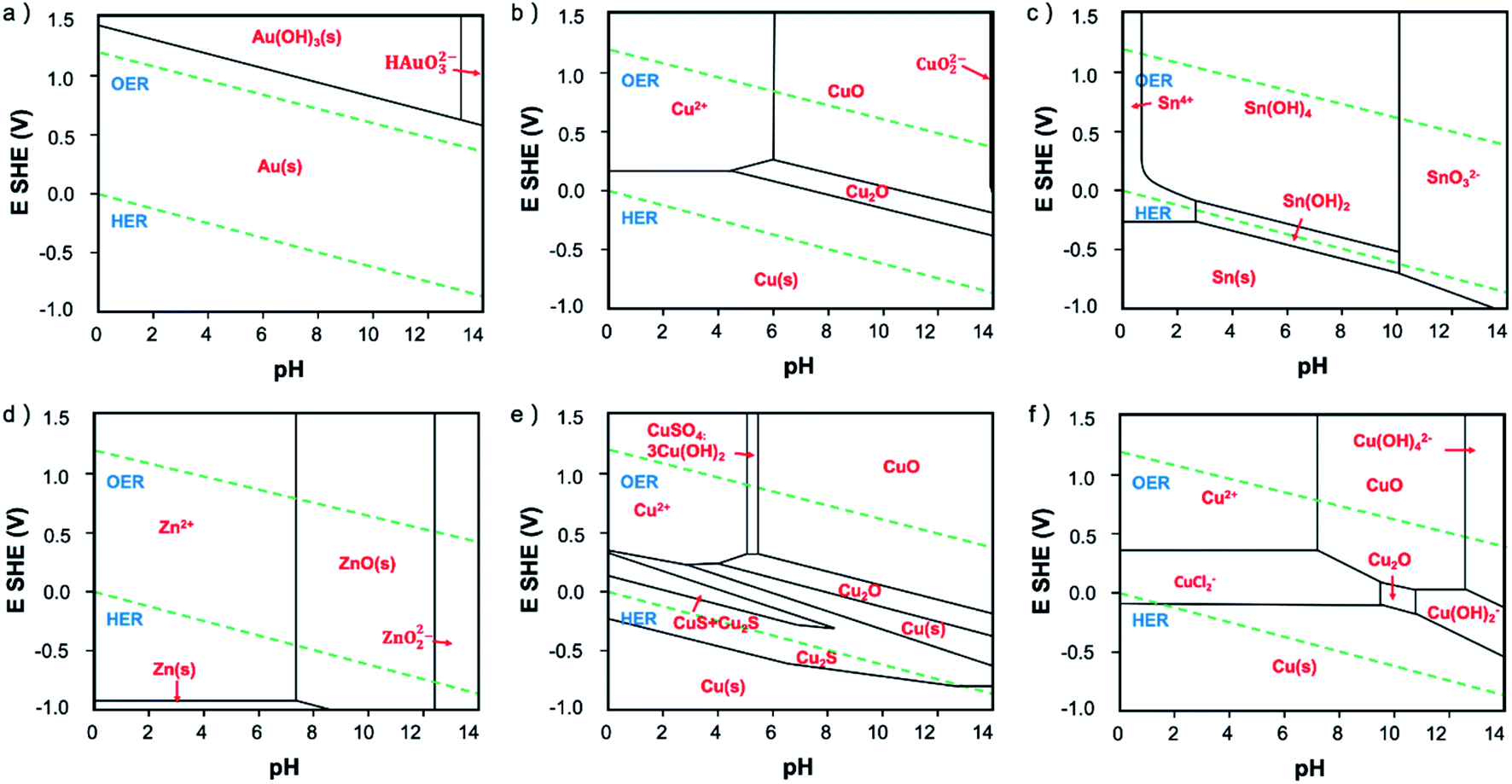 | ||
| Fig. 2 The Pourbaix diagram of (a) Au–H2O, (b) Cu–H2O, (c) Sn–H2O, (d) Zn–H2O, (e) Cu–S–H2O and (f) Cu–Cl–H2O at 25 °C. Au, Cu, Sn and Zn Pourbaix diagram are produced with a total concentration of [M(aq)]tot = 10−4 M. Cu–Cl–H2O Pourbaix diagram is produced with total concentration of [Cu(aq)]tot = 10−6 M, and [Cl(aq)]tot = 0.2 M. Cu–S–H2O Pourbaix diagram is produced with total concentration of [Cu(aq)]tot = 0.001 M and [S(aq)]tot = 0.0009 M. | ||
There are two main obstacles to find out what is the real state of oxygen in MOx pre-catalysts under CO2RR. First, the remnant concentration of O might be too low to be detected by bulk methods such as normal XRD.20,21 Second, the surface of some metal, such as Cu, are very sensitive and vulnerable to the oxidation of air. Thus, the clear signal of oxygen observed in XPS measurement may not really correspond to the remnant of oxygen on the catalyst surface.16,135,136 For instance, Lum et al. reported that there is no Cu2O signal for OD-Cu under CO2RR, however, once the potential was removed, Cu2O peaks began to appear again after 60 s.137 To this end, in situ detection techniques and high-sensitive surface methods are required to further investigate the real composition of the catalyst surface and its influence on CO2RR.
The most common in situ method to detect the oxygen in MOx is APXPS, which can provide clear signal for oxygen on surface or subsurface of catalysts.118 The quasi in situ oxygen K-edge electron energy-loss spectra (EELS)118 is also a powerful method to determine the state of O on surface. In situ X-ray absorption fine structure and near edge structure (XAFS; XANES) have a much higher investigation depth, but they can be still employed to provide surface information by incident mode.21,110 The results showed that the residual O in metal oxides under CO2RR is still strongly related to the SEP of M/Mn+. For metal with very positive SEP, such as Au (SEP: 1.002 V, all SEP are vs. RHE) and Ag (SEP: 0.7996 V), there is no signal of AuOx or AgOx in XRD after CO2RR.18,20 XPS and EDXS exhibited the expected peaks for Au(0) and no peaks related to an Au oxide can be found, indicating that reduction of Au2O3 was complete within the detection limits of these techniques.18 However, the monitoring of surface AgOx structure by operando XAFS with grazing incidence mode (0.1° angle), as shown in Fig. 3a–c, clearly showed that a mixture of both metallic and oxide composition is maintained on the surface of OD-Ag.21 The existence of stable O in OD-Ag was also confirmed by other methods such as Ag M4,5VV Auger signals.138 Cu is the metal that attracts the most attention in CO2RR and has a much negative SEP (0.3419 V) compared with Ag. Based on in situ ambient pressure XPS, quasi in situ oxygen K-edge electron energy-loss spectra (EELS), positron annihilation spectroscopy (PAS) and in situ XANES, a clear signal of O has been observed in many OD-Cu electrocatalyst research and the O signal is found to be relatively stable during CO2RR.118,139,140 This O signal is assumed to be subsurface oxygen in Cu lattice as no copper oxides can be observed in Cu 2p3/2 APXPS spectra (Fig. 3d and e). However, there is no unified opinion on the oxygen content, existence shape and stabilization time of O in OD-Cu.137 It is possible that the state of oxygen in OD-Cu is strongly related to the morphology of catalysts, electrolyte environment and reduction potential. Sn has a more negative SEP (−0.1375 V) compared with Cu; the concentration of O is much higher in OD-Sn and the phase of SnOx can be maintained during CO2RR with low overpotential. The operando Raman spectroscopic survey performed by Broekmann and coworkers clearly exhibited that the practical (kinetic) stability region of SnO and SnO2 well exceeds the thermodynamic stability window of Pourbaix diagram (hydrous Sn(IV) oxide exist for V > 0.1 V; hydrous Sn(II) oxide exist for V = −0.1–0.1 V; Fig. 2 and 4).123
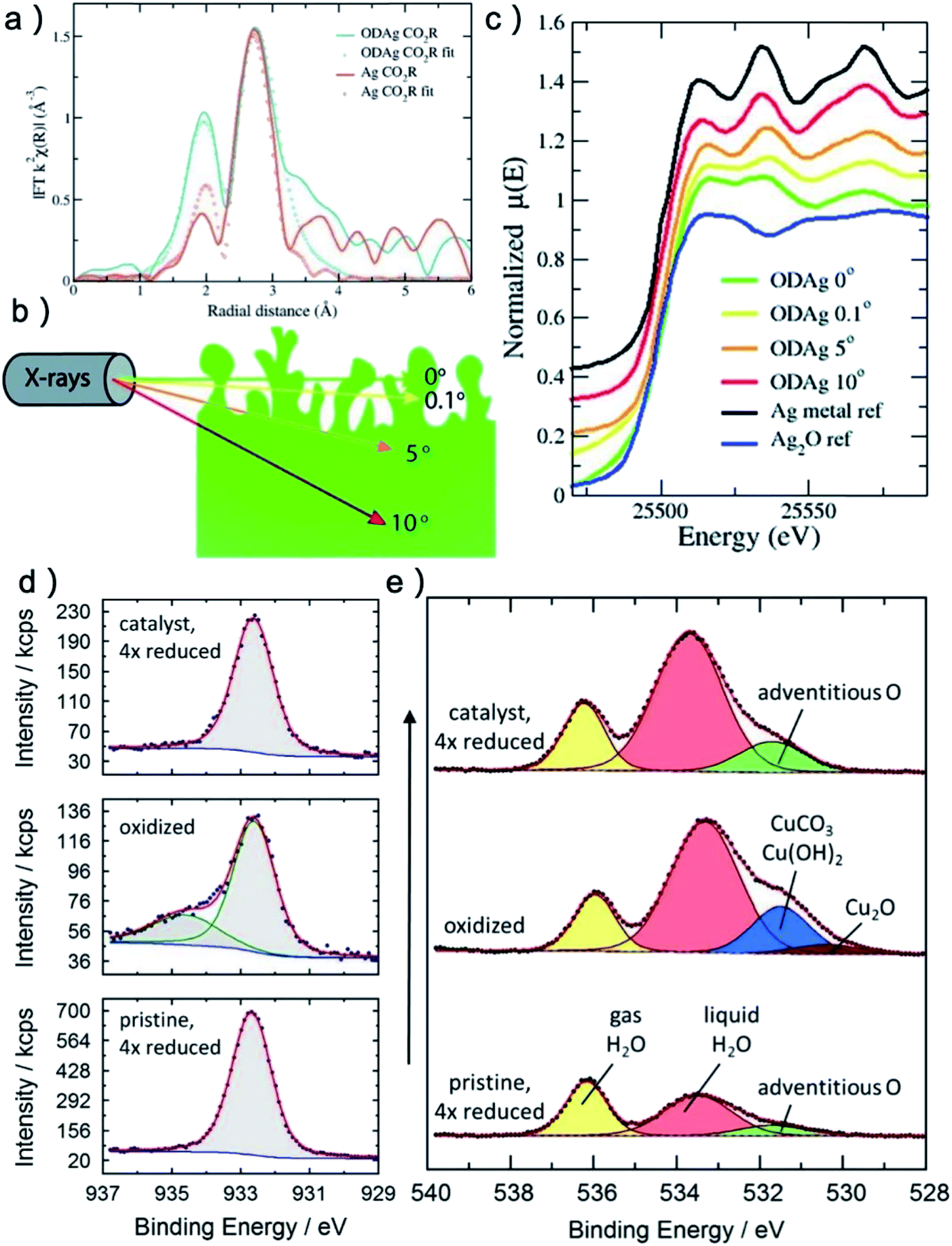 | ||
| Fig. 3 (a) k2-Weighted Fourier transformed EXAFS fluorescence spectra of a Ag catalyst and an oxide-derived Ag catalyst. (b) Schematic illustrations of operando grazing incidence X-rays absorption measurement on OD-Ag. (c) Ex situ normalised XANES spectra of OD-Ag samples. With smaller incident angle, more Ag–O signal can be detected. (a–c) Adapted with permission from ref. 21 Copyright © 2019, Royal Society of Chemistry. (d) In situ Cu 2p3/2 APXPS spectra of oxidized and reduced sample. The oxidized spectrum shows an additional Cu(II) compound while the reduced sample shows no Cu(II). (e) In situ O 1s APXPS spectra of pristine sample, oxidized sample and reduced sample. The reduced sample contains significantly more adventitious oxygen (green) than the pristine sample before oxidation. (d and e) Adapted with permission from ref. 118 Copyright © 2019, American Chemical Society. | ||
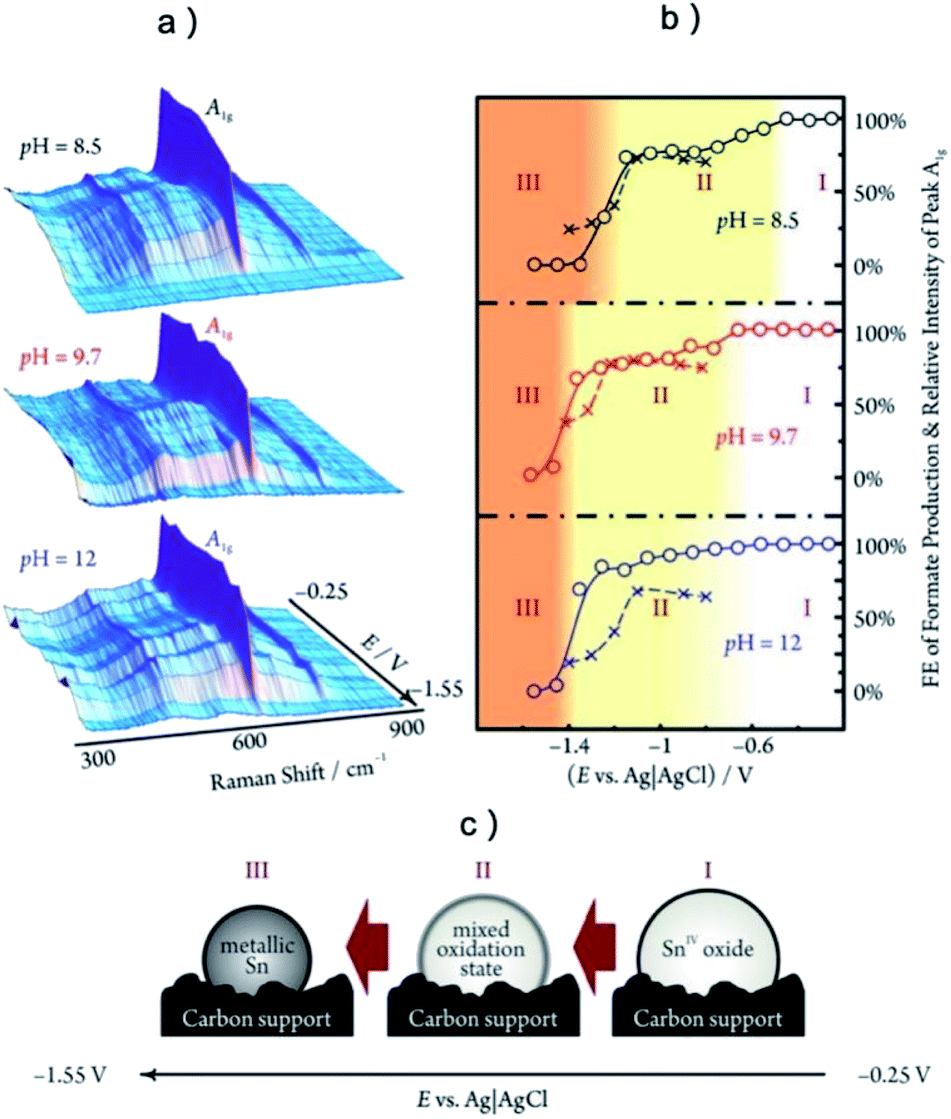 | ||
| Fig. 4 (a) The operando Raman studies of SnO2 during CO2RR at different potential and pH. (b) The relative intensities of the Sn(IV)-related A1g Raman peaks (○, solid line) and the faradaic efficiencies of formate production (×, dashed line) as a function of electrode potential. (c) In region (I), SnO2 remain fully oxidized; in region (II) the SnO2 is partially reduced to SnO; in region (III) SnO2 is fully reduced to metallic Sn. The highest selectivity for formate production existed in a potential range where the SnO2 phase is metastable. (a–c) Adapted with permission from ref. 123 Copyright © 2015, American Chemical Society. | ||
There are also other metal oxides pre-catalysts, such as PbOx (SEP: −0.1262 V), InOx (SEP: −0.34 V), BiOx (SEP: 0.32 V), CdOx (SEP: −0.4030 V), GaOx (SEP: −0.549 V), ZnOx (SEP: −0.7618 V), however, the O state in these pre-catalysts have not been studied by operando surface detection methods. Based on the ex situ measurement or in situ bulk XAFS, the final phase of PbOx, CdOx, GaOx and BiOx after CO2RR are mostly metal phase, however, the residual O in OD-metal can not be excluded.23,49,51,115 Zn is the most active metal among these CO2RR catalysts and a large amount of the oxygen can be maintained in the metal lattice even after CO2RR.116 In most research works, the final crystal phase of OD-Zn after CO2RR is pure metal Zn,97,98 however, Zeng and coworkers reported that ZnO phase could be maintained in ZnO nanosheets,96 indicating that the stability of ZnO is strongly related to the sample morphology and experimental conditions.
The state of oxygen in mixed metal oxides pre-catalysts are more complex because the interaction between metal atoms may stabilize the M–O bond in the pre-catalysts. For instance, the Cu ions in mixed metal oxide of CuInO2 are found to be much more difficult to reduce than that in Cu2O.141 Sn2+ ions were found to be stabilized by many binary structure, such as Bi–SnO,142 Cu–SnOx,143 SnOx/AgOx,144 which are probably due to the activity difference of these two metals that result in the rearranging of the free electrons.
Generally, the stability of metal dichalcogenides under CO2RR is similar to metal oxides. After CO2RR, the XRD detection confirms the disappear of metal dichalcogenides phases for Cu, Au, Bi, Pb, Sn and Ag dichalcogenides pre-catalysts.28,29,146 However, there is clear signal of S for CuS, SnS2 and Ag–Bi–S pre-catalysts after CO2RR in XPS or energy-dispersive X-ray spectroscopy (EDS),71,104,146 indicating that S may not be fully cleaved. For instance, Shao-horn and coworkers reported that for electrodeposited CuxS, only Cu metal and a tiny amount of Cu2O can be observed after CO2RR, however, the S concentration is still one-tenth compared with the as-prepared CuxS,71 as shown in Fig. 5a–c. As a result, the oxidation state of Cu in SD-Cu is much higher than that of the Cu foil. Worth noting that there are reports of MoS2, WS2, MoSe2, WSe2, TiS2, ZnS and S-doped In as efficient CO2RR catalysts,80,147–151 which did not exhibit large composition or morphology change during CO2RR and may be recognized as a new group of catalysts rather than pre-catalysts of metal. However, considering most of the metal–nonmetal catalysts are not really stable under CO2RR conditions, more research work on these catalysts is recommended to clarify the structural evolution behavior under high operation current (>500 mA cm−2) and long operation time (>100 h). For example, the research of Cu–N, Cu–B and Ag–P pre-catalysts revealed that they were also vulnerable to CO2RR,34,35,95 however, the concentration of N, B and P are much higher compared with O in Cu–O and Ag–O after long-term operation.
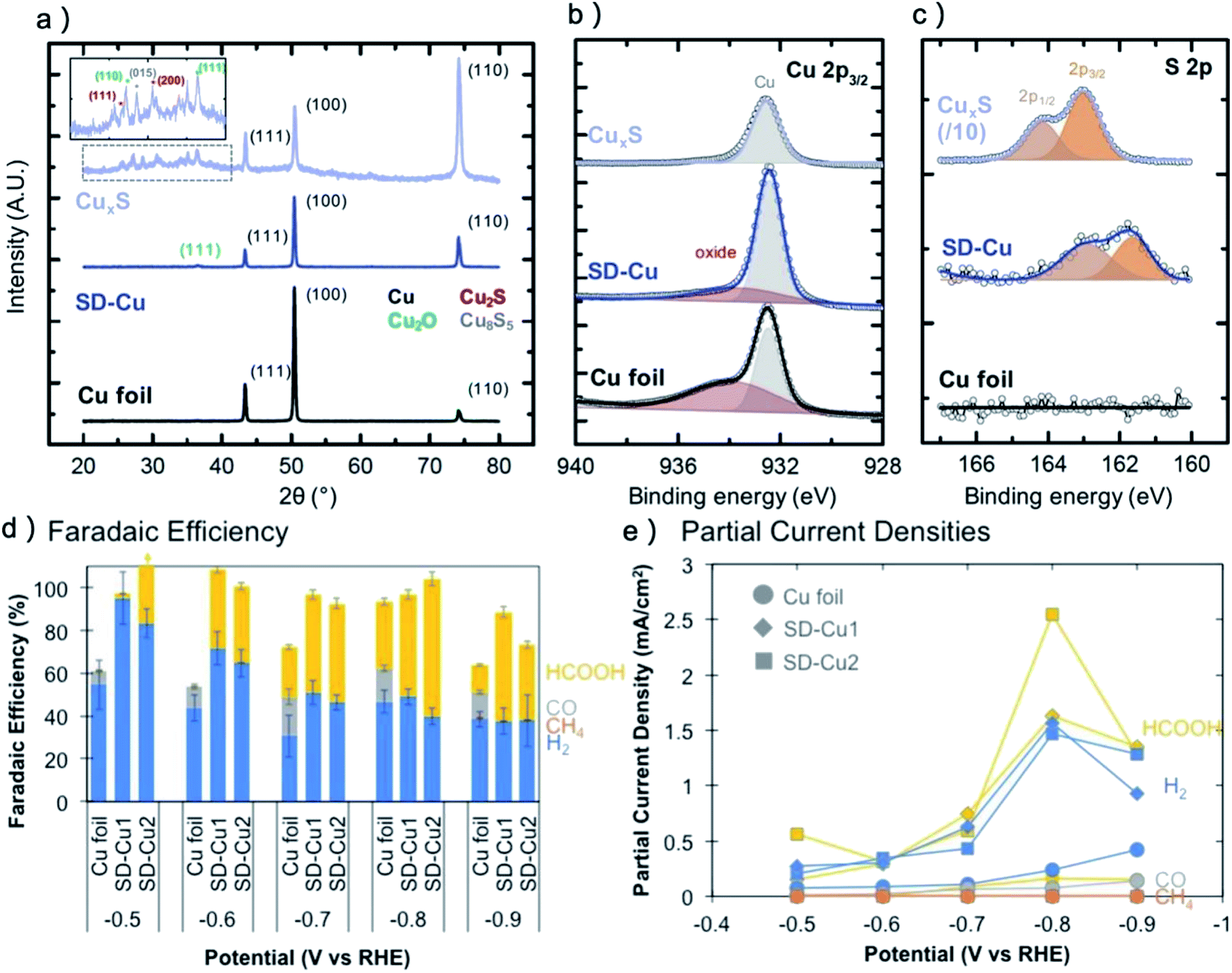 | ||
| Fig. 5 (a) XRD, (b) Cu 2p3/2 XPS, (c) S 2p XPS of CuxS, SD-Cu and reference Cu foil. The sulfur XPS curve of CuxS was reduced for 10-times for comparison. (d) Faradaic efficiencies for all detected products at different potential. (e) Partial current densities for hydrogen and formate. (a–e) Adapted with permission from ref. 71 Copyright © 2018, American Chemical Society. | ||
The halide of Cu, Ag and Bi were generally converted to pure metal phase under CO2RR based on XRD, while clear signal of halide on the surface of reduced catalysts can be observed in XPS or EDX.31,32,69,107,152 For instance, Cu-halide pre-catalysts of Cu(OH)F synthesized by solvothermal method31 exhibited initial surface halogen contents of 6 mol% which remained unchanged at ∼6 mol% in the first hour and decreased progressively to ∼4 mol% after 40 h. The high stability of surface halogen ions is because of the strong bonding of halogen to the electrode surface. In Polyansky's study,32 the bonding between surface Cl− species and the Ag surface atoms is very strong with a high desorption temperature of 700 K. Li and coworkers observed the formation of cuprous halides with well-controlled tetrahedral shapes by simply immersing Cu foil in the aqueous solution of corresponding halide salts (NaCl, KBr, or KI) for 5 min.106
The research works for other metal–nonmetal pre-catalysts, such as MOF, metal salts, V group element compound, are much less compared with the aforementioned categories and mostly focused on Cu, Ag and Bi based pre-catalysts. Based on the limited data, the MOF of Cu, Ag and Bi are all unstable under CO2RR,36,153–155 resulting in the partially reduced MOF-derived catalysts and the formation of small metal clusters. Metal salts such as Ag3PO4, Ag2CO3 and Bi2O2CO3 on the other hand, were observed to be fully reduced during CO2RR based on XRD and XPS.67,82,156
Based on these results, it is clear that the stability of nonmetal elements under CO2RR is related to SEP, bonding strength between cation and anion and the solubility of anion in the electrolyte. Although the leaching of anions from the parent catalysts into electrolytes is significant during CO2RR, the residual anions still play an important role in affecting the electronic structure of the catalysts and determining the adsorption properties of intermediates. Thus, the role of anions existed in derived catalysts should be clarified to guide the design of highly efficient CO2RR catalysts.
3.2. The influence of residual anion ions on CO2RR activity
The pre-catalysts derived metal catalysts usually have various structural parameter changes, thus it is difficult to find direct evidence in the spectrum about the mechanism of how residual nonmetal element alone impact CO2RR. However, an obvious relationship between the CO2RR activity of derived catalysts and the concentration of nonmetal element or M+ ions have been observed, indicating that the existence of nonmetal element could raise the valence state of metal to impact the CO2RR performance. For instance, Gao et al. reported that Cu nanocube sample treated with O2 plasmon maintain a much higher O content (30 at% vs. 14 at%) and C2+ faradaic efficiency (73% vs. 40%) compared with untreated Cu nanocubes.157 KCl was also found to suppress the reduction of Cu2O and maintain a higher Cu+ content compared with normally reduced Cu2O,140 resulting in an impressive 8.7% of FE C3H7OH. This phenomenon also held for Ag–O pre-catalysts as Smith group reported that the faradaic efficiency for CO is proportional with the O/Ag ratio extracted by EXAFS fitting, while there is no clear relationship between CO2RR performance and surface area or Ag–O bonds numbers.21 The situation for SnOx pre-catalysts is slightly different, because partial oxidized SnOx exhibited a better activity compared with both Sn and SnO2, as shown in Fig. 4b.22,52,123 Kanan group reported that catalyst with a SnOx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn0 ratio of 93:7 showed the best HCOOH selectivity of 80%.22
:Sn0 ratio of 93:7 showed the best HCOOH selectivity of 80%.22
The research of other metal–nonmetal pre-catalysts showed that many nonmetal elements can be more stable compared with O under CO2RR and can influence the CO2RR activity by raising the valence value of the metal center. Sargent and coworkers synthesized Cu(B) samples with porous dendritic morphology.38 From XANES, the average oxidation state of copper is increased from 0.25 to 0.78 when the boron concentration increased from 1.3% to 2.2%, and the oxidation state is stable over the course of CO2RR (Fig. 6a–c). A volcano relationship between the oxidation state of copper and the FE of C2 products can be observed and the highest FE of 79% was achieved when Cu's valence value is +0.35, which is much higher than that of the pristine copper (29%) and OD-Cu (37%). The research of Cu–N also showed apparent residue of N after CO2RR which greatly boost the C2 selectivity.34,79 Especially, the investigation of Cu-on-Cu3N catalyst by in situ XAS found that the reducing of Cu3N was alleviated after the initial 60 min, while Cu2O was fully reduced after 1 h, as shown in Fig. 6d–g.34 The stronger interaction between N and Cu on Cu3N pre-catalysts results in a 6.3-fold and 40-fold enhancement in the ratio of C2+/CH4 compared to Cu-on-Cu2O and pure Cu catalysts, respectively. Specifically, the FE for C2H4, C2H5OH, and C3H7OH are 39 ± 2%, 19 ± 1%, and 6 ± 1%, respectively, at −0.95 V.
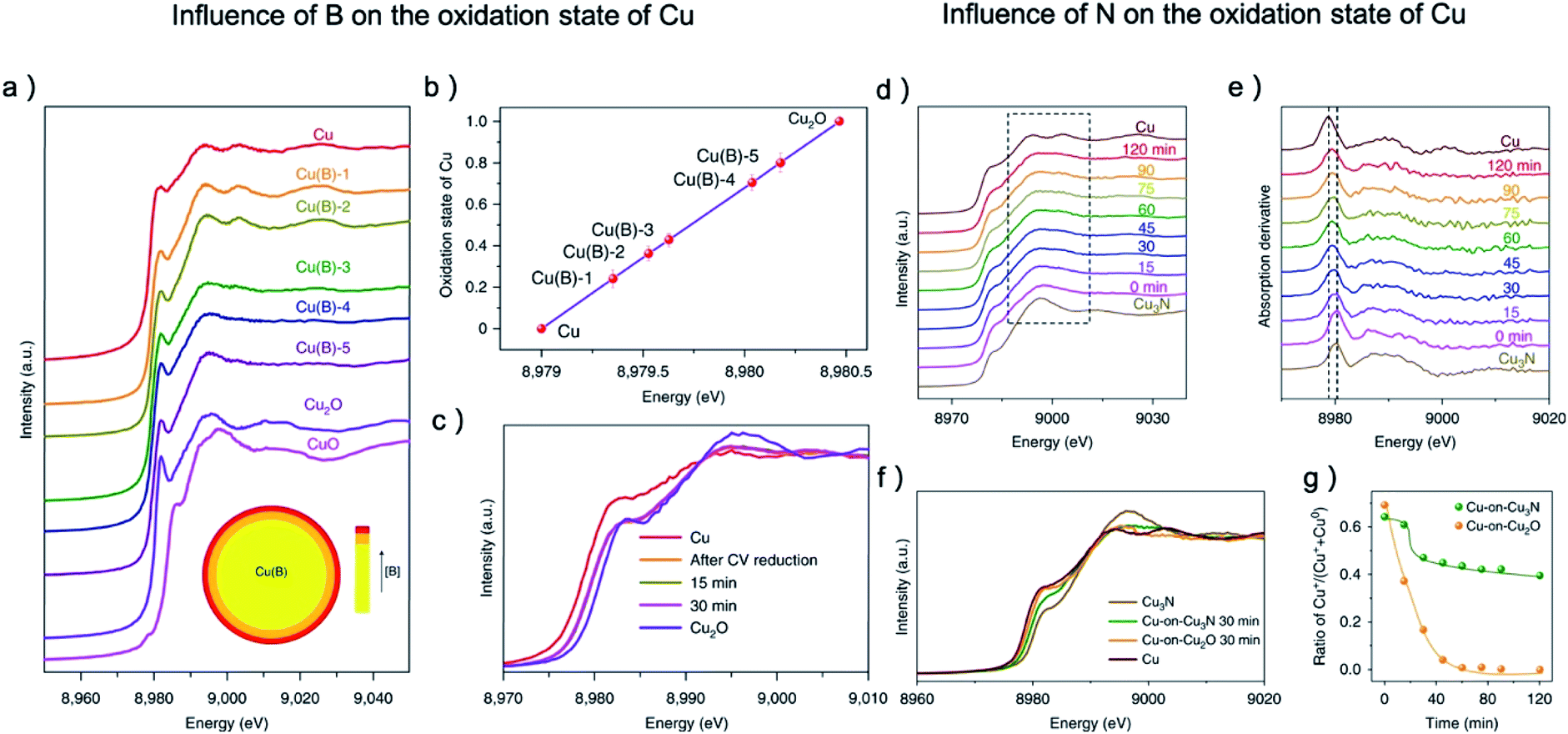 | ||
| Fig. 6 (a) The copper K-edge XANES spectra of Cu(B) samples after being electrochemically reduced, with CuO and Cu2O as reference. (b) The oxidation state of reduced Cu(B) samples calculated from copper K-edge XANES spectra, (c) the copper K-edge XANES spectra of Cu(B) samples after CV reduction, 15 min later and 30 min later in comparison to pristine copper and Cu2O. (d) The copper K-edge XANES spectra and (e) their first derivatives of the Cu-on-Cu3N catalyst as function of reaction time at −0.95 V. The reducing of Cu3N was alleviated after the initial 60 min. (f) In situ Cu K-edge spectra during the initial 30 min on Cu-on-Cu3N and Cu-on-Cu2O. (g) Ratio of Cu+ relative to total Cu for Cu-on-Cu3N and Cu-on-Cu2O during CO2RR at −0.95 V. The reducing of Cu2O was much faster than that of Cu3N. (a–c) Adapted with permission from ref. 38 Copyright © 2018, Springer Nature. (d–g) Adapted with permission from ref. 34 Copyright © 2019, Springer Nature. | ||
The research of metal halide pre-catalysts further revealed that the electronegativity of the nonmetal element is crucial for the concentration of Cu+ in the final derived catalyst. Sargent group synthesized Cu(OH)F, Cu2(OH)3Cl, Cu2(OH)3Br and CuI by solvothermal method.30,31 They found that the average oxidation states of copper in the X–Cu catalysts is proportional with the electronegativity of the halogen,31 as shown in Fig. 7d. The results revealed that the onset potential of C2H4 decreases significantly with the increase of electronegativity of halide ions. The best performance was achieved over the F–Cu catalyst with C2+ FEs of 85.8% at 1600 mA cm−2, as shown in Fig. 7e, and a large amount of surface-bound CHO species, a key intermediate for C–C coupling, can be observed on F–Cu catalyst in comparison to normal copper by in situ ATR-FTIRS (Fig. 7b and c). Notably, the surface area normalized C2+ formation rates also increased in the sequence of Cu < I–Cu < Br–Cu < Cl–Cu < F–Cu, indicating this is an intrinsic difference of CO2RR activity.
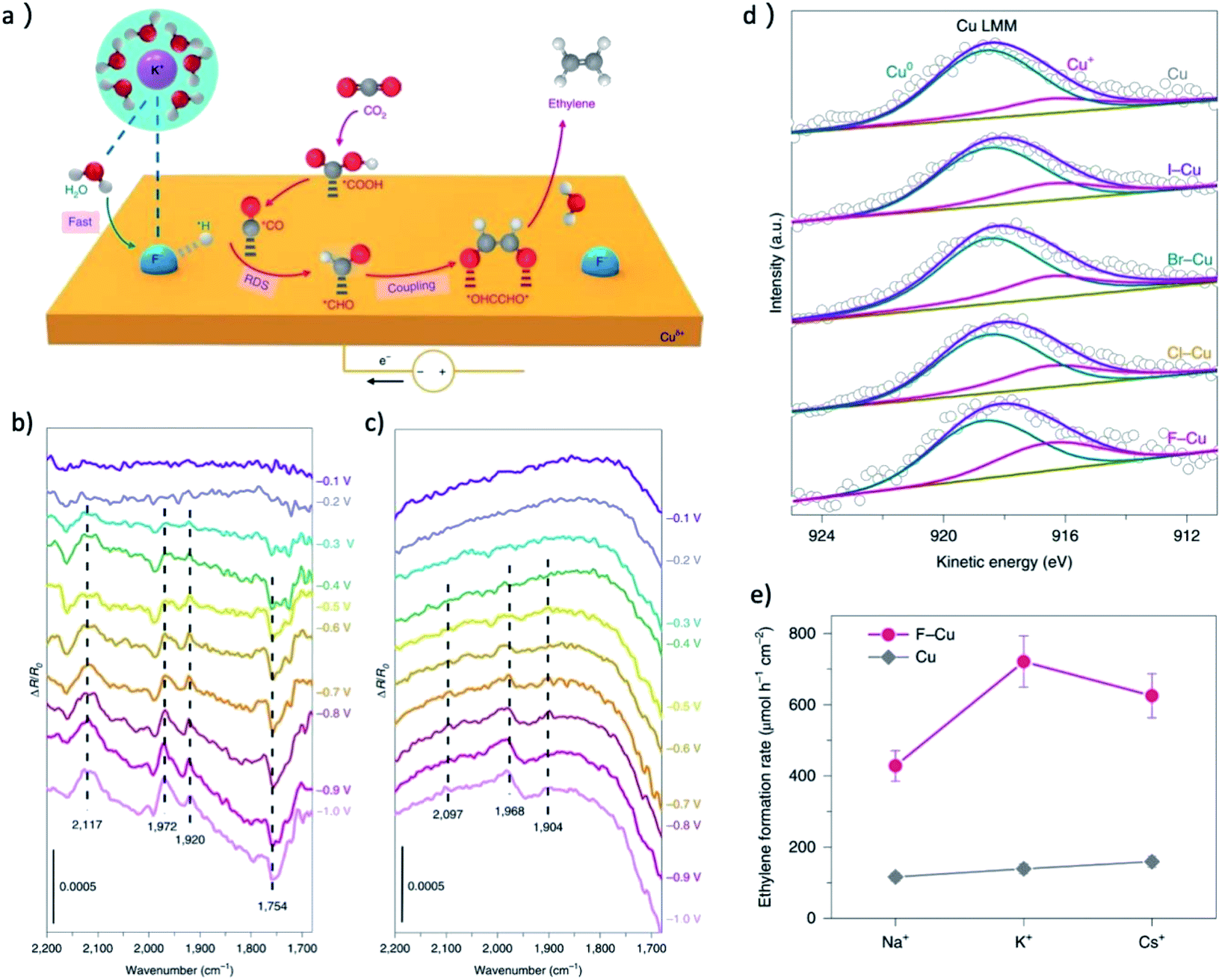 | ||
| Fig. 7 (a) A proposed reaction mechanism for the enhancement of CO2RR to C2H4 on F–Cu. Purple, potassium; blue, fluorine; red, oxygen; grey, carbon; white, hydrogen. (b) In situ ATR-FTIRS recorded at different applied potentials for the F–Cu catalyst in 0.1 M KHCO3 electrolyte. (c) In situ ATR-FTIRS recorded at different applied potentials for the copper catalyst in 0.1 M KHCO3 electrolyte. The band at around 1754 cm−1 could be ascribed to the surface-bound CHO species, a key intermediate for C–C coupling. (d) Copper LMM Auger spectra of the X–Cu catalysts. The concentration of Cu+ was observed to increase with with the electronegativity of the halogen. (e) The formation rate of CO2RR to C2H4 at −0.6 V over F–Cu and Cu in different electrolyte. Adapted with permission from ref. 31 Copyright © 2020, Springer Nature. | ||
The nature of how nonmetal residues influence the performance of metal catalysts is also studied by DFT calculations, which indeed show that the residual nonmetal can benefit the CO2RR on the derived catalyst. Most of the DFT calculations were focusing on the local structure of O in Cu catalysts and its influence on the CO2RR performance. Based on DFT calculations, albeit subsurface oxygen (Osb) is not stable below the Cu surface in the slab model, it is stable below facets of a manually “reduced” Cu nanocube model, which is consistent with the experiment.158 Such disordered structure increases the stability of subsurface oxygen in it. The presence of Osb enhances the adsorption energy of CO on Cu(100), and in turn increase the probability of CO dimerization, which is a rate-determining step toward the production of ethylene. Gu et al. reported the oxygen vacancy-rich CuOx surfaces can provide strong binding affinities to the intermediates of *CO and *COH, but weak affinity to *CH2, thus leading to efficient formation of C2H4.159 Other theoretical calculations claimed that the hydroxy group on the surface of OD-Cu would strongly influence the selectivity of CO2RR.160,161 Electronic structure analysis indicates that the charge transfer from hydroxy groups to coordination-unsaturated Cu sites stabilizes surface-adsorbed COOH*, which is a key intermediate during the CO2RR. For other nonmetal elements such as B, F and S, DFT calculations also suggest that they are quite stable in the subsurface sites and could introduce a strong impact on the adsorption of CO on metal.31,38,146 Notably, Qiao and coworkers systematically calculated the Cu–X (X = B, N, P, S, Cl, Br, or I) catalysts and found that the electronegativity difference between the doping element and oxygen strongly impacted the O affinities of the dopant atom site and Cu site.162 As a result, the ethane selectivity of all Cu–X catalysts were enhanced while the ethanol selectivity were generally suppressed. Cu–X catalysts doped with strong O affinity atoms, such as B, P, N, S, could favor the ethylene pathway due to the bonding between O and X. The influence of nonmetal element on the CO2RR catalytic activity of metal center can also be revealed by the study of metal–organic compound catalysts, which have a clear coordination structure of M–X. For instance, Strasser and coworkers studied metal- and nitrogen-doped porous carbons and discovered that the coordination environment of M–Nx determined the binding energy of *H and *CO on metal center, which in turn decided the selectivity of CO2RR.163
For most of the pre-catalysts, the final derived catalyst generally could suppress the hydrogen evolution, promote the selectivity of the major products and increase the current density, as shown in Table 1 for the CO2RR performance of typical pre-catalysts. However, the selectivity change of CuS is dramatically different compared with that of CuOx as many research works reported CuS have a strong trend to produce HCOOH.71,78,104 In these reports, HCOOH accounted for more than 60% of the total FE and was the only carbon-based product (Fig. 5d and e). The SEIRAS result showed that there is a strong adsorbed CO layer on CuS, unlike in the Cu foil case,71 which might block the CO2-to-CO reaction pathway. DFT calculation further confirmed that additive S would result in strong CO* adsorption and enhance the CO* coverage up to nearly 4 times higher than on a clean Cu surface.164 A synergistic effect between residual S and CO* is the reason for the blocking of other reaction pathways except for HCOOH production. Although a series of research provide repeatable results that CuS pre-catalysts can only produce HCOOH, Sargent group reported that a Cu2S catalyst with abundant vacancies on the surface had faradaic efficiency for C3H7OH and C2H5OH of 8% and 15%,26 indicating that more research is needed to understand the unique situation of Cu–S pre-catalysts.
4. The enhancement of surface area on derived catalysts
The oxidation–reduction process of pre-catalysts was reported to enhance the surface area of final metal catalysts compared with untreated metal catalysts. This can be attributed to the re-construction of catalyst surface and the release of anions during the reduction of M+ to M that leads to the formation of vacancies in the lattice. Compared with the initial planar metal film or untreated metal nanoparticles, several to several hundred times improvements in electrochemically active surface area (ECSA) can be achieved on derived catalysts after CO2RR.16,28,46–48,54,55,66–68,83,87,90,91,99,105,110,155,156,165–169 For instance, in Kanan's report, the surface area of OD-Cu that prepared by thermal annealing and electrochemical reduction is 480 times higher than that of a polycrystalline Cu.16 The partial reduction of SnO2 lead to the formation of small Sn nanoparticles on the surface of nanostructured SnO2, resulting in a large surface area of the porous nanosheet SnOx of 93.6 m2 g−1.90,91 However, in most of the research works of pre-catalysts, the improvement of CO2RR performance can not be explained by the increase of surface area alone due to the significant enhancement of CO2RR at low overpotential and the suppression of hydrogen evolution. For OD-Au reported by Kanan, the roughness factor of 72 can be realized for the resulted agglomerated Au NPs with particle sizes of ∼20–40 nm, which is significantly smaller than the differences in CO2RR current density over the course of electrolysis (500 times difference at −0.4 V).18 Zhou and coworkers showed that the OD-Ag leads to a 5 times larger ECSA enhancement but 21 times increase of CO2RR current density at 0.5 V overpotential compared to pristine Ag.64 The situation is more complex for Cu based pre-catalysts because in many reports the normalized current of CO2RR using ECSA is even less for OD-Cu compared with un-oxidized copper foil,165,170 an optimal roughness and ECSA exists for CO2RR performance indicating that roughness is not the only reason for improved CO2RR.48,114,165One explanation of selectivity and activity enhancement at low overpotential for high roughness catalysts is that they have a strong impact on the solution environment near the electrode surface. The high surface area can benefit the catalytic current at low overpotential but hinder the replenish of consumed proton and CO2 from bulk solution. The high local pH environment could greatly suppress the HER and favor CO2RR. This pH-induced performance improvement is evidenced by increasing the thickness of the porous Au or Ag film, resulting in a 10–30 times increase of CO2RR/HER ratio and >90% CO selectivity.171–173 A finite element numerical model was also applied to establish an accurate 3D geometrical representation of an ordered meso-structured Ag electrode and revealed that roughness factor is crucial to determine the electrolyte composition and pH value near catalyst surface.174 A substantial build-up of OH− and CO32− was observed in the lower part of the film, as the mass transport rate could not keep up with the OH− evolution rate, thus the HER is suppressed and CO2RR is more favored. On Cu catalysts, a high pH value is beneficial to increase the competitiveness of the series of reaction paths of C–C bond coupling, thereby increasing the selectivity of the C2 products.15,175,176 Interestingly, a unique pathway in CO2RR towards C2H6 were observed on many high surface area OD-Cu catalysts with highest FE of 37%, which was seldom observed for Cu foil.113,165 This can be explained by the temporary trapping of C2H4 in the nanoporous structure, resulting in the sequential reduction of C2H4 to C2H6.
The morphology and surface area of the derived catalysts can be controlled by the reduction current, potential and choice of electrolyte. Oh and coworkers reported that pore-like Au nanostructures are produced when thin nanoporous Au(OH)3 is reduced, while pillar-like Au nanostructures are formed by a faster reduction of thicker nanoporous Au(OH)3.62 The origin of these two different morphologies is associated with the electric-field-assisted transport of Au3+ at the Au(OH)3/Au interface. When the induced electric field is high on the tip of Au nanostructure, pillar-like nanostructure is formed. Instead, quasi-isotropic growth at a low electric field leads to the formation of pore-like Au nanostructures. The surface area of electrochemical synthesized OD-Ag can be tuned by changing the scanning speed of CV.177 With higher scanning speed, the particle size of Ag samples would be smaller with high roughness factor. Lee reported that reduce the indium tin oxide nanobranches (ITO BRs) with lower current density from −1.1 mA cm−2 to −0.48 mA cm−2 can provide much smaller metal particles with average particle size decrease from 232 nm to 65 nm.178 Dai and coworkers found that the particle size and roughness of halide-derived Ag can be finely tuned by changing the electrolyte and reducing the current.70 When ethanol is used as electrolyte, the solubility of AgCl is decreased by two magnitudes compared with an aqueous solution. The mobility of Ag+ is restrained, resulted in a much smaller particles size of halide-derived Ag (from 400 nm to 100 nm). Increasing the reducing current could also hinder the reorganization of Ag and further reduce the particle size to 30–50 nm.
The synthesis of pre-catalyst with high surface area can further enhance the roughness of the final derived catalysts and even control the final morphology of derived catalysts.179 Many kinds of nanostructured CuOx and mixed metal oxides were intentionally designed to form nano/microstructured Cu catalysts with high ECSA, resulting in a CO2RR current much higher than that of the planar Cu counterparts.16,47,180,181 Graphene-like ultrathin nanosheet morphology of Bi (Bi-ene) was successfully synthesized by Cao and coworkers from reducing of ultrathin Bi-MOLs.37 The thickness of Bi-ene is ranging from 1.28 to 1.45 nm. The formate formation on Bi-ene starts at −0.58 V and reach near 100% selectivity between −0.83 V and −1.18 V. Similarly, special dendritic or multiple-scale-porous nanostructured Cu and Ag catalysts can be derived from MOF with hollow, nanowires and nanoboxes morphologies.145,153,182 Most interestingly, the halide ions in electrolyte can be employed to react with Cu and tune the morphology of pre-catalyst and derived catalysts.72–74,106 Li and coworkers found that by immersing Cu foil in the aqueous solution of corresponding halide salts (NaCl, KBr, or KI) for 5 min, cuprous halides with well-controlled tetrahedral shapes can be formed, as shown in Fig. 8a–g.106 Specifically, the degree of truncation at both edges and vertices become lower and sharper from Cl− to I− electrolyte, and uniform cubes with a size of around 800 nm, dendrite-like nanostructures and bundles of nanofibers are formed after electroreduction, respectively. These special morphologies may contribute to the high C2H4 selectivity on Cu–Br and Cu–Cl and the unique selectivity toward C2H6 of 30% on Cu–I, as shown in Fig. 8j–m.
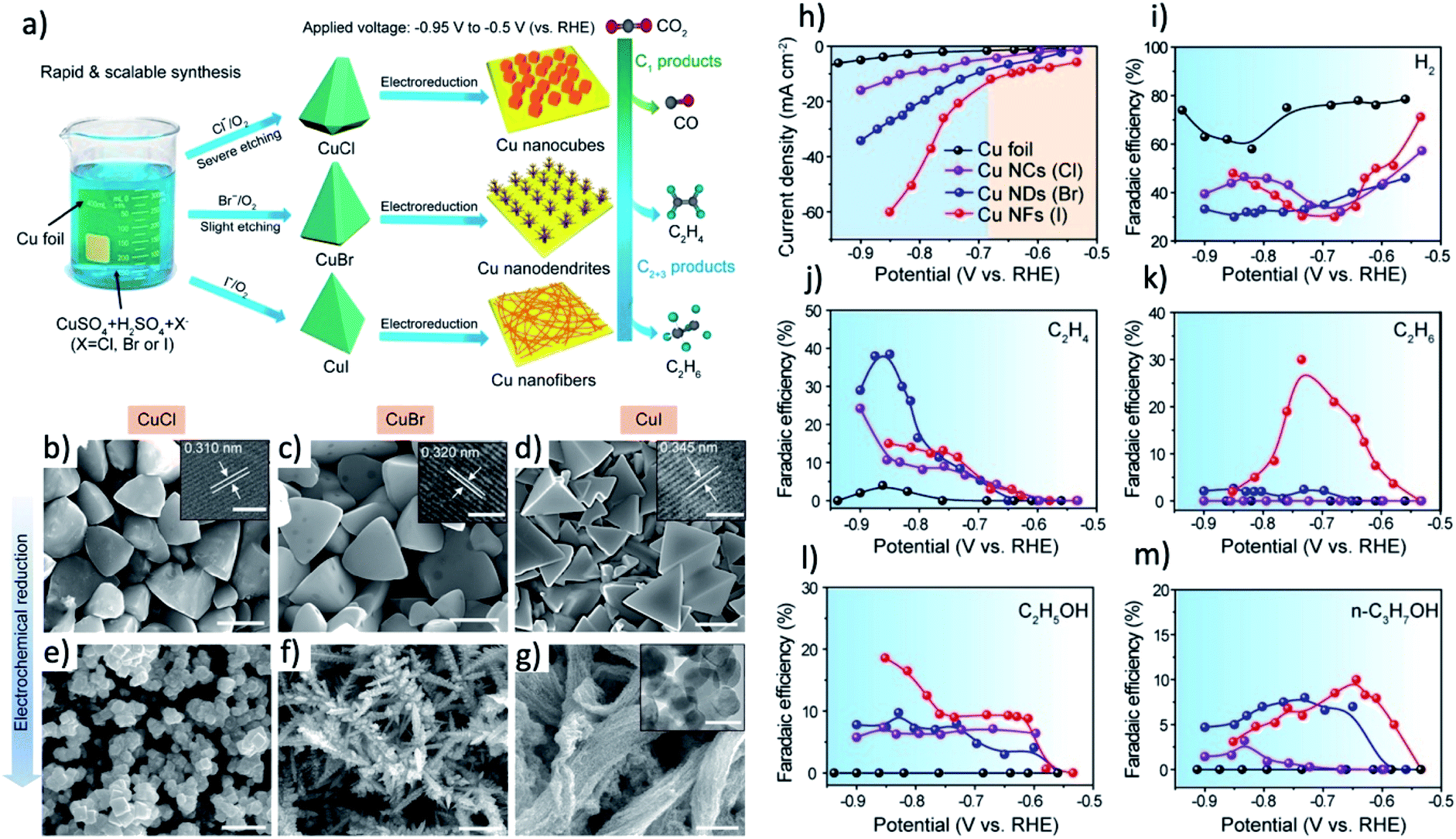 | ||
| Fig. 8 (a) Scheme of the facile synthesis of CuCl, CuBr, and CuI microcrystals and the corresponding Cu nanostructures after electrochemical reduction which lead to different selectivity of CO2 electrochemical reduction. (b–d) SEM images of as-synthesized CuCl, CuBr, and CuI microcrystals (e–g) SEM images of Cu nanocubes, nanodendrites and nanofibers electrochemically reduced from (a)–(c), respectively. Scale bars: 2 μm. (h) CO2RR current densities of Cu foil, Cu nanocubes (Cl), Cu nanodendrites (Br), and Cu nanofibers (I). (i). Faradaic efficiencies of by-product H2. (j–m) Faradaic efficiencies of valuable product of C2H4, C2H6, C2H5OH, and n-C3H7OH. Adapted with permission from ref. 106 Copyright © 2019, American Chemical Society. | ||
4.1. Grain boundaries and undercoordinated surface sites in derived catalysts
Although introducing nanostructures can increase the geometrical surface area for CO2RR, the enhancement of CO2RR selectivity and ECSA normalized CO2RR partial current can not be simply explained by enhanced geometrical surface area. The determination of the real active sites is crucial to understand the underlying mechanism of the unique performance of compound-derived catalysts. In Kanan's research, based on SEM and TEM characterizations, it is clear that the OD-Cu is composed of the stacking of nanoparticles, with interconnecting grain boundaries.16–18 A quantitative relationship between the density of grain boundaries (GBs) and CO2RR activity of OD-Cu and OD-Au were first revealed by Kanan group.183,184 To control the density of GBs, Cu or Au nanoparticles were deposited on carbon nanotube (CNT) by e-beam evaporation and a subsequent annealing process was conducted. The annealing process at high temperatures can gradually decrease the GB density while leaving the general morphology unchanged. For instance, the total GB surface density was 49.5 μm−1 for the as-deposited Au sample and the GB density can be tuned to 28.1, 13.3 and 4.2 μm−1 by annealing at 200, 300, and 400 °C, respectively. It is found that the relationship between the specific jCO and GB surface density was linear across all Cu samples or Au samples. Moreover, at a low overpotential of 200–400 mV, all the normalized jCORRvs. density of GBs curves showed intercepts very close to 0, suggesting that the density of GBs was the only factor in this research to influence the CORR activity.Temperature-programmed desorption (TPD) of CO on OD-Cu indicated the presence of surface sites with strong CO binding strength is correlated to the high CO reduction activity, which can be ascribed to the disordered surfaces at GB.183 It is showed that a high-temperature feature centered at 275 K is observed for OD-Cu, which is distinct from the profile found on polycrystalline Cu.183 Annealing the OD-Cu to 350 °C reduced the area of the high-temperature feature. When plotted the high-temperature feature against electrochemical activity, a linear correlation is obtained between surface area-corrected j and the percentage of strong CO binding sites.
Kanan group performed spatially resolved measurements to elucidate the active regions at GB surface terminations of Au.19,185 The density and profile of GBs are resolved by electron backscatter diffraction (EBSD) in a SEM and a lower potential is chosen to ensure that the CO2-to-CO reduction reaction occurs solely on the GBs. Scanning electrochemical cell microscopy (SECCM) was used to probe the local electrocatalytic activity across GBs, which provide a hopping-mode current profiles with a resolution of 500 nm step size. After that, Kanan group further developed high-resolution SECCM (diameter of droplet ≈ 200 nm) and high-resolution EBSD to probe the physical origin of this enhancement on grain boundaries, as shown in Fig. 9a–c.185 Under Ar saturated electrolyte, a step of current can be observed when scanning from one grain to the neighbouring grain, indicating a difference in HER activity between two grain surfaces. In contrast, under CO2 saturated electrolyte, the currents on two grains are similar, but a much higher current peak is shown for the GB region, as shown in Fig. 9d–g. The results indicated that the GB surface terminations in Au electrodes are more active than the grain surface for CO2RR. They also found that regions of enhanced CO2 electroreduction activity are not correlated with lattice strain but coincident with the geometrical dislocation content. The dislocations can increase the density of undercoordinated sites which can enhance CO2 electroreduction but have little impact on the hydrogen evolution activity. The accumulation of dislocations in the region of grain boundary is the likely origin of grain-boundary-enhanced CO2RR activity observed previously. Another research on electrodeposited copper with in situ XAFS and in situ ECSEM revealed that the formation of reduced copper on copper surface could trigger a surface reconstruction for a rougher surface that contains abundant uncoordinated sites,186 indicating that the effect of uncoordinated sites may actually impact the performance of all metals that vulnerable to oxidation in air or during the open circuit.
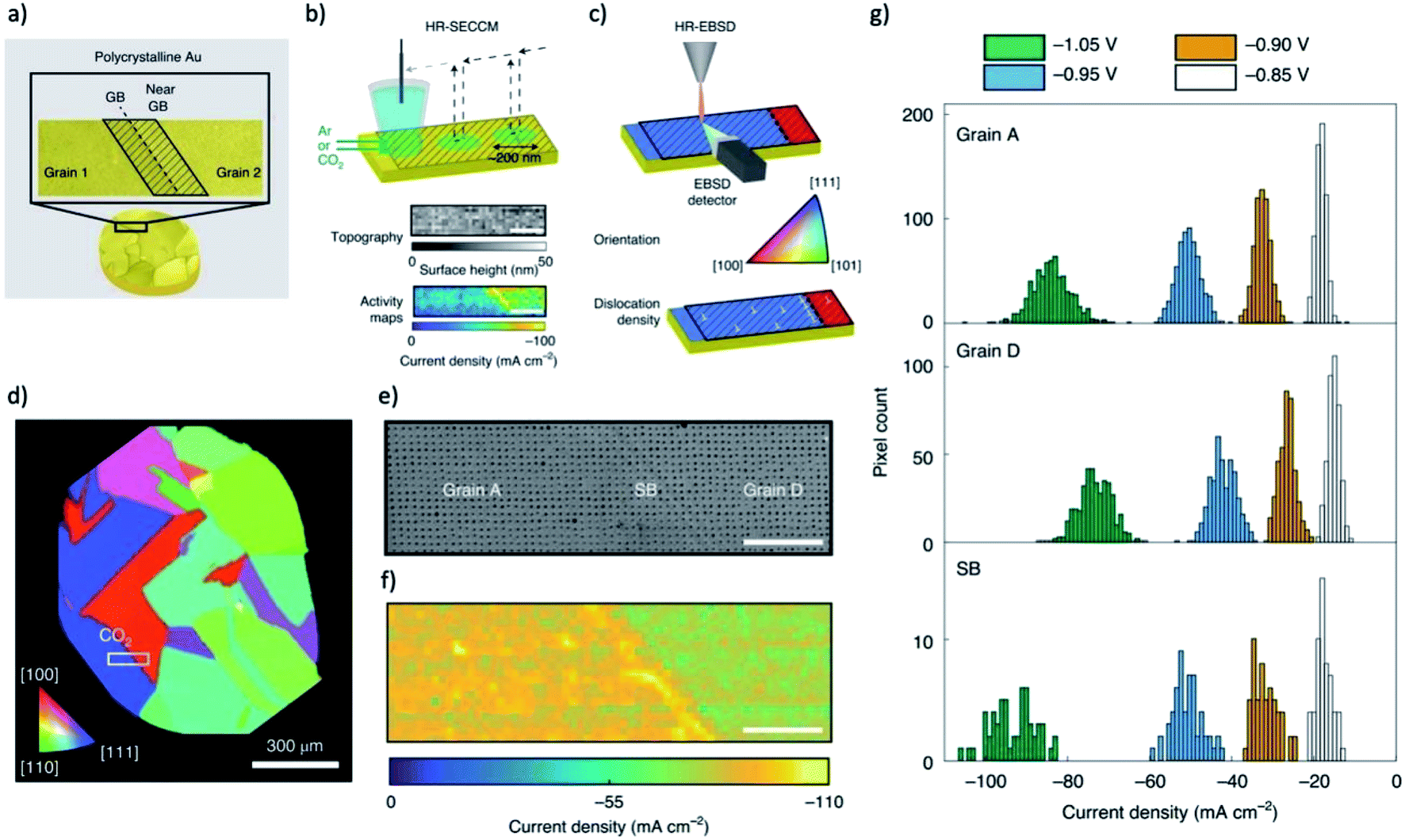 | ||
| Fig. 9 (a) A typical structure of grain boundary probed by correlative SECCM and HR-EBSD measurements. (b) Illustration of voltammetric SECCM and (c) Schematic of HR-EBSD grain mapping on polycrystalline Au surface. (d) An EBSD orientation map of a typical polycrystalline Au sample viewed along the z direction. White rectangle indicates the location of the SECCM scan. (e) SEM map of the region scanned with SECCM, showing residues left from droplet contact points. (f) The current density measured from SECCM at −1.05 V versus Ag/AgCl. (g) Histograms of current densities from all of the pixels in grain and grain boundary regions. The scale bars in (c)–(e) are 5 μm. Adapted with permission from ref. 185 Copyright © 2021, Springer Nature. | ||
Goddard and coworkers calculated the effects of GB on OD-Cu for CORR activity through DFT calculations.187 The results showed that 9% of the surface sites have binding energies larger than three typical facets of (111), (100) and (211). Further calculation for the energy barrier of *OCCOH formation revealed that not all strong CO binding sites were active for C2 formation, but only the strong CO binding sites with at least one under-coordinated neighbor square site adjacent to a subsurface stacking fault could promote C–C coupling.
The GB effect was also observed on other metal oxides, such as SnOx and BiOx. Lou and coworkers reported the ultrathin sub-2 nm SnO2 QWs composed of separate QDs with GBs on the surface which show significantly higher current density as compared to SnO2 NPs.89 Li and coworkers found that by creating more GBs on Sn-NWs with acid etching, the current density would increase by 12 times at −1.0 V,109 while the surface area of acid-etched Sn-NW catalyst is only 6 times higher than untreated Sn-NW. Li and coworkers synthesized a group of Bi/Bi2O3 nanosheets catalysts and found that catalysts with higher GBs actually exhibited higher partial current density and larger FE (>90%) at all applied potential range for formate production compared with samples with larger ECSA.188
Many specially designed pre-catalysts can also create abundant vacancies after reduction. Sargent and coworkers reported Au–S and Pb–S catalysts with obvious vacancies on the surface.29 By decreasing the particle size from 5 nm to 3 nm, the density of vacancies increased from 5% to 20%. After 1.5 h CO2RR, all pre-catalysts were converted into metallic state with much lower metal coordination numbers and enlarged bonding distance compared with commercial metal nanoparticles. This vacancies-induced modification of electric structure significantly increases the current densities of electrosynthesis of formate, carbon monoxide on Pb and Au at low potentials of −0.2, −0.3 V. The same group also reported pre-catalyst of asymmetric paddle-wheel Cu dimer of HKUST-1,36 which can derive into Cu clusters during CO2RR. The in situ EXAFS showed that the average Cu–Cu coordination number of Cu cluster was reduced to 9.5 ± 0.9. The C2H4 FE was further enhanced up to 45% with the current density of 262 mA cm−2 at −1.07 V, which is consistent with the general opinion that undercoordinated edge and corner sites on Cu surfaces are more active for C–C coupling. Li and coworkers studied the reduction of Bi2O3 NTs by EXAFS method, and they found that the coordination number of Bi–Bi is determined to be 2.6 ± 1.8 at −0.24 V, significantly smaller than that of Bi metal foil (CN = 6).23 The FEHCOOH of this low-coordinated Bi catalyst can reach >93% in H-cell and 98% in a flow cell. It is reported that the O2 + H2 plasma-treated Ag showed the highest density of defects and the highest CO evolution activity, while the Ar and H2 plasma-treated samples exhibited much inferior CO evolution activity although they have similar roughness.108
4.2. Surface orientation reconstruction during CO2RR
The surface orientation of metal catalysts is crucial for the tuning of activity and selectivity during CO2RR, particularly on copper catalyst which can drive the further reduction from the key intermediate product of CO to valuable C2+ products. Hori group and other researchers investigated the effect of crystal surface orientation of Cu on the CO2RR and CORR activity, whose results have been confirmed by many other research groups. The results found that the generation of C2H4 is more favored on Cu(100) facet relative to Cu(111) facet.15,189,190 Specifically, the production of CH4 and C2H4 from CO reduction share the same trend on Cu(111), and have a very negative onset potential of −0.8 V, as shown in Fig. 10a. Although these reaction pathways of CO reduction also existed on Cu(100), another low potential reaction pathway for the production of C2H4 from CO reduction on Cu(100) is observed at −0.45 V (Fig. 10b), indicating a highly active structure on the Cu(100) surface for C2H4 evolution. Moreover, the formation of C2H4 can be further enhanced and the evolution of CH4 will be suppressed by introducing (111) steps to the Cu(100) basal plane.189 Jiao and coworkers further found that the Cu(111) facet can manipulate the reaction pathway of CO reduction to acetate rather than C2H4.191 This facet-related performance of CO2RR has also been observed for other metal catalysts. For instance, Luo and coworkers reported that triangular Ag nanoplates with the dominated facet of Ag(100) exhibited enhanced current density and significantly improved faradaic efficiency (96.8%) compared with normal Ag nanoparticles.192 Woo and coworkers selectively synthesized hierarchical hexagonal Zn catalyst and found that Zn (101) facet was favorable to CO formation whereas Zn (002) facet favored the H2 evolution.193 During CO reduction reaction, many experiments also observed that the surface orientation of metal catalysts was evolving during the reduction and greatly influenced the selectivity.194–198 A stepwise surface reconstruction of copper under certain applied potential in the electrolyte is observed by the operando EC-STM of Soriaga group.194 This experimental phenomena trigger the motivation to generate a stepped Cu(S)-[3(100) × (111)], or the Cu (511) surface on Cu electrode through oxidation–reduction cycling, resulting in a FE for ethanol up to 100%.195,196 For the surface facet regulation in CO2RR, Nilsson and coworkers reported that Cu nanocube sample could be synthesized by CV scanning in KHCO3 and KCl mixed electrolyte, and the sample was expected to expose dominated (100) facet and was confirmed to nearly complete suppress methane formation at potentials more negative than −0.6 V.197 Yang and coworkers reported that the ensemble of Cu nanoparticles might go through a structural transformation process during initial electrolysis to form cube-like particles.199 Sargent and coworkers proposed a strategy to preferentially expose and maintain Cu(100) facets by in situ depositing copper under CO2 reduction conditions.198 The resulted copper sample exhibited a high FEC2+ of 90% at 520 mA cm−2. | ||
| Fig. 10 (a) Top: (111) facet of the copper fcc crystal. Middle: The catalytic current measured by cyclic voltammograms for the reduction of a saturated solution of CO (∼1 mM) on Cu(111) in phosphate buffer (pH 7) and NaOH solution (pH 13). Bottom: Products of CO reduction at different potential measured by online electrochemical mass spectrometry. (b) Top: (100) facet of the copper fcc crystal. Middle: The catalytic current measured by cyclic voltammograms for the reduction of a saturated solution of CO (∼1 mM) on Cu(100) in phosphate buffer (pH 7) and NaOH solution (pH 13). Bottom: Products of CO reduction at different potential measured by online electrochemical mass spectrometry. Adapted with permission from ref. 15 Copyright © 2012, American Chemical Society. | ||
The evolution of pre-catalysts during CO2RR is always accompanied by dramatic structure change, thus it is natural to consider it as a potential method to control the exposed facet of resulted catalysts. For the metal oxide pre-catalysts for CO2RR, Cu2O films with [100], [110] and [111] orientation have been prepared to investigate the effect of crystal orientation on selectivity and the results showed that the final performance is not dependent on the initial crystal orientation of the catalysts.113 Sartin et al. also revealed that less than 20% difference in the coverage of adsorbed CO on the shape-controlled Cu2O nanoparticles with different initially exposed crystal planes could be found. The similar coverage for different Cu2O nanoparticles implied that a surface reconstruction occurred during CO2RR, resulting in a similar morphology for the OD-Cu catalysts.200 Cuenya and coworkers synthesized Cu2O and monitored the whole reducing process with liquid cell TEM.201 They found that the fast and extensive restructuring of Cu2O cube to Cu dendrites in 4 min. The experiments of Strasser group also confirmed that the initial cube shape and (100)-rich facet structure of Cu2O had been totally degraded during the initial reduction.202
Although the initial morphology of pre-catalysts may not be effective in regulating the final surface orientation of derived catalysts, there were still a few works reported some useful information of the preferable facet after pre-catalysts reduction. For OD-Cu that favor the production of C2 products, XRD test indicated that the activated Cu catalysts showed a greater abundance of (100) facets after the electrochemical Cu2O reduction, distinct from the (111) orientation that contributes to CH4 products on Cu foil.165Operando ATR-SEIRAS investigations on OD-Cu particles also suggest that they possess distinct CO binding sites aside from those present on the p polycrystalline Cu surface.203 In addition to bands similar to those on polycrystalline Cu (2073, 2089, and 2131 cm−1), a prominent band at 2058 cm−1 is observed in ATR-SEIRAS, corresponding to the CO adsorption band observed on the Cu (100) surface at a similar potential reported by Hori et al.204
For Ag foil and nanoparticle catalysts, (111) facet is the most stable surface orientation, however, Smith group discovered that the ratio (220) over (111) is higher after the oxidation–reduction process on Ag.21 The single crystal facet of (110) has been reported to be more active than (111) facet for CO2RR and could be one of the reasons that OD-Ag has improved selectivity towards CO production.205 Zhou et al. also found that after the Ag foil anodization, the (220) peak intensity became significantly stronger on OD-Ag.64 For OD-Ag samples with different electrochemical anodization potential and total charge, the CO faradaic efficiency of resulted catalysts were substantially increased with enhancing (220)/(111) peak intensity ratio. Although the preferred (220) orientation and thin AgOx layer are strongly related to the high activity and selectivity of OD-Ag, the mechanism of the forming of (220) orientation on OD-Ag sample is not clear and needs to be further investigated.
Bi-halide and Bi-salt pre-catalysts are quite unique because they can form a series of nanosheet morphology with a single exposed facet, which was observed to influence the final morphology of the derived catalysts. For instance, Luo and coworkers reported that by reducing the BiOI nanosheet precursors, Bi catalysts with nanosheet morphology can be obtained.169 Moreover, the exposed facet of (100) or (001) on BiOI nanosheets can determine the final morphology of the Bi nanosheet to be mesoporous Bi nanosheets with interconnected nanoparticles or Bi nanosheets with a smooth surface, respectively. Zou and coworkers reported that the Bi2O2CO3 nanosheet mainly exposed the (001) facets would determine the orientation of the derived Bi nanosheet, resulting in the formation of Bi nanosheets that terminated with (001) facets.105 This Bi nanosheet could exhibit a FEHCOOH of 90% at a low overpotential of 420 mV. Sargent and coworkers studied the BiOBr pre-catalysts by in situ XANES, EXAFS and GIWAXS.152 During the CO2RR, from GIWAXS experiments, the Bi (110) facet is emerged as the dominant facet during CO2RR, as shown in Fig. 11a–e. The CO2RR experiment showed that the BiOBr catalyst exhibit more than two times current density compared with the Bi nanoparticle and the formate FE could reach 99% in the range of −0.8 to −1.0 V (Fig. 11f). DFT calculations in Fig. 11g and h pointed out that the Bi (110) facet can greatly lower the energy barrier of CO2RR and the stepped Bi (110) surface can offer a near-optimal Gibbs free binding energies for HCOO* (−0.01 eV). These results indicated that an atomic-level structural modification of the catalysts surface can be employed as a powerful means to regulate the product distribution for CO2RR.
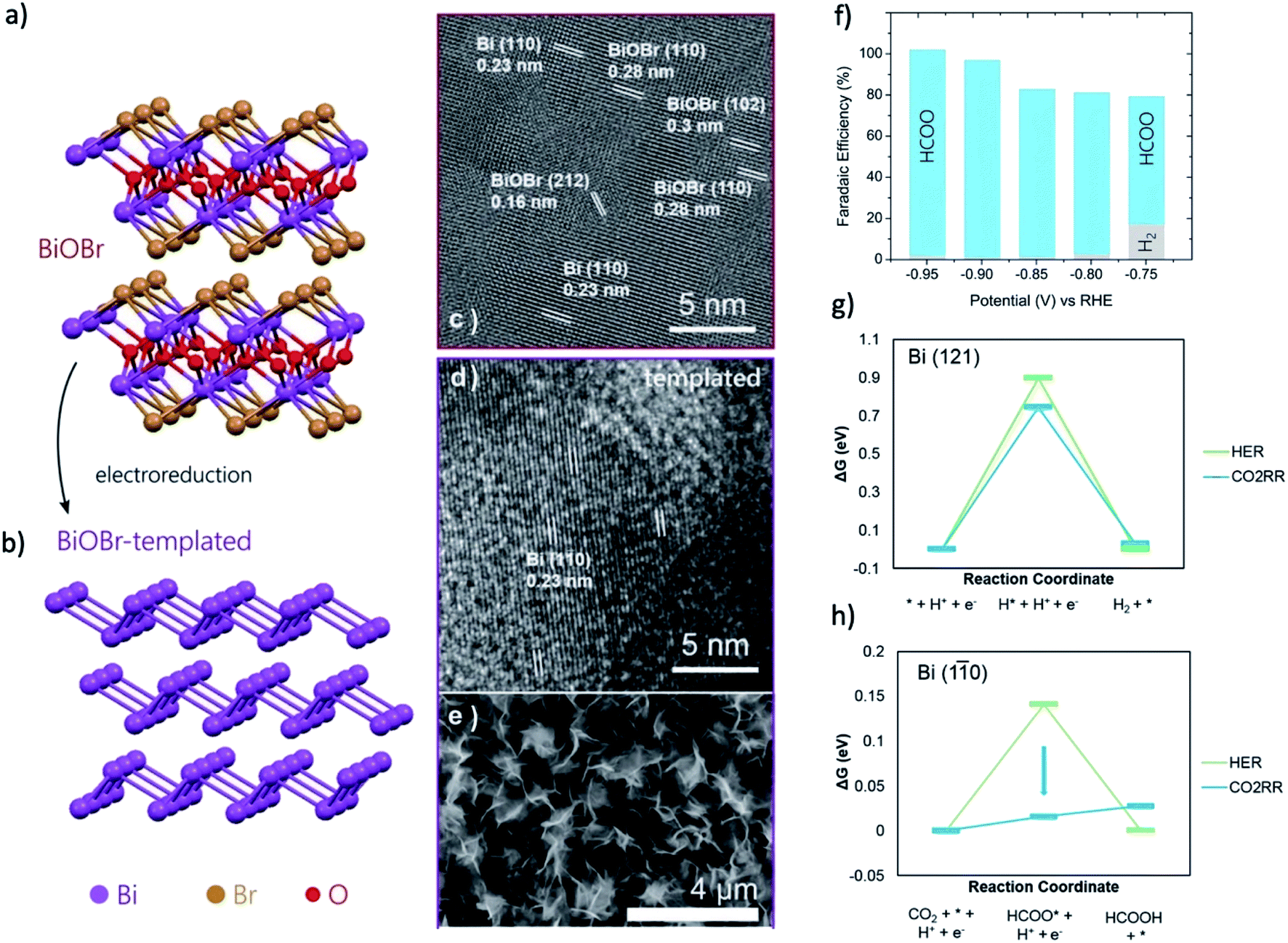 | ||
| Fig. 11 Atomic structure of (a) BiOBr and (b) BiOBr-templated Bi catalyst after electroreduction. HRTEM of BiOBr (c) before and (d) after electroreduction reveal the selective reconstruction into Bi. (e) SEMs of samples after reaction reveal a 2D petal-like layered arrangement. (f) CO2RR product distribution of BiOBr-templated catalyst for different operating potentials. Formate is the only product at <−0.75 V. (g and h) The reaction energy diagram for HER and CO2RR on Bi (121) and Bi (110) facets. Adapted with permission from ref. 152 Copyright © 2018, Wiley-VCH. | ||
5. Stability of the CO2 pre-catalysts
The stability is a universal challenge for all CO2RR catalysts, which originated from the impurities in the solution and the evolution of catalysts.206 The former challenge can be resolved using an ultra-pure electrolyte, chelating agent, or high surface area catalysts with alkaline electrolyte,207–210 while the understanding of the later challenge is still preliminary and the solution is yet to develop.One major evolution process of CO2RR catalysts is the morphology evolution, such as the stepwise surface reconstruction of copper under certain applied potential region,194 and the forming of Cu cube under CV scans197 mentioned above. There are also many experiments found that the high activity morphology suffered from degradation under long-term operation. Cuenya and coworkers reported that for copper cubes deposited on carbon, the Cu(100) facets became rougher and losing the cubic shape, resulting in a suppression of multi-carbon products (i.e., C2H4 and ethanol) versus CH4.211 Buonsanti and coworkers found that at a sufficiently negative potential, the nanoparticle of metal catalysts were reshaped and formed nanocluster to expose the surface facet with a lower adsorption energy of either H- or CO-species on the catalyst surface (Fig. 12a–c). This reshaping was the reason for the unavoidable degradation of the crystals, and small crystals were more vulnerable to degradation (Fig. 12d).212 The pre-catalysts-derived catalysts benefit from a relatively high surface area and nanostructure, which are also vulnerable to the structure evolution under long-term operation. For instance, during CO2RR, the OD-Au seems to be sintered over time, resulting in decreased current density and FE. Specifically, a roughness factor of 72 was reduced to 17 after 8 h operation, indicating that sintering of Au NPs occurred during CO2 reduction electrolysis.18 SEM and PXRD line-broadening analysis of these electrodes after 8 h showed an increase in particle and crystallite size over time, consistent with the observed RF decrease. In another research, ECSA measurements performed after 12 h of CO2 electrolysis reaction showed that the surface areas of OD-Au decrease from 8.3 to 5 m2 g−1, respectively, in good agreement with SEM observations.63 However, it is important to note that FECO for OD-Au stays relatively constant at about 90% even after running for 12 h, thus implying that the feature size alone cannot explain the better performance of OD-Au. The CO2RR efficiency towards CO also degrades slightly after 3 h of reaction on the oxidized Ag samples because of a slight decrease in current towards CO and a growth in the HER current.108 This time-dependent change is likely related to the change in surface structure over the course of the reaction observed by SEM images. Electrochemical surface roughness measurements also indicate that the roughness of the surface decreases by approximately half between 1–3 h of reaction.
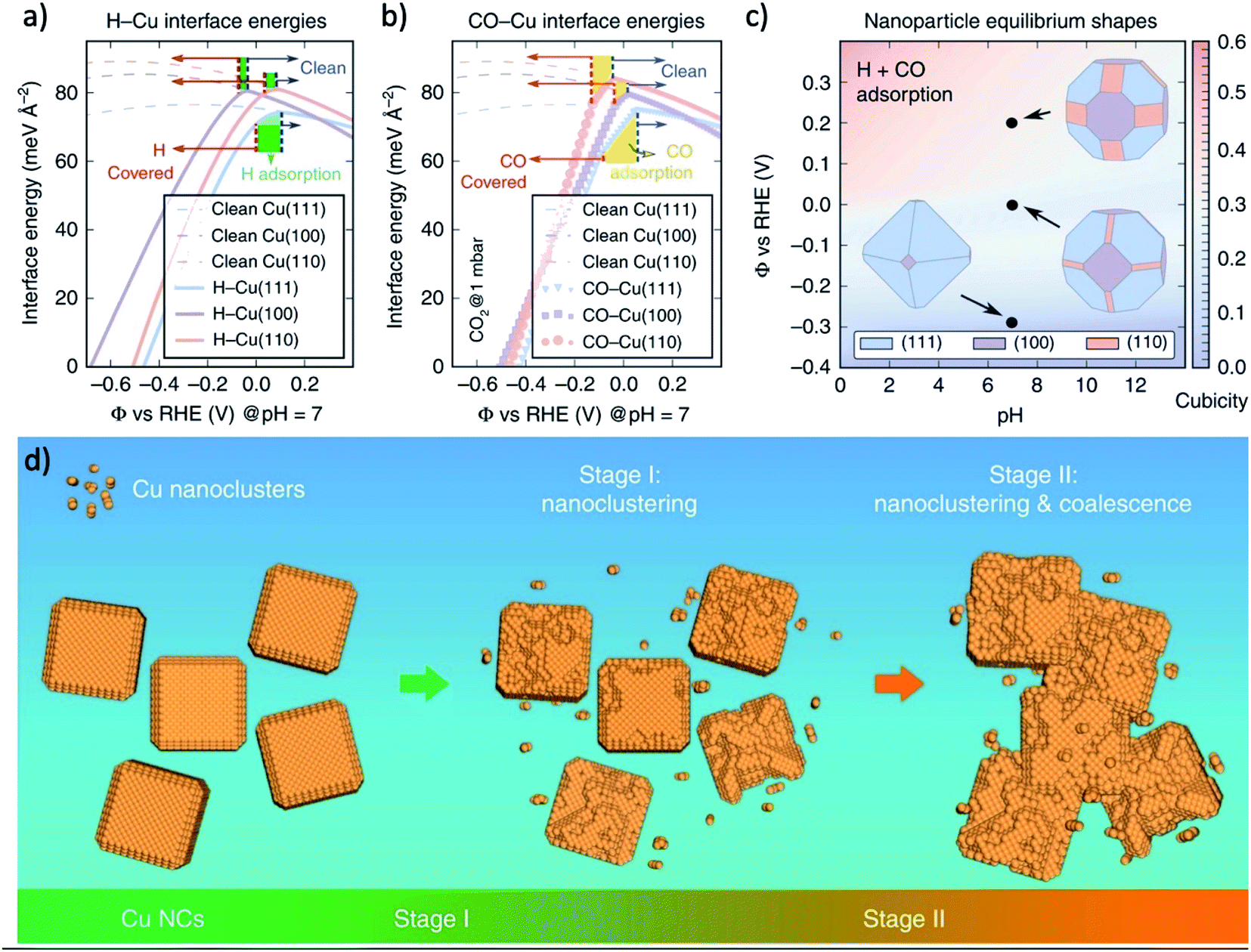 | ||
| Fig. 12 The interface energies of (a) H-covered and (b) CO-covered Cu surfaces of (111), (100) and (110). (c) The equilibrium of Wulff-shape of Cu nanoparticle based on the calculation of interface energies. (d) Schematic illustration of the degradation mechanism of Cu NCs during CO2RR. The degradation mechanism that includes nanoclustering (stage I) followed by a coalescence at a later stage (stage II). Adapted with permission from ref. 212 Copyright © 2018, Springer Nature. | ||
The long-term existence of nonmetal elements in the derived catalysts during CO2RR is highly suspicious due to the relatively high standard electrode potential compared with CO2RR. Obviously, the voltage of catalytic reduction has a great influence on the retention of nonmetallic components, evidenced by the clear transition from SnO2 to Sn when changing the potential from 0.1 V to −0.7 V.123 This phenomenon emphasizes the importance of studying the structure and composition change of pre-catalysts in flow cell which usually adopt much higher voltage and current compared with H-cell. On the other hand, the low conductivity metal compounds pre-catalysts may be in an electrochemically inactive state after the surrounding parts are reduced to metal.179 This could also be part of the reason for the remnant of nonmetal elements under the surface of catalysts. However, based on currently released data, one can not confirm that the nonmetal content will remain at a relatively stable value rather than a continuous decline under the condition of long-term work with even low overpotential.
Currently, the stability of derived catalysts is not properly studied, because most studies do not have long-term test and no structural test after a long-term operation is carefully and detailly characterized. Ager and coworker reported that only a small fraction (<1%) of the original O content remains after CO2RR of 10 min, however, the FE of ethylene could be maintained at 35% for 5 h.137 Gong and coworkers' research, on the other hand, showed that the Cu+ can be stabilized in Cu/CuSiO3 and the FEC2H4 of catalyst remained 50% for 6 h.213 These studies indicated that the derived catalysts can catalyze the CO2RR with a relative stable structure, however, slow structure evolution of derived catalysts cannot be excluded if longer testing period is applied. The prolonged CO2RR test of Han and coworkers on Au sulfide catalysts showed a relatively stable performance for the first 6 h and a clear decline of CO selectivity between 6 and 10 h.214 An even longer test of 72 h had been applied to Ag–Cl pre-catalyst by Polyansky and coworkers.32 They found that the morphology of catalysts was dramatically changed, resulting in 20–30% decrease of electrochemical surface area and current density. Moreover, the relationship between CO2RR selectivity and morphology changes may not be linear. One research indicated that the deactivation of OD-Cu process might affect long-chain products (C3–C4) first and the C2 selectivity could be relatively stable during first several hours.140 It is reported that the Cu2OCl exhibited a stable productivity toward C2H4 and C2H5OH over a period of 7 hours, while the productivity of C3–C4 products are dramatically decreased due to the decreasing relative ratio of Cu+ to Cu0.
The dilemma between short lifetime and high activity of Cu+ ions in CO2 reduction catalyst triggers the efforts to regenerate copper oxide during catalysis. Cuenya and coworkers proposed an alternative pulsed electrolysis strategy to regenerate the Cu(I) ions at a positive potential,24 as shown in Fig. 13a. This regeneration strategy can help to reshape the surface morphology of Cu to nanocube and maintain a high concentration of Cu2O (Fig. 13b and c). Specifically, significantly enhanced selectivity for ethanol (32%) has been found under alternative pulsed electrolysis conditions of 0.6 V and −1.0 V due to the co-existence of Cu(I) and Cu(0) species created by an anodic pulse (Fig. 13d).24 Another research showed that the alternating voltage method is better at providing a large selection of oxygenated hydrocarbons products compared with cyclic voltammetry and chronoamperometric methods.215 Besides anodic pulse, keeping catalyst at open circuit potential can also help the regeneration of copper oxides and prolong the product selectivity towards C2H4.216
 | ||
| Fig. 13 (a) The scheme of regeneration of Cu+ by pulsed electrolysis strategy. (b) Atomic force microscopy images of a Cu(100) electrode after 1 h of alternative pulsed electrolysis of Ea = 0.6 V, Ec = −1.0 V, ta = tc = 1 s. The Cu(100) electrode was transformed to cubic islands with (100) facets. (c) Quasi in situ copper LMM Auger spectra of a Cu(100) electrode after the different pulse protocols. Larger amount of Cu2O can be observed on sample with higher Ea. (d) The bar chart of product selectivity for different pulse protocols. Adapted with permission from ref. 24 Copyright © 2020, Springer Nature. | ||
6. Summary and perspective
The pre-catalyst system showed impressive performance in CO2 reduction. Elements such as O, N, S, and Cl are believed to significantly increase the valence of metals, and the loss of them in the in situ environment will introduce specific structural defects to the catalyst system. Generally, the pre-catalyst strategy does not change the species of products on certain metal catalysts but greatly improve the reaction rate and distribution of products. One universal benefit for all pre-catalysts is the suppression of hydrogen evolution because the rough surface of derived catalysts can maintain a high pH environment during CO2RR to hinder the supply of proton for HER.171–173 The reduction of pre-catalyst can also create undercoordinated reaction sites on the catalysts surface. For instance, the Au–O pre-catalysts can greatly improve the FECO to 99%, which attribute to the enhanced surface area and abundant surface under-coordinated reaction sites.18,19,185 However, the facile control of morphology and the density of grain boundaries for improved CO2RR activity is not yet realized. The concentration of nonmetal element remained in derived catalysts was more systematically studied and was found to be strongly related with the CO2RR activity. For Cu–O and Ag–O pre-catalysts, the sample with higher O content were found to be more efficient for C2+ and CO production, respectively.21,157 For HCOOH producing catalysts, such as Sn, it was found that the partially oxidized metal ions on catalyst surface have much higher activity than pure metal or metal oxides.22,123 S-contained pre-catalyst is the most special one among all other pre-catalysts because it can tune the selectivity of copper to pure HCOOH, and the sulfiding of many non-active metals can result in active CO or HCOOH producing catalysts such as WSe2, TiS2, In–S.78,80,104,147,148 The copper-nonmetal compound, such as Cu–B, Cu–N, Cu-halide,30,31 attracted intense attention recently due to its ability to significantly enhance the FE of C2+ products. Notably, the average oxidation states of copper in the X–Cu catalysts is proportional with the electronegativity of the halogen, and F–Cu catalyst showed the best C2+ FEs and highest surface area normalized C2+ formation rates among all Cu-halide samples. Comparing the CO2RR selectivity of Cu-based pre-catalysts with different nonmetal elements is very important and yet to realize. Generally, the current overall research is still in a very rough stage, lacking guiding theories and systematic rules. Thus, the following challenges should be considered and addressed to push the CO2RR pre-catalysts strategy technique forward.(1) The in situ activation process of the pre-catalyst is very complicated, accompanied by the change of the metal valence and the change of the overall morphology of the catalyst. Therefore, it is just an assumption to attribute the origin of catalyst performance to one of the structure or composition factors. Some studies simply studied a specific element ratio or specific morphology of the pre-catalyst, which is very difficult to obtain an in-depth understanding of the catalytic activity. Other research works were conducted by controlling the ratio of elements, or controlling the changes of certain structural parameters, thus, they can provide more valuable conclusions on the influence of certain parameter. In future research, we should consider separating these structural parameters more clearly. For example, one could control the size and distribution of the pre-catalyst particles on an inert substrate so that it will not form a special macroscopic morphology. With this platform one can focus on the effect of nonmetals on the performance of metal catalysts.
(2) An efficient CO2RR catalyst for large scale application should possess a long lifetime of >1 year even under high reaction rate. However, the composition and morphology of catalysts evolved from the pre-catalysts is more likely to be a metastable state and may keep evolving during electrolysis. Therefore, it is particularly important in the future research to conduct in situ or quasi-in situ tests of long-term CO2RR operation to probe the mechanism of structural evolution. The current research usually focused on the operation period of <5 hours which may not show the effect of structure changes on the performance. Several studies that performed long term stability test such as 10 h or 72 h clearly revealed a structure evolving and the dropping of CO2 reduction efficiency.32,214 Operation-regeneration cycle is confirmed to be a promising approach to prolong the unique CO2 reduction activity on CuOx, however, the regeneration strategy for other pre-catalysts is still lacking. To realize a strategy that can regenerate metal–nonmetal compound pre-catalysts in situ under working conditions would be a game changer in this area.
(3) Considering the pre-catalysts have a high possibility of being transformed to metal phase through an in situ dynamic evolution process, it is obvious that the impurities existed in the electrolyte should also matters and can change the final composition and activity of the catalysts. Indeed, the alkaline electrolyte may etch the stainless flow cell and release various elements to interference the ultimate activity results. On the other hand, the un-purified electrolyte also contains a lot of unexpected impurities, leading to the composition change and deactivation of catalysts. Protecting the catalyst with stable materials217–221 or developing robust catalysts that intrinsically resistant to impurities should be designed to enhance the lifetime and maintain the activity for specific products. Alternatively, introducing chelating agent in electrolyte is another powerful way to minimize the effect of impurities during operation.209 What's more, the optimization of configuration of flow cell to improve operation environment should be contribute to alleviate the pollution, which may can be boosted by modelling and simulation works.
(4) The researches mentioned above were basically focused on the influence of a single nonmetallic element on the final performance of the metal catalyst. Considering the unique effects of different nonmetals on the final properties of metal catalysts, the use of multiple nonmetals to jointly adjust the metal properties or the activation process will enable us to have more refined tools for adjusting the catalyst performance. Furthermore, incorporating the alloy-related research experience and introducing complicated design of the tandem structure will bring about a series of brand-new CO2 reduction catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CL acknowledges financial funding from National Natural Science Foundation of China (No. 22002191).References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- C. Li, Q. Cao, F. Wang, Y. Xiao, Y. Li, J. J. Delaunay and H. Zhu, Chem. Soc. Rev., 2018, 47, 4981–5037 RSC.
- C. Li, J. He, Y. Xiao, Y. Li and J.-J. Delaunay, Energy Environ. Sci., 2020, 13, 3269–3306 RSC.
- C. Feng, M. B. Faheem, J. Fu, Y. Xiao, C. Li and Y. Li, ACS Catal., 2020, 10, 4019–4047 CrossRef CAS.
- Y. Li, W. Cheng, H. Su, X. Zhao, J. He and Q. Liu, Nano Energy, 2020, 77, 105121 CrossRef CAS.
- S. Xu and E. A. Carter, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- J. He, Y. Li, A. Huang, Q. Liu and C. Li, Electrochem. Energy Rev., 2021 DOI:10.1007/s41918-021-00100-y.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer New York, New York, 2008, pp. 89–189, DOI:10.1007/978-0-387-49489-0_3.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- K. J. Schouten, Z. Qin, E. Perez Gallent and M. T. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- R. G. Mariano, K. McKelvey, H. S. White and M. W. Kanan, Science, 2017, 358, 1187–1192 CrossRef CAS PubMed.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- N. J. Firet, M. A. Blommaert, T. Burdyny, A. Venugopal, D. Bohra, A. Longo and W. A. Smith, J. Mater. Chem. A, 2019, 7, 2597–2607 RSC.
- Y. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS PubMed.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Nat. Commun., 2019, 10, 2807 CrossRef PubMed.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- P. Deng, H. Wang, R. Qi, J. Zhu, S. Chen, F. Yang, L. Zhou, K. Qi, H. Liu and B. Y. Xia, ACS Catal., 2019, 10, 743–750 CrossRef.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- Z. Chen, X. Zhang, M. Jiao, K. Mou, X. Zhang and L. Liu, Adv. Energy Mater., 2020, 10, 1903664 CrossRef CAS.
- Y. Zhang, F. Li, X. Zhang, T. Williams, C. D. Easton, A. M. Bond and J. Zhang, J. Mater. Chem. A, 2018, 6, 4714–4720 RSC.
- M. Liu, M. Liu, X. Wang, S. M. Kozlov, Z. Cao, P. De Luna, H. Li, X. Qiu, K. Liu, J. Hu, C. Jia, P. Wang, H. Zhou, J. He, M. Zhong, X. Lan, Y. Zhou, Z. Wang, J. Li, A. Seifitokaldani, C. T. Dinh, H. Liang, C. Zou, D. Zhang, Y. Yang, T.-S. Chan, Y. Han, L. Cavallo, T.-K. Sham, B.-J. Hwang and E. H. Sargent, Joule, 2019, 3, 1703–1718 CrossRef CAS.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- S. He, F. Ni, Y. Ji, L. Wang, Y. Wen, H. Bai, G. Liu, Y. Zhang, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2018, 57, 16114–16119 CrossRef CAS PubMed.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, X. Ma, M. Kim, S. Adhikari, C. Lu, C. Dun, M. Chi, Y. Qiu and S. M. Geyer, Nat. Commun., 2019, 10, 5724 CrossRef CAS PubMed.
- D. H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C. T. Dinh, F. P. Garcia de Arquer, Y. Wang, Z. Liang, A. H. Proppe, C. S. Tan, P. Todorovic, O. Shekhah, C. M. Gabardo, J. W. Jo, J. Choi, M. J. Choi, S. W. Baek, J. Kim, D. Sinton, S. O. Kelley, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS PubMed.
- C. Cao, D. D. Ma, J. F. Gu, X. Xie, G. Zeng, X. Li, S. G. Han, Q. L. Zhu, X. T. Wu and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 15014–15020 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- B. R. Wygant, K. Kawashima and C. B. Mullins, ACS Energy Lett., 2018, 3, 2956–2966 CrossRef CAS.
- A. Bergmann and B. Roldan Cuenya, ACS Catal., 2019, 9, 10020–10043 CrossRef CAS.
- S. W. Chee, T. Lunkenbein, R. Schlögl and B. R. Cuenya, J. Phys.: Condens. Matter, 2021, 33, 153001 CrossRef CAS PubMed.
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- P. Chen, Y. Jiao, Y.-H. Zhu, S.-M. Chen, L. Song, M. Jaroniec, Y. Zheng and S.-Z. Qiao, J. Mater. Chem. A, 2019, 7, 7675–7682 RSC.
- M. Nur Hossain, S. Chen and A. Chen, Appl. Catal., B, 2019, 259, 118096 CrossRef CAS.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- L. Chen, F. Li, C. L. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemElectroChem, 2017, 4, 1402–1410 CrossRef CAS.
- C. I. Shaughnessy, D. T. Jantz and K. C. Leonard, J. Mater. Chem. A, 2017, 5, 22743–22749 RSC.
- C. Yan, L. Lin, D. Gao, G. Wang and X. Bao, J. Mater. Chem. A, 2018, 6, 19743–19749 RSC.
- J. Wu, F. G. Risalvato, S. Ma and X.-D. Zhou, J. Mater. Chem. A, 2014, 2, 1647–1651 RSC.
- C. Chen, X. Sun, L. Lu, D. Yang, J. Ma, Q. Zhu, Q. Qian and B. Han, Green Chem., 2018, 20, 4579–4583 RSC.
- Y. Mi, S. Shen, X. Peng, H. Bao, X. Liu and J. Luo, ChemElectroChem, 2019, 6, 2393–2397 CrossRef CAS.
- M. Ebaid, K. Jiang, Z. Zhang, W. S. Drisdell, A. T. Bell and J. K. Cooper, Chem. Mater., 2020, 32, 3304–3311 CrossRef CAS.
- J. Szlachetko, J. Sa, M. Nachtegaal, U. Hartfelder, J. C. Dousse, J. Hoszowska, D. L. Abreu Fernandes, H. Shi and C. Stampfl, J. Phys. Chem. Lett., 2014, 5, 80–84 CrossRef CAS PubMed.
- L. K. Ono and B. Roldan Cuenya, J. Phys. Chem. C, 2008, 112, 4676–4686 CrossRef CAS.
- G. B. Hoflund, Z. F. Hazos and G. N. Salaita, Phys. Rev. B, 2000, 62, 11126–11133 CrossRef CAS.
- K. Jiang, P. Kharel, Y. Peng, M. K. Gangishetty, H.-Y. G. Lin, E. Stavitski, K. Attenkofer and H. Wang, ACS Sustainable Chem. Eng., 2017, 5, 8529–8534 CrossRef CAS.
- B. Tang and C. Wang, Sci. China: Technol. Sci., 2018, 61, 389–396 CrossRef.
- H. Kim, H. S. Jeon, M. S. Jee, E. B. Nursanto, J. P. Singh, K. Chae, Y. J. Hwang and B. K. Min, ChemSusChem, 2016, 9, 2097–2102 CrossRef CAS PubMed.
- J. Kim, J. T. Song, H. Ryoo, J.-G. Kim, S.-Y. Chung and J. Oh, J. Mater. Chem. A, 2018, 6, 5119–5128 RSC.
- Z. Qi, J. Biener and M. Biener, ACS Appl. Energy Mater., 2019, 2, 7717–7721 CrossRef CAS.
- L. Q. Zhou, C. Ling, M. Jones and H. Jia, Chem. Commun., 2015, 51, 17704–17707 RSC.
- A. H. Shah, Y. Wang, S. Hussain, M. B. Akbar, A. R. Woldu, X. Zhang and T. He, Phys. Chem. Chem. Phys., 2020, 22, 2046–2053 RSC.
- A. Raiyan, S. Das and M. R. Islam, Procedia Eng., 2017, 194, 276–283 CrossRef.
- M. Ma, K. Liu, J. Shen, R. Kas and W. A. Smith, ACS Energy Lett., 2018, 3, 1301–1306 CrossRef CAS PubMed.
- Y. Zhang, X. Zhang, Y. Ling, F. Li, A. M. Bond and J. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13283–13287 CrossRef CAS PubMed.
- W. Qiu, R. Liang, Y. Luo, G. Cui, J. Qiu and X. Sun, Inorg. Chem. Front., 2018, 5, 2238–2241 RSC.
- L. Zhang, Z. Wang, N. Mehio, X. Jin and S. Dai, ChemSusChem, 2016, 9, 428–432 CrossRef CAS PubMed.
- K. R. Phillips, Y. Katayama, J. Hwang and Y. Shao-Horn, J. Phys. Chem. Lett., 2018, 9, 4407–4412 CrossRef CAS PubMed.
- D. Gao, I. T. McCrum, S. Deo, Y.-W. Choi, F. Scholten, W. Wan, J. G. Chen, M. J. Janik and B. Roldan Cuenya, ACS Catal., 2018, 8, 10012–10020 CrossRef CAS.
- D. Gao, F. Scholten and B. Roldan Cuenya, ACS Catal., 2017, 7, 5112–5120 CrossRef CAS.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS.
- A. Basiratnia, J. Rempel, F. Li, A. Pogodaev, T. A. Zienchuk and A. Klinkova, Phys. Chem. Chem. Phys., 2019, 21, 5894–5897 RSC.
- Y. Gao, Q. Wu, X. Liang, Z. Wang, Z. Zheng, P. Wang, Y. Liu, Y. Dai, M. H. Whangbo and B. Huang, Adv. Sci., 2020, 7, 1902820 CrossRef CAS PubMed.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS PubMed.
- T. Shinagawa, G. O. Larrazábal, A. J. Martín, F. Krumeich and J. Pérez-Ramírez, ACS Catal., 2018, 8, 837–844 CrossRef CAS.
- Z. Yin, C. Yu, Z. Zhao, X. Guo, M. Shen, N. Li, M. Muzzio, J. Li, H. Liu, H. Lin, J. Yin, G. Lu, D. Su and S. Sun, Nano Lett., 2019, 19, 8658–8663 CrossRef CAS PubMed.
- W. Ma, S. Xie, X. G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D. Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 CrossRef PubMed.
- B. Qin, Y. Li, H. Wang, G. Yang, Y. Cao, H. Yu, Q. Zhang, H. Liang and F. Peng, Nano Energy, 2019, 60, 43–51 CrossRef CAS.
- H. Yang, N. Han, J. Deng, J. Wu, Y. Wang, Y. Hu, P. Ding, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1801536 CrossRef.
- Y. Qiu, J. Du, C. Dai, W. Dong and C. Tao, J. Electrochem. Soc., 2018, 165, H594–H600 CrossRef CAS.
- F. Li, L. Chen, M. Xue, T. Williams, Y. Zhang, D. R. MacFarlane and J. Zhang, Nano Energy, 2017, 31, 270–277 CrossRef CAS.
- A. Zhang, R. He, H. Li, Y. Chen, T. Kong, K. Li, H. Ju, J. Zhu, W. Zhu and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 10954–10958 CrossRef CAS PubMed.
- R. He, X. Yuan, P. Shao, T. Duan and W. Zhu, Small, 2019, 15, e1904882 CrossRef PubMed.
- H. Zhang, Y. Ma, F. Quan, J. Huang, F. Jia and L. Zhang, Electrochem. Commun., 2014, 46, 63–66 CrossRef CAS.
- Q. Wang, M. Ma, S. Zhang, K. Lu, L. Fu, X. Liu and Y. Chen, ChemPlusChem, 2020, 85, 672–678 CrossRef CAS PubMed.
- S. Liu, J. Xiao, X. F. Lu, J. Wang, X. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2019, 58, 8499–8503 CrossRef CAS PubMed.
- X. Li, S. Dou, J. Wang and X. Wang, Chem.–Asian J., 2020, 15, 1558–1561 CrossRef CAS PubMed.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- J. H. Zhou, K. Yuan, L. Zhou, Y. Guo, M. Y. Luo, X. Y. Guo, Q. Y. Meng and Y. W. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14197–14201 CrossRef CAS PubMed.
- S. Liu, H. Tao, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, ACS Catal., 2018, 8, 1469–1475 CrossRef CAS.
- F. Y. Gao, S. J. Hu, X. L. Zhang, Y. R. Zheng, H. J. Wang, Z. Z. Niu, P. P. Yang, R. C. Bao, T. Ma, Z. Dang, Y. Guan, X. S. Zheng, X. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., Int. Ed., 2020, 59, 8706–8712 CrossRef CAS PubMed.
- Q. Wan, J. Zhang, B. Zhang, D. Tan, L. Yao, L. Zheng, F. Zhang, L. Liu, X. Cheng and B. Han, Green Chem., 2020, 22, 2750–2754 RSC.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- X. Jiang, F. Cai, D. Gao, J. Dong, S. Miao, G. Wang and X. Bao, Electrochem. Commun., 2016, 68, 67–70 CrossRef CAS.
- W. Luo, Q. Zhang, J. Zhang, E. Moioli, K. Zhao and A. Züttel, Appl. Catal., B, 2020, 273, 119060 CrossRef CAS.
- Y. Wang, H. Liu, J. Yu, B. Hu, H. Zhao, P. Tsiakaras and S. Song, Electrochim. Acta, 2019, 328, 135083 CrossRef CAS.
- C. Li, T. Hisatomi, O. Watanabe, M. Nakabayashi, N. Shibata, K. Domen and J.-J. Delaunay, Energy Environ. Sci., 2015, 8, 1493–1500 RSC.
- C. Li, Y. Li and J. J. Delaunay, ACS Appl. Mater. Interfaces, 2014, 6, 480–486 CrossRef CAS PubMed.
- H. Ramezani, D. N. Christodoulides, V. Kovanis, I. Vitebskiy and T. Kottos, Phys. Rev. Lett., 2012, 109, 033902 CrossRef PubMed.
- C. Li, H. Yamahara, Y. Lee, H. Tabata and J. J. Delaunay, Nanotechnology, 2015, 26, 305503 CrossRef PubMed.
- Y. Huang, Y. Deng, A. D. Handoko, G. K. L. Goh and B. S. Yeo, ChemSusChem, 2018, 11, 320–326 CrossRef CAS PubMed.
- M. Zhao, Y. Gu, W. Gao, P. Cui, H. Tang, X. Wei, H. Zhu, G. Li, S. Yan, X. Zhang and Z. Zou, Appl. Catal., B, 2020, 266, 118625 CrossRef CAS.
- H. Wang, E. Matios, C. Wang, J. Luo, X. Lu, X. Hu and W. Li, Nano Lett., 2019, 19, 3925–3932 CrossRef CAS PubMed.
- Y. Zhang, L. Ji, W. Qiu, X. Shi, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 2666–2669 RSC.
- H. Mistry, Y. W. Choi, A. Bagger, F. Scholten, C. S. Bonifacio, I. Sinev, N. J. Divins, I. Zegkinoglou, H. S. Jeon, K. Kisslinger, E. A. Stach, J. C. Yang, J. Rossmeisl and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2017, 56, 11394–11398 CrossRef CAS PubMed.
- B. Kumar, V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara and J. M. Spurgeon, Angew. Chem., Int. Ed., 2017, 56, 3645–3649 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- F. Scholten, I. Sinev, M. Bernal and B. Roldan Cuenya, ACS Catal., 2019, 9, 5496–5502 CrossRef CAS PubMed.
- A. Paracchino, J. C. Brauer, J.-E. Moser, E. Thimsen and M. Graetzel, J. Phys. Chem. C, 2012, 116, 7341–7350 CrossRef CAS.
- R. Kas, R. Kortlever, A. Milbrat, M. T. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- J. Shi, D. Shao, J. Zhang, D. Tan, X. Tan, B. Zhang, B. Han, F. Zhang, L. Liu and X. Cheng, Chem. Commun., 2018, 54, 5450–5453 RSC.
- D. L. T. Nguyen, M. S. Jee, D. H. Won, H. Jung, H.-S. Oh, B. K. Min and Y. J. Hwang, ACS Sustainable Chem. Eng., 2017, 5, 11377–11386 CrossRef CAS.
- D. E. Starr, Z. Liu, M. Havecker, A. Knop-Gericke and H. Bluhm, Chem. Soc. Rev., 2013, 42, 5833–5857 RSC.
- A. Eilert, F. Cavalca, F. S. Roberts, J. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel, L. G. Pettersson and A. Nilsson, J. Phys. Chem. Lett., 2017, 8, 285–290 CrossRef CAS PubMed.
- L. Nguyen, F. F. Tao, Y. Tang, J. Dou and X. J. Bao, Chem. Rev., 2019, 119, 6822–6905 CrossRef PubMed.
- E. J. Crumlin, H. Bluhm and Z. Liu, J. Electron Spectrosc. Relat. Phenom., 2013, 190, 84–92 CrossRef CAS.
- M. Favaro, J. Yang, S. Nappini, E. Magnano, F. M. Toma, E. J. Crumlin, J. Yano and I. D. Sharp, J. Am. Chem. Soc., 2017, 139, 8960–8970 CrossRef CAS PubMed.
- L. Cao, Q. Luo, W. Liu, Y. Lin, X. Liu, Y. Cao, W. Zhang, Y. Wu, J. Yang, T. Yao and S. Wei, Nat. Catal., 2018, 2, 134–141 CrossRef.
- A. Dutta, A. Kuzume, M. Rahaman, S. Vesztergom and P. Broekmann, ACS Catal., 2015, 5, 7498–7502 CrossRef CAS.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- X. Chang, Y. Zhao and B. Xu, ACS Catal., 2020, 10, 13737–13747 CrossRef CAS.
- K. Kuruvinashetti, Y. Zhang, J. Li and N. Kornienko, New J. Chem., 2020, 44, 19953–19960 RSC.
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- N. P. Finkelstein and R. D. Hancock, Gold Bull., 1974, 7, 72–77 CrossRef CAS.
- I. S. Brandt, M. A. Tumelero, S. Pelegrini, G. Zangari and A. A. Pasa, J. Solid State Electrochem., 2017, 21, 1999–2020 CrossRef CAS.
- E. McCafferty, in Introduction to Corrosion Science, Springer New York, New York, 2010, pp. 95–117, DOI:10.1007/978-1-4419-0455-3_6.
- H.-H. Huang, Metals, 2016, 6, 23 CrossRef.
- B. Beverskog and I. Puigdomenech, Pourbaix Diagrams for the System Copper-Chlorine at 5-100 °C, 1998 Search PubMed.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion Engineers, Houston, Texas, 1974 Search PubMed.
- D. Kim, S. Lee, J. D. Ocon, B. Jeong, J. K. Lee and J. Lee, Phys. Chem. Chem. Phys., 2015, 17, 824–830 RSC.
- L. Wang, K. Gupta, J. B. M. Goodall, J. A. Darr and K. B. Holt, Faraday Discuss., 2017, 197, 517–532 RSC.
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS PubMed.
- M. S. Jee, H. Kim, H. S. Jeon, K. H. Chae, J. Cho, B. K. Min and Y. J. Hwang, Catal. Today, 2017, 288, 48–53 CrossRef CAS.
- F. Cavalca, R. Ferragut, S. Aghion, A. Eilert, O. Diaz-Morales, C. Liu, A. L. Koh, T. W. Hansen, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. C, 2017, 121, 25003–25009 CrossRef CAS.
- S. Lee, D. Kim and J. Lee, Angew. Chem., Int. Ed., 2015, 54, 14701–14705 CrossRef CAS PubMed.
- R. Imani, Z. Qiu, R. Younesi, M. Pazoki, D. L. A. Fernandes, P. D. Mitev, T. Edvinsson and H. Tian, Nano Energy, 2018, 49, 40–50 CrossRef CAS.
- X. An, S. Li, A. Yoshida, T. Yu, Z. Wang, X. Hao, A. Abudula and G. Guan, ACS Appl. Mater. Interfaces, 2019, 11, 42114–42122 CrossRef CAS PubMed.
- K. Ye, Z. Zhou, J. Shao, L. Lin, D. Gao, N. Ta, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 4814–4821 CrossRef CAS PubMed.
- Y. W. Choi, F. Scholten, I. Sinev and B. Roldan Cuenya, J. Am. Chem. Soc., 2019, 141, 5261–5266 CrossRef CAS PubMed.
- Q. Zhu, D. Yang, H. Liu, X. Sun, C. Chen, J. Bi, J. Liu, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 8896–8901 CrossRef CAS PubMed.
- X. Zheng, P. De Luna, F. P. García de Arquer, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li, M. Liu, O. Voznyy, C. T. Dinh, T. Zhuang, P. Stadler, Y. Cui, X. Du, P. Yang and E. H. Sargent, Joule, 2017, 1, 794–805 CrossRef CAS.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- A. Aljabour, H. Coskun, X. Zheng, M. G. Kibria, M. Strobel, S. Hild, M. Kehrer, D. Stifter, E. H. Sargent and P. Stadler, ACS Catal., 2019, 10, 66–72 CrossRef.
- P. Abbasi, M. Asadi, C. Liu, S. Sharifi-Asl, B. Sayahpour, A. Behranginia, P. Zapol, R. Shahbazian-Yassar, L. A. Curtiss and A. Salehi-Khojin, ACS Nano, 2017, 11, 453–460 CrossRef CAS PubMed.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Kral, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 CrossRef CAS PubMed.
- C. Li, G. Shen, R. Zhang, D. Wu, C. Zou, T. Ling, H. Liu, C. Dong and X.-W. Du, J. Mater. Chem. A, 2019, 7, 1418–1423 RSC.
- F. P. Garcia de Arquer, O. S. Bushuyev, P. De Luna, C. T. Dinh, A. Seifitokaldani, M. I. Saidaminov, C. S. Tan, L. N. Quan, A. Proppe, M. G. Kibria, S. O. Kelley, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1802858 CrossRef PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef PubMed.
- R. Wang, H. Haspel, A. Pustovarenko, A. Dikhtiarenko, A. Russkikh, G. Shterk, D. Osadchii, S. Ould-Chikh, M. Ma, W. A. Smith, K. Takanabe, F. Kapteijn and J. Gascon, ACS Energy Lett., 2019, 4, 2024–2031 CrossRef CAS.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu, K. Daasbjerg, T. Skrydstrup and N. Lock, Adv. Funct. Mater., 2020, 30, 1910408 CrossRef CAS.
- T. Fan, Q. Wu, Z. Yang, Y. Song, J. Zhang, P. Huang, Z. Chen, Y. Dong, W. Fang and X. Yi, ChemSusChem, 2020, 13, 2677–2683 CrossRef CAS PubMed.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, ACS Nano, 2017, 11, 4825–4831 CrossRef CAS PubMed.
- C. Liu, M. P. Lourenço, S. Hedström, F. Cavalca, O. Diaz-Morales, H. A. Duarte, A. Nilsson and L. G. M. Pettersson, J. Phys. Chem. C, 2017, 121, 25010–25017 CrossRef CAS.
- Z. Gu, N. Yang, P. Han, M. Kuang, B. Mei, Z. Jiang, J. Zhong, L. Li and G. Zheng, Small Methods, 2019, 3, 1800449 CrossRef.
- P. Yang, Z. J. Zhao, X. Chang, R. Mu, S. Zha, G. Zhang and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 7724–7728 CrossRef CAS PubMed.
- L. Qi, S. Liu, W. Gao and Q. Jiang, J. Phys. Chem. C, 2018, 122, 5472–5480 CrossRef CAS.
- X. Zhi, Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, J. Mater. Chem. A, 2021, 9, 6345–6351 RSC.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- D. Liu, Y. Liu and M. Li, J. Phys. Chem. C, 2020, 124, 6145–6153 CrossRef CAS.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- J. Gao, C. Zhu, M. Zhu, Y. Fu, H. Huang, Y. Liu and Z. Kang, ACS Sustainable Chem. Eng., 2019, 7, 3536–3543 CrossRef CAS.
- L. Fan, Z. Xia, M. Xu, Y. Lu and Z. Li, Adv. Funct. Mater., 2018, 28, 1706289 CrossRef.
- M. K. Kim, H. J. Kim, H. Lim, Y. Kwon and H. M. Jeong, Electrochim. Acta, 2019, 306, 28–34 CrossRef CAS.
- D. Wu, J. Liu, Y. Liang, K. Xiang, X. Z. Fu and J. L. Luo, ChemSusChem, 2019, 12, 4700–4707 CrossRef CAS PubMed.
- S. D. Giri, S. M. Mahajani, A. K. Suresh and A. Sarkar, Mater. Res. Bull., 2020, 123, 110702 CrossRef CAS.
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55, 15282–15286 CrossRef CAS PubMed.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- C. Chen, B. Zhang, J. Zhong and Z. Cheng, J. Mater. Chem. A, 2017, 5, 21955–21964 RSC.
- S. Suter and S. Haussener, Energy Environ. Sci., 2019, 12, 1668–1678 RSC.
- F. Calle-Vallejo and M. T. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed.
- J. H. Montoya, C. Shi, K. Chan and J. K. Norskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- M. S. Jee, H. S. Jeon, C. Kim, H. Lee, J. H. Koh, J. Cho, B. K. Min and Y. J. Hwang, Appl. Catal., B, 2016, 180, 372–378 CrossRef CAS.
- W. J. Dong, C. J. Yoo and J.-L. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 43575–43582 CrossRef CAS PubMed.
- J. J. Velasco-Velez, C. H. Chuang, D. Gao, Q. Zhu, D. Ivanov, H. S. Jeon, R. Arrigo, R. V. Mom, E. Stotz, H. L. Wu, T. E. Jones, B. Roldan Cuenya, A. Knop-Gericke and R. Schlogl, ACS Catal., 2020, 10, 11510–11518 CrossRef CAS PubMed.
- A. Dutta, I. Z. Montiel, R. Erni, K. Kiran, M. Rahaman, J. Drnec and P. Broekmann, Nano Energy, 2020, 68, 104331 CrossRef CAS.
- P. Wang, M. Qiao, Q. Shao, Y. Pi, X. Zhu, Y. Li and X. Huang, Nat. Commun., 2018, 9, 4933 CrossRef PubMed.
- S. C. Abeyweera, J. Yu, J. P. Perdew, Q. Yan and Y. Sun, Nano Lett., 2020, 20, 2806–2811 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS PubMed.
- R. G. Mariano, M. Kang, O. J. Wahab, I. J. McPherson, J. A. Rabinowitz, P. R. Unwin and M. W. Kanan, Nat. Mater., 2021, 20, 1000–1006 CrossRef CAS PubMed.
- J. J. Velasco-Velez, R. V. Mom, L. E. Sandoval-Diaz, L. J. Falling, C. H. Chuang, D. Gao, T. E. Jones, Q. Zhu, R. Arrigo, B. Roldan Cuenya, A. Knop-Gericke, T. Lunkenbein and R. Schlogl, ACS Energy Lett., 2020, 5, 2106–2111 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 11642–11645 CrossRef CAS PubMed.
- L. Li, D.-K. Ma, F. Qi, W. Chen and S. Huang, Electrochim. Acta, 2019, 298, 580–586 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J. L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- H. Won da, H. Shin, J. Koh, J. Chung, H. S. Lee, H. Kim and S. I. Woo, Angew. Chem., Int. Ed., 2016, 55, 9297–9300 CrossRef PubMed.
- Y. G. Kim, J. H. Baricuatro, A. Javier, J. M. Gregoire and M. P. Soriaga, Langmuir, 2014, 30, 15053–15056 CrossRef CAS PubMed.
- Y.-G. Kim, A. Javier, J. H. Baricuatro and M. P. Soriaga, Electrocatalysis, 2016, 7, 391–399 CrossRef CAS.
- C. F. Tsang, A. C. Javier, Y.-G. Kim, J. H. Baricuatro, K. D. Cummins, J. Kim, G. Jerkiewicz, J. C. Hemminger and M. P. Soriaga, J. Electrochem. Soc., 2018, 165, J3350–J3354 CrossRef CAS.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2019, 3, 98–106 CrossRef.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS PubMed.
- M. M. Sartin, Z. Yu, W. Chen, F. He, Z. Sun, Y.-X. Chen and W. Huang, J. Phys. Chem. C, 2018, 122, 26489–26498 CrossRef CAS.
- R. M. Aran-Ais, R. Rizo, P. Grosse, G. Algara-Siller, K. Dembele, M. Plodinec, T. Lunkenbein, S. W. Chee and B. R. Cuenya, Nat. Commun., 2020, 11, 3489 CrossRef CAS PubMed.
- T. Moller, F. Scholten, T. N. Thanh, I. Sinev, J. Timoshenko, X. Wang, Z. Jovanov, M. Gliech, B. Roldan Cuenya, A. S. Varela and P. Strasser, Angew. Chem., Int. Ed., 2020, 59, 17974–17983 CrossRef PubMed.
- A. S. Malkani, M. Dunwell and B. Xu, ACS Catal., 2018, 9, 474–478 CrossRef.
- Y. Hori, O. Koga, Y. Watanabe and T. Matsuo, Electrochim. Acta, 1998, 44, 1389–1395 CrossRef CAS.
- N. Hoshi, M. Kato and Y. Hori, J. Electroanal. Chem., 1997, 440, 283–286 CrossRef CAS.
- S. Popovic, M. Smiljanic, P. Jovanovic, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef CAS PubMed.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479–4484 CrossRef CAS.
- J. He, A. Huang, N. J. J. Johnson, K. E. Dettelbach, D. M. Weekes, Y. Cao and C. P. Berlinguette, Inorg. Chem., 2018, 57, 14624–14631 CrossRef CAS PubMed.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed.
- J. Huang, N. Hormann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed.
- Y. Zhang, L. Hu and W. Han, J. Mater. Chem. A, 2018, 6, 23610–23620 RSC.
- C. S. Le Duff, M. J. Lawrence and P. Rodriguez, Angew. Chem., Int. Ed., 2017, 56, 12919–12924 CrossRef CAS PubMed.
- A. Engelbrecht, M. Hämmerle, R. Moos, M. Fleischer and G. Schmid, Electrochim. Acta, 2017, 224, 642–648 CrossRef CAS.
- K. Obata and K. Takanabe, Angew. Chem., Int. Ed., 2018, 57, 1616–1620 CrossRef CAS PubMed.
- D. V. Esposito, ACS Catal., 2017, 8, 457–465 CrossRef.
- N. Y. Labrador, E. L. Songcuan, C. De Silva, H. Chen, S. J. Kurdziel, R. K. Ramachandran, C. Detavernier and D. V. Esposito, ACS Catal., 2018, 8, 1767–1778 CrossRef CAS.
- J. E. Robinson, N. Y. Labrador, H. Chen, B. E. Sartor and D. V. Esposito, ACS Catal., 2018, 8, 11423–11434 CrossRef CAS.
- A. A. Bhardwaj, J. G. Vos, M. E. S. Beatty, A. F. Baxter, M. T. M. Koper, N. Y. Yip and D. V. Esposito, ACS Catal., 2021, 11, 1316–1330 CrossRef CAS.
- P. F. Liu, M. Y. Zu, L. R. Zheng and H. G. Yang, Chem. Commun., 2019, 55, 12392–12395 RSC.
- C. Cao, D.-D. Ma, J.-F. Gu, X. Xie, G. Zeng, X. Li, S.-G. Han, Q.-L. Zhu, X.-T. Wu and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 15014–15020 CrossRef CAS PubMed.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |