
Carbocatalytic ozonation toward advanced water purification
Ya
Liu
ab,
Chunmao
Chen
a,
Xiaoguang
Duan
 b,
Shaobin
Wang
b and
Yuxian
Wang
*a
b,
Shaobin
Wang
b and
Yuxian
Wang
*a
aState Key Laboratory of Heavy Oil Processing, State Key Laboratory of Petroleum Pollution Control, China University of Petroleum-Beijing, Beijing 102249, China. E-mail: yuxian.wang@cup.edu.cn
bSchool of Chemical Engineering and Advanced Materials, The University of Adelaide, Adelaide, SA 5005, Australia
First published on 1st June 2021
Abstract
Carbon-based catalytic ozonation is state-of-the-art technology with high oxidation capabilities for wastewater remediation, taking advantage of the synergies of direct ozone oxidation and the generated reactive oxygen species (ROS). Replacing metal-based materials with active and robust carbonaceous catalysts in catalytic ozonation will lead to outperformed catalytic performance, minimized operational cost, and no secondary contamination. Additionally, the variety of allotropes and maneuverable surface chemistry of carbons facilitate structural and surface engineering, which enables the regulation of reactivity, stability and reaction pathways. This review summarizes the recent development of structural control and surface modification of carbocatalysts and their applications in catalytic ozonation. The structure–performance relations and mechanisms are elucidated by a novel model based on the interaction intensity between reactants and carbon surface. Meanwhile, influences of the water matrix parameters on the catalytic system are unveiled. Finally, we provide directions to the rational design of reaction-oriented carbocatalysts, the methodology for mechanistic explorations, and the implementation of ozone-based AOPs in real wastewater treatment.
 Yuxian Wang | Yuxian Wang received his B.Eng. with first-class honor and PhD from Curtin University in 2013 and 2016, respectively. He then joined the College of Chemical and Environmental Engineering, China University of Petroleum-Beijing as the associate professor. His current research interests include seeking feasible applications of nanomaterial-based advanced oxidation processes (AOPs) in environmental remediation and facilitating mechanistic insights into carbocatalysis with advanced protocols. |
1. Introduction
Advanced oxidation processes (AOPs) exploit reactive oxygen species (ROS), such as hydroxyl radicals (HO˙, E0 = 2.7 V, NHE) and sulfate radicals (SO4˙−, E0 = 2.5–3.1 V, NHE) generated from diverse peroxides and have emerged as powerful techniques in wastewater decontamination.1,2 Among AOPs, ozonation treatments relying on the high oxidation potential of O3 (E0 = 2.05 V, NHE) can directly destruct unsaturated organics via the Criegee reaction and outperform other AOPs in terms of low energy demand and facile implementation at a full-scale.3,4 However, the selectivity and low solubility of O3 in the aqueous medium limited the mineralization efficiency of the ozonation process.5 Catalytic ozonation treatments achieve improved reaction kinetics and mineralization efficiency compared with ozonation treatment. The heterogeneous catalysts in the ozonated solutions accelerate O3 activation into ROS, which possess greater oxidation capacities and are less selective, thus facilitating the interactions of O3 molecules and organic contaminants.6 Additionally, the tunable oxidation regimes in catalytic ozonation improve the adaptability to complicated water matrixes. Various metal-based heterogeneous catalysts have been developed for O3 activation, yet the secondary pollution arising from metal leaching becomes the bottleneck in practical applications.7The past two decades witness a rapid development of carbons to replace conventional metal-based systems in environmental remediations on account of fast reaction kinetics and environmentally benign nature of carbocatalysis.8 As a result, various carbonaceous materials, including bulky carbons (e.g., activated carbon, biochar and carbon fiber) and novel structured nanocarbons (e.g., carbon nanotubes, graphene-based materials and three-dimensional porous nanocarbons), have been applied in heterogeneous catalytic ozonation (Fig. 1).9 A broad range of allotropes with different dimensions and porous structures facilitate the in-depth investigation of the mechanism of dimensional effects on catalytic performance. Additionally, the facilely-tuned surface chemistry together with the large surface area of carbonaceous materials provide an advanced platform to build and maneuver surface engineered active sites such as defects, surface functionalities, heteroatom dopants, and anchored metal species.10,11 Hence, the engineered carbonaceous materials with optimized physiochemical and electronic properties stimulate O3 activation to induce either radical or nonradical-based reaction pathways to destroy the recalcitrant organics.
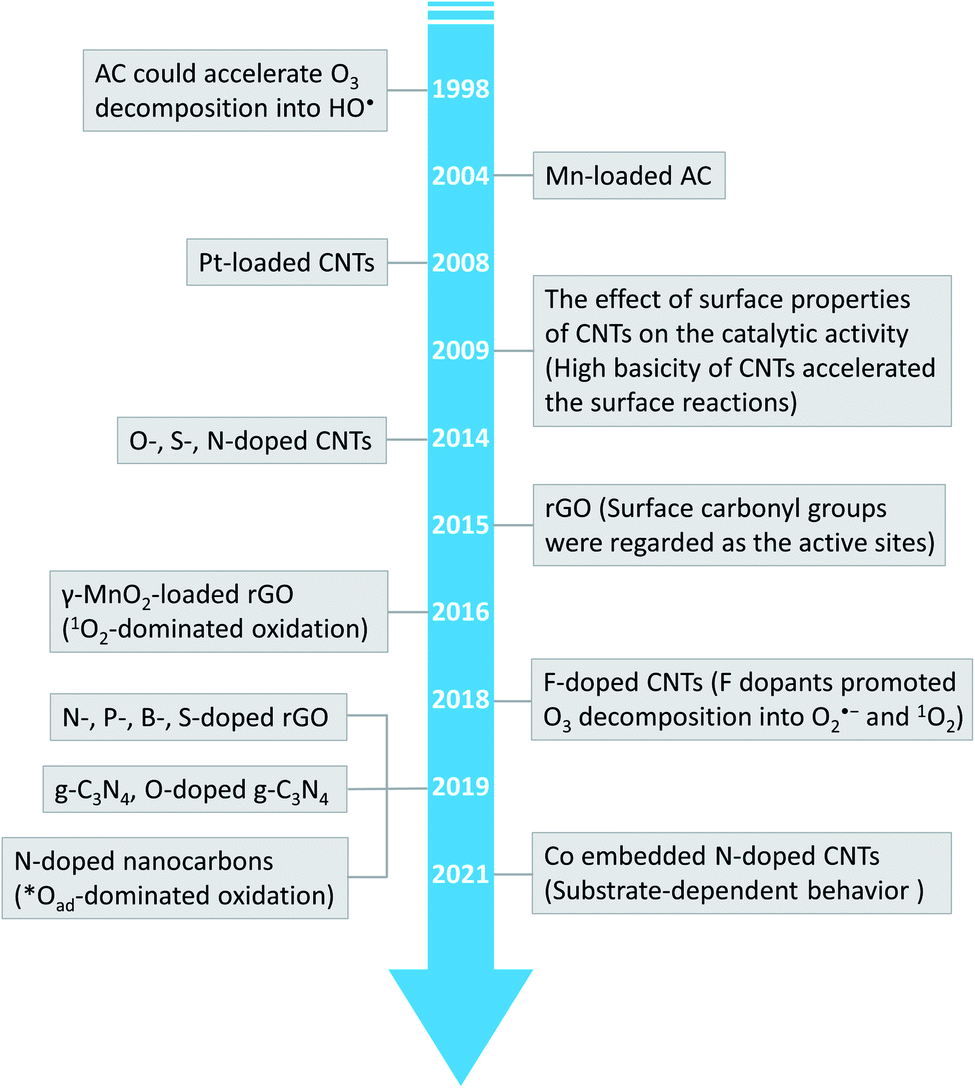 | ||
| Fig. 1 Timeline of the important development of carbon-based catalysts in catalytic ozonation. | ||
The innovative carbocatalysis in AOPs significantly expedites the development of new carbocatalysts and excavation of the underlying mechanisms. Till now, several reviews have been reported to summarize the development of carbocatalysis in wastewater decontamination,9,12–14 while few focused on the application of carbonaceous materials in catalytic ozonation. Recently, two reviews dedicated to the mechanistic investigations on nanocarbon-based catalytic ozonation.15,16 To the best of our knowledge, there is still lack of a review that comprehensively summarizes the recent advances in carbon-based catalytic ozonation treatments and the synthesis/modification strategies for advanced design of carbocatalysts with desirable activity, selectivity, and stability. In addition, the effects of water matrix parameters such as solution pH and background organic/inorganic substances on the performance of carbon-based catalytic ozonation treatments are rarely mentioned.
In this review, apart from showcasing the roadmap of catalytic ozonation treatments using different carbon allotropes, we will discuss the catalytic origin and establish the surface engineering strategies for rational design and fast screen of the highly efficient carbocatalysts. For better elucidation of the mechanistic insights into ROS generation and differentiation of reaction pathways, a novel concept based on the intensity of interactions between O3 and catalysts surface will be proposed. The environmental implications of solution pH, ionic strength, and other background substances such as inorganic anions and natural organic matters (NOMs) on the efficiency of the carbon-based catalytic ozonation treatments will be analyzed. Furthermore, the challenges and future research opportunities for the elaborate design of reaction-oriented carbocatalysts and systematically unravelling the mechanistic insights into carbon-based catalytic ozonation will be envisaged.
2. Synthesis and surface engineering protocols for surface active sites
In general, pristine carbon catalysts are catalytically inactive and their catalytic performances are lower than those of metal-based homogeneous and heterogeneous catalysts. For example, pristine carbon nanotubes (CNTs) obtained by the chemical vapor deposition (CVD) method generally possess a low density of functional groups with C–H terminations on the surface and poor dispersibility in aqueous solution.11,17 Surface chemistry and pore structure are key properties of activated carbon (AC) that directly influence their performances in various applications.18 Thus, surface modification and functionalization are conducive to promoting their catalytic performances in the development of highly effective carbon-based materials. Diverse modification methods have been summarized in several reviews,8,17,19,20 in which all modification principles are dedicated to creation/tailoring of catalytically active sites and electronic structures of the carbocatalysts. To rationally design and synthesize catalysts with improved activity, selectivity, and stability for a target application, it is crucial to unravel the catalytic origin and intrinsic active sites that govern the catalytic activity.2.1 Carbon dimensions
Dimensional effects have a great impact on the catalytic activities of catalysts, especially for carbon materials (Fig. 2a). The pristine graphene network with free-flowing π electrons is beneficial to the adsorption and activation of reactants and the electron transfer process. In the catalytic ozonation processes, the conjugated π system of the graphitic carbon framework facilitates the adsorption and activation of O3 molecules.21 The difference in the electron density between carbocatalysts and O3/organics is of great significance to the strength of charge-transfer interactions.22 Generally speaking, strong activation/binding is more likely to occur when charge density difference is significant.23,24 Moreover, the highly delocalized π system endows sp2 carbon with a faster electron transfer rate, which helps the redox reactions for O3 activation and organics oxidation.25 The delocalized π electrons were also reported to be closely correlative to superoxide radical (O2˙−) formation in catalytic O3 decomposition.26 On the other hand, the carbon framework is able to function as a charge-transfer mediator that induces the direct charge transport from the co-adsorbed organics (the electron donor) to oxidants (the electron acceptor) via the surface region of the carbocatalyst to achieve contaminant destruction.27 In addition, aromatic organics tend to be adsorbed or activated on the basal plane due to the π–π interactions or electrostatic interactions, followed by an electrophilic attack by ROS.16 It is worth mentioning that the π orbitals are perpendicular to the graphene layer in graphene, while in CNTs, the curvature structures induce a bent π system and the redistributed electron density from the inner to the outer surface, thereby affecting the catalytic activity.28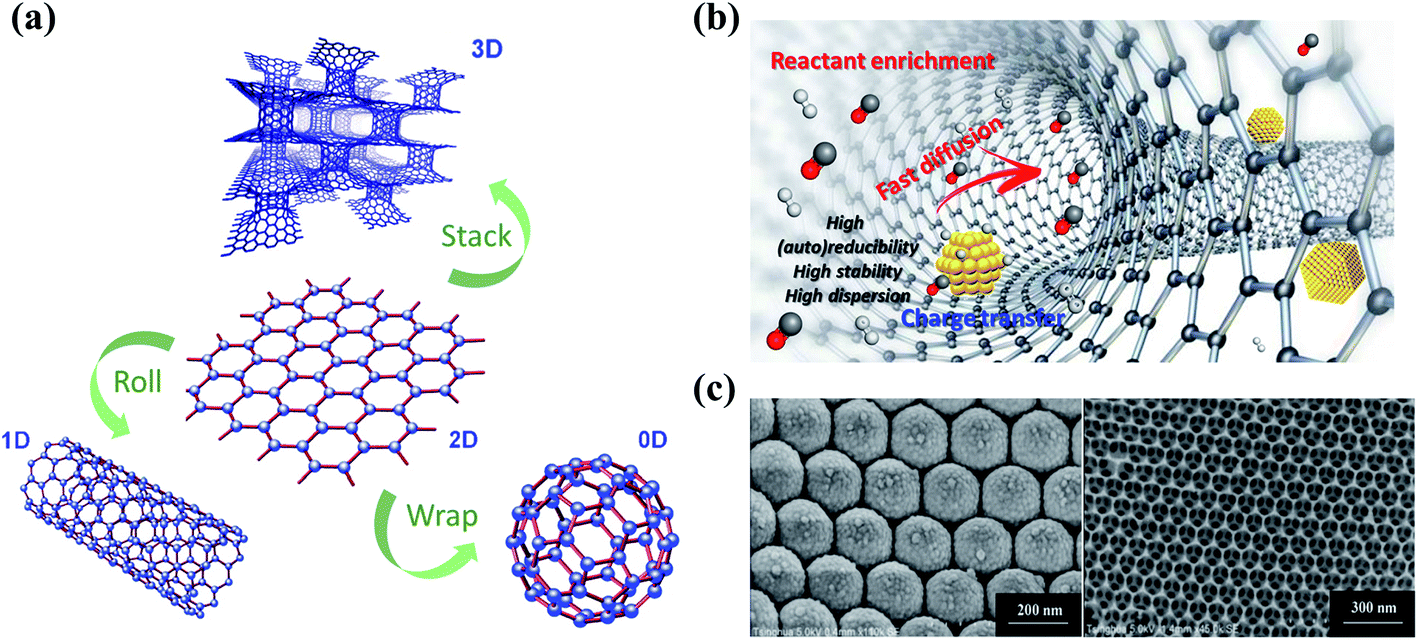 | ||
| Fig. 2 (a) Construction of different dimensional carbon materials based on graphene. Elements are adapted with permission from ref. 32. Copyright 2012, American Chemical Society; adapted with permission from ref. 33. Copyright 2012, Elsevier. (b) Nanoconfinement effects in carbon nanotubes. Reprinted with permission from ref. 28. Copyright 2020, American Chemical Society. (c) HRTEM images of 3DOM structures. Reprinted with permission from ref. 34. Copyright 2015, Wiley-VCH. | ||
Engineering three-dimensional (3D) carbon catalysts with mesoporous and macroporous structures is a promising strategy to improve the catalytic activity by shortening the mass transport/diffusion length, increasing specific surface area (SSA) and the number of exposed active sites, and further accelerating the reaction kinetics. Nanoconfinement effects are also expected in the porous structure, which facilitate both mass and charge transfer (Fig. 2b). 3D hierarchical carbon structures further enhance electron mobility and facilitate the adsorption of reactants in water media.16 In the presence of metal catalysts at a high pyrolysis temperature, 3D hybrid nanostructures can be synthesized by intercalating one-dimensional (1D) wirelike nanocarbons between the stacked two-dimensional (2D) sheets (Fig. 2a).29 The 3D ordered macroporous (3DOM) carbon architecture with an increased number of active sites and enlarged SSA also demonstrates a favorable application in environmental catalysis (Fig. 2c).30,31
2.2 Surface functionalization
The surface chemistry such as surface charge and the electronic properties of the carbon materials is largely dependent on the surface functionalities, especially the surface oxygen functionalities, which have been proposed as the active sites in various carbon-based catalysts (Fig. 3a). Carbonyl groups (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) with high basicity and nucleophilicity are catalytically active. It was found that reduced graphene oxide (rGO)/g-C3N4 with a higher content of carbonyl groups exhibited better catalytic performance toward O3 activation.35,36 The electron-rich carbonyl groups with lone-pair electrons can act as Lewis basic sites to activate O3 by an electron-transfer process. Similarly, carbonyl groups are critical active sites in persulfate activation. It was reported that carbonyl groups located at the boundaries showed a high affinity to interact with peroxymonosulfate (PMS), leading to the significant elongation of the peroxide O–O bond in PMS.37,38 Carboxyl groups (–COOH) can act as Lewis basic sites to facilitate O3 activation due to its similar properties with –CO in catalytic ozonation systems. Qu et al. reported that carboxyl functionalized CNTs (CNTs-COOH) possessed a better activity compared with hydroxyl functionalized CNTs (CNTs-OH), because the high charge density of –COOH was beneficial for the direct oxidation of pollutants by O3.39 Moreover, the deprotonated carboxyl groups reacted with O3 to form electrophilic oxygen species, which served as radical promoters to produce HO˙.40,41 Hydroxyl groups (–OH) as electron-donating groups would reinforce the electron density of the conjugated π system to facilitate the electron-transfer process. Ren and co-workers found that, as compared with carbonyl and carboxyl groups, hydroxyl groups on the CNTs surface were more conducive to persulfate adsorption and electron migration in the peroxodisulfate (PDS)/CNTs system.42 In catalytic ozonation, O3 will electrophilically attack the surface –OH groups to generate ROS (HO˙ and O2˙−).43 Furthermore, –OH groups might be oxidized into –CO groups by O3, and the redox cycle of carbonyl–hydroxyl groups is beneficial to O3 activation.44
O) with high basicity and nucleophilicity are catalytically active. It was found that reduced graphene oxide (rGO)/g-C3N4 with a higher content of carbonyl groups exhibited better catalytic performance toward O3 activation.35,36 The electron-rich carbonyl groups with lone-pair electrons can act as Lewis basic sites to activate O3 by an electron-transfer process. Similarly, carbonyl groups are critical active sites in persulfate activation. It was reported that carbonyl groups located at the boundaries showed a high affinity to interact with peroxymonosulfate (PMS), leading to the significant elongation of the peroxide O–O bond in PMS.37,38 Carboxyl groups (–COOH) can act as Lewis basic sites to facilitate O3 activation due to its similar properties with –CO in catalytic ozonation systems. Qu et al. reported that carboxyl functionalized CNTs (CNTs-COOH) possessed a better activity compared with hydroxyl functionalized CNTs (CNTs-OH), because the high charge density of –COOH was beneficial for the direct oxidation of pollutants by O3.39 Moreover, the deprotonated carboxyl groups reacted with O3 to form electrophilic oxygen species, which served as radical promoters to produce HO˙.40,41 Hydroxyl groups (–OH) as electron-donating groups would reinforce the electron density of the conjugated π system to facilitate the electron-transfer process. Ren and co-workers found that, as compared with carbonyl and carboxyl groups, hydroxyl groups on the CNTs surface were more conducive to persulfate adsorption and electron migration in the peroxodisulfate (PDS)/CNTs system.42 In catalytic ozonation, O3 will electrophilically attack the surface –OH groups to generate ROS (HO˙ and O2˙−).43 Furthermore, –OH groups might be oxidized into –CO groups by O3, and the redox cycle of carbonyl–hydroxyl groups is beneficial to O3 activation.44
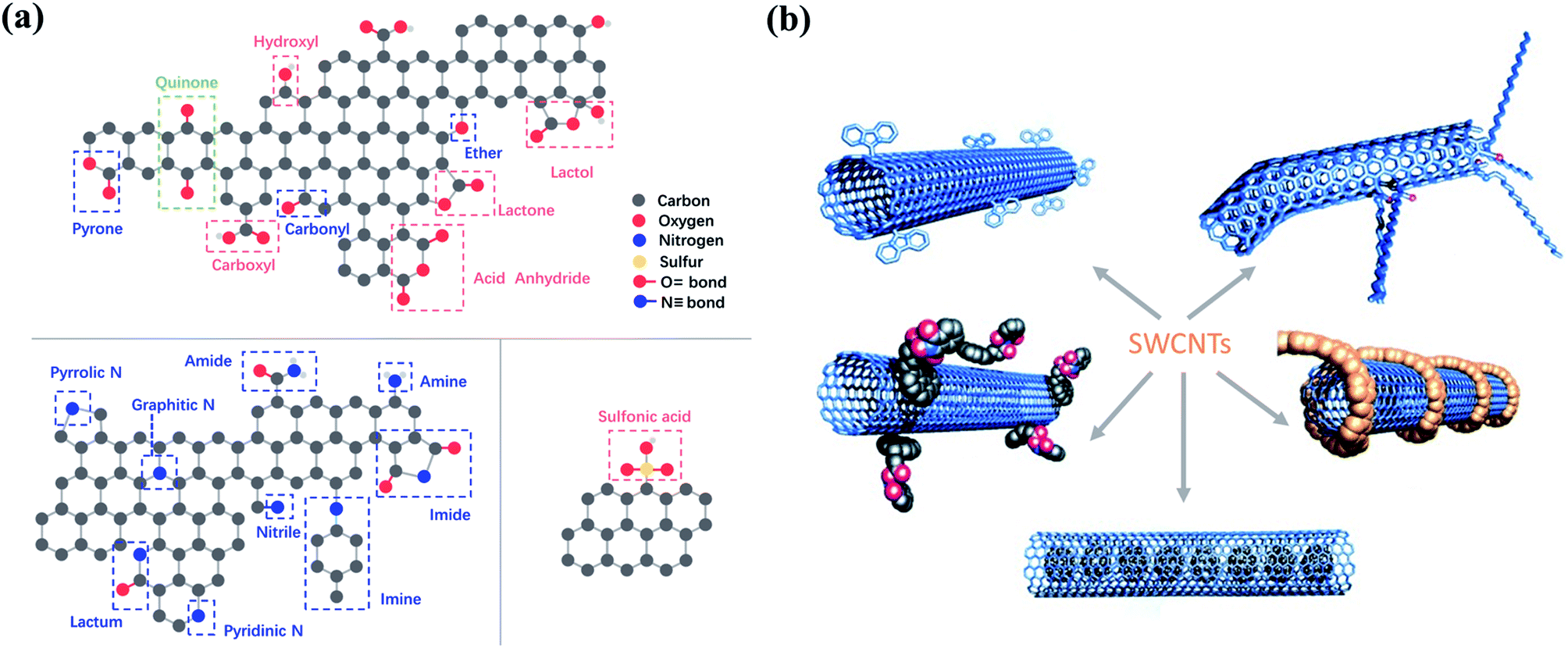 | ||
| Fig. 3 (a) Possible oxygen-, nitrogen- and sulfur-containing functional groups on carbon surface. Reproduced with permission from ref. 49. Copyright 2013, Royal Society of Chemistry. (b) Functionalization strategies for CNTs. Adapted with permission from ref. 50. Copyright 2002, Wiley-VCH. | ||
Tailoring the type and amount of the surface functionalities is a practical step to enhance the catalytic performance (Fig. 3b). Thus, surface engineering is a crucial way to manipulate the surface functionalities, which can be categorized into surface oxidation and reduction modifications.45 Surface oxidative modification generally employs oxidants (e.g., HNO3, KMnO4, H2SO4, H2O2, and O3) to introduce a large number of oxygen-containing groups (e.g., –COOH, –OH, –NO2, –SO3H, etc.) on the carbon surface, thereby increasing the acidic property. During the surface reduction modification, carbon materials are treated at a high temperature in a reducing atmosphere or are reduced in alkaline solutions, leading to the formation of basic surface functionalities (e.g., –CO, –NH2, etc.). Noteworthly, the carbon structure can be entirely damaged by HNO3 at its boiling point.46 For surface-oxidized multi-walled CNTs (MWCNTs), the increased oxygen-containing functionalities (–CO) on MWCNTs facilitated the adsorption of reaction intermediates and thus accelerated the reaction via tuning the electronic structures of the adjacent carbon atoms.47 Graphene modified with amine groups (–NH2) demonstrated better catalytic activity compared with unmodified graphene for persulfates activation.48 Nevertheless, excessive surface functionalities are not desirable.38 Overabundant surface oxygen groups hindered the catalytic activity due to increased electrostatic repulsions and stereo-hindrance effect.42 Moreover, the extra surface oxygen moieties decrease the reductive degree of the carbon lattice for charge transfer and occupy the defective sites or edges, which are also crucial active sites for oxidant activation. Therefore, fabricating carbon materials with optimized oxygen contents to enhance the catalytic performance is highly imperative for future studies. In addition, unveiling the roles of the individual oxygen-containing groups on carbocatalysts is challenging since it is hard to regulate single type of surface functional groups without influencing others.
2.3 Defects engineering
Defect engineering has been demonstrated as an effective strategy to tailor the electronic properties of carbon catalysts. Nanocarbons are inherently produced with edges and topological defects (Fig. 4a). These defective sites with unpaired electrons disturb the homogeneity of carbon skeleton and tune the electronic structures of basal planes (Fig. 4b).51 The catalytic activity of edges originates from the presence of dangling bonds, which are high-energy sites and possess high reactivity toward the reactants to promote the radical chain oxidations. The carbon atoms at the edging sites obtain unpaired electrons in a “localized state”, which strongly interact with the small molecules to form an attached complex.52 It was revealed that the electron transfer at graphene edges was much faster than that on the honeycomb basal plane.53 With the merits, edge sites have been reported to promote O3 adsorption and subsequent activation.25,52,54,55 On the other hand, topological defects (e.g., vacancies and non-hexagonal carbon rings) can break the conjugated π network and redistribute the electrons.53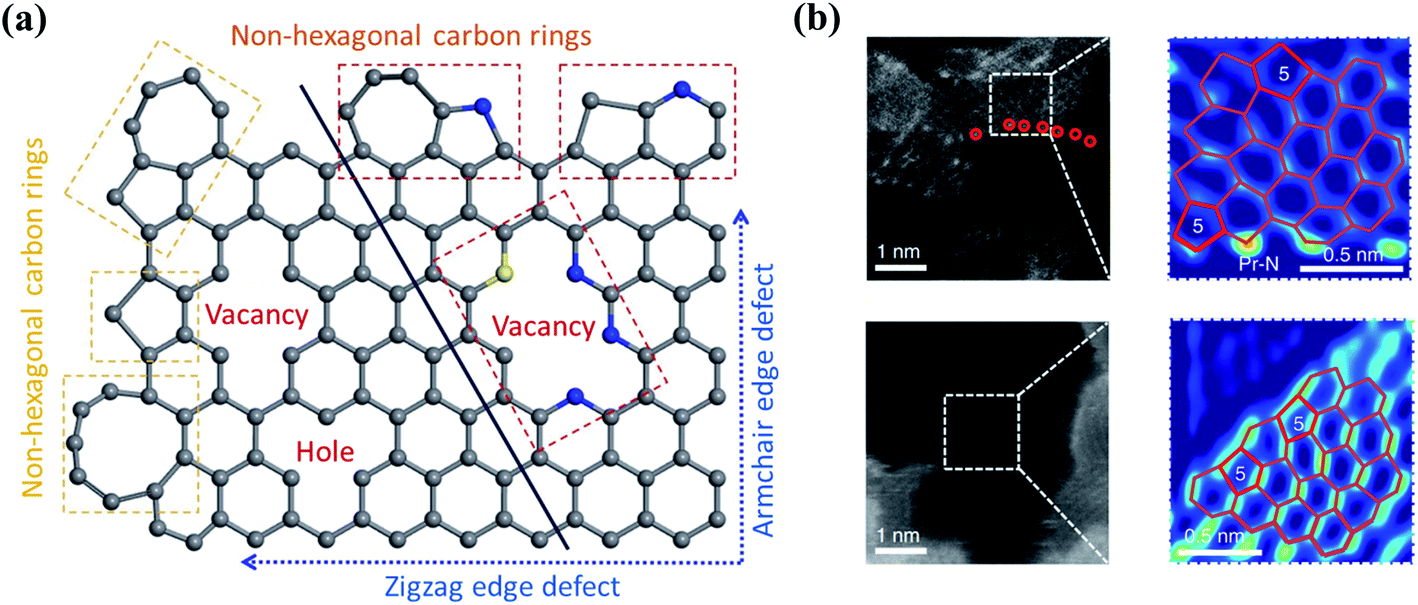 | ||
| Fig. 4 (a) Defect-engineered nanocarbon. Adapted with permission from ref. 61. Copyright 2021, Springer. (b) The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of carbon defects. Reprinted with permission from ref. 62. Copyright 2019, Nature. | ||
Defects in a carbon framework can be further created either by downsizing carbons or by removal of the template materials. A novel catalyst for oxygen reduction reaction (ORR) based on graphene quantum dots (GQDs) supported on graphene nanoribbons (GNRs) was developed through a one-step reduction of methylbenzene and hexabromobenzene by Na.56 Such a treatment resulted in abundant surface and edge defects on the GQD/GNR surfaces or at their interface, which served as active sites to promote charge transfer. During the synthesis of N-doped graphene, both edge and topological defects can be produced by high-temperature pyrolysis for decomposition of the N-rich precursors.57 Meanwhile, topological defects can be created through the direct carbonization of carbon/nitrogen precursors on templates of MgO/Zn-metal–organic frameworks (MOFs).58 Additionally, microwave reduction is an efficient method to synthesize rGO with abundant structural defects, attributed to the microwave absorbing property of graphene oxide (GO) and the severe and uniform heating ambience provided by microwave irradiation.59 Nevertheless, these carbocatalysts were fabricated under high temperatures or using template precursors, in which an accurate control of the generated defects was difficult. Plasma technology for etching the surface of nanocarbons has been proposed as an effective strategy to prepare defect-rich electrocatalysts in a controllable manner by altering treatment time and plasma power.60 It is worth mentioning that, although nanocarbons with high-defect levels obtained good reactivities in AOPs, they usually show poor stabilities, because the unsaturated dangling bond and long-paired electrons on defects would induce the electrophilic attack of the oxidants. Moreover, defects might also be changed or recombined under the working conditions.60 Hence, manipulating the defective level is required to balance the trade-off between reactivity and stability.
2.4 Heteroatom doping
Introduction of heteroatoms (e.g., B, N, F, P or S) to the carbon skeleton results in charge/spin redistribution and structural disorder because of the differences in electronegativities and atomic radius (Fig. 5a).10,63,64 As a result, the heteroatom dopants and the C atoms adjacent to the dopants with variations in electronic properties are often regarded as the active sites favoring the interaction with O3.65 N-dopants alter the charge distribution and the density of states (DoS) of the doping area, owing to the great difference in electronegativity between N and C (3.07 vs. 2.55); N dopants also endow the adjacent C atoms with a highly positive charge density, facilitating the adsorption and activation of the reactants. Moreover, the intrinsic chemical and electronic coupling induced by N-doping synergistically promoted proton adsorption and the reduction kinetics.66 Meanwhile, heteroatom doping results in the redistribution of spin density.64 Although the similar electronegativities of S to C weakened the intramolecular charge transfer, the higher spin density of S-doped graphene than that of pristine graphene (0.39 vs. 0) favored adsorption of reactants.67 Co-doping of the heteroatoms can further improve carbocatalysis due to the synergy between different dopants. The performances of binary (N, P-carbon), ternary (N, P, F-carbon) and quaternary (S, N, B, P-carbon) doped carbon catalysts have well outperformed the single doping counterparts.68–70 In a N, F co-doped system, F dopants tailored the N-doping-induced charge transfer and decreased the work function, which accelerated charge transfer for redox reactions.71 Meanwhile, in N, S co-doped graphene, S atoms could activate the C atoms adjacent to N, which was advantageous for the interaction with PMS to lower the energy barrier for ROS generation.67,72 Additionally, the synergistic effect arising from co-doping dramatically enlarged the positively charged regions. Nevertheless, overloading of the S dopants restrained the synergistic effect due to the redistribution of electrons/spins and reduced active regions (Fig. 5b–e).67 Therefore, the doping species, level and configuration need to be carefully controlled.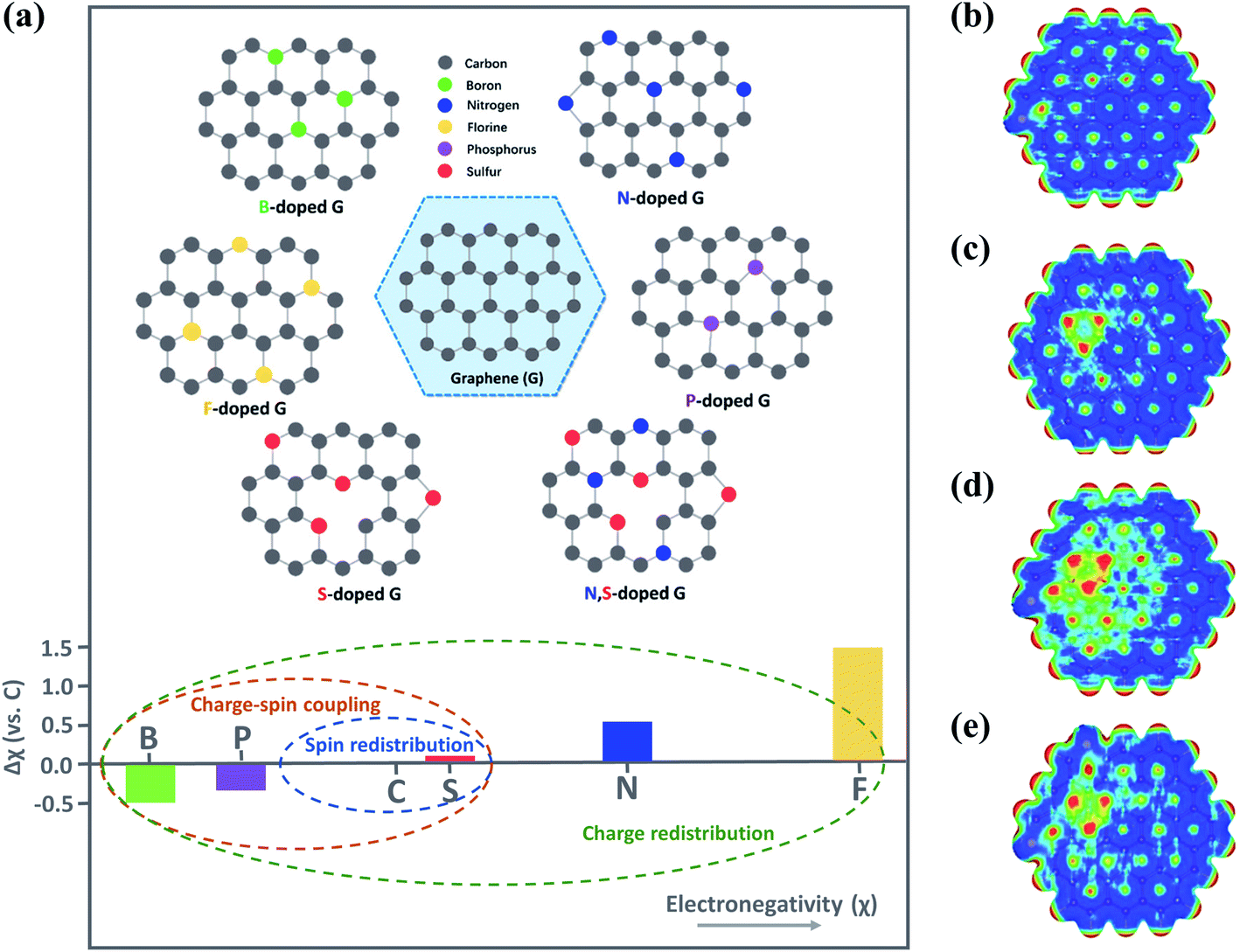 | ||
| Fig. 5 (a) Illustration on heteroatom-doped carbon materials. Reproduced with permission from ref. 64. Copyright 2018, Wiley-VCH. Reprinted with permission from ref. 16. Copyright 2020, American Chemical Society. Electrostatic potential mapping from charge density matrix for (b) S-rGO, (c) N-rGO, (d) S, N-rGO, and (e) S, S, N-rGO. Reprinted with permission from ref. 67. Copyright 2015, Wiley-VCH. | ||
Typically, heteroatom doping could be achieved by two strategies, direct/in situ synthesis and post-treatment.11,73 Direct/in situ synthesis strategy starts from molecular carbon and dopant precursors; the formation of graphitic carbon framework and heteroatom doping occur concurrently at the elevated temperature by CVD, solvothermal, segregation growth, or arc-discharge approaches.73 Homogeneous doping could be attained by precisely controlling the direct/in situ synthesis parameters. The post-treatment method is to pyrolyze the derived carbon materials with heteroatom-rich resources by a hydrothermal treatment, pyrolysis, ball milling method or plasma treatment.
Multiple factors can affect the catalytic performances of heteroatom-doped carbon catalysts. First, the dopant types and the bonding configurations determine the catalytic activity. N doping introduces graphitic, pyridinic, and pyrrolic Ns in the carbon framework, among which the electron-donating graphitic N is more favorable for O3 adsorption. According to density functional theory (DFT) simulation, graphitic N showed a superior catalytic activity to pyridinic and pyrrolic Ns.74 Altering the annealing temperature and ambience could manipulate the types and compositions of the N-dopants because of the different thermal stability of N species.75,76 Second, the content of heteroatom dopants can also influence doping performance. Although increasing the N content in N-doped CNTs was reported to enhance the electrocatalytic activity,77 the excessive doping amount might hinder the catalytic activity because of the resulted unfavorable electronic structure and poor structural robustness.78 Therefore, it is significant to optimize the doping content by tuning the amount of heteroatom-containing precursors, substrate structures and treatment parameters. Third, catalytic activity may vary with the dopant location even with the same doping element and configuration. It was suggested that N doping at graphene edges demonstrated the best catalytic activity because the edges obtained a much faster electron-migration rate than the basal plane in graphene.74,79 Employing the heteroatom-containing precursors with well-controlled locations of heteroatom atoms is a promising strategy to control the location of the heteroatom dopants and to establish structure–property relationships in specific applications.19 Noteworthily, the type, content, and location of dopants in heteroatom-doped carbon catalysts are difficult to be precisely manipulated owing to the complexity of heteroatom dopants and the variety in the surface properties. Novel doping strategies need to be developed, and combined experimental and theoretical approaches will help unveil the chemical nature of the dopants.
2.5 Compositing and doping with metals
Compositing with metal species is another appealing strategy to enhance the performance of carbocatalysis because of the interfacial coupling and charge transfer between the dissimilar counterparts. The defects, heteroatom dopants, and surface oxygen functional groups on the carbon surface provide unique sites for anchoring the metal species owing to the strong binding strength and accelerated electron transfer. Metal species with electron-donating d-orbital electrons can improve electron conductivity and electron-transfer capacity of carbon materials by regulating their electronic properties such as spin density and work functions.80–82 Therefore, the strong couplings between the metal species and carbon support significantly change the interface charge density and the coordination environment of active sites, ultimately regulating the catalytic activity.The redox couples of metal cations (Men+/Men+1) facilitate O3 decomposition into ROS via creating the electron-transfer environment during catalytic ozonation.83 Moreover, the surface of metal oxides is commonly covered by hydroxyl groups, including the inherent hydroxyl groups and the hydroxyl groups arising from the surface hydroxylation process, which act as the Lewis acid sites to accelerate O3 decomposition or induce a substitution reaction with O3 to form surface-adsorbed active species (Fig. 6a).16,84,85 Oxygen vacancies (OVs) are typical defects in metal oxides which contain unsaturated coordination sites and unpaired electrons.86 OVs can enhance the mobility of the surface oxygen and thus facilitate electron transfer in catalytic reactions. Electronic properties of metal/carbon composites can be regulated with the existence of OVs. The abundant OVs within the surface in NiCo2O4-loaded hollow carbon spheres (HCS) increased DoS near the Fermi level and decreased work function and also optimized the valence state of active sites, resulting in a remarkable enhancement in ORR activity.87 Meanwhile, OVs of TiO2 resulted in inter-bandgap states and a lower flat band potential in a CNT/TiO2 hybrid, promoting the photocatalytic activity.88 Recently, the generation of high-valent metal species has been successfully verified in various kinds of AOPs.89–91 In the Fe(II)/O3 system, the involvement of high-valent iron-oxo species (Fe(IV)-oxo) formed from the reaction of Fe(II) with O3 was identified by 18O isotope-labeling technique.92 The generated high-valent metal-oxo species are highly reactive and capable of oxidizing the organic contaminants.
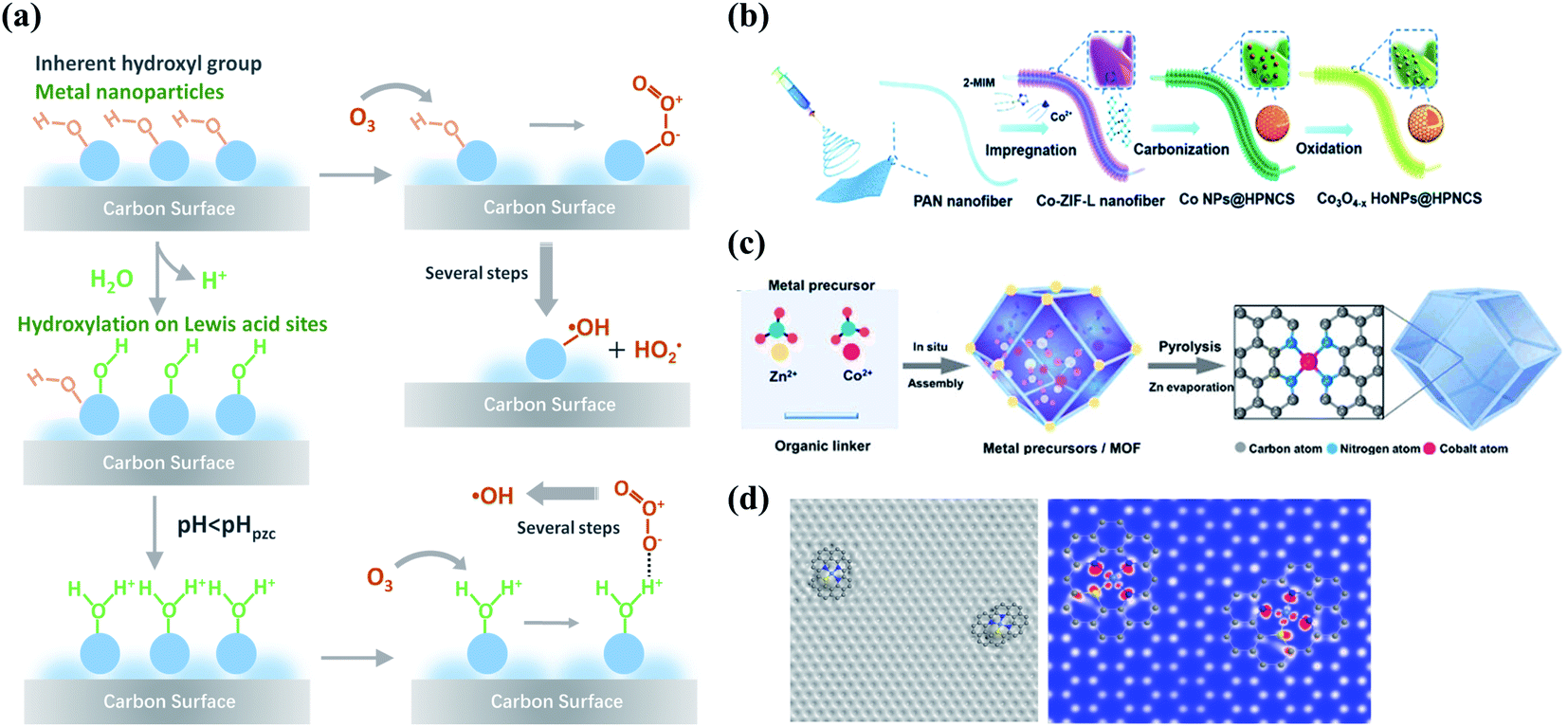 | ||
| Fig. 6 (a) Schematic illustration of the interactions between O3 and the surface hydroxyl groups on the metal sites of carbon materials. Reproduced with permission from ref. 16. Copyright 2020, American Chemical Society. (b) Preparation process of Co nanoparticles embedded carbon nanoflakes. Reprinted with permission from ref. 106. Copyright 2019, Wiley-VCH. (c) The formation procedure of Co-SA. Reprinted with permission from ref. 107. Copyright 2021, Wiley-VCH. (d) Simulated HRTEM and STM images of TM–Nx architecture in atomically disperse S|NiNx species embedded in porous carbon (PC) nanosheets. Reprinted with permission from ref. 108. Copyright 2019, Nature. | ||
Impregnation has been widely employed for the synthesis of metal-loaded carbon catalysts, whereas the loading rate should be below the threshold of 30 wt% to attain the well-distributed metal particles.93 To achieve both high metal loading and optimal particle size distribution, MOFs can be utilized as templates.94,95 Compared with metal-loaded carbon composites, encapsulation of metal clusters/nanoparticles within carbon layers by forming a core@shell structure successfully avoid undesirable metal leaching (Fig. 6b). Meanwhile, the encapsulated metal species will modulate the electronic state of outer-sphere graphitic layers via an electron tunnelling effect at the carbon–metal interfaces and enhance electron-transfer capability.96,97 Additionally, the size of anchored transition metals (TM) can be downsized to an atomic level to form single-atom catalysts (SAC), which can be synthesized by a variety of approaches, such as CVD, atomic layer deposition, pyrolysis, wet chemical method, and electron/ion irradiation (Fig. 6c).98,99 Extensive attention has been given to single-atom transition metal (TM) species coordinated N-doped nanocarbons (TM–Nx–C) because of their high catalytic activities stemmed from the unique TM–N coordination (Fig. 6d).100–102 Such TM–N coordinations have been regarded as the principal active centers, because the hybridizations between the d-orbitals of TM and the 2p-orbitals of nitrogen increased the electron densities near the Fermi level and the work function of carbon basal plane.16 The inexpensive precursors, such as inorganic salts, carbon, and nitrogen sources, have been extensively employed for the preparation of TM–Nx–C catalysts.103–105
3. Catalytic ozonation with different carbon allotropes
3.1 Bulky carbons
By virtue of its high surface area, large porosity, plentiful surface functionalities and low costs, activated carbon (AC) has been widely used to remove micropollutants from water. Combined AC with ozonation was found to be an attractive method for wastewater decontamination owing to the synergistic effect between adsorption and catalytic oxidation (summarized in Table 1). The rich porosity and maneuverable surface functionalities expedite the interactions of both organic pollutants and O3 on the surface of AC. Moreover, it was reported that AC or carbon black could act as the initiator for radical chain reactions to accelerate O3 decomposition into HO˙.109,110 In AC-initiated catalytic ozonation, variations in solution pH and surface chemistry of AC will exert a profound influence on the catalytic performance.| Catalysts | Synthesis/modification methods | Target pollutants | Active sites | ROS | Ref. |
|---|---|---|---|---|---|
| AC | Commercially purchased | Oxalic acid, gallic acid, 1,3,6-naphthalenetrisulphonic acid, diethyl phthalate | Delocalized-π system, surface acid/basic groups | HO˙, activated surface–O3 complex | 40, 111, 117 and 160 |
| GAC | Commercially purchased | P-Nitrophenol, chemical industrial wastewater | Surface basic groups | HO˙ | 113–115 |
| ACHNO3 | Modification with HNO3 | Oxalic acid, oxamic acid, benzenesulfonic acid, sulfanilic acid | Surface basic groups | HO˙ | 112 and 161 |
| AC-NO2 | Modification with HNO3/ammonia | Oxalic acid | Surface basic group (–NH2, pyrone groups), acid oxygenated surface groups | HO˙ | 118 |
| AC-NH3 | |||||
| AC-N2 | Thermal treatment under N2/H2/NH3 atmosphere | Oxalic acid | Nitrogen-containing groups, basal planes, basic oxygen-containing groups | HO˙ | 120 |
| AC-H2 | |||||
| AC-NH3 | |||||
| AC-NH3 | Modification with ammonia/ammonium carbonate/urea | Naphthalenesulphonic acid | Nitrogen-containing groups, basic oxygen-containing groups | HO˙ | 119 |
| AC-(NH4)2CO3 | |||||
| AC-urea | |||||
| Biochar | Pyrolysis of pistachio hull | Reactive red 198 | Surface functional groups | HO˙, activated surface–O3 complex | 125 |
| Biochar | Pyrolysis of sludge | Phenol, oxalic acid | Surface carbonyl groups | O2˙− | 124 and 126 |
| Biochar | Pyrolysis of sludge, treatment with HCl/NaOH | Petroleum refinery wastewater | Functional C groups, Si–O structures, metallic oxides | HO˙ | 122 |
| CNTs | Commercially purchased | P-Chlorobenzoic acid, oxalic acid | Graphene basal plane, surface oxygen groups, defects | HO˙ | 21 and 55 |
| MWCNTs | Pretreatment with O3 | Oxalic acid | Surface basic groups | HO˙, surface-oxygenated radicals | 129 |
| CNTs-COOH | Pretreatment with HNO3/H2SO4 | Indigo | Surface carboxylic groups | HO˙ | 39 |
| CNT-N2 | Pretreatment with HNO3 followed by thermal treatment under N2/H2 atmosphere | Oxalic acid | Surface basic groups | HO˙ | 130 |
| CNT-H2 | |||||
| MWCNT-HNO3 | Pretreatment with HNO3/H2O2, thermal treatment in the gas phase with 5% O2 in N2 | Oxalic acid, oxamic acid | Surface basic groups | HO˙, surface-oxygenated radicals | 131 |
| MWCNT-H2O2 | |||||
| MWCNT-O2 | |||||
| N-doped CNTs | Ball milling of CNTs with urea or melamine followed by thermal annealing | Oxalic acid | Doped N functionalities | — | 136 |
| N/S-doped CNTs | Liquid-phase oxidation with HNO3/H2SO4 | Oxalic acid, phenol | Doped N/S functionalities | HO˙ | 135 |
| F-doped CNTs | Mixing of CNTs with hydrofluoric acid | Oxalic acid | Doped F, surface basic groups, delocalized-π system | O2˙−, 1O2 | 138 |
| GO | Modified Hummer's method, commercially purchased | 5-Chloro-2-methyl-4-isothiazolin-3-one, N,N-diethyl-m-toluamide | Surface oxygen groups | HO˙ | 139, 140 and 148 |
| rGO | Thermal reduction of GO under N2 atmosphere | P-Hydroxybenzoic acid | Surface carbonyl groups | O2˙−, 1O2 | 35 |
| rGO | Thermal reduction/chemical reduction/hydrothermal method | Oxalic acid, 4-nitrophenol | Defect sites | HO˙, O2˙−, 1O2 | 142 |
| rGO | Microwave reduction method, mixing of GO with ammonium nitrate followed by microwave irradiation | 4-Nitrophenol | Edging structures, dangling bonds, doped N | HO˙, O2˙−, 1O2 | 59 |
| N-doped rGO | |||||
| N-doped rGO | Ball milling of GO with urea or melamine followed by thermal annealing | Oxalic acid | Doped N functionalities | HO˙ | 145 |
| N/P-doped rGO | Mixing of GO with ammonium nitrate/ammonium phosphate monobasic followed by thermal annealing | Sulfamethoxazole | Defective sites, dopants | HO˙ | 146 |
| N/P/B/S-doped rGO | Mixing of GO with melamine/H3PO4/H3BO3/mercaptoacetic acid followed by thermal annealing/hydrothermal treatment | Benzotriazole, p-chlorobenzoic acid | Surface hydroxyl groups, the free electron in heteroatoms, delocalized π electron | HO˙, O2˙−, 1O2 | 25 |
| Layered N-doped nanocarbon | Pyrolysis of β-cyclodextrin and melamine under N2 atmosphere | Oxalic acid | Pyridinic N and C atoms adjacent to graphitic N | HO˙, *Oad | 74 |
| Nanocarbon with built-in nanotubes | Pyrolysis of α-cyclodextrin and cobalt nitrate hexahydrate under Ar atmosphere | Oxalic acid | Defective sites | HO˙, *Oad | 159 |
| N-doped hollow sphere carbon | Pyrolysis of polydopamine using a template-etching method | Ketoprofen | Doped N functionalities | HO˙, electron transfer mechanism | 158 |
| g-C3N4 | Pyrolysis of melamine/urea | 4-Chlorobenzoic acid, benzotriazole | N vacancies | HO˙, O2˙− | 36 |
| O-doped g-C3N4 | Pyrolysis of melamine followed by hydrogen peroxide hydrothermal treatment | Atrazine | Surface oxygen groups, N vacancies | HO˙, O2˙− | 155 |
| O-doped g-C3N4 | Pyrolysis of melamine and oxalic acid | P-Chlorobenzoic acid, atrazine | Surface carbonyl groups | HO˙ | 154 and 156 |
Solution pH can be a critical factor affecting the ozonation activation pathways, yet the mechanism is still controversial. Beltrán et al. applied AC in catalytic ozonation of oxalic acid (OA) at acid pH and proved that catalytic degradation of OA at pH 2.5 could be ascribed to a HO˙-based radical pathway occurring in the bulk solution.111 In another study, O3 mainly adsorbed on the surface of AC without generating free radicals at acidic pH to near neutral pH (pH 2 to 6), while it tended to react with the surface hydroxyl groups on the AC to produce HO˙ in bulk solution when solution pH > 6.110 Faria et al. reported that surface oxidation was the dominant degradation route of OA at pH 3 since the addition of HO˙ scavenger resulted in a marginal inhibition on the destruction rate of OA.112 At alkaline pH, both surface oxidations and bulk reactions relying on HO˙ were responsible for OA destruction. Additionally, the greater degradation efficiency of OA was achieved at acidic pH than at basic pH, though a higher pH promoted the decomposition of O3 into more reactive species.
Variations in solution pH not only affect the decomposition rate of O3, but also alter the surface charge of AC as well as the dissociation of organic molecules in aqueous solutions. Surface of AC is positively charged when solution pH is lower than pH at the point of zero charge (pHpzc) of AC, and its surface becomes negatively charged vice versa. Meanwhile, the organic molecules could dissociate to anionic species at a solution pH higher than its pKa. Hence, the resulted electrostatic forces (attraction or repulsion) between the surface of AC and the organic molecules at different solution pHs affect their interactions, which influence the catalytic performance and oxidation pathways.112 The principal reactions on AC at different pH conditions are schematically illustrated in Fig. 7a and b.113
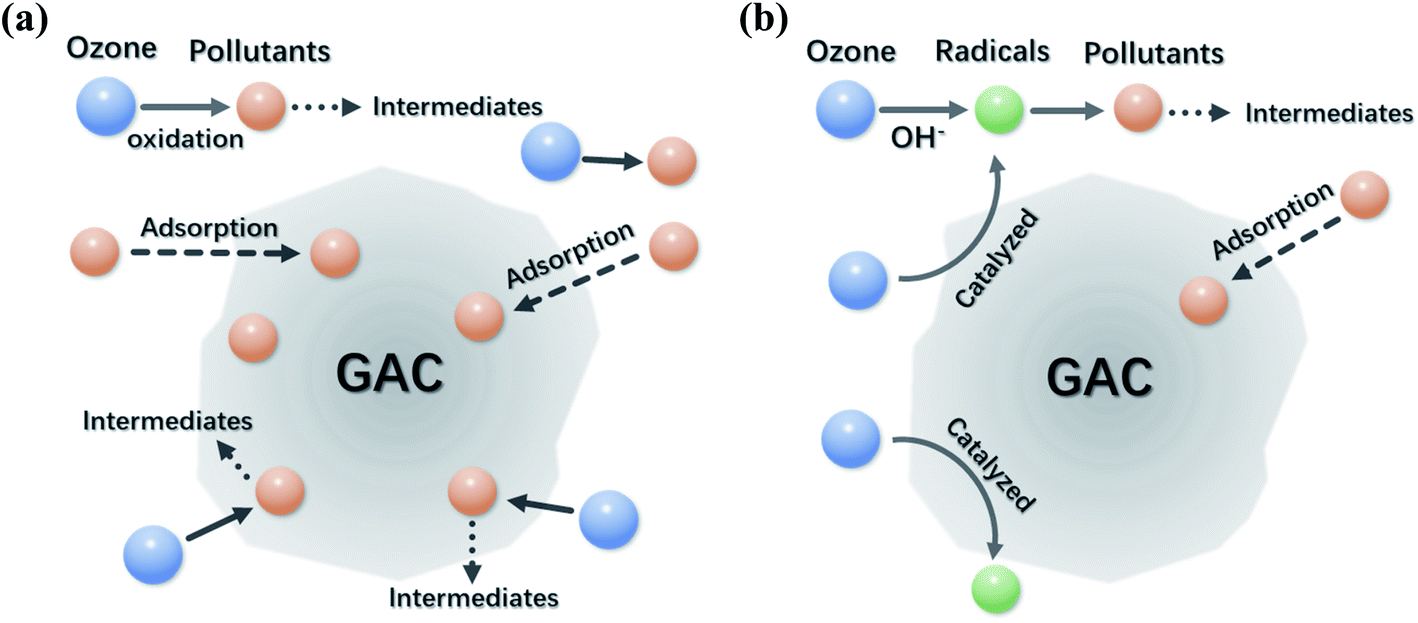 | ||
| Fig. 7 Schematic illustrations of the main reactions on GAC surface under (a) acidic conditions and (b) basic conditions. Reproduced with permission from ref. 113. Copyright 2008, American Chemical Society. | ||
The activity of AC can also be correlated with the types and contents of surface functionalities. Increasing the amounts of acidic surface oxygen functionalities with electron-withdrawing ability can be accounted for the activity loss of AC after the catalytic ozonation reaction.114 The increased amount of acidic functionalities reduces the affinity to the electrophilic interaction with O3 molecules and prevents O3 decomposition in solution. Acid or alkali surface modifications are effective in optimizing the catalytic activity of AC. Faria and co-workers modified AC by nitric acid treatment (ACHNO3), and the increased surface acidity by introducing carboxyl groups on AC surface led to the decrease in catalytic ozonation activity.112 The delocalized π electron system of basic groups on AC surface favored OH− adsorption and acted as catalytic centers to catalyze HO˙ generation.110,115,116 Also, pyrone-like and chromene-like surface oxygen functionalities react actively with O3via Criegee mechanism to yield hydrogen peroxide (H2O2), which reacts with the AC surface or with O3 itself for HO˙ generation.113,114,117 It was also observed that the adsorption capacity of basic AC was superior to more acidic ones because of the increased electronic attraction resulted from the higher pHpzc of alkaline AC.112 In addition, surface basic oxygen functionalities facilitate the occurrence of surface-based oxidations. However, the generated HO˙ around the high basicity active sites was difficult to diffuse into the bulk solution owing to its short lifetime and rapid reaction with surface-adsorbed organics. Additionally, the surface basic oxygen functionalities on AC tended to dissociate into cationic species with positive charges, which impeded the electrophilic attack of O3 for HO˙ generation. Thus, the basicity of AC may indirectly weaken HO˙-dominated oxidation in bulk solution.118 Despite of its electron-withdrawing character, the deprotonated acid oxygenated surface groups can enhance the formation of O2˙− and O3˙− as a consequence of its reaction with O3, which function as radical initiators to generate HO˙ through the radical chain reactions.40 Surface nitrogen-containing functionalities also influenced the catalytic activity of AC. ACs with various types and concentrations of nitrogenated functional groups were prepared, and urea-treated AC with enriched pyrrole groups and low concentrations of pyridine and pyridone groups demonstrated the best catalytic ozonation activity among ACs treated by different nitrogenating agents.119 The pyrrole groups extensively increased the electronic density of the graphitic layers in AC, which served as the catalytic active sites and promoted O3 decomposition into HO˙.120 On the contrary, the π-deficient pyridine groups diminished the electronic density of AC basal plane. This is in line with another study of Cao's group, where the pyrrole groups mainly accelerated HO˙ generation in the bulk solution but the pyridine groups had a detrimental effect on the surface reaction.120
Biochar is a type of bulky carbon prepared from the pyrolysis of biomass or solid wastes, which possesses the merits such as abundant precursors with low costs, bulk level preparation and manipulatable surface chemistry. Thus, biochar as a highly modifiable green platform has drawn widespread attention in environmental remediation.121–124 The properties of biochar are highly dependent on different factors such as feedstock type and pyrolysis temperature. Moussavi and Khosravi converted pistachio hull into biochar via thermal pyrolysis (500 °C for 2 h).125 The pistachio hull-derived biochar had a strongly basic surface (pHpzc = 11.5) and a macroporous structure in favor of adsorption of O3 and dye molecules. Therefore, a higher decolorization efficiency of reactive red 198 was observed in the catalytic ozonation process with pistachio hull-derived biochar than that with AC. Pyrolysis temperature has certain effects on the morphological, physicochemical, and catalytic capacity of biochar. For instance, Zhang et al. produced biochar using raw sludge obtained from coking wastewater at different pyrolysis temperatures (300, 500, 700, 900 °C) for catalytic ozonation of phenol.126 Raising the pyrolysis temperature enhanced the graphitization level of the biochar and stabilized the carbon structure and the inherently integrated metal components. Substantial reduction in total organic carbon (TOC) releasing and metal ions (e.g., Co2+, Fe3+, Ni2+, Cu2+ and Mn2+) leaching were observed when pyrolysis temperature was 900 °C. In addition, high-temperature pyrolysis also favored the formation of a porous structure with an increased surface area by promoting the evaporation of the volatile matters in biochar feedstock. The increasing amount of oxygen-bonded carbon groups formed at elevated pyrolysis temperature endows the biochar with the ability to remove contaminants, since the functionality will facilitate the adsorption of O3 within the surface region and participate in the electron-transfer process.
3.2 Nanocarbons
Although bulky carbonaceous materials are cost-effective and facile to prepare, their microporous structures limit the diffusion of both reactants and products in catalytic reactions. Thus, bulky carbons usually demonstrate poor activity and reusability in catalytic ozonation. The mixed mesoporous or microporous structures in nanocarbons such as carbon nanotubes (CNTs), graphene allotropes, carbon nitride (g-C3N4), and 3D hierarchical carbons shorten the length of mass diffusion, facilitate the exposure of active sites, and accelerate the reaction kinetics. Additionally, the surface chemistry of nanocarbons can be easily regulated. The introduced defects or heteroatoms will further improve the catalytic activity. Herein, nanocarbons with well-defined structures and chemistry are promising platforms to gain insights into catalytic ozonation as well as other carbon-based AOPs. Table 1 summarizes carbonaceous materials in catalytic ozonation for the removal of organics.The effects of oxidized surface of CNTs on the catalytic activity are debatable, which can be favorable,55 unfavorable129,130 or marginal.131,132 One obstacle in the clarification of this argument is that it is difficult to alter the surface chemistry without changing the morphology or dispersity of CNTs after a prolonged exposure in the highly oxidative environment during catalytic ozonation processes; additionally, it is challenging to directly correlate the catalytic activity with one or more surface properties.133,134 To distinguish the influence of CNTs surface chemistry and CNTs aggregation on catalytic activity, Oulton et al. investigated the effects of MWCNTs surface properties and suspension stability on HO˙ production in a catalytic ozonation system by functionalizing MWCNTs with HNO3.55 It was found that the oxidized MWCNTs possessed a higher rate of HO˙ formation compared with the pristine MWCNTs. Dynamic light scattering and sedimentation studies indicated that oxidized MWCNTs exhibited a lower degree of aggregation than pristine MWCNTs, and the better dispersion favored O3 decomposition and HO˙ formation. Moreover, surface oxygen functionalities (e.g., –COOH, –CO, and –OH) resulting from HNO3-pretreatment were also responsible for HO˙ formation.
Tailoring the surface chemistry and the electrochemical properties of CNTs by substituting carbon atoms with metal-free heteroatoms (e.g., N, S, and F) can be an alternative strategy to promote catalytic activity. Restivo et al. reported that the introduction of N and S species led to improved catalytic efficiency of CNTs in catalytic ozonation.135 Soares et al. further prepared N-doped CNTs by a simple ball-milling method.136 The elevated catalytic activity and the high stability were observed for as-prepared N-CNTs, which may be primarily attributed to the pyridine N groups. However, compared with non-doped ball-milled CNTs, no significant improvement in the mineralization degree of the organic contaminants was observed for N-doped CNTs.136,137 It was suggested that ball milling led to the breakage of nanotubes and the decrease in the entanglement, thus resulting in the increase in surface area and more exposure of active sites; however, N-doping decreased the surface area. F atoms are the most electronegative elements with an extraordinary electron-withdrawing ability. Wang and co-workers found that F-doped CNTs obtained a significantly improved catalytic ozonation activity in OA destruction than the undoped CNTs, and outperformed N-CNTs and several metal oxides (e.g., ZnO, Al2O3, Fe2O3, and MnO2).138 The covalent C–F bonds in F-CNTs formed in sp3 hybridization maintained the integrity of the delocalized π networks, which were beneficial to O3 activation via an electron-transfer process.
GO139–141 and rGO35,36,59,142 have been widely employed to activate O3 for the degradation of various contaminants. rGO displayed much higher catalytic performance than GO ascribed to the higher defective level and a proper amount of surface oxygen functionalities. By the purification of the graphite from the anode of waste lithium-ion battery (LIB), Wang et al. synthesized defects-rich rGO (LIB-rGO).142 The as-prepared LIB-rGO outperformed rGO obtained from commercial graphite powder with a mediocre defective level in catalytic ozonation for 4-nitrophenol degradation. DFT simulations validated that the peroxide bond in O3 would be broken to generate surface-adsorbed atomic oxygen (*Oad) and free peroxide species (*O2 free) when O3 molecules were placed on vacancies, zigzag, and armchair edges of rGO, giving rise to the adsorption energies of −3.88, −5.69, and −4.64 eV, respectively (Fig. 8c–e). In contrast, the peroxide bond in O3 only slightly prolonged when placed above the graphene basal plane, resulting in adsorption energy of −0.32 eV (Fig. 8b). The more negative adsorption energy of O3 on defective sites than that on the basal plane demonstrated the vital role of structural defects, especially the edging sites.142 Additionally, O3 interacting with different oxygen functional groups (–OH, –CO, –COOH, and C–O–C) of graphene were also performed by DFT calculations. Among different configurations, –OH groups on the zigzag edge of graphene exhibited the highest affinity toward O3 as it obtained the most negative adsorption energy with O3 (−1.00 eV). Moreover, the longest peroxide bond (lO–O) in O3 (1.360 Å) also indicated the greatest possibility for O3 decomposition on surface hydroxyl groups.142 However, as discussed above, the number of surface oxygen functionalities should be manipulated at a proper level, otherwise, the overabundant surface oxygen functionalities, such as the ones on GO, can impede the catalytic activity by affecting the electron-transfer process and inhibiting the adsorption of both O3 and organics due to the steric effect.
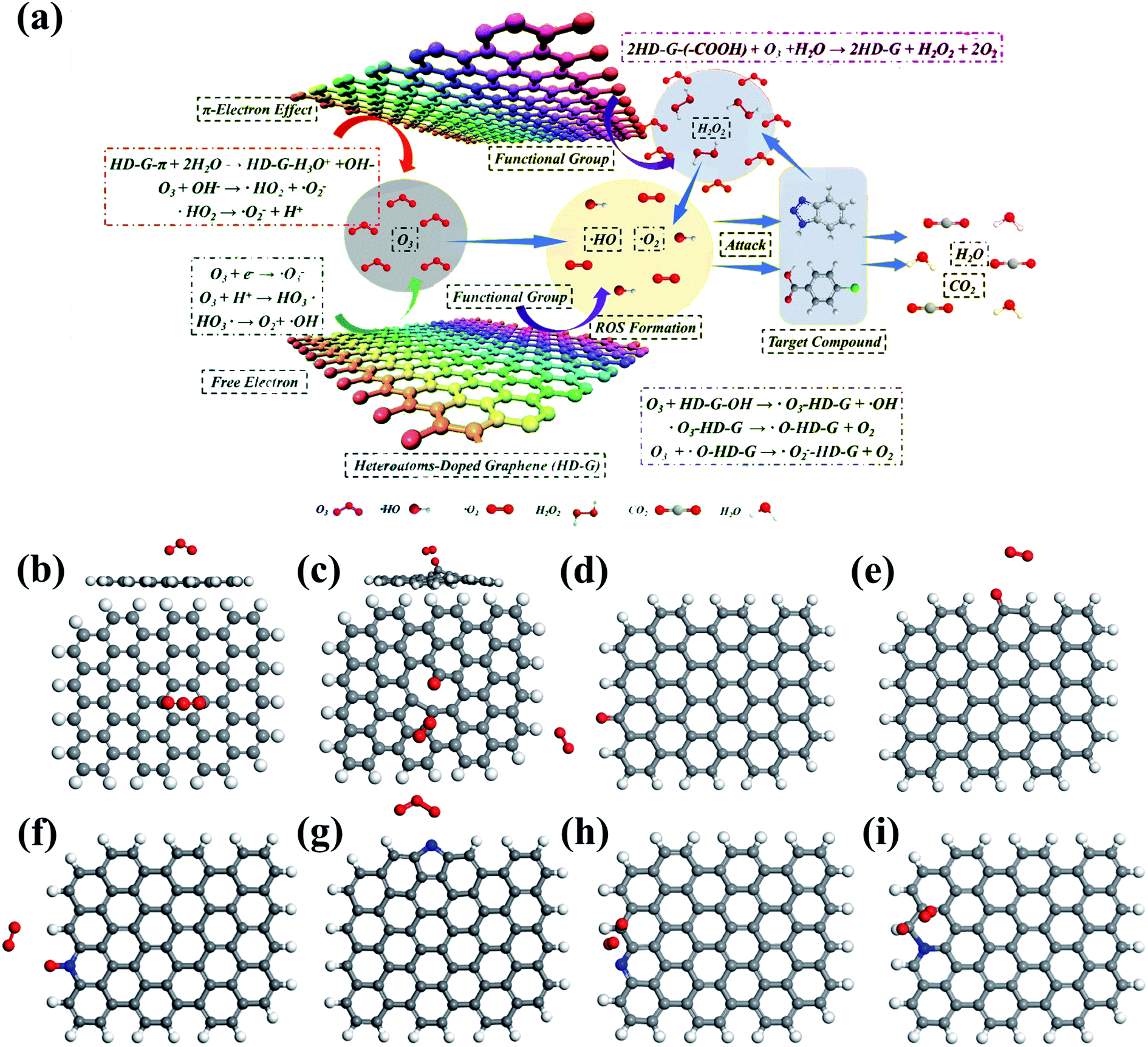 | ||
| Fig. 8 (a) Mechanism of catalytic ozonation by heteroatom-doped graphene. Adapted with permission from ref. 25. Copyright 2019, American Chemical Society. DFT simulations of O3 adsorption on the (b) basal plane, (c) vacancy, (d) zigzag edge, and (e) armchair edge of graphene. Reprinted with permission from ref. 142. Copyright 2018, Elsevier. DFT simulations of O3 adsorption on (f) pyridinic N, (g) pyrrolic N, C with high charge density adjacent to (h) pyridinic N, and (i) edge graphitic N. Reprinted with permission from ref. 74. Copyright 2019, Elsevier. | ||
Doping of metal-free heteroatoms in the graphene structure can further improve the catalytic activity and stability by regulating the chemical and electronic properties of rGO.143,144 N-rGO was prepared by a simple ball milling method using urea (rGO-U) or melamine (rGO-M) as nitrogen precursors.145 The higher SSA (102 vs. 47 m2 g−1) and a greater amount of doped N (9.3% vs. 7.5%) endowed rGO-M with higher catalytic activity than rGO-U. Furthermore, metal-free doping also created a large number of defective sites.146 Song and co-workers prepared N-, P-, B-, and S-doped rGO and the catalytic ozonation activity for wastewater decontamination was in the order of N-rGO > P-rGO > B-rGO > rGO > S-rGO.25 They suggested that the unpaired electron/free electrons and delocalized π electrons from defective sites in heteroatom-doped rGO participated in the electron-transfer process, in which unpaired electrons were easily captured by O3 to generate HO˙ and the delocalized π electrons expedited O2˙− formation. Furthermore, surface –OH and –COOH groups also acted as the active centers for ROS evolution in this study (Fig. 8a). DFT simulations revealed insights into the activity improvement induced by heteroatom doping.74 It was found that N-doping redistributed the charge density of the graphene basal plane and enabled the pyridinic and graphitic N-doping sites as well as C atoms adjacent to graphitic N to be electron-rich zones, which acted as active sites for catalyzing O3 dissociation (Fig. 8f–i). The chemisorbed O3 molecules on the active sites tended to dissociate into *Oad and *O2 free based on in situ Raman spectrum and DFT simulations. With a high oxidation potential of 2.43 V, *Oad could directly attack the adsorbed OA molecules via nonradical oxidation, and it was able to react with the ambient H2O to generate surface-adsorbed HO˙.
Despite of their high catalytic activities, graphene-based materials in catalytic ozonation reactions often demonstrated poor stability and became passivated after the long-term operation. In Song's study, the increase of TOC was observed in both 4-chlorobenzoic acid and benzotriazole degradation using GO as the catalyst.36 The soluble GO would react with O3 and ROS during catalytic ozonation, leading to the destruction of its structure and the change in surface chemistry. O3 would attack GO first by oxidation and follow by decomposition. In the oxidation stage, oxygen-containing groups were generated on the surface of GO, and the decomposition stage would give rise to the decay of carbon framework and release of CO2/CO.147 Du et al. reported that O3 was only capable of oxidizing functionalized aromatic rings in GO; the C–O–C/C–OH groups could be oxidized to CO and O–CO by O3, which could lead to the opening of the aromatic ring.148 The combination of HO˙ and O3 could oxidize both functionalized and unfunctionalized aromatic carbon rings. Previous studies on applications of GO in other oxidative systems, such as UV/H2O2,149 UV/PS,149 and photo-Fenton process150 also reported the similar observations, indicating that GO was not an appropriate candidate for AOPs-based technologies. Though rGO exhibited better structural robustness than GO in catalytic ozonation,36 it would still experience gradual deactivation with the increase of cyclic numbers,35,54,142 which was mainly ascribed to the change of surface chemistry and the blockage of active sites by the degradation intermediates. Herein, the stability and recyclability of graphene-based materials should be evaluated for applications in AOPs, and the treatment time needs to be optimized to avoid the increase of TOC caused by graphene decomposition.
3D nanocarbons with porous structures and large SSA are conducive to the maximum exposure of active sites and the mass transfer in heterogeneous catalytic processes. Ma's group prepared in situ N-doped hollow sphere carbons (NHCs) via the facile pyrolysis of polydopamine using a template-etching method, and principally figured out the specific role of three N species including pyrrolic, pyridinic, and graphitic Ns in catalytic ozonation (Fig. 9a).158 The extraordinary catalytic ozonation activity of NHCs for ketoprofen destruction was attributed to the synergy between HO˙ radical-based oxidation and a nonradical intra-electron-transfer pathway. Pyrrolic and pyridinic Ns at edges and structural vacancies functioned as the “radical-generation” region to initiate HO˙-based oxidation, while the graphitic N served as the “electron-mobility” region to accelerate the electron transfer from the adsorbed organics to the activated O3via a nonradical oxidation pathway (Fig. 9b).158 Metal-free 3D mesoporous graphitic carbons with built-in nanotubes (CPGs) were synthesized via a facile one-pot co-pyrolysis method using cost-efficient precursors (Fig. 9c).159 The CPGs showed superior catalytic ozonation activities in OA destruction compared with CNTs, rGO, and LaMnO3 perovskite catalysts, which could be ascribed to the nanoconfinement effect brought by the presence of few-layer (5–10) built-in nanotubes within the bulk structure of graphitic carbons.
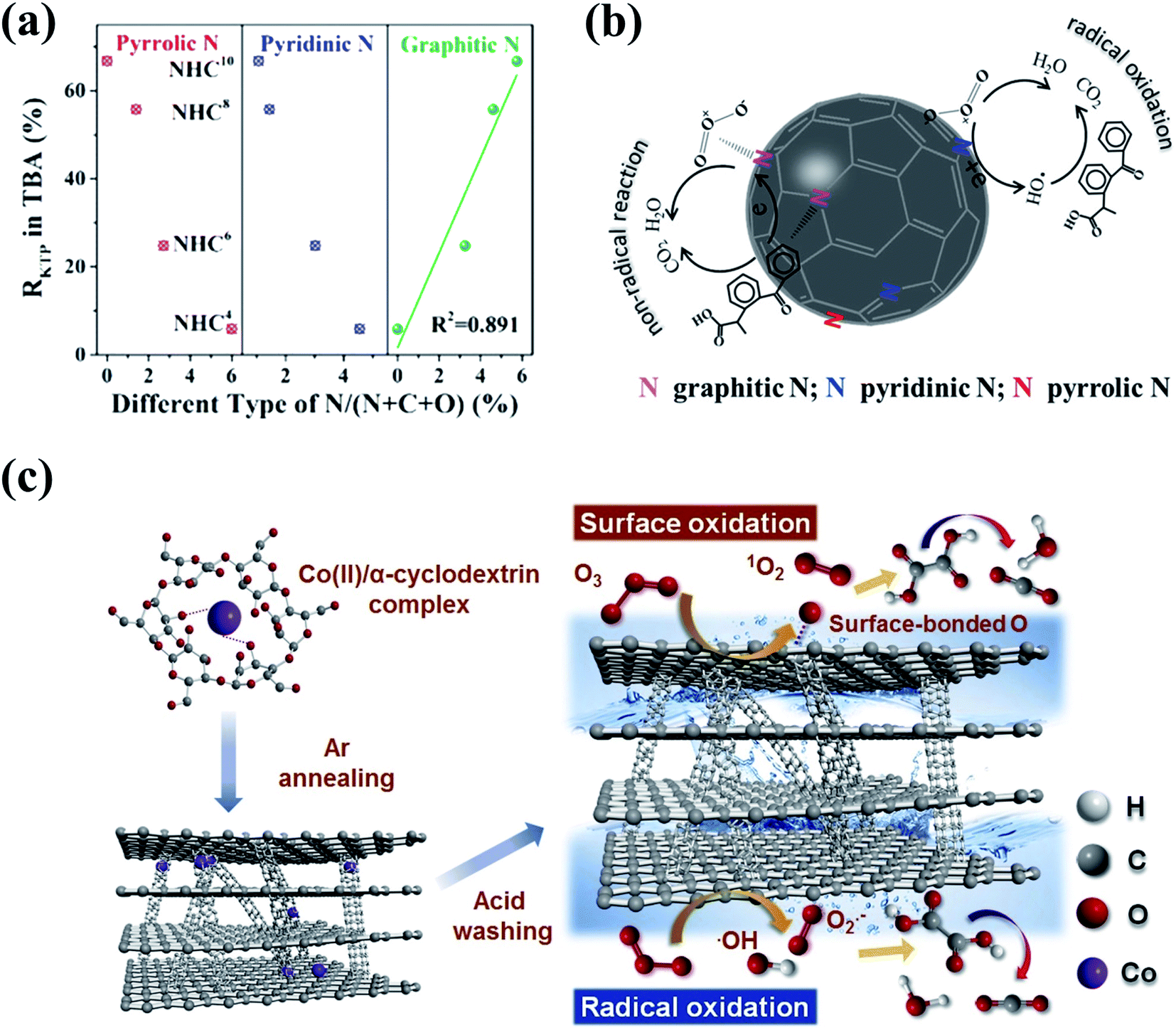 | ||
| Fig. 9 (a) Relationships between the removal rates of ketoprofen and the content of different N species of NHCs. (b) Mechanism of ketoprofen degradation by catalytic ozonation on NHCs. Reprinted with permission from ref. 158. Copyright 2019, American Chemical Society. (c) Schematic diagram of catalytic ozonation of OA with CPGs. Reprinted with permission from ref. 159. Copyright 2020, Elsevier. | ||
3.3 Carbon-based nanocomposites
Heterogeneous metal-based catalysts show desirable activities, but generally suffer from poor stability due to metal leaching and agglomeration. Anchoring metal species on supports can significantly improve the stability and dispersion of metal species, thereby suppressing the metal leaching and facilitating the exposure of active metal sites (Fig. 10a). Moreover, the electron conductivity and electron-transfer capacity can be enhanced due to the synergistic effect between the metal species and the supports, which further contributes to the catalytic activity. Carbonaceous materials have been exploited as versatile catalyst supports because of their adjustable dimensional architecture, SSA, and surface chemistry which facilitate the anchoring and dispersion of metal species.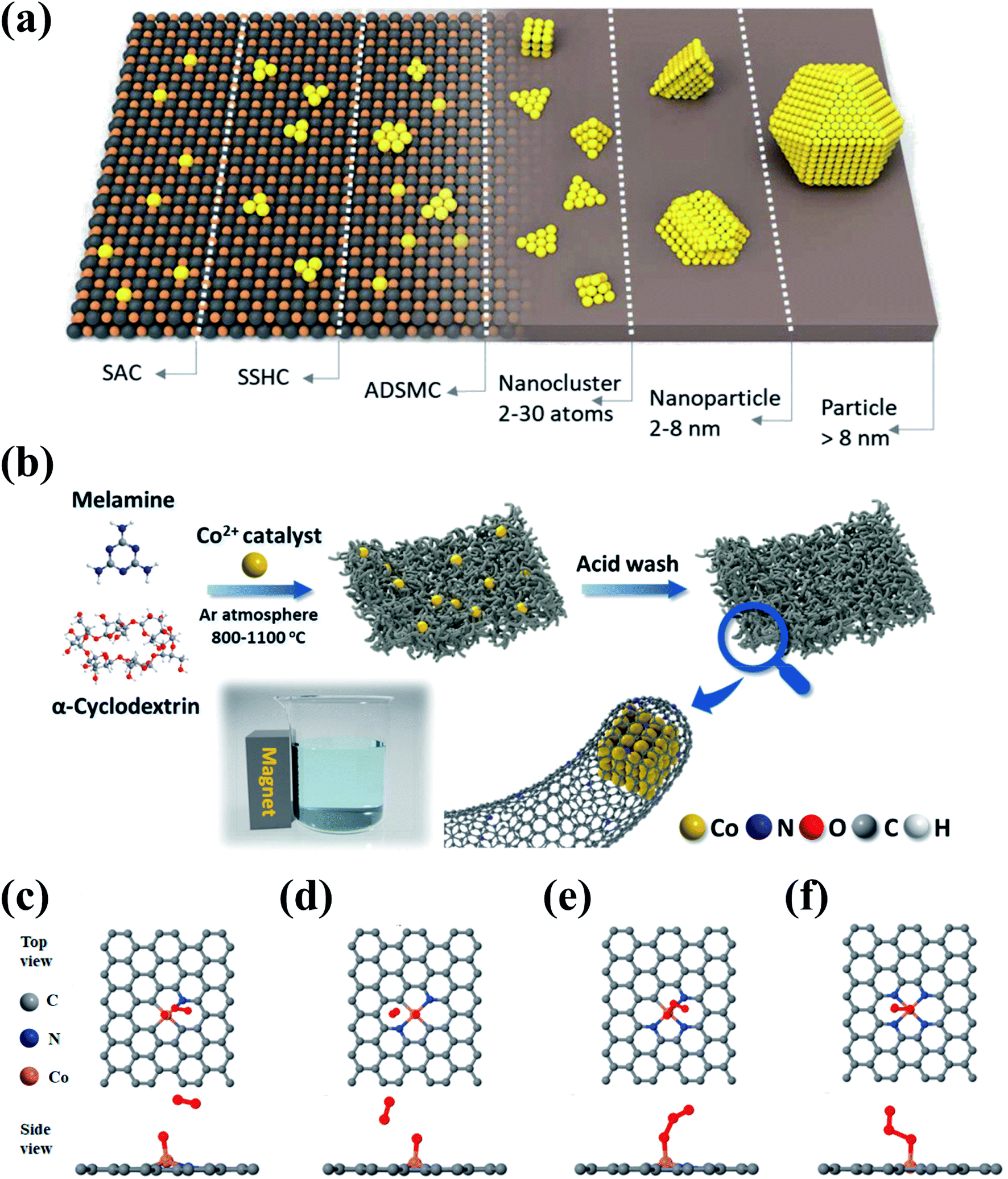 | ||
| Fig. 10 (a) Schematic illustration of the various types of metal–carbon nanocomposites. (SSHC and ADSMC are defined as single site heterogeneous catalyst and atomically dispersed supported metal catalyst, respectively.) Reprinted with permission from ref. 195. Copyright 2019, Wiley-VCH. (b) Schematic illustration of the synthesis route for Co–N@CNTs. DFT simulations for mimicking the adsorption of O3 onto (c) Co–N1, (d) Co–N2, (e) Co–N3, and (f) Co–N4. Reprinted with permission from ref. 96. Copyright 2021, American Chemical Society. | ||
Transition metals have been recognized as the catalytic active sites in ozone activation owing to their multivalence states originated from their 3d orbitals, which accelerate the electron-transfer process via redox reactions. Different transition metals have been applied to modify carbon materials, such as Mn, Fe, Co, and Zn, as well as their oxides (summarized in Table 2). Ma et al. anchored manganese oxides (MnOx) on granular active carbon (GAC) to enhance the ozonation of nitrobenzene, and higher catalytic treatment efficiency was observed at low pH (2.74–3.52) than that at high pH (6.72–9.61) because the low pH condition benefited the adsorption of nitrobenzene on the catalyst.162 AC derived from sewage sludge (SBAC) was also used to load Mn.163,164 The organic degradation pathways were different in these studies which were either attributed to HO˙-based degradation or the surface electron transfer among the loaded metal oxides and the adsorbed organics and O3.163,164 However, the multivalence of Mn stimulated the electron-transfer in both processes. Great efforts have also been put into the hybridization of rGO and MnO2 for catalytic ozonation.83,165,166 2D γ-MnO2/rGO nanohybrids (MnO2/rGO) were fabricated and used for catalytic ozonation by Wang and co-workers.83 The redox reactions between Mn3+ and Mn4+via single electron transfer acted as the active sites for the catalytic reaction. The electron transfer process was further promoted by the tight interaction between MnO2 and rGO.
| Catalysts | Synthesis/modification methods | Target pollutants | Active sites | ROS | Ref. |
|---|---|---|---|---|---|
| Supported manganese oxides | |||||
| MnOx/GAC | Impregnation method | Nitrobenzene, oxalic acid | Mn species | HO˙, surface adsorbed ROS | 162 and 163 |
| MnO2/biochar | Impregnation method | Piggery bio-treatment effluents | Surface hydroxyl groups | HO˙ | 196 |
| MnOx/biochar | Impregnation method | Atrazine | Lewis acid sites | HO˙, O2˙− | 197 |
| MnOx/MWCNT | Impregnation method | Ciprofloxacin | Mnn+/Mnn+2 | HO˙ | 198 |
| MnO2–NH2/GO | Hydrothermal method | Cephalexin | Mn3+/Mn4+ | O2˙− | 188 |
| α-MnO2/rGO | Hydrothermal method | Bisphenol-A | — | HO˙ | 165 |
| β-MnO2/rGO | Hydrothermal method | Dichloroacetic acid | — | HO˙ | 166 |
| γ-MnO2/rGO | Hydrothermal method | 4-Nitrophenol | Mn(III)/Mn(IV) | O2˙−, 1O2 | 83 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Supported iron oxides | |||||
| Fe/AC | Dipping method, commercially purchased | Dibutyl phthalate, formate | Fe species | HO˙ | 169 and 172 |
| FeOx/biochar | Impregnation method | Atrazine | Lewis acid sites | HO˙, O2˙− | 197 |
| Fe0/CNTs | Electrophoresis deposition followed by calcinations | Methylene blue | Fe(III)/Fe(IV) | HO˙ | 199 |
| Fe3O4/MWCNTs | Hydrothermal method | Bisphenol A | Surface oxygen groups | HO˙ | 173 |
| Fe3O4/GO | Precipitation method | p-Chlorobenzoic acid, ibuprofen | — | HO˙ | 141 |
|
|||||
| Supported cobalt oxides | |||||
| Co/AC | Impregnation method | Nitrobenzene | Co species | HO˙ | 200 |
| Co–N@CNTs | Two-step carbonization method | Oxalic acid, phenol | Two pyridinic N-coordinated Co (Co–N2) | *Oad, 1O2 | 96 |
|
|||||
| Supported zinc oxides | |||||
| GAC@ZnO | Deep-coating method | Metronidazole | Zn species | HO˙ | 201 |
| Zn/CNTs | Thermal solvent treatment process using zinc power, MWCNTs and 40% polyethylene glycol followed by pyrolysis under N2 atmosphere | 4-Chloro-3-methyl phenol | Zinc oxide (hydroxide) | HO˙, O2˙− | 174 |
| ZVZ/g-C3N4 | Pyrolysis of zero-valent zinc (ZVZ) and g-C3N4 | Atrazine | Surface hydroxyl groups, ZVZ/ZnO | HO˙, O2˙−, 1O2 | 175 |
| ZnO/g-C3N4 | Mixing and calcination method | Atrazine | Surface hydroxyl groups, Zn species | HO˙, O2˙−, 1O2 | 94 |
|
|||||
| Supported copper oxides | |||||
| CuO/AC | Impregnation method | Heavy oil refinery wastewater | Cu species | HO˙ | 202 |
| CuO/g-C3N4 | Reflux condensation method | Oxalic acid | Oxygen vacancies, surface hydroxyl groups | HO˙, O2˙− | 203 |
|
|||||
| Supported platinum oxides | |||||
| Pt/CNT | Incipient wetness impregnation method | Oxalic acid | Ptred/Ptox | HO˙ | 176 |
|
|||||
| Supported cerium oxides | |||||
| Ce/AC | Precipitation method | Textile dyes, dimethyl phthalate, p-toluenesulfonic acid | Ce3+ species | HO˙ | 177, 178 and 180 |
| CeO2/AC | Impregnation method | Oxalic acid, aniline | Ce3+ species | HO˙ | 179 and 181 |
| CeO2@AC | Urea-mediated hydrothermal method | Oxalic acid | Oxygen vacancies | HO˙ | 204 |
| CeO2/AC, CeO2/MWCNT | Impregnation method | Erythromycin | Ce3+ species | HO˙, surface-oxygenated radicals | 182 |
| CeO2/OCNT | Pretreatment of CNTs with HNO3, and hydrothermal treatment of OCNT and Ce(NO3)3·6H2O followed by calcinations | Phenol | Surface hydroxyl groups, Ce3+/Ce4+ | HO˙ | 183 |
| CeO2/rGO | Hydrothermal method | N,N-Diethyl-m-toluamide | Ce3+/Ce4+ | HO˙ | 205 |
| Ce/g-C3N4 | Pyrolysis of Ce(NO3)3·6H2O and g-C3N4 | Oxalic acid | Ce(III), surface hydroxyl groups | HO˙ | 96 |
|
|||||
| Supported bimetallic oxides | |||||
| Fe2O3–CeO2/AC | Impregnation method | Sulfamethoxazole | Metal oxides | HO˙ | 206 |
| MnO2–Co3O4/AC | Impregnation–precipitation method | Incineration leachate | — | HO˙ | 192 |
| Fe–Ni/AC | Impregnation method | 2,4-Dichlorophenoxyacetic acid | Iron–nickel | HO˙ | 207 |
| Fe–Mn/GAC | Impregnation–desiccation method | Methyl orange | Metal species | HO˙ | 208 |
| MnFe2O4/rGO | Electrospinning method | Di-n-butyl phthalate | Surface hydroxyl groups, Mn2+/Mn3+ | HO˙ | 193 and 194 |
| LaCoO3/g-C3N4 | Pyrolysis of LaCoO3 and g-C3N4 | Benzotriazole | N vacancies, –C–O–Co bonds, Co2+/Co3+ | HO˙, O2˙−, 1O2 | 97 |
Mn/CNTs composites were developed to intensify catalytic ozonation by uniting the merits of both homogeneous and heterogeneous reactions.167 The reversible desorption and adsorption of Mn2+ on the surface of CNTs could be regulated by the spontaneous variations of solution pH during pollutant mineralization. At the first stage, organic acids as the degradation intermediates would gradually accumulate and lower the solution pH. CNTs surface was positively charged when solution pH < pHpzc of CNTs, leading to the desorption of Mn2+ by the electrostatic repelling forces. The dissolved Mn2+ in the solution allowed homogeneously catalytic ozonation. With the mineralization of organic acid intermediates, solution pH gradually increased and Mn2+ re-adsorbed on the electronegative CNTs surface when solution pH was higher than the pHpzc of the catalyst, thereby achieving the recovery of Mn2+. However, during this process, Mn2+ was inevitably oxidized into MnO2 during catalytic ozonation and resulted in the decrease of catalytic efficiency.
Iron-based catalysts have been widely employed because of their comparatively low price and high catalytic performance. In combination with Fe-loaded AC for catalytic ozonation, the treatment efficiency for various refractory pollutants such as crystal violet,168 dibutyl phthalate,169 and organic contaminants in heavy oil refining wastewater170 and biologically pretreated coal gasification wastewater171 were greatly enhanced. In Yuan's study, an iron oxide-impregnated AC catalyst (JBX) was only effective to activate O3 for formate/oxalate degradation at pH 3, while the alkaline environment (pH 7.3 and 8.5) inhibited the catalytic activity.172 As revealed, the positively charged protonated iron oxide surface sites at acidic pH obtained a higher affinity toward O3 than the deprotonated sites at basic pH. Fe3O4/MWCNTs samples with the similar surface area but different surface oxygen functional groups were also fabricated to investigate the effect of oxygen-containing groups on catalytic ozonation activity.173 Fe3O4/MWCNTs with the maximum amount of surface oxygen-containing groups exhibited the highest adsorption capability and catalytic activity, because surface oxygen functionalities could combine with organics via hydrogen bonding and induce O3 decomposition.
The built-in electric field at the Zn-CNTs interface facilitated O3 adsorption and subsequent conversion into H2O2, which further evolved into HO˙ for organics degradation.174 Additionally, the formed zinc oxide and zinc hydroxide with small particle sizes and large SSA could also promote the decomposition of O3 to yield HO˙. The introduction of zero-valent zinc (ZVZ) modified the electronic structure of pristine g-C3N4, and the redox couple of ZnO/ZVZ was favorable to the electron-transfer process.175 Loading ZnO on g-C3N4 also induced a higher density of defective sites.94 Meanwhile, the formed Zn–N bonds between ZnO and g-C3N4 adjusted the electron density of the nanocomposites near the Fermi level and optimized the work function for promoting the generation of photoinduced electrons. Platinum (Pt) has also been loaded on the carbonaceous supports as the ozonation catalysts.176 CNTs supported Pt catalyst (Pt/CNTs) was found to be more active than Pt/AC in the ozonation of OA, because of the higher accessibility of Pt particles locating on the outer surface of CNTs.176 It was reported that the redox couple of Pt was decisive for HO˙ generation.
As a typical rare earth oxide, cerium oxide (CeO2) is more active than other rare earth oxides due to its special structure of 4f orbit and high oxygen mobility.177 A coupling effect of AC and CeO2 was reported to enhance the catalytic ozonation efficiency in the treatment of textile effluents,178 pharmaceutical wastewater177 as well as the removal of individual or mixed micropollutants.179,180 The free electrons on the basal plane of AC were beneficial for the formation of Ce3+ species and thus promoted the redox reactions involving the cerium redox pair (Ce3+/Ce4+).178,181 The catalytic activity and selectivity of Ce/AC depend on the cerium precursors (Ce3+ or Ce4+). For oxalic acid destruction, the catalysts obtained from the “Ce4+” precursor possessed the highest mineralization efficiency, whereas aniline mineralization was favored by the catalysts prepared from the “Ce3+” precursor.179 Gonçalves et al. compared the catalytic performance between CeO2/MWCNTs and CeO2/AC for the ozonation of erythromycin (ERY).182 The synergic effect between carbon materials and CeO2 was more evident in CeO2/MWCNTs because of the higher dispersion of CeO2 on MWCNTs than that on AC. Additionally, ERY with large molecular weight tended to adsorb and react on the mesoporous surface of CeO2/MWCNTs rather than on the microporous surface of CeO2/AC. The dispersion of CeO2 on CNTs can be regulated by chemical treatment to increase the surface functionalities of CNTs. The well-dispersed CeO2 was beneficial to the exposure of active sites and decrease the mass-transfer resistance.183 The abundant π–π electrons on the surface-functionalized CNTs facilitated the mobility of electron and thus accelerated the redox cycle of Ce3+/Ce4+ for O3 activation.
The bonding strength between metal species and carbon supports is a decisive factor influencing the activity and stability of carbon-supported catalysts. Previous studies indicated that the presence of strong chemical bonds between graphene and active components could lead to the improvement of electrochemical performance as well as cyclic stability of constituents.184,185 Therefore, reinforcing the interface bonding force can be a viable strategy to improve both the activity and the long-term stability of carbon–metal nanocomposites. The introduction of surface functionalities on the carbon supports provides strong binding sites for metal species and helps to stabilizing the structure of catalysts.186,187 Given that the hybrid structure of MnO2–GO was not stable enough because of the weak physical adsorption and electrostatic interaction between MnO2 and GO, Xu et al. synthesized a novel structured catalyst via amino-functionalizing the hybrids of MnO2 and GO (MnO2–NH2–GO).188 The amino-functionalization endowed MnO2–NH2–GO higher stability than MnO2–GO via strengthening the covalent bonding between MnO2 and GO to prevent their decoupling. The formed strong covalent bonds bridged by NH2 would accelerate the charge transfer.
In addition to the surface functionalization, the introduction of heteroatoms into the carbon lattice alters the electronic and adsorption properties in the local region and increases the reactivity of adjacent carbon atoms, which possess efficient localized grounds for bonding with metal species.189,190 Taking advantage of Co3O4 and N modification, the N-doped graphene (NG) supported Co3O4 nanocrystals (Co3O4/NG) showed outstanding catalytic activity and stability compared with its sole counterparts.191 Co3O4/NG obtained a smaller particle size and better dispersion of Co3O4 compared with Co3O4/G because of NH3–Co coordination. The increased surface π electronic density and the basic active centers (e.g., pyridinic-N and pyrrolic-N) brought by N dopants promoted electron transfer between the π orbitals of graphene and the d orbitals of Co atoms, leading to the enhanced phenol adsorption and O3 activation. Recently, Wang et al. reported the remarkable catalytic activity and chemical stability of N-doped carbon nanotubes with encapsulated Co (Co–N@CNTs).96 The embedded Co nanoparticles endowed Co–N@CNTs with magneticity, which was favorable for magnetic separation and recycling (Fig. 10b). DFT calculations suggested that pyridinic N–Co manifested more negative adsorption energy than other types of N–Co coordination, and Co coordinated with two pyridinic N atoms (Co–N2) demonstrated the greatest affinity toward O3 adsorption to induce surface-based nonradical oxidation (Fig. 10c–f). Meanwhile, the high DoS of Co–N2 at the Fermi level facilitated the electron transfer from Co–N2 to O3 and the redox cycle at the active sites.
Additionally, the activity and durability of carbocatalysts can also be improved through bimetallic loading. Li and co-workers used MnO2–Co3O4/AC in catalytic ozonation and observed the increased biodegradability (BOD5/COD) of incineration leachate containing abundantly refractory humic-like and fulvic-like components, compared with AC loaded with single metal.192 The relationship between the concentration of surface hydroxyl sites on rGO–MnFe2O4 and the removal rate of di-n-butyl phthalate was in positive linearity.193,194 The hydroxyl groups on the surface of rGO–MnFe2O4 hybrids bonded with MnFe2O4 and C atoms to form chemisorbed oxygen groups (Mn–O–H, C–O–H, or C–OOH), which acted as the active sites for catalytic ozonation.194 In a LaCoO3/g-C3N4 activated O3 system, the charge transfer between electron-rich centers created by –C–O–Co or nitrogen vacancies and electron-poor centers promoted HO˙ generation.97
4. Catalytic mechanisms in carbon-driven ozonation
4.1 Correlations of O3@carbon interaction intensity and reaction pathways
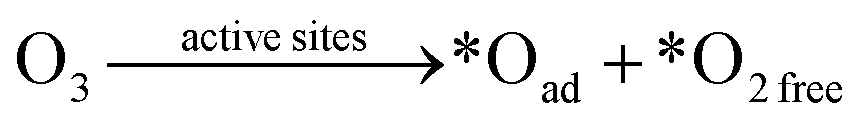
Carbon-based ozonation processes typically involve free radicals (such as HO˙ and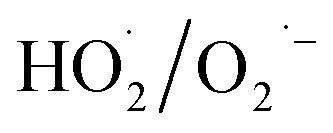 ) and nonradical (surface-adsorbed activated species and singlet oxygen (1O2)) oxidation pathways.15,16 The adsorption of O3 on the catalysts surface is a vital process for O3 activation and the subsequent ROS formation. The intensity of interactions between the active sites and the reactants (O3 and organics) governs the oxidation pathways (Fig. 11). Defects and active sites with high electron density could induce strong interactions with O3, resulting in the dissociation of O3 into surface-adsorbed atomic oxygen (*Oad) and a free peroxide species (*O2 free) (eqn (1)). *Oad with a high oxidation potential of 2.43 V will elevate the surface potential, which facilitates the activation of the ambient water molecules to generate HO˙ and triggers the subsequent radical chain reactions (Fig. 11).74,96,142 *Oad also favors the hydrogen abstraction reaction with surrounding water molecules or H+, and such a process is governed by the solution pH. The as-formed *Oad–H might further activate O3 molecules for HO˙ formation. In situ characterizations with the isotopic substitution of 16O3 or H216O by 17O3 or H217O209,210 and the DFT simulations on thermodynamics feasibility of the reactions between *Oad and H2O via calculating the free energies of intermediates and the energy barriers of transition states can be performed in the future studies to unveil the origin of HO˙.211,212 The HO˙-based ozonation obtains fast reaction rates toward almost all kinds of organics because of the high oxidizing capability and nonselectivity of HO˙. Nevertheless, the short half-life of HO˙ (t1/2 ≤ 1 μs) limits the effective mass transfer and long-range attack, making HO˙ only effective around the surface region of catalysts.213 Additionally, *Oad can induce nonradical oxidation. The high oxidation potential makes it capable of mineralizing recalcitrant organics such as aliphatic acids. The strong interactions between the active sites and O3 could form a positively charged zone on catalyst surface. Even if organics cannot adsorb on the surface, organics within this activated zone can be captured and oxidized (Fig. 11).
) and nonradical (surface-adsorbed activated species and singlet oxygen (1O2)) oxidation pathways.15,16 The adsorption of O3 on the catalysts surface is a vital process for O3 activation and the subsequent ROS formation. The intensity of interactions between the active sites and the reactants (O3 and organics) governs the oxidation pathways (Fig. 11). Defects and active sites with high electron density could induce strong interactions with O3, resulting in the dissociation of O3 into surface-adsorbed atomic oxygen (*Oad) and a free peroxide species (*O2 free) (eqn (1)). *Oad with a high oxidation potential of 2.43 V will elevate the surface potential, which facilitates the activation of the ambient water molecules to generate HO˙ and triggers the subsequent radical chain reactions (Fig. 11).74,96,142 *Oad also favors the hydrogen abstraction reaction with surrounding water molecules or H+, and such a process is governed by the solution pH. The as-formed *Oad–H might further activate O3 molecules for HO˙ formation. In situ characterizations with the isotopic substitution of 16O3 or H216O by 17O3 or H217O209,210 and the DFT simulations on thermodynamics feasibility of the reactions between *Oad and H2O via calculating the free energies of intermediates and the energy barriers of transition states can be performed in the future studies to unveil the origin of HO˙.211,212 The HO˙-based ozonation obtains fast reaction rates toward almost all kinds of organics because of the high oxidizing capability and nonselectivity of HO˙. Nevertheless, the short half-life of HO˙ (t1/2 ≤ 1 μs) limits the effective mass transfer and long-range attack, making HO˙ only effective around the surface region of catalysts.213 Additionally, *Oad can induce nonradical oxidation. The high oxidation potential makes it capable of mineralizing recalcitrant organics such as aliphatic acids. The strong interactions between the active sites and O3 could form a positively charged zone on catalyst surface. Even if organics cannot adsorb on the surface, organics within this activated zone can be captured and oxidized (Fig. 11).
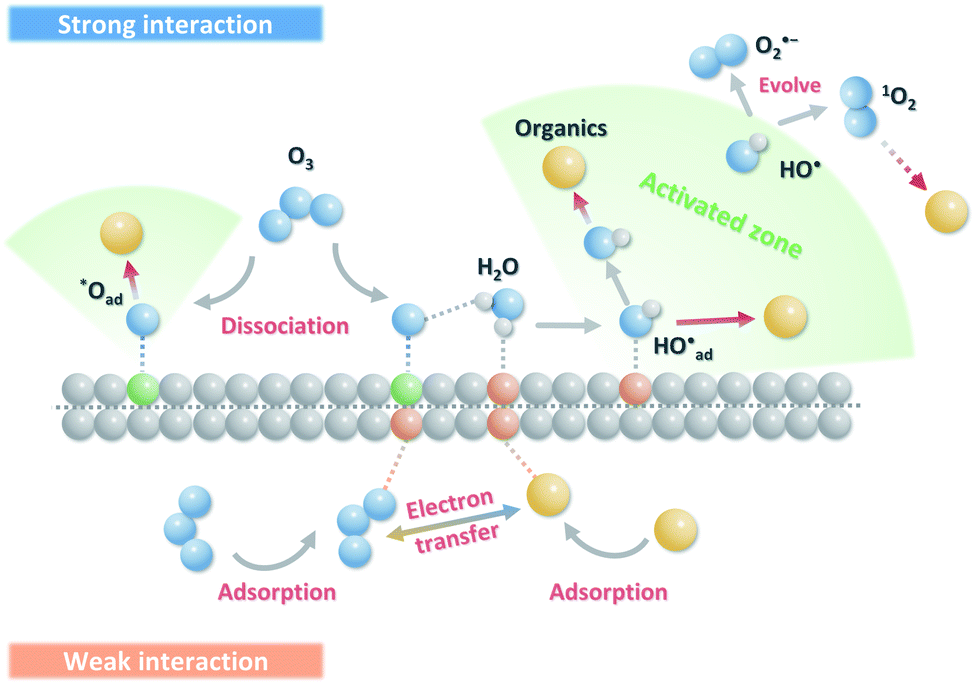 | ||
| Fig. 11 Schematic illustration of the interactions of O3 on carbon surface for ROS generation. | ||
During the radical chain reactions, 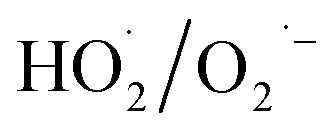 and 1O2 can be produced as side products (eqn (2)–(7)). With a mild oxidation potential of 0.89–1.7 V,
and 1O2 can be produced as side products (eqn (2)–(7)). With a mild oxidation potential of 0.89–1.7 V, 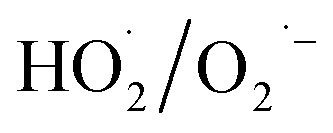 favors nucleophilic attack of certain organics (e.g., phenolics) via nucleophilic substitution. Singlet oxygen, as one of nonradical ROS, also possesses a mild oxidation potential (0.81 V). The reaction rates of 1O2 with organic compounds range from 104 to 107 M−1 s−1, which are much inferior to those with free radicals.2
favors nucleophilic attack of certain organics (e.g., phenolics) via nucleophilic substitution. Singlet oxygen, as one of nonradical ROS, also possesses a mild oxidation potential (0.81 V). The reaction rates of 1O2 with organic compounds range from 104 to 107 M−1 s−1, which are much inferior to those with free radicals.2
 | (1) |
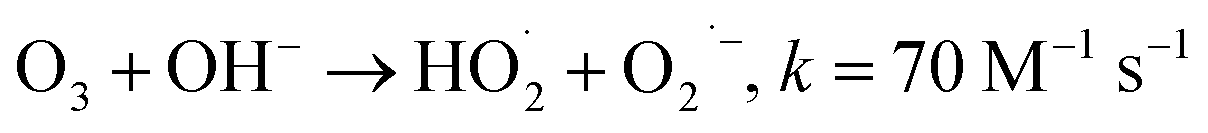 | (2) |
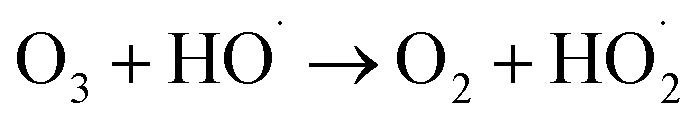 | (3) |
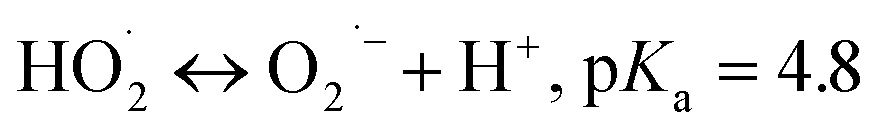 | (4) |
 | (5) |
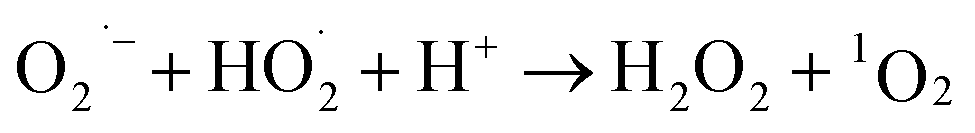 | (6) |
| O2˙− + HO˙ → 1O2 + OH− | (7) |
Surface active sites on carbon-based catalysts with moderate electron density would lead to weak interactions with O3, which is conducive to forming a surface–O3 complex. The activated complex would potentially elevate the redox potential of the carbon framework, making it capable of oxidizing the adsorbed organics via an intramolecular electron-transfer process (Fig. 11).158,214,215 In this process, the carbon substrate acts both as a catalyst and an electron tunnel for electron transport. A similar activated surface–oxidant complex was reported in persulfate/nanocarbon systems, where the formed carbon–persulfate complex demonstrated a mild oxidation potential (<1.2 V) with particular selectivity toward electron-rich organics.216,217 Although PDS obtains a similar oxidation potential to O3 (2.01 vs. 2.05 V), the more complicated molecular structure of PDS dissipates the energy transferred from the adsorption site to the O–O bond and hinders its further activation to a higher-energy state. Moreover, O3 presents a higher oxidation potential than PMS (2.05 vs. 1.75 V). Therefore, a greater oxidation ability of the activated surface–O3 complex is expected than that of activated persulfates (PDS and PMS) complex, which has been proved by higher efficiency of aliphatic acids mineralization.218,219
4.2 Analytical approaches for ROS identification
Tracing the occurrence of ROS and confirming their contributions during catalytic ozonation processes are indispensable for mechanistic studies. Due to the diversity of ROS generated from O3 activation, it is difficult to precisely evaluate the species, quantity, and roles of the reactive species by a single strategy. Therefore, multi-strategies would be encouraged to obtain an in-depth understanding of their generation and contributions.:2:2:1, hyperfine splitting constants: aN = aβH = 14.9 G) can be easily discriminated by EPR, while the identification of  is challenging in catalytic ozonation. This is because the oxidation potential of
is challenging in catalytic ozonation. This is because the oxidation potential of 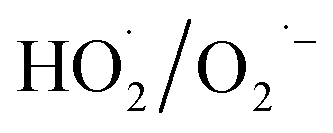 is relatively weak and their production often accompany by other ROS such as HO˙ and 1O2.35,220 Additionally, the reaction rate of DMPO with O2˙− is extremely lower than that with HO˙ (2–170 M−1 s−1vs. 1.9–4.3 × 109 M−1 s−1),221 and the characteristic signals for DMPO–HOO˙ adducts (aN = 14.2 G, aHβ = 11.4 G, and aγ1H = 1.2 G) and DMPO–HO˙ adducts normally overlap. In order to identify the DMPO–HOO˙ adducts, adopting absolute ethanol as the reaction medium instead of ultrapure water can prolong the half-life time (t1/2) of O2˙− and scavenge HO˙ in the meantime.35 However, carbon-centered radicals, as the byproducts of the quenching reaction, can also be captured by DMPO, which interfere with the distinguish of O2˙−.222 Spin-fittings can thus be performed for the further differentiation. Furthermore, DMPO–HOO˙ adducts are unstable (t1/2 = 45 s) and tend to convert into DMPO–HO˙ adducts.222 In some cases, the characteristic signal of 5,5-dimethyl-2-oxo-pyrroline-1-oxyl (DMPOX) with the peak intensity ratio of 1:2:1:2:1:2:1 (aN = 7.3 ± 0.1 G, aH = 3.9 ± 0.1 G) is observed, which is attributed to the oxidation of DMPO by O3 or surface-activated species (Fig. 12a).223 DMPO–HO˙ can also be oxidized into DMPOX via electron extraction by HO˙.224 Compared with DMPO, BMPO is a better candidate for O2˙− spin trapping. The higher stability and longer half-life of BMPO–HOO˙ adducts (t1/2 = 23 min) facilitate the identification of
is relatively weak and their production often accompany by other ROS such as HO˙ and 1O2.35,220 Additionally, the reaction rate of DMPO with O2˙− is extremely lower than that with HO˙ (2–170 M−1 s−1vs. 1.9–4.3 × 109 M−1 s−1),221 and the characteristic signals for DMPO–HOO˙ adducts (aN = 14.2 G, aHβ = 11.4 G, and aγ1H = 1.2 G) and DMPO–HO˙ adducts normally overlap. In order to identify the DMPO–HOO˙ adducts, adopting absolute ethanol as the reaction medium instead of ultrapure water can prolong the half-life time (t1/2) of O2˙− and scavenge HO˙ in the meantime.35 However, carbon-centered radicals, as the byproducts of the quenching reaction, can also be captured by DMPO, which interfere with the distinguish of O2˙−.222 Spin-fittings can thus be performed for the further differentiation. Furthermore, DMPO–HOO˙ adducts are unstable (t1/2 = 45 s) and tend to convert into DMPO–HO˙ adducts.222 In some cases, the characteristic signal of 5,5-dimethyl-2-oxo-pyrroline-1-oxyl (DMPOX) with the peak intensity ratio of 1:2:1:2:1:2:1 (aN = 7.3 ± 0.1 G, aH = 3.9 ± 0.1 G) is observed, which is attributed to the oxidation of DMPO by O3 or surface-activated species (Fig. 12a).223 DMPO–HO˙ can also be oxidized into DMPOX via electron extraction by HO˙.224 Compared with DMPO, BMPO is a better candidate for O2˙− spin trapping. The higher stability and longer half-life of BMPO–HOO˙ adducts (t1/2 = 23 min) facilitate the identification of 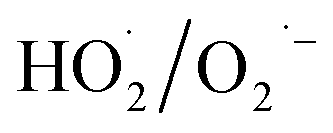 against HO˙.225
against HO˙.225
 | ||
| Fig. 12 (a) Structures and simulated EPR signals of DMPO–HO˙ and DMPO–HOO˙ adducts formed by trapping HO˙ and O2˙− with DMPO and DMPOX formed by oxidation of DMPO with O3. (b) Structures and EPR signals of BMPO–HO˙ and BMPO–HOO˙ adducts formed by trapping HO˙ and O2˙− with BMPO. Adapted with permission from ref. 16. Copyright 2020, American Chemical Society. (c) Chemical structures of four sterically hindered amines. (d) EPR signals of nitroxide radicals formed by trapping 1O2 with four sterically hindered amines. Reproduced with permission from ref. 230. Copyright 2017, Royal Society of Chemistry. Reproduced with permission from ref. 231. Copyright 2011, Royal Society of Chemistry. | ||
Singlet oxygen can be detected by EPR in aqueous solution by four different sterically hindered amines as trapping reagents, including 2,2,6,6-tetramethylpiperidine (TEMP), 2,2,6,6-tetramethyl-4-piperidone (4-oxo-TEMP), 2,2,6,6-tetramethyl-4-piperidinol (4-hydroxy-TEMP) and 2,2,5,5-tetramethyl-3-pyrroline-3-carboxamide (TPC) (Fig. 12c), which can be oxidized by 1O2 to generate nitroxide radicals.226 The EPR signals of nitroxide radicals generated from each amine demonstrate distinctive triplet peaks (Fig. 12d). Interestingly, 4-hydroxy-TEMP can be oxidized into two different EPR signals: 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPOL) and 4-oxo-2,2,6,6-tetramethyl-1-piperidinyloxy (4-oxo-TEMPO). Among the four sterically hindered amines, 4-hydroxy-TEMP and TPC have a proper sensitivity and a wide detectable range for the yield of 1O2.226 It is noteworthy that other reactive species (e.g., HO˙, 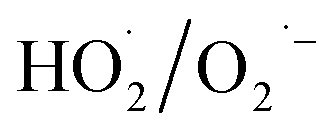 ) can also oxidize TEMP to produce similar 3-line characterization signals as 1O2.221,227–229 Introducing radical scavengers to minimize the effects of other radicals would cause the competitive reactions and further mislead the results. Additionally, TEMP-based spin trapping agents are alkaline and might induce the base activation of O3. Therefore, the dosage of TEMP and solution pH should be precisely controlled.
) can also oxidize TEMP to produce similar 3-line characterization signals as 1O2.221,227–229 Introducing radical scavengers to minimize the effects of other radicals would cause the competitive reactions and further mislead the results. Additionally, TEMP-based spin trapping agents are alkaline and might induce the base activation of O3. Therefore, the dosage of TEMP and solution pH should be precisely controlled.
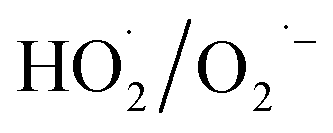 , HO˙ and O3, (9.8 × 108, 1.2 × 109 and 2.5 × 108 M−1 s−1, respectively).236 Meanwhile, the low solubility of p-BQ hinder its contact with O2˙−. NaN3 is a strong reductant which would only quench 1O2 (2 × 109 M−1 s−1), but react with O3 with a mediocre constant (2.5 × 106 M−1 s−1).237 Moreover, N3− is a strong alkaline agent, which would influence solution pH and cause base activation of O3. Taking advantage of the kinetic solvent isotope effect (KSIE), the contribution of 1O2 can be better evaluated via using D2O instead of H2O as the solvent, because the lifetime and steady-state concentration of 1O2 would increase in D2O owing to the lower reaction rate of 1O2 in D2O than that in H2O (kd(H2O) = 2.4–3.2 × 105 s−1; kd(D2O) = 1.4–2.3 × 104 s−1).238–240 The KSIE effect using D2O as the reaction medium can also be employed in EPR technique for 1O2 identification, and augmented signal intensity is expected for a 1O2-based system. Nevertheless, similar effects can also be caused by other ROS because of their reduced reaction rates in D2O prolonged the life time.241 Therefore, careful attention should be paid to the explanation of the experimental results. Potassium iodine (KI) and dimethyl sulfoxide (DMSO) have been used as scavengers for surface-adsorbed ROS in other AOPs systems.96,242 However, KI can react with O3 (1 × 108 M−1 s−1) and other oxidative species with high reaction rates, leading to the depletion of both the quenchers and dissolved O3. DMSO obtains an inferior reaction rate with O3 (0.4162 M−1 s−1), whereas it can fast react with both HO˙ and surface-adsorbed ROS. Hence, the type and concentration of the scavengers should be carefully selected, in view of the undesired competition reactions with ozone and other ROS as well as the impact on solution properties. In a complex system involving various types of ROS, the scavenger might become unspecific to the desired ROS; thus comparing the reaction rates between scavengers and different oxidants is essential to determine the contribution of the specific ROS. In addition, the combination of scavengers with higher selectivity is necessary to comprehensively evaluate the contribution of each ROS during pollutants degradation.
, HO˙ and O3, (9.8 × 108, 1.2 × 109 and 2.5 × 108 M−1 s−1, respectively).236 Meanwhile, the low solubility of p-BQ hinder its contact with O2˙−. NaN3 is a strong reductant which would only quench 1O2 (2 × 109 M−1 s−1), but react with O3 with a mediocre constant (2.5 × 106 M−1 s−1).237 Moreover, N3− is a strong alkaline agent, which would influence solution pH and cause base activation of O3. Taking advantage of the kinetic solvent isotope effect (KSIE), the contribution of 1O2 can be better evaluated via using D2O instead of H2O as the solvent, because the lifetime and steady-state concentration of 1O2 would increase in D2O owing to the lower reaction rate of 1O2 in D2O than that in H2O (kd(H2O) = 2.4–3.2 × 105 s−1; kd(D2O) = 1.4–2.3 × 104 s−1).238–240 The KSIE effect using D2O as the reaction medium can also be employed in EPR technique for 1O2 identification, and augmented signal intensity is expected for a 1O2-based system. Nevertheless, similar effects can also be caused by other ROS because of their reduced reaction rates in D2O prolonged the life time.241 Therefore, careful attention should be paid to the explanation of the experimental results. Potassium iodine (KI) and dimethyl sulfoxide (DMSO) have been used as scavengers for surface-adsorbed ROS in other AOPs systems.96,242 However, KI can react with O3 (1 × 108 M−1 s−1) and other oxidative species with high reaction rates, leading to the depletion of both the quenchers and dissolved O3. DMSO obtains an inferior reaction rate with O3 (0.4162 M−1 s−1), whereas it can fast react with both HO˙ and surface-adsorbed ROS. Hence, the type and concentration of the scavengers should be carefully selected, in view of the undesired competition reactions with ozone and other ROS as well as the impact on solution properties. In a complex system involving various types of ROS, the scavenger might become unspecific to the desired ROS; thus comparing the reaction rates between scavengers and different oxidants is essential to determine the contribution of the specific ROS. In addition, the combination of scavengers with higher selectivity is necessary to comprehensively evaluate the contribution of each ROS during pollutants degradation.
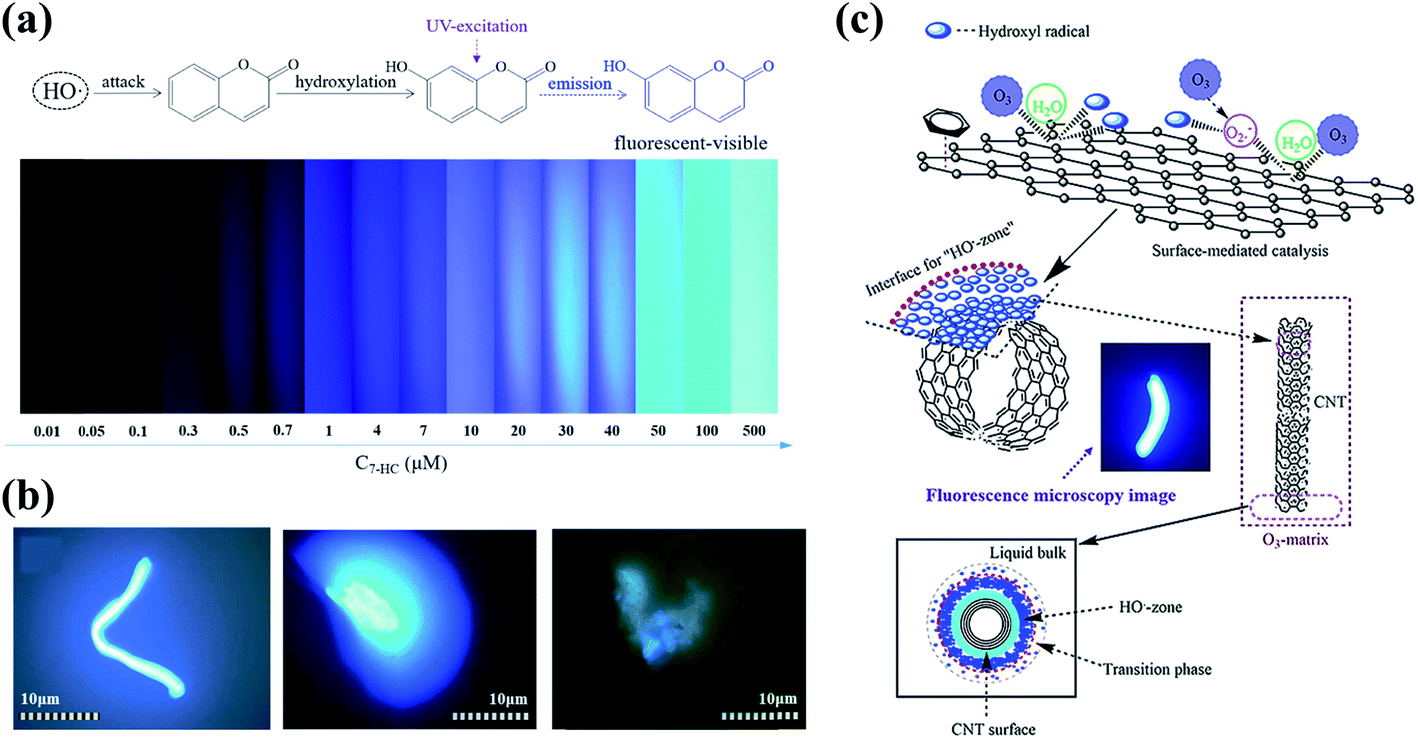 | ||
| Fig. 13 (a) Colorimetric references of 7-hydroxycoumarin solutions with the changes to the 7-hydroxycoumarin concentration for FMI. (b) Fluorescence microscopy images of the produced HO˙ during the catalytic ozonation on the surface of the carbonaceous materials. (c) Schematic illustration for the generation of interphase “HO˙ zone” on the surface of CNTs. Reprinted with permission from ref. 259. Copyright 2017, Elsevier. | ||
4.3 Selectivity of the ROS toward organic pollutants
The free and surface-confined HO˙ can non-selectively destroy most of organic contaminants at a high reaction rate (108 to 1010 M−1 s−1) because of its strong oxidizing capability, while it might not be the dominant ROS responsible for the abatement of some certain electrophilic organic compounds such as CCl4 (kHO˙ < 2 × 106 M−1 s−1), perfluorooctanesulfonic acid (PFOS, kHO˙ < 104 M−1 s−1), and perfluorooctanoic acid (PFOA, kHO˙ < 104 M−1 s−1).270,271 O2˙− with a mild oxidation potential of 0.89–1.7 V can oxidize less recalcitrant organics. Particularly, O2˙− exhibits higher reaction rates with some halogenated hydrocarbons such as CCl4, PFOS, and PFOA (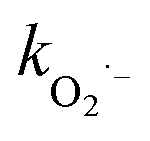 = 107–108 M−1 s−1) than HO˙ due to its strong nucleophilicity.244,245 Additionally, the halogenated hydrocarbons are recalcitrant for O3 attack (kO3 < 0.1 M−1 s−1), which have been used as the chemical probes for quantifying the concentrations of HO˙ and
= 107–108 M−1 s−1) than HO˙ due to its strong nucleophilicity.244,245 Additionally, the halogenated hydrocarbons are recalcitrant for O3 attack (kO3 < 0.1 M−1 s−1), which have been used as the chemical probes for quantifying the concentrations of HO˙ and 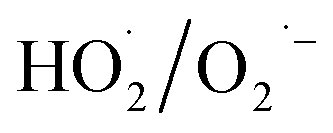 generated in catalytic ozonation systems.244,245 Nonradical-based oxidations in catalytic ozonation reactions also demonstrated a substrate-dependent behavior.96,142 The surface-confined nonradical species (including activated surface–O3 complex and *Oad) with high oxidation potentials tend to attack aliphatic compounds with saturated carbon bonds rather than organics with unsaturated carbon bonds. Similar to O2˙−, 1O2 obtains a moderate oxidation capacity (E0 = 0.81 V, NHE), which exhibits high selectivity in the decomposition of organics with unsaturated carbon bonds and electron-rich functionalities (e.g., phenols, sulfides, and anilines) via electrophilic addition and electron abstraction, but not reactive to saturated alcohols (e.g., methanol, ethanol and tertiary butanol).272 Some studies revealed that O2˙− or 1O2 played a predominant role in the degradation of phenolic compounds during the catalytic ozonation processes, even though the existence of HO˙ was confirmed by EPR spectra and quenching tests.142,220,273 It is worth noting that, although studies have reported that O2˙− or 1O2 were primarily responsible for the TOC abatement, O2˙− and 1O2 with mild oxidation potentials are substrate-dependent and cannot completely mineralize saturated compounds.274–276 Therefore, these conclusions are questionable and require a careful reevaluation. In addition, it is also quite challenging to justify the contribution of O2˙−/1O2 in the carbon-catalyzed ozonation system since the formation of O2˙−/1O2 is often accompanied by other ROS.
generated in catalytic ozonation systems.244,245 Nonradical-based oxidations in catalytic ozonation reactions also demonstrated a substrate-dependent behavior.96,142 The surface-confined nonradical species (including activated surface–O3 complex and *Oad) with high oxidation potentials tend to attack aliphatic compounds with saturated carbon bonds rather than organics with unsaturated carbon bonds. Similar to O2˙−, 1O2 obtains a moderate oxidation capacity (E0 = 0.81 V, NHE), which exhibits high selectivity in the decomposition of organics with unsaturated carbon bonds and electron-rich functionalities (e.g., phenols, sulfides, and anilines) via electrophilic addition and electron abstraction, but not reactive to saturated alcohols (e.g., methanol, ethanol and tertiary butanol).272 Some studies revealed that O2˙− or 1O2 played a predominant role in the degradation of phenolic compounds during the catalytic ozonation processes, even though the existence of HO˙ was confirmed by EPR spectra and quenching tests.142,220,273 It is worth noting that, although studies have reported that O2˙− or 1O2 were primarily responsible for the TOC abatement, O2˙− and 1O2 with mild oxidation potentials are substrate-dependent and cannot completely mineralize saturated compounds.274–276 Therefore, these conclusions are questionable and require a careful reevaluation. In addition, it is also quite challenging to justify the contribution of O2˙−/1O2 in the carbon-catalyzed ozonation system since the formation of O2˙−/1O2 is often accompanied by other ROS.
5. Impacts of water matrix
5.1 Solution pH
Solution pH is a crucial factor in catalytic ozonation processes, affecting the surface properties of catalysts, the nature of organic contaminants, and the generation and transformation of ROS, thereby influencing the catalytic performances. The surface charge of the catalyst can be affected by solution pH. The catalyst surface will be negatively charged when the solution pH is greater than pHpzc of the catalysts, which favors the electrophilic adsorption of O3. In contrast, the interaction of O3 with the positively charged surface is weak. Additionally, the charge and molecular states of organics are closely related to their pKa values, which are also affected by solution pH. The states of organics will subsequently influence the electrostatic interactions with the surface of the catalysts and the affinities toward ROS. The opposite charge between the catalysts surface and the organics will induce electrostatic attraction and facilitate surface reactions,277,278 while the deprotonated organics are more likely to be electrophilically attacked by ROS.279 Additionally, the electrochemical parameters of organics, such as ionization potential, half-wave potential, and Hammett constant, will vary with solution pH and closely relate with the redox capacity of the oxidation system.23,159,280,281 For example, the half-wave potential of phenol declined with the increased pH, leading to a higher degradation rate. However, when solution pH was further elevated beyond the pKa of phenol, the phenolate would form and demonstrate strong recalcitrance for degradation.216 The variations in surface interactions and affinities toward ROS at different pHs might result in different reaction mechanisms. Altering solution pH might change the adsorption capacity of organics and alter the reaction pathway from a nonradical pathway to radical-dominated oxidation.112Solution pH can also influence the conversion of reactive species and their redox potentials. O3 molecules can be activated by alkaline solution into HO˙ (k = 70 M−1 s−1) (eqn (2)).282 However, deprotonation of HO˙ takes place at pH > 11.9, and the formed conjugate base radical (O˙−) is less reactive than HO˙ and thus reduces the treatment efficiency (eqn (8)).221 For O2˙−, it can be oxidized with 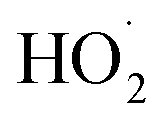 in acidic solutions to generate 1O2 (eqn (6)), while HO2˙− will deprotonate into O2˙− when solution pH is over 4.8, thereby changing from electrophilic attack to nucleophilic attack (eqn (4)).6 The reduction potentials of ROS are pH-dependent, because the concentrations of H+/OH− affect the activity of the generated ROS and their conjugated reductive pairs.283 Generally, an increased H+ concentration demonstrates an improved activity of ROS, resulting in an increase in reductive potential as the solution pH decreases.221,283
in acidic solutions to generate 1O2 (eqn (6)), while HO2˙− will deprotonate into O2˙− when solution pH is over 4.8, thereby changing from electrophilic attack to nucleophilic attack (eqn (4)).6 The reduction potentials of ROS are pH-dependent, because the concentrations of H+/OH− affect the activity of the generated ROS and their conjugated reductive pairs.283 Generally, an increased H+ concentration demonstrates an improved activity of ROS, resulting in an increase in reductive potential as the solution pH decreases.221,283
| HO˙ + OH− → O˙− + H2O, pKa = 11.9 | (8) |
5.2 Inorganic anions
Inorganic anions such as Cl−, CO32−, HCO3−, and SO42− ubiquitously exist in practical wastewater and natural water systems, which can potentially affect O3 activation even at low concentrations. Inorganic anions are commonly considered as radical scavengers to consume ROS and generate less reactive radicals (eqn (9)–(14)), resulting in the decrease in the overall degradation efficiency.272,284 The scavenging capacity can be affected by solution pH. For instance, Cl− can scavenge HO˙ to produce Cl˙. However, the scavenging effect was only effective within an acidic pH range and was limited under neutral conditions, because the intermediate of the radical chain reaction (ClOH˙−) would rapidly revert to HO˙ rather than forming Cl˙ (eqn (9) and (10)).285 A similar effect was observed when HCO3− was involved. The elevated solution pH would deprotonate HCO3− into CO32−, resulting in an increased scavenging effect because of the higher reaction rate of HO˙ with CO32− (3.9 × 108 M−1 s−1) than that with HCO3− (8.5 × 106 M−1 s−1) (eqn (11) and (12)).232,286 CO32− also increases the stability of O3 by removing the dominant chain carrier radicals, such as O3˙−,287 while SO42− and NO3− showed negligible effects on O3 decomposition.288 The presence of inorganic anions might also result in the improved selectivity of oxidative systems, because the generated reactive halogen species or CO3˙− are more selective than HO˙ and favor the attack of electron-rich organics.2,289| Cl− + HO˙ ↔ ClOH˙−, k = 4.3 × 109 M−1 s−1 | (9) |
| ClOH˙− + H+ ↔ Cl˙ + H2O, k = 2.1 × 1010 M−1 s−1 | (10) |
| CO32− + HO˙ → CO3˙− + OH−, k = 3.9 × 108 M−1 s−1 | (11) |
| HCO3− + HO˙ → CO3˙− + H2O, k = 8.5 × 106 M−1 s−1 | (12) |
| HCO3− + O2˙− → CO3˙− + HO2−, k = 1.5 × 106 M−1 s−1 | (13) |
| PO43− + HO˙ → PO4˙2− + OH−, k = 1.1 × 109 M−1 s−1 | (14) |
The effect of inorganic anions on catalytic ozonation processes also depends on their concentrations. It was reported that the Cl− concentration can influence the scavenging products and thus affect the catalytic ozonation efficiency.35,159 At low concentrations, Cl− would quench HO˙ (E0 = 2.7 V, NHE) to form the less reactive Cl˙ (E0 = 2.4 V, NHE). In contrast, a high concentration of Cl− favored the transformation of Cl˙ into reactive chlorine species (Cl2 and HClO), which could couple with HO˙ for organics degradation. However, opposite conclusions were derived in another study.290 The discrepancies in these studies might be explained by the combined effects of ionic strength, which influences the solubility and mass transfer of O3, the reaction rates of ROS, and the interactions of O3 and organics on catalyst surface. It was reported that the solubility and mass transfer of O3 decrease as ionic strength grows, especially when Cl−, CO32− and PO43− are present, while SO42− gives rise to an insignificant effect.291 Moreover, positive effects of ionic strength on reaction rates were observed in various reaction systems.292–294 Studies suggested that the reaction rates between oxidants and organics raised with the increase in ionic strength.295 The ionic strength created by different ions might also influence reaction rates. The reaction rate between O3 and Cl− improved with the increase in the ionic strength caused by NaClO4 or KNO3, whereas simply increasing the concentration of Cl− resulted in the decrease of the corresponding reaction rate.296 Additionally, increasing the ionic strength reduces the zeta potential of catalyst surface via compressing the thickness of the electric double layer and therefore affects the weak interactions (via electrostatic bonding) of O3 and organics on catalyst surface, such as the activated surface–O3 complex.297 However, marginal influence occurs to strong interactions, i.e., the *Oad-induced oxidation model. Therefore, variations in ionic strength can be utilized to distinguish the different interaction models of O3 on the catalyst surface. Ionic strength also affects the adsorption capacity of the organics on catalysts. As ionic strength increases, the adsorption capacity decreases when the electrostatic forces between the catalyst surface and organics are attractive.298
Inorganic anions may compete on the surface-active sites of catalysts with O3 or organics, thus inhibiting the oxidation performance. The positively charged catalysts can provide suitable adsorption sites for inorganic anions. PO43− as a strong Lewis base shows the strong inhibitory effect by occupying surface Lewis acid sites and impeding O3 activation.299,300 Notably, the induced alkalinity with the presence of CO32− and PO43− as basic anions would promote the base activation of O3 and affect the surface charge of the catalyst.287,290 Therefore, special attention should be paid to the variation in solution pH with the presence of basic anions. In the most of current studies, individual effects of inorganic anions on the activation of O3 have been well evaluated. Nevertheless, considering the complexity in compositions of the practical wastewater, the synergistic effects of various inorganic anions cannot be overlooked.
5.3 Natural organic matters
NOMs are pervasive constituents in natural water, and are known to decrease the efficiency of AOPs-based treatments by competing with organic contaminants for HO˙ consumption (1.6–3.3 × 108 M−1 s−1).301 NOMs with electron-rich sites could also deplete the dissolved O3via direct O3 attack, thus lowering the utilization efficiency of O3.302 Furthermore, NOMs containing carboxyl and phenolic hydroxyl groups would adsorb on the surface of carbonaceous materials and block the active sites, giving rise to inhibited interactions between the carbocatalysts and O3/organics.303 For HO˙-dominant oxidation pathway in catalytic ozonation, the inhibitory effects would continue until most of NOMs are mineralized because of the non-selectivity of HO˙.304 The nonradical pathways based on the surface oxidations are highly selective to the aliphatic acids with small molecular weight; therefore, NOMs may have a less impact on nonradical-based oxidation.159 Nevertheless, debates on the effect of NOMs on catalytic ozonation still exist. Some studies reported that NOMs could act as initiators and promotors for radical chain reactions of O3 decomposition to HO˙, which enhanced the degradation rate.305,306 Specific moieties, such as aromatic and aliphatic unsaturated constituents in NOMs, could directly activate O3 and accelerate the radical chain reactions to generate more HO˙.307 Moreover, the presence of humic acid (HA), a major constituent of NOMs, could introduce oxygen functional groups on Fe3O4/MWCNTs and thus accelerate HO˙ production.1736. Conclusions and future perspectives
In summary, this review presents the current application of carbon-based catalysts for O3 activation and degradation of organic pollutants. The engineering principles for regulating the active sites and electronic structures of carbocatalysts have been discussed, which will provide directions and general guidelines for developing high-performance carbocatalysts. This review also puts forward new insights into ROS generation and the reaction pathways based on the interaction intensity model between O3 and catalyst surface. In addition, the effect of the water matrix factors, including pH, inorganic anions and NOMs, on catalytic ozonation processes are highlighted. Based on our review, future directions to material design, ROS identification and mechanism regulation aiming at enhancing the application potential of carbon-based catalytic ozonation in real wastewaters are proposed as follows:(1) Encapsulating metal species beneath graphitic carbons (metal@C) has been proven as an effective strategy to increase both the reactivity and stability of the composites. Additionally, the magneticity endowed by the encapsulated metal species facilitates the separation of the nano-scaled catalysts. Apart from the dimensional effects, surface properties of the graphitic carbons and the types of the encapsulated metals are critical factors governing the electron-transfer ability and the catalytic activity of the nanocomposites. Therefore, it is promising to design metal@C catalysts with various numbers of coated graphitic shells, defective levels, and heteroatom dopants and manage the types and exposed crystal facets of the encapsulated metal species. The electron hybridizing, distribution, and transfer process at the metal–carbon heterojunctions can be evaluated by the DFT simulations which guide the theoretical design and screening the metal@C catalysts.
(2) In the current stage, intensive research attentions have been paid to identifying the types of ROS and elucidating ROS evolution mechanisms. However, the interactions of reactants on the active sites of the carbocatalysts are normally transient and thus hinder the unravelling of the in-depth insights. Future mechanistic studies require combined theoretical and experimental methodologies. The development of in situ, cryo-, or time-resolved characterization techniques with the isotopic substitutions will help unravel the interactions of the O3 and/or ambient molecules on catalyst surface and differentiate the types of generated ROS. Additionally, evaluating the existence and effects of other ROS apart from HO˙ in catalytic ozonation is challenging because of the high oxidation capacity and nonselectivity of HO˙. Therefore, it is crucial to develop probes or sensors that selectively react with targeted ROS, especially those with mild oxidation potentials.
(3) Water matrix parameters such as pH, inorganic anions and NOMs can significantly affect the interactions of O3 and organics on the catalyst surface and the reactivities of the generated ROS. HO˙-based radical pathway demonstrates a highly oxidative and nonselective behavior, yet the treatment efficiency would be decreased in the presence of inorganics anions and NOMs as radical scavengers. Nonradical pathways arising from the weak interactions between O3 and catalyst surface is vulnerable to be influenced by solution pH and ionic strength. Nonradical oxidation induced by strong interactions are typically recalcitrant toward the ionic strength and NOMs, while solution pH might influence the catalytic performance by affecting the surface charges. Therefore, developing reaction-oriented carbonaceous materials that can precisely regulate the reaction pathways based on the characters of different types of wastewater is highly desirable. DFT calculations of the interactions and electron properties at the atomic level can be employed to guide the design of on-demand active sites on carbocatalysts to trigger specific reaction pathways for different application scenarios.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciate the financial supports from the National Natural Science Foundation of China (No. 21978324), Beijing Natural Science Foundation (No. 8192039), and Science Foundation of China University of Petroleum, Beijing (No. 2462020YXZZ034). Partial support from the Australian Research Council (DE210100253) is also acknowledged.References
- B. C. Hodges, E. L. Cates and J.-H. Kim, Nat. Nanotechnol., 2018, 13, 642–650 Search PubMed.
- J. Lee, U. Von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 Search PubMed.
- H. G. Kjaergaard, T. Kurtén, L. B. Nielsen, S. Jørgensen and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 2525–2529 Search PubMed.
- R. Criegee, Angew. Chem., Int. Ed., 1975, 14, 745–752 Search PubMed.
- J. Nawrocki and B. Kasprzyk-Hordern, Appl. Catal., B, 2010, 99, 27–42 Search PubMed.
- B. Kasprzyk-Hordern, M. Ziółek and J. Nawrocki, Appl. Catal., B, 2003, 46, 639–669 Search PubMed.
- J. Nawrocki, Appl. Catal., B, 2013, 142, 465–471 Search PubMed.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 Search PubMed.
- J. Wang and H. Chen, Sci. Total Environ., 2020, 704, 135249 Search PubMed.
- L. Zhang, C.-Y. Lin, D. Zhang, L. Gong, Y. Zhu, Z. Zhao, Q. Xu, H. Li and Z. Xia, Adv. Mater., 2019, 31, 1805252 Search PubMed.
- L.-H. Zhang, Y. Shi, Y. Wang and N. R. Shiju, Adv. Sci., 2020, 7, 1902126 Search PubMed.
- J. Xiao, Y. Xie, J. Rabeah, A. Brückner and H. Cao, Acc. Chem. Res., 2020, 53, 1024–1033 Search PubMed.
- X. Duan, H. Sun and S. Wang, Acc. Chem. Res., 2018, 51, 678–687 Search PubMed.
- D. T. Oyekunle, X. Zhou, A. Shahzad and Z. Chen, J. Mater. Chem. A, 2021, 9, 8012–8050 Search PubMed.
- G. Yu, Y. Wang, H. Cao, H. Zhao and Y. Xie, Environ. Sci. Technol., 2020, 54, 5931–5946 Search PubMed.
- Y. Wang, X. Duan, Y. Xie, H. Sun and S. Wang, ACS Catal., 2020, 10, 13383–13414 Search PubMed.
- D. S. Su, G. Wen, S. Wu, F. Peng and R. Schlögl, Angew. Chem., Int. Ed., 2017, 56, 936–964 Search PubMed.
- Q. Ma, Y. Yu, M. Sindoro, A. G. Fane, R. Wang and H. Zhang, Adv. Mater., 2017, 29, 1605361 Search PubMed.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- C. Hu, Y. Xiao, Y. Zou and L. Dai, Electrochem. Energy Rev., 2018, 1, 84–112 Search PubMed.
- S. Zhang, X. Quan, J.-F. Zheng and D. Wang, Water Res., 2017, 122, 86–95 Search PubMed.
- H. Li, X. Miao, J. Zhang, J. Du, S. Xu, J. Tang and Y. Zhang, Chem. Eng. J., 2020, 381, 122680 Search PubMed.
- J. Xiao, Y. Xie, Q. Han, H. Cao, Y. Wang, F. Nawaz and F. Duan, J. Hazard. Mater., 2016, 304, 126–133 Search PubMed.
- Y. Yao, Y. Xie, B. Zhao, L. Zhou, Y. Shi, Y. Wang, Y. Sheng, H. Zhao, J. Sun and H. Cao, Chem. Eng. J., 2020, 382, 122708 Search PubMed.
- Z. Song, M. Wang, Z. Wang, Y. Wang, R. Li, Y. Zhang, C. Liu, Y. Liu, B. Xu and F. Qi, Environ. Sci. Technol., 2019, 53, 5337–5348 Search PubMed.
- J. Xiao, J. Rabeah, J. Yang, Y. Xie, H. Cao and A. Brückner, ACS Catal., 2017, 7, 6198–6206 Search PubMed.
- H. Lee, H.-i. Kim, S. Weon, W. Choi, Y. S. Hwang, J. Seo, C. Lee and J.-H. Kim, Environ. Sci. Technol., 2016, 50, 10134–10142 Search PubMed.
- I. C. Gerber and P. Serp, Chem. Rev., 2019, 120, 1250–1349 Search PubMed.
- G. K. Dimitrakakis, E. Tylianakis and G. E. Froudakis, Nano Lett., 2008, 8, 3166–3170 Search PubMed.
- H. Arandiyan, Y. Wang, H. Sun, M. Rezaei and H. Dai, Chem. Commun., 2018, 54, 6484–6502 Search PubMed.
- Y.-z. Liu, R.-t. Guo, C.-p. Duan, G.-l. Wu, Y.-f. Miao, J.-w. Gu and W.-g. Pan, Chemosphere, 2021, 262, 127886 Search PubMed.
- X. Wan, Y. Huang and Y. Chen, Acc. Chem. Res., 2012, 45, 598–607 Search PubMed.
- S. Sihn, V. Varshney, A. K. Roy and B. L. Farmer, Carbon, 2012, 50, 603–611 Search PubMed.
- H. Arandiyan, H. Dai, K. Ji, H. Sun, Y. Zhao and J. Li, Small, 2015, 11, 2366–2371 Search PubMed.
- Y. Wang, Y. Xie, H. Sun, J. Xiao, H. Cao and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 9710–9720 Search PubMed.
- Z. Song, Y. Zhang, C. Liu, B. Xu, F. Qi, D. Yuan and S. Pu, Chem. Eng. J., 2019, 357, 655–666 Search PubMed.
- X. Duan, H. Sun, Z. Ao, L. Zhou, G. Wang and S. Wang, Carbon, 2016, 107, 371–378 Search PubMed.
- Y. Wang, Z. Ao, H. Sun, X. Duan and S. Wang, Appl. Catal., B, 2016, 198, 295–302 Search PubMed.
- R. Qu, B. Xu, L. Meng, L. Wang and Z. Wang, Water Res., 2015, 68, 316–327 Search PubMed.
- T. F. de Oliveira, O. Chedeville, H. Fauduet and B. Cagnon, Desalination, 2011, 276, 359–365 Search PubMed.
- C. D. Vecitis, T. Lesko, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. A, 2010, 114, 4968–4980 Search PubMed.
- W. Ren, L. Xiong, G. Nie, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2019, 54, 1267–1275 Search PubMed.
- Y. Pi, J. Schumacher and M. Jekel, Water Res., 2005, 39, 83–88 Search PubMed.
- F. Bernat-Quesada, J. C. Espinosa, V. Barbera, M. Álvaro, M. Galimberti, S. Navalon and H. García, ACS Sustainable Chem. Eng., 2019, 7, 17443–17452 Search PubMed.
- A. Bhatnagar, W. Hogland, M. Marques and M. Sillanpää, Chem. Eng. J., 2013, 219, 499–511 Search PubMed.
- I. Gerber, M. Oubenali, R. Bacsa, J. Durand, A. Gonçalves, M. F. R. Pereira, F. Jolibois, L. Perrin, R. Poteau and P. Serp, Chem.–Eur. J., 2011, 17, 11467–11477 Search PubMed.
- X. Lu, W.-L. Yim, B. H. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901–2907 Search PubMed.
- H. Chen and K. C. Carroll, Environ. Pollut., 2016, 215, 96–102 Search PubMed.
- F. de Clippel, M. Dusselier, S. Van de Vyver, L. Peng, P. A. Jacobs and B. F. Sels, Green Chem., 2013, 15, 1398–1430 Search PubMed.
- A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853–1859 Search PubMed.
- Y. Jiang, L. Yang, T. Sun, J. Zhao, Z. Lyu, O. Zhuo, X. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707–6712 Search PubMed.
- X. Duan, Z. Ao, L. Zhou, H. Sun, G. Wang and S. Wang, Appl. Catal., B, 2016, 188, 98–105 Search PubMed.
- W. Yuan, Y. Zhou, Y. Li, C. Li, H. Peng, J. Zhang, Z. Liu, L. Dai and G. Shi, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- X. Duan, H. Sun, J. Kang, Y. Wang, S. Indrawirawan and S. Wang, ACS Catal., 2015, 5, 4629–4636 Search PubMed.
- R. Oulton, J. P. Haase, S. Kaalberg, C. T. Redmond, M. J. Nalbandian and D. M. Cwiertny, Environ. Sci. Technol., 2015, 49, 3687–3697 Search PubMed.
- H. Jin, H. Huang, Y. He, X. Feng, S. Wang, L. Dai and J. Wang, J. Am. Chem. Soc., 2015, 137, 7588–7591 Search PubMed.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 Search PubMed.
- X. Zhao, X. Zou, X. Yan, C. L. Brown, Z. Chen, G. Zhu and X. Yao, Inorg. Chem. Front., 2016, 3, 417–421 Search PubMed.
- Y. Wang, H. Cao, C. Chen, Y. Xie, H. Sun, X. Duan and S. Wang, Chem. Eng. J., 2019, 355, 118–129 Search PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 Search PubMed.
- J. Zhang, J. Zhang, F. He, Y. Chen, J. Zhu, D. Wang, S. Mu and H.-Y. Yang, Nano-Micro Lett., 2021, 13, 65 Search PubMed.
- Y. Jia, L. Zhang, L. Zhuang, H. Liu, X. Yan, X. Wang, J. Liu, J. Wang, Y. Zheng, Z. Xiao, E. Taran, J. Chen, D. Yang, Z. Zhu, S. Wang, L. Dai and X. Yao, Nat. Catal., 2019, 2, 688–695 Search PubMed.
- Z. Zhao, M. Li, L. Zhang, L. Dai and Z. Xia, Adv. Mater., 2015, 27, 6834–6840 Search PubMed.
- K. Gao, B. Wang, L. Tao, B. V. Cunning, Z. Zhang, S. Wang, R. S. Ruoff and L. Qu, Adv. Mater., 2019, 31, 1805121 Search PubMed.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 Search PubMed.
- K. Qu, Y. Zheng, X. Zhang, K. Davey, S. Dai and S. Z. Qiao, ACS Nano, 2017, 11, 7293–7300 Search PubMed.
- X. Duan, K. O'Donnell, H. Sun, Y. Wang and S. Wang, Small, 2015, 11, 3036–3044 Search PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., 2016, 128, 2270–2274 Search PubMed.
- J. Zhang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 13296–13300 Search PubMed.
- P. Huang, H. Li, X. Huang and D. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 21083–21088 Search PubMed.
- J. Zhou, J. Lian, L. Hou, J. Zhang, H. Gou, M. Xia, Y. Zhao, T. A. Strobel, L. Tao and F. Gao, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 Search PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 Search PubMed.
- Y. Wang, L. Chen, C. Chen, J. Xi, H. Cao, X. Duan, Y. Xie, W. Song and S. Wang, Appl. Catal., B, 2019, 254, 283–291 Search PubMed.
- S. Indrawirawan, H. Sun, X. Duan and S. Wang, J. Mater. Chem. A, 2015, 3, 3432–3440 Search PubMed.
- D. Li, X. Duan, H. Sun, J. Kang, H. Zhang, M. O. Tade and S. Wang, Carbon, 2017, 115, 649–658 Search PubMed.
- Z. Mo, S. Liao, Y. Zheng and Z. Fu, Carbon, 2012, 50, 2620–2627 Search PubMed.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 Search PubMed.
- A. Shen, Y. Zou, Q. Wang, R. A. Dryfe, X. Huang, S. Dou, L. Dai and S. Wang, Angew. Chem., 2014, 126, 10980–10984 Search PubMed.
- G. Pacchioni and H.-J. Freund, Chem. Soc. Rev., 2018, 47, 8474–8502 Search PubMed.
- G. Pacchioni, Phys. Chem. Chem. Phys., 2013, 15, 1737–1757 Search PubMed.
- T. Binninger, T. J. Schmidt and D. Kramer, Phys. Rev. B, 2017, 96, 165405 Search PubMed.
- Y. Wang, Y. Xie, H. Sun, J. Xiao, H. Cao and S. Wang, J. Hazard. Mater., 2016, 301, 56–64 Search PubMed.
- L. Zhao, Z. Sun and J. Ma, Environ. Sci. Technol., 2009, 43, 4157–4163 Search PubMed.
- J. Bing, C. Hu, Y. Nie, M. Yang and J. Qu, Environ. Sci. Technol., 2015, 49, 1690–1697 Search PubMed.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, ACS Catal., 2017, 7, 6493–6513 Search PubMed.
- H. Yuan, J. Li, W. Yang, Z. Zhuang, Y. Zhao, L. He, L. Xu, X. Liao, R. Zhu and L. Mai, ACS Appl. Mater. Interfaces, 2018, 10, 16410–16417 Search PubMed.
- A. Moya, A. Cherevan, S. Marchesan, P. Gebhardt, M. Prato, D. Eder and J. J. Vilatela, Appl. Catal., B, 2015, 179, 574–582 Search PubMed.
- Z. Wang, J. Jiang, S. Pang, Y. Zhou, C. Guan, Y. Gao, J. Li, Y. Yang, W. Qiu and C. Jiang, Environ. Sci. Technol., 2018, 52, 11276–11284 Search PubMed.
- S. Liang, L. Zhu, J. Hua, W. Duan, P.-T. Yang, S.-L. Wang, C. Wei, C. Liu and C. Feng, Environ. Sci. Technol., 2020, 54, 6406–6414 Search PubMed.
- Y. Zong, Y. Shao, Y. Zeng, B. Shao, L. Xu, Z. Zhao, W. Liu and D. Wu, Environ. Sci. Technol., 2021, 55, 7634–7642 Search PubMed.
- O. Pestovsky, S. Stoian, E. L. Bominaar, X. Shan, E. Münck, L. Que, Jr and A. Bakac, Angew. Chem., Int. Ed., 2005, 44, 6871–6874 Search PubMed.
- Y. Chen, J. Wei, M. S. Duyar, V. V. Ordomsky, A. Y. Khodakov and J. Liu, Chem. Soc. Rev., 2021, 50, 2337–2366 Search PubMed.
- C. Zhang, X. Guo, Q. Yuan, R. Zhang, Q. Chang, K. Li, B. Xiao, S. Liu, C. Ma and X. Liu, ACS Catal., 2018, 8, 7120–7130 Search PubMed.
- X. Sun, A. I. O. Suarez, M. Meijerink, T. Van Deelen, S. Ould-Chikh, J. Zečević, K. P. De Jong, F. Kapteijn and J. Gascon, Nat. Commun., 2017, 8, 1–8 Search PubMed.
- Y. Wang, N. Ren, J. Xi, Y. Liu, T. Kong, C. Chen, Y. Xie, X. Duan and S. Wang, ACS ES&T Engg, 2021, 1, 32–45 Search PubMed.
- X. Duan, J. Kang, W. Tian, H. Zhang, S.-H. Ho, Y.-A. Zhu, Z. Ao, H. Sun and S. Wang, Appl. Catal., B, 2019, 256, 117795 Search PubMed.
- S. Weon, D. Huang, K. Rigby, C. Chu, X. Wu and J.-H. Kim, ACS ES&T Engg, 2021, 1, 157–172 Search PubMed.
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 Search PubMed.
- C. Chu, J. Yang, X. Zhou, D. Huang, H. Qi, S. Weon, J. Li, M. Elimelech, A. Wang and J.-H. Kim, Environ. Sci. Technol., 2021, 55, 1242–1250 Search PubMed.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. D. Kim, E. L. Samuel, Z. Peng, Z. Zhu, F. Qin and J. Bao, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong and Z. Deng, Angew. Chem., 2016, 128, 10958–10963 Search PubMed.
- X. Chen, D.-D. Ma, B. Chen, K. Zhang, R. Zou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2020, 267, 118720 Search PubMed.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 4585 Search PubMed.
- X. Zhang, S. Zhang, Y. Yang, L. Wang, Z. Mu, H. Zhu, X. Zhu, H. Xing, H. Xia, B. Huang, J. Li, S. Guo and E. Wang, Adv. Mater., 2020, 32, 1906905 Search PubMed.
- D. Ji, L. Fan, L. Tao, Y. Sun, M. Li, G. Yang, T. Q. Tran, S. Ramakrishna and S. Guo, Angew. Chem., Int. Ed., 2019, 58, 13840–13844 Search PubMed.
- X. Mi, P. Wang, S. Xu, L. Su, H. Zhong, H. Wang, Y. Li and S. Zhan, Angew. Chem., Int. Ed., 2021, 60, 4588–4593 Search PubMed.
- Y. Hou, M. Qiu, M. G. Kim, P. Liu, G. Nam, T. Zhang, X. Zhuang, B. Yang, J. Cho, M. Chen, C. Yuan, L. Lei and X. Feng, Nat. Commun., 2019, 10, 1392 Search PubMed.
- U. Jans and J. Hoigné, Ozone: Sci. Eng., 1998, 20, 67–90 Search PubMed.
- F. J. Beltrán, J. Rivas, P. Álvarez and R. Montero-de-Espinosa, Ozone: Sci. Eng., 2002, 24, 227–237 Search PubMed.
- F. J. Beltrán, F. J. Rivas, L. A. Fernández, P. M. Álvarez and R. Montero-de-Espinosa, Ind. Eng. Chem. Res., 2002, 41, 6510–6517 Search PubMed.
- P. Faria, J. Órfão and M. Pereira, Appl. Catal., B, 2008, 79, 237–243 Search PubMed.
- L. Gu, X. Zhang and L. Lei, Ind. Eng. Chem. Res., 2008, 47, 6809–6815 Search PubMed.
- P. M. Alvarez, J. F. Garcia-Araya, F. J. Beltrán, I. Giráldez, J. Jaramillo and V. Gómez-Serrano, Carbon, 2006, 44, 3102–3112 Search PubMed.
- L. Lei, L. Gu, X. Zhang and Y. Su, Appl. Catal., A, 2007, 327, 287–294 Search PubMed.
- P. C. Faria, J. J. Órfão and M. F. R. Pereira, Ind. Eng. Chem. Res., 2006, 45, 2715–2721 Search PubMed.
- J. Rivera-Utrilla and M. Sánchez-Polo, Appl. Catal., B, 2002, 39, 319–329 Search PubMed.
- H. Cao, L. Xing, G. Wu, Y. Xie, S. Shi, Y. Zhang, D. Minakata and J. C. Crittenden, Appl. Catal., B, 2014, 146, 169–176 Search PubMed.
- J. Rivera-Utrilla and M. Sánchez-Polo, Langmuir, 2004, 20, 9217–9222 Search PubMed.
- L. Xing, Y. Xie, H. Cao, D. Minakata, Y. Zhang and J. C. Crittenden, Chem. Eng. J., 2014, 245, 71–79 Search PubMed.
- Y.-d. Chen, R. Wang, X. Duan, S. Wang, N.-q. Ren and S.-H. Ho, Water Res., 2020, 187, 116390 Search PubMed.
- C. Chen, X. Yan, Y. Xu, B. A. Yoza, X. Wang, Y. Kou, H. Ye, Q. Wang and Q. X. Li, Sci. Total Environ., 2019, 651, 2631–2640 Search PubMed.
- C. Zhao, B. Shao, M. Yan, Z. Liu, Q. Liang, Q. He, T. Wu, Y. Liu, Y. Pan and J. Huang, Chem. Eng. J., 2021, 416, 128829 Search PubMed.
- G. Wen, Z.-H. Pan, J. Ma, Z.-Q. Liu, L. Zhao and J.-J. Li, J. Hazard. Mater., 2012, 239, 381–388 Search PubMed.
- G. Moussavi and R. Khosravi, Bioresour. Technol., 2012, 119, 66–71 Search PubMed.
- F. Zhang, K. Wu, H. Zhou, Y. Hu, P. Sergei, H. Wu and C. Wei, J. Environ. Manage., 2018, 224, 376–386 Search PubMed.
- A. Gonçalves, J. J. Órfão and M. F. R. Pereira, Appl. Catal., B, 2013, 140, 82–91 Search PubMed.
- X. Fan, J. Restivo, J. J. Órfão, M. F. R. Pereira and A. A. Lapkin, Chem. Eng. J., 2014, 241, 66–76 Search PubMed.
- Z.-Q. Liu, J. Ma, Y.-H. Cui and B.-P. Zhang, Appl. Catal., B, 2009, 92, 301–306 Search PubMed.
- Z.-Q. Liu, J. Ma, Y.-H. Cui, L. Zhao and B.-P. Zhang, Appl. Catal., B, 2010, 101, 74–80 Search PubMed.
- A. G. Gonçalves, J. L. Figueiredo, J. J. Órfão and M. F. Pereira, Carbon, 2010, 48, 4369–4381 Search PubMed.
- A. G. Gonçalves, J. J. Órfão and M. F. Pereira, Catal. Commun., 2013, 35, 82–87 Search PubMed.
- I. D. Rosca, F. Watari, M. Uo and T. Akasaka, Carbon, 2005, 43, 3124–3131 Search PubMed.
- B. Smith, K. Wepasnick, K. E. Schrote, H.-H. Cho, W. P. Ball and D. H. Fairbrother, Langmuir, 2009, 25, 9767–9776 Search PubMed.
- J. Restivo, R. P. Rocha, A. M. Silva, J. J. Órfão, M. F. Pereira and J. L. Figueiredo, Chin. J. Catal., 2014, 35, 896–905 Search PubMed.
- O. Soares, R. Rocha, A. Gonçalves, J. Figueiredo, J. Órfão and M. Pereira, Appl. Catal., B, 2016, 192, 296–303 Search PubMed.
- A. S. G. Santos, C. A. Orge, O. S. G. Soares and M. F. R. Pereira, J. Water Process. Eng., 2020, 38, 101573 Search PubMed.
- J. Wang, S. Chen, X. Quan and H. Yu, Chemosphere, 2018, 190, 135–143 Search PubMed.
- J.-N. Liu, Z. Chen, Q.-Y. Wu, A. Li, H.-Y. Hu and C. Yang, Sci. Rep., 2016, 6, 1–9 Search PubMed.
- B. Ye, M.-Y. Lee, W.-L. Wang, A. Li, Z.-Y. Liu, Q.-Y. Wu and H.-Y. Hu, J. Hazard. Mater., 2020, 394, 122563 Search PubMed.
- L. Jothinathan and J. Hu, Water Res., 2018, 134, 63–73 Search PubMed.
- Y. Wang, H. Cao, L. Chen, C. Chen, X. Duan, Y. Xie, W. Song, H. Sun and S. Wang, Appl. Catal., B, 2018, 229, 71–80 Search PubMed.
- X.-K. Kong, C.-L. Chen and Q.-W. Chen, Chem. Soc. Rev., 2014, 43, 2841–2857 Search PubMed.
- S. Osumi, S. Saito, C. Dou, K. Matsuo, K. Kume, H. Yoshikawa, K. Awaga and S. Yamaguchi, Chem. Sci., 2016, 7, 219–227 Search PubMed.
- R. P. Rocha, A. Gonçalves, L. Pastrana-Martínez, B. Bordoni, O. Soares, J. Órfão, J. L. Faria, J. L. Figueiredo, A. Silva and M. F. R. Pereira, Catal. Today, 2015, 249, 192–198 Search PubMed.
- R. Yin, W. Guo, J. Du, X. Zhou, H. Zheng, Q. Wu, J. Chang and N. Ren, Chem. Eng. J., 2017, 317, 632–639 Search PubMed.
- F. Yang, M. Zhao, Z. Wang, H. Ji, B. Zheng, D. Xiao, L. Wu and Y. Guo, RSC Adv., 2014, 4, 58325–58328 Search PubMed.
- T. Du, A. S. Adeleye, T. Zhang, N. Yang, R. Hao, Y. Li, W. Song and W. Chen, Environ. Sci.: Nano, 2019, 6, 2484–2494 Search PubMed.
- Z. Wang, L. Sun, X. Lou, F. Yang, M. Feng and J. Liu, J. Colloid Interface Sci., 2017, 507, 51–58 Search PubMed.
- C.-Z. Zhang, T. Li, Y. Yuan and J. Xu, Chemosphere, 2016, 153, 531–540 Search PubMed.
- G. Liao, D. Zhu, L. Li and B. Lan, J. Hazard. Mater., 2014, 280, 531–535 Search PubMed.
- J. Xiao, Y. Xie, F. Nawaz, S. Jin, F. Duan, M. Li and H. Cao, Appl. Catal., B, 2016, 181, 420–428 Search PubMed.
- J. Xiao, Y. Xie, F. Nawaz, Y. Wang, P. Du and H. Cao, Appl. Catal., B, 2016, 183, 417–425 Search PubMed.
- J. Zhang, B. Xin, C. Shan, W. Zhang, D. D. Dionysiou and B. Pan, Appl. Catal., B, 2021, 292, 120155 Search PubMed.
- X. Yuan, R. Xie, Q. Zhang, L. Sun, X. Long and D. Xia, Sep. Purif. Technol., 2019, 211, 823–831 Search PubMed.
- F. Zhang, J. Liao, J. Lu and J. Niu, Sep. Purif. Technol., 2021, 256, 117806 Search PubMed.
- J. Xiao, Q. Han, Y. Xie, J. Yang, Q. Su, Y. Chen and H. Cao, Environ. Sci. Technol., 2017, 51, 13380–13387 Search PubMed.
- Z. Sun, L. Zhao, C. Liu, Y. Zhen and J. Ma, Environ. Sci. Technol., 2019, 53, 10342–10351 Search PubMed.
- Y. Wang, J. Xi, X. Duan, W. Lv, H. Cao, C. Chen, Z. Guo, Y. Xie and S. Wang, J. Hazard. Mater., 2020, 384, 121486 Search PubMed.
- F. J. Beltrán, J. F. García-Araya and I. Giráldez, Appl. Catal., B, 2006, 63, 249–259 Search PubMed.
- P. Faria, J. Órfão and M. Pereira, Appl. Catal., B, 2008, 83, 150–159 Search PubMed.
- J. Ma, M. Sui, T. Zhang and C. Guan, Water Res., 2005, 39, 779–786 Search PubMed.
- Y. Huang, Y. Sun, Z. Xu, M. Luo, C. Zhu and L. Li, Sci. Total Environ., 2017, 575, 50–57 Search PubMed.
- H. Zhuang, H. Han, B. Hou, S. Jia and Q. Zhao, Bioresour. Technol., 2014, 166, 178–186 Search PubMed.
- G. Li, Y. Lu, C. Lu, M. Zhu, C. Zhai, Y. Du and P. Yang, J. Hazard. Mater., 2015, 294, 201–208 Search PubMed.
- G. Li, K. Li, A. Liu, P. Yang, Y. Du and M. Zhu, Sci. Rep., 2017, 7, 1–7 Search PubMed.
- J. Wang, X. Quan, S. Chen, H. Yu and Y. Chen, Environ. Sci.: Nano, 2019, 6, 1932–1940 Search PubMed.
- J. Wu, H. Gao, S. Yao, L. Chen, Y. Gao and H. Zhang, Sep. Purif. Technol., 2015, 147, 179–185 Search PubMed.
- Y. Huang, C. Cui, D. Zhang, L. Li and D. Pan, Chemosphere, 2015, 119, 295–301 Search PubMed.
- C. Chen, H. Chen, X. Guo, S. Guo and G. Yan, J. Ind. Eng. Chem., 2014, 20, 2782–2791 Search PubMed.
- S. Hou, S. Jia, J. Jia, Z. He, G. Li, Q. Zuo and H. Zhuang, J. Environ. Manage., 2020, 267, 110615 Search PubMed.
- Y. Yuan, G. Xing, S. Garg, J. Ma, X. Kong, P. Dai and T. D. Waite, Water Res., 2020, 177, 115785 Search PubMed.
- Y. Huang, W. Xu, L. Hu, J. Zeng, C. He, X. Tan, Z. He, Q. Zhang and D. Shu, Catal. Today, 2017, 297, 143–150 Search PubMed.
- Y. Liu, A. Zhou, Y. Liu and J. Wang, Chemosphere, 2018, 191, 54–63 Search PubMed.
- X. Yuan, W. Qin, X. Lei, L. Sun, Q. Li, D. Li, H. Xu and D. Xia, Chemosphere, 2018, 205, 369–379 Search PubMed.
- Z.-Q. Liu, J. Ma and Y.-H. Cui, Carbon, 2008, 46, 890–897 Search PubMed.
- Q. Dai, J. Wang, J. Chen and J. Chen, Sep. Purif. Technol., 2014, 127, 112–120 Search PubMed.
- P. Faria, J. Órfão and M. Pereira, Appl. Catal., B, 2009, 88, 341–350 Search PubMed.
- A. Gonçalves, J. Silvestre-Albero, E. V. Ramos-Fernandez, J. C. Serrano-Ruiz, J. J. Órfão, A. Sepúlveda-Escribano and M. F. R. Pereira, Appl. Catal., B, 2012, 113, 308–317 Search PubMed.
- L. Li, W. Ye, Q. Zhang, F. Sun, P. Lu and X. Li, J. Hazard. Mater., 2009, 170, 411–416 Search PubMed.
- P. Faria, J. Órfão and M. Pereira, Catal. Commun., 2008, 9, 2121–2126 Search PubMed.
- A. G. Gonçalves, J. J. Órfão and M. F. R. Pereira, Chem. Eng. J., 2014, 250, 366–376 Search PubMed.
- J. Wang, X. Quan, S. Chen, H. Yu and G. Liu, J. Hazard. Mater., 2019, 368, 621–629 Search PubMed.
- Y. Jiang, M. Wei, J. Feng, Y. Ma and S. Xiong, Energy Environ. Sci., 2016, 9, 1430–1438 Search PubMed.
- X. Meng, C. Yu, X. Song, J. Iocozzia, J. Hong, M. Rager, H. Jin, S. Wang, L. Huang and J. Qiu, Angew. Chem., 2018, 130, 4772–4776 Search PubMed.
- S. Shrestha, Y. Liu and W. E. Mustain, Catal. Rev., 2011, 53, 256–336 Search PubMed.
- B. Zhang and D. S. Su, ChemCatChem, 2015, 7, 3639–3645 Search PubMed.
- J. Xu, Y. Li, M. Qian, J. Pan, J. Ding and B. Guan, Appl. Catal., B, 2019, 256, 117797 Search PubMed.
- Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 Search PubMed.
- S. Campisi, C. E. Chan-Thaw and A. Villa, Appl. Sci., 2018, 8, 1159 Search PubMed.
- Q. Bao, K. San Hui and J. G. Duh, J. Environ. Sci., 2016, 50, 38–48 Search PubMed.
- C. Li, F. Jiang, D. Sun and B. Qiu, Chem. Eng. J., 2017, 325, 624–631 Search PubMed.
- Y. Yu, H. An, Y. Zhao, J. Feng, T. Wei, S. Yu, Y. Ren and Y. Chen, Sep. Purif. Technol., 2021, 259, 118097 Search PubMed.
- Y. Ren, H. Zhang, H. An, Y. Zhao, J. Feng, L. Xue, T. Luan and Z. Fan, J. Colloid Interface Sci., 2018, 526, 347–355 Search PubMed.
- J. Wu, L. Xiong, B. Zhao, M. Liu and L. Huang, Small Methods, 2020, 4, 1900540 Search PubMed.
- Z. Luo, D. Wang, W. Zeng and J. Yang, Sci. Total Environ., 2020, 734, 139448 Search PubMed.
- S.-Q. Tian, J.-Y. Qi, Y.-P. Wang, Y.-L. Liu, L. Wang and J. Ma, Water Res., 2021, 193, 116860 Search PubMed.
- M. Sui, S. Xing, L. Sheng, S. Huang and H. Guo, J. Hazard. Mater., 2012, 227, 227–236 Search PubMed.
- S. Zhang, D. Wang, X. Quan, L. Zhou and X. Zhang, Sep. Purif. Technol., 2013, 116, 351–359 Search PubMed.
- A. Abdedayem, M. Guiza, F. J. R. Toledo and A. Ouederni, Sep. Purif. Technol., 2017, 184, 308–318 Search PubMed.
- N. Nasseh, F. S. Arghavan, S. Rodriguez-Couto, A. H. Panahi, M. Esmati and T. J. A-Musawi, Adv. Powder Technol., 2020, 31, 875–885 Search PubMed.
- W. Wang, H. Yao and L. Yue, Environ. Sci. Pollut. Res., 2020, 27, 7199–7210 Search PubMed.
- J. Liu, J. Li, S. He, L. Sun, X. Yuan and D. Xia, Sep. Purif. Technol., 2020, 234, 116120 Search PubMed.
- J. Feng, X. Zhang, J. Fu and H. Chen, Catal. Commun., 2018, 110, 28–32 Search PubMed.
- B. Ye, Z. Chen, X. Li, J. Liu, Q. Wu, C. Yang, H. Hu and R. Wang, Front. Environ. Sci. Eng., 2019, 13, 1–9 Search PubMed.
- J. Akhtar, N. S. Amin and A. Aris, Chem. Eng. J., 2011, 170, 136–144 Search PubMed.
- X. Lü, Q. Zhang, W. Yang, X. Li, L. Zeng and L. Li, RSC Adv., 2015, 5, 10537–10545 Search PubMed.
- S. Tang, D. Yuan, Q. Zhang, Y. Liu, Z. Liu and H. Huang, Environ. Sci. Pollut. Res., 2016, 23, 18800–18808 Search PubMed.
- L. Khachatryan, C. A. McFerrin, R. W. Hall and B. Dellinger, Environ. Sci. Technol., 2014, 48, 9220–9226 Search PubMed.
- R. J. Singh, H. Karoui, M. R. Gunther, J. S. Beckman, R. P. Mason and B. Kalyanaraman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6675–6680 Search PubMed.
- K. Qian, H. Chen, W. Li, Z. Ao, Y.-n. Wu and X. Guan, Environ. Sci. Technol., 2021, 55, 7034–7043 Search PubMed.
- Y. Zong, X. Guan, J. Xu, Y. Feng, Y. Mao, L. Xu, H. Chu and D. Wu, Environ. Sci. Technol., 2020, 54, 16231–16239 Search PubMed.
- T. Olmez-Hanci and I. Arslan-Alaton, Chem. Eng. J., 2013, 224, 10–16 Search PubMed.
- Q. Dai, J. Wang, J. Yu, J. Chen and J. Chen, Appl. Catal., B, 2014, 144, 686–693 Search PubMed.
- R. C. Martins and R. M. Quinta-Ferreira, Appl. Catal., B, 2009, 90, 268–277 Search PubMed.
- W. Ren, L. Xiong, X. Yuan, Z. Yu, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2019, 53, 14595–14603 Search PubMed.
- W. Ren, G. Nie, P. Zhou, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2020, 54, 6438–6447 Search PubMed.
- R. Andreozzi, A. Insola, V. Caprio, R. Marotta and V. Tufano, Appl. Catal., A, 1996, 138, 75–81 Search PubMed.
- F. J. Beltrán, F. J. Rivas and R. Montero-de-Espinosa, Water Res., 2005, 39, 3553–3564 Search PubMed.
- F. Nawaz, H. Cao, Y. Xie, J. Xiao, Y. Chen and Z. A. Ghazi, Chemosphere, 2017, 168, 1457–1466 Search PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 Search PubMed.
- H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599–606 Search PubMed.
- J. Xiao, Q. Han, H. Cao, J. Rabeah, J. Yang, Z. Guo, L. Zhou, Y. Xie and A. Brückner, ACS Catal., 2019, 9, 8852–8861 Search PubMed.
- M. J. Davies, B. C. Gilbert, J. K. Stell and A. C. Whitwood, J. Chem. Soc., Perkin Trans. 2, 1992,(3), 333–335 Search PubMed.
- X. Chen, W.-D. Oh and T.-T. Lim, Chem. Eng. J., 2018, 354, 941–976 Search PubMed.
- K. Nakamura, K. Ishiyama, H. Ikai, T. Kanno, K. Sasaki, Y. Niwano and M. Kohno, J. Clin. Biochem. Nutr., 2011, 49, 87–95 Search PubMed.
- P. Bilski, K. Reszka, M. Bilska and C. Chignell, J. Am. Chem. Soc., 1996, 118, 1330–1338 Search PubMed.
- R. Poupko and I. Rosenthal, J. Phys. Chem., 1973, 77, 1722–1724 Search PubMed.
- T. A. Konovalova, J. Lawrence and L. D. Kispert, J. Photochem. Photobiol., A, 2004, 162, 1–8 Search PubMed.
- J. Cattani, V. Subramaniam and M. Drescher, Phys. Chem. Chem. Phys., 2017, 19, 18147–18151 Search PubMed.
- D. L. Marshall, M. L. Christian, G. Gryn'ova, M. L. Coote, P. J. Barker and S. J. Blanksby, Org. Biomol. Chem., 2011, 9, 4936–4947 Search PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 Search PubMed.
- J. Xiao, Y. Xie, H. Cao, Y. Wang, Z. Guo and Y. Chen, Carbon, 2016, 107, 658–666 Search PubMed.
- T. Turan-Ertas and M. D. Gurol, Chemosphere, 2002, 47, 293–301 Search PubMed.
- Y. Wang, L. Chen, H. Cao, Z. Chi, C. Chen, X. Duan, Y. Xie, F. Qi, W. Song and J. Liu, Appl. Catal., B, 2019, 245, 546–554 Search PubMed.
- E. Mvula and C. Von Sonntag, Org. Biomol. Chem., 2003, 1, 1749–1756 Search PubMed.
- A. Jawad, X. Lu, Z. Chen and G. Yin, J. Phys. Chem. A, 2014, 118, 10028–10035 Search PubMed.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1995, 24, 663–677 Search PubMed.
- C. A. Davis, K. McNeill and E. M.-L. Janssen, Environ. Sci. Technol., 2018, 52, 9908–9916 Search PubMed.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1758 Search PubMed.
- O. Núñez, L. Madriz, D. Carvajal, J. Tatá and R. Vargas, J. Phys. Org. Chem., 2019, 32, e3952 Search PubMed.
- S. Zhu, X. Huang, F. Ma, L. Wang, X. Duan and S. Wang, Environ. Sci. Technol., 2018, 52, 8649–8658 Search PubMed.
- Y. Pi, J. Schumacher and M. Jekel, Ozone: Sci. Eng., 2005, 27, 431–436 Search PubMed.
- Y. Guo, J. Zhan, G. Yu and Y. Wang, Water Res., 2021, 194, 116927 Search PubMed.
- Y. Guo, Y. Zhang, G. Yu and Y. Wang, Appl. Catal., B, 2021, 280, 119418 Search PubMed.
- G.-X. Huang, J.-Y. Si, C. Qian, W.-K. Wang, S.-C. Mei, C.-Y. Wang and H.-Q. Yu, Anal. Chem., 2018, 90, 14439–14446 Search PubMed.
- L. Zhu, J. Ji, J. Liu, S. Mine, M. Matsuoka, J. Zhang and M. Xing, Angew. Chem., Int. Ed., 2020, 59, 13968–13976 Search PubMed.
- J.-F. Jen, M.-F. Leu and T. C. Yang, J. Chromatogr. A, 1998, 796, 283–288 Search PubMed.
- D. Wu, M. Liu, D. Dong and X. Zhou, Microchem. J., 2007, 85, 250–256 Search PubMed.
- C. A. Martínez-Huitle, M. A. Quiroz, C. Comninellis, S. Ferro and A. De Battisti, Electrochim. Acta, 2004, 50, 949–956 Search PubMed.
- C. Tai, J.-F. Peng, J.-F. Liu, G.-B. Jiang and H. Zou, Anal. Chim. Acta, 2004, 527, 73–80 Search PubMed.
- P. L. Gai and E. D. Boyes, Microsc. Res. Tech., 2009, 72, 153–164 Search PubMed.
- L. Linxiang, Y. Abe, Y. Nagasawa, R. Kudo, N. Usui, K. Imai, T. Mashino, M. Mochizuki and N. Miyata, Biomed. Chromatogr., 2004, 18, 470–474 Search PubMed.
- P. Zhou, F. Cheng, G. Nie, Y. Yang, K. Hu, X. Duan, Y. Zhang and S. Wang, Green Energy Environ., 2020, 5, 414–422 Search PubMed.
- J. Wang, Y. Guo, B. Liu, X. Jin, L. Liu, R. Xu, Y. Kong and B. Wang, Ultrason. Sonochem., 2011, 18, 177–183 Search PubMed.
- F. Auchère and F. Rusnak, J. Biol. Inorg Chem., 2002, 7, 664–667 Search PubMed.
- S. Miyamoto, G. R. Martinez, M. H. Medeiros and P. Di Mascio, J. Am. Chem. Soc., 2003, 125, 6172–6179 Search PubMed.
- G. Louit, S. Foley, J. Cabillic, H. Coffigny, F. Taran, A. Valleix, J. P. Renault and S. Pin, Radiat. Phys. Chem., 2005, 72, 119–124 Search PubMed.
- S. Zhang, X. Quan, J. Zheng and D. Wang, Water Res., 2017, 122, 86–95 Search PubMed.
- K. Zhang, P. Sun, M. C. A. Faye and Y. Zhang, Carbon, 2018, 130, 730–740 Search PubMed.
- H. Wang, W. Guo, R. Yin, J. Du, Q. Wu, H. Luo, B. Liu, F. Sseguya and N. Ren, Chem. Eng. J., 2019, 362, 561–569 Search PubMed.
- M. T. Pinho, H. T. Gomes, R. S. Ribeiro, J. L. Faria and A. M. Silva, Appl. Catal., B, 2015, 165, 706–714 Search PubMed.
- M. Martin-Martinez, M. F. F. Barreiro, A. M. Silva, J. L. Figueiredo, J. L. Faria and H. T. Gomes, Appl. Catal., B, 2017, 219, 372–378 Search PubMed.
- D. T. Sawyer and J. S. Valentine, Acc. Chem. Res., 1981, 14, 393–400 Search PubMed.
- R. Baum, Chem. Eng. News, 1984, 62, 20–26 Search PubMed.
- A. A. Frimer, T. Farkash-Solomon and G. Aljadeff, J. Org. Chem., 1986, 51, 2093–2098 Search PubMed.
- D. R. Kearns, Chem. Rev., 1971, 71, 395–427 Search PubMed.
- M. J. Thomas and C. S. Foote, Photochem. Photobiol., 1978, 27, 683–693 Search PubMed.
- F. E. Scully, Jr and J. Hoigné, Chemosphere, 1987, 16, 681–694 Search PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 Search PubMed.
- M. H. Plumlee, K. McNeill and M. Reinhard, Environ. Sci. Technol., 2009, 43, 3662–3668 Search PubMed.
- J. Wang and S. Wang, Chem. Eng. J., 2020, 401, 126158 Search PubMed.
- Y. Wang, Y. Xie, H. Sun, J. Xiao, H. Cao and S. Wang, Catal. Sci. Technol., 2016, 6, 2918–2929 Search PubMed.
- R. Luo, M. Li, C. Wang, M. Zhang, M. A. N. Khan, X. Sun, J. Shen, W. Han, L. Wang and J. Li, Water Res., 2019, 148, 416–424 Search PubMed.
- C. Zhu, Y. Zhang, Z. Fan, F. Liu and A. Li, J. Hazard. Mater., 2020, 393, 122395 Search PubMed.
- X. Cheng, H. Guo, Y. Zhang, X. Wu and Y. Liu, Water Res., 2017, 113, 80–88 Search PubMed.
- F. Qi, B. Xu, Z. Chen, L. Zhang, P. Zhang and D. Sun, Chem. Eng. J., 2010, 165, 490–499 Search PubMed.
- J.-S. Park, H. Choi and J. Cho, Water Res., 2004, 38, 2285–2292 Search PubMed.
- J. Hoigné and H. Bader, Water Res., 1983, 17, 185–194 Search PubMed.
- M. Heyrovský and S. Vavřička, J. Electroanal. Chem. Interfacial Electrochem., 1972, 36, 203–221 Search PubMed.
- S. Zhu, X. Li, J. Kang, X. Duan and S. Wang, Environ. Sci. Technol., 2018, 53, 307–315 Search PubMed.
- B. Legube and N. K. V. Leitner, Catal. Today, 1999, 53, 61–72 Search PubMed.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1637–1755 Search PubMed.
- S. Giannakis, K.-Y. A. Lin and F. Ghanbari, Chem. Eng. J., 2021, 406, 127083 Search PubMed.
- U. Von Gunten, Water Res., 2003, 37, 1469–1487 Search PubMed.
- Z. Zuo, Z. Cai, Y. Katsumura, N. Chitose and Y. Muroya, Radiat. Phys. Chem., 1999, 55, 15–23 Search PubMed.
- A. Nemes, I. Fabian and R. van Eldik, J. Phys. Chem. A, 2000, 104, 7995–8000 Search PubMed.
- R. Huang, H. Yan, L. Li, D. Deng, Y. Shu and Q. Zhang, Appl. Catal., B, 2011, 106, 264–271 Search PubMed.
- K. Zhang and K. M. Parker, Environ. Sci. Technol., 2018, 52, 9579–9594 Search PubMed.
- W. Yang and T. Wu, Ind. Eng. Chem. Res., 2019, 58, 3468–3477 Search PubMed.
- J. Sotelo, F. Beltran, F. Benitez and J. Beltran-Heredia, Water Res., 1989, 23, 1239–1246 Search PubMed.
- M. Mekic, M. Brigante, D. Vione and S. Gligorovski, Atmos. Environ., 2018, 185, 237–242 Search PubMed.
- Y. Huang, F. Yang, L. Ai, M. Feng, C. Wang, Z. Wang and J. Liu, Chemosphere, 2017, 179, 331–336 Search PubMed.
- M. Mekic, G. Loisel, W. Zhou, B. Jiang, D. Vione and S. Gligorovski, Environ. Sci. Technol., 2018, 52, 12306–12315 Search PubMed.
- H. Herrmann, Chem. Rev., 2003, 103, 4691–4716 Search PubMed.
- A. Levanov, I. Kuskov, A. Zosimov, E. Antipenko and V. Lunin, Kinet. Catal., 2003, 44, 740–746 Search PubMed.
- T. Zhang, Y. Chen, Y. Wang, J. Le Roux, Y. Yang and J.-P. Croue, Environ. Sci. Technol., 2014, 48, 5868–5875 Search PubMed.
- Y. S. Al-Degs, M. I. El-Barghouthi, A. H. El-Sheikh and G. M. Walker, Dyes Pigm., 2008, 77, 16–23 Search PubMed.
- M. Sui, L. Sheng, K. Lu and F. Tian, Appl. Catal., B, 2010, 96, 94–100 Search PubMed.
- T. Zhang, C. Li, J. Ma, H. Tian and Z. Qiang, Appl. Catal., B, 2008, 82, 131–137 Search PubMed.
- O. S. Keen, G. McKay, S. P. Mezyk, K. G. Linden and F. L. Rosario-Ortiz, Water Res., 2014, 50, 408–419 Search PubMed.
- H. El-taliawy, M. Ekblad, F. Nilsson, M. Hagman, N. Paxeus, K. Jönsson, M. Cimbritz, J. la Cour Jansen and K. Bester, Chem. Eng. J., 2017, 325, 310–321 Search PubMed.
- I. Michael-Kordatou, C. Michael, X. Duan, X. He, D. Dionysiou, M. Mills and D. Fatta-Kassinos, Water Res., 2015, 77, 213–248 Search PubMed.
- M. Nihemaiti, D. B. Miklos, U. Hübner, K. G. Linden, J. E. Drewes and J.-P. Croué, Water Res., 2018, 145, 487–497 Search PubMed.
- D. Wang, H. Xu, J. Ma, S. Giannakis, X. Lu, H. Chi, S. Song and J. Qi, Water Res., 2019, 149, 136–148 Search PubMed.
- Y. Yang, J. Jiang, X. Lu, J. Ma and Y. Liu, Environ. Sci. Technol., 2015, 49, 7330–7339 Search PubMed.
- M.-O. Buffle and U. von Gunten, Environ. Sci. Technol., 2006, 40, 3057–3063 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |