
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombination of pose and rank consensus in docking-based virtual screening: the best of both worlds†
Valeria
Scardino
ab,
Mariela
Bollini
c and
Claudio N.
Cavasotto
 *bde
*bde
aMeton AI, Inc., Wilmington, DE 19801, USA
bAustral Institute for Applied Artificial Intelligence, Universidad Austral, Pilar, Buenos Aires, Argentina
cCentro de Investigaciones en BioNanociencias (CIBION), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Ciudad de Buenos Aires, Argentina
dComputational Drug Design and Biomedical Informatics Laboratory, Instituto de Investigaciones en Medicina Traslacional (IIMT), Universidad Austral-CONICET, Pilar, Buenos Aires, Argentina
eFacultad de Ciencias Biomédicas, and Facultad de Ingeniería, Universidad Austral, Pilar, Buenos Aires, Argentina. E-mail: CCavasotto@austral.edu.ar; cnc@cavasotto-lab.net
First published on 2nd November 2021
Abstract
The use of high-throughput docking (HTD) in the drug discovery pipeline is today widely established. In spite of methodological improvements in docking accuracy (pose prediction), scoring power, ranking power, and screening power in HTD remain challenging. In fact, pose prediction is of critical importance in view of the pose-dependent scoring process, since incorrect poses will necessarily decrease the ranking power of scoring functions. The combination of results from different docking programs (consensus scoring) has been shown to improve the performance of HTD. Moreover, it has been also shown that a pose consensus approach might also result in database enrichment. We present a new methodology named Pose/Ranking Consensus (PRC) that combines both pose and ranking consensus approaches, to overcome the limitations of each stand-alone strategy. This approach has been developed using four docking programs (ICM, rDock, Auto Dock 4, and PLANTS; the first one is commercial, the other three are free). We undertook a thorough analysis for the best way of combining pose and rank strategies, and applied the PRC to a wide range of 34 targets sampling different protein families and binding site properties. Our approach exhibits an improved systematic performance in terms of enrichment factor and hit rate with respect to either pose consensus or consensus ranking alone strategies at a lower computational cost, while always ensuring the recovery of a suitable number of ligands. An analysis using four free docking programs (replacing ICM by Auto Dock Vina) displayed comparable results.
Introduction
The experimental evaluation of chemical libraries for activity against a target of pharmaceutical interest through high-throughput screening has been long used in the drug discovery pipeline; however, this is both a time and resource consuming technique.1 Computational methods are today valuable and established tools in all drug discovery endeavors, saving time, resources, and costs.2–4Among in silico methods in drug discovery, molecular docking has been widely used during the last three decades.4–6 In protein-molecule docking, the optimal position, orientation and conformation (pose) of the molecule within the binding site is assessed (“docking stage”), and an estimation of its binding energy calculated. High-throughput docking (HTD) allows the screening of large chemical libraries (from thousands to millions of molecules) to generate a hit-list enriched with potential binders, which will be then advanced for biochemical and biological evaluation. To be computationally efficient, HTD involves several approximations at different levels,7 and the binding free energy calculation is later replaced by a docking score, which is a measure of the probability that the molecule will bind to the target. Thus, the docking stage is followed in this case by the “scoring stage”.7,8
In spite of its undoubted success, HTD is not without challenges, since its performance depends on the energy representation of the system, the degree of target flexibility,4,9–11 and the consideration of water molecules within the binding site.4,12,13 A recent extensive comparison of docking programs showed that, in agreement with earlier works,14,15 they perform better in terms of docking accuracy (docking stage) than in terms of scoring power, ranking power, and screening power (scoring stage).16 We would like to stress that pose prediction is nevertheless of the utmost importance in molecular docking, since incorrect poses will result in meaningless scores, which would thus reduce the ranking capacity of scoring functions. The performance of HTD using different docking programs has been further evaluated on several systems,17–19 and many inconsistencies have been found, such as different performances across programs, also showing that the effectiveness of each scoring function is system dependent.18,20,21 Several efforts have been conducted to improve the reliability at the scoring stage, such as machine-learning-based scoring functions,22,23 and quantum mechanical-base scoring.24–29
The combination of several docking programs (consensus scoring) has been shown to improve the performance of HTD.20,30–33 In 2013, Houston and Walkinshaw proposed for the first time a consensus docking procedure that used several docking programs to increase the reliability of the predicted poses.33 Tuccinardi et al. later used ten docking protocols to evaluate pose consensus on database enrichment,32 and later extended their analysis to 36 benchmark targets of the DUD database.31 They obtained comparable results to Arciniega and Lange's Docking Data Feature Analysis (DDFA), an approach for carrying out virtual screening analysis based on artificial neural networks which was among the best performing methods at the time.34 To obtain good hit rates with their pose consensus strategy, molecules with at least seven matching poses between programs should be selected; in general, the best results were obtained with ten matching poses, which could represent a high computational cost. However, and more importantly, the number of ligands retrieved in most of those cases was very small, with the risk of being zero in some cases.31
It should be highlighted that in consensus scoring (or consensus ranking), for the sake of robustness, it would desirable that scores for a given molecule be combined only when the poses assessed by the different docking programs are similar. We thus present a new strategy that combines both pose and ranking consensus to overcome the limitations of each strategy when used in a stand-alone fashion, and thus increase the performance of HTD campaigns. This method, named Pose/Ranking Consensus (PRC) is consistent with theory in the sense that scores (or ranks) obtained with different programs are only combined when poses are coincident. Using four docking programs (ICM, rDock, Auto Dock 4, and PLANTS) we performed an exhaustive search to look for the best way of combining pose and rank requirements, and evaluated this new method over a wide range of targets that correspond to diverse protein families sampling different binding site properties. Our results show a consistent and improved performance compared to either pose consensus alone, or consensus scoring (ranking) alone strategies. This method is simple to use, simpler than machine learning consensus scoring methods, and displays an excellent performance also using free software programs.
Methods
Target systems preparation
The 34 targets listed in Table 1 were downloaded from the PDB. Water molecules and co-factors were deleted, except in the following cases (cf. Table S1†): (i) within 8 Å of the native ligand: Ca2+ in PA2GA and NRAM; Zn2+ in LKHA4 and ACE; Zn2+ and Ca2+ in HDAC2; Zn2+ and Mg2+ in PDE5A; nicotinamide-adenine-dinucleotide phosphate (napd) in ALDR and DHI1; dihydro nicotinamide-adenine-dinucleotide phosphate (nadph) in DYR; flavine mononucleotide (fmn) in PYRD. In the case of water molecules, they were conserved within 4 Å of the native ligand in the following cases: for HSP90a, water molecules 2059, 2121, 2123, and 2236; FA7, 2440; FABP4, 303, 623, 634, 665; LKHA4, 1099, 1322; UROK, 6 and 61; PDE5A, 38 and 75 (in this case, a cluster of nine neighboring water molecules in contact with those two were also included). The structure of the Dopamine D3 receptor was in the antagonist bound conformation, and that of β2 adrenergic receptor was in the agonist bound conformation. In the case of the HMDH, XIAP, HIVRT and DHI1, two protomers (chains a and b) were included in the docking calculations.| Receptor | Receptor code | Receptor | Receptor code |
|---|---|---|---|
| Thymidine kinase | KITH | Tyrosine-protein kinase ABL | ABL1 |
| Phospholipase A2 | PA2GA | Protein-tyrosine phosphatase 1B | PTN1 |
| Coagulation factor VII | FA7 | Inhibitor of apoptosis protein 3 | XIAP |
| Hexokinase type IV | HXK4 | Androgen receptor | ANDR |
| Cyclin-dependent kinase 2 | CDK2 | Renin | RENI |
| Cyclooxygenase-1 | COX1 | Glutamate receptor ionotropic, AMPA 2 | GRIA2 |
| Fatty acid-binding protein 4 | FABP4 | Aldose reductase | ALDR |
| Heat shock protein 90 alpha | HSP90a | Dihydrofolate reductase | DYR |
| Estrogen receptor alpha | ESR1 | Dihydroorotate dehydrogenase | PYRD |
| Neuraminidase | NRAM | 11-Beta-hydroxysteroid dehydrogenase 1 | DHI1 |
| β2 Adrenergic receptor (agonist bound) | ADRB2 | Angiotensin-converting enzyme | ACE |
| HMG-CoA reductase | HMDH | Progesterone receptor | PRGR |
| Dopamine D3 receptor (antagonist bound) | DRD3 | Human immunodeficiency virus type 1 reverse transcriptase | HIVRT |
| Histone deacetylase 2 | HDAC2 | Purine nucleoside phosphorylases | PNPH |
| Leukocyte function associated antigen-1 | LFA1 | Protein kinase C beta | KPCB |
| Leukotriene A4 hydrolase | LKHA4 | Insulin-like growth factor I receptor | IGF1R |
| Urokinase-type plasminogen activator | UROK | Phosphodiesterase 5A | PDE5A |
Receptors were prepared with the ICM program35 (version 3.8-7c; MolSoft, San Diego, CA 2020), in a similar fashion as in other works.25 Missing residues and hydrogen atoms were added followed by a local energy minimization of the system. Polar and water hydrogens within the binding site were optimized using a Monte Carlo simulation in the torsional space. Glutamate and asparte side chains were assigned a −1 charge, and lysine and arginine were assigned a +1 charge. Asparagine and glutamine were inspected for possible flipping and adjusted if necessary. Histidine tautomers were assigned according to their most favorable hydrogen bonding pattern.
Docking libraries
Docking chemical libraries were prepared for each target by merging a set of actives and their corresponding matching decoys according to similar physico-chemical properties and structural dissimilarity, which has been shown to ensure unbiased calculations in docking simulations.36,37 The number of actives, decoys and sources for each target are shown in Table S2.† For all molecules, chirality and protonation states were inherited from the corresponding original databases.Docking calculations
For protein-molecule docking, five programs were used in total: ICM,35 Auto Dock 4,38 rDock,39 PLANTS,40 and Auto Dock Vina.41 The latter was used for the free software evaluation only, replacing ICM. These programs have different search algorithms and scoring functions as described in previous works.30,40 For all the HTD runs, the top scored conformation of each molecule was selected. The box center and dimensions were determined with ICM in such a way that all molecules in the chemical library would fit within the binding site, and then used for all programs. In rDock, the docking cavity was automatically built using the reference ligand method, which defines a docking volume of a given size around the binding mode of a known ligand.Auto Dock Tools utilities38 were used to prepare the input files for Auto Dock 4, where the Lamarckian genetic algorithm was used for a 20-run search for each compound using 1.75 million of energy evaluation. For PLANTS, the ChemPLP scoring function was used and speed 1 was set as search speed. For rDock, a radius of 8.0 Å ± 2.0 Å from a reference ligand binding mode was used to represent the cavity. For Vina, an exhaustiveness value of 8 was set. For ICM, a thoroughness of 2 was used for the search algorithm. All the other parameters for every software remained at their default values. On average, each program took between 13 and 130 seconds per core per molecule, with ICM being the fastest and Auto Dock 4 the slowest program.
Exponential consensus ranking
In the Exponential Consensus Ranking (ECR),30 the consensus rank ECR(i) for each molecule i is calculated as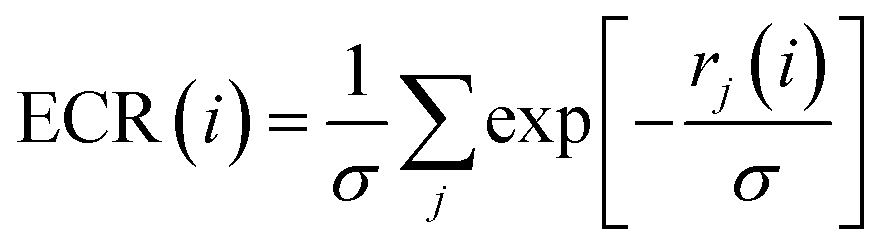 | (1) |
Pose consensus approach
From the four HTD campaigns, four binding modes were obtained for each molecule in the database, which correspond to the 4 docking programs used. The RMSD among all combinations of these poses was calculated using the ICM software, which allowed for the calculation of the static deviation between molecules. Poses were considered to match if they were within 2.0 Å RMSD. A molecule was considered to have three matching poses (MPs) if the three corresponding combinations of two poses matched. For four matching poses, the six corresponding combinations of two poses must be coincident.Evaluation metrics
The enrichment factor (EF) is defined as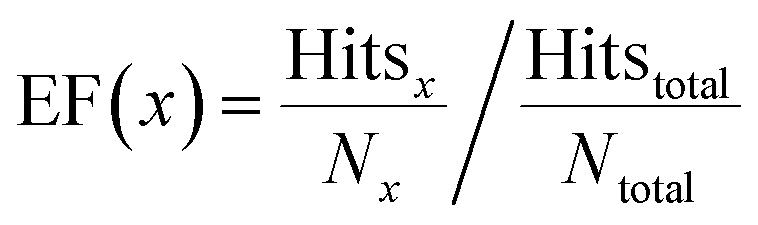 | (2) |
The hit rate (HR) was calculated as
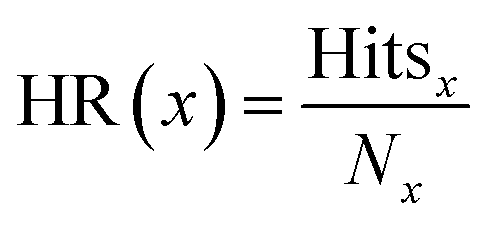 | (3) |
Results and discussion
For the HTD campaigns, we selected a benchmark set of 34 targets from diverse protein families, exhibiting different binding site properties, and including the presence of co-factors and water molecules (cf. Table S1†). The chemical libraries used are described in the Methods section (cf. Table S2†). Four docking programs were used, AutoDock 4, ICM, rDock and PLANTS, which have different search algorithms and scoring functions. Auto Dock Vina was also evaluated, but we selected only the best four performing programs to develop a method with the lowest computational cost for a future prospective campaign. For each docking program, the pose corresponding to the best score for each molecule was selected, and the ranking was established according to that score. On average, ICM presented the best performance, followed by rDock. None of the programs performed the best over all the systems evaluated.As starting point, we calculated the Exponential Consensus Ranking (ECR).30 This consensus method combines results from several docking programs using an exponential distribution for each individual rank. In a previous work, it demonstrated a higher performance than other traditional consensus strategies and individual programs. In this work we extended the analysis of the ECR to 34 targets using four instead of the original six programs. Our results confirmed its better performance when compared to individual programs. On average, it showed at least a 1.4-fold increase for the enrichment (average ratio over all targets between the ECR EF1 and an individual program EF1) (cf.Table 2).
| Average | ICM | rDock | Auto Dock 4 | PLANTS |
|---|---|---|---|---|
a Average value calculated as 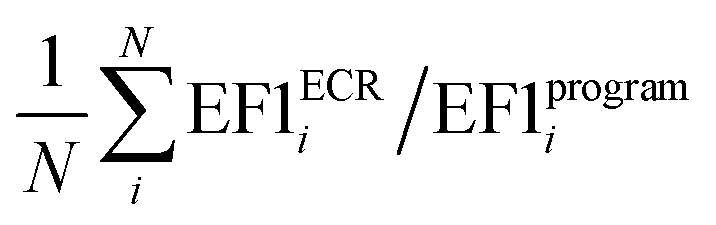 , where N is the number of targets.
b This value does not include XIAP and ANDR, which had EF = 0, and therefore make the average fold increase ≫ 100.
c This value does not include these five targets: HXK4, NRAM, XIAP, DYR and PNPH, which had EF = 0, and therefore make the average fold increase ≫ 100.
d This value does not include FABP4 and PYRD, which had EF = 0, and therefore make the average fold increase ≫ 100. , where N is the number of targets.
b This value does not include XIAP and ANDR, which had EF = 0, and therefore make the average fold increase ≫ 100.
c This value does not include these five targets: HXK4, NRAM, XIAP, DYR and PNPH, which had EF = 0, and therefore make the average fold increase ≫ 100.
d This value does not include FABP4 and PYRD, which had EF = 0, and therefore make the average fold increase ≫ 100.
|
||||
| EF1 | 23.5 | 10.5 | 5.8 | 9.9 |
| Fold increasea | 1.4 | 3.4b | 7.4c | 3.2d |
Pose consensus alone is not enough to guarantee high enrichment
Initially, we evaluated the performance of a pose consensus alone strategy using the four docking programs on the 34 benchmarking targets. Table 3 shows the enrichment factor (EF) for each target, calculated on the subset of molecules that meet the selection criteria according to the number of matching poses (MPs) between programs. Poses were considered to match if they are within 2.0 Å RMSD. Consistent with earlier works,31,32 in general, the EF increases as the number of coincident poses requested was increased. However, the number of ligands in some cases was already low when considering four coincident poses. For example, in XIAP only 1 ligand was present in the subset of molecules selected, and similar numbers were observed for ACE and IGF1R. An extended version of Table 3 including the Active/Selected (A/S) molecule rate can be found in Table S3 of the ESI.† Furthermore, it can be seen from these results that a solely pose consensus strategy with four docking programs is not enough to obtain acceptable EFs.| Receptor | 2 MPs | 3 MPs | 4 MPs |
|---|---|---|---|
| KITH | 1.4 | 2.6 | 4.7 |
| PA2GA | 1.6 | 3.4 | 4.7 |
| FA7 | 1.6 | 3.4 | 3.2 |
| HXK4 | 1.3 | 1.4 | 1.7 |
| CDK2 | 1.2 | 1.9 | 3.3 |
| COX1 | 1.1 | 1.3 | 1.5 |
| FABP4 | 1.2 | 1.5 | 1.5 |
| HSP90a | 1.0 | 1.3 | 2.2 |
| ESR1 | 1.2 | 1.9 | 4.1 |
| NRAM | 1.8 | 4.7 | 5.6 |
| ADRB2 | 1.2 | 1.2 | 0.4 |
| HMDH | 2.3 | 5.2 | 7.1 |
| DRD3 | 1.1 | 1.1 | 0.9 |
| HDAC2 | 1.2 | 2.1 | 1.8 |
| LFA1 | 0.9 | 1.4 | 2.8 |
| LKHA4 | 1.5 | 1.9 | 1.8 |
| UROK | 1.5 | 3.8 | 9.9 |
| ABL1 | 1.4 | 1.6 | 2.8 |
| PTN1 | 1.3 | 1.9 | 1.3 |
| XIAP | 1.4 | 4.1 | 7.5 |
| ANDR | 1.2 | 1.9 | 4.6 |
| Renin | 1.8 | 9.3 | 28.9 |
| GRIA2 | 1.4 | 3.5 | 7.7 |
| ALDR | 1.2 | 2.0 | 4.0 |
| DYR | 1.3 | 1.7 | 2.5 |
| PYRD | 1.3 | 2.6 | 3.4 |
| DHI1 | 1.1 | 1.7 | 2.7 |
| ACE | 1.3 | 5.0 | 5.6 |
| PRGR | 1.2 | 1.8 | 3.5 |
| HIVRT | 1.4 | 1.8 | 3.8 |
| PNPH | 1.2 | 2.3 | 5.6 |
| KPCB | 1.3 | 2.6 | 6.6 |
| IGF1R | 1.4 | 2.3 | 1.6 |
| PDE5A | 1.9 | 4.8 | 14.2 |
| Average | 1.4 | 2.7 | 4.8 |
Combining pose and rank consensus outperforms previous strategies
We observed that adding a ranking filter to pose consensus enhanced the performance of the latter. To further explore this fact, various possible combinations of the number of required MPs and ranking thresholds were considered, and three general options were initially explored: (A) pose consensus with at least two programs, selecting only among those molecules with the two corresponding ranks in the top 5, 10, or 20%; (B) pose consensus with at least three programs, selecting only among those molecules with the three corresponding ranks in the top 5, 10, or 20%; (C) pose consensus with the four programs, selecting only among those molecules with the four corresponding ranks in the top 15, 20, or 25%. These three options were evaluated in terms of minimum, maximum, and average EF values for the 34 benchmark targets (see Table S4†); among the ones that showed high averages, those with higher minimum values and EFs closer to the average were preferred, in order to prioritize strategies that work well across all targets. Strategies that exhibited the best EFs in those specific targets that displayed low performance in the four programs were also prioritized. The average number of actual ligands (actives) retrieved in each strategy was also considered. For option A (two MPs), the best results were obtained with a 5% rank cutoff. For option B (three MPs), similar results were obtained with 5% rank cutoff, but 10% was preferred in order to obtain a larger number of ligands. For option C (four MPs), the best results were obtained with a 20% rank cutoff. Option C marginally showed the best performance among the three options, followed by option B. It was observed, however, that in option C (and to a lesser extent also in option B), there are very few molecules that meet the requirements, and in the case of ACE, for example, no molecule was selected. In Table 4 the best performance for each option is presented.| Receptor | Option A | Option B | Option C | |||
|---|---|---|---|---|---|---|
| A/Sa | EF | A/Sa | EF | A/Sa | EF | |
| a Number of actives and selected molecules for each target. b Average (A)/average (S). | ||||||
| KITH | 24/38 | 14.3 | 3/4 | 17.0 | 1/3 | 7.6 |
| PA2GA | 26/48 | 22.8 | 9/10 | 37.9 | 3/3 | 42.1 |
| FA7 | 84/107 | 27.5 | 33/35 | 33.1 | 1/1 | 35.1 |
| HXK4 | 17/72 | 9.2 | 0/10 | 0.0 | 0/2 | 0.0 |
| CDK2 | 23/56 | 12.2 | 17/47 | 10.8 | 11/22 | 14.9 |
| COX1 | 11/210 | 1.8 | 11/134 | 2.8 | 9/54 | 5.7 |
| FABP4 | 22/65 | 17.3 | 14/30 | 23.8 | 5/7 | 36.5 |
| HSP90a | 21/84 | 10.1 | 5/23 | 8.8 | 0/5 | 0.0 |
| ESR1 | 44/136 | 17.0 | 30/70 | 22.5 | 19/24 | 41.7 |
| NRAM | 20/58 | 10.0 | 10/12 | 24.2 | 0/1 | 0.0 |
| ADRB2 | 63/147 | 17.2 | 9/38 | 9.5 | 1/10 | 4.0 |
| HMDH | 30/69 | 22.8 | 6/12 | 26.2 | 3/3 | 52.4 |
| DRD3 | 11/140 | 3.1 | 2/29 | 2.8 | 0/6 | 0.0 |
| HDAC2 | 24/84 | 16.2 | 9/19 | 26.8 | 1/5 | 11.3 |
| LFA1 | 15/121 | 7.7 | 11/46 | 14.9 | 3/11 | 17.0 |
| LKHA4 | 36/166 | 12.2 | 12/43 | 15.7 | 1/10 | 5.6 |
| UROK | 47/80 | 36.3 | 35/38 | 56.9 | 15/19 | 48.7 |
| ABL1 | 42/164 | 15.4 | 20/58 | 20.7 | 2/18 | 6.7 |
| PTN1 | 33/129 | 14.5 | 16/63 | 14.4 | 2/21 | 5.4 |
| XIAP | 7/13 | 28.2 | 1/2 | 26.2 | 1/1 | 52.4 |
| ANDR | 28/172 | 8.8 | 16/45 | 19.3 | 7/12 | 31.6 |
| Renin | 15/21 | 48.2 | 8/9 | 60.0 | 3/3 | 67.5 |
| GRIA2 | 34/129 | 20.0 | 15/37 | 30.8 | 11/12 | 69.6 |
| ALDR | 68/188 | 20.8 | 46/97 | 27.3 | 26/48 | 31.2 |
| DYR | 39/144 | 20.4 | 12/39 | 23.1 | 1/8 | 9.4 |
| PYRD | 38/120 | 18.7 | 25/42 | 35.1 | 7/14 | 29.5 |
| DHI1 | 31/334 | 5.5 | 21/128 | 9.8 | 8/60 | 7.9 |
| ACE | 30/94 | 19.5 | 7/17 | 25.2 | 0/0 | 0.0 |
| PRGR | 33/300 | 6.0 | 31/150 | 11.2 | 25/54 | 25.1 |
| HIVRT | 47/214 | 12.5 | 17/76 | 12.7 | 5/22 | 13.0 |
| PNPH | 44/133 | 22.7 | 29/62 | 32.0 | 9/22 | 28.0 |
| KPCB | 47/74 | 41.5 | 25/33 | 49.5 | 15/16 | 61.3 |
| IGF1R | 20/43 | 29.7 | 7/13 | 34.3 | 1/1 | 63.7 |
| PDE5A | 61/201 | 21.3 | 27/52 | 36.4 | 15/22 | 47.8 |
| Average | 33/122b | 18.0 | 16/45b | 23.6 | 6/15b | 25.7 |
Next, we considered a combination of the three options A, B, and C, in the following fashion: if a molecule had a maximum of two MPs, the corresponding ranks obtained with those two programs should be within the top 5%; with a maximum of three MPs, those corresponding three ranks should be within the top 10%; with four MPs, the four ranks ought to be in the top 20%. While this strategy (named option D) showed a slightly less average EF than options B and C (20.0 vs. 25.7), there were no cases where actual ligands could not be found. Therefore, it was preferred over each individual option. We explored other combinations of ranking thresholds which are presented in Table S5,† but 5%, 10% and 20% for two, three and four MPs, respectively, was the best choice (similar results were also obtained with values of 5%, 10% and 25%).
The best of both worlds: the PRC method
If the selected molecules were sorted by ECR, and only those in the top 1.5% were selected, an even better performance was obtained (Table 5). This approach was named the Pose/Ranking Consensus (PRC). We also evaluated threshold values between 0.5% and 2%, with 1.5% showing the best results.| Receptor | Option D | PRC | |||
|---|---|---|---|---|---|
| A/Sa | EF | A/Sa | EF | HR | |
| a Number of actives and selected molecules for each target. b Average (A)/average (S). | |||||
| KITH | 15/23 | 14.8 | 13/15 | 19.7 | 0.87 |
| PA2GA | 16/30 | 22.4 | 12/16 | 31.5 | 0.75 |
| FA7 | 64/73 | 30.7 | 44/45 | 34.3 | 0.98 |
| HXK4 | 15/50 | 11.6 | 9/23 | 15.2 | 0.39 |
| CDK2 | 14/33 | 12.6 | 11/17 | 19.3 | 0.65 |
| COX1 | 11/111 | 3.4 | 8/47 | 5.8 | 0.17 |
| FABP4 | 20/37 | 27.6 | 20/25 | 40.9 | 0.80 |
| HSP90a | 11/39 | 11.4 | 8/21 | 15.4 | 0.38 |
| ESR1 | 33/80 | 21.7 | 33/53 | 32.8 | 0.62 |
| NRAM | 14/29 | 14.0 | 9/19 | 13.8 | 0.47 |
| ADRB2 | 53/101 | 21.1 | 35/60 | 23.4 | 0.58 |
| HMDH | 25/55 | 23.8 | 14/30 | 24.5 | 0.47 |
| DRD3 | 7/77 | 3.6 | 6/48 | 5.0 | 0.13 |
| HDAC2 | 23/68 | 19.2 | 21/43 | 27.7 | 0.49 |
| LFA1 | 9/59 | 9.5 | 8/43 | 11.6 | 0.19 |
| LKHA4 | 29/130 | 12.6 | 18/69 | 14.7 | 0.26 |
| UROK | 46/70 | 40.5 | 46/50 | 56.8 | 0.92 |
| ABL1 | 36/125 | 17.3 | 33/75 | 26.4 | 0.44 |
| PTN1 | 31/128 | 13.7 | 24/57 | 23.9 | 0.42 |
| XIAP | 4/8 | 26.2 | 3/6 | 26.2 | 0.5 |
| ANDR | 12/94 | 6.9 | 10/40 | 13.5 | 0.25 |
| Renin | 15/20 | 50.6 | 14/17 | 55.6 | 0.82 |
| GRIA2 | 25/83 | 22.9 | 20/52 | 29.2 | 0.38 |
| ALDR | 55/135 | 23.5 | 51/81 | 36.3 | 0.63 |
| DYR | 34/112 | 22.8 | 24/70 | 25.8 | 0.34 |
| PYRD | 31/80 | 22.9 | 30/51 | 34.7 | 0.59 |
| DHI1 | 23/245 | 5.6 | 21/136 | 9.2 | 0.15 |
| ACE | 27/86 | 19.2 | 22/59 | 22.8 | 0.37 |
| PRGR | 36/185 | 10.5 | 30/94 | 17.3 | 0.32 |
| HIVRT | 36/169 | 13.5 | 28/97 | 16.5 | 0.29 |
| PNPH | 29/87 | 22.8 | 26/51 | 34.9 | 0.51 |
| KPCB | 43/65 | 43.2 | 42/51 | 53.8 | 0.82 |
| IGF1R | 20/38 | 33.6 | 20/33 | 38.6 | 0.61 |
| PDE5A | 46/153 | 21.1 | 41/98 | 29.3 | 0.42 |
| Average | 27/85b | 20.0 | 22/50b | 26.1 | 0.50 |
Fig. 1 shows a schematic representation of this Pose/Ranking Consensus (PRC) pipeline. Starting from the binding poses and ranks obtained with the four docking programs, a pose/ranking filtering approach is carried out. For this, the maximum number of MPs (1–4) is assessed for each molecule, coupled with identifying those programs where the poses matched. Then, the ones with four MPs are identified and filtered according to the 20% rank threshold in the corresponding programs. The same is performed for three MPs (10% rank threshold), and two MPs (5% rank threshold). In parallel, the ECR method is calculated onto the whole database. The molecules that pass the pose/ranking filters are ordered by their corresponding ECR, previously calculated, and the ones in the top 1.5% are finally selected.
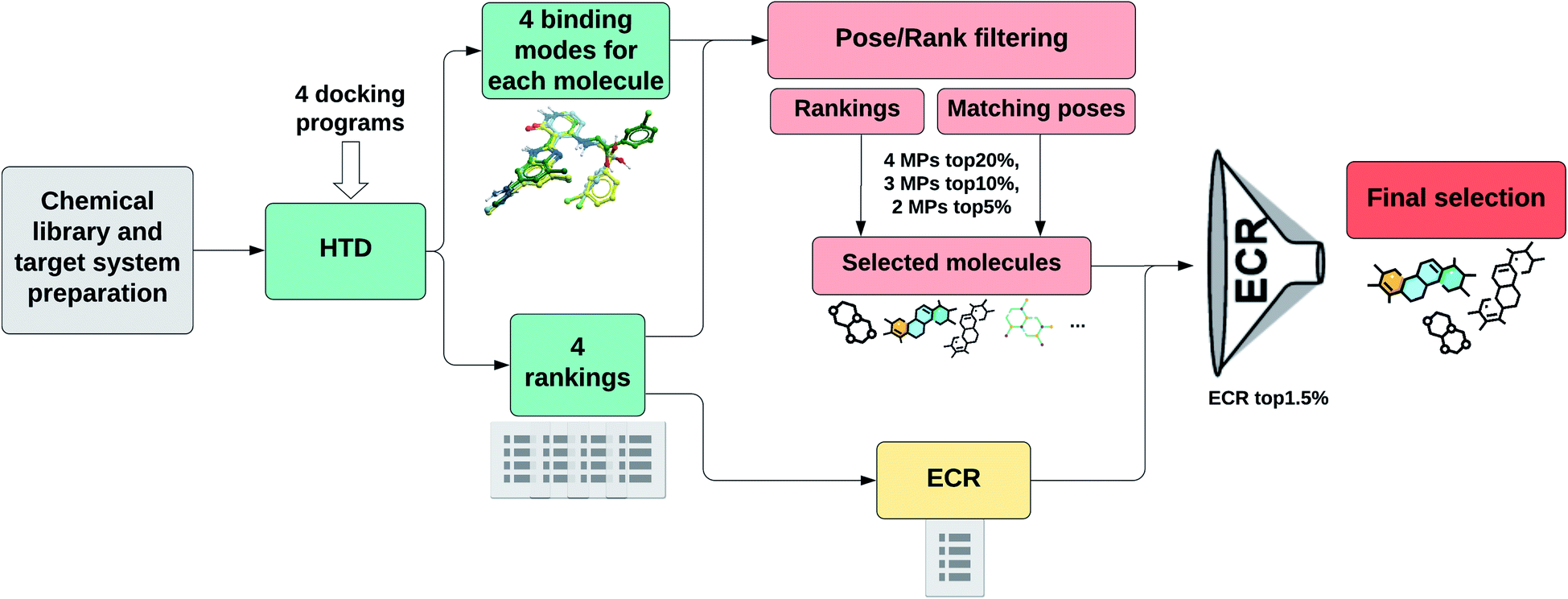 | ||
| Fig. 1 PRC pipeline for high-throughput docking. | ||
In Table 5, we show the performance of the PRC method in terms of EF and number of actual ligands (actives) retrieved for each target. EF values of option D selection strategy are also presented. The last column shows the hit rate (probability of finding an actual ligand within the selected pool of molecules) in the PRC selected compounds. It can be readily noticed from these results that both the pose/ranking filtering and ECR threshold requirements are important to achieve high EF values. The PRC showed the best performance in almost every target evaluated, with the exception of one case (NRAM) where the difference was minimal.
As can be seen from Table 5, our method results in very high enrichment values, with an appropriate number of ligands. The latter could be critical in a prospective scenario, where the number of actual ligands might be scarce. When viewed in terms of probability, an average hit rate of 50% is achieved on the subsets of molecules selected. The maximum value (98%) was obtained for FA7 where 44 out of 45 selected molecules were ligands. DRD3 showed the lowest hit rate value (13%) and the lowest number of ligands retrieved (6). In 2016, Tuccinardi et al. achieved an average hit rate of 45% (vs. 50% with PRC), which they required to be at the level of the best performing methods.31 We note, however, that the results they report correspond to the maximum hit rate that can be obtained for each target, which depends on the number of MPs used, and therefore is not directly applicable in a prospective analysis.
This novel method achieves very high EF values, greatly surpassing previous pose consensus techniques and ranking consensus techniques, including the ECR, as it is shown in the next section. The results are especially higher for those targets that have a poor performance in the four docking programs (and ECR), reaching EFs of more than triple the values of EF1 ECR (see below and Table 6).
| Receptor | ECR EF1 | PRC EF | Fold increase |
|---|---|---|---|
| a Average of the fold increase values. | |||
| KITH | 12.5 | 19.7 | 1.58 |
| PA2GA | 25.4 | 31.5 | 1.24 |
| FA7 | 34.5 | 34.3 | 0.99 |
| HXK4 | 5.5 | 15.2 | 2.76 |
| CDK2 | 18.5 | 19.3 | 1.04 |
| COX1 | 3.4 | 5.8 | 1.71 |
| FABP4 | 40.5 | 40.9 | 1.01 |
| HSP90a | 4.9 | 15.4 | 3.14 |
| ESR1 | 35.1 | 32.8 | 0.93 |
| NRAM | 4.5 | 13.8 | 3.07 |
| ADRB2 | 24.5 | 23.4 | 0.96 |
| HMDH | 17.1 | 24.5 | 1.43 |
| DRD3 | 3.2 | 5.0 | 1.56 |
| HDAC2 | 13.6 | 27.7 | 2.04 |
| LFA1 | 10.9 | 11.6 | 1.06 |
| LKHA4 | 15.2 | 14.7 | 0.97 |
| UROK | 44.5 | 56.8 | 1.28 |
| ABL1 | 25.3 | 26.4 | 1.04 |
| PTN1 | 29.5 | 23.9 | 0.81 |
| XIAP | 20.2 | 26.2 | 1.30 |
| ANDR | 9.0 | 13.5 | 1.50 |
| Renin | 17.4 | 55.6 | 3.20 |
| GRIA2 | 19.8 | 29.2 | 1.47 |
| ALDR | 33.5 | 36.3 | 1.08 |
| DYR | 26.1 | 25.8 | 0.99 |
| PYRD | 26.3 | 34.7 | 1.32 |
| DHI1 | 8.8 | 9.2 | 1.05 |
| ACE | 14.3 | 22.8 | 1.59 |
| PRGR | 9.2 | 17.3 | 1.88 |
| HIVRT | 15.1 | 16.5 | 1.09 |
| PNPH | 37.1 | 34.9 | 0.94 |
| KPCB | 45.3 | 53.8 | 1.19 |
| IGF1R | 18.3 | 38.6 | 2.11 |
| PDE5A | 17.1 | 29.3 | 1.71 |
| Average | 20.3 | 26.1 | 1.50a |
Performance of the PRC method compared to ECR in view of traditional metrics
To further evaluate the performance of the PRC method we compared the improvement against the ECR for every target. We chose the ECR as a comparison method since it presented a better performance than other traditional ranking consensus strategies (such as RbR or RbV).30Table 6 shows the EFs of the PRC method compared to those of ECR at 1% (EF1). We chose EF1 as it is a standard metric, widely used in virtual screening. The fold increase (the ratio between PRC EF and ECR EF1) is also presented for a clearer comparison of the results. It can be noticed that 27 out of 34 targets showed an increase in the EF. The remaining seven targets showed similar results in both strategies. On average, the PRC method had a 1.50-fold increase over ECR EF1. The improvements are especially noticeable in targets with low EF1 both on individual programs and on ECR; for example, EF values are increased by a factor of three in PRC for HSP90a, neuraminidase and renin. Regarding DRD3 (the worst performer in PRC), the four docking programs performed poorly on this target, and our method displayed a noticeably higher EF than ECR EF1. Table S6 in ESI† shows the same comparison (PRC vs. ECR) in terms of hit rates. On average, the ECR has a hit rate of 41% (vs. 50% in PRC).We also analyzed for each target the ECR EF when selecting the same number of molecules from the top as those returned by the PRC. It should be noted that this is not a measure of practical value in prospective HTD, as this threshold is never known beforehand. However, the PRC in this case also surpassed the ECR, showing, on average, a 1.33-fold increase; moreover, our method showed an eight times higher EF in HXK4, and still showed 3-fold increase values in the worst performing targets.
Taking into account that the ECR already represents an improvement of the results over previous consensus strategies and to individual programs, these results show that the PRC method allows for significantly higher hit rates and EF values, with a minimal computational cost, and can therefore reach better results in future prospective HTD campaigns.
Performance of the PRC method using only free available docking programs
In some cases, it may happen that only free docking programs are available. Therefore, we present the results using only free and accessible programs. For this task, we replaced ICM with Auto Dock Vina, which was the other available software. Table 7 shows the results obtained after applying the PRC pipeline (Fig. 1) using Auto Dock 4, rDock, PLANTS and Auto Dock Vina for the HTD. For 2 MPs, we evaluated the possibility of excluding the combination of Auto Dock 4 and Auto Dock Vina, as we saw that there were many molecules that met this requirement. This exclusion allowed better results, and so it was maintained for the free programs procedure. The results of a solely pose consensus approach using free docking programs are presented in Table S7.† Option D selection strategy is also displayed in Table 7 as a reference. The maximum hit rate (77%) was obtained for PA2GA where 10 out of 13 molecules selected were active. For HSP90a, all the individual programs performed poorly, and no actual ligands could be found. On average, a hit rate of 34% was obtained. It can be noted that the best results were also obtained when combining the pose/ranking filters with the ECR threshold (PRC). However, option D is shown as a good alternative for targets that do not perform well in none of the programs used, as it is the case of HXK4, HSP90a and NRAM.| Receptor | Option D | PRC | |||
|---|---|---|---|---|---|
| A/Sa | EF | A/Sa | EF | HR | |
| a Number of actives and selected molecules for each target. b Average (A)/average (S). | |||||
| KITH | 3/17 | 4 | 3/9 | 7.6 | 0.33 |
| PA2GA | 11/16 | 28.9 | 10/13 | 32.4 | 0.77 |
| FA7 | 27/40 | 23.7 | 22/29 | 26.6 | 0.76 |
| HXK4 | 2/31 | 2.5 | 1/22 | 1.8 | 0.05 |
| CDK2 | 17/34 | 14.9 | 12/22 | 16.3 | 0.55 |
| COX1 | 11/236 | 1.6 | 5/91 | 1.9 | 0.05 |
| FABP4 | 13/64 | 10.4 | 12/23 | 26.7 | 0.52 |
| HSP90a | 2/48 | 1.7 | 0/31 | 0 | 0 |
| ESR1 | 36/159 | 11.9 | 31/80 | 20.4 | 0.39 |
| NRAM | 4/33 | 3.5 | 2/25 | 2.3 | 0.08 |
| ADRB2 | 23/119 | 7.8 | 20/73 | 11.1 | 0.27 |
| HMDH | 16/41 | 20.4 | 7/21 | 17.5 | 0.33 |
| DRD3 | 9/151 | 2.4 | 6/70 | 3.4 | 0.09 |
| HDAC2 | 22/72 | 17.3 | 16/38 | 23.9 | 0.42 |
| LFA1 | 10/86 | 7.3 | 9/53 | 10.6 | 0.17 |
| LKHA4 | 46/181 | 14.3 | 31/74 | 23.6 | 0.42 |
| UROK | 46/89 | 31.9 | 43/62 | 42.8 | 0.69 |
| ABL1 | 22/172 | 7.7 | 19/85 | 13.4 | 0.22 |
| PTN1 | 28/80 | 19.8 | 23/43 | 30.3 | 0.53 |
| XIAP | 2/11 | 9.5 | 2/9 | 11.7 | 0.22 |
| ANDR | 14/164 | 4.6 | 13/97 | 7.3 | 0.13 |
| Renin | 9/18 | 33.7 | 9/13 | 46.7 | 0.69 |
| GRIA2 | 25/95 | 19.9 | 16/51 | 23.8 | 0.31 |
| ALDR | 45/187 | 13.9 | 38/90 | 24.3 | 0.42 |
| DYR | 24/179 | 10.1 | 20/104 | 14.5 | 0.19 |
| PYRD | 28/88 | 18.8 | 24/50 | 28.3 | 0.48 |
| DHI1 | 25/317 | 4.7 | 20/169 | 7.1 | 0.12 |
| ACE | 16/87 | 11.3 | 11/49 | 13.7 | 0.22 |
| PRGR | 36/313 | 6.2 | 21/149 | 7.6 | 0.14 |
| HIVRT | 32/269 | 6.7 | 14/150 | 5.3 | 0.09 |
| PNPH | 24/90 | 18.3 | 20/52 | 26.3 | 0.38 |
| KPCB | 41/92 | 29.1 | 39/56 | 45.5 | 0.70 |
| IGF1R | 20/61 | 20.9 | 20/42 | 30.4 | 0.48 |
| PDE5A | 44/210 | 14.7 | 39/140 | 19.5 | 0.28 |
| Average | 27/140b | 13.4 | 22/78b | 18.4 | 0.34 |
In Table 8 we compare the results of the PRC and ECR for free docking programs. Better results were obtained in 28 of the 34 targets with an average 1.62-fold increase of the PRC method over the ECR EF1. Of the remaining six, ANDR is the one that shows the highest decrease. In this target, the docking programs did not perform well, with rDock showing zero EF1. For HSP90a, neither the ECR nor the PRC exhibited good results. In option D it achieved a slightly better EF than the ECR EF1, and it may be a better selection strategy for cases where individual performances in terms of scoring are very poor. Regarding ESR1 and ABL1, while they still show acceptable EF values, they performed slightly worse than ECR. This was also the case for ESR1 with the previous procedure (Table 6). It should be noted, anyway, that the number of selected molecules for this target (80) is higher than 1% of its database (67).
| Receptor | ECR EF1 | PRC EF | Fold increase |
|---|---|---|---|
| a Average of the fold increase values. | |||
| KITH | 2.3 | 7.6 | 3.22 |
| PA2GA | 16.7 | 32.4 | 1.94 |
| FA7 | 24.1 | 26.6 | 1.1 |
| HXK4 | 0.8 | 1.8 | 2.23 |
| CDK2 | 12.8 | 16.3 | 1.27 |
| COX1 | 1 | 1.9 | 1.95 |
| FABP4 | 19.4 | 26.7 | 1.38 |
| HSP90a | 0 | 0 | 1 |
| ESR1 | 23.9 | 20.4 | 0.85 |
| NRAM | 0.5 | 2.3 | 5.12 |
| ADRB2 | 10.8 | 11.1 | 1.03 |
| HMDH | 7.6 | 17.5 | 2.30 |
| DRD3 | 3.1 | 3.4 | 1.11 |
| HDAC2 | 16.3 | 23.9 | 1.46 |
| LFA1 | 12.3 | 10.6 | 0.86 |
| LKHA4 | 18.2 | 23.6 | 1.30 |
| UROK | 30.9 | 42.8 | 1.39 |
| ABL1 | 18.2 | 13.4 | 0.74 |
| PTN1 | 33.4 | 30.3 | 0.91 |
| XIAP | 6.1 | 11.7 | 1.92 |
| ANDR | 10.5 | 7.3 | 0.70 |
| Renin | 12.5 | 46.7 | 3.74 |
| GRIA2 | 14.7 | 23.8 | 1.62 |
| ALDR | 25.3 | 24.3 | 0.96 |
| DYR | 10.4 | 14.5 | 1.39 |
| PYRD | 20.0 | 28.3 | 1.42 |
| DHI1 | 6.1 | 7.1 | 1.16 |
| ACE | 5.0 | 13.7 | 2.74 |
| PRGR | 4.1 | 7.6 | 1.85 |
| HIVRT | 5.3 | 5.3 | 1 |
| PNPH | 25.4 | 26.3 | 1.04 |
| KPCB | 37.9 | 45.5 | 1.20 |
| IGF1R | 17.6 | 30.4 | 1.73 |
| PDE5A | 12.9 | 19.5 | 1.51 |
| Average | 13.7 | 18.4 | 1.62a |
A very noticeable improvement of PRC over ECR can be seen for KITH, HXK4, NRAM, HMDH, renin and ACE, where EF values of more than double the ECR EF1 were obtained. The average fold increase was even higher than in the previous case (1.62 vs. 1.50), therefore confirming the applicability of PRC method even when only free docking programs were available.
Conclusions and perspective
A new method combining both pose and ranking consensus (PRC) is presented and evaluated in 34 diverse protein targets, displaying an improved performance with respect to either pose consensus alone, or consensus scoring alone approaches. Our method is especially robust in the sense that scores (and ranks) are only combined when poses are coincident within a 2 Å threshold. In the PRC method we used four docking programs to build consensus strategies (ICM, rDock, Auto Dock 4, and PLANTS), and performed a comprehensive analysis of the optimal way of combining pose and rank requirements, which greatly improved the results compared to individual programs, and also to previous consensus strategies. It should be noted that high hit rates were obtained with low computational cost, yielding an appropriate number of ligands. It was observed that PRC greatly improves the results even when only free available docking programs are used (replacing ICM by Auto Dock Vina).In spite of the obvious success, we would like to point out two facts related to this methodology: (i) it is still dependent on the performance of the individual programs on the target. If no program managed to perform well, the PRC method would still improve the results obtained, but in a limited way; (ii) option D (cf.Table 5) is a good alternative in a prospective case when it is suspected that a little number of actual ligands might be present in the query database, or when the target belongs to a family of proteins that does not usually perform well in HTD campaigns, since it will likely retrieve more ligands. While (i) is a common limitation to all consensus strategies, PRC shows itself as a promising tool to by-pass it. In a follow-up contribution, we will evaluate the dependence of the method on the relationship between the number of ligands and decoys in the database for each target.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Agency for the Promotion of Science and Technology (ANPCyT) (PICT-2017-3767). CNC thanks Molsoft LLC (San Diego, CA) for providing an academic license for the ICM program. The authors thank the Centro de Cálculo de Alto Desempeño (Universidad Nacional de Córdoba) for granting the use of their computational resources.References
- S. S. Phatak, C. C. Stephan and C. N. Cavasotto, Expert Opin. Drug Discovery, 2009, 4, 947–959 CrossRef CAS.
- W. L. Jorgensen, Acc. Chem. Res., 2009, 42, 724–733 CrossRef CAS PubMed.
- G. Schneider, Nat. Rev. Drug Discovery, 2017, 17, 97–113 CrossRef PubMed.
- F. Spyrakis and C. N. Cavasotto, Arch. Biochem. Biophys., 2015, 583, 105–119 CrossRef CAS.
- A. Ciancetta and S. Moro, in In Silico Drug Discovery and Design: Theory, Methods, Challenges, and Applications, ed. C. N. Cavasotto, CRC Press, Taylor & Francis Group, Boca Raton, FL, 2015, ch. 7, pp. 189–213 Search PubMed.
- A. Sulimov, D. Kutov, I. Ilin, D. Zheltkov, E. Tyrtyshnikov and V. Sulimov, SAR QSAR Environ. Res., 2019, 30, 733–749 CrossRef CAS PubMed.
- C. N. Cavasotto and A. J. Orry, Curr. Top. Med. Chem., 2007, 7, 1006–1014 CrossRef CAS PubMed.
- I. A. Guedes, F. S. S. Pereira and L. E. Dardenne, Front. Pharmacol., 2018, 9, 1089 CrossRef CAS PubMed.
- C. N. Cavasotto and N. Singh, Curr. Comput.-Aided Drug Des., 2008, 4, 221–234 CrossRef CAS.
- P. Cozzini, G. E. Kellogg, F. Spyrakis, D. J. Abraham, G. Costantino, A. Emerson, F. Fanelli, H. Gohlke, L. A. Kuhn, G. M. Morris, M. Orozco, T. A. Pertinhez, M. Rizzi and C. A. Sotriffer, J. Med. Chem., 2008, 51, 6237–6255 CrossRef CAS PubMed.
- C. N. Cavasotto, M. G. Aucar and N. S. Adler, Int. J. Quantum Chem., 2019, 119, e25678 CrossRef.
- A. Amadasi, J. A. Surface, F. Spyrakis, P. Cozzini, A. Mozzarelli and G. E. Kellogg, J. Med. Chem., 2008, 51, 1063–1067 CrossRef CAS PubMed.
- P. Cozzini, M. Fornabaio, A. Mozzarelli, F. Spyrakis, G. E. Kellogg and D. J. Abraham, Int. J. Quantum Chem., 2006, 106, 647–651 CrossRef CAS.
- C. N. Cavasotto and R. A. Abagyan, J. Mol. Biol., 2004, 337, 209–225 CrossRef CAS PubMed.
- O. Slater and M. Kontoyianni, Expert Opin. Drug Discovery, 2019, 14, 619–637 CrossRef CAS PubMed.
- M. Su, Q. Yang, Y. Du, G. Feng, Z. Liu, Y. Li and R. Wang, J. Chem. Inf. Model., 2019, 59, 895–913 CrossRef CAS PubMed.
- S. S. Çınaroğlu and E. Timuçin, J. Chem. Inf. Model., 2019, 59, 3846–3859 CrossRef PubMed.
- Z. Wang, H. Sun, X. Yao, D. Li, L. Xu, Y. Li, S. Tian and T. Hou, Phys. Chem. Chem. Phys., 2016, 18, 12964–12975 RSC.
- W. Xu, A. J. Lucke and D. P. Fairlie, J. Mol. Graphics Modell., 2015, 57, 76–88 CrossRef CAS PubMed.
- A. Kukol, Eur. J. Med. Chem., 2011, 46, 4661–4664 CrossRef CAS.
- J. B. Cross, D. C. Thompson, B. K. Rai, J. C. Baber, K. Y. Fan, Y. Hu and C. Humblet, J. Chem. Inf. Model., 2009, 49, 1455–1474 CrossRef CAS PubMed.
- P. J. Ballester, Drug Discovery Today: Technol., 2019, 32–33, 81–87 CrossRef PubMed.
- J. C. Pereira, E. R. Caffarena and C. N. Dos Santos, J. Chem. Inf. Model., 2016, 56, 2495–2506 CrossRef CAS PubMed.
- M. G. Aucar and C. N. Cavasotto, Methods Mol. Biol., 2020, 2114, 269–284 CrossRef CAS PubMed.
- C. N. Cavasotto and M. G. Aucar, Front. Chem., 2020, 8, 246 CrossRef CAS PubMed.
- S. M. Eyrilmez, C. Kopruluoglu, J. Rezac and P. Hobza, ChemPhysChem, 2019, 20, 2759–2766 CrossRef CAS PubMed.
- A. V. Sulimov, D. K. Kutov, I. S. Ilin and V. B. Sulimov, Biomed. Khim., 2019, 65, 80–85 CAS.
- C. N. Cavasotto and J. I. Di Filippo, Mol. Inf., 2021, 40, e2000115 CrossRef PubMed.
- C. N. Cavasotto, N. S. Adler and M. G. Aucar, Front. Chem., 2018, 6, 188 CrossRef PubMed.
- K. Palacio-Rodriguez, I. Lans, C. N. Cavasotto and P. Cossio, Sci. Rep., 2019, 9, 5142 CrossRef PubMed.
- G. Poli, A. Martinelli and T. Tuccinardi, J. Enzyme Inhib. Med. Chem., 2016, 31, 167–173 CrossRef CAS.
- T. Tuccinardi, G. Poli, V. Romboli, A. Giordano and A. Martinelli, J. Chem. Inf. Model., 2014, 54, 2980–2986 CrossRef CAS PubMed.
- D. R. Houston and M. D. Walkinshaw, J. Chem. Inf. Model., 2013, 53, 384–390 CrossRef CAS.
- M. Arciniega and O. F. Lange, J. Chem. Inf. Model., 2014, 54, 1401–1411 CrossRef CAS PubMed.
- R. Abagyan, M. Totrov and D. Kuznetsov, J. Comput. Chem., 1994, 15, 488–506 CrossRef CAS.
- E. A. Gatica and C. N. Cavasotto, J. Chem. Inf. Model., 2012, 52, 1–6 CrossRef CAS PubMed.
- N. Huang, B. K. Shoichet and J. J. Irwin, J. Med. Chem., 2006, 49, 6789–6801 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- S. Ruiz-Carmona, D. Alvarez-Garcia, N. Foloppe, A. B. Garmendia-Doval, S. Juhos, P. Schmidtke, X. Barril, R. E. Hubbard and S. D. Morley, PLoS Comput. Biol., 2014, 10, e1003571 CrossRef PubMed.
- O. Korb, T. Stutzle and T. E. Exner, J. Chem. Inf. Model., 2009, 49, 84–96 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05785e |
| This journal is © The Royal Society of Chemistry 2021 |