
Magnesium bromide (MgBr2) as a catalyst for living cationic polymerization and ring-expansion cationic polymerization†
Yuji
Daito
a,
Reina
Kojima
a,
Naoyuki
Kusuyama
a,
Yasuhiro
Kohsaka
 bc and
Makoto
Ouchi
*a
bc and
Makoto
Ouchi
*a
aDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: ouchi.makoto.2v@kyoto-u.ac.jp
bResearch Initiative for Supra-Materials (RISM), Shinshu University, Japan
cFaculty of Textile Science and Technology, Shinshu University, 3-15-1 Tokida, Ueda, Nagano 386-8567, Japan
First published on 30th December 2020
Abstract
Magnesium bromide (MgBr2) was found to be an effective catalyst for the ring-expansion cationic polymerizations of isobutyl vinyl ether (IBVE) initiated by a “cyclic” hemiacetal ester (HAE) bond-based initiator leading to the syntheses of cyclic poly(IBVE)s. Crucial to the achievement is the choice of the quencher (e.g., DMF or DMSO) instead of methanol as well as the realization of living cationic polymerization with an “acyclic” HAE-based initiator under magnesium catalysis. The advantage of MgBr2 over a conventional catalyst (i.e., SnBr4) is that several types of cyclic molecules are available as initiators, including a methacrylate type vinylidene group-embedded cyclic compound. The reactive group-carrying initiator allowed the pin-point incorporation of a polar group (e.g., glycerol) into the resultant cyclic polymer via a thiol-Michael addition reaction while retaining the cyclic topology.
Introduction
In cationic addition polymerization of vinyl monomers, various metal halide Lewis acids (MXns) are often used as a catalyst in conjunction with a protonic acid (HB), and the resultant counteranion (BMXn−) derived from the two components greatly affects the propagation behaviors of the growing carbocationic species.1 The extensive investigations of the binary system have allowed the living cationic polymerization of various monomers such as vinyl ether, isobutylene, and styrene derivatives, and thus the methodology to control cationic polymerization has been systematically developed.2 The selection of a Lewis acid is crucial to realize the living polymerization, and particularly this is the case even for achieving more advanced control of primary characteristics, such as tacticity,3–7 regioselectivity,8 and sequence.9–11In response to increasing interest in the physical properties and functions of topologically unique polymers, the precise syntheses of cyclic polymers have attracted attention.12–15 One efficient and reliable approach is “ring-expansion polymerization (REP)”.16,17 The notable feature is that dilution conditions are not necessarily required, which is completely different from the cyclization approach using linear telechelic chains. Most of the REP systems developed earlier were based on the ring-opening polymerizations of cyclic monomers, but recently the ring-expansion addition polymerization of acyclic monomers (i.e., vinyl monomers)18–28 has also been achieved through molecular design on the basis of precision polymerization systems. For instance, we have reported the ring-expansion cationic polymerization of vinyl ethers (Fig. 1A).29–35 The preliminary key for the achievement is the control of the reversible activation of an in-chain hemiacetal ester (HAE) bond in a cyclic initiator (1) on the basis of the living cationic polymerization of vinyl ethers with the acetic acid adduct initiator [2, an adduct of acetic acid to isobutyl vinyl ether (IBVE)].36,37 The side reaction that could happen in cationic polymerization, i.e., β-proton elimination, is unfavorable for the synthesis of cyclic polymers (Fig. 1B). In addition, the counteranion exchange reaction between the acetate anion and the halogen atom in the Lewis acid must be avoided because the reaction causes the generation of halogen-capped “linear” dormant species. Our previous study has revealed typical Lewis acid catalysts that are known to be useful for catalyzing living cationic polymerization, such as EtAlCl2, SnCl4, and TiCl4, in conjunction with 1 resulting in the generation of a linear polymer carrying the methoxy terminal. Probably, a counteranion exchange reaction occurred during the polymerization, and the chlorine terminal was replaced with methoxy on quenching with methanol to deactivate the Lewis acid catalyst.30 On the other hand, tin(IV) bromide (SnBr4) specifically allowed the syntheses of cyclic polymers even after quenching by methanol. The result confirmed that SnBr4-catalyzed living cationic polymerization of IBVE with the acyclic HAE-based initiator (2) affords polyIBVEs carrying the HAE-bonded terminal after methanol quenching.37 To date, the applicable catalyst for the cationic REP has been limited to SnBr4 in combination with 1. An emergence of a versatile catalyst enabling the construction of various ring-based architectures in conjunction with some types of cyclic initiators has been in demand.
 | ||
| Fig. 1 (A) SnBr4-catalyzed ring-expansion cationic polymerization of vinyl ether with a cyclic HAE-based initiator (1); ideal propagation and termination (a) and unfavorable side reactions (b and c). (B) SnBr4-catalyzed living cationic polymerization of IBVE with an acyclic HAE-based initiator 2 affording an HAE terminal structure. | ||
In this report, we thus focused on magnesium bromide (MgBr2) as a catalyst for ring-expansion cationic polymerization. The magnesium ion (Mg2+) is categorized as a “hard” acid showing relatively high oxygen affinity, and hence we expected that it can work as a catalyst for activation of the HAE bond via the coordination of the carbonyl group. Indeed, some reports on organic reactions38 and polymerizations39,40 with MgBr2 showed the promotion of the reaction, as well as enhancement in the selectivity, via coordination of the carbonyl group. As for cationic polymerization, the study on the use of magnesium catalysts dates back to 1963,41,42 and, to the best of our knowledge, there have been no examples of living cationic polymerization with a magnesium catalyst. Therefore, prior to the study of ring-expansion cationic polymerization with the “cyclic” initiator (1), we examined the potential of MgBr2-catalyzed living cationic polymerization with the “acyclic” initiator (2).
Results and discussion
Model experiments with acyclic initiator 2
Fig. 2 shows the results of the cationic polymerization of IBVE with MgBr2 as a catalyst in conjunction with 2 as an initiator. A solution of MgBr2 in diethyl ether (Et2O) was added to a toluene solution containing other components for the initiation of the polymerization. 2,6-Di-tert-butyl-4-methylpyridine (DTBMP) was used as a proton trap to align the optimized conditions in our previous study for ring-expansion cationic polymerization with SnBr4.29 Indeed, the proton trap addition was helpful to ensure the reproducibility of the results. As shown in Fig. 2A–C, the magnesium catalytic system enabled highly controlled living cationic polymerization: the first order kinetic plots were almost linear regardless of the injection ratio ([monomer]0/[initiator]0); the molecular weight linearly increased in proportion to conversion depending on the injection ratio; the SEC curves shifted to a higher molecular weight while keeping very narrow molecular weight distributions (Mw/Mn < 1.06). The structure of the obtained poly(IBVE) after quenching with methanol (92% conversion for the 304 mer condition, Mn = 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mw/Mn = 1.06 from SEC with PSt calibration) was characterized by 1H NMR (Fig. 2D). The peak (f) derived from the methine proton neighboring to the methoxy group at the ω-terminal was clearly observed, and the integration ratio to another minor peak (a) from the methyl protons of the initiator at the α-terminal was almost 1:3. The peak from the methine proton of the HAE bond end group was not observed. Thus, in this case, methoxy capping took place by methanol for the deactivation of MgBr2. The quenching reaction should be noted in the ring-expansion polymerization for the syntheses of cyclic polymers. MALDI-TOF-MS of the lower molecular weight sample (65% conversion for the 38 mer condition, Mn = 4200, Mw/Mn = 1.04 from SEC with PSt calibration) also supported the formation of a well-defined polymer carrying the methoxy-capped acetal terminal with the lower molecular weight sample (Fig. S1†).
000, Mw/Mn = 1.06 from SEC with PSt calibration) was characterized by 1H NMR (Fig. 2D). The peak (f) derived from the methine proton neighboring to the methoxy group at the ω-terminal was clearly observed, and the integration ratio to another minor peak (a) from the methyl protons of the initiator at the α-terminal was almost 1:3. The peak from the methine proton of the HAE bond end group was not observed. Thus, in this case, methoxy capping took place by methanol for the deactivation of MgBr2. The quenching reaction should be noted in the ring-expansion polymerization for the syntheses of cyclic polymers. MALDI-TOF-MS of the lower molecular weight sample (65% conversion for the 38 mer condition, Mn = 4200, Mw/Mn = 1.04 from SEC with PSt calibration) also supported the formation of a well-defined polymer carrying the methoxy-capped acetal terminal with the lower molecular weight sample (Fig. S1†).
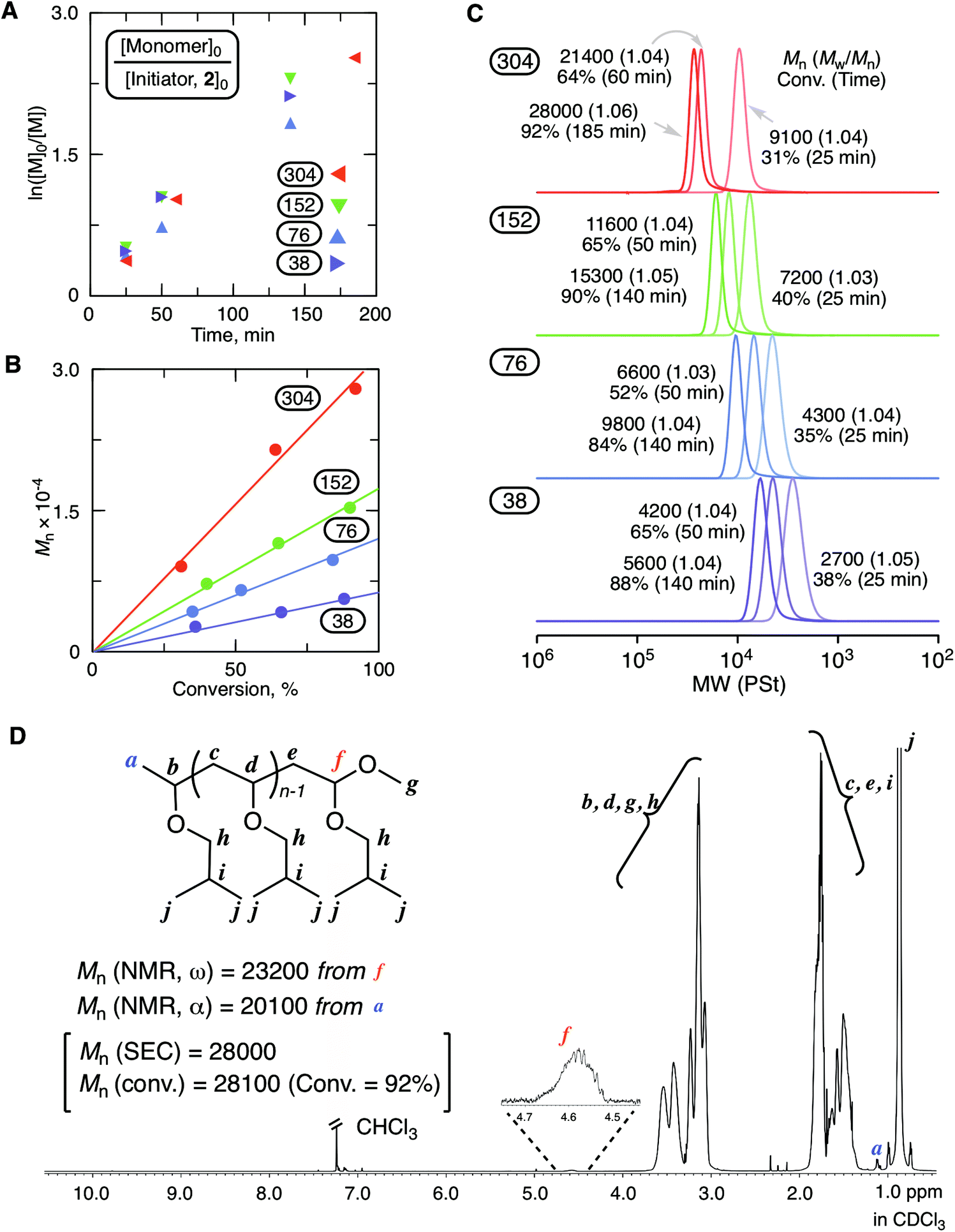 | ||
| Fig. 2 MgBr2-catalyzed living cationic polymerizations of IBVE with 2 as an initiator: [IBVE]0/[2]0/[MgBr2]0/[DTBMP]0 = 1520, 760, 380, or 190/5.0/10/0.15 mM in toluene/Et2O (90/10 vol%) at 0 °C. (A) The first order kinetic plots, (B) the relationship between Mns of the obtained poly(IBVE) and conversion, (C) SEC curves of the obtained poly(IBVE)s, and (D) 1H NMR spectrum of poly(IBVE) obtained at 92% conversion for [monomer]0/[initiator]0 = 304. | ||
Other magnesium compounds, such as diethyl etherate (MgBr2·OEt2), MgCl2, MgI2, and Mg(OTf)2, were also tested as catalysts for cationic polymerization with 2 (Fig. S2†). The etherate complex allowed living polymerization similar to MgBr2. The polymerization with MgCl2 proceeded heterogeneously due to the low solubility, and the molecular weights of the obtained polymers were not controlled. When iodide was used, the polymerization proceeded very slowly. The molecular weight distributions were narrow at the earlier stage but became broader as polymerization proceeded. In particular, a tailing peak was observed in the lower molecular weight region, which is presumably due to the occurrence of a chain-transfer reaction via β-proton elimination. Magnesium triflate was incapable of catalyzing the polymerization at all. Thus, MgBr2 and etherate were found specifically to be appropriate as catalysts for the HAE-based initiating system. In the following polymerizations, MgBr2 was used as a catalyst.
Cationic polymerization with cyclic initiator 1
The excellent catalyst performance of MgBr2 in living cationic polymerization with an acyclic initiator encouraged us to use the catalyst for ring-expansion cationic polymerization with the cyclic counterpart (1) (Fig. 3A). Given the fact that a methoxy-capped polymer was formed by methanol quenching in the acyclic initiator model system, it was predicted that methanol was not suitable as the quencher to synthesize cyclic polymers. Thus, we studied other Lewis bases as a quencher for the deactivation of MgBr2. Here, from our previous studies,29 some signs were observed in SEC analyses when cyclic polymers are formed: SEC curves are multimodal; the curves turn unimodal via an acidolysis treatment with trifluoroacetic acid (TFA) while giving higher peak molecular weights. These signs are due to the generation of fused cyclic chains via a counteranion exchange or chain transfer reaction between cyclic polymers and transformation of the cyclic chains into linear chains by irreversible cleavage of the HAE bonds in the chains via the acidolysis treatment. Consequently, when methanol was used as the quencher, the SEC curve was unimodal similar to the case with 2, and the position of the SEC curve did not shift at all via acidolysis. This trend indicated that the obtained polymer was not cyclic but linear (Fig. 3B). On the other hand, when DMF or DMSO was used as the quencher, multimodal SEC curves peculiar to the formation of a cyclic polymer via ring-expansion cationic polymerization were obtained. As expected, the multimodal peak turned unimodal via acidolysis with TFA, and the main peak shifted to a higher molecular weight. The SEC curve after the acidolysis was very narrow (Mw/Mn < 1.05), and the molecular weight distribution was comparable to the model polymerization with the acyclic initiator 2. The structures of the obtained polymers after quenching with DMF, DMSO, and methanol were characterized by 1H NMR and MALDI-TOF-MS. A peak from a methine proton of the HAE bond (j) was clearly detected when DMF or DMSO was used as the quencher. On the other hand, the peak was not observed in the case with the methanol quencher, and instead a peak from a methine proton of methoxy-capped acetal (j′) was observed. In all cases, the apparent molecular weights calculated from the integration ratio of the minor peak to the repeating unit peaks agreed well with those from the injection ratio and conversion, suggesting the quantitative introduction of the HAE or acetal bond. These structures depending on the quencher were also supported by MALDI-TOF-MS (Fig. S3†). The mass of the peak series with the interval of the IBVE repeating unit agreed with the molecular weight of the ideal cyclic structure in the case with DMF and DMSO as a quencher. A minor peak series that cannot be identified was also observed for the DMSO quencher, but they were not from the linear chain. On the other hand, the main peak series in the case with methanol quencher clearly supported the formation of a different type of polymer that is definitely a linear chain capped with the methoxy group. As predicted from the result of the model polymerization with 2, methanol likely worked as a capping agent via reaction with the carbocationic species. It is assumed that a methoxy capping reaction took place prior to the full deactivation of MgBr2 due to the relatively robust nature of MgBr2. On the other hand, in the case of SnBr4, the catalyst is probably deactivated very quickly upon methanol addition, and thus the HAE bond is maintained. Thus, methanol was found to be unavailable as the quencher for the syntheses of cyclic polymers with the MgBr2 catalyst. DMF was used as the quencher for the following experiments of ring-expansion cationic polymerization with MgBr2. We also confirmed that DMF was available as a quencher for the syntheses of cyclic polymers even with SnBr4 as the catalyst.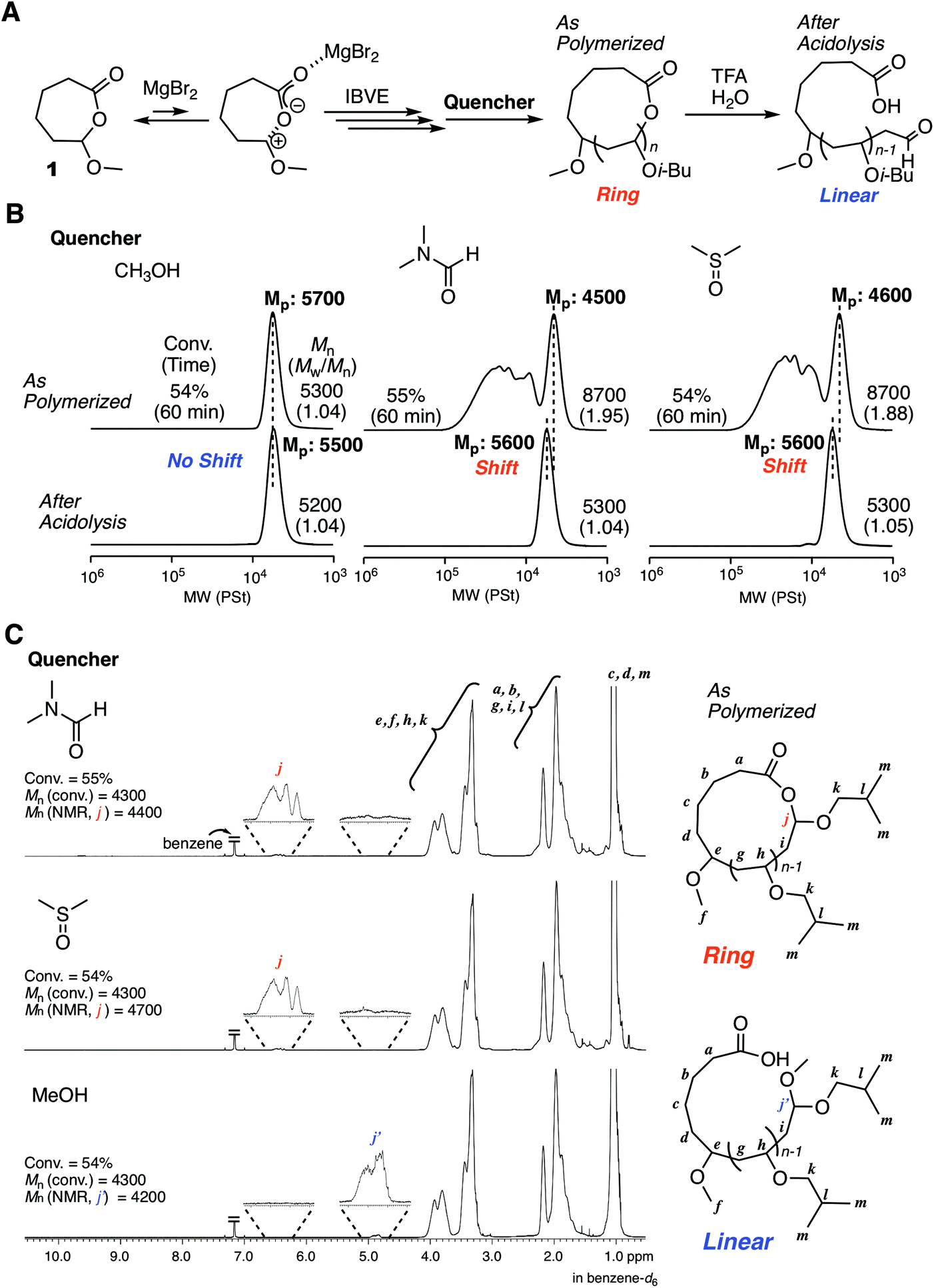 | ||
| Fig. 3 Effects of a quencher for the cationic polymerization of IBVE with 1 and MgBr2. (A) Scheme; (B) SEC curves of the obtained poly(IBVE)s with methanol, DMF, and DMSO as the quencher and those after acidolysis with TFA/H2O. (C) 1H NMR spectra of poly(IBVE)s: [IBVE]0/[1]0/[MgBr2]0/[DTBMP]0 = 380/5.0/10/0.15 mM in toluene/Et2O (90/10 vol%) at 0 °C. Polymerization was terminated by the addition of a quencher (methanol, DMF, or DMSO). | ||
Fig. 4 shows some SEC curves of the polymers with different conversions quenched by DMF in comparison with those obtained with SnBr4. The curves consisted of unimodal and multimodal peaks and shifted to higher molecular weights without spreading to lower molecular weights as polymerization proceeded. Thus, the polymerization was basically controlled without β-proton elimination. The SEC curves were broad due to the ring fusion, and the trend was very similar to the case with SnBr4. However, a careful analysis of the SEC curves revealed differences in the ratio of the higher molecular weight peak derived from ring fusion: MgBr2 seemed to give a slightly greater ratio than SnBr4. The ratio of the fused ring to the whole cyclic polymer, which was estimated from the integration ratio of the peaks, was plotted against conversion for comparison between the two catalytic systems. As a result, the system with MgBr2 tended to give a higher ratio of the fused ring than with SnBr4, and the difference was obvious at the earlier polymerization stage. To confirm the peculiar feature of MgBr2, the evaluation of the interaction for the cyclic initiator was attempted by 1H NMR. However, the spectrum of the mixture of 1 and MgBr2 was too complicated for the evaluation due to the ring-opening polymerization of 1. Therefore, 3 was newly prepared as an “inactive” HAE bond-embedded cyclic molecule for the evaluation. Herein, two carbons in the 6-membered cyclic HAE structure are shared with benzene, and thus 3 is too stable to be opened by the Lewis acid activation. Indeed, 3 was incapable of initiating the cationic polymerization at all. In the spectrum of the 1:2 mixture of 3 and MgBr2, the peak from the methylene protons of the HAE bond apparently shifted to a lower magnetic field. On the other hand, such a peak shift was not observed with SnBr4. Here, for the rigorous comparison of the catalytic activity in cationic polymerization between MgBr2 and SnBr4, the cationic polymerization with 1/SnBr4 was performed under the conditions same as those with MgBr2 ([Lewis acid]0 = 10 mM in the presence of 10 vol% Et2O). The polymerization proceeded much faster, and the polymerization was not controlled enough resulting in the generation of linear polymers, which was found through the acidolysis experiment (Fig. S4†). From the result, the catalytic activity of MgBr2 was found to be essentially lower than that of SnBr4, while the optimized conditions were required for the control of the ring-expansion polymerization according to the Lewis acid catalyst. Nevertheless, the model reaction experiments indicated that MgBr2 interacts with the HAE bond more strongly than SnBr4. Presumably, due to the higher oxophilicity or poor solubility of MgBr2, the degree of interaction with the HAE bond is higher, and thus the apparent concentration of the initiator or the generating polymer chain becomes higher, and thus ring fusion is likely to occur more frequently than with SnBr4.
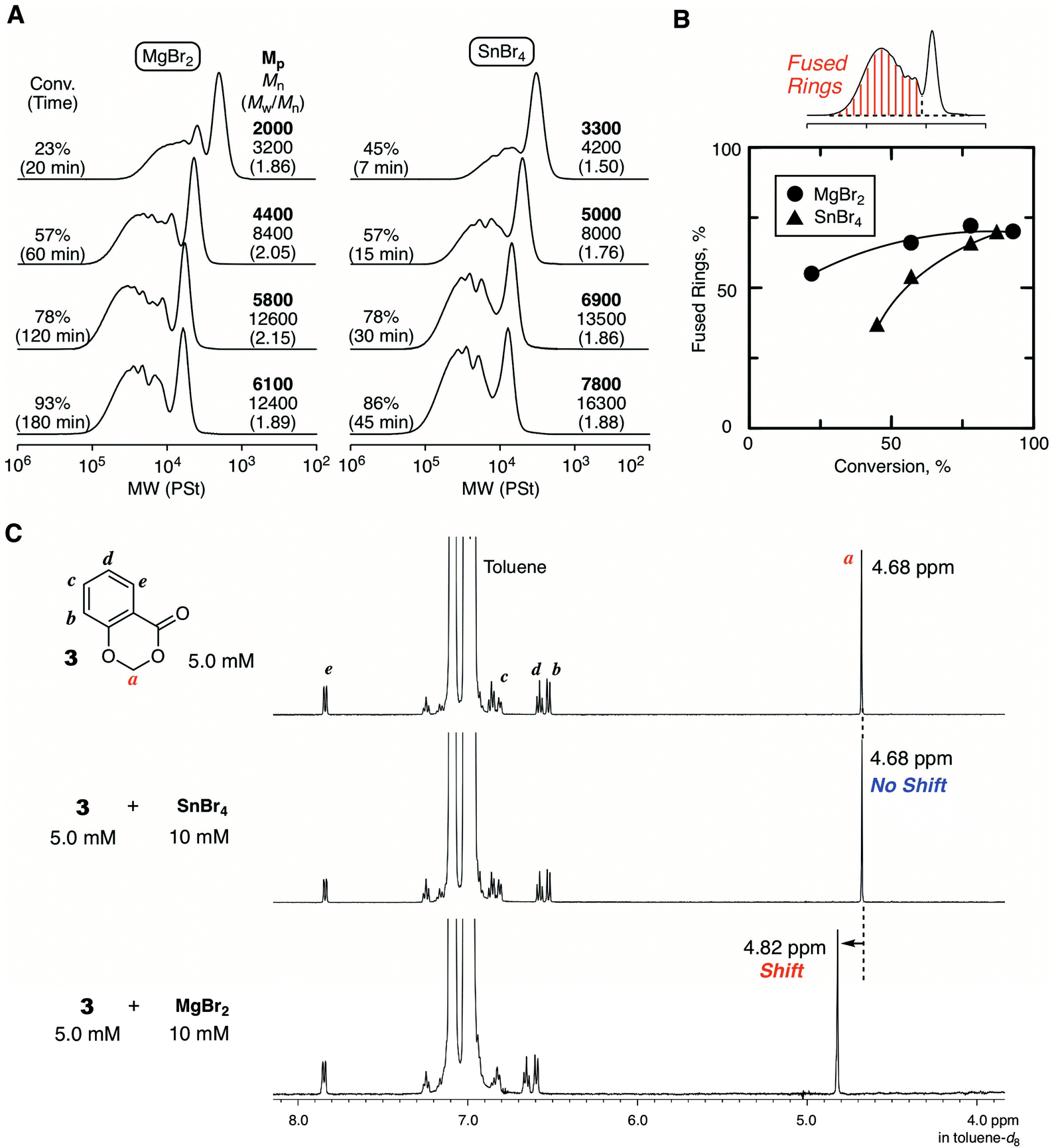 | ||
| Fig. 4 Comparison between MgBr2 and SnBr4 in the ring-expansion cationic polymerization of IBVE with 1: (A) SEC curves of the obtained poly(IBVE)s (B) ratios of the fused ring estimated by peak separation. (C) 1H NMR analyses of the mixture of MgBr2 or SnBr4 with 3 as the model compound for the evaluation of the interaction with the HAE bond. | ||
Ring-expansion cationic polymerization with various cyclic initiators
Another type of HAE-embedded cyclic compound was also studied as the initiator. When a 6-membered cyclic molecule carrying the HAE bond (4)43 was used as the initiator in conjunction with SnBr4, the polymerization was apparently decelerated in comparison with 1 (Fig. 5A). In the SEC analyses of the obtained polymers, multimodal peaks shifted to higher molecular weights as the polymerization proceeded, indicating the progression of ring-expansion polymerization. However, the ratio of the peak at lower molecular weight, which is probably derived from the non-fused or single ring, was low, and the peak eventually became quite minor for the whole peak. In addition, the SEC curves of the polymers after acidolysis were relatively broad (Mw/Mn > 1.3). It is supposedly considered that the initiation was not regulated and the polymerization was accompanied by some side reactions. On the other hand, MgBr2 allowed almost the same rate as with 1, although a slower initiation reaction of 4 was predicted than that of 1, because the ring strain is lower. Presumably, the higher interaction of MgBr2 with the HAE bond led to ring fusion at the earlier stage, and therefore the effects of the ring strain were negligible. The SEC curves of the polymers quenched by DMF were peculiar to ring-expansion cationic polymerization. Furthermore, they turned very narrow (Mw/Mn < 1.1) via acidolysis, and the peak top shifted to a higher molecular weight, indicating that the cyclic chains including the fused rings were converted into linear ones of narrow molecular weight distribution. The cyclic structure of the resultant polymer was supported by 1H NMR and MALDI-TOF-MS (Fig. S5†). Note that the specific peak to the moiety in the initiator 4 (methylene protons neighboring the carbonyl group), which was not detected when 1 was used (see Fig. 3C), was clearly observed at around 2.5 ppm with a reasonable integration ratio. Thus, MgBr2 was found to work as a catalyst to control ring-expansion cationic polymerization with 4 that was not available as the initiator in conjunction with SnBr4.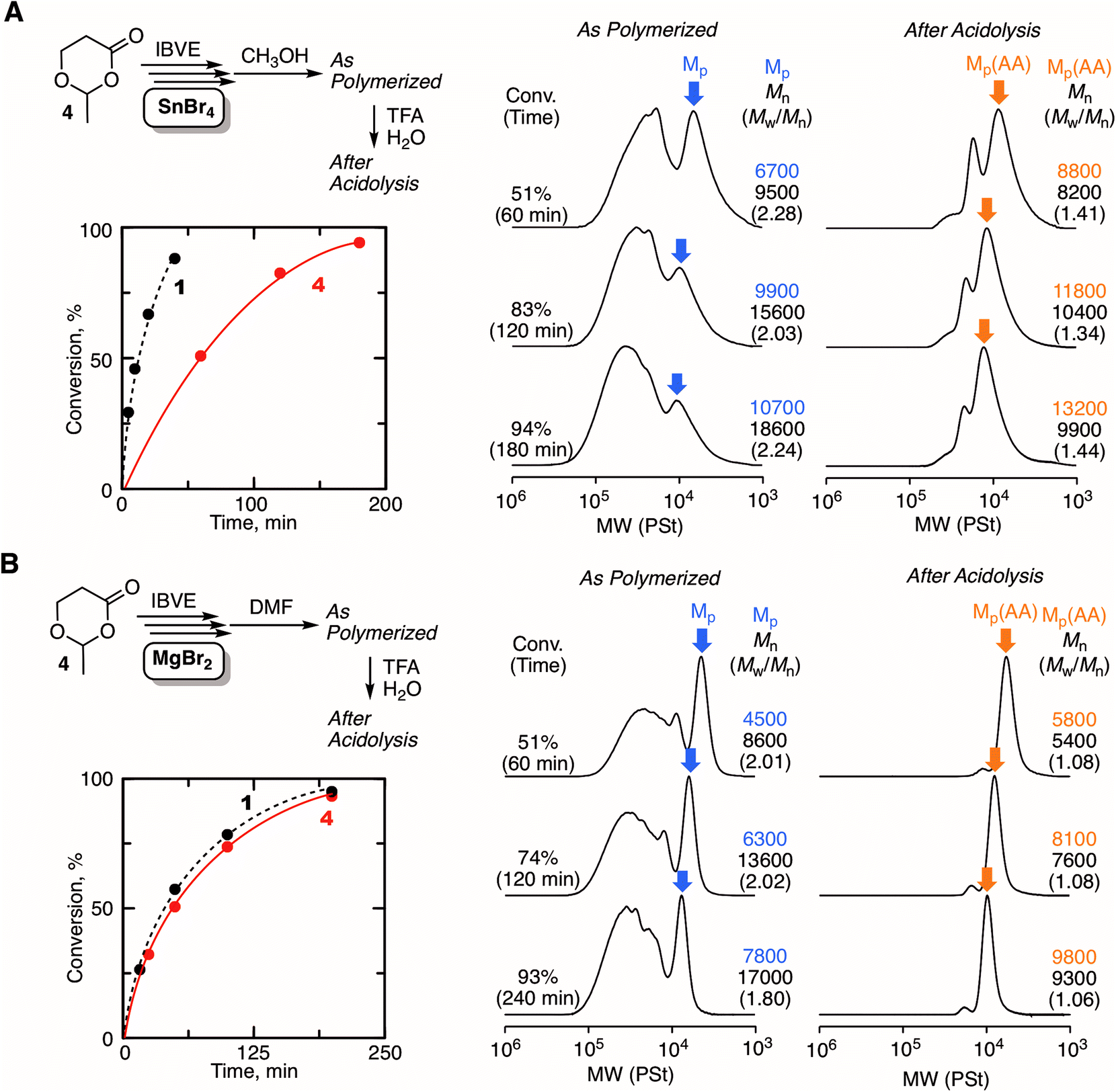 | ||
| Fig. 5 Comparison between SnBr4 (A) and MgBr2 (B) as a catalyst for the ring-expansion cationic polymerization of IBVE with 4: [IBVE]0/[4]0/[SnBr4 or MgBr2]0/[DTBMP]0 = 380/5.0/5.0 or 10/0.15 mM in toluene (for SnBr4) or toluene/Et2O (90/10 vol%) (for MgBr2) at 0 °C. | ||
MgBr2 might contribute to expanding the scope of the applicable initiator for ring-expansion cationic polymerization. Particularly, the use of an initiator carrying a reactive site is of great interest toward the modification of the resultant cyclic polymer via post-polymerization. Thus, a 6-membered cyclic molecule 5, in which the methacrylate-type vinylidene group is embedded along with a HAE bond, was used as the initiator in conjunction with MgBr2. The compound has been studied as a methacrylate-type cyclic monomer for chain-growth polymerization.44 In contrast to SnBr4 resulting in obviously poor control (Fig. S6†), MgBr2 gave the typical results of ring-expansion polymerization for the SEC analysis similar to those with cyclic initiators such as 1 and 4 (Fig. S7†). The structure of the thus obtained polymer (conv. 49%, 6 hours, Mn = 8000, Mw/Mn = 1.95, Fig. 6A) was characterized by 1H NMR (Fig. 6B). The minor peak based on the methine proton (a) in the HAE bond around 6.5 ppm was observed as usual, and those from the vinylidene group (b) seemed to overlap at the same area. However, the mass of the peak series observed by MALDI-TOF-MS certainly supported the generation of the ideal cyclic polymer carrying the methacrylate vinylidene group in the chain (Fig. 6C). The vinylidene group is available not only as a monomer for radical or anionic polymerization, but also as a reactive site for thiol-Michael addition. Herein, the latter reaction was studied, and thioglycerol was selected as the model reactant for the introduction of the glycerol group. An excess of thioglycerol was added into a THF solution of the polymer in the presence of Et3N. The structure of the thus obtained polymer was then analyzed for the evaluation of whether the glycerol group was introduced without damaging the cyclic structure. The SEC curve after the reaction was almost the same as before, indicating that the HAE bond was not cleaved during the reaction or the workup process. Meanwhile, a clear change was observed in the 1H NMR spectrum: several minor peaks around 6–7 ppm turned simpler, and the peak shape resembled that usually observed from the methine (b′) of the HAE bond in cyclic polyIBVE. Instead, other minor peaks appeared around 2.5–3 ppm, which was likely attributed to the methylene and methine protons (c, d, and e) introduced via the reaction. The molecular weights calculated with the integration ratios of the minor peaks almost agreed with the calculated value from the conversion. Thus, it can be concluded that the introduction of the vinylidene group as well as the glycerol group was almost quantitative.
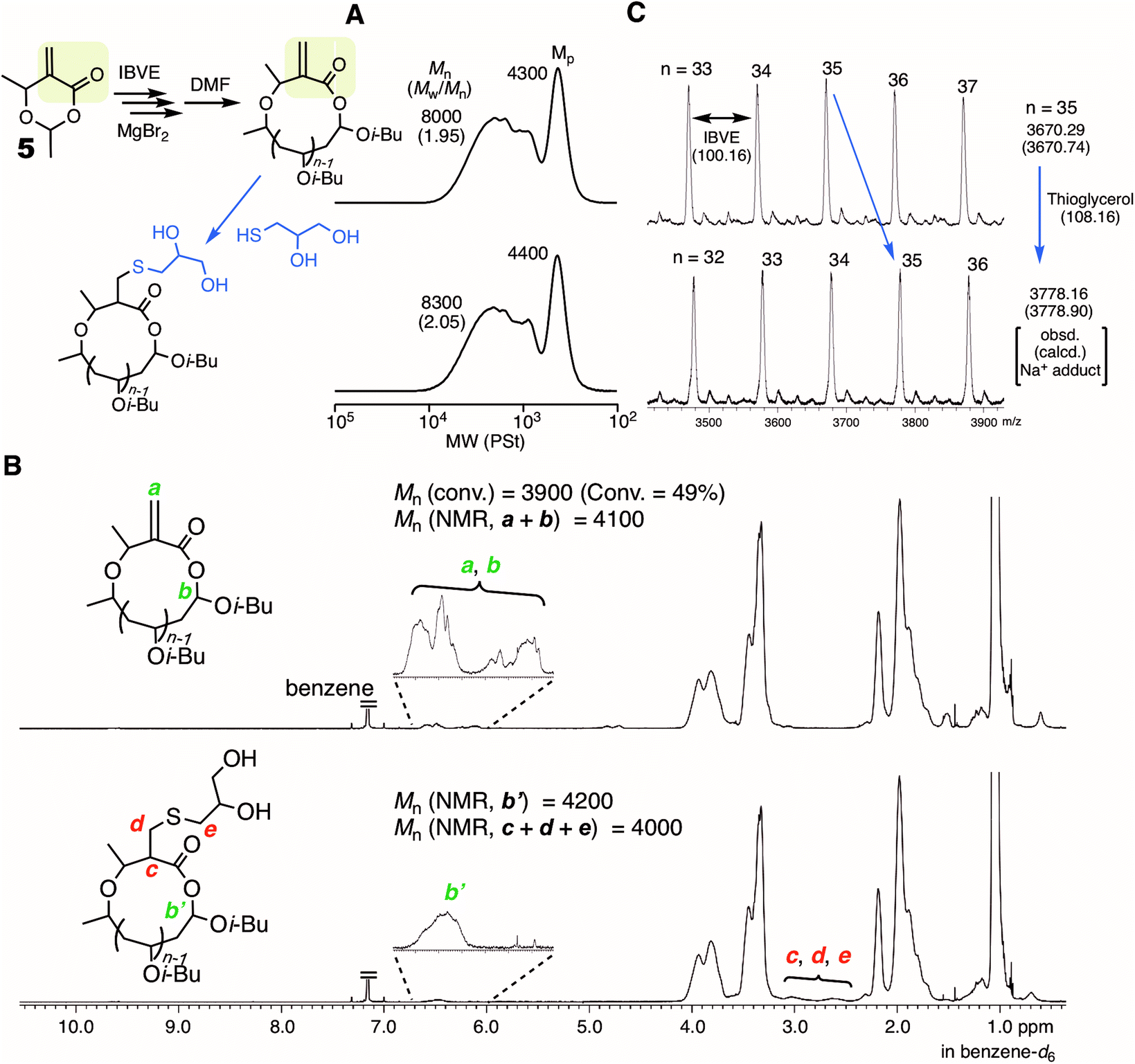 | ||
| Fig. 6 Ring-expansion cationic polymerization of IBVE with 5 and MgBr2, and the post-polymerization reaction of the vinylidene group with thioglycerol. (A) SEC; (B) 1H NMR; (C): MALDI-TOF-MS [IBVE]0/[5]0/[MgBr2]0/[DTBMP]0 = 380/5.0/5.0 or 10/0.15 mM in toluene/Et2O (90/10 vol%) at 0 °C. The sample was obtained at 49% conversion (1 hour). See the ESI† for the conditions of the post-polymerization reaction. | ||
Conclusion
MgBr2 was useful as a catalyst for synthesizing cyclic poly(IBVE)s under the control of reversible activation of the HAE bond in ring-expansion cationic polymerization. The key was the precise control of the model polymerization with an acyclic HAE-based initiator and judicious selection of the quencher (DMF or DMSO) to terminate the polymerization while keeping the cyclic dormant species. In contrast to SnBr4 that is a conventional catalyst useful only for the 7-membered initiator, MgBr2 allowed the generation of cyclic polymers from other types of cyclic initiators including the methacrylate-embedded one useful for pinpoint modification via post-polymerization. Thus, the new initiation system with MgBr2 is promising for the construction of ring-based architectures made of poly(vinyl ether)s.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant 17H06453 (M. O.) and 16K05792 (Y. K.).References
- M. Sawamoto, Prog. Polym. Sci., 1991, 16, 111–172 CrossRef CAS.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS.
- M. Ouchi, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 6407–6411 CrossRef CAS.
- H. Watanabe, A. Kanazawa and S. Aoshima, ACS Macro Lett., 2017, 6, 463–467 CrossRef CAS.
- A. J. Teator and F. A. Leibfarth, Science, 2019, 363, 1439–1443 CrossRef CAS.
- A. J. Teator, T. P. Varner, P. E. Jacky, K. A. Sheyko and F. A. Leibfarth, ACS Macro Lett., 2019, 8, 1559–1563 CrossRef CAS.
- H. Watanabe, T. Yamamoto, A. Kanazawa and S. Aoshima, Polym. Chem., 2020, 11, 3398–3403 RSC.
- M. Ouchi, M. Kamigaito and M. Sawamoto, Macromolecules, 2001, 34, 6586–6591 CrossRef CAS.
- A. Kanazawa and S. Aoshima, ACS Macro Lett., 2015, 4, 783–787 CrossRef CAS.
- S. Matsumoto, A. Kanazawa, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2017, 139, 7713–7716 CrossRef CAS.
- M. Mimura, A. Kanazawa and S. Aoshima, Macromolecules, 2019, 52, 7572–7583 CrossRef CAS.
- Y. Tezuka, Polym. J., 2012, 44, 1159–1169 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS.
- M. Romio, L. Trachsel, G. Morgese, S. N. Ramakrishna, N. D. Spencer and E. M. Benetti, ACS Macro Lett., 2020, 9, 1024–1033 CrossRef CAS.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041–2044 CrossRef CAS.
- Y. A. Chang and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2892–2902 CrossRef CAS.
- C. D. Roland, H. Li, K. A. Abboud, K. B. Wagener and A. S. Veige, Nat. Chem., 2016, 8, 791–796 CrossRef CAS.
- W. J. Niu, S. A. Gonsales, T. Kubo, K. C. Bentz, D. Pal, D. A. Savin, B. S. Sumerlin and A. S. Veige, Chem, 2019, 5, 237–244 CAS.
- Z. H. Miao, T. Kubo, D. Pal, B. S. Sumerlin and A. S. Veige, Macromolecules, 2019, 52, 6260–6265 CrossRef CAS.
- A. Narumi, S. Zeidler, H. Barqawi, C. Enders and W. H. Binder, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3402–3416 CrossRef CAS.
- A. Narumi, S. Hasegawa, R. Yanagisawa, M. Tomiyama, M. Yamada, W. H. Binder, M. Kikuchi and S. Kawaguchi, React. Funct. Polym., 2016, 104, 1–8 CrossRef CAS.
- A. Narumi, T. Kobayashi, M. Yamada, W. H. Binder, K. Matsuda, M. S. A. Shaykoon, K. Enomoto, M. Kikuchi and S. Kawaguchi, Polymers, 2018, 10, 638 CrossRef.
- A. Narumi, M. Yamada, Y. Unno, J. Kumaki, W. H. Binder, K. Enomoto, M. Kikuchi and S. Kawaguchi, ACS Macro Lett., 2019, 8, 634–638 CrossRef.
- Y. Hosoi, A. Takasu, S. Matsuoka and M. Hayashi, J. Am. Chem. Soc., 2017, 139, 15005–15012 CrossRef CAS.
- K. Naruse, A. Takasu and M. Higuchi, Macromol. Chem. Phys., 2020, 221, 2000004 CrossRef CAS.
- Y. Oga, Y. Hosoi and A. Takasu, Polymer, 2019, 186, 122019 CrossRef.
- Y. Muramatsu, Y. Oga, A. Takasu and M. Higuchi, Polym. J., 2020, 52, 1253–1261 CrossRef CAS.
- H. Kammiyada, A. Konishi, M. Ouchi and M. Sawamoto, ACS Macro Lett., 2013, 2, 531–534 CrossRef CAS.
- H. Kammiyada, M. Ouchi and M. Sawamoto, Macromol. Symp., 2015, 350, 105–116 CrossRef CAS.
- H. Kammiyada, M. Ouchi and M. Sawamoto, Polym. Chem., 2016, 7, 6911–6917 RSC.
- H. Kammiyada, M. Ouchi and M. Sawamoto, Macromolecules, 2017, 50, 841–848 CrossRef CAS.
- H. Kammiyada, M. Ouchi and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3082–3089 CrossRef CAS.
- M. Ouchi, H. Kammiyada and M. Sawamoto, Polym. Chem., 2017, 8, 4970–4977 RSC.
- H. Kubota, S. Yoshida and M. Ouchi, Polym. Chem., 2020, 11, 3964–3971 RSC.
- S. Aoshima and T. Higashimura, Macromolecules, 1989, 22, 1009–1013 CrossRef CAS.
- T. Hashimoto, T. Iwata, A. Minami and T. Kodaira, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 3173–3185 CrossRef CAS.
- J. A. Marshall, Chem. Rev., 1996, 96, 31–48 CrossRef CAS.
- A. Matsumoto and S. Nakamura, J. Appl. Polym. Sci., 1999, 74, 290–296 CrossRef CAS.
- S. Naumann, P. B. V. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS.
- K. Iwasaki, H. Fukutani and S. Nakano, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 1937–1946 CrossRef CAS.
- K. Iwasaki, S. Nakano, Y. Tsuchida and H. Fukutani, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 2371–2381 CrossRef CAS.
- A. E. Neitzel, M. A. Petersen, E. Kokkoli and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 1156–1160 CrossRef CAS.
- Y. Kohsaka, Y. Matsumoto, T. Y. Zhang, Y. Matsuhashi and T. Kitayama, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 955–961 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01584a |
| This journal is © The Royal Society of Chemistry 2021 |