
Pairing Suzuki–Miyaura cross-coupling and catalyst transfer polymerization
Michael V.
Bautista
 ,
Anthony J.
Varni
,
Josué
Ayuso-Carrillo
,
Matthew C.
Carson
and
Kevin J. T.
Noonan
*
,
Anthony J.
Varni
,
Josué
Ayuso-Carrillo
,
Matthew C.
Carson
and
Kevin J. T.
Noonan
*
Department of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213-2617, USA. E-mail: noonan@andrew.cmu.edu
First published on 22nd January 2021
Abstract
Suzuki–Miyaura catalyst transfer polymerization (SM CTP) is a versatile method to prepare conjugated polymers with control over molecular weight, sequence, and dispersity. This perspective aims to highlight some of the progress in using Suzuki–Miyaura coupling to prepare well-defined conjugated polymers via a chain-growth mechanism. We detail some of the advantages and challenges of this coupling to make aromatic polymers from monomers bearing two functional groups. The advances in arene borylation over the last twenty years are briefly highlighted, as these strategies should serve to diversify monomer scope in the future. The proposed mechanism for transmetalation in Suzuki–Miyaura polymerization is discussed, as it is different from the more typical Kumada coupling. We describe the versatility of the organoboron group used for this reaction and how it can be used tune polymerization behavior. Finally, some of the advances in catalyst design to prepare conjugated polymers using SM CTP are noted.
Introduction
Conjugated polymers are defined by a pattern of alternating single and double bonds along the backbone, and most commonly are comprised of aromatic rings. These π-electron frameworks have desirable optical and electronic properties that can be tuned by the choice of aromatic repeat unit(s) along the backbone and by the solid-state organization of the material. Molecular weight and molecular weight distribution are particularly important parameters that can influence solid-state packing and charge transport.1–3 Establishing such relationships4–7 has been made possible by advances in the synthesis of benchmark materials, such as poly(3-hexylthiophene) (P3HT).8Given the importance of molecular weight, precise methods to ensure reproducibility and specificity in conjugated polymer synthesis are desirable. Conjugated macromolecules are typically prepared using cross-coupling chemistry, and the polymerization often proceeds by a step-growth mechanism.9–16 Preparing conjugated polymers by a chain-growth process (known as catalyst-transfer polymerization or CTP) enables improved control over the molecular weight and molecular weight distribution of the resultant sample. Moreover, controlled polymerization offers the possibility to build backbone compositions that are inaccessible using step-growth techniques (e.g. block copolymers).17–24 The chain-growth mechanism relies on catalyst complexation to the growing polymer chain during the reaction, as shown in Fig. 1.17–24
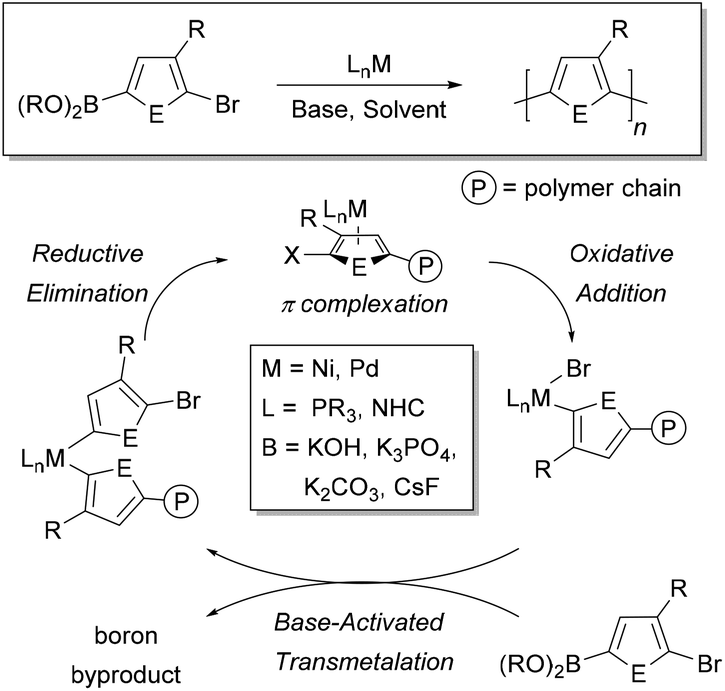 | ||
| Fig. 1 General mechanism of Suzuki–Miyaura catalyst-transfer polymerization with a 3-substituted heteroarene monomer (where E = element). M refers to metal, L refers to the ancillary ligand bound to the metal centre and B refers to the inorganic base. PR3 and NHC represent a tricoordinate phosphine and N-heterocyclic carbene, respectively. | ||
To date, the number of aromatic building blocks which can be polymerized via a chain-growth mechanism is limited.17–24 Herein, we highlight the opportunities and challenges of Suzuki–Miyaura coupling as a method to expand the choice of aromatic repeat units in CTP. Yokozawa reported the first Suzuki–Miyaura CTP (SM CTP) reaction with a 9,9-dialkylfluorene monomer in 2007,25 not long after the discovery of controlled polymerization of 3-alkylthiophenes using Kumada coupling.26–30 Since that time, SM CTP has been used to prepare alkyl and ester-functionalized polythiophenes, polyfluorenes, ester-functionalized polyfurans, poly(para-phenylenes), poly(meta-phenylenes), poly(ortho,para-alternating-phenylenes) poly (benzo[1,2,3]triazoles), poly(3,6-phenanthrenes) and poly(phenylene-vinylenes).31–59
In this perspective, we specifically highlight the combination of Suzuki–Miyaura coupling with controlled polymerization to prepare well-defined polyaromatics. To this end, we first compare Suzuki polymerization to direct arylation and Kumada polymerization. We then describe the typical methods used to prepare difunctional organoboron monomers for SM CTP. We briefly discuss the mechanism of this reaction, and how transmetalation differs from the more conventional Kumada CTP. Next, we highlight the versatility of the boron coupling partner and discuss the impact of protodeboronation. Finally, we showcase some of the catalysts that have been used in SM CTP to date.
Why use Suzuki–Miyaura coupling for chain-growth polymerization?
The Suzuki–Miyaura reaction has become ubiquitous in the organic chemist's toolbox. It is used to prepare organic molecules, therapeutics, and advanced materials. It involves cross-coupling of an aryl halide (or pseudo-halide) and an organoboron reagent to form a new C–C bond.60,61 The mild reaction conditions, non-toxic byproducts and the tolerance to pendant functional groups on the arene makes this method highly attractive for the polymerization of aromatics.Monomers bearing two different functional groups (e.g., H–Ar–Br or (HO)2B–Ar–Br) are required for chain-growth polymerization,17–24 and Table 1 highlights advantages and drawbacks of Suzuki–Miyaura coupling as compared to other polymerization approaches for this type of monomer. Deprotonative polymerization via Kumada coupling has been used extensively for the preparation of poly(3-alkylthiophenes).17–24 The active monomer is formed by deprotonation with lithium diisopropylamide (LDA) followed by magnesiation (Table 1),62,63 or by activation of a carbon–halogen bond with a Grignard reagent.27 The resultant Grignard monomer is polymerized rapidly in excellent yield, most often using a Ni catalyst. This method offers precise control over side chain orientation and molecular weight, but the nucleophilicity of the organomagnesium monomer limits the choice of aromatic ring and side group in this reaction.
|
|
||
|---|---|---|
| Suzuki–Miyaura polymerization | Deprotonative polymerization | Direct arylation polymerization |
| Advantages | Advantages | Advantages |
| 1. Functional group tolerance | 1. Highly reactive transmetalating group | 1. Functional group tolerance |
| 2. Mild rxn conditions | 2. Mild rxn conditions | 2. Atom economy |
| 3. Isolable monomer | 3. Chain-growth | 3. High Mw |
| 4. Chain-growth | ||
| Challenges | Challenges | Challenges |
| 1. Extra synthetic step | 1. Functional group tolerance | 1. Harsh conditions |
| 2. Atom economy | 2. Atom economy | 2. Achieving chain-growth |
| 3. Defects possible | ||
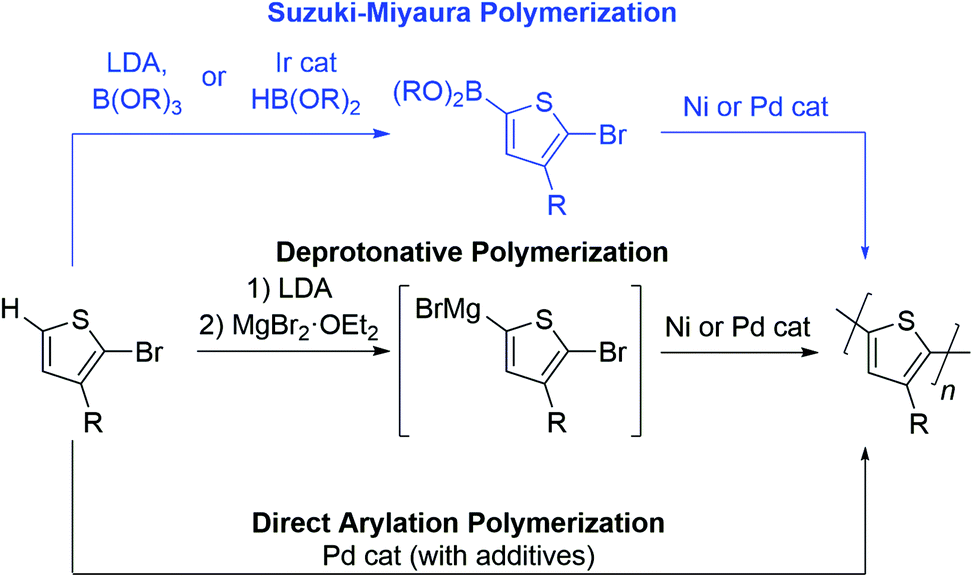
Suzuki–Miyaura polymerization has the same benefits as the Kumada reaction (controllable side group orientation and rapid reaction rates under basic conditions) but is more functional group tolerant than Kumada coupling. The reduced nucleophilicity of the organoboron makes Suzuki–Miyaura coupling an excellent alternative for polymerization of more sensitive substrates (e.g. with ester or amide side groups), though the additional synthetic step adds time and expense relative to the other two methods (Table 1). In addition to functional group tolerance, the boron group serves as a tool to tailor polymerization chemistry. More specifically, the –OR groups bound to the boron attenuate monomer reactivity and therefore can be used to change polymerization rate. As will be discussed below, masking group strategies can also enable precise feeding of monomers into polymerization to access block copolymers in one-pot. As such, the boron group is unique in that it can be changed to improve polymerization behaviour and access different polymer compositions.
Direct arylation polymerization (DArP), has rapidly become one of the most important methods to construct polyaromatics via coupling of a C–Br and C–H bond.12–15 This atom economical approach to synthesizing conjugated polymers can be optimized in most instances to afford high molecular weight materials and is compatible with a diverse set of monomers. While this method has many advantages, chain-growth polymerization is difficult due to the harsh reaction conditions required, and backbone defects can arise when multiple C–H sites are present on the monomer.12–15
Preparation of monomers with a halogen and boron group for SM CTP
The most common borylation strategies are highlighted in Fig. 2. Metalation of a C–H or C–X bond with LDA or isopropylmagnesium chloride (iPrMgCl) produces a metalated arene that can be quenched with a borate ester to afford the desired monomer (Fig. 2, Method A).64 This strategy is highly effective for the preparation of aryl boronic esters, but the nucleophilicity of the organometallic reagents can limit the choice of arene and R group, similar to Kumada polymerization.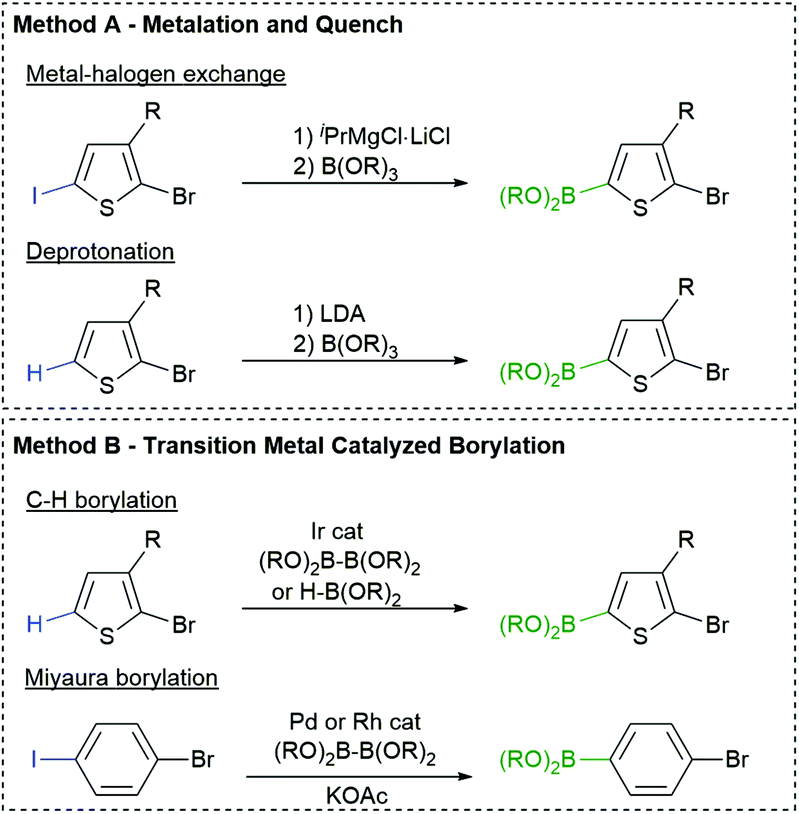 | ||
| Fig. 2 Common methods used to synthesize halogenated aryl boronic esters. Abbreviations: iPrMgCl = isopropylmagnesium chloride and KOAc = potassium acetate. Generic borate esters are represented as B(OR)3 and diboron reagents as (RO)2B–B(OR)2. | ||
Fortunately, aromatics can also be borylated using transition metals and boron reagents via C–H65 and C–X66–68 activation (Fig. 2, Method B). These strategies are operationally simple and highly functional group tolerant, making them attractive for monomer synthesis. Our group and others have utilized Ir borylation to prepare Suzuki-type monomers for polymerization.35,46,69 The iridium borylation is highly regioselective and tolerates chloro and bromo substituents on the arene70 or heteroarene.71
Miyaura borylation can also be used to borylate the C–I bond in bromoiodoarenes using bis(pinacolato)diboron (B2pin2) and potassium acetate (KOAc) (Fig. 2, Method B).72 Our group has recently expanded on this work with [Rh(COD)Cl]2 as the catalyst rather than Pd(dppf)Cl2. This is beneficial for the selective borylation of the C–I bond in heteroarenes (e.g. bromoiodothiophenes).73 The chemo- and regioselectivity of these transition metal catalysed borylation reactions is highly desirable for monomer synthesis.
Mechanistic considerations for SM CTP
A cross-coupling polymerization proceeds via oxidative addition, transmetalation, and reductive elimination to build the polymer chain (Fig. 1). Coordination of the M(0) catalyst to the π-system of the growing chain during polymerization has been proposed as the key intermediate which brings about a controlled polymerization.26–30 This proposed intermediate is consistent with NMR studies that identify a Ni π-complex as the first irreversible step in Kumada cross-coupling.74 The metal polymer π-complex, which forms after each reductive elimination event, promotes intramolecular oxidative addition of the halide end-group, such that the catalyst acts effectively as an initiator for the reaction (Fig. 1). Though the π-complex has not been directly observed during polymerization, studies have provided indirect support for this proposed intermediate.75–77CTP has some of the characteristics of a living polymerization including: linear increase of molecular weight with conversion, narrow molecular weight distributions, well-defined end-groups, and reactive chain-ends that can be used to synthesize block copolymers.17–24 Dissociation of the catalyst from the growing chain, catalyst disproportionation, or loss of ancillary ligand from the metal will result in deviations from ideal behaviour.17–24 The active catalyst in these reactions is typically a Ni or Pd complex with an electron-rich ancillary ligand (L) such as a phosphine, diphosphine, or N-heterocyclic carbene (Fig. 1). This metal species must promote efficient cross-coupling and formation of the π-complex to promote a chain-growth mechanism. Computational efforts have helped provide additional insight into the metal polymer π-complex.78–81 Further efforts with computation are likely to help predict optimal catalyst and monomer pairings for future exploration.
The key difference between Kumada and Suzuki–Miyaura cross-coupling is the transmetalation step. In Kumada coupling, no additives are needed to transfer the organic group to the metal catalyst. However, Suzuki–Miyaura coupling requires inorganic bases and water to promote effective transfer of the organic group to the metal. In THF with base and water present, the Suzuki–Miyaura reaction is typically biphasic.82 The aqueous phase acts as a reservoir for the inorganic base, with the coupling reaction proceeding in the organic phase. Importantly, transmetalation is proposed to be the rate-determining step in Suzuki–Miyaura reactions when oxidative addition is fast (e.g. with Ar–Br and Ar–I substrates).83 Our analysis of the rate-limiting step in Ni-catalyzed Suzuki–Miyaura CTP is consistent with this proposal, where the catalyst resting state is the LnM(Ar)Br complex (Fig. 1).41 Importantly, the rate-determining step may change depending on the choice of electrophile in the reaction (e.g. Ar–Cl).
The specific role of the base in transmetalation has been debated, with the two proposed pathways shown in Fig. 3. The oxo-metal pathway proceeds by ligand exchange of the halide for hydroxide in the oxidatively added catalyst. The Lewis acidic boron then coordinates to the M–OH to transfer the aryl group to the metal (Fig. 3, top). Alternatively, the boronate pathway proceeds by transmetalation of an anionic (hydroxy)borate with the metal halide bond (Fig. 3, bottom). The oxo-metal route has been identified as the kinetically favoured pathway in a number of mechanistic investigations.83–87 Denmark has also demonstrated that both boronic acids and esters can participate in the transmetalation step.86
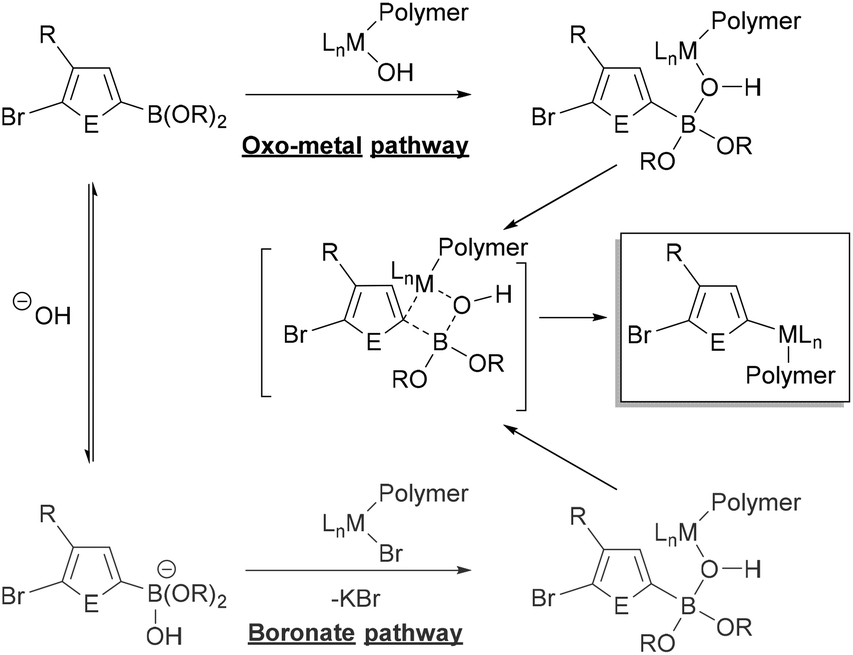 | ||
| Fig. 3 Simplified generic mechanism of transmetalation in Suzuki–Miyaura coupling via the “oxo-metal” and “boronate” pathways with the 3-substituted heteroarene monomer (E = element).82 Metal-hydroxo complexes can be dimeric,83–87 though not shown explicitly here. | ||
A variety of inorganic bases can promote transfer of the organic group to the metal. Phase-transfer agents such as 18-crown-6 have also been used in polymerization to aid in extraction of the base into the organic phase.47,50,59 The amount of base used in polymerizations can span a wide range (1–20 equiv.), which can impact initiation and polymerization rates.25,31–59 Amatore, Jutand and Le Duc demonstrated that the base (OH− or F−) plays three roles in Suzuki–Miyaura coupling.84,88 First, it enables formation of the M–OH or M–F to promote transmetalation via the oxo-metal pathway. Second, it facilitates reductive elimination through formation of a 5-coordinate metal intermediate. Finally, it also behaves as an antagonist as it retards catalytic turnover by conversion of the boronic ester to an anionic boronate. This anionic boronate is linked with monomer stability, another key consideration for polymerization which is discussed in a later section. Ultimately, the optimal ratios of base, water, and solvent will depend on the organoboron monomer and catalyst. These parameters should be optimized with novel monomers and catalysts.
Organoborons for SM CTP
A selection of boron derivatives have been used in SM CTP as shown at the top of Fig. 4, with boronic esters being the most common. One of the first reports describing Suzuki–Miyaura polymerization of an AB-type monomer appeared in 1998 where the thiophene monomer was functionalized with neopentyl boronic ester.124 Generally, Bpin functionalized thiophenes are used most often in Suzuki–Miyaura polymerization.125,126 Remarkably, choice of the OR groups on the boron centre can impact the rate of transmetalation. Denmark has noted that 4-fluorophenylboronic acid couples ∼4.5 times faster than the corresponding pinacol ester, and ∼4 times slower than its catechol counterpart.86 This result highlights how the boron group can potentially be used as a tool in polymerization to control monomer polymerization rates.86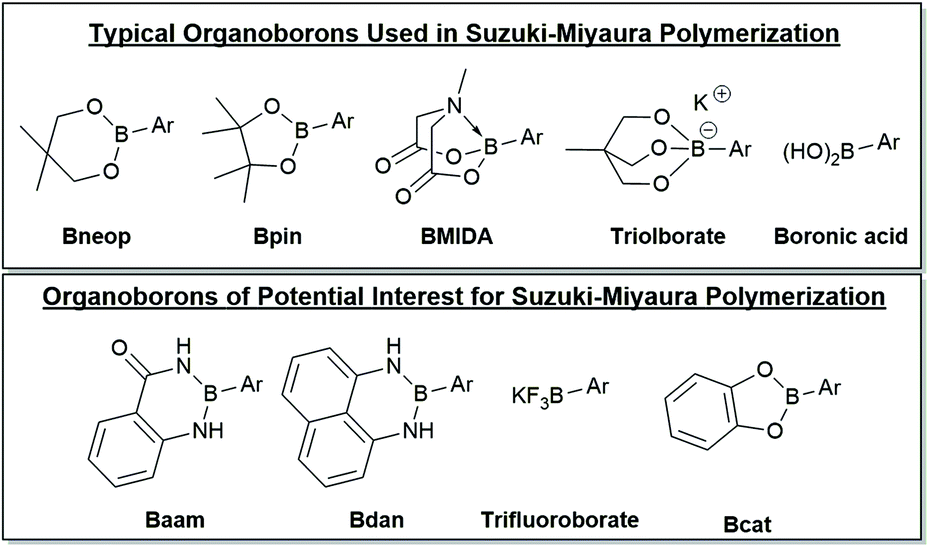 | ||
| Fig. 4 Selection of organoborons for cross-coupling. The top group of derivatives have been employed in Suzuki–Miyaura polymerization and the bottom group should be of interest for polymerization. Unsubstituted six-membered boronic esters have also been used as the coupling partner in SM CTP.51 | ||
Masking groups have also become important derivatives for Suzuki–Miyaura polymerization. A well-known class of masked borons are derived from N-methyliminodiacetic acid (BMIDA in Fig. 4).89 MIDA boronates are typically air-stable, crystalline solids making them highly attractive in synthesis.90 Though the MIDA boronate cannot be used directly in coupling due to the tetrahedral boron center, it is hydrolysed to the corresponding boronic acid under basic conditions, so it can still be used directly in Suzuki–Miyaura coupling (Fig. 5).89 This unmasking strategy was first utilized in polythiophene synthesis by Ingleson and coworkers with Mn values approaching 15 kg mol−1, and molecular weight distributions ranging from 2–3.5.91 Choi and coworkers then reported a controlled polymerization of a thiophene MIDA boronate with good control over Mn and dispersity (17.6 kg mol−1, Đ = 1.16).31,39 Notably, polymerizing a thienyl-Bpin monomer resulted in lower yields as compared to the MIDA monomer due to protodeboronation.31,39 Using the thienyl-MIDA monomer also narrowed dispersities and improved end-group fidelity, which was attributed to the lower concentration of active monomer for polymerization.31,39
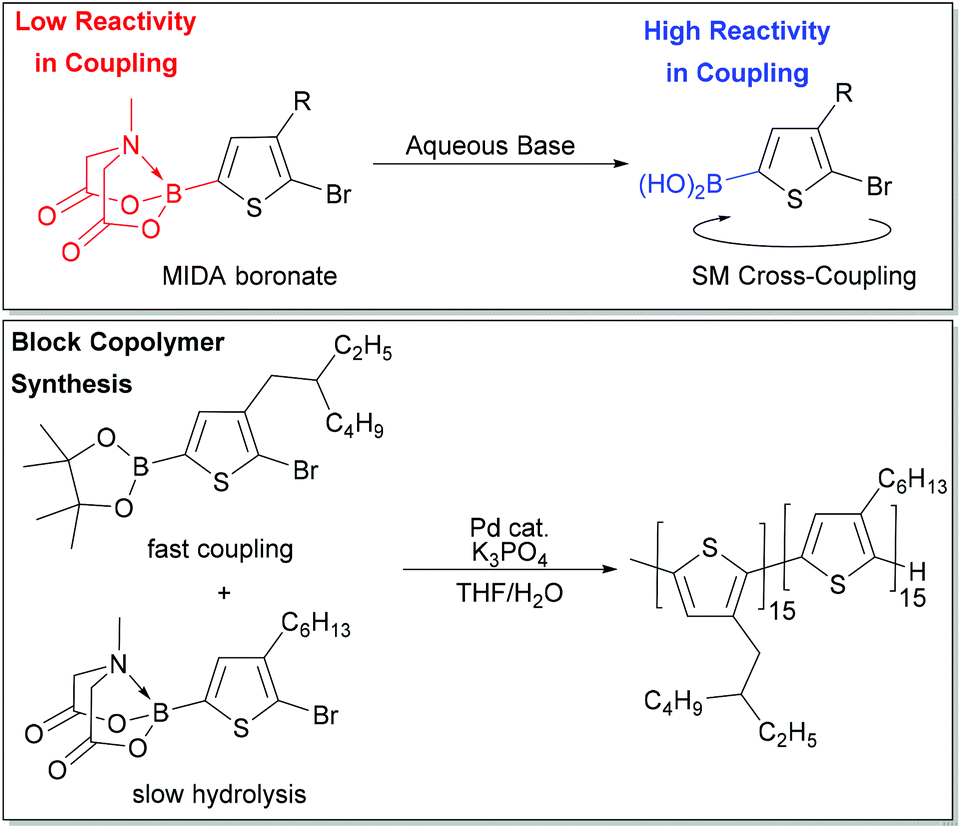 | ||
| Fig. 5 Top – Unmasking of the MIDA boronate in the presence of base to form the corresponding boronic acid. Bottom – One-pot synthesis of P3EHT-b-P3HT from two monomers which hydrolyse at different rates. | ||
Choi and coworkers also exploited reactivity differences between Bpin and BMIDA moieties to build a polythiophene block copolymer by combination of two monomers in one pot (Fig. 5). The Bpin monomer is consumed first due to its higher reactivity, while the BMIDA monomer hydrolyses slowly, and is consumed after the Bpin monomer. This experiment represents a unique entry to block copolymers for SM CTP, where monomer reactivity can be tuned by identity of the boron group. One-pot block copolymerization of more complex monomer scaffolds could be possible using this strategy.
Pre-activated anionic boronates such as the triolborate are bench stable boron complexes that are highly efficient coupling partners for Suzuki–Miyaura reactions (Fig. 4). These moieties can participate in coupling without base present in the reaction mixture,92 which is highly beneficial given the intricate role of hydroxide in most coupling reactions. SM CTP using triolborates has been reported for the synthesis of P3HT and polyfluorene.32,50 High molecular weight polyfluorenes were prepared and graft copolymers with polystyrene could be produced.32 Use of the triolborate monomer was beneficial to reduce the water loading during the graft copolymerization, as the polystyrene is not soluble in this solvent.32
Many other organoborons should be of interest for SM CTP.90 A few of the possibilities are shown in Fig. 4, though this is by no means exhaustive. Catechol-based boronic esters (Bcat) have not been used in polymerization, though these have been employed in small molecule cross-coupling.90 Diaminonaphthalene boronamides93,94 (Bdan) and anthranilamide borons (Baam)95,96 are both known as remarkably stable organoboron moieties. For the most part, these have only been used as protecting groups, though recent work has shown some of these derivatives can participate in coupling directly.97,98 The trifluoroborate salts99 (BF3K) have not yet been explored for chain-growth polymerizations, but these derivatives are typically compatible with environmentally friendly solvents such as water and alcohol. Pairing trifluoroborates with water-soluble conjugated polymers100 could be an exciting new strategy to build polyaromatics in environmentally benign media.
Protodeboronation in SM CTP
Monomer stability is an important consideration in Suzuki–Miyaura polymerization (Fig. 6). Protodeboronation, where a C–B bond is cleaved to form a C–H bond, can occur during coupling.101–104 This monomer deactivation proceeds from the hydroxyboronate as shown in Fig. 6. The identity of the aromatic ring, the substituents appended to the ring, and the substitution pattern of groups, all impact the rate of protodeboronation.101–104 To better illustrate this, one can compare the half-lives of a few aryl boronic acids.101,102 For example, 2-thienylboronic acid protodeboronates much faster (t1/2 < 2 h) than phenyl boronic acid (t1/2 ∼ 6 months) at a pH of 13 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O:1,4-dioxane at 70 °C. Substituents on the phenyl ring, particularly ortho-substituents, impact organoboron stability, as demonstrated by the 4-fluorophenylboronic acid, 2-fluorophenylboronic acid and 2,6-difluorophenylboronic acid half-lives of 4 months, 19 h, and 5 s respectively (at pH 13 in 1:1 H2O:dioxane at 70 °C).101
:1 H2O:1,4-dioxane at 70 °C. Substituents on the phenyl ring, particularly ortho-substituents, impact organoboron stability, as demonstrated by the 4-fluorophenylboronic acid, 2-fluorophenylboronic acid and 2,6-difluorophenylboronic acid half-lives of 4 months, 19 h, and 5 s respectively (at pH 13 in 1:1 H2O:dioxane at 70 °C).101
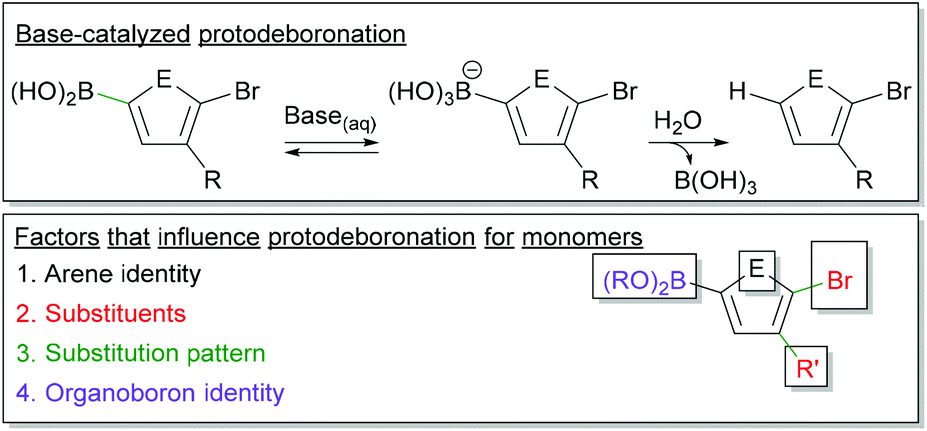 | ||
| Fig. 6 Top – Simplified mechanism for the base-catalysed protodeboronation. Bottom – Factors which impact protodeboronation of aryl boronic esters. | ||
Monomers for SM CTP are typically comprised of an aromatic ring bearing a boronic ester, a halide and at least one solubilizing group (Fig. 6). Heterocyclic rings are common building blocks for conjugated polymers, so protodeboronation will be a larger concern with these derivatives. The halogen, which is required for polymerization, will also accelerate this side reaction.101,102 More detailed studies are needed on protodeboronation of Suzuki-type monomers used in CTP, especially since these monomers often have boronic esters as opposed to boronic acids which will likely impact the rate of this side reaction. Empirically, we have noted adjusting base strength can help lower the rate of this side reaction, as isolated yields of ester-functionalized thiophenes and furans were increased when using CsF rather than K3PO4.35,46 Regardless, protodeboronation remains a challenging aspect of cross-coupling for unstable boronic acids and esters.101,102
SM CTP examples with palladium and nickel catalysts
For a chain-growth mechanism to be operative, a catalyst must promote efficient coupling as well as effective π-complexation to the growing polymer chain. As the electronic properties of conjugated polymers are influenced by the aromatic repeat unit (and by the choice of side-groups), the metal catalyst is critical to achieving a controlled polymerization. Fortunately, Suzuki–Miyaura coupling has been catalysed by nickel and palladium complexes, which ensures the steric/electronic parameters of the catalyst can be easily modulated.Precatalyst selection
Formation of the active M(0) catalyst for polymerization can be accomplished from a variety of M(II) precatalysts or by simply combining a M(0) precursor with the desired ligand. Precatalyst activation is critical in CTP, as this initiation event influences the molecular weight distribution of the final polymer.105,106 In the top of Fig. 7, simplified catalyst structures are shown where the metal, the ancillary ligand, and the reactive ligands are represented by M, L and X/X′ respectively. Palladacycle precatalysts that have been developed for small molecule coupling107,108 have also been used in SM CTP (Fig. 7).31,33,39,40,44,45 Recent advances with allyl or indenyl precatalysts may prove to be beneficial for SM CTP in the future.109–112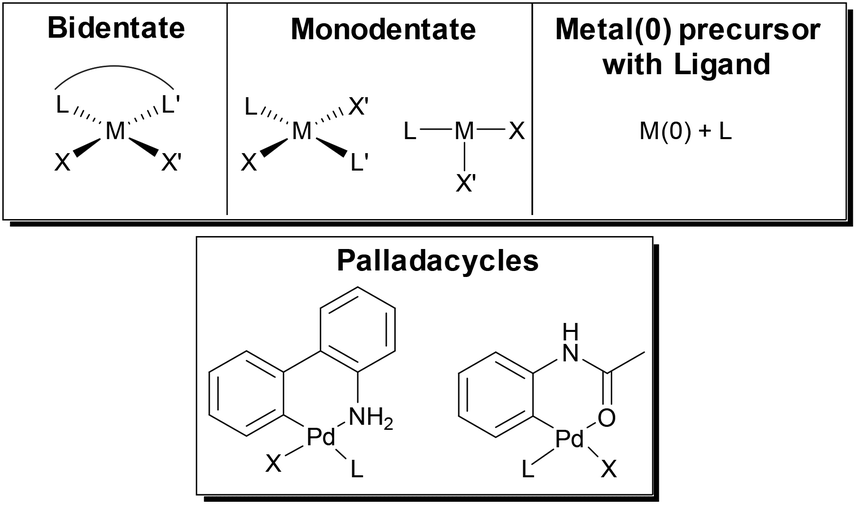 | ||
| Fig. 7 General scheme of precatalysts for Suzuki–Miyaura cross-coupling polymerization. The metal can be Ni or Pd, the ancillary ligand (L) can be a phosphine, diphosphine or N-heterocyclic carbene and the X/X′ groups can be acetates, halides, tosylate, mesylate or a combination of an aryl group and a halide. | ||
The externally initiated catalysts with a ligand, a halide and an arene (LM(Ar)Br) produce well-defined chain-ends and narrow molar mass distributions in SM CTP. These soluble catalysts are rapidly converted to the active M(0) catalyst as only one transmetalation and reductive elimination is required to enter the catalytic cycle.25 As such, these are the most common catalysts used in SM CTP. Interestingly, they are often targeted even from palladacycle precatalysts.31,33,39,40
Ancillary ligand selection
Another important feature to consider when choosing a catalyst for SM CTP is the ancillary ligand. The ligands that have been employed in SM CTP to date are illustrated in Fig. 8, along with the respective homopolymers synthesized. The figure is organized with ligands shown on top and homopolymers on bottom. Ligands have been paired with either Pd or Ni (which is noted), and Fig. 8 is colour coded to reference pairings of catalysts and polymers. Generally, electron rich monophosphines have been used in most SM CTP polymerizations, where the active catalyst is a 12 electron L-Pd(0) complex. Geng and coworkers also reported on an NHC-Pd for thiophene and fluorene polymerization (Pink Squares, Fig. 8).51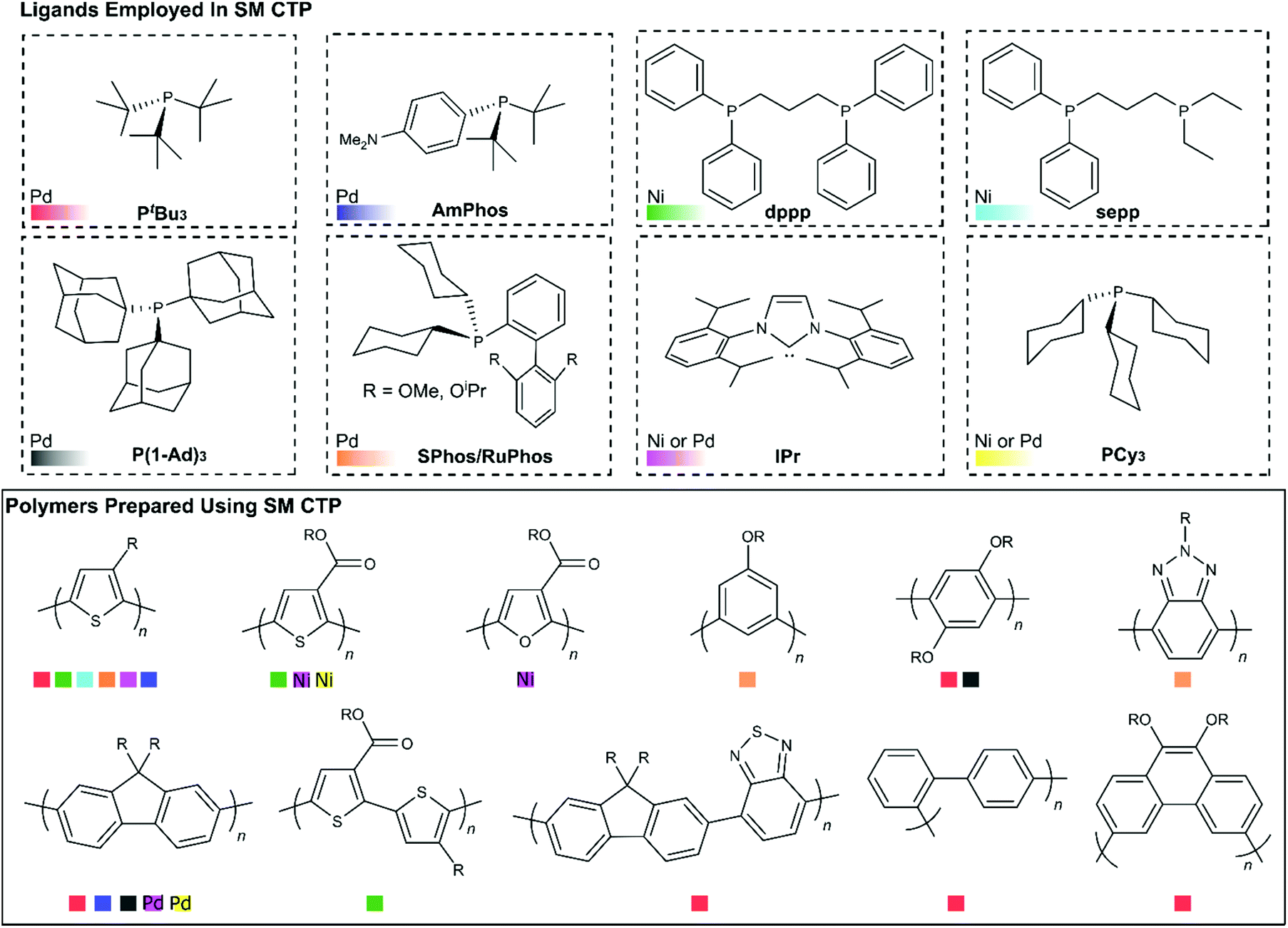 | ||
| Fig. 8 Top – Ligands used for Suzuki–Miyaura cross-coupling polymerization. Ligand abbreviations: PtBu3 = tri-tert-butylphosphine, P(1-Ad)3 = tri-1-adamantylphosphine, AmPhos = di-tert-butyl(4-dimethylaminophenyl)phosphine, SPhos = 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl, RuPhos = 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl, dppp = 1,3-Bis(diphenylphosphino)propane, IPr = 1,3-bis(2,6-diisopropylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene, sepp = 1,3-splitdiethyldiphenylphosphinopropane, and PCy3 = tricyclohexylphosphine. Bottom – Homopolymers prepared using SM CTP. For some of the polymers shown, chain-growth character was noted during polymerization, but may not have been classified as SM CTP. | ||
The most common catalyst system used in SM CTP is tBu3P-Pd (Red Squares, Fig. 8) with a range of precatalyst variations. This catalyst system has been successful for the controlled polymerization of polythiophenes, polyfluorenes, and various polyphenylenes.25,36–38,43,45,48–50,52–55,57–59 The success of tBu3P-Pd has led to investigations to prepare more exotic polymers such as poly(3,6-phenanthrene),54 poly(fluorene-alt-benzothiadiazole),58 and poly(pyridyl-alt-thiophene).113 For poly(fluorene-alt-benzothiadiazole), Huck and coworkers were able to bring about controlled chain-growth polymerization of this large monomer system, though molecular weights were modest (3–7.3 kg mol−1). For the phenanthrene and pyridyl-thiophene monomers, chain-growth was limited in both instances.54,113 Together, these studies highlight how monomer changes can impact controlled polymerization behaviour.
Hu and coworkers have recently demonstrated that the electron releasing bulky tri(1-adamantyl)phosphine (P(1-Ad)3) (Grey Squares, Fig. 8), can be combined with Pd to prepare polyfluorenes, poly(p-phenylenes) and poly(m-phenylenes) with modest molecular weights and narrow molecular weight distributions (Mn = 2.5–11.4 kg mol−1, Đ = 1.12–1.19).44 Yokozawa and coworkers discovered that AmPhos (Blue Squares, Fig. 8) can be employed to prepare polythiophenes and polyfluorenes.40 Interestingly, Yokozawa also noted that block copolymers of thiophene and fluorene could be prepared irrespective of which monomer was polymerized first. Triarylamine-based Pd complexes with PEt3, PCy3, and P(o-tolyl)3 ligands have also been used to prepare polyfluorenes using SM CTP (Yellow Squares, Fig. 8).47 The authors noted that the Cy3P-Pd and (o-tolyl)3P-Pd produced chain-growth behaviour to some extent.47
Recently, dialkylbiarylphosphine palladium catalysts have been explored in SM CTP (Orange Squares, Fig. 8). These catalysts are highly modular, with a rich history in Suzuki–Miyaura cross-coupling.114 The electron releasing alkyl groups on the phosphine ensure facile oxidative addition of aryl halides to Pd and the biaryl substituent promotes rapid reductive elimination around the metal center. Furthermore, the tunability of the alkyl groups and biaryl structure enables precise optimization of the steric and electronic parameters of the phosphine. In CTP, two dialkylbiaryl ligands have received considerable attention, namely SPhos and RuPhos.31,33,39,69,115,116 The first example of these ligands being used in SM CTP of 3-alkylthiophenes appeared in 2006 from Higgins.69 Though chain-growth polymerization was not the focus of that work, it is clear from the data obtained that the obtained P3HT was highly regioregular, with a narrow molecular weight distribution. Schluter also noted that the polymerization of m-phenylene organoboron monomers with an SPhosPd catalyst showed characteristics of a chain-growth polymerization.56
Following this work, Choi recently described an approach to synthesize poly(3-alkylthiophenes) via polymerization of MIDA boronates with externally initiated RuPhos and SPhos catalysts.31,39 The authors noted that additional phosphine in the reaction mixture relative to catalyst was key to achieving a controlled polymerization. In our own work, we have also noted the importance of additional equivalents of phosphine and diphosphine ligand in chain-growth polymerizations (with both Pd and Ni).33,34,41 Choi also noted that dialkylbiarylphosphines result in superior control as compared to PtBu3 in the polymerization of 3-alkylthiophenes.
Building from these reports, our group recently reported on the chain-growth polymerization of benzotriazole.33 SPhos, RuPhos, and DavePhos Pd catalysts were used to prepare polybenzotriazole with relatively narrow dispersities and high molecular weights under mild conditions.33 In that work, we also noted that the benzotriazole monomer, which bears a pinacol boronic ester, partially hydrolysed during polymerization. A polybenzotriazole-poly(3-hexylthiophene) block copolymer could be synthesized by sequential addition of the benzotriazole and thiophene monomers. Block copolymerization of electronically distinct monomers is challenging,117 and speaks to the potential of dialkylbiarylphosphines for SM CTP.
Beyond Pd catalysts, Ni-mediated polymerizations are also of interest due to the low cost, relative abundance and exceptional reactivity of nickel catalysts for Suzuki-cross coupling.118–121 Our group has shown NHC-Ni complexes (Light Purple Squares, Fig. 8) are highly effective for alkyl- and ester-functionalized thiophenes.34,41,46 We have also noted that diphosphine catalysts can also be highly effective in 3-alkylthiophene polymerizations (Green/Light Blue Squares, Fig. 8) with fast reaction times and high molecular weights.34,41,46
In our work, we discovered how critical additional water was for fast turnover in Ni-catalysed SM CTP, and how water can lead to hydrolysis and deactivation of dihalide precatalysts.41 Fortunately, the hydrolysis event was serendipitous, as it revealed the importance of added ligand in the chain-growth polymerization of thiophenes with diphosphine ligands. With both Ni(dppp) and Ni(sepp) catalysts, additional ligand in the reaction mixture was necessary to achieve good end-group fidelity and narrow dispersities in the polymerization of 3-alkylthiophene. Further analysis into the precise role of the additional ligand is needed, as additional ligand has been shown to be beneficial in palladium reactions as well.31,33,40
Future opportunities & challenges
Though the chain-growth polymerization of thiophene was reported over 15 years ago, synthesis of conjugated polymers by CTP is still centered on the polymerization of monomers such as thiophene and fluorene. The limited choice of aromatic monomers for chain-growth polymerization is, in part, a consequence of the limitations of conventional synthetic methods to make difunctional monomers. Organolithium and organomagnesium reagents are still common strategies to install boryl groups on arenes, but these approaches are not always compatible with complex arene scaffolds. The extensive exploration of arene borylation with transition metals should serve as inspiration to make novel organoboron monomers for CTP.65–68The different borylation methods can offer monomers bearing two different functional groups with a wide range of conjugated frameworks, and perhaps enable preparation of well-defined polymers from ring systems that have been used in conjugated polymer devices (Fig. 9). As novel boron monomers are evaluated in CTP, an improved understanding of the detrimental effects of protodeboronation are needed. Monitoring loss of the boron group should be possible using NMR techniques, and it is anticipated this kind of information can be used to inform better practices for monomer design. Moreover, an improved understanding of the transmetalation step in SM CTP with different organoborons would be highly valuable for the community. Understanding how monomers transmetalate, and how different organoborons impact the rate, would provide valuable information to enable further improvements in polymerization.
 | ||
| Fig. 9 Common building blocks in conjugated polymers that have rarely been explored in CTP. | ||
One general challenge in the controlled synthesis of conjugated polymers is achieving high-molecular weight materials. Most SM CTP examples have a degree of polymerization (DP) less than 100, presumably due to a combination of issues with catalyst stability, undesirable side reactions, and polymer solubility. Realization of low dispersity, high-molecular weight polymers could be particularly relevant with respect to mechanical properties of aromatic polymers.122 For example, Schluter noted that poly(m-phenylenes) of sufficient molecular weight are high Tg polymers with outstanding toughness and good processability.123 Schluter also illustrated the importance of catalyst choice and polymerization conditions to improve the molecular weight of poly(m-phenylenes) preparaed using SM CTP.56 Goodson and coworkers also demonstrated how the choice of organoboron and palladium catalyst are key to achieving high molecular weights in step-growth polycondensation reactions.127
Despite the higher complexity of SM CTP relative to Kumada-Corriu CTP, it remains one of the most popular and versatile cross-coupling reactions. Deeper understanding of the interplay between aromatic building block, organoboron moiety, and choice of catalyst are likely to lead to exciting advances in this field. Further insights into transmetalation and role of additives will pave the way to more diverse well-defined polymers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the NSF (CHE-1455136) for supporting their exploration of new synthetic methods in conjugated polymer synthesis.Notes and references
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Frechet, Adv. Mater., 2003, 15, 1519–1522 CrossRef CAS.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu, J. M. J. Frechet and M. F. Toney, Macromolecules, 2005, 38, 3312–3319 CrossRef CAS.
- P. Kohn, S. Huettner, H. Komber, V. Senkovskyy, R. Tkachov, A. Kiriy, R. H. Friend, U. Steiner, W. T. S. Huck, J. U. Sommer and M. Sommer, J. Am. Chem. Soc., 2012, 134, 4790–4805 CrossRef CAS.
- K. Gu and Y. L. Loo, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1559–1571 CrossRef CAS.
- B. Kuei and E. D. Gomez, Soft Matter, 2017, 13, 49–67 RSC.
- R. Zhang, B. Li, M. C. Iovu, M. Jeffries-El, G. Sauvé, J. Cooper, S. Jia, S. Tristram-Nagle, D. M. Smilgies, D. N. Lambeth, R. D. McCullough and T. Kowalewski, J. Am. Chem. Soc., 2006, 128, 3480–3481 CrossRef CAS.
- F. P. V. Koch, J. Rivnay, S. Foster, C. Müller, J. M. Downing, E. Buchaca-Domingo, P. Westacott, L. Yu, M. Yuan, M. Baklar, Z. Fei, C. Luscombe, M. A. McLachlan, M. Heeney, G. Rumbles, C. Silva, A. Salleo, J. Nelson, P. Smith and N. Stingelin, Prog. Polym. Sci., 2013, 38, 1978–1989 CrossRef CAS.
- P. Sista and C. K. Luscombe, Progress in the Synthesis of Poly (3-hexylthiophene), in P3HT Revisited – From Molecular Scale to Solar Cell Devices, ed. S. Ludwigs, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 1–38. DOI:10.1007/12_2014_278.
- J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schlüter, Macromol. Rapid Commun., 2009, 30, 653–687 CrossRef CAS.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS.
- M. Wakioka and F. Ozawa, Asian J. Org. Chem., 2018, 7, 1206–1216 CrossRef CAS.
- N. S. Gobalasingham and B. C. Thompson, Prog. Polym. Sci., 2018, 83, 135–201 CrossRef CAS.
- J. R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS.
- T. Bura, J. T. Blaskovits and M. Leclerc, J. Am. Chem. Soc., 2016, 138, 10056–10071 CrossRef CAS.
- A. E. Rudenko and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 135–147 CrossRef CAS.
- P. O. Morin, T. Bura and M. Leclerc, Mater. Horiz., 2016, 3, 11–20 RSC.
- M. A. Baker, C. H. Tsai and K. J. T. Noonan, Chem. – Eur. J., 2018, 24, 13078–13088 CrossRef CAS.
- L. Verheyen, P. Leysen, M.-P. Van Den Eede, W. Ceunen, T. Hardeman and G. Koeckelberghs, Polymer, 2017, 108, 521–546 CrossRef CAS.
- T. Yokozawa and Y. Ohta, Chem. Rev., 2016, 116, 1950–1968 CrossRef CAS.
- A. K. Leone and A. J. McNeil, Acc. Chem. Res., 2016, 49, 2822–2831 CrossRef CAS.
- R. Grisorio and G. P. Suranna, Polym. Chem., 2015, 6, 7781–7795 RSC.
- Z. J. Bryan and A. J. McNeil, Macromolecules, 2013, 46, 8395–8405 CrossRef CAS.
- K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424–2434 RSC.
- T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595–5619 CrossRef CAS.
- A. Yokoyama, H. Suzuki, Y. Kubota, K. Ohuchi, H. Higashimura and T. Yokozawa, J. Am. Chem. Soc., 2007, 129, 7236–7237 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542–17547 CrossRef CAS.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649–8656 CrossRef CAS.
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS.
- E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, Macromol. Rapid Commun., 2004, 25, 1663–1666 CrossRef CAS.
- J. Lee, H. Park, S.-H. Hwang, I.-H. Lee and T.-L. Choi, Macromolecules, 2020, 53, 3306–3314 CrossRef CAS.
- S. Kobayashi, K. Fujiwara, D.-H. Jiang, T. Yamamoto, K. Tajima, Y. Yamamoto, T. Isono and T. Satoh, Polym. Chem., 2020, 11, 6832–6839 RSC.
- M. V. Bautista, A. J. Varni, J. Ayuso-Carrillo, C.-H. Tsai and K. J. T. Noonan, ACS Macro Lett., 2020, 9, 1357–1362 CrossRef CAS.
- M. A. Baker, J. Ayuso-Carrillo, M. R. M. Koos, S. N. MacMillan, A. J. Varni, R. R. Gil and K. J. T. Noonan, Polym. J., 2020, 52, 83–92 CrossRef CAS.
- A. J. Varni, A. Fortney, M. A. Baker, J. C. Worch, Y. Qiu, D. Yaron, S. Bernhard, K. J. T. Noonan and T. Kowalewski, J. Am. Chem. Soc., 2019, 141, 8858–8867 CrossRef CAS.
- M. Nojima, T. Kamigawara, Y. Ohta and T. Yokozawa, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 297–304 CrossRef CAS.
- S. Huber and S. Mecking, Macromolecules, 2019, 52, 5917–5924 CrossRef CAS.
- H.-H. Zhang, Y.-X. Zhu, W. Wang, J. Zhu, P. V. Bonnesen and K. Hong, Polym. Chem., 2018, 9, 3342–3346 RSC.
- K.-B. Seo, I.-H. Lee, J. Lee, I. Choi and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 4335–4343 CrossRef CAS.
- K. Kosaka, T. Uchida, K. Mikami, Y. Ohta and T. Yokozawa, Macromolecules, 2018, 51, 364–369 CrossRef CAS.
- M. A. Baker, S. F. Zahn, A. J. Varni, C. H. Tsai and K. J. T. Noonan, Macromolecules, 2018, 51, 5911–5917 CrossRef CAS.
- T. Hardeman and G. Koeckelberghs, Polym. Chem., 2017, 8, 3999–4004 RSC.
- R. Grisorio and G. P. Suranna, ACS Macro Lett., 2017, 6, 1251–1256 CrossRef CAS.
- J. Dong, H. Guo and Q.-S. Hu, ACS Macro Lett., 2017, 6, 1301–1304 CrossRef CAS.
- H.-H. Zhang, W. Peng, J. Dong and Q.-S. Hu, ACS Macro Lett., 2016, 5, 656–660 CrossRef CAS.
- Y. Qiu, J. C. Worch, A. Fortney, C. Gayathri, R. R. Gil and K. J. T. Noonan, Macromolecules, 2016, 49, 4757–4762 CrossRef CAS.
- Z. Zhang, P. Hu, X. Li, H. Zhan and Y. Cheng, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1457–1463 CrossRef CAS.
- H.-H. Zhang, C.-H. Xing, Q.-S. Hu and K. Hong, Macromolecules, 2015, 48, 967–978 CrossRef CAS.
- H.-H. Zhang, Q.-S. Hu and K. Hong, Chem. Commun., 2015, 51, 14869–14872 RSC.
- K. Kosaka, Y. Ohta and T. Yokozawa, Macromol. Rapid Commun., 2015, 36, 373–377 CrossRef CAS.
- A. Sui, X. Shi, H. Tian, Y. Geng and F. Wang, Polym. Chem., 2014, 5, 7072–7080 RSC.
- M. Nojima, Y. Ohta and T. Yokozawa, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2643–2653 CrossRef CAS.
- R. Grisorio, P. Mastrorilli and G. P. Suranna, Polym. Chem., 2014, 5, 4304–4310 RSC.
- M. Verswyvel, C. Hoebers, J. De Winter, P. Gerbaux and G. Koeckelberghs, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5067–5074 CrossRef CAS.
- H.-H. Zhang, C.-H. Xing and Q.-S. Hu, J. Am. Chem. Soc., 2012, 134, 13156–13159 CrossRef CAS.
- B. Hohl, L. Bertschi, X. Zhang, A. D. Schluter and J. Sakamoto, Macromolecules, 2012, 45, 5418–5426 CrossRef CAS.
- T. Yokozawa, R. Suzuki, M. Nojima, Y. Ohta and A. Yokoyama, Macromol. Rapid Commun., 2011, 32, 801–806 CrossRef CAS.
- E. Elmalem, A. Kiriy and W. T. S. Huck, Macromolecules, 2011, 44, 9057–9061 CrossRef CAS.
- T. Yokozawa, H. Kohno, Y. Ohta and A. Yokoyama, Macromolecules, 2010, 43, 7095–7100 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- N. Miyaura, Metal-Catalyzed Cross-Coupling Reactions of Organoboron Compounds with Organic Halides, in Metal–Catalyzed Cross–Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, Germany, 2nd edn, 2004, vol. 1, pp. 41–123 Search PubMed.
- R. D. McCullough, R. D. Lowe, M. Jayaraman and D. L. Anderson, J. Org. Chem., 1993, 58, 904–912 CrossRef CAS.
- S. Tamba, K. Shono, A. Sugie and A. Mori, J. Am. Chem. Soc., 2011, 133, 9700–9703 CrossRef CAS.
- D. G. Hall, Boronic acids: Preparations and applications in organic synthesis, medicine and materials, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2nd edn, 2011 Search PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS.
- K. Kubota, H. Iwamoto and H. Ito, Org. Biomol. Chem., 2017, 15, 285–300 RSC.
- N. Primas, A. Bouillon and S. Rault, Tetrahedron, 2010, 66, 8121–8136 CrossRef CAS.
- T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 3, 271–280 CrossRef CAS.
- I. A. Liversedge, S. J. Higgins, M. Giles, M. Heeney and I. McCulloch, Tetrahedron Lett., 2006, 47, 5143–5146 CrossRef CAS.
- T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS.
- G. A. Chotana, V. A. Kallepalli, R. E. Maleczka and M. R. Smith, Tetrahedron, 2008, 64, 6103–6114 CrossRef CAS.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS.
- A. J. Varni, M. V. Bautista and K. J. T. Noonan, J. Org. Chem., 2020, 85, 6770–6777 CrossRef CAS.
- N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 15258–15259 CrossRef CAS.
- Z. J. Bryan and A. J. McNeil, Chem. Sci., 2013, 4, 1620–1624 RSC.
- H. Komber, V. Senkovskyy, R. Tkachov, K. Johnson, A. Kiriy, W. T. S. Huck and M. Sommer, Macromolecules, 2011, 44, 9164–9172 CrossRef CAS.
- T. Beryozkina, V. Senkovskyy, E. Kaul and A. Kiriy, Macromolecules, 2008, 41, 7817–7823 CrossRef CAS.
- C. R. Bridges, T. M. McCormick, G. L. Gibson, J. Hollinger and D. S. Seferos, J. Am. Chem. Soc., 2013, 135, 13212–13219 CrossRef CAS.
- A. A. Pollit, S. Ye and D. S. Seferos, Macromolecules, 2020, 53, 138–148 CrossRef CAS.
- J. A. Bilbrey, A. N. Bootsma, M. A. Bartlett, J. Locklin, S. E. Wheeler and W. D. Allen, J. Chem. Theory Comput., 2017, 13, 1706–1711 CrossRef CAS.
- K. Mikami, M. Nojima, Y. Masumoto, Y. Mizukoshi, R. Takita, T. Yokozawa and M. Uchiyama, Polym. Chem., 2017, 8, 1708–1713 RSC.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS.
- B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS.
- C. Amatore, A. Jutand and G. Le Duc, Chem. – Eur. J., 2011, 17, 2492–2503 CrossRef CAS.
- A. F. Schmidt, A. A. Kurokhtina and E. V. Larina, Russ. J. Gen. Chem., 2011, 81, 1573 CrossRef CAS.
- A. A. Thomas, A. F. Zahrt, C. P. Delaney and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 4401–4416 CrossRef CAS.
- A. A. Thomas and S. E. Denmark, Science, 2016, 352, 329–332 CrossRef CAS.
- C. Amatore, A. Jutand and G. Le Duc, Angew. Chem., Int. Ed., 2012, 51, 1379–1382 CrossRef CAS.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- J. Ayuso Carrillo, M. J. Ingleson and M. L. Turner, Macromolecules, 2015, 48, 979–986 CrossRef.
- Y. Yamamoto, M. Takizawa, X.-Q. Yu and N. Miyaura, Angew. Chem., Int. Ed., 2008, 47, 928–931 CrossRef CAS.
- H. Noguchi, T. Shioda, C. M. Chou and M. Suginome, Org. Lett., 2008, 10, 377–380 CrossRef CAS.
- H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758–759 CrossRef CAS.
- M. Koyanagi, N. Eichenauer, H. Ihara, T. Yamamoto and M. Suginome, Chem. Lett., 2013, 42, 541–543 CrossRef CAS.
- H. Ihara, M. Koyanagi and M. Suginome, Org. Lett., 2011, 13, 2662–2665 CrossRef CAS.
- H. Yoshida, M. Seki, S. Kamio, H. Tanaka, Y. Izumi, J. Li, I. Osaka, M. Abe, H. Andoh, T. Yajima, T. Tani and T. Tsuchimoto, ACS Catal., 2020, 10, 346–351 CrossRef CAS.
- Y. Mutoh, K. Yamamoto and S. Saito, ACS Catal., 2020, 10, 352–357 CrossRef CAS.
- G. A. Molander, J. Org. Chem., 2015, 80, 7837–7848 CrossRef CAS.
- J. Liu and B. Liu, Strategies to Bring Conjugated Polymers into Aqueous Media, in Conjugated Polymers for Biological and Biomedical Applications, ed. B. Liu, Wiley-VCH, Weinheim, Germany, 2018, pp. 1–33. DOI:10.1002/9783527342747.ch1.
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS.
- P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS.
- H. G. Kuivila, J. F. Reuwer and J. A. Mangravite, Can. J. Chem., 1963, 41, 3081–3090 CrossRef CAS.
- J. Lozada, Z. Liu and D. M. Perrin, J. Org. Chem., 2014, 79, 5365–5368 CrossRef CAS.
- S. R. Lee, Z. J. Bryan, A. M. Wagner and A. J. McNeil, Chem. Sci., 2012, 3, 1562–1566 RSC.
- S. R. Lee, J. W. G. Bloom, S. E. Wheeler and A. J. McNeil, Dalton Trans., 2013, 42, 4218–4222 RSC.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC.
- L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392–6395 CrossRef CAS.
- P. R. Melvin, A. Nova, D. Balcells, W. Dai, N. Hazari, D. P. Hruszkewycz, H. P. Shah and M. T. Tudge, ACS Catal., 2015, 5, 3680–3688 CrossRef CAS.
- A. J. DeAngelis, P. G. Gildner, R. Chow and T. J. Colacot, J. Org. Chem., 2015, 80, 6794–6813 CrossRef CAS.
- L. L. Hill, J. L. Crowell, S. L. Tutwiler, N. L. Massie, C. C. Hines, S. T. Griffin, R. D. Rogers, K. H. Shaughnessy, G. A. Grasa, C. C. C. Johansson Seechurn, H. Li, T. J. Colacot, J. Chou and C. J. Woltermann, J. Org. Chem., 2010, 75, 6477–6488 CrossRef CAS.
- M. S. Viciu, R. F. Germaneau, O. Navarro-Fernandez, E. D. Stevens and S. P. Nolan, Organometallics, 2002, 21, 5470–5472 CrossRef CAS.
- Y. Tokita, M. Katoh, Y. Ohta and T. Yokozawa, Chem. – Eur. J., 2016, 22, 17436–17444 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS.
- M. Verswyvel, J. Steverlynck, S. H. Mohamed, M. Trabelsi, B. Champagne and G. Koeckelberghs, Macromolecules, 2014, 47, 4668–4675 CrossRef CAS.
- M. Verswyvel, P. Verstappen, L. De Cremer, T. Verbiest and G. Koeckelberghs, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5339–5349 CrossRef CAS.
- C. R. Bridges, H. Yan, A. A. Pollit and D. S. Seferos, ACS Macro Lett., 2014, 3, 671–674 CrossRef CAS.
- S. Saito, M. Sakai and N. Miyaura, Tetrahedron Lett., 1996, 37, 2993–2996 CrossRef CAS.
- K. Inada and N. Miyaura, Tetrahedron, 2000, 56, 8657–8660 CrossRef CAS.
- M. Ueda, A. Saitoh, S. Oh-tani and N. Miyaura, Tetrahedron, 1998, 54, 13079–13086 CrossRef CAS.
- S. Saito, S. Oh-tani and N. Miyaura, J. Org. Chem., 1997, 62, 8024–8030 CrossRef CAS.
- D. Pei, Z. Wang, Z. Peng, J. Zhang, Y. Deng, Y. Han, L. Ye and Y. Geng, Macromolecules, 2020, 53, 4490–4500 CrossRef CAS.
- R. Kandre, K. Feldman, H. E. H. Meijer, P. Smith and A. D. Schluter, Angew. Chem., Int. Ed., 2007, 46, 4956–4959 CrossRef CAS.
- S. Guillerez and G. Bidan, Synth. Met., 1998, 93(2), 123–126 CrossRef CAS.
- M. Jayakannan, X. Lou, J. L. J. Van Dongen and R. A. J. Janssen, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1454–1462 CrossRef CAS.
- W. Li, Y. Han, B. Li, C. Liu and Z. Bo, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4556–4563 CrossRef CAS.
- J. Murage, J. W. Eddy, J. R. Zimbalist, T. B. McIntyre, Z. R. Wagner and F. E. Goodson, Macromolecules, 2008, 41, 7330–7338 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |