DOI:
10.1039/D0PY00793E
(Paper)
Polym. Chem., 2021,
12, 113-121
Nano-assemblies with core-forming hydrophobic polypeptide via polymerization-induced self-assembly (PISA)†
Received
2nd June 2020
, Accepted 18th August 2020
First published on 24th August 2020
Abstract
The aim of this study is to produce self-assembled structures with hydrophobic polypeptide cores via Reversible Addition–Fragmentation chain Transfer (RAFT) – mediated Polymerisation-Induced Self-Assembly (PISA). Hydrophilic poly(glycerol monomethacrylate) macromolecular chain transfer agents (PGMA mCTAs) were used to polymerize the self-assembling peptide monomers, resulting in the formation of diblock copolymer nano objects. Methacrylamide derivatives containing self-assembling tripeptides MAm-GFF (MAm-Gly-Phe-Phe-NH2) and MAm-FGD (MAm-Phe-Gly-Asp-NH2) were used as hydrophobic monomers. The self-assembling behaviours of these monomers mainly derive from the interactions of the phenylalanine residues, however their difference in hydrophobicity required different polymerization conditions. MAm-GFF was polymerized in the presence of organic solvent (ethanol or acetonitrile), under either dispersion or emulsion polymerization, while MAm-FGD was polymerized under aqueous dispersion conditions. PGMA-b-P(MAm-FGD) obtained from aqueous PISA typically formed fibrous structures while a range of morphologies such as fibre-, flake-, and leaf-like or spherical vesicles were obtained for PGMA-b-P(MAm-GFF) depending on the copolymer composition and solvent used. In all cases the peptides self-assembling core had a crucial influence on the final morphologies.
Introduction
Self-assembled nanostructures based on polypeptide-polymer hybrids are interesting biomaterials with potential significant impacts in applied biology and medicine. Compared to pure polymeric nanoparticles, these structures could present increased biocompatibility and biofunctionality typical of the peptide sequences,1–3 in addition to the robustness and functional diversity conferred by the polymer chains. Moreover, the range of self-assembled peptide-containing structures is more diverse than that of polymer nano-objects.4–6 They can form more complex supramolecular assemblies such as nano-fibers, ribbons, tapes, tubes, wires or rods.6–15 In particular, since the molecular self-organization of these structures is mainly attributed to weak interactions such as electrostatic forces, hydrophobic interactions, π–π stacking and hydrogen bonding,16 these structures can largely vary in both shape and size depending on internal or external stimuli (pH, temperature, ionic strength, solvents…).17–19 The diversity and versatility of peptide-based nanostructures allow them to be potentially used in different biomedical fields such as biosensors, tissue engineering, or drug delivery.4,20–23
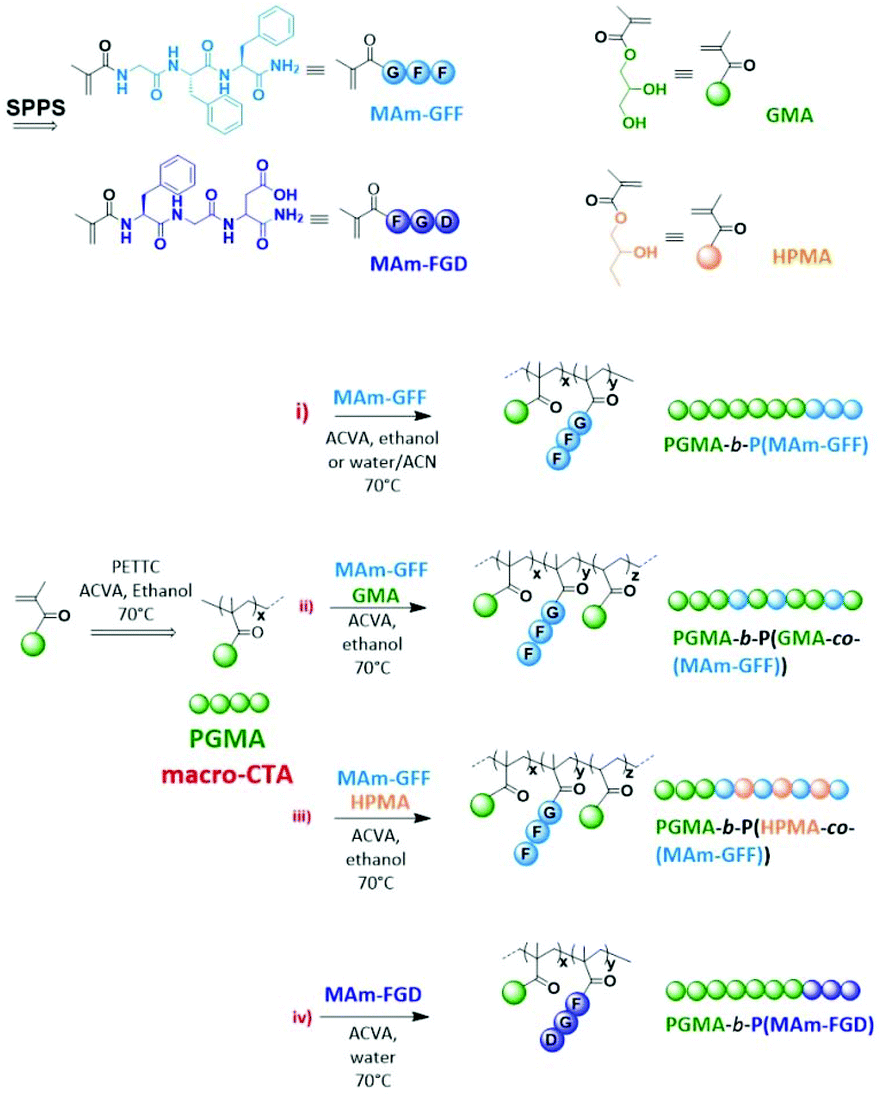
Besides the conventional self-assembly strategies using purified and isolated amphiphilic block copolymers, polymerization-induced self-assembly (PISA) has been shown to be a powerful polymer self-assembly method over the last decade and as such has received considerable interest.24,25 PISA strategy is fast, eliminating time-consuming steps such as isolation/purification and redispersion of the amphiphilic polymers since it proceeds via the simultaneous formation and self-assembly of the amphiphilic block copolymers.26 While the conventional post-polymerization methods provide dilute suspensions of nanoparticles (ca. 1% w/w), PISA generates nanoparticles at much higher concentrations (up to 50% w/w as reported in recent studies).27,28 Moreover, PISA works well under either dispersion29–33 or emulsion polymerization conditions34–37 and in a wide range of solvents.38–45 Although the use of PISA is drastically growing due to its advantages, most reports show relatively simple self-assembled morphologies (spheres, worms and vesicles).25 Inspired by the properties of peptide-based nanostructures as well as PISA approach, we have recently reported the first example of PISA nano-objects derived from self-assembling peptide (SAP) containing polymer.46 The SAP monomer used in this study (MAm-GFF), was a methacrylamide-functionalized GFF tripeptide (GFF = Glycine-Phenylalanine-Phenylalanine). The tripeptide moieties were incorporated along with glycerol monomethacrylate (GMA) in the stabilizing block which was successfully chain extended under PISA conditions with poly(2-hydroxypropyl methacrylate) (PHPMA). The resulting P(GMA65-stat-(MAm-GFF)7)-b-PHPMA28 diblock copolymer formed fibrous structures in water. Herein, the same peptide methacrylamide was used but as precursor of the core-forming block. PISA formulations with core-forming block composed of copolymer of this SAP methacrylamide and a solvophilic monomer (GMA or HPMA) were also examined. Moreover, a new tripeptide-containing methacrylamide, (MAm-FGD) based on the phenylalanine-glycine-aspartic acid SAP sequence was also studied. As for MAm-GFF, the self-assembly property of MAm-FGD is also attributed to the phenylalanine residue. However MAm-FGD is slightly less hydrophobic than MAm-GFF at physiological pH, and thus allows the use of different PISA conditions. All the synthetic approaches of this work are summarized in Scheme 1.
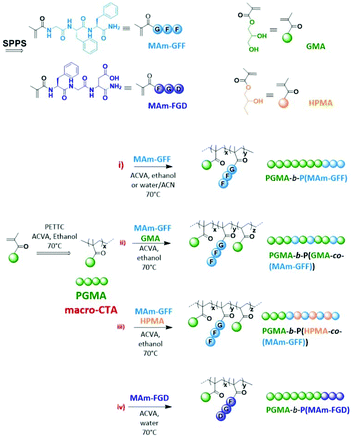 |
| | Scheme 1 Schematic representation of the synthesis of the monomers and block copolymers. | |
Experimental section
Materials
Fmoc-amino acid derivatives (Fmoc-Phe-OH, Fmoc-Gly-OH, Fmoc-Asp(tBu)-OH), HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate) and polystyrene Fmoc Rink Amide resins (100–200 mesh) with a loading of 0.94 mmol g−1 were purchased from Iris Biotech GmbH. Methacrylic acid, piperidine, N,N-diisopropylethylamine (DIEA), trifluoroacetic acid (TFA), triisopropylsilane (TIS), glycerol monomethacrylate (GMA), 2-hydroxypropyl methacrylate (HPMA), 4,4′-azobis(4-cyanopentanoic acid) (ACVA) and all organic solvents were purchased from Sigma-Aldrich. The NMR deuterated solvents (DMSO-d6, methanol-d4 and D2O) were purchased from Eurisotop. The RAFT agent (4-cyano-4-(2-phenylethanesulfanylthiocarbonyl)-sulfanylpentanoic acid) (PETTC) and the peptide monomer MAm-GFF was prepared according to our previous report.46
Synthesis of methacrylamide-functionalized peptide monomer MAm-FGD
MAm-FGD was synthesized on the resin using the same protocol employed to make MAm-GFF in our previous report46 but with Fmoc-Phe-OH, Fmoc-Gly-OH and Fmoc-Asp(But)-OH as the amino acids. At the end, the tripeptide was cleaved from the resin with a TFA/TIS/H2O (95/2.5/2.5) mixture for 2 h at room temperature. It was recovered by precipitation in cold diethyl ether, taken up in water and freeze-dried. The obtained crude white powder was used for polymerisations without any further purification. Its purity and molecular structure were verified by LC-MS (Fig. S2†) and 1H NMR spectroscopy (Fig. S1†). 1H NMR in DMSO δ (ppm): 1.85 (s, 3H, –CH3); 2.66–2.73 (m, 2H, –CH–CH2–Ph); 2.90–3.10 (dd, 2H, –CH–CH2–COOH); 3.70 (m, 2H, –NH–CH2CO–) 4.41 (m, 2H, –NH–CHCO–CH2–); 5.38 (s, 1H, vinyl); 5.71 (s, 1H, vinyl); 7.10–7.28 (m, 5H, phenyl); 8.08–8.20 (m, 3H, –NH–).
RAFT homopolymerization of GMA
A representative synthesis of the PGMA macromolecular chain transfer agents (mCTAs) proceeded as follows: a round bottom Schlenk flask equipped with a stirrer bar was charged with GMA (30 eq.), PETTC (1 eq.), ACVA initiator (0.1 eq.) and a volume of ethanol adjusted for the reaction to proceed at a solids content of 50% w/w. The flask was subsequently sealed with a rubber septum, placed in an ice-bath, degassed by bubbling nitrogen for 30 min and immersed in a preheated oil bath at 70 °C. After 2 h, the polymerization was quenched by immersing the flask in ice-cold water and opening the reaction to air. GMA conversion was determined by 1H NMR in methanol-d4, from the ratio of the integral of the residual vinyl 1H signals at 5.65 ppm (1H) or 6.15 ppm (1H) to the signals at 3.5–4.3 (5H). The mixture reaction was then dialyzed using a MWCO = 1 kDa membrane against a 9/1 mixture of water and ethanol during two days and freeze-dried. The final product was a light yellow powder. The mean DP was determined by 1H NMR and the molar mass distribution was determined using DMAc SEC. All the 1H NMR spectra and SEC chromatograms of the PGMA mCTAs are provided in Fig. S3–S6.†
Preparation of diblock PGMAx-b-P(MAm-GFF)yvia RAFT-PISA using PGMAx as mCTA
PGMAx mCTA (1 eq.), MAm-GFF (y eq.), and ACVA (0.2 eq.) were charged into a 10 mL glass vial and stirred with the chosen polymerization solvent. The vial was sealed, submerged in an ice-bath, degassed for 30 minutes and put into an oil-bath set previously at 70 °C. After 24 h, reaction was cooled down, opened to the air and the MAm-GFF conversion was determined by 1H NMR (comparison of the integral of the residual vinyl signals at 5.42 (1H) or 5.85 (1H) to that of the phenyl group at 7.2–7.4 (10H)). Other aliquots were also taken for SEC, DLS and TEM analyses.
Preparation of diblock PGMA18-b-P((MAm-GFF)9-co-GMA48) via RAFT-PISA using PGMA18 as mCTA
The procedure to prepare PGMA18-b-P((MAm-GFF)9-co-GMA48) using PGMA18 as mCTA was as follows: PGMA18 mCTA (1 eq.), MA-GFF (10 eq.) GMA (48 eq.) and ACVA (0.2 eq.) were stirred with pure ethanol into a 10 mL glass vial. The vial was subsequently sealed with a rubber septum, submerged in an ice-bath, degassed by bubbling nitrogen for 30 min and immersed in a preheated oil bath at 70 °C. After 24 h, the reaction was quenched by cooling and exposing to air. MAm-GFF conversion was determined as described above while GMA conversion was always 100% (no remaining GMA-vinyl signals). Other aliquots were also taken for SEC, DLS and TEM analyses.
Preparation of diblock PGMA18-b-P((MAm-GFF)9-co-HPMA61) and PGMA18-b-P((MAm-GFF)19-co-HPMA50) via RAFT-PISA using PGMA18 as mCTA
The procedures were as follow: PGMA18 mCTA, MA-GFF, HPMA and ACVA at ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:61:0.2 (to prepare PGMA18-b-P((MAm-GFF)9-co-HPMA61) and 1:20:50:0.2 (to prepare PGMA18-b-P((MAm-GFF)19-co-HPMA50)) were stirred with ethanol into a 10 mL glass vial. The vial was subsequently sealed with a rubber septum, submerged in an ice-bath, degassed by bubbling nitrogen for 30 min and immersed in a preheated oil bath at 70 °C. After 24 h, the reaction was quenched by exposing to air. MAm-GFF conversion was determined as same as described above while HPMA conversion was always 100% (no remaining HPMA-vinyl signals). Other aliquots were also taken for SEC, DLS and TEM analyses.
:10:61:0.2 (to prepare PGMA18-b-P((MAm-GFF)9-co-HPMA61) and 1:20:50:0.2 (to prepare PGMA18-b-P((MAm-GFF)19-co-HPMA50)) were stirred with ethanol into a 10 mL glass vial. The vial was subsequently sealed with a rubber septum, submerged in an ice-bath, degassed by bubbling nitrogen for 30 min and immersed in a preheated oil bath at 70 °C. After 24 h, the reaction was quenched by exposing to air. MAm-GFF conversion was determined as same as described above while HPMA conversion was always 100% (no remaining HPMA-vinyl signals). Other aliquots were also taken for SEC, DLS and TEM analyses.
Preparation of diblock PGMAx-b-P(MAm-FGD)yvia RAFT-PISA using PGMAx as mCTA
The typical procedure of RAFT-PISA to prepare diblock PGMAx-b-P(MAm-FGD)y using PGMAx as mCTA was carried out as described below. PGMAx mCTA (1 eq.), MAm-FGD (y eq.), ACVA (0.2 eq.) and water (to reach 10% w/w of solids content) were charged into a 10 mL glass vial. The vial was sealed, degassed for 30 minutes and put into an oil-bath set previously at 70 °C. After 24 h, reaction was cooled down, opened to the air and the MAm-FGD conversion was determined by 1H NMR (comparison of its residual vinyl signals at 5.42 (1H) or 5.85 (1H) to the phenyl group at 7.2–7.4 (5H)). Other aliquots were also taken for SEC, DLS and TEM analyses.
Characterization
Liquid chromatography – mass spectrometry (LC-MS).
LC-MS analyses were performed on Waters Alliance 2695 HPLC system, coupled to a Water Micromass ZQ spectrometer (electrospray ionization mode, ESI+). The peptide monomer MAm-GFF was dissolved in a minimum amount of DMF and diluted with acetonitrile/water (50/50 v/v) mixture containing 0.1% TFA. UV detection was performed at 214 nm.
Proton nuclear magnetic resonance spectroscopy (1H NMR).
1H NMR spectra were recorded on a 300 MHz Bruker Avance-300 spectrometer, processed and analyzed with MestReNova 9.0.
Size exclusion chromatography (SEC).
Polymer molar mass distributions were analysed using GPC Varian 390-LC system with PL1113-6300 ResiPore 300 × 7.5 columns connected with 390LC PL0390-0601 refractive index detector (RI). The mobile phase was DMAc with 0.1% w/w LiBr adjusted at a flow rate of 1 mL min−1 while the columns were thermostated at 70 °C. The calibration was done with near-monodisperse poly(methyl methacrylate) (PMMA) standards ranging from 550 to 1568000 g mol−1 (EasiVial-Agilent).
Dynamic light scattering (DLS).
The hydrodynamic radii RH were analysed by dynamic light scattering at 90° using Litesizer TM 500 Anton Paar. All measurements were performed at 20 °C and with samples diluted to 0.1% w/w.
Transmission electron microscopy (TEM).
TEM images were acquired using either JEOL 1200 EXII-120 kV or JEOL 1400 P+ – 120 kV. To prepare the TEM grid, 10 μL of 100-fold diluted sample from PISA suspension (0.1% w/w) was deposited onto the grid for 60 s and then blotted with filter paper to remove excess solution. Afterward, the sample-loaded grid was stained with 7 μL of 1% ammonium molybdate solution for 20 s and also removed the excess stain with filter paper. The grid was allowed to dry under the hood for 5 minutes.
To prepare the TEM grids at 70 °C, the same protocol was followed but glass Pasteur pipettes pre-warmed at 70 °C was used to quickly load one drop of sample incubated at 70 °C onto a grid that was kept in oven chamber set at 70 °C during the whole procedure.
Results and discussion
PGMA macromolecular chain transfer agents (PGMA mCTAs)
The homopolymerizations of GMA were performed in ethanol under solution polymerization conditions. Although almost full conversion could be reached in 5 h as shown in previous studies,47 in this work, the polymerizations of GMA were stopped at about 60% conversion to ensure good end-group fidelity. The DP of the resulting PGMA mCTAs (18, 40 and 83 for mCTAs 1, 2, and 3 respectively, Fig. S3–5†) were determined by 1H NMR using the integrals of the backbone signals of PGMA (0.8–2 ppm, 5H) and of those of the PETTC end-group (7.2–7.4 ppm, 5H). The PETTC RAFT agent efficiency was estimated to be 90%, 85% and 78% for mCTAs 1, 2 and 3 respectively. All three mCTAs had narrow molar mass distributions (Đ = 1.16, 1.21 and 1.27) as judged by SEC in DMAc. SEC traces of these PGMA mCTAs are shown in Fig. S6.†
PISA using the GFF-functionalized methacrylamide (MAm-GFF)
The polymerizations of MAm-GFF using PGMA mCTAs to prepare self-assembled structures with P(MAm-GFF) as core-forming block were performed in different media containing organic solvents since MAm-GFF is only sparingly soluble in pure water. Common good solvents for hydrophobic peptides are DMF, DMSO, acetonitrile and ethanol. DMF and DMSO could not be used here, as they are both good solvent for P(MAm-GFF). Direct visual observation suggested that in ethanol, MAm-GFF was soluble up to 0.4% w/w at ambient temperature (ca. 20 °C) and up to 5% w/w at 70 °C. At higher concentrations (7%–10% w/w), it formed emulsion at both 20 and 70 °C. Furthermore, a mixture of water/acetonitrile was also chosen to study as the reaction medium. It was found that in water/acetonitrile 1/2 v/v mixture, MAm-GFF could be dispersed up to 10% w/w at both 20 °C and 70 °C, whereas in water/acetonitrile 1/1 v/v mixture, a 10% w/w MAm-GFF formed an emulsion at 20 °C and a solution at 70 °C. TEM images of MAm-GFF-derived structures prepared in the above media at low concentration (0.1% w/w) are presented in Fig. 1.
 |
| | Fig. 1 Representative TEM images of MAm-GFF-derived structures prepared from solutions at c = 0.1% w/w in: water/acetonitrile 1/2 v/v (left); water/acetonitrile 1/1 v/v (centre); and ethanol (right). | |
In the water/acetonitrile 1/2 v/v mixture, only ill-defined aggregates were observed (Fig. 1, left), suggesting that MAm-GFF was completely solubilized. However, in mixture of water/acetonitrile 1/1 v/v or in ethanol, MAm-GFF self-assembled into relatively well-defined vesicular morphologies, (Fig. 1, centre) and bundles of short fibres (Fig. 1, right). Note that this bundling is to a large extent the effect of drying. As discussed already in our previous work,46 the self-assembly of MAm-GFF is attributed to the combination of hydrogen bonding and aromatic π-stacking interactions between the FF moieties. The final morphologies thus are not only determined by MAm-GFF molecular structure but also by the solvent quality (polarity and hydrogen bonding properties). This was indeed illustrated here: changing the solvent from ethanol to water/acetonitrile 1/1 v/v mixture provides two types of distinct structures: fibres and vesicles.
These preliminary results on the solution properties of MAm-GFF allowed its use in PISA formulation as the core-forming building block (Scheme 1).
PISA in ethanol
Solids content effect.
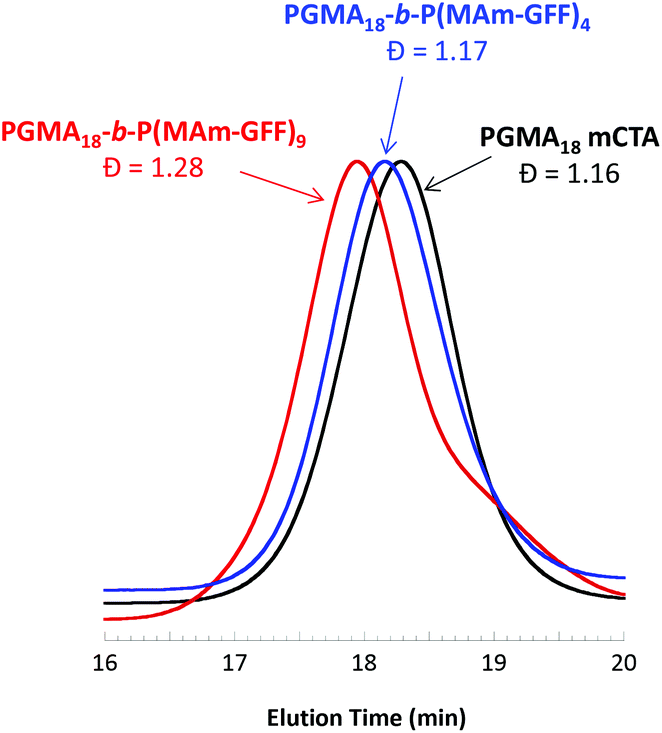
A series of polymerisation in ethanol using mCTA 1 (PGMA18) was carried out with an initial ratio of 1:MAm-GFF:ACVA of 1:5:0.2, for 24 h at 70 °C, at various solids contents (Table S1†). At 5% w/w, the reaction medium was visually a clear solution while above 10% w/w, the reaction mixture was turbid. Surprisingly, only 45% MAm-GFF conversion could be attained at 5% w/w solids whereas at 11.2% w/w solids, MAm-GFF conversion reached 87%. This trend was confirmed by a third experiment carried out at 12.5% w/w solids (targeting a DP of P(MAm-GFF) of 10) which reached 92% MAm-GFF conversion. The DMAc SEC traces (Fig. 2) of the resulting PGMA18-b-P(MAm-GFF)x diblock copolymers confirmed the relative good control of the RAFT polymerizations. However, a significant shoulder can be seen on the trace of the PGMA18-b-P(MAm-GFF)9 block copolymer (Fig. 2, red trace). The origin of this shoulder is unclear but it is thought to be an artefact since it does not appear in any other GPC traces apart from that of PGMA40-b-P(MAm-GFF)15 (Fig. S12,† blue trace). Mn and Đ values are given in Table S1.†
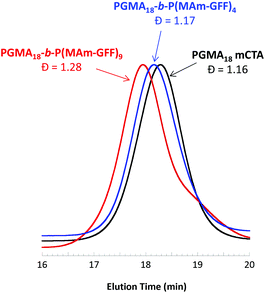 |
| | Fig. 2 DMAc SEC chromatograms (refractive index detector) for mCTA 1 (PGMA18) and the diblock copolymers PGMA18-b-P(MAm-GFF)4 and PGMA18-b-P(MAm-GFF)9 prepared via RAFT-mediated PISA at 70 °C in ethanol at 11.2% w/w and 12.5% w/w respectively. | |
In all cases, at 70 °C, the final reaction mixtures were turbid yellow suspensions. However, upon cooling, both suspensions were unstable. They became more turbid and yellow solids were observed on the wall of the vial at ambient temperature, even under continuous stirring, as shown in Fig. S7.† However, after dilution (100-fold) of the suspensions at 70 °C for TEM and DLS analyses, the samples remained stable at 20 °C. TEM images of the PGMA18-b-P(MAm-GFF)4 diblock copolymer revealed the formation of flake-like objects about 50 nm wide and 100–200 nm long. In comparison, the PGMA18-b-P(MAm-GFF)9 diblock copolymer formed micrometric branched bundles of fibres (Fig. 3). TEM samples were prepared at 70 °C to ensure that the morphologies were not affected by the change of temperature. Indeed no significant difference was observed between the samples prepared at 20 °C (Fig. 3) and 70 °C (Fig. S8†). In agreement with TEM results, DLS experiments indicated the presence of nano-objects in PGMA18-b-P(MAm-GFF)4 suspension with narrow size distribution (PDI = 0.187). Meanwhile, the DLS histogram of PGMA18-b-P(MAm-GFF)9 showed the existence of large structures at the micrometric scale. The size distribution in this case was broad, probably due to its highly anisotropic morphology as seen with TEM. DLS data are presented in Fig. S9.† The significant change in both size and shape between the two diblocks (PGMA18-b-P(MAm-GFF)4 and PGMA18-b-P(MAm-GFF)9) indicated that the length of the hydrophobic SAP block P(MAm-GFF) effectively determines the final morphology of the PGMA-b-P(MAm-GFF) block copolymer. This result confirms the interplay and synergy between SAP and PISA in the formation of these morphologies.
 |
| | Fig. 3 Representative TEM images at different magnification of the PGMA18-b-P(MAm-GFF)4 (top row) and PGMA18-b-P(MAm-GFF)9 (bottom row) diblock copolymers morphologies prepared by RAFT-PISA in ethanol at 11.2% and 12.5% solids content respectively. Samples were diluted 100-fold at 70 °C. | |
Effect of the mCTA length
To overcome the instability, observed at 20 °C for the previous diblock copolymer morphologies, stabilized by a relatively short PGMA18, longer hydrophilic mCTAs: PGMA40 (2) and PGMA83 (3) were used. All the PISA formulations were carried out at 12% w/w solids in ethanol. The polymerization led to yellow turbid suspensions after 24 h. In these polymerizations, MAm-GFF conversions reached 90% or higher as judged by 1HNMR and SEC analyses. However, the resulting diblock copolymers (PGMA40-b-P(MAm-GFF)8, PGMA83-b-P(MAm-GFF)10 and PGMA83-b-P(MAm-GFF)6) suspensions again lost their colloidal stability upon cooling from 70 °C to 20 °C. Increasing the length of the PGMA block did not improve the stability of PGMA-b-P(MAm-GFF) self-assembled objects. Here, the balance between the hydrophobic and hydrophilic parts in the PGMA-b-P(MAm-GFF) system was not the dominant element in the self-assembly in contrast to the case of PGMA-b-PHPMA which have been thoroughly described.48 The morphologies observed here were likely more affected by the self-assembling properties of the polypeptide part.
The diblock copolymer morphologies were observed by TEM using 100-fold diluted samples. The dilution was carried out at 70 °C. Well-defined morphologies were observed for all PISA formulations as shown in Fig. 4. In all cases, despite the composition difference, the self-assembled morphologies were large (several μm) dendritic structures similar to the structures observed for PGMA18-b-P(MAm-GFF)9, and composed of fibrous elements (Fig. 4). These fibrous substructures were better defined for the diblocks prepared from the longer PGMA83 mCTA. Increasing the length of the P(MAm-GFF) block from 6 to 10 for these PGMA83-derived block copolymers led to even better defined fibrous substructures (Fig. 4). When PGMA40 was used as the stabilizing block, these substructures lost much of their fibre-like aspect.
 |
| | Fig. 4 Representative TEM images of the PGMA40-b-P(MAm-GFF)8, PGMA83-b-P(MAm-GFF)6 and PGMA83-b-P(MAm-GFF)10 diblock copolymers morphologies prepared RAFT PISA in emulsion in ethanol at 12% solids. The samples were diluted to 0.1% w/w at 70 °C. | |
Effect of the composition of the core-forming block
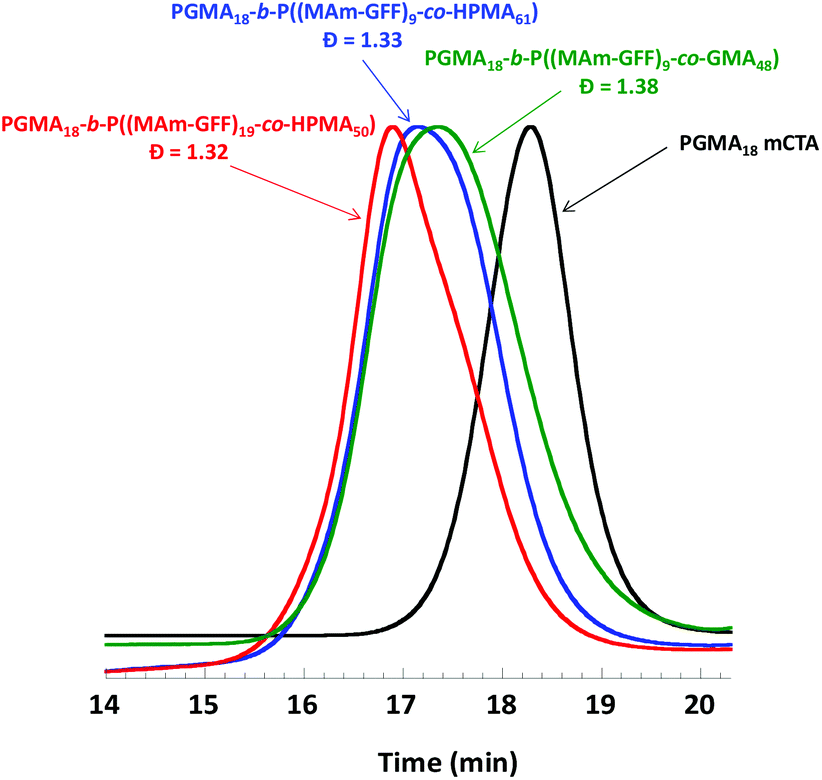
The effect of the composition of the solvophobic block was also examined by replacing the P(MAm-GFF) segment by a copolymer of HPMA (or GMA) and MAm-GFF. GMA and HPMA are highly soluble in ethanol. Three PISA formulations based on mCTA 1 were examined: 1:GMA:MAm-GFF = 1:48:10, 1:HPMA:MAm-GFF = 1:61:10, and 1:HPMA:MAm-GFF = 1:50:20. The addition of GMA or HPMA increased the solubility of MAm-GFF in the reaction medium, and thus the PISA could be carried out at higher solids contents (15% w/w) which led to higher monomer conversions. The SEC chromatograms of the resulting diblock copolymers shown in Fig. 5 suggest that the PISA copolymerizations proceeded with relatively good control (the trace are monomodal and shifted towards higher molar masses). However, these chromatograms were also slightly broad suggesting a subpar blocking efficiency.
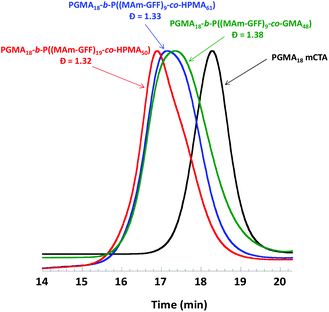 |
| | Fig. 5 DMAc SEC chromatograms (refractive index detector) for mCTA 1 (PGMA18) and the three diblock copolymers prepared by RAFT PISA in emulsion in ethanol. | |
As in the case of the previous formulations described above, the suspension of PGMA18-b-P((MAm-GFF)9-co-GMA48) diblock copolymer was turbid and stable at 70 °C but rapidly lost colloidal stability in a few minutes after cooling down (Fig. S10†). TEM images (Fig. S11†) of the sample diluted at 70 °C showed large clusters of fibrous structures. In contrast, the PGMA18-b-P((MAm-GFF)9-co-HPMA61) and PGMA18-b-P((MAm-GFF)19-co-HPMA50) diblock copolymers suspensions were colloidally stable at both 70 °C and 20 °C. The TEM images of these morphologies (samples prepared at 20 °c) show that PGMA18-b-P((MAm-GFF)19-co-HPMA50) self-assembled into vesicles while PGMA18-b-P((MA-GFF)9-co-HPMA61) self-assembled into a mixture of short worms and vesicles (Fig. 6). It is worth noticing here that these vesicles are undoubtedly the results of the self-assembling properties of MAm-GFF since PGMA-b-PHPMA block copolymers are completely soluble in ethanol.
 |
| | Fig. 6 Representative TEM images of the PGMA18-b-P((MAm-GFF)9-co-HPMA61) diblock copolymer morphology (top row) and of the PGMA18-b-P((MAm-GFF)19-co-HPMA50) diblock copolymer morphology (bottom row) prepared by RAFT PISA in dispersion in ethanol at 15% w/w solids. The samples were diluted to 0.15% w/w at 20 °C. | |
Solvent effect
The effect of the solvent on the PISA of PGMA-b-P(MAm-GFF) diblock copolymer was investigated, and ethanol was replaced by water/acetonitrile mixtures. The following PISA formulation based on mCTA 2 was examined: 2:MAm-GFF:ACVA = 1:20:0.2 in 1/1 v/v and 1/2 v/v water/acetonitrile mixtures. The polymerizations were carried out at 10 w/w solids under RAFT dispersion polymerization conditions at 70 °C. The MAm-GFF conversions reached 69% and 72% in 1/1 v/v and 1/2 v/v water/acetonitrile mixtures respectively. The successful formation of the diblock copolymers by chain extension of mCTA 2 with P(MAm-GFF) was confirmed by SEC in DMAc as shown in Fig. S12.† The resulting diblock copolymer morphologies were well-dispersed, giving rather clear colloidal suspensions that remained colloidaly stable even upon cooling to 20 °C. TEM images (Fig. 7) showed that PGMA40-b-(MAm-GFF)15 formed monodisperse 220 nm spherical particles in water/acetonitrile 1/1 v/v. In water/acetonitrile 1/2 v/v the PISA of PGMA40-b-(MAm-GFF)15 led to polydisperse vesicles with diameters ranging between 50 and 400 nmn (Fig. 7). In contrast to the PISA experiments carried out in ethanol which resulted in unstable dispersions and fibrous structures, no fibrous structures were observed for PGMA-b-P(MAm-GFF) in the two water/acetonitrile binary mixtures examined. Nevertheless, these experiments cast some light on the colloidal instability observed in ethanol. In ethanol the fibres formed at 70 °C readily aggregated upon cooling regardless of the length of the PGMA steric stabilizer, but upon dilution, the self-assembled objects recovered colloidal stability. In contrast in water/acetonitrile binary mixtures, similar diblock copolymers formed spherical colloidally stable self-assembled objects. These phenomena are likely ascribed to the GFF-GFF interactions (H-bonding, pi–pi stacking) which are much stronger at 20 °C than at 70 °C. The fibre/ribbon morphologies observed in ethanol are also much more likely to allow such core–core interactions, at the edge of the ribbon/platelet, than the spherical aggregates formed in water/acetonitrile binary mixture. The recovery of colloidal stability triggered by dilution is consistent with this interpretation. The water/acetonitrile binary mixtures tested here were also much better solvents of MA-GFF than ethanol and prevented such GFF–GFF interactions responsible of the fibrous structures in ethanol (Fig. 1). Finally, the difference in colloidal stabilities observed in ethanol for the PGMA18-b-P((MAm-GFF)9-co-GMA48) and for the PGMA18-b-P((MAm-GFF)9-co-HPMAx) (x = 50 or 61) diblock copolymers morphologies is likely caused by a difference of solubility of MA-GFF in the two reaction media. MA-GFF is most probably insoluble in ethanol/GMA mixtures and soluble in ethanol/HPMA mixtures.
 |
| | Fig. 7 Representative TEM images of the PGMA40-b-P(MAm-GFF)15 diblock copolymer morphology in 1/1 v/v (top row) and (1/2 v/v) (bottom row) water/acetonitrile mixtures prepared by RAFT PISA in dispersion at 10% w/w solids at 70 °C. The samples were diluted to 0.1% w/w. | |
PISA using GFD-functionalized methacrylamide (MAm-FGD)
In order to perform PISA under aqueous dispersion conditions, a new peptide-functionalized methacrylamide, MAm-FGD, was designed. This monomer, reported for its self-assembling properties, similar to MAm-GFF contains a phenylalanine residue, however it is less hydrophobic as it also contains a polar aspartic acid unit instead of a second phenylalanine moiety. Indeed, MAm-FGD was soluble in water up to 10% w/w. The TEM images (Fig. 8) of the MAm-FGD aqueous solution at 0.1% w/w revealed the presence of long (several μm) shard–like structures with width in the 100–200 nm range that sometimes aggregated into bundles. These TEM images confirmed that a single phenylalanine unit can also drive self-assembly and lead to the formation of fibrous structures, as demonstrated by Gazit et al.49
 |
| | Fig. 8 Representative TEM images of self-assembled structures of MAm-FGD in water at 0.1% w/w. | |
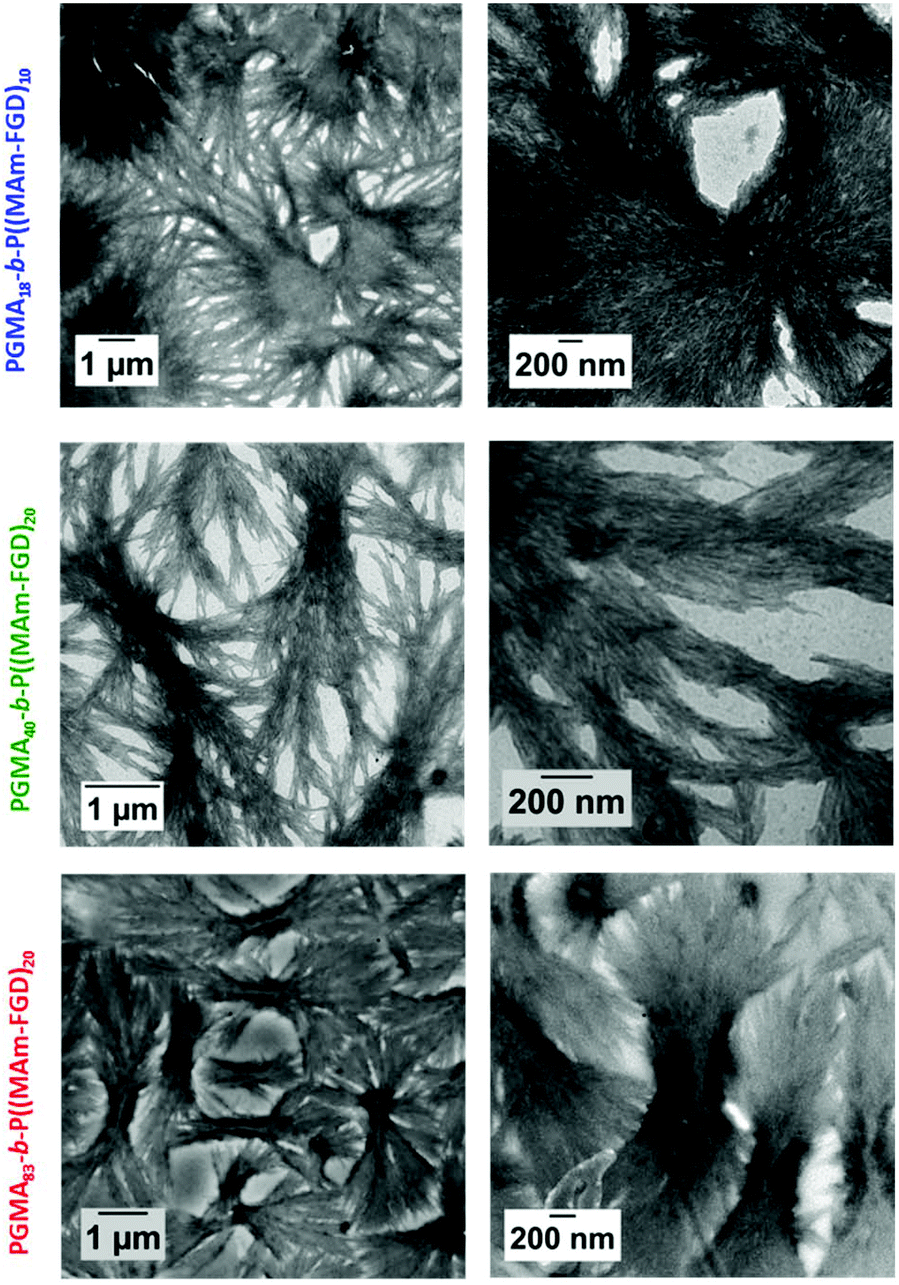
Polymerizations of MAm-FGD were performed using mCTAs 1, 2 and 3 at 10% w/w solids in water. In all cases, 100% MAm-FGD conversion were achieved and a yellow turbid suspension was obtained. Three diblock copolymers were thus prepared: PGMA18-b-P(MAm-FGD)10, PGMA40-b-P(MAm-FGD)20 and PGMA83-b-P(MAm-FGD)20. All three samples formed colloidally stable suspensions at both 70 °C and 20 °C. TEM images of these suspensions revealed the formation of large dendritic and bow-tie shaped fibrous spherulitic structures (Fig. 9). Although the overall shape is similar for all the compositions, indicating similar organization, a closer look at their internal structures reveals slight differences as seen in images at higher magnifications. From PGMA18-b-P(MAm-FGD)10, PGMA40-b-P(MAm-FGD)20 to PGMA83-b-P(MAm-FGD)20, it seems that the fibers become shorter and the structures tend to be less packed.
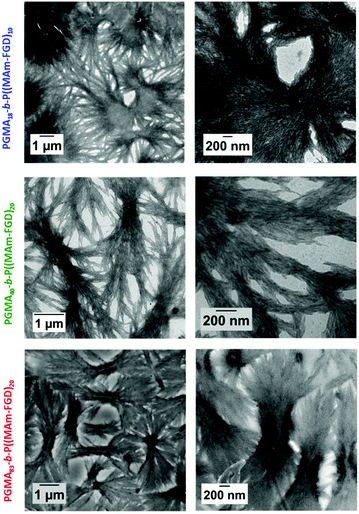 |
| | Fig. 9 Representative TEM images at different magnification of the PGMA18-b-P(MAm-FGD)10 (top row), PGMA40-b-P(MAm-FGD)20, (medium row) and PGMA83-b-P(MAm-FGD)20 (bottom row) diblock copolymer morphologies prepared by RAFT PISA aqueous dispersion at 10% w/w solids. The samples were diluted to 0.1% w/w. | |
Conclusions
Nanostructures constituted of a solvophilic PGMA corona and peptide-containing solvophobic cores were successfully prepared via RAFT-mediated polymerization-induced-self-assembly using SAP-functionalized methacrylamide. The self-assembly of these polymer-polypeptide formulations proved to be much more diverse compared to the polymeric nano-objects reported in previous PISA works. PISA formulations based on PGMA-b-P(MAm-GFF) or PGMA-b-P(MAm-FGD) formed large spherulitic bundles of fibrous structures in ethanol and water respectively. In contrast, only spherical particles and vesicles were observed for PGMA-b-P(MAm-GFF) synthesized via PISA in dispersion in water/acetonitrile mixtures. As observed in our previous work46 the introduction of only few SAP residues was necessary to greatly influence the self-assembled morphology. This observation further strengthens our hypothesis that PISA and SAP motifs can synergistically contribute to the formation of nano- and micrometric morphologies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
PDRA fellowship managed by the French national research agency (ANR): “investissement d'Avenir” LabEX CheMISyst, grant number ANR-10-LABX-05-01. The authors also thank CNRS for funding this work via the “Osez l'interdisciplinarité” programme awarded to MS.
Notes and references
- J. Shu, B. Panganiban and T. Xu, Annu. Rev. Phys. Chem., 2013, 64, 631–657 CrossRef CAS PubMed.
- V. Mikhalevich, I. Craciun, M. Kyropoulou, C. G. Palivan and W. Meier, Biomacromolecules, 2017, 18, 3471–3480 CrossRef CAS PubMed.
- E. Radvar and H. Azevedo, Macromol. Biosci., 2018, 19, 1800221 CrossRef PubMed.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed.
- D. Mandal, A. Shirazi and K. Parang, Org. Biomol. Chem., 2014, 12, 3544–3561 RSC.
- J. Ryu and C. Park, Biotechnol. Bioeng., 2010, 105, 221–230 CrossRef CAS PubMed.
- C. Görbitz, Chem. Commun., 2006, 22, 2332–2334 RSC.
- S. Marchesan, A. Vargiu and K. Styan, Molecules, 2015, 20, 19775–19788 CrossRef CAS PubMed.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Nano Lett., 2004, 4, 581–585 CrossRef CAS.
- M. Reches and E. Gazit, Phys. Biol., 2006, 3, S10–S19 CrossRef CAS PubMed.
- R. Huang, W. Qi, R. Su, J. Zhao and Z. He, Soft Matter, 2011, 7, 6418 RSC.
- C. Guo, Y. Luo, Z. Ruhong and G. Wei, Nanoscale, 2014, 6, 2800–2811 RSC.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- H. Arakawa, K. Takeda, S. Higashi, A. Shibata, Y. Kitamura and M. Ikeda, Polym. J., 2020, 1–8, DOI:10.1038/s41428-019-0301-5..
- S. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS PubMed.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
-
A. Mendes, E. Baran, R. L. Reis and H. Azevedo, Wiley interdisciplinary reviews. Nanomed. Nanobiotechnol, 2013, 6, 582–612 Search PubMed.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- Z. Fan, L. Sun, Y. Huang, Y. Wang and M. Zhang, Nat. Nanotechnol., 2016, 11, 388–394 CrossRef CAS PubMed.
- M. Şardan Ekiz, G. Cinar, M. Khalily and M. Guler, Nanotechnology, 2016, 27, 402002 CrossRef PubMed.
- L. Adler-Abramovich, M. Reches, V. Sedman, S. Allen, S. Tendler and E. Gazit, Langmuir, 2006, 22, 1313–1320 CrossRef CAS PubMed.
- M. Lansalot and J. Rieger, Macromol. Rapid Commun., 2019, 40, 1800885 CrossRef PubMed.
- N. Penfold, J. Yeow, C. Boyer and S. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- C. Ferguson, R. Hughes, B. Pham, B. Hawkett, R. Gilbert, A. Serelis and C. Such, Macromolecules, 2002, 35, 9243 CrossRef CAS.
- V. Cunningham, A. Alswieleh, K. Thompson, M. Williams, G. Leggett, S. Armes and O. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- M. Derry, L. Fielding and S. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. Ryan and S. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- A. Blanazs, A. Ryan and S. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- S. Boissé, J. Rieger, B. Khaled, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- I. Chaduc, A. Crepet, O. Boyron, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS PubMed.
- M. Derry, L. Fielding and S. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. Armes, Polym. Chem., 2014, 5, 3466–3475 RSC.
- M. Semsarilar, N. Penfold, L. Jones and S. Armes, Polym. Chem., 2015, 6, 1751–1757 RSC.
- X. Zhang, J. Rieger and B. Charleux, Polym. Chem., 2012, 3, 1502–1509 RSC.
- L. Fielding, J. Lane, M. Derry, O. Mykhaylyk and S. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- A. Lopez-Oliva, N. Warren, A. Rajkumar, O. Mykhaylyk, M. Derry, K. Doncom, M. Rymaruk and S. Armes, Macromolecules, 2015, 48, 150522153344002 CrossRef.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 CrossRef CAS.
- D. Zhou, R. Kuchel, S. Dong, F. Lucien, S. Perrier and P. Zetterlund, Macromol. Rapid Commun., 2018, 40, 1800335 CrossRef PubMed.
- T. P. T. Dao, L. Vezenkov, G. Subra, M. Amblard, M. In, J.-F. Le Meins, F. Aubrit, M.-A. Moradi, V. Ladmiral and M. Semsarilar, Macromolecules, 2020 DOI:10.1021/acs.macromol.0c01260.
- V. Ladmiral, M. Semsarilar, I. Cantón and S. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- N. Warren and S. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel and E. Gazit, Nat. Chem. Biol., 2012, 8, 701–706 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, SEC curves, DLS data, digital photos taken, Summary Table for all obtaining products. See DOI: 10.1039/d0py00793e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 c,
Vincent
Ladmiral
c,
Vincent
Ladmiral