
All poly(ionic liquid) block copolymer nanoparticles from antagonistic isomeric macromolecular blocks via aqueous RAFT polymerization-induced self-assembly†
Jérémy
Depoorter
a,
Xibo
Yan
 ab,
Biao
Zhang
ac,
Guillaume
Sudre
d,
Aurélia
Charlot
a,
Etienne
Fleury
a and
Julien
Bernard
*a
ab,
Biao
Zhang
ac,
Guillaume
Sudre
d,
Aurélia
Charlot
a,
Etienne
Fleury
a and
Julien
Bernard
*a
aUniv Lyon, INSA Lyon, CNRS, IMP UMR 5223, F-69622, Villeurbanne, France. E-mail: julien.bernard@insa-lyon.fr
bSchool of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China
cFrontiers Science Center for Flexible Electronics (FSCFE), Shaanxi Institute of Flexible Electronics (SIFE) & Shaanxi Institute of Biomedical Materials and Engineering (SIBME), Northwestern Polytechnical University (NPU), Xi'an, 710072 Shaanxi, China
dUniv Lyon, Université Claude Bernard Lyon 1, CNRS, IMP UMR 5223, F-69621, Villeurbanne, France
First published on 6th July 2020
Abstract
We describe herein a straightforward methodology for designing all-poly(ionic liquid) block copolymer nanoparticles with various morphologies by aqueous RAFT polymerization-induced self-assembly relying on the use of a couple of isomeric ionic liquid monomers giving rise to macromolecular building blocks with antagonistic solution behavior in water.
Introduction
Combining the positive aspects of ionic liquids and polymers, e.g. liquid-like ionic conductivity and chemical and thermal stability on one hand, and mechanical properties and processability on the other hand, poly(ionic liquid)s (PILs) have progressively arisen as a promising class of polymers with a multitude of applications in the domains of catalysis, energy storage and conversion, electrochemical devices, separation, and so on.1 Poly(ionic liquid) systems have now reached a high degree of complexity and sophistication in terms of their composition, architecture and functionality. Access to this broad range of macromolecular materials has been achieved by exploiting step-growth polymerization processes,2 chain-growth polymerization processes such as conventional radical polymerization,3 controlled/living polymerization techniques,4 or Ring Opening Metathesis Polymerization (ROMP),5 and/or post-polymerization modification strategies and by combining diverse polymer-bound cationic species, e.g. imidazolium,6 pyridinium,7 phosphonium,8 ammonium,9 triazolium,10 cyclopropenium11 or pyrolidinium12 species with halides, aromatic anions, inorganic fluorides or per-fluorinated sulfonimides as mobile anionic counterparts or, more rarely, poly-anionic species with mobile cations.13Efforts have been recently made to design precisely defined poly(ionic liquid)-containing block copolymers capable of self-assembling in solution4a,14 or in bulk.15 In the latter case, the influence of the composition, morphology, orientation of the domains or process has been investigated in detail to conceive nanostructured materials with long-range order and enhanced ionic conductivity. The majority of these studies rely on block copolymers composed of one poly(ionic liquid) block and one neutral block. In spite of attracting great interest, reports on all-poly(ionic liquid) amphiphilic block copolymers remain rather rare. To our knowledge, the only examples of all-poly(ionic liquid) amphiphilic block copolymers have been developed by Detrembleur and Taton.16–18 These authors explored the sequential organometallic-mediated radical polymerization of N-vinyl-3-alkylimidazolium-based monomers (VRImX) with methyl, ethyl or butyl groups and bromide (Br−) or bis(trifluoromethylsulfonyl)imide (TFSI−) as counterions to confer a hydrophilic or organophilic character to the resulting blocks (and promote phase separation). Unfortunately, fast ionic exchanges resulted in the formation of PIL-b-PIL with a random distribution of Br− and TFSI− along the chain.16 To address this issue, the same authors further evolved the substitution pattern of the N-vinyl imidazolium-based ionic liquid monomers (introducing triethylene glycol, octyl or perfluorooctyl groups in the 3-position of the imidazolium ring) to generate all-poly(ionic liquid) amphiphilic block copolymer nanoparticles by Polymerization-Induced Self-Assembly (PISA) in water.17,18 However, no morphological transition from spheres to fibers or vesicles was observed for all-poly(ionic liquid) block copolymer systems (whatever the solid content and composition) possibly due to the low incompatibility of the two blocks.
In this communication, we report on the preparation of amphiphilic block copolymer nanoparticles from ionic liquid monomers. At the core of our strategy is the selection of a pair of water-soluble isomeric imidazolium-based monomers, 1-[2-acryloylethyl]-3-dodecylimidazolium bromide (AEDIBr) and 1-[2-acryloyldodecyl]-3-ethylimidazolium bromide (ADEIBr, see Fig. 1), whose resulting homopolymers display disparate solution behaviour. Capitalizing on the opposite solubility for these two homopolymers, we demonstrate here that this couple of isomers can be easily combined to design precisely defined poly(ionic liquid)-b-poly(ionic liquid) amphiphilic block copolymers using aqueous dispersion RAFT-mediated PISA. Using PADEIBr as a water soluble macro-chain transfer agent and tuning the length of the PAEDIBr block and the solid content, we show that poly(ionic liquid)-b-poly(ionic liquid) nanospheres, nanorods or vesicles, can be generated in high yields. To the best of our knowledge, the preparation of nanoparticles with diverse morphologies from all-poly(ionic liquid) diblock copolymers is unprecedented.
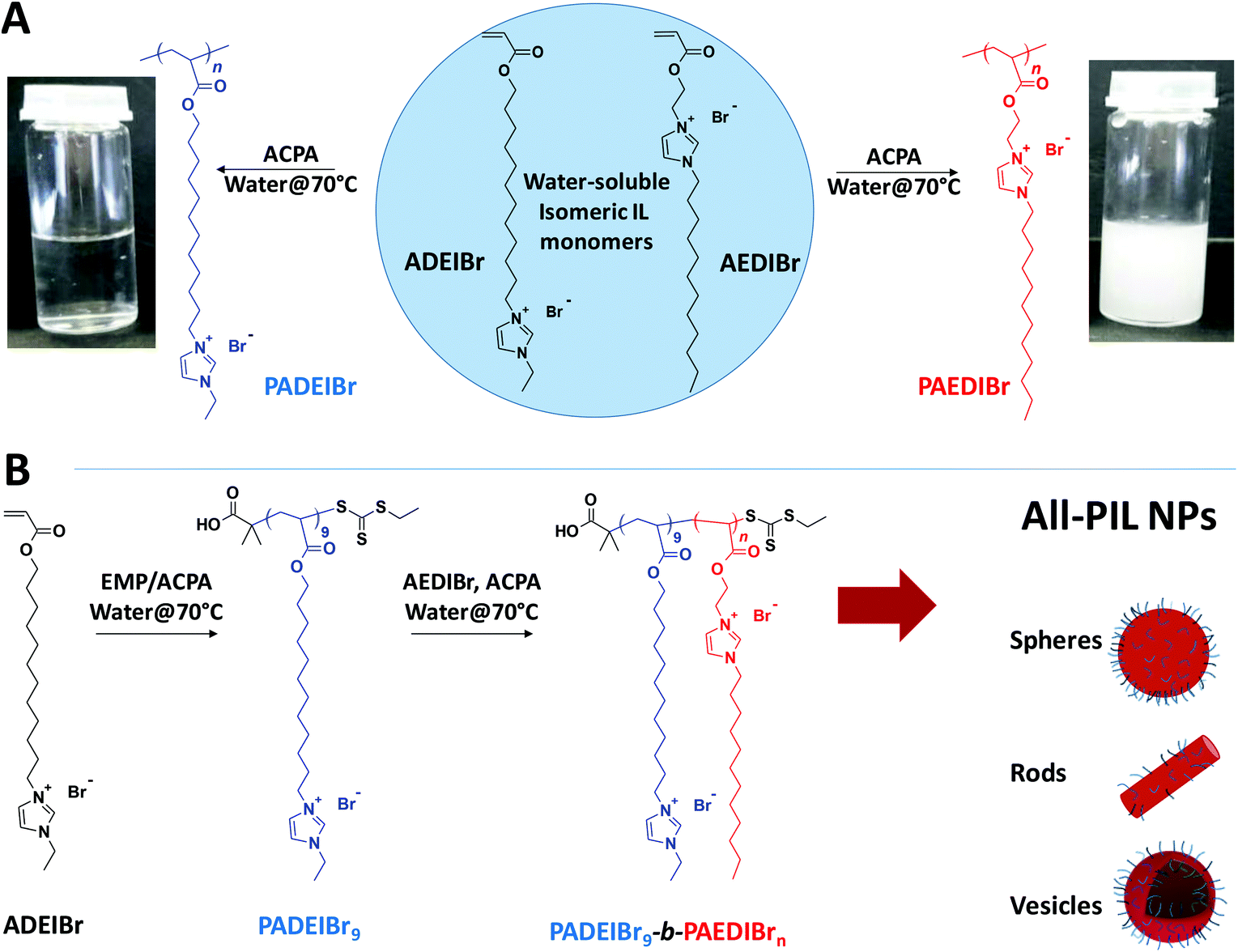 | ||
| Fig. 1 Route to PIL block copolymer nanoparticles by PISA. (A) Synthesis of isomeric PIL homopolymers with antagonistic solution behavior in water (polymer solution at 40% wt). (B) Generation of all-PIL nanoparticles by RAFT dispersion polymerization of AEDIBr. | ||
Experimental section
Materials
Anhydrous acryloyl chloride (97%), Aliquat 336, 2-bromoethanol (95%), 12-bromo-1-dodecanol (99%), anhydrous trimethylamine (≥99%), 1-ethylimidazole (≥95%), 1-dodecylimidazole (98%), sodium bicarbonate (NaHCO3), magnesium sulfate (99%), aluminium oxide (activated, neutral, Brockmann I, 50–300 mesh, 58 pore size), silica nanoparticles (60 Å, 70–230 mesh), 4,4′-azobis (4-cyanovaleric acid) (ACPA, ≥98%), N-isopropylacrylamide (97%), DMSO (anhydrous, 99%), bis(trifluoromethylsulfonyl) imide lithium salt (LiTFSI), ethanethiol (97%), carbon disulfide (≥99%), acetone, chloroform, hexane, cyclohexane, sodium hydroxide, dichloromethane, diethyl ether, hydrochloric acid, acetonitrile (anhydrous, 99.8%), anhydrous N,N-dimethylformamide (DMF), anhydrous tetrahydrofuran (THF, ≥99%), deuterium oxide (D2O), dimethyl sulfoxide-d6 (DMSO-d6), chloroform-d, and methanol-d4 were purchased from Aldrich and used as received. Dialysis membranes (Mw cut-off: 1000 Da) were purchased from Spectrum Laboratories. S-Ethyl-S′-(α,α′-dimethyl-α′′-acetic acid) trithiocarbonate (EMP) was prepared as previously described.19Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700000 g mol−1). Omnisec software was used for the analysis of the results.
700000 g mol−1). Omnisec software was used for the analysis of the results.
Note that, prior to SEC analyses, bromine counter-anions must be exchanged with bis(trifluoromethyl sulfonyl)imide ones as follows. Typically, the polymer material (10 mg) was first dissolved in 1 mL deionized water and an excess of LiTFSI (∼40 mg) in 1 mL of water was added dropwise. The solution was left under stirring for 1 h. Water was eliminated and the polymer deposited at the bottom of the flask was washed five times with water to remove the residual lithium bromide. The resulting TFSI-containing polymers were finally dried at 60 °C for one night under vacuum before SEC analysis.
Preparation of ionic liquid monomers (ILM)
1H NMR (400 MHz, DMSO-d6, δ): 6.29–6.34 (1H, dd, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.18–6.12 (1H, dd, CH2CH), 5.93–5.90 (1H, m, CH2CH), 4.11–4.08 (2H, t, –OCOCH2), 3.51–3.48 (2H, t, BrCH2CH2−), 1.77–1.88(2H, t, –(CH2)10–CH2), 1.59–1.62 (2H, t, –OCH2–CH2–(CH2)10), 1.26–1.37 (20H, s, OC2H4–(CH2)8–C2H4).
CH), 6.18–6.12 (1H, dd, CH2CH), 5.93–5.90 (1H, m, CH2CH), 4.11–4.08 (2H, t, –OCOCH2), 3.51–3.48 (2H, t, BrCH2CH2−), 1.77–1.88(2H, t, –(CH2)10–CH2), 1.59–1.62 (2H, t, –OCH2–CH2–(CH2)10), 1.26–1.37 (20H, s, OC2H4–(CH2)8–C2H4).
1H NMR (400 MHz, DMSO-d6, δ): 9.21 (1H, m, N–CH–N), 7.82–7.80 (2H, dd, N–CHCH–N), 6.34–6.30 (1H, dd, CH2CH), 6.21–6.14 (1H, dd, CH2CH), 5.96–5.93 (1H, m, CH2CH), 4.21–4.10 (6H, m, –(CH2)11–CH2O– & CH2–N), 1.78–1.81 (2H, t, (CH2)10–CH2), 1.59–1.61 (2H, t, OCH2–CH2), 1.45–1.41 (3H, t, CH3–CH2N), 1.25–1.29 (16H, s, –(CH2)8–); 13C NMR (400 MHz, DMSO-d6, δ): 166 (OCOCH2), 136 (N–CH–N), 131 (CH2CH), 129 (CH2CH), 123 (N–CHCH–N), 123.2 (N–CHCH–N), 65 (CH2O–), 49 (N–CH2), 45 (N–CH2), 29 (–(CH2)8–), 26 (O(CH2)9–CH2), 16 (NCH2–CH3).
1H NMR (400 MHz, DMSO-d6, δ): 6.41–6.36 (1H, dd, CH2CH), 6.25–6.18 (1H, dd, CH2CH), 6.02–5.99 (1H, dd, CH2CH), 4.45–4.43 (2H, t, –OCOCH2), 3.74–3.71 (2H, t, BrCH2CH2−).
1H NMR (400 MHz, DMSO-d6, δ): 9.32 (1H, s, N–CH–N), 7.85–7.84 (2H, s, N–CHCH–N), 6.37–6.32 (1H, dd, CH2CH), 6.2–6.13 (1H, dd, CH2CH), 6.00–5.97 (1H, dd, CH2CH), 4.54–4.49 (4H, m, N–CH2CH2O–), 4.21–4.18 (2H, t, N–CH2–(CH2)11), 1.80–1.76 (2H, s, NCH2–CH2–(CH2)10), 1.24–1.10 (18H, s, N–C2H4–(CH2)9) 0.88–0.84 (3H, t, NCH2–CH3); 13C NMR (400 MHz, DMSO-d6, δ): 165. (OCOCH2), 137 (N–CH–N), 132 (CH2CH), 128 (CH2CH), 122 (N–CHCH–N), 123 (N–CHCH–N), 62. (N–CH2–CH2O–), 48 (N–CH2), 32 (NCH2–CH2), 29 (NC2H4–(CH2)8), 22 (NC9H18–CH2), 14 (NC11H22–CH3).
Polymerization of ILMs
RAFT polymerization of AEDIBr in DMF
2.5 mg of EMP (1.1 × 10−5 mol), 0.6 mg of ACPA (2.24 × 10−6 mol), 465 mg of 1-[2-acryloylethyl]-3-dodecylimidazolium bromide (AEDIBr, 1.12 × 10−3 mol) were dissolved in DMF. The final monomer concentration was fixed at 40 wt%. After mixing, the yellow solution was deoxygenated by five consecutive freeze–pump–thaw cycles. After the last cycle, the Schlenk tube was filled with nitrogen, warmed to room temperature and placed in an oil bath at 70 °C. An initial sample (t = 0) was collected with a syringe. Aliquots were further withdrawn at regular time intervals to determine conversion via1H NMR, molar masses and dispersity via SEC in DMF/LiTFSI after ionic exchange. The reaction was quenched by plunging the tubes into liquid nitrogen. The samples were purified by dialysis (Mw cut-off = 1000 Da) against water, and dried by freeze-drying.| Entrya | Convb (%) | DPPAEDIBrc |
M
n SECd (kg mol−1) |
Đ | Morphologye |
|---|---|---|---|---|---|
| a The notation D9E200-5 refers to a PADEIBr9-mediated polymerization of AEDIBr targeting a degree of polymerization equal to 200 and a solid content of 5 wt%, respectively. All the polymerizations were performed in water for 3 h at 70 °C using PADEIBr9 as the macroCTA (1 mg; 2.4 × 10−6 mol L−1) and ACPA initiator ([macroCTA]0/[ACPA]0 = 5). b Determined by 1H NMR. c Estimated from monomer conversion. d Determined by SEC (in DMF/LiTFSI after ionic exchange, PS standards). e Morphologies observed by TEM; S, R and V refer to spherical nanoparticles, rods and vesicles, respectively. f A minor population of spherical nanoparticles was also detected by TEM in these formulations. | |||||
| D9-E200-5 | 99 | 198 | 48 | 1.16 | S |
| D9-E400-5 | 99 | 396 | 49 | 1.15 | S |
| D9-E600-5 | 98 | 588 | 51 | 1.18 | S |
| D9-E800-5 | 95 | 760 | 94 | 1.52 | S |
| D9-E200-15 | 97 | 194 | 49 | 1.23 | S |
| D9-E400-15 | 98 | 396 | 53 | 1.21 | S |
| D9-E600-15 | 98 | 588 | 70 | 1.56 | Rf |
| D9-E800-15 | 99 | 792 | 133 | 1.19 | V |
| D9-E200-25 | 97 | 194 | 61 | 1.20 | S |
| D9-E400-25 | 99 | 398 | 69 | 1.62 | S |
| D9-E600-25 | 98 | 588 | 96 | 1.30 | Rf |
| D9-E800-25 | 99 | 792 | 157 | 1.09 | V |
Results and discussion
In order to design precisely defined amphiphilic all-poly(ionic liquid) block copolymers through aqueous dispersion RAFT polymerization, we first selected a couple of water-miscible imidazolium-based acrylate isomers, i.e. 1-[2-acryloyldodecyl]-3-ethylimidazolium bromide (ADEIBr) and 1-[2-acryloylethyl]-3-dodecylimidazolium bromide (AEDIBr) whose homopolymerizations in aqueous medium give rise to water-soluble and non-water-soluble polymer chains, respectively (see Fig. 1). The contrasting solution behavior of the two isomeric PILs in water probably arises from the position of the C12 segments either inserted between the main chain and the imidazolium ring (PADEIBr) or at the extremity of the pendant group (PAEDIBr) leading to disparate accessibility, mobility and propensity to self-organize in aqueous solution.As a prerequisite to perform PISA with success, we first investigated in detail the aqueous RAFT homopolymerization of ADEIBr to identify conditions ensuring the synthesis of water-soluble macroRAFT agents with good control and high chain-end fidelity. With this aim, ADEIBr was polymerized at 70 °C in the presence of S-ethyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (EMP), a chain transfer agent which has been previously shown to efficiently mediate the polymerization of imidazolium-based acrylates in water,14f and 4,4′-azobis(4-cyanovaleric acid) (ACPA) as an initiator ([ADEIBr]:[EMP]:[ACPA] = 100:1:0.2, [AEDIBr] = 3.6 × 10−3 mol L−1, 40% wt). Aliquots were withdrawn every 15 minutes and monomer conversion was analyzed by 1H NMR from integration of vinylic peaks (at 5.93 and 6.30 ppm) and proton peaks from alkyl chains at 1.80 ppm (see Fig. S9†). Consistent with our previous study on aqueous RAFT polymerization of imidazolium-based acrylates,14f high conversions (above 70%) were achieved in 60 minutes (including a period of inhibition of 5–10 minutes). Linear pseudo-first order kinetics were observed in this lapse of time (see Fig. 2A). As revealed by SEC analyses performed in DMF/bis(trifluoromethylsulfonyl)imide lithium salt after anion exchange of the bromide counterions with TFSI, the molar mass of PADEIBr chains gradually increased with conversion (Fig. 2B). The SEC peaks were narrowly distributed with no trace of peak tailing and the dispersity was maintained around 1.20 up to high conversions. A good agreement between experimental molar mass evaluated by 1H NMR (from integration of the EMP chain end and polymer backbone protons) and theoretical values (calculated from ADEIBr conversion) was observed (at 83% of conversion, Mn th = 35 kg mol−1, Mn RMN = 28 kg mol−1).
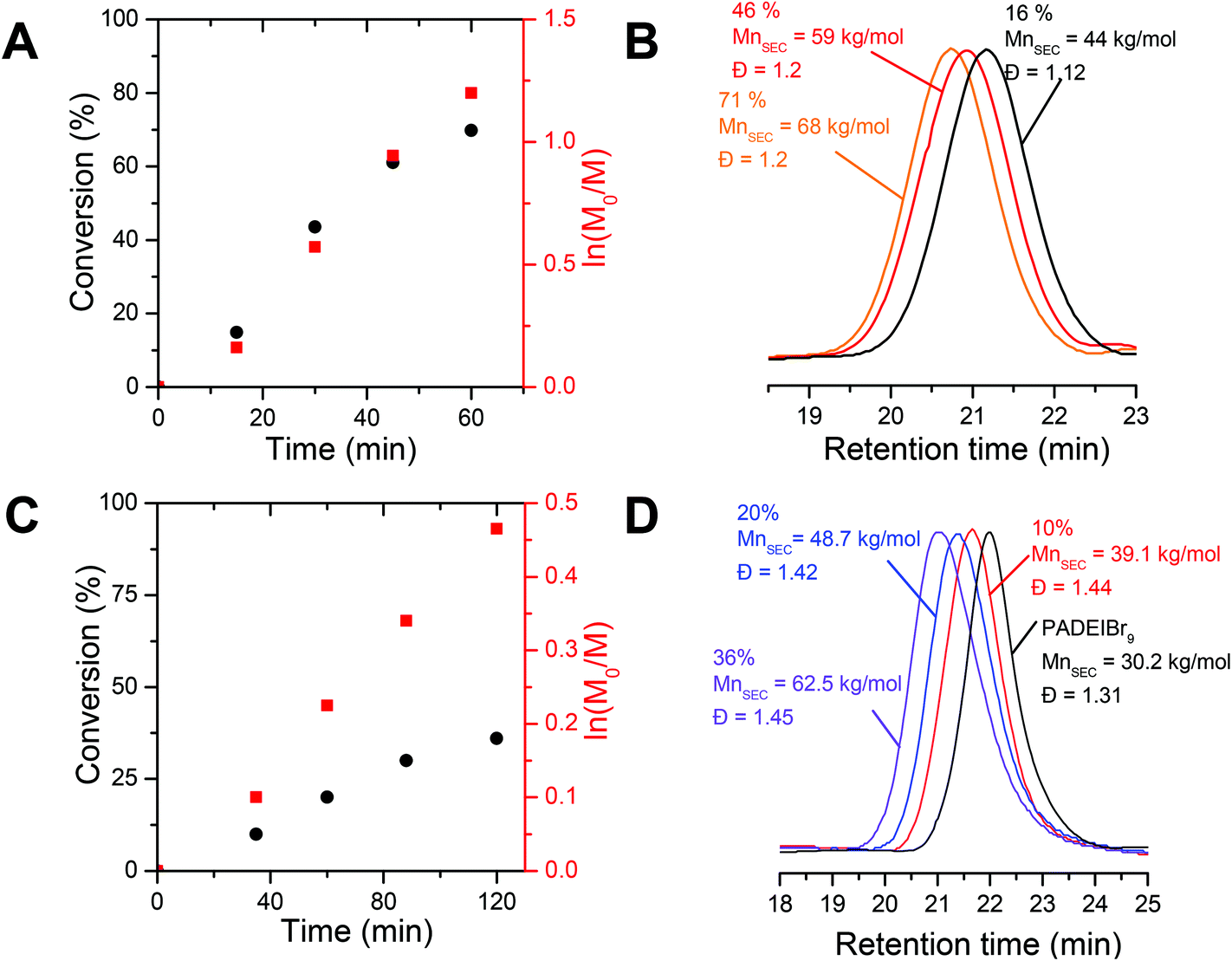 | ||
| Fig. 2 (A) Monomer conversion vs. time curve (black circles) obtained for the aqueous RAFT polymerization of ADEIBr 40% w/w at 70 °C using EMP as a CTA and ACPA as an initiator ([ADEIBr]:[EMP]:[ACPA] = 100:1:0.2) and corresponding semilogarithmic plots (red squares). (B) SEC trace evolution for the aqueous RAFT polymerization of ADEIBr (analyses in DMF/LiTFSI as the eluent after ionic exchange of bromide anions by TFSI). (C) Monomer conversion vs. time curve (black circles) obtained for the chain extension of PADEIBr9 with NIPAAm 15% w/w in DMSO at 70 °C ([NIPAAm]:[MacroCTA]:[ACPA] = 400:1:0.2) and corresponding semilogarithmic plots (red squares). (D) SEC trace evolution for the chain extension of PADEIBr9 with NIPAAm (analyses in DMF/LiTFSI as the eluent after ionic exchange of bromide anions by TFSI). | ||
To assess the chain-end fidelity of the resulting PILs, we next investigated the ability to chain-extend a PIL macroRAFT agent (PADEIBr9, generated under the following polymerization conditions, i.e. [ADEIBr]:[EMP]:[ACPA] = 20:1:0.2, [ADEIBr] = 0.49 mol L−1, 15% wt, Mn NMR = 3.9 kg mol−1 and Mn SEC = 30.2 kg mol−1 after anion exchange, Đ = 1.31) with NIPAAm in DMSO at 70 °C (15% wt NIPAAm, ([NIPAAm]:[MacroCTA]:[ACPA] = 400:1:0.2, see Fig. 2C and S10†). After 2 h of polymerization at 70 °C, the SEC trace (Fig. 2D) clearly shifted towards high molecular weight (PADEIBr9-b-PNIPAAm180, Mn NMR = 25 kg mol−1, Mn SEC = 62.5 kg mol−1, Đ = 1.45) and the peak corresponding to the PADEIBr precursor was no longer observed confirming that the macroRAFT agent efficiently participates in the chain-extension process.
As a control experiment, AEDIBr was also successfully homopolymerized at 70 °C in DMF (instead of water) in the presence of EMP ([AEDIBr]/[EMP]/[ACPA] = 100/1/0.2 (conversion >80% in 2 h see ESI Fig. S11†)).
Encouraged by the aptitude of RAFT-made PADEIBr to grow precisely defined block copolymers through chain extension under homogeneous conditions, we further explored the aqueous RAFT dispersion polymerization of AEDIBr in the presence of PADEIBr9 as a macro-RAFT agent and a stabilizing block. First, to prove beyond doubt that the (expected) formation of nanoparticles was due to the synthesis/self-assembly of PIL block copolymers and not the precipitation polymerization of AEDIBr from water as described by Yuan et al. with long alkyl (C14–18) vinylimidazoliums in water,20 polymerization of AEDIBr was performed in water in the presence of RAFT agent-free PADEIBr9 (15 wt%). In contrast to macroRAFT-mediated polymerizations of AEDIBr vide infra, this resulted in rapid aggregation and formation of a coagulum at the bottom of the tube.
Next, using PADEIBr9 as a macro-RAFT agent, hydrophobic core-forming PAEDIBr degrees of polymerization ranging from 200 to 800 were targeted at final solid contents ranging between 5 and 25 wt% and [macroCTA]0/[ACPA]0 = 5. The polymerization temperature was fixed to 70 °C. The initially transparent polymerization medium rapidly turned milky, consistent with the growth of a PAEDIBr block becoming insoluble in water and promoting micellization. Proton NMR analyses assessed that full conversion of AEDIBr was attained within 3 hours under such conditions of polymerization (see Table 1). The resulting latexes were stable for at least 10 days.
DMF/LiTFSI SEC analyses of the resulting all-poly(ionic liquid) block copolymers (after ion exchange) revealed a progressive increase of the molar masses with the conversion and dispersities which showed values between 1.15 and 1.30 with the exception of D9-E800-5, D9-E600-15 and D9-E400-25 (SEC chromatograms given in Fig. S12†). The evolution of the morphology of the nanoparticles is summarized in Table 1 and illustrated in Fig. 3.
 | ||
| Fig. 3 TEM pictures of all-PIL block copolymer nanoparticles obtained by chain extension of PADEIBr9 with AEDIBr at 15 wt% solid content (targeted DPs = 200, 400, 600 and 800). | ||
Chain-extension experiments employing PADEIBr9 as a stabilizing block and setting the all-poly(ionic liquid) block copolymer concentration at 5 wt% exclusively resulted in the generation of spherical nanoparticles (see Fig. S13†) irrespective of the PAEDIBr block DP (which ranged from 200 to 800). To gain further insight into the influence of the solid content on the morphology adopted by the block copolymers, the solid content was further increased to 15 and 25 wt%. Again, the chain extension of PADEIBr9 targeting PAEDIBr blocks with degrees of polymerization up to 400 gave rise to spherical nanoparticles with dimensions gradually increasing with the DP. However, in contrast to PISA experiments performed at 5 wt%, growth of hydrophobic PAEDIBr blocks with higher DPs afforded the generation of all poly(ionic liquid) block copolymer nanoparticles with different morphologies. Targeting PAEDIBr DPs of 600 or 800 gave rise to peculiar 100–200 nm long rod-like PIL nanoparticles and vesicular self-assemblies (see TEM pictures of the nanoparticles at 15 wt% and 25 wt% solid contents in Fig. 3 and 4, respectively, and DLS of spherical nanoparticles and vesicles in Fig. S14†). Interestingly, TEM analyses of D9-E800-25 dispersions revealed the formation of vesicles with non-spherical shapes (see Fig. 4B).
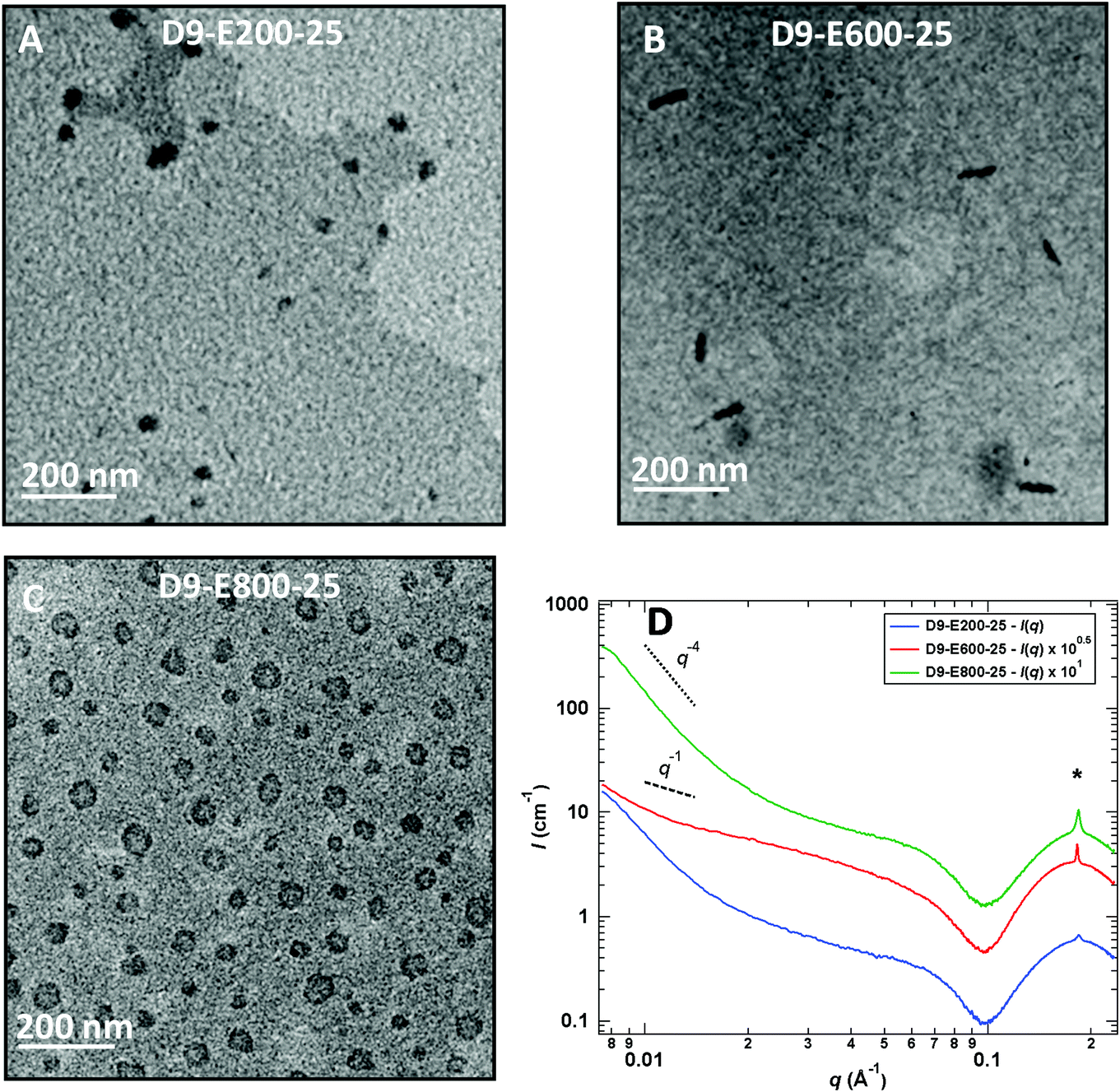 | ||
| Fig. 4 TEM images of all-PIL block copolymer spherical nanoparticles (A, D9-E200-25), rod-like nanoparticles (B, D9-E600-25) and non-spherical vesicles (C, D9-800-25) obtained by chain extension of PADEIBr9 with AEDIBr at 25 wt% solid content (DP = 800). (D) SAXS patterns of the corresponding aqueous dispersions of PIL NPs at a concentration of 5 g L−1; the curves have been vertically shifted for clarity. | ||
These observations are consistent with the study of Yuan and co-workers on the generation of anisotropic nanoparticles by precipitation polymerization of C14–18 alkyl vinylimidazolium bromide20 and the work of Zhu's research group on the preparation of rigid straight rods by PISA involving C12-vinylimidazolium bromide.14h Accordingly, we made the assumption that the generation of these non-spherical vesicles (D9-E800-25) and stiff rod-like nanoparticles (D9-E600-15 and D9-E600-25) stems from the self-organization of the C12 alkyl pendant groups in the course of the PISA process. To shed light on the internal organization of PEADIBr-b-PAEDIBr NPs, the resulting dispersions were further analyzed by small-angle X-ray scattering (SAXS, see Fig. 4D) at a concentration of 5 g L−1. At low q values (q < 2 10−2 Å−1), the intensity scattered by D9-E200-25 and D9-E800-25 varies as follows: I(q) ∝ q−4, which is consistent with either spherical or vesicular objects. The intensity scattered by D10-E600-25 does not follow the same trend and seems closer to I(q) ∝ q−1, which confirms the formation of a majority of rod-like objects. For 10−1 Å−1 < q < 2.5 10−1 Å−1, a strong oscillation accompanied by a sharp peak at its maximum are observed. This sharp peak at a q value of ∼1.8 × 10−1 Å−1 is consistent with the ordering of the alkyl pendant groups within the NPs.21 The intensity of this peak increases with increasing contents of PAEDIBr and the d-spacing (∼34.4 Å) roughly corresponds to a bilayer of the AEDIBr motif. It has to be noted that the formation of exotic morphologies solely occurs when the intensity of the sharp peak at q ∼ 1.8 × 10−1 Å−1 is rather high, which suggests that the auto-assembly of the AEDIBr units drives the formation of rods and/or vesicles. Finally, the region between 5 × 10−2 Å−1 < q < 2.5 × 10−1 Å−1, the plateau followed by an oscillation, can be explained by the presence of bilayered structures (core–shell micelles or rods, or vesicles) as already described by Manet et al.,22 Yuan et al.20 and Rymaruk et al.23
Conclusions
In summary, we selected here a pair of ionic liquid isomeric imidazolium-based acrylates, AEDIBr and ADEIBr, giving rise to macromolecular blocks with antagonistic solution behavior in water, i.e. PADEIBr is a water-soluble homopolymer contrary to PAEDIBr. Exploiting this rather unique combination of isomeric building blocks, we showed that aqueous dispersion polymerization of AEDIBr mediated by a short PADEIBr block as a macroCTA affords the preparation of precisely defined all-PIL amphiphilic block copolymers. By tuning the conditions of the PISA process, a range of original all-PIL block copolymer nanostructures including spherical nanoparticles or peculiar rod-like nanoparticles and vesicles were formed.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the French Ministry of Research for J. D.'s PhD grant. Dr B. Z and Dr X. Y. acknowledge CSC for a PhD grant. The authors acknowledge Isabelle Morfin (LIPhy, Grenoble, France) for her help with X-ray scattering measurements and Pierre Alcouffe and the Centre technologique des microstructures (CTμ, Université Lyon 1) for their help with microscopy analyses.References
- (a) J. Y. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS; (b) W. Qian, J. Texter and F. Yan, Frontiers in Poly(Ionic Liquid)s : Syntheses and Applications, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- (a) B. P. Mudraboyina, M. M. Obadia, I. Allaoua, R. Sood, A. Serghei and E. Drockenmuller, 1,2,3-Triazolium-Based Poly(ionic liquid)s with Enhanced Ion Conducting Properties Obtained through Click Chemistry and Polyaddition Strategy, Chem. Mater., 2014, 26, 1720–1726 CrossRef CAS; (b) J. Lee, V. M. Lau, Y. Ren, C. M. Evans, J. S. Moore and N. R. Sottos, Effect of Polymerized Ionic Liquid Structure and Morphology on Shockwave Energy Dissipation, ACS Macro Lett., 2019, 8, 535–539 CrossRef CAS; (c) Q. Zhao, C. Shen, K. P. Halloran and C. M. Evans, ACS Macro Lett., 2019, 8, 658–663 CrossRef.
- J. E. Bara, C. J. Gabriel, E. S. Hatakeyama, T. K. Carlisle, S. Lessmann, R. D. Noble and D. L. Gin, Improving CO2 selectivity in polymerized room-temperature ionic liquid gas separation membranes through incorporation of polar substituents, J. Membr. Sci., 2008, 321, 3–7 CrossRef CAS.
- (a) K. Vijayakrishna, S. K. Jewrajka, A. Ruitz, R. Marcilla, J. A. Pomposo, D. Mecerreyes, D. Taton and Y. Gnanou, Synthesis by RAFT and Ionic Responsiveness of Double Hydrophilic Block Copolymers Based on Liquid Ionic Monomer Units, Macromolecules, 2008, 41, 6299–6308 CrossRef CAS; (b) H. Mori, M. Yahagi and T. Endo, RAFT Polymerization of N-Vinylimidazolium and Synthesis of Thermoresponsive Ionic Liquid Block Copolymers, Macromolecules, 2009, 42, 8082–8092 CrossRef CAS; (c) J. Texter, V. A. Vasantha, R. Crombez, R. Maniglia, L. Slater and T. Mourey, Triblock Copolymer Based on Poly(propylene oxide) and Poly(1-[11-acryloylundecyl]-3-methyl-imidazolium bromide), Macromol. Rapid. Commun., 2012, 33, 69–74 CrossRef CAS PubMed; (d) H. He, D. Luebke, H. Nulwala and K. Matyjaszewski, Synthesis of Poly(ionic liquid)s by Atom Transfer Radical Polymerization with ppm of Cu Catalyst, Macromolecules, 2014, 47, 6601–6609 CrossRef CAS; (e) C. M. Stancik, A. R. Lavoie, J. Schütz, P. A. Achurra, P. Lindner, A. P. Gast and R. M. Waymouth, Micelles of Imidazolium-Functionalized Polystyrene Diblock Copolymers Investigated with Neutron and Light Scattering, Langmuir, 2004, 20, 596–605 CrossRef CAS PubMed; (f) G. Sudre, S. Inceoglu, P. Cotanda and N. P. Balsara, Influence of Bound Ion on the morphology and Conductivity of Anion-Conducting Block Copolymers, Macromolecules, 2013, 46, 1519–1527 CrossRef CAS.
- (a) E. F. Wiesenauer, J. P. Edwards, V. F. Scalfani, T. S. Bailey and D. L. Gin, Synthesis and Ordered Phase Separation of Imidazolium-Based Alkyl-Ionic Diblock Copolymers Made via ROMP, Macromolecules, 2011, 44, 5075–5078 CrossRef CAS; (b) T. Suga, K. Sakata, K. Aoki and H. Nishide, Synthesis of Pendant Radical- and Ion-Containing Block Copolymers via Ring-Opening Metathesis Polymerization for Organic Resistive Memory, ACS Macro Lett., 2014, 3, 703–707 CrossRef CAS.
- (a) J. C. Salamone, S. C. Israel, P. Taylor and B. Snider, Synthesis and Homopolymerization of Vinyl Imidazolium Salts, Polymer, 1973, 14, 639–644 CrossRef CAS; (b) H. Ohno, Design of Ion Conductive Polymer Based on Ionic Liquids, Macromol. Symp., 2007, 249–250, 551–556 CrossRef; (c) F. Yan and J. Texter, Solvent-Reversible Poration in Ionic Liquid Copolymers, Angew. Chem., Int. Ed., 2007, 46, 2440–2443 CrossRef CAS PubMed.
- (a) K.-I. Seno, S. Kanaoka and S. Aoshima, Synthesis and LCST-Type Phase Separation Behavior Inorganic Solvents of Poly(vinyl ethers) with Pendant Imidazolium or Pyridinium Salts, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5724–5733 CrossRef CAS; (b) J. Yuan, A. G. Marquez, J. Reinacher, C. Giordano, J. Janek and M. Antonietti, Nitrogen-Doped Carbon Fibers and Membranes by Carbonization of Electrospun Poly(Ionic Liquid)s, Polym. Chem., 2011, 2, 1654–1657 RSC.
- (a) S. T. Hemp, M. Q. Zhang, M. H. Allen, S. J. Cheng, R. B. Moore and T. E. Long, Comparing ammonium and phosphonium polymerized ionic liquids: thermal analysis, conductivity, and morphology, Macromol. Chem. Phys., 2013, 214, 2099–2107 CrossRef CAS; (b) S. Liang, M. V. O'Reilly, U. H. Choi, H.-S. Shiau, J. Bartels, Q. Chen, J. Runt, K. I. Winey and R. H. Colby, High Ion Content Siloxane Phosphonium Ionomers with Very Low Tg, Macromolecules, 2014, 47, 4428–4437 CrossRef CAS; (c) P. Cotanda, G. Sudre, M. A. Modestino, X. C. Chen and N. P. Balsara, High Anion Conductivity and Low Water Uptake of Phosphonium Containing Diblock Copolymer Membranes, Macromolecules, 2014, 47, 7540–7547 CrossRef CAS; (d) A. R. Schultz, P. M. Lambert, N. A. Chartrain, D. M. Ruohoniemi, Z. Zhang, C. Jangu, M. Zhang, C. B. Williams and T. E. Long, 3D Printing Phosphonium Ionic Liquid Networks with Mask Projection Microstereolithography, ACS Macro Lett., 2014, 3, 1205 CrossRef CAS.
- (a) J. Tang, Y. Shen, M. Radosz and W. Sun, Isothermal carbon dioxide sorption in poly(ionic liquid)s, Ind. Eng. Chem. Res., 2009, 48, 9113–9118 CrossRef CAS.
- (a) R. Sood, B. Zhang, A. Serghei, J. Bernard and E. Drockenmuller, Triethylene glycol-based poly(1,2,3-triazolium acrylate)s with enhanced ionic conductivity, Polym. Chem., 2015, 6, 3521–3528 RSC; (b) W. Zhang and J. Yuan, Poly(1-vinyl-1,2,4-triazolium) Poly(Ionic Liquid)s : Synthesis and the Unique Behavior in Loading Metal Ions, Macromol. Rapid. Commun., 2016, 37, 1124–1129 CrossRef CAS PubMed.
- J. L. Freyer, S. D. Brucks, G. S. Gobieski, S. T. Russell, C. E. Yozwiak, M. Sun, Z. Chen, Y. Jiang, J. S. Bandar, B. R. Stockwell, T. H. Lambert and L. M. Campos, Clickable Poly(ionic liquids) : A Materials Platform for Transfection, Angew. Chem., Int. Ed., 2016, 55, 12382–12386 CrossRef CAS PubMed.
- J. Qin, J. Guo, Q. Xu, Z. Zheng, H. Mao and F. Yan, Synthesis of Pyrrolidinium-Type Poly(ionic liquid) Membranes for Antibacterial Applications, ACS Appl. Mater. Interfaces, 2017, 9, 10504–10511 CrossRef CAS PubMed.
- (a) A. S. Shaplov, P. S. Vlasov, M. Armand, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, O. V. Okatova, G. M. Pavlov, C. Wandrey, I. A. Godovikov and Y. S. Vygodskii, Design and synthesis of new anionic “polymeric ionic liquids” with high charge delocalization, Polym. Chem., 2011, 2, 2609–2618 RSC; (b) Y. Kohno and H. Ohno, Key factors to prepare polyelectrolytes showing temperature-sensitive lower critical solution temperature-type phase transitions in water, Aust. J. Chem., 2012, 65, 91–94 CrossRef CAS.
- (a) C. M. Stancik, A. R. Lavoie, P. A. Achurra, R. M. Waymouth and A. P. Gast, A Neutron Scattering Study of the Structure and Water Partitioning of Selectively Deuterated Copolymer Micelles, Langmuir, 2004, 20, 8975–8987 CrossRef CAS PubMed; (b) Z. Wang, H. Lai and P. Wu, Influence of PIL Segment on Solution Properties of Poly(N-isopropylacrylamide)-b-Poly(Ionic Liquid) Copolymer Micelles, Thermal Phase Behavior and Microdynamics, Soft Matter, 2012, 8, 11644–11653 RSC; (c) H. He, K. Rahimi, M. Zhong, A. Mourran, D. R. Luebke, H. B. Nulwala, M. Möller and K. Matyjaszewski, Cubosomes from Hierarchical Self-Assembly of Poly(ionic liquid), Nat. Commun., 2017, 8, 14507 CrossRef PubMed; (d) S. Chen, A. Funtan, F. Gao, B. Cui, A. Meister, S. S. P. Parkin and W. H. Binder, Synthesis and Morphology of Semifluorinated Polymeric Ionic Liquids, Macromolecules, 2018, 51, 8620–8628 CrossRef CAS; (e) W. Zhang, Z. Kochovski, Y. Lu, B. V. K. J. Schmidt, M. Antonietti and J. Yuan, Internal Morphology-Controllable Self-Assembly in Poly(Ionic Liquid) Nanoparticles, ACS Nano, 2016, 10, 7731–7737 CrossRef CAS PubMed. A few examples of PISA have also been reported: (f) B. Zhang, X. Yan, P. Alcouffe, A. Charlot, E. Fleury and J. Bernard, Aqueous RAFT Polymerization of Imidazolium-type ionic liquid monomers- En route to poly(ionic liquid)-based nanoparticles through RAFT polymerization-induced self-assembly, ACS Macro Lett., 2015, 4, 1008–1011 CrossRef CAS; (g) C. Liu, S. Wang, H. Zhou, C. Gao and W. Zhang, Thermoresponsive poly (ionic liquid): controllable RAFT synthesis, thermoresponse, and application in dispersion RAFT polymerization, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 945–954 CrossRef CAS; (h) Q. Zhang, M. Fu, C. Wang, J. Wang and S. Zhu, Preparation of Poly(Ionic Liquid) Nanoparticles through RAFT/MADIX Polymerization-Induced Self-Assembly, Polym. Chem., 2017, 8, 5469 RSC; (i) Y. Yang, J. Zheng, S. Man, X. Sun and Z. An, Synthesis of Poly(Ionic Liquid)-based Nano-Objects with Morphological Transitions via RAFT Polymerization-Induced Self-Assembly in Ethanol, Polym. Chem., 2018, 9, 824 RSC.
- (a) K. M. Meek and Y. A. Elabd, Polymerized Ionic Liquid Block Copolymers for electrochemical Energy, J. Mater. Chem. A, 2015, 3, 24187–24194 RSC; (b) R. L. Weber, Y. Ye, A. L. Schmitt, Y. A. Banik, Y. A. Elabd and M. K. Mahanthappa, Effect of Nanoscale Morphology on the Conductivity of Polymerized Ionic Liquid Block Copolymers, Macromolecules, 2011, 44, 6509–6517 CrossRef; (c) V. F. Scalfani, E. F. Wiesenauer, J. R. Ekblad, J. P. Edwards, D. L. Gin and T. S. Bailey, Morphological Phase Behavior of Poly(RTIL)-Containing Diblock Copolymer Melts, Macromolecules, 2012, 45, 4262–4276 CrossRef CAS; (d) J.-H. Choi, Y. Ye, Y. A. Elabd and K. I. Winey, Network Structure and Strong Microphase Separation for High Ion Conductivity in Polymerized Ionic Liquid Block Copolymers, Macromolecules, 2013, 46, 5290–5300 CrossRef CAS; (e) S. Hanif, B. Oschmann, D. Spetter, M. N. Tahir, W. Tremel and R. Zentel, Block Copolymer from ionic liquids for the preparation of thin carbonaceous shells, Beilstein J. Org. Chem., 2017, 13, 1693–1701 CrossRef CAS PubMed.
- D. Cordella, A. Kermagoret, A. Debuigne, C. Jérôme, D. Meccereyes, M. Isik, D. Taton and C. Detrembleur, All-Poly(ionic liquid)-Based Block Copolymers by Sequential Controlled Radical Copolymerization of Vinylimidazolium Monomers, Macromolecules, 2015, 48, 5230–5243 CrossRef CAS.
- D. Cordella, A. Debuigne, C. Jérôme, Z. Kochovski, D. Taton and C. Detrembleur, One-Pot Synthesis of Double Poly(Ionic Liquid) Block Copolymers by Cobalt-Mediated Radical Polymerization-Induced Self-Assembly (CMR-PISA) in Water, Macromol. Rapid. Commun., 2016, 37, 1181–1187 CrossRef CAS PubMed.
- D. Cordella, F. Ouhib, A. Aqil, T. Defize, C. Jérôme, A. Serghei, E. Drockenmuller, K. Aissou, D. Taton and C. Detrembleur, Fluorinated Poly(ionic liquid) Diblock Copolymers Obtained by Cobalt-Mediated Radical Polymerization-Induced Self-Assembly, ACS Macro Lett., 2017, 6, 121–126 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Direct Synthesis of Thermally Responsive DMA/NIPAM Diblock and DMA/NIPAM/DMA Triblock Copolymers via Aqueous, Room Temperature RAFT Polymerization, Macromolecules, 2006, 39, 1724–1730 CrossRef CAS.
- J. Yuan, S. Soll, M. Dreschler, A. H. E. Müller and M. Antonietti, Self-Assembly of Poly(ionic liquid)s: Polymerization, Mesostructure Formation and Directional Alignment in One Step, J. Am. Chem. Soc., 2011, 133, 17556–17559 CrossRef CAS PubMed.
- M. Summers, J. Eastoe, S. Davis, Z. Du, R. M. Richardson, R. K. Heenan, D. Steytler and I. Grillo, Polymerization of Cationic Surfactant Phases, Langmuir, 2001, 17, 5388–5397 CrossRef CAS.
- S. Manet, A. Lecchi, M. Impéror-Clerc, V. Zholobenko, D. Durand, C. L. P. Oliveira, J. S. Pedersen, I. Grillo, F. Meneau and C. Rochas, Structure of Micelles of a Nonionic Block Copolymer Determined by SANS and SAXS, J. Phys. Chem. B, 2011, 39, 11318–11329 CrossRef PubMed.
- M. J. Rymaruk, C. T. O'Brien, S. L. Brown, C. N. Williams and S. P. Armes, RAFT Dispersion Polymerization of Benzyl Methacrylate in Silicone Oil Using a Silicone-Based Methacrylic Stabilizer Provides Convenient Access to Spheres, Worms, and Vesicles, Macromolecules, 2020, 53, 1785–1794 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00698j |
| This journal is © The Royal Society of Chemistry 2021 |