
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePISA: construction of self-organized and self-assembled functional vesicular structures
Samuel
Pearce
 a and
Juan
Perez-Mercader
*ab
a and
Juan
Perez-Mercader
*ab
aDepartment of Earth and Planetary Sciences and Origins of Life Initiative, Harvard University, Cambridge, MA 02138, USA. E-mail: jperezmercader@fas.harvard.edu
bSanta Fe Institute, Santa Fe, NM 87501, USA
First published on 2nd September 2020
Abstract
The ability of living organisms such as cells and bacteria to create intricate and precise structures through the self-organization, segregation and assembly of their building blocks from simpler molecules found in their environment is vital for their morphologies and functions. The encapsulation of the essential ingredients for life within a semi-permeable membrane is widely accepted to have been a key event in the origin of life. Within this membrane a state far-from-equilibrium is maintained, which provides energetic conditions enabling complex functionalities such as adaptation and reproduction through internal metabolic processes. Polymerization-induced self-assembly (PISA) is a rapidly-developing method to produce polymersomes, or polymer vesicles, which can be considered as a synthetic analogy to vesicles with a lipid bilayer. Though to express the range of functions that systems that exhibit the basic functionalities of early life is indeed a challenge, PISA offers a particularly robust “boot up” mechanism which solves some of the challenges associated with the emergence of life. Recent advances in the exploitable chemistries applied to PISA expand the scope of complex nano-to-micron scale vesicular structures autonomously generated from a homogeneous mixture in mild conditions. This could ultimately lead to the achievement of synthesizing objects capable of mimicking basic behaviors and functions of natural life. Herein, we discuss the principles, syntheses and recent advances in the controlled formation of functional vesicle structures produced by PISA, while highlighting the exciting opportunities that can be explored in this emerging field.
 Samuel Pearce | Samuel Pearce received his MChem degree at the University of Sussex in 2014 and his Ph.D. at the University of Bristol, UK in February 2019. He joined the group of Dr Perez-Mercader at Harvard University in November 2019. His research interests encompass materials chemistry, nanoscience and self-assembly. His current research focuses on the development of polymerization-induced self-assembly systems to create artificial structures which behave in a similar manner to natural living systems. |
 Juan Perez-Mercader | Juan Perez-Mercader earned his Ph.D. in Physics from the City College of New York. A Profesor de Investigacion in Spain’s National Research Council (CSIC), in 1998 he became the founding director of Spain’s Centro de Astrobiologia in association with the NASA Astrobiology Institute. In 2010 he joined Harvard as a Senior Research Fellow in the Department of Earth and Planetary Sciences and Harvard Origins of Life Initiative. He is also an External Professor at the Santa Fe Institute. He has worked in many areas of Science, including Theoretical High Energy Physics, General Relativity, Non-equilibrium Processes, Self-organization, Complexity, Astrobiology and the development of In-situ Instrumentation for Life-Detection Outside the Earth. He was the architect of Spain’s contribution to the NASA Mars Program and its participation in the Curiosity Rover. After joining Harvard his work has focused on understanding the practical role of chemistry and non-equilibrium physics in the emergence of life to generate in the laboratory ex novo synthetic artificial, non-biochemistry based forms of life. He is also active in the development of autonomous (native) chemical computation. (Picture Copyright, Harcourt Studio, Paris 2006.) |
1. Introduction
PISA can be viewed as a framework that uniquely integrates polymer chemistry and non-equilibrium physics into a unified controllable molecular environment where the above feed on one another. Its basic research and applications span many areas, from materials science or medicine, to autonomous and programmable materials and molecular soft-robots.1 But given its capacity to implement the directed self-assembly of predesigned self-organized molecular aggregates, PISA also opens new doors for the autonomous creation of biomimetic objects. We are used to seeing biological materials self-assemble into functional structures and systems with the ability to adapt by the acquisition of new functionalities to solve “problems” encountered by the living system, and therefore to evolve.2–4 Hence, among the most interesting basic functionalities that one may choose to implement are the ones shared by all extant living systems (LS), i.e., functionalities that allow LS to (a) process information, (b) synthesize its own parts from simpler components (for example, found in its environment), (c) self-reproduce and (d) evolve.2,5 Natural life as we know it of course uses biochemistry to implement the above. It comes as a nice surprise from PISA to see how non-biological chemistry is actually capable of generating rudimentary forms of basic functionalities provided by the architecture of natural living systems.Indeed, one important and attractive feature of PISA is that it enables pathways for the synthesis of out-of-equilibrium chemical systems which (in at least a limited way) are capable of executing functions analogous to some that natural living systems carry out. In other words, PISA-generated systems can be designed to be artificial mimics of natural living systems121: they offer a great “chassis” on which to implement interesting chemistry-based and chemically-programmed functional architectures.
The generation of active vesicular structures from an initially homogeneous mixture of chemicals through their autonomous self-assembly during a controlled chemical reaction and without external intervention, as is done by PISA, actually solves one of the most fundamental issues in the development of functionality: the selective encapsulation of the required molecules in small volumes where they will not easily escape, and within which the action of the so-called chemical “arithmetic demon” can be greatly suppressed.6,7 This selective encapsulation increases the likelihood of chemical reactions and maintains the local concentration of their subsequent products by preventing their diffusion into the bulk solution. At the same time, it generates an important free-energy gradient between the inside of the vesicles and their environment.
PISA is a versatile process compatible with a variety of chemistries,8 which brings closer the creation of simple (vesicular) life-mimics in the lab and potentially bring us closer to understanding the (largely postulated9,10) first steps that life on Earth may have taken in the form of protocells as it evolved from abiotic to biotic activity. Perhaps, in this way, PISA could also be used to inform the potential presence of life in other locations of the Universe where “life” (as we can imagine) may not be based on the biochemistry we see on Earth.
The formation of inanimate, i.e. not involving extant natural life and biochemistry, information-containing material structures by the self-assembly of simpler components is an extraordinary process to contemplate. This is so, perhaps, because in some sense these processes counter our common intuition, for we are used to seeing unassisted transitions usually proceeding from ordered states to less well-ordered states of matter.11,12 We know that PISA with its use of chemical fuels (monomers and activation reagents), dissipates energy, such as heat or light, to generate order. In fact, since self-assembly in PISA refers to an out-of-equilibrium polymerization process where a chemical fuel is used, PISA provides an excellent vehicle and arena for the implementation of these processes with non-biochemical materials and for their use in the synthesis of autonomously controlled functional structures. Indeed, closer examination tells us that we only need to adjust our intuition to the fact that these “order generating” processes take place in dissipative systems, and that their nature is (or can be) mostly transient.
During dissipative processes, transient patterns or structures can form and re-organize. Given the appropriate conditions, this can be accompanied by the structure acquiring more forms of doing work and the generation (or deletion) of ways in which these patterns or structures and their internal components interact within or with their external environment to perform some kind of work. That is, new functions can be generated or destroyed during these processes. But self-organization and self-assembly in PISA are both dissipative processes in their nature and we can imagine that PISA could, with appropriate control, generate new functions.
To appreciate and identify the nature of the material processes that we have in mind, it is important to briefly recall some features of these two allied concepts: self-organization and self-assembly.13 Loosely speaking, these processes occur in sufficiently complex matter. Such processes usually involve a large number of individual molecules interacting in a cooperative manner to produce ordered architectures at molecular and longer length scales. Self-organization is an out-of-equilibrium phenomenon, while self-assembly has been generally considered as the exclusive result of an equilibrium process. Specifically, self-organization is considered to be a dissipative, non-equilibrium generation of order at scales longer than the ones involving direct interactions among the self-organizing components or molecules. To maintain an ordered state, the injection or input of usable energy into the self-organized system is required. On the other hand, as noted above, self-assembly is generally understood as “a non-dissipative structural order” at a “macroscopic” level promoted by collective interactions taking place at the “microscopic level”.14–16 The latter interactions do not change the “character” of the components when they are integrated into the self-assembled structure, because chemical reactions are processes in which atoms and ions change partners with their attendant consequences, both in the structure and composition of matter and in the accompanying energy changes.17,18
However there is a third logical possibility, which is seen in nature and plays a fundamental role in bringing together the self-organization and self-assembly of collective material structures. These phenomena fall under the class of Dissipative Self-Assembled (DSA) structures and correspond to material organizations combining self-organization and self-assembly via a dissipative process.14 These processes require a source of energy capable of producing the self-organizing components that would, eventually, self-assemble. However, these processes can also affect the energy landscape at the self-assembled scale and thereby make the self-assembly depend on the out-of-equilibrium conditions. DSA phenomena have a lot in common with the bases of metabolism in chemical biology but, significantly, they are not restricted to biochemistry.
We may think of this self-assembly process as controlled by the length of the amphiphile blocks as an order parameter in phase transitions:19,20 changing the order parameter changes the cooperative properties of the self-assembled objects/structures. This, of course, is well known and has been described by many authors3,21 who introduced the packing parameter, P, for the amphiphile as a “de facto” order parameter controlling the geometries attained by self-assembled amphiphiles.1,22,23 The packing parameter can be considered as a convenient rationalization of the relationship between the ratios of the amphiphilic moieties by volume and the observed morphologies. Thus we see how by changing the value of P, we can actually enact a sequence of interesting structures, including vesicles. However, it must be remembered that the free energy of the system determines the phenomena observed, of which the primary contributions are the degree of chain-stretching within the solvophobic region, the repulsive interactions between the solvophilic moieties such as steric or electrostatic repulsions, and the interfacial energy between the solvophobic region and solvent media.24 In fact, it is here where PISA polymerization chemistry meets condensed matter physics and enables both the self-organized synthesis of amphiphiles and their self-assembly into structures. These, in turn evolve in a morphology sequence controlled by the chemical nature of the PISA reaction and the environment into which it takes place. Furthermore, since chemical reactions can be thought of as events of molecular recognition25 one can begin to envisage26–29 how PISA can potentially enable an extraordinary scenario for molecules recognizing molecules and use the sequences into which these molecules react to build material structures which can accomplish tasks or functions in a programmed fashion.
Prominent among the structures generated in PISA are the so-called polymersomes.30 These are vesicular objects consisting of a polymer membrane enclosing a lumen. Before the advent of PISA, lipid-based vesicles (liposomes) and polymersomes have been studied and applied in many areas of science and technology, with applications ranging from studies of the origin of DNA/RNA-based life to medicine and cosmetics. Without PISA, the fabrication of vesicles is usually achieved using pre-prepared amphiphiles via numerous methods which involve careful physical tuning of the constituent parts and their environments. These multistep processes are by no means autonomous and include, but are not confined to, microfluidics, extrusion and solvent-switch techniques.31
The lumen of the PISA produced vesicles can in principle contain active chemical reactions, including instances of the PISA reaction itself, and the polymer membrane of the vesicle provides an already mentioned free-energy gradient between the lumen and the environment within which the vesicle exists. This membrane contains defects and is not perfectly impermeable which can allow materials to be exchanged between the lumen and the bulk solution. The size of these polymersomes ranges between 100's nm and 10's of microns, depending on the actual details of the PISA chemistry and of course the solvent in which the chemistry takes place. Once the vesicles form, the RAFT polymerization reaction goes on both inside and outside the vesicle. Monomers which make up the solvophobic contribution are therefore consumed at different rates and their concentration in both sides of the membrane will change. This generates a compensating change in the free-energy values and gradient generated when the vesicle assembly first took place. Superimposed on this is the hydrodynamic evolution of the vesicles.3,32,33 As a consequence of the above, there begin to appear new local and global minima for the free-energy which lead to changes in morphology. All this involves simultaneous interaction at different time and space scales and, therefore, provides a number (if not more) of new possibilities for the distribution and dissipation of the energy and entropy produced in the full (open and out-of-equilibrium) system. Depending on the detailed chemical nature of the reaction components, conditions and products, the essential gradient of free-energy keeps changing as the PISA reaction goes on and may generate conditions that lead to internal order within the PISA-generated structure. In other words, PISA can lead to new functionalities by the dynamical competition between internal and external components acting as vehicles to organize the interior of the, now evolving, initially generated structure.
Some protocell systems exist which exhibit complex functions related to natural life, including lipid-based vesicles capable of division34 or systems based on coacervation35 which are of substantial interest in origins of life scenarios. These include coacervates capable of maintaining concentration gradients through membrane formation (via the addition of amphiphilic molecules),36 forming secondary structures37 and promoting complex biological processes.38 The introduction of PISA in protocell research has opened up a completely new set of scenarios, as the autonomous amphiphile synthesis and vesicular self-assembly provided by PISA addresses the significant challenge surrounding the property (b) mentioned above and offers a means for the autonomous transformation of external energy and chemical fuels into a system-level “boot-up”39 solution from a homogeneous mixture to one of the chicken and egg problems in the origin of life: the “concentration” problem. The concentration of certain key chemicals inside a vesicle/compartment can be substantially greater than in the surrounding solution. Thus, their probability of reacting increases and makes possible otherwise unlikely sequential chemical processing and synthesis due to the presence of the aforementioned “arithmetic demon”6,7 which plagues origins of life scenarios. Remarkably, besides generating functional vesicles with applications to many areas of science and technology, PISA has the potential to solve in a single shot the metabolism-first, information-first and “boot-up” process in the origin of life.40,41
To explore the application of PISA for producing synthetic functional vesicles, this review is structured as follows: during this introduction we have discussed, in a qualitative way, some of the non-equilibrium aspects of PISA. To make the review somewhat self-contained, in section 2 we will briefly discuss the principles behind PISA, followed by section 3 where we will briefly touch on some relevant methods for producing vesicles by PISA. In section 4 we discuss PISA-produced vesicles which exhibit emergent phenomena and complex behavior. This is followed by a section on responsive and controllable vesicle architectures for some areas of PISA related research. We will end with section 6 where we offer some conclusions and a perspective outlook regarding the use of PISA to synthesize objects featuring intrinsic functionalites.
2. Principles behind polymerization-induced self-assembly
2.1. The significance of controlled polymerizations
To produce well-defined and controllable assemblies in solution, it is essential that the amphiphiles are consistent in nature. Living polymerization methods yield block copolymers of low polydispersity and controllable molecular weight, which act as well-defined amphiphilic building blocks. This has allowed for the systematic study of the solution-phase self-assembly of amphiphilic block copolymers.24 In particular, controlled radical polymerizations such as nitroxide-mediated polymerization (NMP), atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain-transfer (RAFT) polymerizations have provided robust protocols to produce block copolymers of controlled molecular weight and low polydispersity. Additionally, if the living character of the polymerization is maintained during PISA, consumption of monomeric reagents can continuously occur and keep the system in a state away from thermodynamic equilibrium.2.2. Amphiphilic block copolymer solution-based self-assembly and the challenge of scale
In contrast to self-assembled constructs of molecular amphiphiles, in which the structures are highly dynamic and amphiphiles readily exchange between the structures and solution, structures comprising BCP amphiphiles are not labile with regards to the exchange of individual molecules.42 This means that the structures formed in solution by amphiphilic block copolymers are primarily under kinetic, rather than thermodynamic, control.43 The traditional method of BCP self-assembly in solution involves an approach in which a pre-prepared BCP is molecularly dissolved, followed by a switch which results in the aggregation of the core-forming block, for instance by changing the pH or solvent conditions.44 Eisenberg and coworkers published a series of important early reports of this process, in which BCPs with a short solvophilic block were used to produce a remarkable array of well-defined “crew cut” structures.45Considering the previously discussed packing parameter, in practical terms the obtained morphologies for a given BCP in solution are dependent on the relative solubility of the respective co-blocks and so changes in the solvent medium can drastically alter the morphology observed.46 More recently the use of corona-forming blocks which exhibit variable solubility profiles dependent on conditions such as temperature or pH have been used to alter the effective packing parameter, which results in a switch between observed morphologies. This gives rise to a range of triggerable, dynamic behaviors of BCP vesicles following initial assembly. Due to the complexity of the solution-phase self-assembly behavior of BCPs and its dependence on concentration, post-polymerization solvent-switch methods are often carried out under highly dilute conditions (<1% w/w). Additionally, controlled BCP assemblies produced by the traditional approach are often difficult to replicate on a large scale, limiting their commercial potential.
2.3. Morphological evolution during PISA
Chain-extension of a solvophobic block from a solvophilic polymer using a controlled polymerization method in selective solvent media, results in the precipitation of the solvophobic block in situ to produce BCP aggregates in solution (provided that a critical length of the solvophobic chain is reached). This process is the driving force behind PISA, which produces consistent micellar architectures at relatively high concentration (typically between 10–50% w/w).47,48 Although a range of different controlled radical polymerization methods can be applied to the PISA process, it is RAFT polymerization that is the most prevalent method of choice.49,50In a standard PISA experiment, the degree of polymerization of the initial solvophilic block is pre-determined and so the solvophilic volume remains constant. Initial boot-up of the PISA process occurs by input of an energy stimulus which initiates the polymerization reaction and the consumption of monomer that forms the solvophobic block begins. During the polymerization of the solvophobic block, the free-energy-gradient is maintained as the continued polymerization keeps the system in a state far-from-equilibrium. During this process, the volume fraction of the solvophobic block continually increases until full conversion of monomer is achieved.33 Mechanistic studies into the PISA process have shown that for a system in which vesicles are targeted, the observed morphologies evolve during polymerization according to the packing parameter, and the mean curvature of the structures is continually reduced in situ (Fig. 1A).51 At the point of aggregation, spherical micelles are obtained, which subsequently fuse to form short worm-like micelle structures. Long, linear worm-like micelles are then formed but rapidly branch with multiple junction points. These junction points swell and merge, producing “octopi” structures. The bilayers reach a critical point during the polymerization and “wrap-up” to form jellyfish structures, which finally enclose the remaining worm-like micelles to form vesicles (Fig. 1A). Interestingly, Zhang and coworkers showed that this process can be reversed by the polymerization of a third solvophilic coblock from pre-formed vesicles, as the volume fraction of the solvophobic block is continuously reduced.52
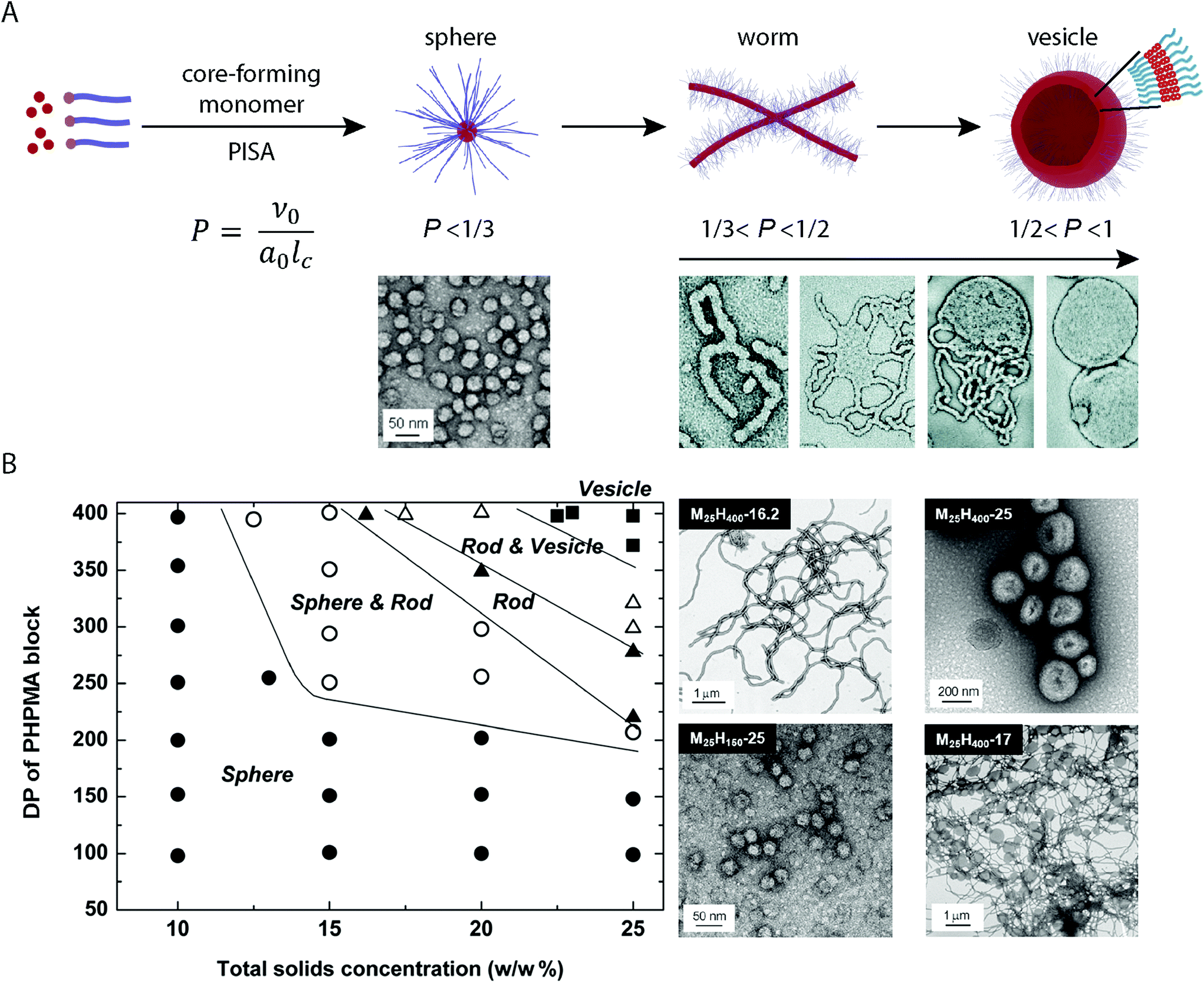 | ||
| Fig. 1 (A) Basic mechanism of the structural evolution from spheres to vesicles during the PISA process. Adapted with permission from ref. 33 and 51. (B) Constructed morphological phase diagram for a poly(2-(methacryloyloxy)ethylphosphorylcholine) (PMPC)-b-PHPMA BCP in aqueous solution. Adapted with permission from ref. 33. Copyright 2011 American Chemical Society. | ||
2.4. Morphological control and the construction of phase diagrams
Given that morphologies can be observed to change in situ during the PISA process, phase-pure BCP nanostructures can be targeted by careful control over the DPn of the core-forming block and concentration in solution.33,53 Through systematic studies, it is possible to map the experimental conditions with the observed morphologies, for a given BCP in specific solvent media. These morphological roadmaps, termed phase diagrams, allow for the predictable and reproducible PISA synthesis of BCP nanostructures in solution (Fig. 1B). The phase diagrams differ greatly between different PISA systems, so these must be constructed for each individual case.53 To obtain vesicles, generally high DPns of the core-forming block and relatively high concentrations are required. As with conventional solution-phase BCP self-assembly, it is the relative solubility of the respective coblocks in solution that determines the obtained morphologies. O'Reilly and coworkers demonstrated a method to predict the compatibility of core-forming blocks for aqueous PISA, based upon the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value per unit surface area of the core-forming block.54 This included the correlation between differences in the obtained morphologies and hydrophobicity of the core-forming block. Recently, Tan and coworkers showed that a seemingly minor difference between using poly(poly(ethylene glycol) methyl ether methacrylate) (PPEGMA)- and poly(poly(ethylene glycol) methyl ether acrylate) (PPEGA) as mCTAs in the aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA) by photo-initiated RAFT resulted in very different assemblies being obtained under analogous conditions.55 Higher order morphologies were obtained when a PPEGMA-based mCTA was used whereas nanoscale micelles were obtained when HPMA was polymerized from a PPEGA-based mCTA. Other reports include the temperature56 or light-triggered57 modification of the corona-forming block solubility, resulting in a range of morphologies being accessible with a single BCP in solution. These serve as examples by which the packing parameter can be subsequently modified, altering the “phase space” post-, (or during), polymerization.
P value per unit surface area of the core-forming block.54 This included the correlation between differences in the obtained morphologies and hydrophobicity of the core-forming block. Recently, Tan and coworkers showed that a seemingly minor difference between using poly(poly(ethylene glycol) methyl ether methacrylate) (PPEGMA)- and poly(poly(ethylene glycol) methyl ether acrylate) (PPEGA) as mCTAs in the aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA) by photo-initiated RAFT resulted in very different assemblies being obtained under analogous conditions.55 Higher order morphologies were obtained when a PPEGMA-based mCTA was used whereas nanoscale micelles were obtained when HPMA was polymerized from a PPEGA-based mCTA. Other reports include the temperature56 or light-triggered57 modification of the corona-forming block solubility, resulting in a range of morphologies being accessible with a single BCP in solution. These serve as examples by which the packing parameter can be subsequently modified, altering the “phase space” post-, (or during), polymerization.
2.5. Giant vesicles
Due to the significance of BCP vesicles in applied research, synthetic methods to produce such structures using PISA is an interesting and important subject. Though the basic mechanism for vesicle formation and subsequent increase in membrane thickness58 is relatively well-understood in a qualitative sense, the intricacies involved with controlling the chemical, physical topological distribution, and size over multiple length scales, is less well-understood. Detailed studies into the in situ changes of BCP vesicles during the continued polymerization of the core-forming block in oil or in aqueous solution showed that the overall diameter of the vesicle remained constant as the membrane thickness increased.58 This results in the reduction of the interior lumen volume until the membrane becomes unstable and destroys the BCP vesicle.58 This growth mechanism can be explained by the reduction in free energy associated with minimizing the surface area of the vesicle.The implication of this mechanism of formation and growth is that predicting and controlling the diameter of obtained vesicles produced by PISA can be difficult. It is possible to obtain vesicles of low size polydispersity by using two trithiocarbonate mCTA molecules of different chain length, PMAA62 and PMAA171.59 RAFT polymerization of benzylmethacrylate (BzMA) from the two mCTA molecules in ethanol resulted in small, uniform vesicles of ca. 199 nm in diameter, whereas larger, polydisperse vesicles of 351 nm in diameter were obtained when PMAA62 alone was used (Fig. 2A).59 This was due to the spontaneous organization of the coronae, in which the outer corona consisted mainly of the longer stabilizing PMAA171 block, whereas the shorter PMAA62 chains were located within the lumen of the vesicle. D'Agosto and coworkers displayed that a degree of control over the vesicle size could be implemented by adjusting the topology of the solvophilic block (Fig. 2B).60 Hydrophilic poly(N-acryloylmorpholine)-based (PNAM23-stat-poly(ethylene glycol)3 acrylate (PEGA)) copolymers were prepared, in which the PEG-grafted monomer units were either distributed throughout the corona-forming block or confined to the end furthest from, or at, the core–corona interface. Vesicles, rather than spherical objects, were obtained following PISA by emulsion RAFT polymerization of styrene, due to the presence of PEG-grafted macromonomers. Additionally, the specific location of the PEG-grafted macromonomers along the PNAM backbone could influence the size of the polymer vesicles. It was shown that larger vesicles were obtained when the PEG-grafted macromonomers were confined at the core–corona interface, as the steric penalty associated with the brush polymer lowered the structural curvature.60 Yuan and coworkers observed a similar effect by changing the topology of the core-forming block through brush density.61 RAFT statistical copolymerization of stearyl methacrylate (SMA) and BzMA from a poly(N,N-dimethyl-aminoethyl methacrylate) (PDMA) mCTA in ethanol resulted in the formation of vesicles which featured diameters ranging from 200 to 1500 nm, dependent on the relative volume fractions of SMA and BzMA. The largest vesicles were observed from polymerization of a 1:1 molar ratio of SMA:BzMA, as this caused the maximum disruption to the packing efficiency in the vesicle hydrophobic membrane.
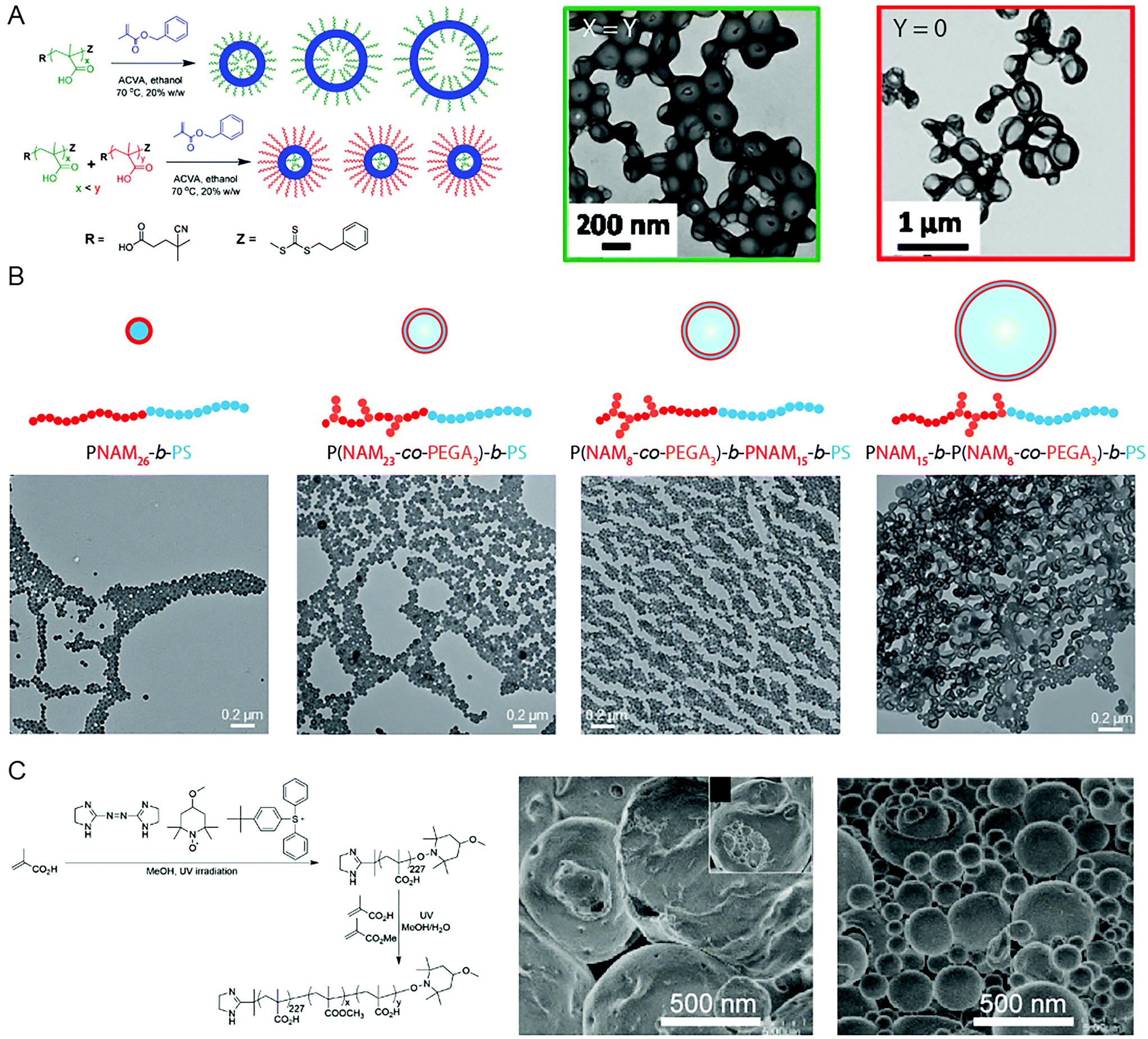 | ||
| Fig. 2 (A) Control of vesicle size and uniformity by selection of the molecular weight and ratio of two mCTA reagents. Adapted with permission from ref. 59. Copyright 2014 American Chemical Society. (B) Vesicles of controlled size by manipulation of the mCTA topology and brush location. Adapted with permission from ref. 60. Copyright 2016 Wiley-VCH. (C) Giant vesicles comprising PAA-b-(PAA-stat-PMMA) produced by PISA in a methanol/water solvent system. Adapted with permission from ref. 66. Copyright 2014 Springer Nature. | ||
Though some degree of control over the vesicle size has been demonstrated, there are relatively few reports of using PISA to produce vesicles over multiple micron scales. To obtain structures through PISA relevant to artificial biology, such as artificial protocells, the ability to produce vesicles over the relevant length scales of 1–100 μm is essential. Such structures are termed giant vesicles (GVs) or giant unilamellar vesicles (GUVs).62 GVs have been produced by conventional BCP self-assembly protocols63,64 and PISA can also be applied to the formation of such structures. Yoshida published early examples of giant vesicles in a series of reports (Fig. 2C), dating back to 2013.65 In this first example, UV-initiated NMP was used to produce a poly(methacrylic acid) (PMAA) stabilizing block in methanol, from which a poly(methyl methacrylate) (PMMA)-r-MAA random copolymer was polymerized in a MeOH/H2O 3/1 v/v selective solvent mixture. SEM imaging showed the resulting giant vesicles to be relatively uniform, exhibiting an average diameter of 4.74 μm with a polydispersity index of 1.3.65 The giant vesicles were sensitive to temperature, dissociating into small vesicles at 35 °C, followed by membrane and fiber-like aggregates at 40 °C.65 Another report documented the contortion and subsequent fission of the giant vesicle morphology with increasing polymerization of the PMMA-r-PMAA core-forming block.66 In all cases random copolymers comprising hydrophobic alkyl methacrylate and hydrophilic PMAA were used as the solvophobic coblock, which suggests that partial solvation of the core-forming block may be important to obtain the giant vesicle morphology.65,66
GVs have been produced by photo-initiated RAFT PISA under aqueous dispersion conditions, in which the mechanism of growth could be analyzed in situ by optical and fluorescence microscopy.40,67 In contrast to previous reports of the vesicle growth mechanism58 the continued polymerization of HPMA from a PEG43 mCTA resulted in the growth of giant vesicles of over 10 μm in diameter, until their eventual collapse. Associated with the variation of osmotic pressure each side of the membrane during irradiation with blue light, GV growth was driven by the influx of water through pore-defects, followed by the rapid expulsion of the lumen during collapse. This could occur in a cyclic manner, due to the presence of unreacted mCTA molecules and residual HPMA, observed as “phoenix” behavior: a name that underscores the property of these systems which experience growth, the collapse to start to grow and collapse again (sometimes for tens of cycles), as if it were from “their own ashes”.40 The light-induced formation and emergent properties of these giant vesicles appears to be a general phenomena68 and will be subject to further discussion. Additionally, RAFT polymerization mediated by oscillatory reactions have been used to produce giant vesicles.26,27,69 These represent a significant advance, as the giant vesicles could encapsulate the oscillatory reaction, which can be used as a method to transfer and manipulate chemical information, thus acting to mimic the simplest functions of early life.
3. Methods for producing vesicles by PISA
3.1 RAFT polymerization; thermal-, photo- and redox-initiated
To “boot up” the PISA process for the emergent fabrication of vesicles, an initial energy input is required. In the case of RAFT-mediated PISA, the energy is needed for the radical generation to initiate the polymerization.70 The most obvious and well-used method to input energy into the system is through heating the reaction mixture which contains a thermally-sensitive initiator (i.e. the radical source), such as α,α′-azobis(isobutyronitrile) (AIBN).48 Thermal initiators provide an efficient source of radicals but temperatures of around 70 °C required to activate the polymerization limit the scope of compatible materials and applications, leading to much interest in the exploration of alternative initiation methods.71 The ability to initiate PISA with alternative forms of energy allows us to avoid significant challenges associated with temperature dependent behavior or sensitivity when forming complex functional and adaptive vesicles.Energy from light irradiation in the UV and visible range can be used to provide energy to the system, while having the freedom to hold the reaction at a desired temperature. Zhang and coworkers produced a range of morphologies, including vesicles, through the aqueous dispersion RAFT of HPMA from a PEG113 trithiocarbonate mCTA, using sodium phenyl-2,4,6-trimethylbenzoylphosphinate (SPTP) as an initiator under 405 nm light at 25 °C.72 As the SPTP photoinitiated RAFT polymerization of HPMA is efficient over a range of temperatures, a morphological phase-diagram of PEG113-b-PHPMAn between temperature and DPn of PHMPA could be constructed, which included large compound vesicles of multiple microns in diameter.73 Differences between using a thermal and photo-initiator for PISA were highlighted by O'Reilly and coworkers, who observed that higher-order structures (i.e. worms and vesicles) were favored when photo-powered PISA, was implemented, due to the loss of the trithiocarbonate end group.74 Through appropriate choice of CTA and wavelength of irradiating light, the photo-PISA reaction can be initiated by the macro RAFT agent itself, without any added initiator or catalyst, providing a clean route to obtain BCP vesicles.75
Alternatively, it is possible for redox processes to initiate PISA. Using hydrogen peroxide to activate the enzyme horseradish peroxidase, radicals were produced by oxidation of acetyl acetone at room temperature. Aqueous dispersion polymerization of HPMA from a PEG trithiocarbonate CTA produced BCP objects ranging from spheres to vesicles.76 This method could be used to load protein molecules into the vesicles in situ, utilizing the mild conditions under which PISA was conducted. Belousov–Zhabotinsky (BZ) redox oscillatory reactions have also been employed to initiate RAFT–PISA at room temperature, by the polymerization of butyl acrylate and acrylonitrile monomers from PEG-based mCTAs in the aqueous BZ reaction media.26,27,41
3.2. Oxygen–tolerant systems for producing vesicles by PISA
As RAFT polymerizations rely on propagating radical species, most RAFT-based PISA protocols which use initiator molecules as radical sources are sensitive to oxygen and require thorough deoxygenation through nitrogen/argon displacement or multiple freeze–pump–thaw cycles. This can introduce limitation to the potential throughput and limit the scope of applications for vesicles produced using PISA-based protocols. Oxygen–tolerant living radical polymerizations have been the subject of intense interest in recent years77 and similar protocols can be applied to RAFT-mediated PISA. Boyer and coworkers reported the use of a tris(bipyridine)ruthenium(II) chloride photoredox catalyst to activate the RAFT polymerization by photoinduced electron transfer (PET) under 460 nm light irradiation (Fig. 3A).78 The continuous generation of radicals under irradiation allows for PISA without prior degassing of the solution,67,79 while also providing additional temporal control over the polymerization by “ON/OFF” irradiation of the catalyst. Other photoredox catalysts, e.g. 5,10,15,20-tetraphenyl21H,23H-porphine zinc (ZnTPP) can catalyze PET–RAFT under lower energy light irradiation to fabricate vesicles of various chemical composition by PISA.80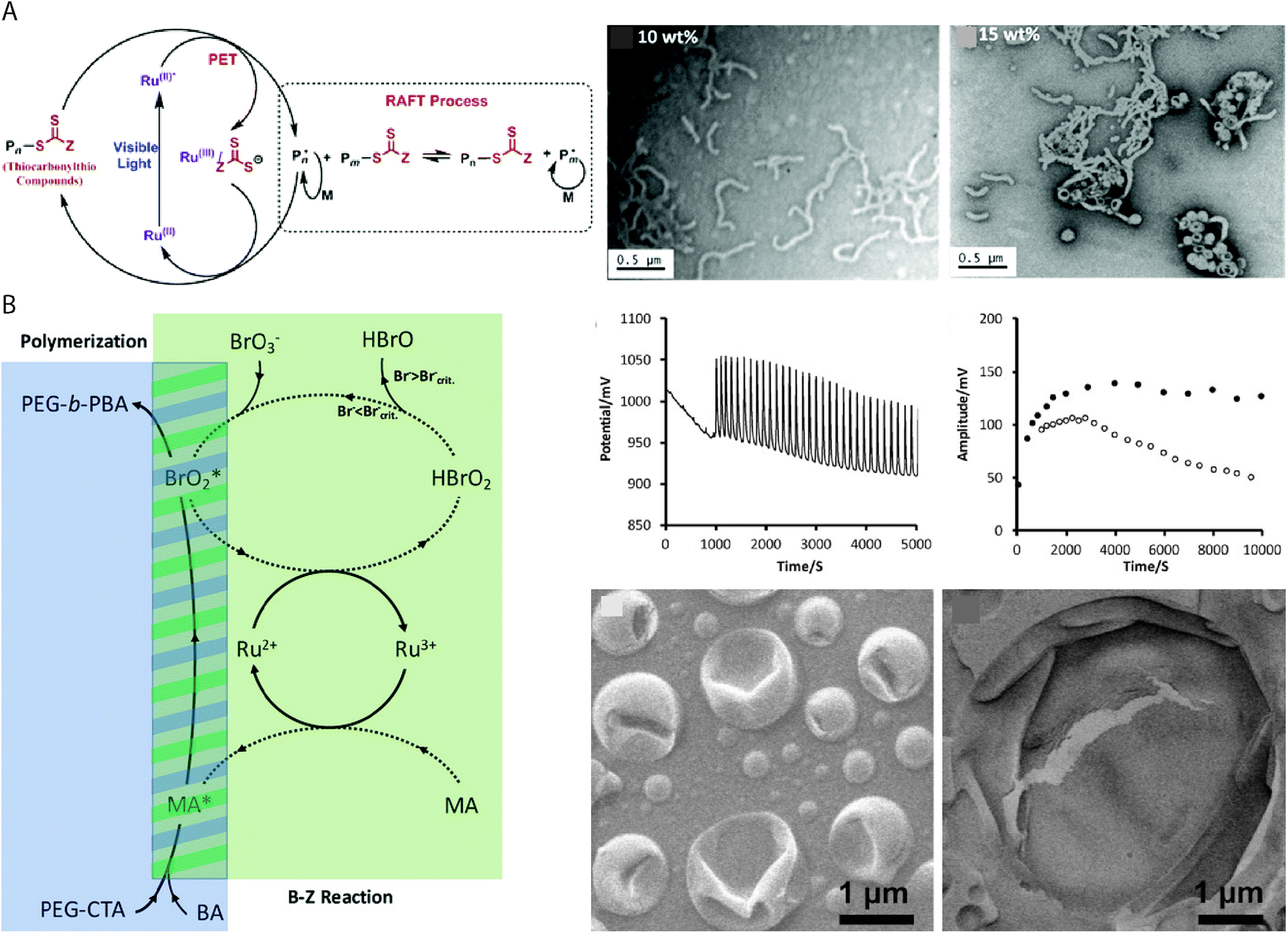 | ||
| Fig. 3 (A) Proposed mechanism of ruthenium-catalyzed PET RAFT, with TEM images of the POEGMAn-b-PBzMAm BCP structures produced by ethanolic PET–RAFT PISA at 10% and 15% w/w concentrations. Reproduced with permission from ref. 78. Copyright American Chemical Society 2015. (B) Mechanism of BZ-induced polymerization, producing BrO2* and organic radicals for polymerization. Oscillation profile of the BZ reaction, measured amplitude of redox oscillations and SEM images of the giant polymer vesicles. Reproduced with permission from ref. 26. Copyright 2017 Wiley-VCH. | ||
An early example of redox-initiated polymerization to drive vesicle formation, involved the emulsion polymerization of butyl acrylate in aqueous media using cerium(IV) ions at 40 °C for 3 hours.81 Importantly, no prior treatment such as degassing or removal of inhibitors was required for the polymerization reaction to occur. In this reaction, the alcohol functional group of a PEG34 macromolecule was activated by oxidation using the cerium(IV) ions, allowing for the radical polymerization of n-butyl acrylate to occur. Giant vesicles with an average diameter of 3.5 μm were obtained, in contrast to the nanoscale latex particles usually obtained by aqueous emulsion polymerizations. As mentioned previously, the redox oscillatory BZ reaction can be applied to PISA as a vehicle for using chemicals as a fuel to produce the radicals for RAFT polymerization.26,27,82 The BZ reaction consists of the oxidation of an organic species, typically malonic acid, by the bromate species of a strong acid, in the presence of a redox catalyst e.g. tris(bipyridine)ruthenium(II) chloride that can oscillate between two redox states. The radicals are sourced from the BrO2* and the organic species, though studies suggest that it is the organic species specifically which initiates the polymerization (Fig. 3B).26 The oscillatory nature of the reaction means that radicals are constantly generated, which results in “polymerization through” the oxygen in the system,83 allowing the PISA reaction to be carried out under open-to-air conditions. Giant vesicles produced in the reaction entrap the oscillating chemical reaction, which can be used in chemical communication or as a chemical Turing machine.25 Alternatively, a semibatch pH oscillator has been used to initiate RAFT PISA of BA τ a dithioester PEG mCTA.69 The radicals produced by the bromate–sulfite pH oscillations at 40 °C initiated the aqueous dispersion polymerization, resulting in the formation of giant vesicles after an incubation time of ca. 150 min. The giant vesicles were observed to be dynamic in the system with budding and division events, which were attributed to the far-from-equilibrium state maintained by continuous addition of sulfite necessary to maintain the pH oscillations.69
3.3. In-flow, high-throughput fabrication applied to PISA using thermal-, photo- and redox-initiated processes
PISA is an established method to obtain reproducible BCP assemblies on larger scales and at higher concentration than post-polymerization BCP self-assembly approaches. Most PISA experiments are conducted in batch, which is not ideal for industrial applications or in the case of processes which rely on the system maintaining a state far-from equilibrium. In-flow PISA set ups have been reported using thermal-initiated RAFT polymerization to produce spheres, worms and vesicles in aqueous solution (Fig. 4A).84 Warren and coworkers reported the sequential aqueous RAFT polymerization of dimethylacrylamide (DMAm), followed by diacetone acrylamide (DAAm) in the reactor yielded PDMAm-b-PDAAm BCP micelles on >100 g scales. It was observed that the polymerization rates were significantly higher in-flow compared to batch syntheses, attributed to the enhanced heat transfer to the polymerization solution in the tubular reaction. By careful control over the DPn of the mCTA, solids concentration and residence time in the reactor, pure BCP vesicles could be obtained.84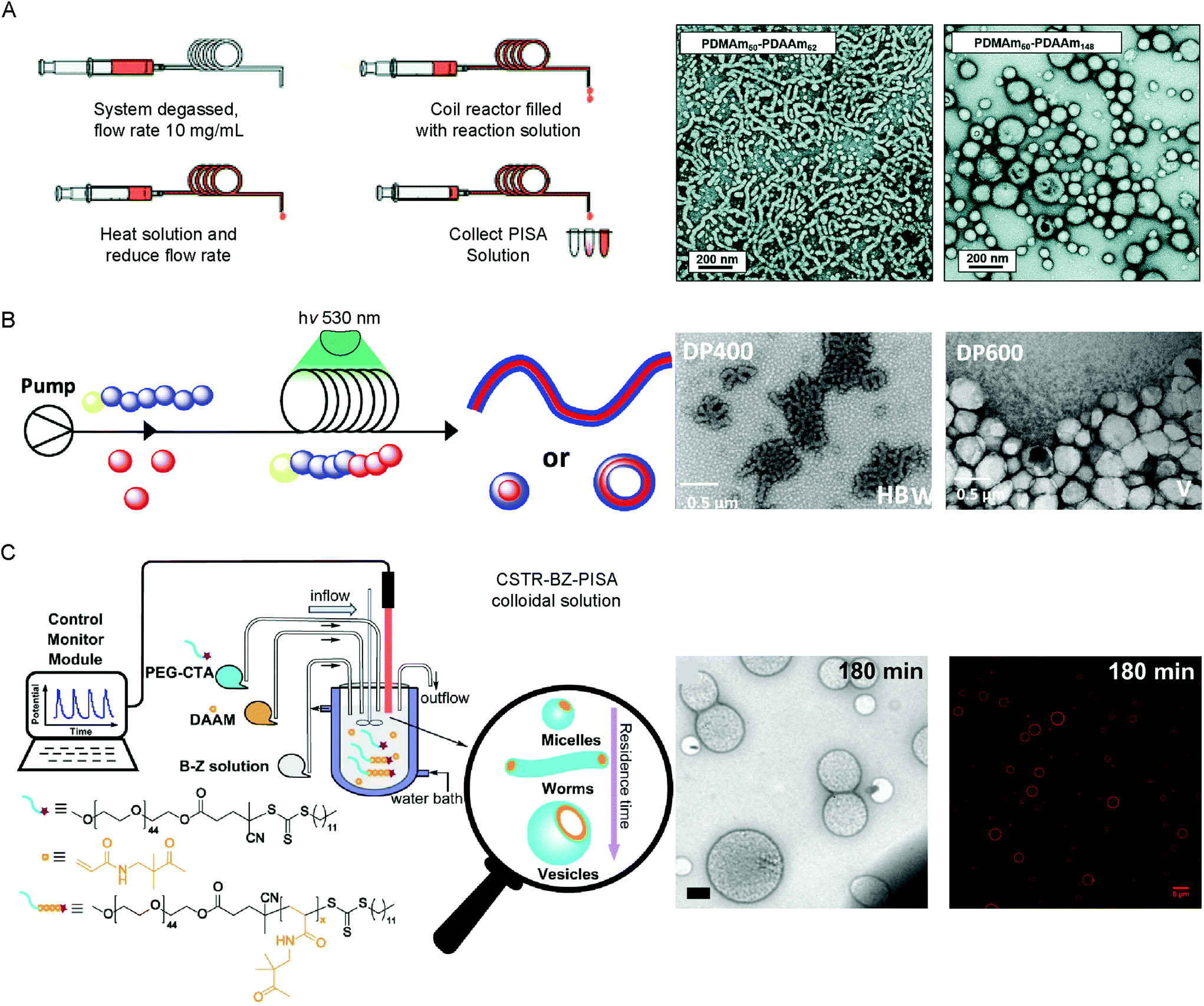 | ||
| Fig. 4 Methods used to produce vesicles by PISA in flow by (A) thermal-initiated RAFT polymerization of DAAm from PDMAm50 mCTA. Adapted with permission from ref. 84. Copyright 2019 Royal Society of Chemistry. (B) Photo-initiated PET–RAFT of diaceteone-stat-dimethyl acrylamide from a PDMA trithiocarbonate mCTA. Reproduced with permission from ref. 86. Copyright 2019 American Chemical Society. (C) BZ-initiated PISA of DAAm from a PEG44 trithiocarbonate mCTA using a CSTR in flow. Scale bar = 500 nm (TEM image) and 5 μm (fluorescence image). Reproduced with permission from ref. 29. Copyright 2019 Springer Nature. | ||
Continuous reactors for photo-initiated RAFT have been used to produce BCP structures from spherical micelles to vesicles by PISA. This approach was first reported by Boyer and coworkers, using a trithiocarbonate PEG mCTA to initiate the aqueous polymerization of HPMA under 460 nm light irradiation at 37 °C.85 Excellent control over the obtained morphology of the BCP structures could be achieved by optimization of the residence time in the reactor, greatly increasing the scalability of the PISA process. To further expand this approach, BCP vesicles were produced through an air–tolerant PET–RAFT procedure using an Eosin Y/triethanol amine catalyst system for the aqueous dispersion polymerization of PDMAA-b-(poly(diaceteone-stat-dimethyl acrylamide) PDMAA-b-(PDAAM-stat-PDMAA) copolymers using a trithiocarbonate CTA (Fig. 4B).86 The flow reactor was set up to allow the initial polymerization of the PDMAA mCTA, before DAAM monomer was introduced to the system, following a pre-determined incubation time. The reduction in optical path lengths within the tubular flow reactors allows for the uniform irradiation at higher intensity, accelerating the photo-controlled polymerization rate.87 Recently, an alternative approach was reported using an air–tolerant in-flow PISA system based on the oscillatory BZ redox reaction (Fig. 4C).29 A continuously stirred tank reactor (CSTR) was used to ensure the homogeneity of the BZ oscillations, such as the amplitude, periodicity and shape, preventing stationary space-periodic structures from occurring. Polymerization of DAAM from a trithiocarbonate PEG mCTA using BZ–RAFT–PISA yielded spherical micelles, worms, nano- and giant-sized vesicles, dependent on the residence time, BZ oscillation periodicity and the DPn of the DAAM core-forming block. The continuous nature of the flow-chemistry meant that the state of the encapsulated BZ oscillations (i.e. the chemical properties) are known and can therefore be tuned.29
4. Vesicles produced by PISA which exhibit emergent phenomena and complex behavior
4.1. Vesicle birth and death; phoenix and dye sensitive behavior of vesicles
Vesicles are formed in the last morphological transition during a conventional PISA process, after which the membrane continues to increase in thickness until a critical point is reached where the bilayer is no longer stable.58 This does not mean that the vesicle morphology is not dynamic, as the continued consumption of the ingredients of the polymerization reaction maintain a “driving force” to keep the system in a far-from-equilibrium state.Dynamic behavior of giant vesicles was observed during the Ru(bpy)32+ catalyzed photo-PISA of PEG44-b-PHPMAn in aqueous media (Fig. 5A).40,67 Initial observation of the growth then collapse of giant vesicles between 8 hours 15 minutes and 13 hours 45 minutes reaction times during the polymerization reaction, was observed by Szymanski and Perez-Mercader using fluorescence microscopy (Fig. 5A).67 This demonstrated that the far-from-equilibrium state of the system was maintained after giant vesicle formation. Giant vesicles of ca. 10 μm in diameter produced by photo-PISA could be easily imaged by optical fluorescence microscopy (Fig. 5A), using rhodamine 6G as a fluorescent probe. Additionally, the dynamic behavior of the structures due to the oxygen–tolerant polymerization of residual HPMA monomers under 450 nm light irradiation could be monitored. Acceleration of the giant vesicle growth was driven by the RAFT polymerization as the bilayer volume continually increased. During this growth period, the outer reaction solution enters the vesicle lumen through pore-defects in the vesicle membrane. The growth rate decays exponentially until a critical diameter is reached and local monomer depletion has occurred. The vesicle then reaches a maximum diameter at a critical point where the forces contributed by the membrane surface tension, the increase in core-forming polymer produced by the PISA process and the osmotic pressure between the vesicle interior and exterior become unbalanced, resulting in the rupture of the vesicle and its destructive collapse back into a droplet. This growth, collapse and regrowth of giant vesicles could occur multiple times in a process termed “phoenix” behavior (Fig. 5B).40 The maximum diameter of the giant vesicles decreased following each cycle as material is continually consumed (Fig. 5B).40 The mechanism behind “phoenix” behavior is currently not well understood, though subsequent studies found that judicious selection of the wavelength of the irradiated light, coupled with the appropriate fluorescent probe was important.68 Merging of giant vesicles stained with rhodamine 6G was observed at higher solids concentration under 540–590 nm irradiation, demonstrating the transient nature of these structures in a non-equilibrium state.
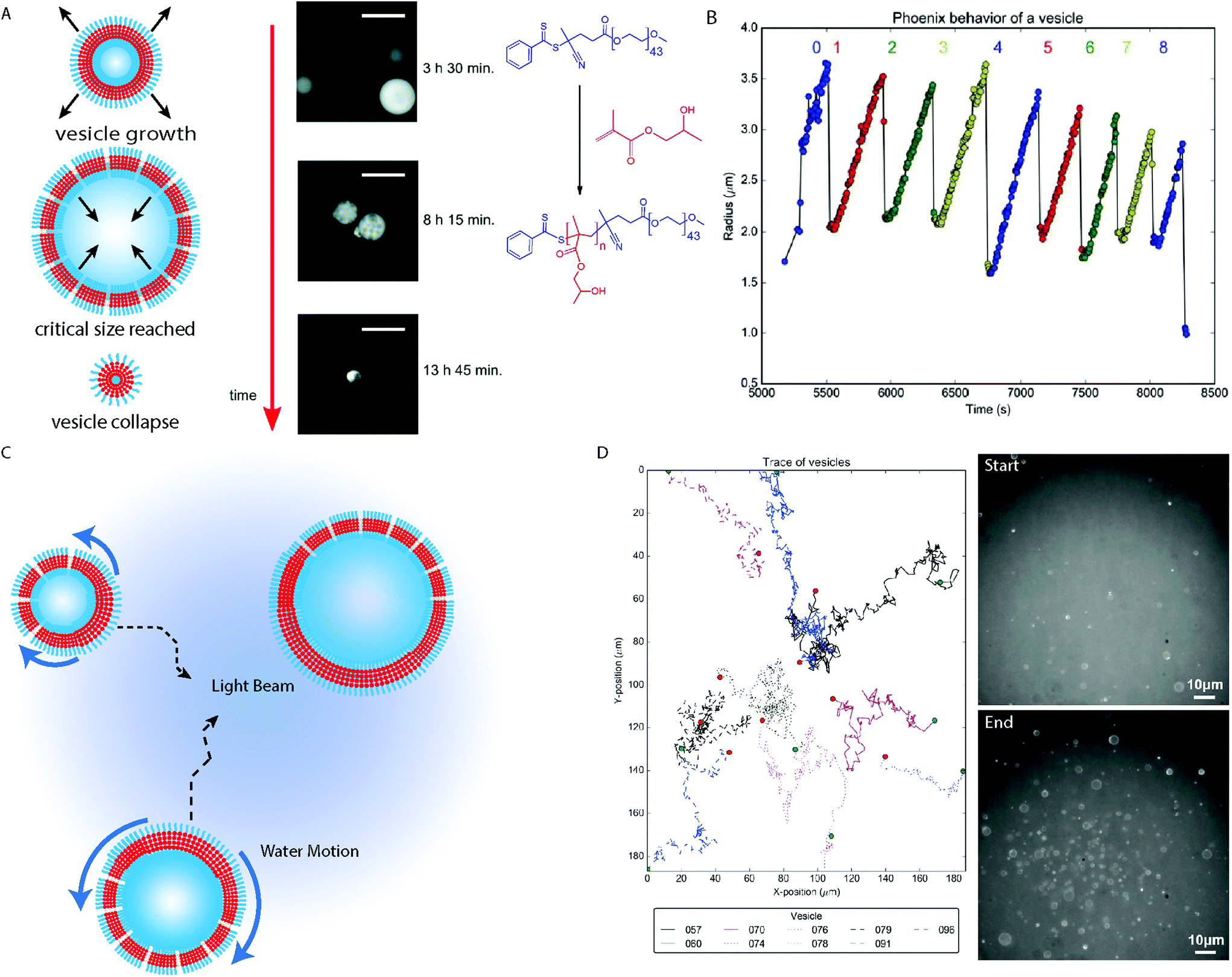 | ||
| Fig. 5 (A) Cartoon depiction of PEG-b-PHPMA vesicles with fluorescence microscopy images. Scale bars = 20 μm. Adapted with permission from ref. 67. Copyright 2016 Royal Society of Chemistry. (B) Trace displaying the change in polymersome radius when exposed to the blue light during microscopy analysis. Reproduced with permission from ref. 40. (C) Cartoon schematic depicting the Marangoni effect on vesicles formed by PISA under blue light irradiation used to activate the Ru(bpy)32+ PET–RAFT catalyst in a fluorescence microscope. (D) Path tracking of the BCP vesicles during the PISA experiment. PEG-b-PHPMA vesicles before (top) and after (bottom) analysis of the phoenix behavior within the microscope. Reproduced with permission from ref. 40. Copyright 2017 Springer Nature. | ||
Another interesting phenomena observed during the in situ microscopy analysis was the population growth of the phoenix vesicles observed during the experiment.40 To this there are two contributions: firstly, phototaxis of the giant vesicles towards the area under blue light irradiation from the surrounding regions (Fig. 5C and D). Irradiation of a specific area with blue light in the fluorescence microscope results in a reaction gradient and consumption rate of the constituent molecules, as the polymerization reaction is most efficient within the beam center, which decreases with the light intensity towards the surrounding area. In the absence of blue light irradiation, the vesicles move randomly due to Brownian motion. The reason for the phototaxis of the objects can be rationalized by the Marangoni effect, as tension is induced within the membrane of the vesicle due to the vesicle surface closest to the center of the beam experiencing a greater rate of reaction. This creates anisotropy across the membrane of the vesicle, as the regions experiencing the fastest reaction have the greatest volume of polymer within the membrane and so have a lower surface tension (Fig. 5C). The surface tension gradient within the vesicle membrane results in aqueous solution moving from the region of lowest surface tension nearest the beam center, to the region of higher surface tension, where the membrane is thinner and has a greater defect density. This produces currents within the vesicles aqueous environment pushing the giant vesicles towards the area of highest light intensity.40
The second contribution to the population growth is contributed by the conversion of droplets comprising mainly HPMA monomers stabilized by PEG-b-PHPMA amphiphiles to GVs.40 Consumption of the HPMA monomer in the beam causes a modest increase in objects, until the RAFT polymerization undergoes an exponential phase in HPMA consumption, resulting in a sharp increase in the vesicle population which continues in a non-linear manner. As the PEG mCTA and HPMA monomer are consumed by the PISA process, the rate of population growth slows and the system reaches a maximum vesicle population number. Thus it was observed that, due to the reaction gradient across the beam, a cumulative effect of monomer droplet to vesicle morphological evolution and the Marangoni-driven phototaxis of the giant vesicles to the beam center results in the dynamical phase transition observed during the analysis.40 Currently, there are many aspects to these behaviors which are unknown and need to be explored, which is the subject of ongoing investigation by our group. In principle, a parallel between the behavior exhibited by this system can be drawn with some aspects of a proto-living system, such as the spontaneous formation of a membrane from a chemically active homogenous media, metabolism and possible forms of self-replication and system selection.
4.2. Vesicle division
Living cells exist in a non-equilibrium steady state or homeostasis and the interaction between their semi-permeable membranes is of critical importance for the transfer of chemical information and biological processes involved in cellular replication. To this end, the division of polymersomes produced by PISA would further demonstrate its applicability for protolife research. Vesicles comprising PEG-b-poly(acrylonitrile) (PAN) produced by the chemically oscillating BZ reaction were observed to grow, bud then divide during the PISA process.27 The vesicle reproduction initially appeared as a smaller bleb on the side of the parent vesicle, which separates upon reaching a critical size, visualized by SEM imaging.27 PEG-b-PBA vesicles produced by PISA initiated by a chemically-fuelled pH oscillator also exhibited similar behavior.69 The nature of the oscillatory systems means that far-from-equilibrium conditions are maintained, and so the chemical environment within the interior lumen of the vesicle will be different to the outer medium, therefore chemical gradients and varying reaction rates compromises the membrane stability and so emergent phenomena result.4.3. Multi-compartment vesicles for synthetic cell-like structures
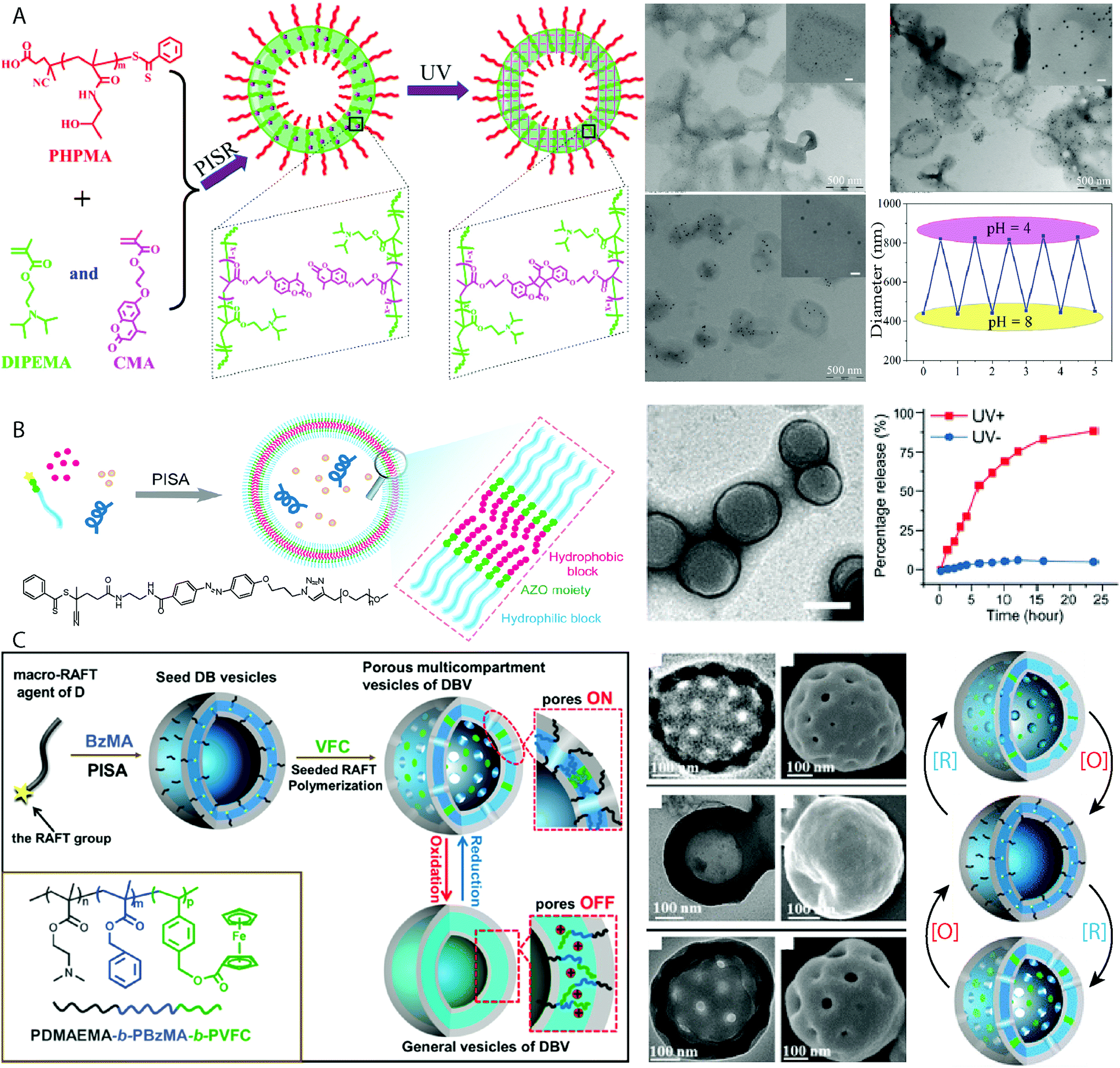
Compartmentalization is a vital aspect of the structure of eurkaryotic cells for their functionality, allowing for chemical reactions and gradients to be maintained. PISA can be used to fabricate multicompartment BCP assemblies using a variety of strategies. Framboidal compartmentalized vesicles have been produced by a “seeded” RAFT–PISA approach,88 which involved the polymerization of a third coblock, BzMA, from pre-assembled PGMA-b-PHPMA diBCP assemblies. The immiscibility between PHPMA and PBzMA resulted in the formation of PBzMA nanodomains within the wall of the vesicle, which increased in size with increasing DPn.88 A subsequent report showed that this strategy could be employed to produce pH-responsive framboidal vesicles with a PHPMA-b-PDPA core, which disassembled upon acidification of the aqueous media.89 Considering the mechanism of vesicle formation during the PISA process, the continued chain extension of an immiscible third coblock within the bilayer to form nanodomains within the membrane is an effective strategy. Yuan and coworkers systematically investigated this approach, producing a series of multicompartment BCP structures comprising PDMA-b-PBzMA-b-PFHEMA.90 Seeded RAFT–PISA was conducted using spherical, worm-like and vesicular PDMA-b-PBzMA seed structures, before chain extension of the PFHEMA block. A wide-variety of compartmentalized structures were obtained including framboidal and core–shell-corona type vesicles, clearly imaged by bright-field TEM using different staining protocols.90 Complex architectures with a porous inner structure could be achieved by the photo-initiated PISA synthesis of ABC/BC PEG-b-PHPMA-b-PBzMA/PHPMA-b-PBzMA triBCP/BCP blends.91 Polymerization of BzMA from PEG-b-PHPMA/PHPMA vesicles resulted in the formation of framboidal vesicles, with nanoscale PBzMA domains.Studies in recent years clearly show the practicality of applying PISA to produce multicompartment structures, which can offer a route to produce more complex protocell architectures. Most reports rely on immiscibility between respective coblocks within a multiBCP and the living nature of RAFT polymerization has aided such strategies. A more substantial challenge is to produce controllable structures with multiple, discrete hollow compartments. Such structures would be more akin to eukaryotic cells, which can host multiple complex chemical processes in parallel.
4.4. Oscillating vesicles
Control over contortion and motion is an important ability exhibited by biological microorganisms. Cells can expend energy and exploit osmosis to achieve such behavior. It is possible to induce swelling-deswelling behavior in synthetic giant vesicles using osmotic pressure gradients.92 When the concentration of the internal cargo is greater than the outer media, influx of water causes the vesicle to swell to a critical limit. Upon reaching a critical membrane tension, membrane defects and pore formation become energetically favorable, resulting in the release of a fraction of the interior vesicle lumen. This process occurs in a cyclic manner, observed as cycles of swelling and bursting. Autonomous oscillating vesicles have been developed by Yoshida and coworkers, using BCPs functionalized with the redox-active Ru(bpy)32+ catalyst within the core-forming block.93 When coupled to the BZ oscillatory redox reaction, the vesicles oscillated between swollen and contracted states, with the redox state of the Ru(bpy)32+/Ru(bpy)33+ catalyst.93 Although not formed through BCP self-assembly mechanisms, complex polymer–protein PNIPAM-b-PAA/bovine serum albumin-based conjugates were observed to oscillate through reversible contractions in response to externally-applied changes in temperature.94 The complex proteinosomes could be integrated with enzymatic reactions for modulating the chemomechanical response of the material. Van Hest and coworkers produced pH-responsive vesicles capable of self-regulating their size through a feedback-induced mechanism.95 The system was driven by an increase in pH through the enzymatic conversion of urea to ammonia. Depletion of the urea fuel led to shrinking of the polymersome, resulting in a switch from an “ON” to “OFF” state of an internal reaction within the vesicle lumen. Currently exploration of similar complex systems using PISA as a driving force or fabrication tool for oscillatory systems is in its infancy. The development of new synthetic protocols using PISA could result in the production of vesicles capable of similar complex adaptive and responsive behaviors.5. Responsive and controllable vesicle structures for applied research
5.1. Functionalizable and cross-linkable vesicles
As the field of PISA matures, more strategies for its application in the synthesis of “smart” structures which respond to changes in environment, or exhibit stimuli-dependent functions have emerged in recent years.71 Living cells are capable of emergent behavior such as adaptivity, self-regulation, self-replication, contortion and motion, which are reliant on a multitude of complex chemical process occurring in a coherent manner. There are relatively few examples of the utilization of PISA to produce vesicles with adaptive or self-regulatory properties, though reports featuring vesicles capable of increasingly complex functions are emerging.96While the composition of BCP vesicles is less transient in comparison to small molecule or lipid bilayer-based analogues, the vesicle structure will degrade, or dissolution of the structures will result, following changes in conditions such as solvent composition, addition of surfactants or pH, in the absence of full life-like behaviors.97 The self-assembled BCP morphology can be made resilient to such stresses through intra-core cross-linking, which can be performed in situ or using multi-pot post-polymerization techniques. For a detailed summary of the cross-linkage of BCP assemblies produced by PISA, the reader is referred to a recent perspective by Armes, Boyer and coworkers, particularly for the cross-linking of worm-like micelles.71 Herein an overview of the primary strategies associated with crosslinking BCP vesicles is discussed. Post-polymerization strategies usually involve a 2-step process which consists of the PISA synthesis of the vesicular structures, followed by an additional procedure to induce cross-linking. Chemical cross-linking by covalent bonds can be easily achieved by introducing appropriate functionality within the vesicle core. For instance, vesicles with a poly(glycidyl methacrylate) (PGlyMA) core-forming block can be easily cross-linked by a range of bifunctional nucleophilic reagents. This approach was used by Chambon, Armes and coworkers, by preparing vesicles comprising PGMA55-(PHPMA247-stat-PGlyMA82) which were subsequently cross-linked using several reagents with terminal amine functional groups.98 This allowed the vesicles to maintain their structural integrity in the presence of surfactants, which caused destruction of uncross-linked vesicles. Although this strategy makes the vesicle morphology robust, it is important to consider the implications of chemical covalent cross-linking on the properties of the membrane bilayer. Using colorimetric assays, Varlas, O'Reilly and coworkers showed that the membranes of PEG113-b-P(HPMA320-co-GlyMA80) vesicles exhibited lower permeabilities, which decreased with increasing hydrophobicity of the cross-linking reagent.99 Alternatively, ketone-containing polymers can be used as cross-linkable core-forming blocks. For instance, vesicles with a 2-(acetoacetoxy)ethyl methacrylate (AEMA) were cross-linked using keto-alkoxyamine click chemistry or functionalized with Ag nanoparticles, which showed the range of ways that keto-functionality within the vesicle bilayer could be exploited.100
Cross-linking does not necessarily rely on addition of more reagents to form covalent bonds. Through appropriate choice of monomers, energy from UV light can be used to cross-link the vesicle bilayer.101 Boyer and coworkers used an photo-orthogonal strategy whereby PEG113-b-PHPMA was synthesized by photo-RAFT polymerization under 595 nm irradiation using a zinc meso-tetra(N-methyl-4-pyridyl) porphine tetrachloride (ZnTMPyP) catalyst. Copolymerization with a small amount of 7-[4-(trifluoromethyl)coumarin] methacrylamide (TCMAm) monomers in the reaction mixture allowed for post-polymerization cross-linking of the structures by UV irradiation specifically, without adversely affecting the obtained morpholgies.102
The in situ cross-linking of vesicles is possible using bifunctional divinyl monomers, although this can be challenging as cross-linkage can compete with the primary polymerization reaction. An and coworkers avoided this issue by using an asymmetric cross-linker, allyl acrylamide, which exhibited two different vinyl reactivities.103 This resulted in the less reactive vinyl group incurring cross-linking at the latter stages of polymerization, producing vesicles with up to 5 mol% loading of cross-linkable monomer. Subsequently, it was reported that these vesicles could seed the continued polymerization of a third hydrophilic block, providing enhanced colloidal stability to the cross-linked structures.104 A similar principle can be applied to symmetric cross-linkers,105 through the copolymerization of 2-(diisopropylamino)ethyl methacrylate (DIPEMA) and cystamine–bismethacrylamide (CBMA) using a dithioester PEG macro-CTA. As the vinyl groups of the cross-linker are less reactive to the polymerization than the DIPEMA monomer, cross-linking was delayed to the latter stages of polymerization, allowing for controlled polymerization to occur beforehand.105 It is important to note that while interesting for applications and for the extension of functionality of PISA-produced objects, techniques involving the covalent post-polymerization cross-linking of vesicles require multiple processes and are not achieved from homogenous solution in a 1-pot technique as most other PISA syntheses are conducted.
5.2. Thermoresponsive and pH-responsive vesicles
The ability to react or adapt to changes in the immediate surrounding environment is of great importance to cellular life. This can include the local chemical environment, such as pH, or the physical environment such as temperature. To obtain adaptive synthetic structures using PISA, application of responsive materials or molecules and adaptive chemistries in PISA would provide a tool to achieving such goals. Such strategies can be used to implement triggered behavior such as changeable porosity or morphological transitions. For instance, Armes and coworkers produced pH-responsive assemblies by utilizing CTA molecules functionalized with morpholine and carboxylic acid terminal groups.106,107 COOH-b-PGMA43-b-PHPMAn vesicles were observed to undergo vesicle-to-sphere and vesicle-to-worm morphological transitions with PHPMA DPns of 175 and 200 respectively upon raising the pH of the aqueous solution from 3.5 to 6.106 These transitions were, however, slow over 12 h, and dependent on the DPn of the PHPMA block as no transitions were observed for vesicles with a PHPMA DPn of 250. The slow kinetics of morphological changes can be explained by the limited effect that ionization of the end terminus has on the packing parameter of vesicles with a long PHPMA core-forming block.The response rate can be increased by using a pH-responsive coblock, for instance Guragain and Perez-Mercader produced vesicles by aqueous photoRAFT PISA comprising PEG17-b-PDMAEMA45-b-PHPMAn.108 The central PDMAEMA coblock is protonated during initial polymerization of HPMA monomer at pH 3.5. Micelle-to-worm/vesicle transitions were observed upon increasing the pH to 8, which was attributed to decreased solubility of the neutral PDMAEMA central block and loss of electrostatic repulsion between monomer units.108 Zhang and coworkers produced vesicles comprising PPEGMA9-b-(PDMAEMA40/80-co-PHPMA200) by photoPISA at 25 °C, which were sensitive to the bubbling of CO2 through the aqueous solution.109 This caused a decrease in the solution pH as the CO2 dissolved to form carbonic acid, which protonated the PDMAEMA repeating units increasing the hydrophilicity of the core-forming block, resulting in the complete dissolution of the vesicle structures into polymer chains. In another example of using a weakly basic repeating unit in the polymer micelle core, Mable et al. reported rapid pH-triggered morphological transitions exhibited by PGMA-b-PHPMA-b-PDPA (DPA = 2-(diiso-propylamino)ethyl methacrylate) framboidal vesicles at pH = 8.89 The PDPA coblock is hydrophobic at high pH, resulting in colloidally-stable framboidal vesicles being produced. Once the pH was lowered to pH = 3, the PDPA block is rapidly protonated, resulting in dissolution of the vesicles, which was observed by the previously turbid solution becoming clear following acidification.89 Time resolved SAXS analysis of the vesicle following acidification showed that the vesicle structures dissolved within 1 second, with the swelling of the bilayer observed within 50 ms. To prevent vesicle dissolution upon an environmental change, core cross-linking can be utilized, as discussed previously. Pan and coworkers produced PEG113-b-PDPA90 vesicles cross-linked by a divinyl monomer CBMA which exhibited reversible pH responsivity.105 The average diameter of the vesicles was 320 nm at pH = 8, which increased to 560 nm at pH = 4, due to swelling of the core-forming block as the DPA block is protonated. The swelling/deswelling cycle could be repeated for three consecutive cycles, without dissolution of the cross-linked vesicles.105
Morphological transitions can be induced by changing temperature, without changing the chemistry of the media. Recently Ratcliffe, Armes and coworkers reported that a single block copolymer PHPMAC-b-PHPMA (PHPMAC = poly(N-(2-hydroxypropyl) methacrylamide) could produce spheres, worms and vesicles in identical media, by simply changing the solution temperature.110 After initially synthesizing PHPMAC41-b-PHPMA180 by aqueous thermal RAFT PISA at 70 °C at 10% w/w solids content, during cooling the physical appearance was observed to change from an opaque white turbid liquid, to a gel or less turbid fluid. Closer inspection by TEM analysis revealed the solution contained vesicles at 50 °C, worms at 22 °C and spherical micelles at 5 °C, which was confirmed by SAXS measurements. 1H NMR and computational experiments suggested that the degree of hydration within the PHPMA core increases as the temperature is reduced, the degree of chain-stretching within the PHPMA coblock is reduced resulting in structural transitions.110 The thermal sensitivity of PHPMA is well known; HOOC-b-PGMA43-b-PHPMA175 vesicles were observed to transition to worms after the solution temperature was lowered from 20 °C to 5 °C,106 whereas morpholine-capped PGMA43-b-HPMAn vesicles did not exhibit a change in morphology when cooled from 20 °C to 4 °C at pH = 7.107 However, at a constant pH = 3 the morpholine-capped PGMA43-b-HPMAn vesicles transitioned to spheres, indicating that the protonation of the morpholine group was necessary to induce structural transitions.107
Vesicles formed by PISA which are sensitive to alternative stimuli other than pH and temperature have also been produced. Deng, Armes and coworkers used dynamic covalent chemistry to trigger vesicle-to-worm structural transition of PGMA45-b-PHPMAn vesicles, by reaction of the alcohol functional groups on the PGMA block with 3-aminophenylboronic acid (APBA) under aqueous conditions at pH = 10.111 The formation of phenylborate ester groups along the hydrophilic PGMA polymer chains increases their effective volume, causing the structural transition to occur. The vesicle lumens were pre-loaded with silica nanoparticles, which could be released from the interior into the bulk solution upon addition of APBA. Faster release kinetics of the silica nanoparticles were observed at higher pH or APBA concentrations.111 This method also worked with vesicles which possessed a longer PHPMA block of 300 repeating units, which did not previously exhibit thermal sensitivity, extending the scope for vesicles as a delivery vector.111 UV light has been used to trigger worm-vesicle structural transitions by including UV-sensitive azo-groups within the hydrophobic segment.57 PDMA32-b-P(BzMA60-co-AzoMA6) (AzoMA = 4-phenyl-azophenyl methacrylate) worms-like micelles were initially produced by thermal RAFT PISA in ethanol at 15% w/w, then were subsequently diluted to 0.5% w/w. After UV irradiation for 1 h, vesicles structures were observed, due to the trans–cis conversion of the azo groups. This process could be reversed and the vesicles were observed to revert back to worm-like structures following 60 minutes of visible light irradiation, with intermediate structures observed during the irradiation time.57 Such structural transitions are interesting phenomena which are important when considering the next generation of smart objects, which could provide triggered structural changes of relevance to the development of complex and functional vesicles.
5.3. Vesicles with customizable porosity-triggered release
The lipid bilayer that encapsulates a cell is a sophisticated semi-permeable barrier that can maintain its chemical content against concentration gradients. A particularly interesting property of the cell membrane is its ability to undergo stimuli-responsive or triggered molecular-specific uptake/release. Producing smart vesicles capable of selective or triggered porosity is valuable for understanding how biological behaviors function. One strategy to achieve vesicles with triggered porosity was reported by Pan and coworkers, in which vesicles composed of PHPMA-b-(DIPEMA-co-PCMA) (PCMA = (2-methacryloyloxy-ethoxy)-4-methylcoumarin) prepared by PISR (polymerization-induced self-assembly and reorganization) in an ethanol/water mixture (Fig. 6A).112 The vesicle bilayer was cross-linked by UV irradiation through light-triggered dimerization of the coumarin functional groups. The pore size of these vesicles could be controlled by the degree of bilayer cross-linking, whereby the pore sizes decreased with continued exposure to UV light. Large Au nanoparticles of 15 nm diameter could diffuse into vesicles that had been exposed to 15 minutes irradiation, whereas this was limited to Au nanoparticles of 5 nm diameter after the vesicles were irradiated for 2 h.112 The tertiary amine moiety of the DIPEMA also allowed for the control over membrane porosity by pH, as the tertiary amine groups become protonated allowing the cross-linked membrane to swell, observed by the release of encapsulated fluorescein dye molecules.112 Though this method achieved control over the vesicle porosity, release and encapsulation relied on passive diffusion and stimulated release could not be quickly switched on or off. Xu and Hong also produced vesicles with switchable porosity under pH control by incorporating DIPEMA repeating units in the bilayer of PEG44-b-P(DIPEMAx-co-BzMAy) vesicles.113 At molar ratios of DIPEMA:BzMA ≥0.5 complete vesicle dissolution was observed following a change in pH from pH 7 to pH 4. Encapsulation of a hydrophilic rhodamine B dye molecule within the vesicles allowed for studies into the release kinetics of entrapped cargo following pH reduction. It was observed that sustained release without degradation of the vesicle morphology was observed for PEG44-b-P(DIPEMA0.25-co-BzMA0.75)80 specifically. On/off switching modes of release by returning the media to pH = 7 was not investigated as vesicles with a higher DIPEMA content disintegrated in the absence of cross-linking.113
 | ||
| Fig. 6 (A) UV cross-linked PHPMA-b-(DIPEMA-co-PCMA) vesicles with controllable porosity exemplified by the selective uptake of Au nanoparticles of 5 nm, 10 nm and 15 nm diameters respectively. TEM images and graph depicting the reversible increase and decrease in vesicle diameter following cyclic reduction and increase in pH. Adapted with permission from ref. 112. Copyright 2017 American Chemical Society. (B) Vesicles which exhibit UV-switchable pores using an azo-benzene linker between the hydrophilic PEG and hydrophobic PHPMA block, with TEM images displaying the vesicle morphology and graph displaying the release of rhodamine 6G during UV irradiation. Scale bar = 200 nm. Adapted with permission from ref. 114. Copyright 2019 American Chemical Society. (C) Vesicles with redox-switchable pores by reversible chemical oxidation and reduction of a ferrocene-functionalized coblock. Scale bars = 100 nm. Adapted with permission from ref. 115. Copyright 2015 American Chemical Society. | ||
Cheng and Perez-Mercader produced vesicles with switchable porosity under temporal control using UV-light by using a PEG macroCTA functionalized with an azo-benzene group to initiate photo RAFT of HPMA under aqueous dispersion conditions (Fig. 6B).114 The UV-activated switch between the cis and trans isomers of the azobenzene functional group opened and closed pores in the vesicle membrane. Repeated photoswitching of the PEG-b-PHPMA vesicles was observed by UV-vis spectroscopy, with no degradation of the vesicular morphology observed by TEM and DLS analysis. Loading of the vesicle with a dye molecule, rhodamine 6G and macromolecular FTIC-dextran showed the selective release of rhodamine 6G upon UV irradiation with no detectable release FTIC-dextran.114 FTIC-dextran release was achieved by exploiting the chemosensitivity of the azo group through cleavage using sodium dithionite. This resulted in irreversible disassembly of the vesicles and release of their interior lumen contents. Shi and Zhang achieved the synthesis of pore-switchable vesicles under redox control by using a two-step PISA-based protocol (Fig. 6C).115 Initially vesicles comprising PDMAEMA35-b-PBzMA274 were produced using thermal RAFT dispersion PISA at 70 °C. These vesicles were used to seed the growth of a redox-active poly(4-vinylbenzyl ferrocenecarboxylate) (PVFC) third coblock, which resulted in phase separation within the membrane between the immiscible PBzMA and PVFC domains. This phase separation produced pores of 30 nm diameter within the vesicles when the ferrocene moieties were in a neutral state. Oxidation of the ferrocene units using FeCl3 resulted in the closure of the pores, which reform upon reduction using SnCl2. This process can be repeated at least twice, demonstrating the redox-operated on/off switchability of the porous vesicles.115
These examples show how responsive or triggerable porosity can be achieved using relatively simple mechanisms. It should be noted that these functions usually result from the selection of the comprising BCP blocks and are not necessarily a consequence of the PISA process and alternative routes to functional vesicle structures exist via other methods.116 By integrating such processes with further functions such as release/uptake of initiator signaling molecules, biomimetic functions could later be achieved using similar principles using PISA-fabricated vesicular structures.
5.4. Information-containing vesicles using oscillatory chemical reactions
Eukaryotic cells are multi-compartment structures capable of maintaining a constant state far-from-equilibrium. They include multiple compartments which are capable of merging and delivering vital signals and chemicals for various functions. These signals are reliant on the ability of life to convert chemical energy to the distribution of information and mechanisms of computation. One synthetic example of a system which maintains an independent and dynamical far-from-equilibrium state is the BZ reaction. As discussed previously, the oscillatory reaction produces radicals which can initiate the RAFT polymerization which drives PISA as well as being the intermediate vehicles of chemical information. BZ-mediated PISA was used to generate giant vesicles from individual molecular parts, which themselves encapsulate the oscillatory reaction media.26,27,29 Recently Cheng and Perez-Mercader produced multicompartment and transient giant vesicles by BZ-mediated PISA synthesis of poly(butyl acrylate), using a Ru(bpy)32+-functionalized PEG macro-CTA (Fig. 7A).41 Ru(bpy)32+ is a redox catalyst for the BZ reaction that can also act as a photoredox catalyst for PET–RAFT polymerization, when activated by blue light.78 The multicompartment giant vesicles were characterized using fluorescence microscopy, cryo-TEM and cryo-SEM (Fig. 7B), which displayed the polymer-rich and reaction media-rich domains. Control experiments with an unfunctionalized PEG mCTA revealed no multicompartment structures, as the freely available Ru(bpy)32+ catalyst concentration in solution is not reduced by the self-assembly process. The interior compartments remain stable in the absence of light, though under 472 nm light irradiation they were observed to change in configuration and merge together due to activation of the Ru(bpy)32+ catalyst (Fig. 7C). This maintained a far-from-equilibrium state which produced the dynamic nature of the multicompartment vesicles.41 Inside cells, compartments are known to merge together for the transmission of genetic or chemical information, through non-equilibrium processes. The BZ reaction media within the lumen of the protocells can manage information, as careful configuration can allow BZ to act as a chemical Turing machine capable of processing information carried by chemistry.25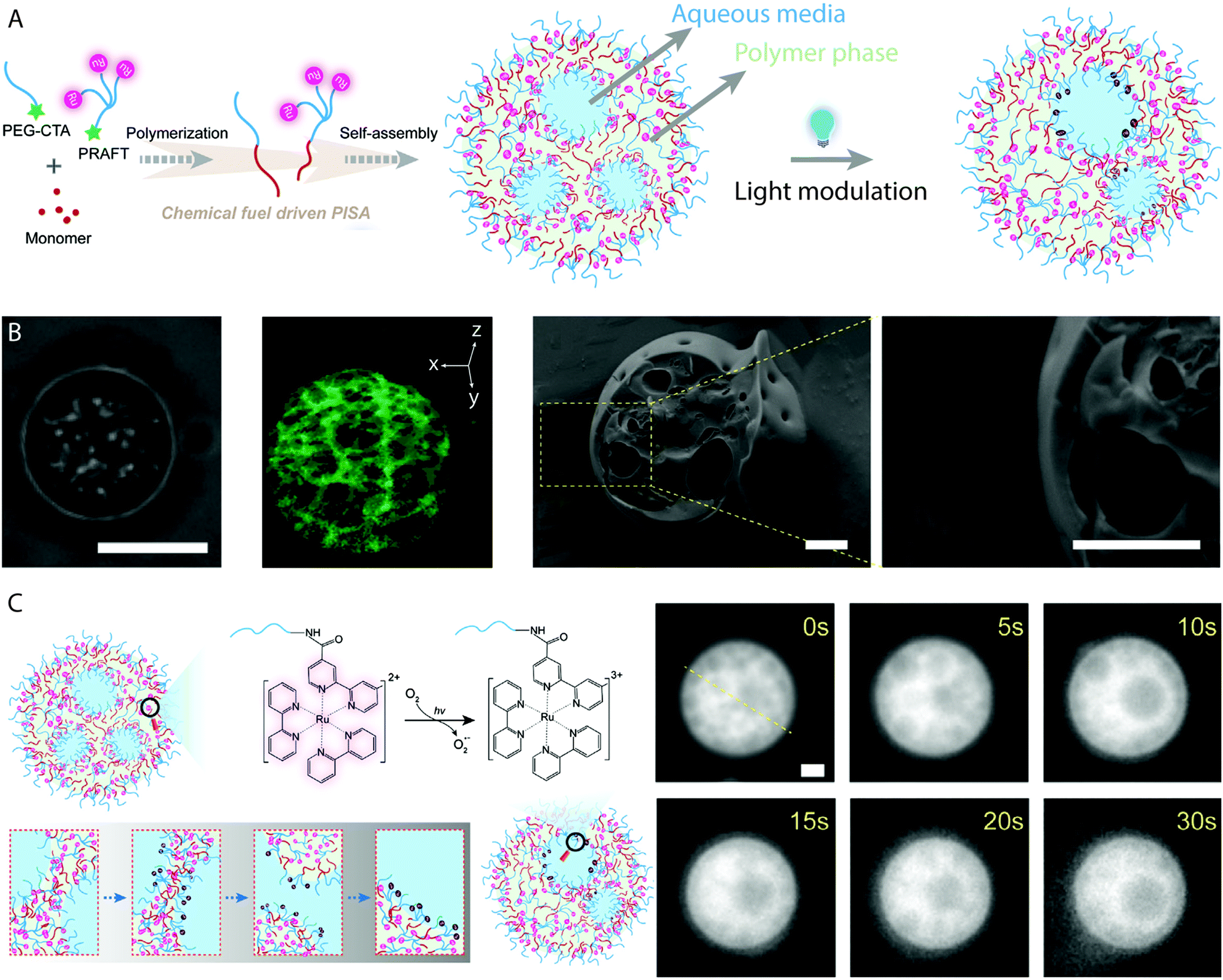 | ||
| Fig. 7 (A) Cartoon schematic representing the generation of protocell-like transient multicompartment vesicles by BZ-mediated PISA with a Ru(bpy)32+-functionalized macro-CTA. (B) Bright-field microscopy image, 3D reconstruction laser confocal microscopy image and cryo-SEM image of multicompartment vesicles. Scale bars = 5 μm (optical image) and 1 μm (cryo-SEM image). (C) Cartoon schematic depicting the mechanism of compartment reorganization and merging under 470 nm light irradiation, which corresponding fluorescence microscopy images. Scale bar = 1 μm. Adapted with permission from ref. 41. Copyright 2020 Elsevier. | ||
6. Conclusions
As the field of PISA matures, the development of new protocols expands the range of tools available to produce vesicles with multiple capabilities. By integration of the PISA process with other complementary chemistries, it is possible to generate new structures capable of maintaining a collective state far-from equilibrium. When such a far-from-equilibrium state is maintained, new structures capable of complex functions can result. Essentially, to summarize what we understand by “function” in a naïve way, is to consider it as the capacity of doing some “useful” work (i.e. leading to at least maintain the system or machine operational) using free-energy with a minimum generation of local (system) entropy. PISA provides an excellent experimental bench to study and perhaps even understand “how collective, macroscopic variables that embody [the simplest primary mimics of] biological function emerge from the many microscopic degrees of freedom”.117Complex functions in this context can result in the object possessing the simple properties or ability to exhibit behaviors in parallel to living objects of similar size. This includes response to environment, handling of information, maintenance of structure, metabolism reproduction or a combination of chemical changes or responses favorable to the environment in which the object finds itself. There are many examples in the literature which look to utilize similar principles, albeit non-PISA based, directly to applied research, for instance developing vesicles with controllable release mechanisms for drug delivery.96
The application of PISA is therefore well-suited to achieve key milestones in fields using polymer chemistry for artificial biology. For instance, the synthesis of information-containing polymers using PISA is a challenge which, to the best of our knowledge, has not been subject to investigation and new reports detailing the RAFT synthesis of sequence-controlled polymers118,119 promise potential in this area. The integration of PISA with chemical reactions which contain autocatalytic components has already been demonstrated.26,27,29,41,69 The versatility of RAFT polymerization could allow for other chemical reactions to be integrated with the PISA process, which offers another interesting route for the construction of complex systems. We believe that the dissipative nature of PISA can unlock a new subsection of systems chemistry by similar strategies and lead to a completely new generation of evolving adaptive materials.
But besides this already important help in combining the “emergence of functionality” in vesicles, there is another added bonus brought about by the introduction of a free-energy gradient when a membrane and therefore vesicles are formed: now there is a gradient of free energy between the interior and exterior of the vesicle, which can be used to actually process information. Akin to processes thought to have occurred in early life, PISA itself continuously requires and consumes energy from an external source to initiate then propagate the polymerization reaction. During the polymerization, consumption of molecular reagents, including monomer, drives the formation of structures of which the shape and conformation changes in response to the free energy landscape between the objects and their surrounding environment. The continuous input of energy induces dynamic behaviors of individual objects and the overall population through a dissipative-self-assembly mechanism.40
Intricate biological processes require the constant consumption of energy to drive the essential chemical reactions that maintain their living state. For instance, chemiosmosis is the process whereby H+ ions move across the semipermeable cell membrane, driven by the transfer of electrons between donor and acceptor species through synchronous organoredox reactions,120 vital for the synthesis of ATP and respiration. Within the membrane of synthetic polymer vesicles, solvophobic polymer chains are held in similarly close proximities, over which transfer of excitons or electrons is possible. The ability of the vesicle to allow osmosis of the PISA solution through its membrane continually changes when kept in a far-from-equilibrium state, observed for example as “phoenix” behavior.40 The propagating radical electron species are still active within the membrane during photo-polymerization and alongside the continuous generation of photoelectrons by the Ru(bpy)32+ catalyst produce a local electron-rich environment. The highly reactive electron species can push (or force) chemical reactions and processes, providing further internal osmotic instabilities favoring anisotropy in amphiphile packing and hence increase local defects. Vesicle collapse upon reaching a critical state can allow the system to reduce its free energy, release accumulated high energy chemical species and refresh the cycle. Through these contributions, an albeit simplified, parallel between PISA systems and natural processes governing function can be drawn. Further investigations regarding similar phenomena will shed important understanding of these dynamic processes and perhaps even our understanding of potential early life scenarios.121
Combining PISA-supporting polymerization methods with compatible chemistries capable of leading to modifications of the vesicle membrane parameters, integrating additional time scales or spatially segregated or concomitant changes to physico-chemical state variables, provides a unique and robust scenario/tool combination where one can construct systems capable of increasing in complexity and functional capabilities. Therefore, we can extend the chemist's toolbox to generate new materials and system-based solutions which were previously unachievable.
Here we have presented our view of this extraordinary transdisciplinary area of science and highlighted some of the progress in a selected set of problems associated with functionality, its design and implementation. We expect that when the design and implementation of function is complemented with fully adaptive chemistry, many more developments will be awaiting for us to make.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. P. would like to thank Drs Chenyu Lin, Sai Krishna-Katla, Gong Cheng, Marta Dueñas-Díez and Thomas Draper for useful discussions regarding PISA and BZ chemistry. In addition to the above, J. P.-M. thanks former members of his group Drs Albertsen, Bastakoiti, Pereira de Souza, Ren and Szymanski for many enlightening discussions on PISA and on oscillatory chemistry and Drs Guo, Guragain, Poros-Tarcali and Srivastava for useful discussions on PISA. JP-M also thanks his colleagues in the Harvard Origins of Life Initiative, Profs. Stein Jacobsen, Andrew Knoll, Dimitar Sasselov and Jack Szostak and the students in the Origins of Life Consortium for many discussions. Discussions with Prof. C. Boyer, the late Prof. Murray Gell-Mann, Prof. Stephen Mann and Prof. Geoffrey West are gratefully acknowledged. This work was funded by Repsol, S. A. and we are very grateful for all their support. The funders had no role in study design, decision to publish, or preparation of the manuscript.References
- M. Lansalot and J. Rieger, Macromol. Rapid Commun., 2019, 40, 1800885 CrossRef.
- J. Morris, D. Hartl, R. Lue, M. Michael, A. Knoll, A. Berry and A. Beiwener, Biology: How Life Works, W. H. Freeman, 3rd edn, 2019 Search PubMed.
- D. Boal, Mechanics of the cell, Cambridge University Press, 2nd edn, 2012 Search PubMed.
- D. Botstein, Decoding the language of genetics, Cold Spring Harbour Laboratory Press, 1st edn, 2015 Search PubMed.
- F. Crick, Life Itself: Its Origin and Nature, Simon and Schuster, 1st edn, 1981 Search PubMed.
- J. C. Gilbert and S. F. Martin, Experimental Organic Chemistry: A Miniscale & Microscale Approach, Cengage Learning, 6th edn, 2012 Search PubMed.
- F. Serratosa, Organic Chemistry in Action, Elsevier Science, 1st edn, 1990 Search PubMed.
- G. Cheng and J. Pérez-Mercader, Macromol. Rapid Commun., 2019, 40, 1800513 CrossRef.
- I. A. Chen and P. Walde, Cold Spring Harbor Perspect. Biol., 2010, 2, a002170 Search PubMed.
- J. Maynard Smith and S. Eörs Szathmary, The Major Transitions in Evolution, Oxford University Press, Reprint edn, 1995 Search PubMed.
- G. Nicolis and I. Prigogine, Self-organization in nonequilibrium systems: from dissipative structures to order through fluctuations, Wiley, 1st edn, 1977 Search PubMed.
- G. Lebon, D. Jou and J. Casas-Vázquez, Understanding non-equilibrium thermodynamics: Foundations, applications, frontiers, Springer, 2008 Search PubMed.
- J. D. Halley and D. A. Winkler, Complexity, 2008, 14, 10–17 CrossRef.
- S. A. P. Van Rossum, M. Tena-Solsona, J. H. Van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519 RSC.
- A. J. Bissette and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 12800–12826 CrossRef CAS.
- G. J. M. Koper, J. Boekhoven, W. E. Hendriksen, J. H. Van Esch, R. Eelkema, I. Pagonabarraga, J. M. Rubí and D. Bedeaux, Int. J. Thermophys., 2013, 34, 1229–1238 CrossRef CAS.
- M. B. Adelnik, B. A. Fisher, C. Larive, C.-J. Li, K. Matyjaszewski, A. A. Spector, M. A. White and G. Wnek, McGraw-Hill Concise Encyclopedia of Chemistry, McGraw-Hill, 1st edn, 2004 Search PubMed.
- R. P. Feynman, The Feynman Lectures on Physics, Vol. I: The New Millennium Edition: Mainly Mechanics, Radiation and Heat, Basic Books, Millen edn, 1963 Search PubMed.
- R. C. Desai and R. Kapral, Dynamics of Self-Organized and Self-Assembled Structures, Cambridge University Press, 1st edn, 2009 Search PubMed.
- P. M. Chaikin and T. C. Lubensky, Principles of condensed matter physics, Cambridge University Press, 7th edn, 2013 Search PubMed.
- J. Israelachvili, Colloids Surf., A, 1994, 91, 1–8 CrossRef CAS.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, 3rd edn, 2011 Search PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. Dueñas-Díez and J. Pérez-Mercader, iScience, 2019, 19, 514–526 CrossRef.
- B. P. Bastakoti and J. Perez-Mercader, Angew. Chem., Int. Ed., 2017, 56, 12086–12091 CrossRef CAS.
- B. P. Bastakoti and J. Perez-Mercader, Adv. Mater., 2017, 29, 1704368 CrossRef.
- R. Srivastava, M. Dueñas-Díez and J. Pérez-Mercader, React. Chem. Eng., 2018, 3, 216–226 RSC.
- L. Hou, M. Dueñas-Díez, R. Srivastava and J. Pérez-Mercader, Commun. Chem., 2019, 2, 1 CrossRef.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- L. Jin, A. E. Engelhart, K. P. Adamala and J. W. Szostak, J. Visualized Exp., 2018, 132, e57324 Search PubMed.
- U. Seifert, K. Berndl and R. Lipowsky, Phys. Rev. A, 1991, 44, 1182–1202 CrossRef CAS.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS.
- T. F. Zhu, K. Adamala, N. Zhang and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9828–9832 CrossRef CAS.
- A. I. Oparin, The Origin of Life, Macmillan, New York, 3rd edn, 1938 Search PubMed.
- T. Y. Dora Tang, C. Rohaida Che Hak, A. J. Thompson, M. K. Kuimova, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2014, 6, 527–533 CrossRef CAS.
- S. Koga, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2011, 3, 720–724 CrossRef CAS.
- M. M. K. Hansen, L. H. H. Meijer, E. Spruijt, R. J. M. Maas, M. V. Rosquelles, J. Groen, H. A. Heus and W. T. S. Huck, Nat. Nanotechnol., 2016, 11, 191–197 CrossRef CAS.
- “boot v4.”, O. E. D. Online, June 2020.
- A. N. Albertsen, J. K. Szymański and J. Pérez-Mercader, Sci. Rep., 2017, 7, 41534 CrossRef CAS.
- G. Cheng and J. Perez-Mercader, Chem, 2020, 6, 1160–1171 CAS.
- S. Jain and F. S. Bates, Macromolecules, 2004, 37, 1511–1523 CrossRef CAS.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- N. S. Cameron, M. K. Corbierre and A. Eisenberg, Can. J. Chem., 1999, 77, 1311–1326 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- L. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777–1779 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- W. M. Wan, X. L. Sun and C. Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- F. Huo, S. Li, X. He, S. A. Shah, Q. Li and W. Zhang, Macromolecules, 2014, 47, 8262–8269 CrossRef CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS.
- Y. Zhang, L. Yu, X. Dai, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 1102–1109 CrossRef CAS.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- Q. Ye, M. Huo, M. Zeng, L. Liu, L. Peng, X. Wang and J. Yuan, Macromolecules, 2018, 51, 3308–3314 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef CAS.
- J. Lesagedelahaye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef CAS.
- M. Huo, Z. Xu, M. Zeng, P. Chen, L. Liu, L. T. Yan, Y. Wei and J. Yuan, Macromolecules, 2017, 50, 9750–9759 CrossRef CAS.
- H. G. Döbereiner, Curr. Opin. Colloid Interface Sci., 2000, 5, 256–263 CrossRef.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- Y. Hu and J. Pérez-Mercader, Colloids Surf., B, 2016, 146, 406–414 CrossRef CAS.
- E. Yoshida, Colloid Polym. Sci., 2013, 291, 2733–2739 CrossRef CAS.
- E. Yoshida, Colloid Polym. Sci., 2014, 292, 1463–1468 CrossRef CAS.
- J. K. Szymanski and J. Perez-Mercader, Polym. Chem., 2016, 7, 7211–7215 RSC.
- K. Ren and J. Perez-Mercader, Polym. Chem., 2018, 9, 3594–3599 RSC.
- J. Guo, E. Poros-Tarcali and J. Perez-Mercader, Chem. Commun., 2019, 55, 9383–9386 RSC.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- Y. Zhang, J. He, X. Dai, L. Yu, J. Tan and L. Zhang, Polym. Chem., 2019, 10, 3902–3911 RSC.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 CrossRef CAS.
- J. Tan, Q. Xu, X. Li, J. He, Y. Zhang, X. Dai, L. Yu, R. Zeng and L. Zhang, Macromol. Rapid Commun., 2018, 39, 1700871 CrossRef.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- J. Yeow, J. Xu and C. Boyer, ACS Macro Lett., 2015, 4, 984–990 CrossRef CAS.
- K. Ren and J. Perez-Mercader, Polym. Chem., 2017, 8, 3548–3552 RSC.
- G. Ng, J. Yeow, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 2841–2851 RSC.
- J. K. Szymański and J. Pérez-Mercader, Langmuir, 2014, 30, 11267–11271 CrossRef.
- B. P. Bastakoti, S. Guragain and J. Perez-Mercader, Chem. – Eur. J., 2018, 24, 10621–10624 CrossRef CAS.
- K. Matyjaszewski and T. P. Davis, Handbook of Radical Polymerization, Wiley Subscription Services, Inc., A Wiley Company, 1st edn, 2003 Search PubMed.
- S. Parkinson, N. S. Hondow, J. S. Conteh, R. A. Bourne and N. J. Warren, React. Chem. Eng., 2019, 4, 852–861 RSC.
- N. Zaquen, J. Yeow, T. Junkers, C. Boyer and P. B. Zetterlund, Macromolecules, 2018, 51, 5165–5172 CrossRef CAS.
- N. Zaquen, H. Zu, Ak. M. N. B. P. H. A. Kadir, T. Junkers, P. B. Zetterlund and C. Boyer, ACS Appl. Polym. Mater., 2019, 1, 1251–1256 CrossRef CAS.
- N. Zaquen, Ak. M. N. B. P. H. A. Kadir, A. Iasa, N. Corrigan, T. Junkers, P. B. Zetterlund and C. Boyer, Macromolecules, 2019, 52, 1609–1619 CrossRef CAS.
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC.
- C. J. Mable, L. A. Fielding, M. J. Derry, O. O. Mykhaylyk, P. Chambon and S. P. Armes, Chem. Sci., 2018, 9, 1454–1463 RSC.
- M. Huo, M. Zeng, D. Li, L. Liu, Y. Wei and J. Yuan, Macromolecules, 2017, 50, 8212–8220 CrossRef CAS.
- J. Tan, Q. Xu, Y. Zhang, C. Huang, X. Li, J. He and L. Zhang, Macromolecules, 2018, 51, 7396–7406 CrossRef CAS.
- K. Oglęcka, P. Rangamani, B. Liedberg, R. S. Kraut and A. N. Parikh, eLife, 2014, 3, 3695 CrossRef.
- R. Tamate, T. Ueki and R. Yoshida, Adv. Mater., 2015, 27, 837–842 CrossRef CAS.
- P. Gobbo, A. J. Patil, M. Li, R. Harniman, W. H. Briscoe and S. Mann, Nat. Mater., 2018, 17, 1145–1153 CrossRef CAS.
- H. Che, S. Cao and J. C. M. Van Hest, J. Am. Chem. Soc., 2018, 140, 5356–5359 CrossRef CAS.
- Y. Altay, S. Cao, H. Che, L. K. E. A. Abdelmohsen and J. C. M. Van Hest, Biomacromolecules, 2019, 20, 4053–4064 CrossRef CAS.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2012, 28, 1196–1205 CrossRef CAS.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- W. Zhou, Q. Qu, W. Yu and Z. An, ACS Macro Lett., 2014, 3, 1220–1224 CrossRef CAS.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Biomacromolecules, 2017, 18, 1210–1217 CrossRef CAS.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 CrossRef CAS.
- Q. Qu, G. Liu, X. Lv, B. Zhang and Z. An, ACS Macro Lett., 2016, 5, 316–320 CrossRef CAS.
- L. Zhang, Q. Lu, X. Lv, L. Shen, B. Zhang and Z. An, Macromolecules, 2017, 50, 2165–2174 CrossRef CAS.
- M. Chen, J. W. Li, W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromolecules, 2019, 52, 1140–1149 CrossRef.
- J. R. Lovett, N. J. Warren, S. P. Armes, M. J. Smallridge and R. B. Cracknell, Macromolecules, 2016, 49, 1016–1025 CrossRef CAS.
- N. J. W. Penfold, J. R. Lovett, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2017, 8, 272–282 RSC.
- S. Guragain and J. Perez-Mercader, Polym. Chem., 2018, 9, 4000–4006 RSC.
- J. Tan, X. Zhang, D. Liu, Y. Bai, C. Huang, X. Li and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1600508 CrossRef.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58, 18964–18970 CrossRef CAS.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 15086–15095 CrossRef CAS.
- X. F. Xu, C. Y. Pan, W. J. Zhang and C. Y. Hong, Macromolecules, 2019, 52, 1965–1975 CrossRef CAS.
- G. Cheng and J. Pérez-Mercader, Chem. Mater., 2019, 31, 5691–5698 CrossRef CAS.
- P. Shi, Y. Qu, C. Liu, H. Khan, P. Sun and W. Zhang, ACS Macro Lett., 2016, 5, 88–93 CrossRef CAS.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- W. Bialek, Biophysics: Searching for principles, Princeton University Press, 2012 Search PubMed.
- A. Aerts, R. W. Lewis, Y. Zhou, N. Malic, G. Moad and A. Postma, Macromol. Rapid Commun., 2018, 39, 1800240 CrossRef.
- Z. Huang, N. Corrigan, S. Lin, C. Boyer and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1947–1955 CrossRef CAS.
- A. M. Morelli, S. Ravera, D. Calzia and I. Panfoli, Open Biol., 2019, 9, 180221 CrossRef CAS.
- J. Pérez-Mercader, Artif. Life Conf. Proc., 2020, 32, 483–490 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |