
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of tetracyclic spiroindolines by an interrupted Bischler–Napieralski reaction: total synthesis of akuammicine†
Matteo
Faltracco
 and
Eelco
Ruijter
*
and
Eelco
Ruijter
*
Department of Chemistry & Pharmaceutical Sciences, Amsterdam Institute of Molecular & Life Sciences (AIMMS), Vrije Universiteit Amsterdam, De Boelelaan 1108, 1081 HZ Amsterdam, The Netherlands. E-mail: e.ruijter@vu.nl
First published on 27th October 2021
Abstract
Judicious substrate design allows interruption of the classical Bischler–Napieralski reaction, providing access to a range of diversely substituted tetracyclic spiroindolines. These complex polycyclic scaffolds are valuable building blocks for the construction of indole alkaloids, as showcased in a concise total synthesis of (±)-akuammicine.
Cascade reactions allow the rapid generation of molecular complexity through multiple sequential bond-forming (and bond-breaking) events in a one-pot process. These exceptional advantages over traditional stepwise procedures make them particularly suited to efficiently access complex molecules such as natural products.1 Consequently, the use of cascade reactions in total synthesis is a well-established and prolific field that is constantly expanding in both methodology and scope, covering polyketides,2 alkaloids,3 terpenes,4 and steroids.5 Owing to their abundance, structural variety, and diverse biological activities, indole alkaloids occupy a prominent position among natural products (Fig. 1).6 As a result, they have attracted considerable interest from the synthetic community, leading to numerous novel approaches to this compound class in recent years.7
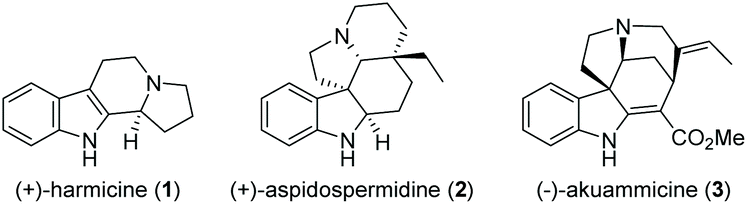 | ||
| Fig. 1 Representative indole alkaloids. | ||
Synthetic approaches toward β-carboline alkaloids (such as harmicine (1), Fig. 1) continue to rely on the classical Pictet- Spengler and Bischler–Napieralski reactions and their variations. Discovered as early as 1893, the Bischler–Napieralski reaction8 (together with its contemporary variations) is still an object of intensive study in natural product synthesis.9
Following our interest in indole alkaloids and related com-pounds,10 we recently discovered that reaction of styrylacetamides 4 under typical Bischler–Napieralski conditions (POCl3, MeCN, Δ) leads to near-quantitative formation of carbazoles 5 instead of the expected dihydro-β-carbolines (Scheme 1A).11 Our ensuing mechanistic investigation revealed a highly complex cascade pathway (Scheme 2) which proceeds via several intriguing intermediates.11 In particular, the tetracyclic spiroindoline 10 caught our interest, owing to its wide-spread presence in indole alkaloids.7i Unfortunately, our efforts to isolate 10 (R1 = H) were futile, frustrated by an elimination step (Scheme 2A) that always follows. Indeed, we only succeeded in diverting the cascade towards a different carbazole product if R3 = Br (6, Scheme 1A). This strongly suggests that interrupting the cascade at the stage of tetracyclic scaffold 10 is challenging due to the high thermodynamic driving force for the system to evolve towards aromatic products. Moreover, the intermediate 10 could revert back to 9 by retro-Mannich reaction and subsequently undergo Plancher rearrangement, irreversibly leading to β-carbolines.12
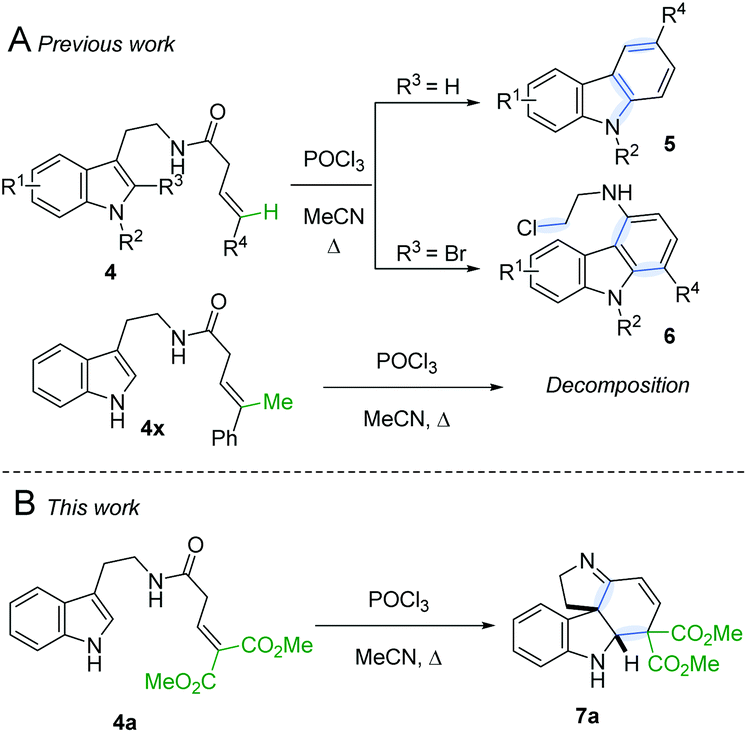 | ||
| Scheme 1 A. Previously reported carbazole formation B. Cascade interrupted at the tetracyclic intermediate. | ||
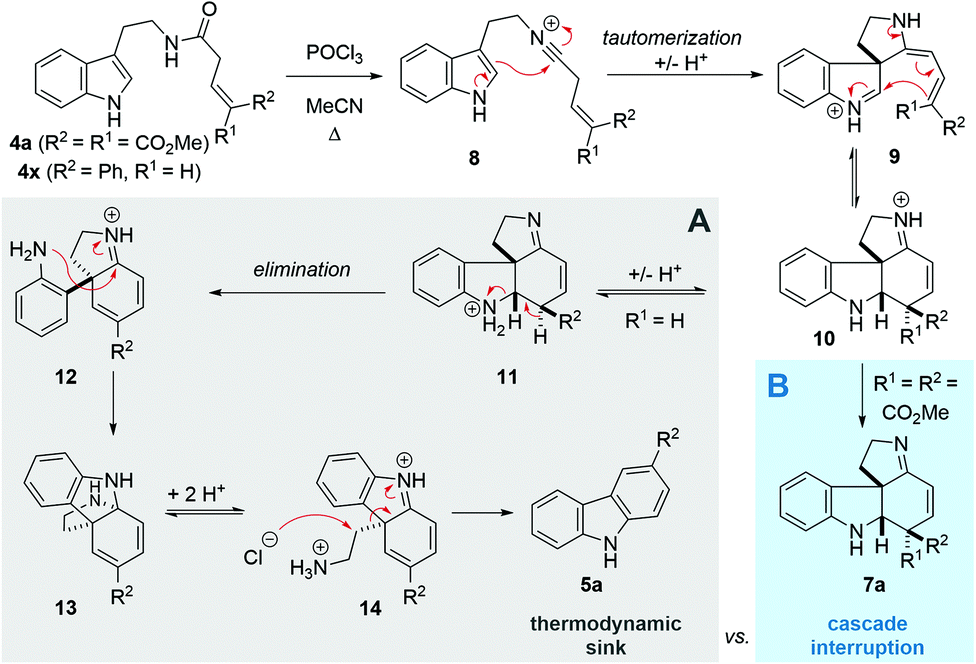 | ||
| Scheme 2 Previous cascade mechanism towards carbazoles (A) and interruption at the tetracyclic intermediate (B). | ||
A potentially viable strategy to achieve the desired interruption would be to employ a γ,γ-disubstituted vinylacetamide as the substrate, as the elimination cannot occur in this case. Since the introduction of a methyl group at this position only led to decomposition (Scheme 1A), and given our success in employing ester R4 substituents,11 we identified malonate-derived tryptamide 4a as a promising substrate. To our delight, upon subjecting 4a to the cyclization conditions (Scheme 1B), we were able to isolate imine 7a in 27% yield after basic workup. Optimization of the conditions (Table 1) showed that only POCl3 is able to promote the reaction (entries 1–5). Conveniently, we could avoid the strictly anhydrous conditions required for (both reactions and storage of) the highly reactive Tf2O that is often employed for related transformations.9a–d
|
|
||||
|---|---|---|---|---|
| Entrya | Reagent | Solvent | T (°C) | Yieldb (%) |
| a Reaction conditions 4a (0.2 mmol), reagent (0.3 mmol) solvent (1 mL). b Determined by 1H NMR using 2,5-dimethylfuran as an internal standard. c Isolated yield. d Performed with 0.4 mmol of POCl3. e Performed with 0.5 mmol of POCl3. f Additional POCl3 (0.1 mmol) added after 30 min (1 h total reaction time). g Additional POCl3 (0.1 mmol) added after 30 min and 1 h (1.5 h total reaction time). h Additional POCl3 (0.1 mmol) added after 30 min, 1 h and 1.5 h (2 h total reaction time). i Non-anhydrous MeCN was used. | ||||
| 1 | POCl3 | MeCN | Reflux | 27c |
| 2 | Tf2O/2-Cl-Py | MeCN | Reflux | — |
| 3 | Tf2O/3-CN-Py | MeCN | Reflux | — |
| 4 | Ac2O | MeCN | Reflux | — |
| 5 | TFAA | MeCN | Reflux | — |
| 6 | POCl3 | Toluene | 90 | 16 |
| 7d | POCl3 | MeCN | Reflux | 20 |
| 8e | POCl3 | MeCN | Reflux | 18 |
| 9f | POCl3 | MeCN | Reflux | 37 |
| 10g | POCl3 | MeCN | Reflux | 58c |
| 11h | POCl3 | MeCN | Reflux | 48 |
| 12g | POCl3 | MeCNi | Reflux | 58c |
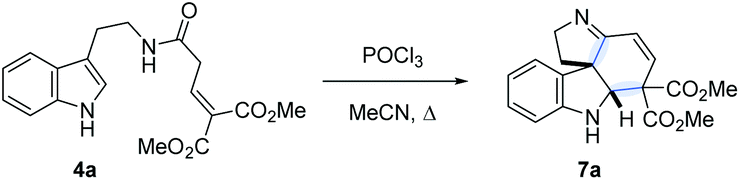
Small adjustments of POCl3 stoichiometry and reaction time (entries 6–10) allowed us to increase the isolated yield of 7a to 58%. The reaction proceeded with nearly identical efficiency on a 1.5 g scale.
We then focused on the generality of the process, subjecting differently substituted amide precursors to the optimized conditions (Scheme 3). Substituents on the benzenoid ring do not significantly affect the reactivity: reaction of the 5-F- and 5-MeO-substituted tryptamides afforded the corresponding tetracyclic scaffolds 7b and 7d in nearly identical yield (51–52%). The best result was obtained using the Cl-substituted amide 4c, producing the tetracyclic product 7c in 66% yield. Ethyl esters are also well tolerated, affording the desired product 7e in the same yield as the benchmark product 7a. The introduction of an N1-substituent on the indole ring proved to be beneficial, possibly because of the more reactive iminium ion intermediate (cf.9, Scheme 2) favoring the cyclization step. Substituents on the R5 position are also tolerated: amide 4g, derived from tryptophanol, was converted to 7g in good yield (54%) as a single diastereomer. Replacing the ethylene linker with an ortho-phenylene linker also furnished the desired spiro product 7h, albeit in lower yield (24%). Gratifyingly, the homotryptamine derivative 4i was smoothly converted to the corresponding tetracycle 7i in 55% yield. Interestingly, when substituents are present on the C2 position, a different type of product was observed (15j–l). Indeed, when R3 ≠ H, the C2-position of the indole becomes too hindered to undergo ring closure. In this case, enamine 9 can only undergo cycloaromatization (after E/Z isomerization) by attack on one of the two ester moieties to give the corresponding 2-pyridones 15. Curiously, when R3 = Me, we observed the highest yield (15j, 89%), possibly owing to the higher stability of the product. On the other hand, the bromide-substituted product 15k was obtained in lower yield. This is hardly surprising, considering the typical lability of imidoyl bromides. Similarly, the intriguing, but rather strained polycycle 15l was isolated in 59% yield. Indeed, for both 15k and 15l decomposition was already observed during purification, accounting for the lower yields.
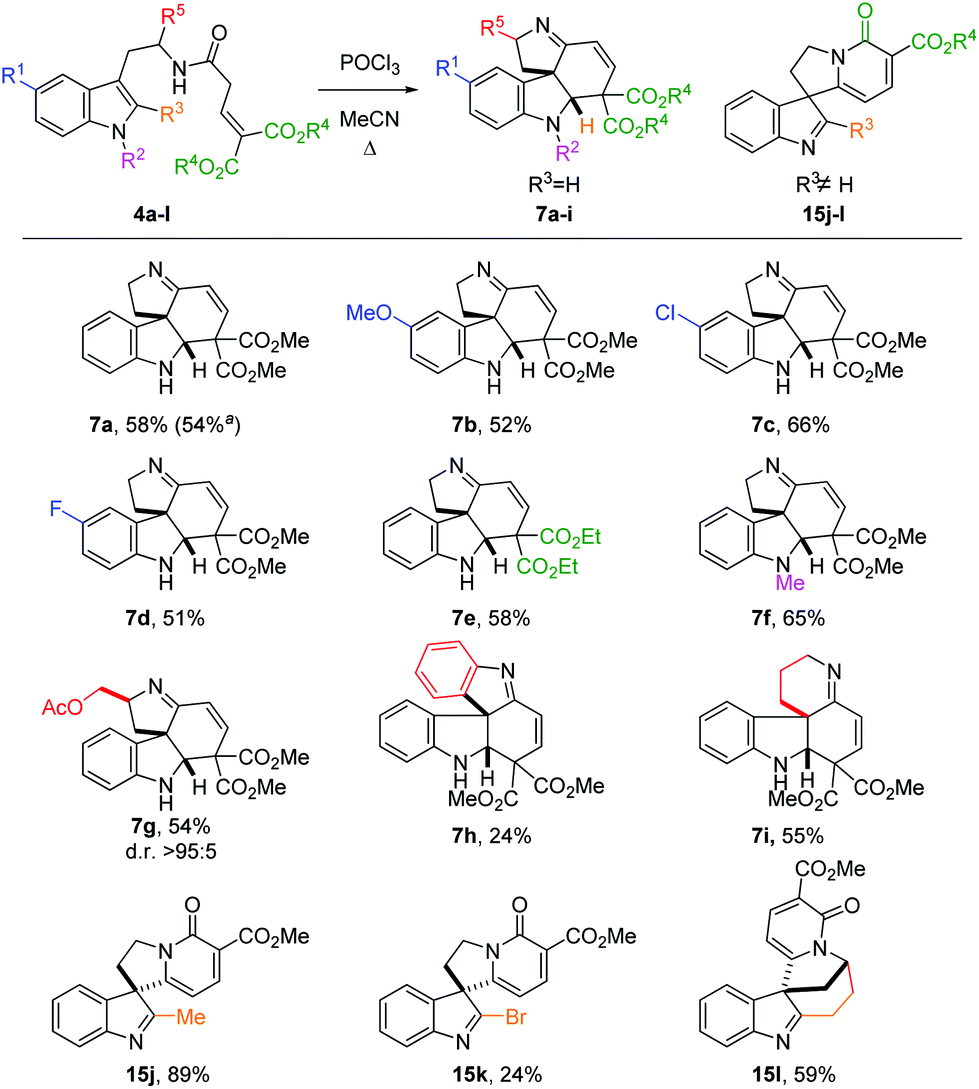 | ||
| Scheme 3 Scope of the cyclization. Reaction conditions: 4 (0.2 mmol), POCl3 (0.3 mmol), MeCN (1.0 mL), 1.5 h, reflux. Additional portions of POCl3 (0.1 mmol) were added after 30 min and 1 h. aReaction performed on 4.49 mmol scale. | ||
Once we established the scope and limitation of this trans-formation, we set out to investigate the utility of these valuable intermediates in the total synthesis of indole alkaloids. Notably, we envisioned the transformation of 7a to akuammicine (Scheme 4). Unfortunately, 7a proved recalcitrant towards selective 1,2-reduction under various conditions (NaBH4, LiBH4, NaBH3CN, NaHB(OAc)3, or Et3SiH, both in the presence and absence of acidic promotors). Presumably, the conjugated imine is too rigid and/or sterically congested to undergo efficient 1,2-reduction. To circumvent this issue, we first performed a conjugate addition with thiophenol to give imine 16, which could then be smoothly reduced by treatment with NaBH4 to give the desired amine 17 in 86% yield. Subsequent alkylation with 18 afforded the tertiary amine 19. In order to reinstall the double bond, the thioether was first oxidized and then eliminated in a two-step procedure, producing the olefin 20 in 81% yield. Subsequently, the use of KOH (1.0 equiv.) in a MeOH/THF/H2O mixture at 0 °C afforded 21via one-pot saponification, decarboxylation and double bond migration.13 Finally, known intermediate 21 was converted to (±)-akuammicine (3) by intramolecular Heck reaction as reported previously.14
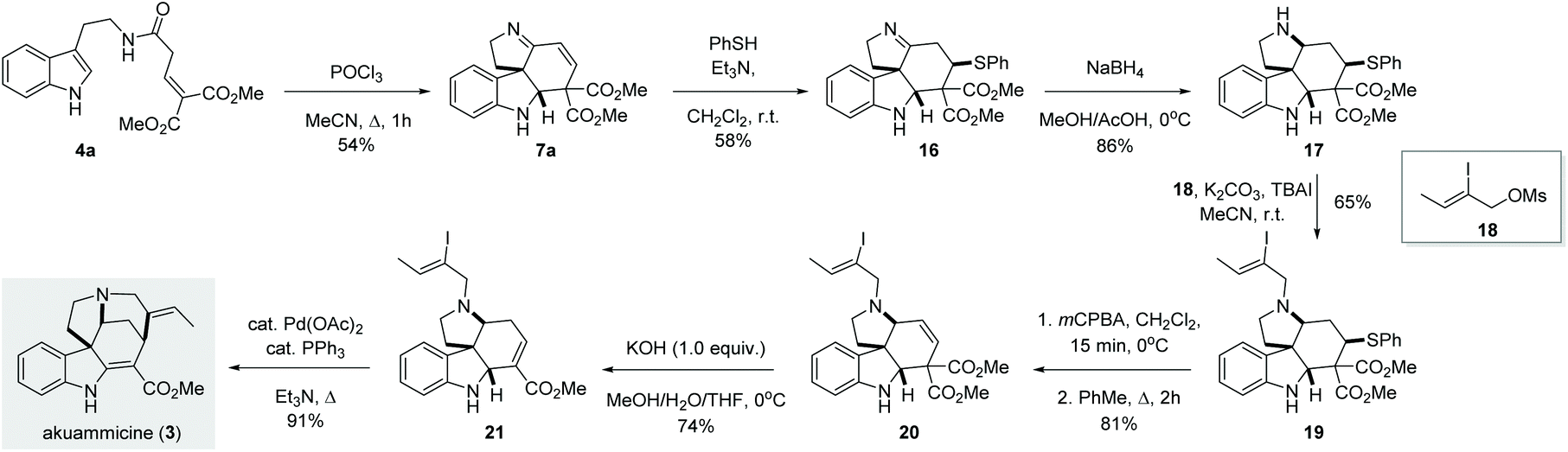 | ||
| Scheme 4 Total synthesis of (±)-akuammicine. | ||
In conclusion, we report an alternative, interrupted variation of our previously serendipitously discovered diverted Bischler–Napieralski cascade reaction. The reaction generates complex polycyclic scaffolds in a single step and is compatible with a wide range of substituents, allowing straightforward access to highly functionalized and versatile intermediates. Moreover, tetracyclic indoline 7a could be converted in only six steps to akuammicine, constituting a very short and efficient total synthesis of this Strychnos-type alkaloid. The scope and variability of the interrupted Bischler–Napieralski cyclization likely allow access to various other natural products as well.
Conflicts of interest
The authors declare no competing financial interest.Notes and references
- (a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed; (b) Y. Jiang, R. E. McNamee, P. J. Smith, A. Sozanschi, Z. Tong and E. A. Anderson, Chem. Soc. Rev., 2021, 50, 58–71 RSC.
- (a) H. C. Hailes, S. Handa, P. F. Leadlay, I. C. Lennon, S. V. Ley and J. Staunton, Tetrahedron Lett., 1994, 35, 315–318 CrossRef CAS; (b) S. V. Ley, D. S. Brown, J. A. Clase, A. J. Fairbanks, I. C. Lennon, H. M. I. Osborn, E. S. E. Stokes and D. J. Wadsworth, J. Chem. Soc., Perkin Trans. 1, 1998, 2259–2276 RSC; (c) R. Hofs, M. Walker and A. Zeeck, Angew. Chem., Int. Ed., 2000, 39, 3258–3261 CrossRef CAS; (d) C. D. Vanderwal, D. A. Vosburg, S. Weiler and E. J. Sorensen, J. Am. Chem. Soc., 2003, 125, 5393–5407 CrossRef CAS PubMed.
- (a) G. I. Elliott, J. Velcicky, H. Ishikawa, Y. Li and D. L. Boger, Angew. Chem., Int. Ed., 2006, 45, 620–622 CrossRef CAS PubMed; (b) L. F. Tietze and Y. Zhou, Angew. Chem., Int. Ed., 1999, 38, 2045–2047 CrossRef CAS; (c) J. Cassayre, F. Gagosz and S. Z. Zard, Angew. Chem., Int. Ed., 2002, 41, 1783–1785 CrossRef CAS; (d) C. H. Heathcock, M. M. Hansen, R. B. Ruggeri and J. C. Kath, J. Org. Chem., 1992, 57, 2544–2553 CrossRef CAS; (e) F. von Nussbaum, Angew. Chem., Int. Ed., 2003, 42, 3068–3071 CrossRef CAS PubMed.
- (a) E. J. Corey and S. Lin, J. Am. Chem. Soc., 1996, 118, 8765–8766 CrossRef CAS; (b) D. W. C. MacMillan, L. E. Overman and L. D. Pennington, J. Am. Chem. Soc., 2001, 123, 9033–9044 CrossRef CAS PubMed; (c) E. Lee, C. H. Yoon, Y. Sung, Y. K. Kim, M. Yun and S. Kim, J. Am. Chem. Soc., 1997, 119, 8391–8392 CrossRef CAS; (d) D. J. Kerr, A. C. Willis and B. L. Flynn, Org. Lett., 2004, 6, 457–460 CrossRef CAS PubMed; (e) B. L. Ashfeld and S. F. Martin, Org. Lett., 2005, 7, 4535–4537 CrossRef CAS PubMed.
- M. B. Gravestock, W. S. Johnson, B. E. McCarry, R. J. Parry and B. E. Ratcliffe, J. Am. Chem. Soc., 1978, 4274–4282 CrossRef CAS.
- (a) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC; (b) M. Ishikura, K. Yamada and T. Abe, Nat. Prod. Rep., 2010, 27, 1630–1680 RSC; (c) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC; (d) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2015, 32, 1389–1471 RSC.
- (a) B. M. Trost and M. Osipov, Chem. – Eur. J., 2015, 21, 16318–16343 CrossRef CAS PubMed; (b) W. Zi, Z. Zuo and D. Ma, Acc. Chem. Res., 2015, 48, 702–711 CrossRef CAS PubMed; (c) M. Ito, Y. K. Tahara and T. Shibata, Chem. – Eur. J., 2016, 22, 5468–5477 CrossRef CAS PubMed; (d) M. M. Heravi, S. Rohani, V. Zadsirjan and N. Zahedi, RSC Adv., 2017, 7, 52852–52887 RSC; (e) H. Liu and Y. Jia, Nat. Prod. Rep., 2017, 34, 411–432 RSC; (f) J. Song, D.-F. Chen and L.-Z. Gong, Natl. Sci. Rev., 2017, 4, 381–396 CrossRef CAS; (g) M. S. Kirillova, F. M. Miloserdov and A. M. Echavarren, Org. Chem. Front., 2018, 5, 273–287 RSC; (h) B. P. Pritchett and B. M. Stoltz, Nat. Prod. Rep., 2018, 35, 559–574 RSC; (i) J. M. Saya, E. Ruijter and R. V. A. Orru, Chem. – Eur. J., 2019, 25, 8916–8935 CrossRef CAS PubMed.
- A. Bischler and B. Napieralski, Chem. Ber., 1893, 26, 1903–1908 CrossRef.
- (a) S. Wang, R. Si, Q. Zhuang, X. Guo, T. Ke, X. Zhang, F. Zhang and Y. Tu, Angew. Chem., Int. Ed., 2020, 59, 21954–21958 CrossRef CAS PubMed; (b) M. Mewald, J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2014, 53, 11634–11639 CrossRef CAS PubMed; (c) K. L. White and M. Movassaghi, J. Am. Chem. Soc., 2016, 138, 11383–11389 CrossRef CAS PubMed; (d) T. Kang, K. L. White, T. J. Mann, A. H. Hoveyda and M. Movassaghi, Angew. Chem., Int. Ed., 2017, 56, 13857–13860 CrossRef CAS PubMed; (e) P. Magnus, A. H. Payne and L. Hobson, Tetrahedron Lett., 2000, 41, 2077–2081 CrossRef CAS; (f) P. Magnus, L. Gazzard, L. Hobson, A. H. Payne and V. Lynch, Tetrahedron Lett., 1999, 40, 5135–5138 CrossRef CAS.
- (a) C. de Graaff, L. Bensch, S. J. Boersma, R. C. Cioc, M. J. van Lint, E. Janssen, N. J. Turner, R. V. A. Orru and E. Ruijter, Angew. Chem., Int. Ed., 2015, 54, 14133–14136 CrossRef CAS PubMed; (b) J. M. Saya, E. D. H. van Wordragen, R. V. A. Orru and E. Ruijter, Eur. J. Org. Chem., 2019, 5156–5160 CrossRef CAS PubMed; (c) J. M. Saya, T. R. Roose, J. J. Peek, B. Weijers, T. J. S. de Waal, C. M. L. Vande Velde, R. V. A. Orru and E. Ruijter, Angew. Chem., Int. Ed., 2018, 57, 15232–15236 CrossRef CAS PubMed.
- M. Faltracco, S. Ortega-Rosales, E. Janssen, R. C. Cioc, C. M. L. Vande Velde and E. Ruijter, Org. Lett., 2021, 23, 3100–3104 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, S. M. Dalby, S. Li, T. Suzuki and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2009, 48, 7616–7620 CrossRef CAS PubMed; (b) W. Zhang, X. Chen, Y. An, J. Wang, C. Zhuang, P. Tang and F. Chen, Chem. – Eur. J., 2020, 26, 10439–10443 CrossRef CAS PubMed; (c) Y. Liu, Q. Wang, Y. Zhang, J. Huang, L. Nie, J. Chen, W. Cao and X. Wu, J. Org. Chem., 2013, 78, 12009–12017 CrossRef CAS PubMed; (d) G. He, B. List and M. Christmann, Angew. Chem., Int. Ed., 2021, 60, 13591–13596 CrossRef CAS PubMed.
- Several attempts to promote Krapcho decarboxylation of 18 (using LiCl, NaCl or KCl, with or without water, in both DMF and DMSO) only led to decomposition, most likely involving retro-Mannich reaction and Plancher rearrangement. The same result was observed when the hydrolysis was performed with LiOH.
- (a) A. Y. Hong and C. D. Vanderwal, J. Am. Chem. Soc., 2015, 137, 7306–7309 CrossRef CAS PubMed; (b) For an earlier application in the synthesis of dehydrotubifoline, see: V. H. Rawal, C. Michoud and R. Monestel, J. Am. Chem. Soc., 1993, 115, 3030–3031 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and 1H and 13C NMR spectra. See DOI: 10.1039/d1ob01966j |
| This journal is © The Royal Society of Chemistry 2021 |