
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of [Pd2L4][BF4]4 cages for binding of n-octyl glycosides and nitrate (L = isophthalamide or dipicolinamide linked dipyridyl ligand)†
Brian J. J.
Timmer
 ,
Eduard O.
Bobylev
and
Tiddo J.
Mooibroek
*
,
Eduard O.
Bobylev
and
Tiddo J.
Mooibroek
*
Van ‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands. E-mail: t.j.mooibroek@uva.nl
First published on 12th July 2021
Abstract
Two dipyridyl ligands were synthesized, where the pyridyl donor fragments were separated by an isophthalamide (1) or a dipicolinamide moiety (2). Both ligands formed [Pd2(Ligand)4][BF4]4 complexes in CD2Cl2 containing 5% dmso-d6. It was found that while [Pd2(1)4][BF4]4 readily binds to n-octyl glycosides and to nitrate anions, [Pd2(2)4][BF4]4 did not. The difference in binding properties could be rationalized based on the reduced flexibility and size of the [Pd2(2)4]2+ cage and/or stronger interior binding of a BF4− counter anion.
Introduction
It is well-known that cage complexes of the type [M2L4]4+ are readily prepared when the metal (M) is a divalent Pd2+ ion and the ligand (L) is a dipyridyl ligand.1 Such complexes typically exhibit a hollow interior that is suitable to host smaller molecules.1a,2 Recently, it was shown that such [M2L4]4+ complexes could be used to bind carbohydrates.3 In particular, it was shown that a dipyridyl ligand such as 1 in Fig. 1 formed a [Pd2L4]2+ cage4 that could bind to n-octyl-β-D-glucoside.3c As is highlighted in red in Fig. 1, the amides in a cage derived from 1 experience a steric clash with the isophthalamide's central C–H fragment. We wondered what the effect would be of C–H → N adjustment of 1 to the dipicolinamide analogue 2, where the amides should be preorganized by intramolecular N–H⋯N hydrogen bonds (also highlighted in red).5 Herein, we report on the synthesis of 1 and 2 and their [Pd2(Ligand)4][BF4]4 complexes and shown that the C–H → N adjustment is detrimental to the binding properties of the cage.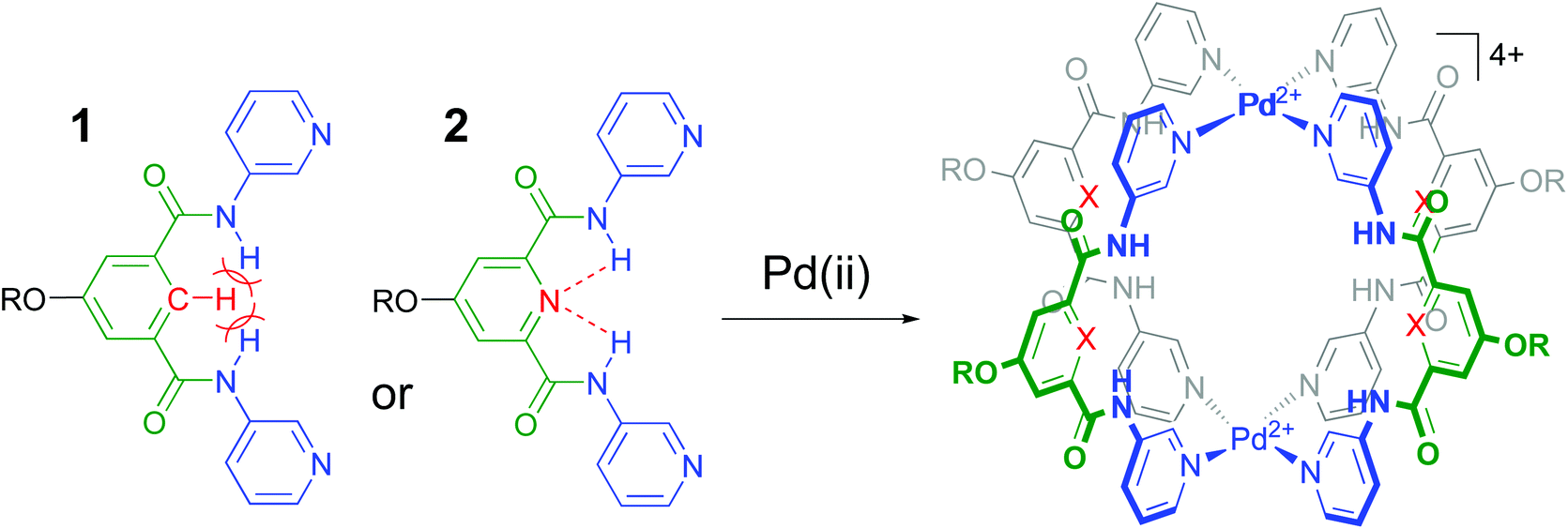 | ||
| Fig. 1 Ligands 1 and 2 used in this study to make [Pd2(Ligand)4]4+ complexes. R = solubility group = –(CH2)2O-p-Ph-C(p-tBu-Ph)3. See section S2† for synthetic details and see Fig. S52† for a model of the steric clash present in 1. | ||
Results and discussion
As is detailed in the ESI (section S2†), ligands 1 and 2 could easily be prepared according to known chemistry adapted from literature procedures.3c,6 Instead of the –C(O)NHCH2C((CH2)2t-Bu)3 solubilizing group used in a previous version of 1,3c we opted for the –(CH2)2O-p-Ph-C(p-tBu-Ph)3 group6a due to the more facile synthesis, particularly to obtain the dipicolinamide derived ligand. Stepwise addition of 0.5 equivalents of [Pd(OSMe2)4[BArF]2 to a solution of 1 in CD2Cl2 containing 5% dmso-d6 led to the formation of single species on NMR, most likely [Pd214][BArF]4 (Fig. S32,† BArF = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate). Following the same procedure with ligand 2 however, gave complicated spectra that did not resolve to a neat spectrum, not even after standing for a week (Fig. S33†). Repeating the procedure but with [Pd(NCMe)4[BF4]2 gave clearly resolved spectra for both 1 and 2 after standing for 7 days and these final spectra are shown in Fig. 2. The difference in complex formation is likely due to a templating effect of BF4−.7 For the dipyridyl ligand 1 (Fig. 2a), the inwards facing s3NH, p2 and s4 displayed significant downfield shifts that are characteristic of [Pd2(Ligand)4]4+ formation.3c,d With ligand 2 (Fig. 2b) the inwards facing s3NH and p2 underwent an upfield shift upon Pd-coordination. With both ligands, the resonances belonging to the CH's of the solubilizing groups remained unperturbed. Moreover, DOSY-NMR showed that the complexes had a larger diffusion constant than their parent ligand, which is consistent with formation of [Pd2(Ligand)4]2+ complexes. A thorough NMR spectroscopic evaluation of the complexes was also consistent with the formation of [Pd214]4+ and [Pd224]4+ and high resolution mass spectroscopy of both complexes was congruent with the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio of Pd to ligand (see Fig. S23 and S32†).
:4 molar ratio of Pd to ligand (see Fig. S23 and S32†).
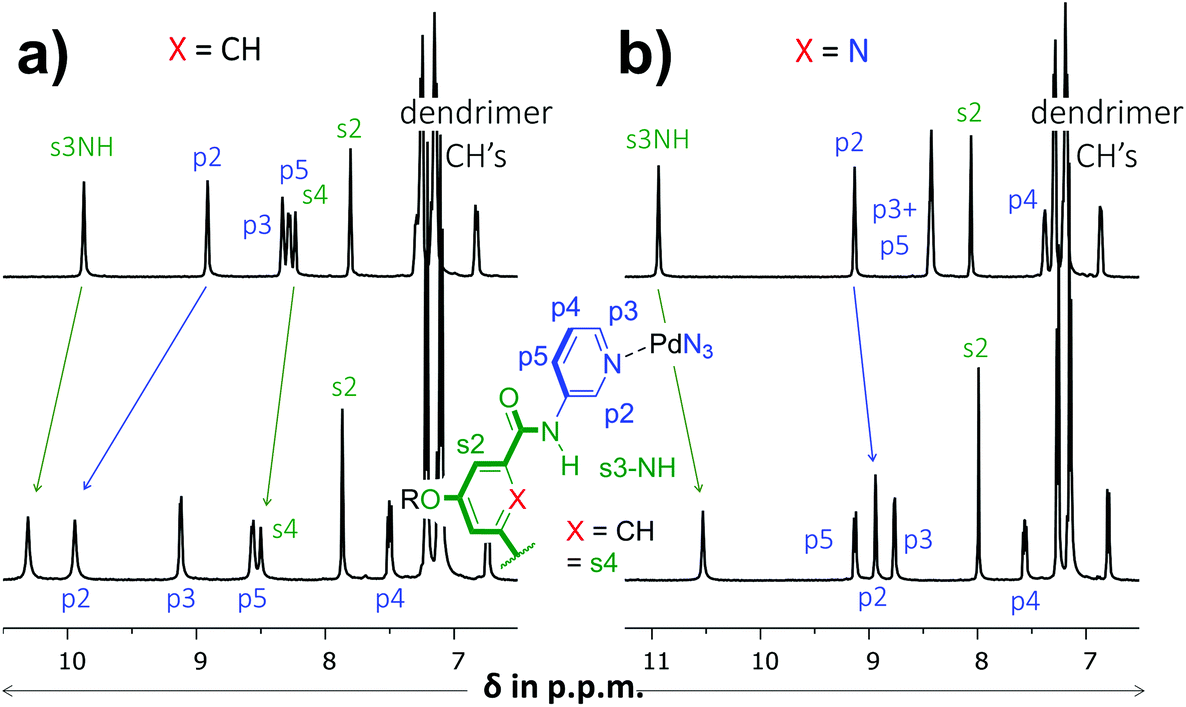 | ||
| Fig. 2 Partial 1H NMR spectra involving dipyridyl ligands 1 (a) and 2 (b). The top spectra are of pure ligand and the bottom spectra are of the [Pd2(Ligand)4[BF4]4 complexes. The solvent is CD2Cl2 containing 5% dmso-d6 and further details are given in the ESI.† | ||
The binding affinities of both complexes for carbohydrates 3–6 listed in Table 1 was probed by 1H-NMR titration experiments in CD2Cl2 containing 5% dmso-d6.
| Host → Guest ↓ |
K
a of 1:1 bindinga (M−1) |
|
|---|---|---|
| [Pd214]4+ | [Pd224]4+ | |
|
a Binding constants were obtained by fitting observed chemical shift differences with HypNMR8 as is detailed in section S3 of the ESI.†
b ‘i.p.s.’ stands for the relatively ‘insignificant peak-shifts’ that were observed in the concentration range of 0–25 mM glycoside.
c Incorporating the higher concentration ranges, the affinities could only be modelled with a more complicated stoichiometry than simple 1:1 binding, but the 1:1 stoichiometries were still dominant or representative of the binding strength of the cages for nitrate anions. Details are provided in the text and in Fig. S42† for [Pd214]4+ and Fig. S48† for [Pd224]4+.
|
||
| 3 | 541 | i.p.s.b |
| 4 | 262 | |
| 5 | 447 | |
| 6 | 262 | |
| NO 3 − | 1862c | 159c |

|
||
As is illustrated in Fig. 3 for the titrations with n-octyl-β-D-mannoside 3, significant peak shifting was observed for [Pd214]4+ (Fig. 3a) in the concentration range to 25 mM of 3. Shifting of peaks cannot result from the dilution of the complex, as dilution studies in the concentration range used during titrations (0.64–0.27 mM) revealed that all resonances remained stationary (see Fig. S36†). The resonances that shift most are the inwards pointing s3NH, p2 and s4. The peak shifts could be analyzed using HypNMR, as is shown in the left-hand side of Fig. 3a. From this plot it is evident that all resonances display clear saturation behavior around 10 mM of 3. Fitting these shifts with HypNMR8 to a 1:1 binding model gave Ka = 541 ± 2.9 M−1 with a reasonable accuracy (r2 = 0.9862). Similar spectra and fits were obtained by titrations of [Pd214]4+ with the other carbohydrates. Moreover, NOESY spectroscopy of solutions of [Pd214]4+ containing galactoside 3 or glucoside 5 were consistent with carbohydrate binding to the interior of the cage (see Fig. S43 and S47†). Mass spectroscopic analysis of a solution of [Pd214]4+ with glucoside 5 supports a 1:1 binding stoichiometry (see Fig. S44†).
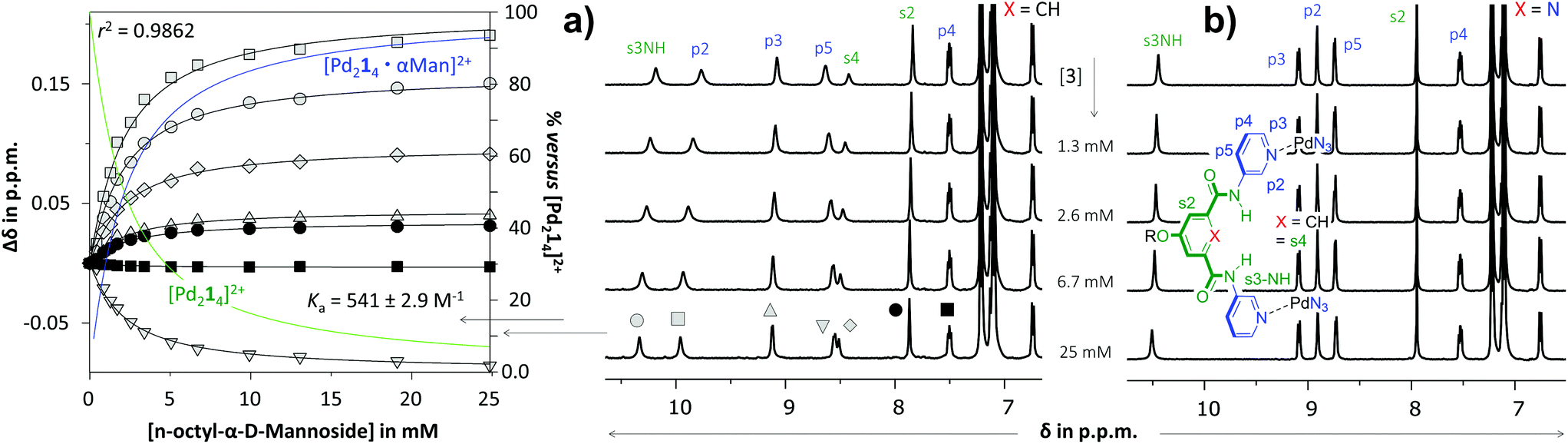 | ||
| Fig. 3 Partial 1H NMR spectra of titration experiments with n-octyl-β-D-mannoside 3 added to a solution of [Pd214][BF4]4 (a) or [Pd224][BF4]4 (b). The peak shifting observed in the titration with [Pd214][BF4]4 were fitted with HypNMR8 as shown in the left. The solvent is CD2Cl2 containing 5% dmso-d6. Further details are provided in section S40 of the ESI.† | ||
In sharp contrast to the titrations with [Pd214]4+, the spectrum of [Pd224]4+ (Fig. 3b) remained unperturbed when adding 3 to a concentration of 25 mM. Particularly surprising was the complete absence of any shifting of the inwards facing p2, which is very characteristic for binding to the interior of these type of M2L4 cages.3c,d Very similar titration data could be collected with carbohydrates 4–6 (see Table 1).
The twofold selectivity of [Pd214]4+ for β-glucoside 5 over β-galactoside 6 is consistent with an earlier report of the same cage with an alternative solubilizing group (measured in 10% dmso-d6 in CD2Cl2).3c Surprisingly, the cage binds strongest to α-mannoside 3 (Ka = 541 M−1), while the affinity for α-glucoside 4 is the same as that measured for β-galactoside 6 (Ka = 261 M−1). These data thus shown that [Pd214]4+ binds well with all carbohydrates in the order 3 > 5 > 4 = 6.
Another noticeable observation from Table 1 is the lack of binding of [Pd224]4+ for all four carbohydrates. This made us wonder if the interior of [Pd224]4+ was capable of binding at all. To this end, a titration was conducted with (n-Bu)4N+NO3−. As is shown in Fig. 4, very significant peak shifting was observed which appear to saturate around 13 mM of NO3−.
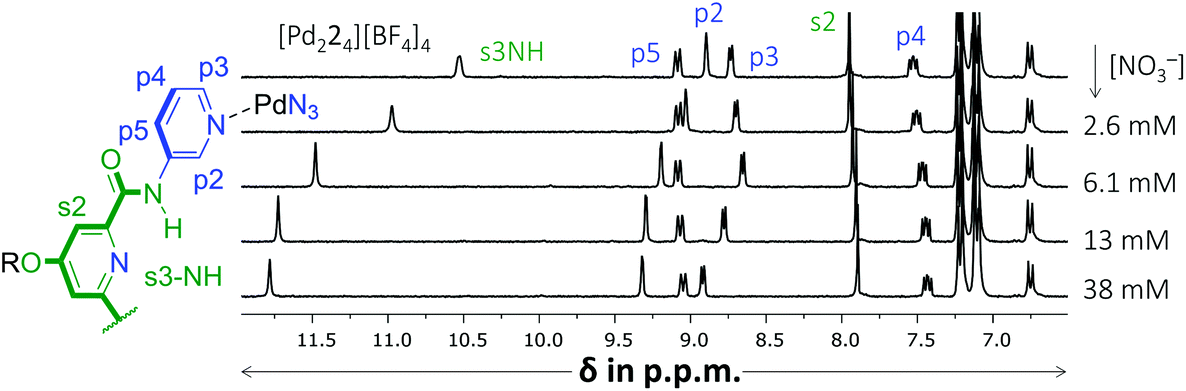 | ||
| Fig. 4 Partial 1H NMR spectra of a binding study with (n-Bu)4N+NO3− added to [Pd224][BF4]4. The solvent is CD2Cl2 containing 5% dmso-d6. See Fig. S52† for details. | ||
Interestingly, while s3NH and p2 only shifted downfield, the resonance of p3 initially shifted upfield, but then downfield. Such behavior is evidence of a binding stoichiometry that is more complex than simple 1:1 binding. As is detailed in the ESI (Fig. S52†), these shifts could be modelled to a 1:3 binding model with a Ka1:1 = 159, Ka1:2 = 63 M−1 and Ka1:3 = 31 M−1 (r2 = 0.9968). Such a stoichiometry is consistent with a nitrate anion binding to the interior of [Pd224]4+ (s3NH and p2 shifts) as well as with both exterior sites involving p3, in close proximity to both [Pd(pyridyl)4]2+ environments. It has indeed been noted that such a binding mode, involving four charge assisted [C–H]+⋯nitrate interaction, is common in [Pd(pyridyl)4]2+ complexes.5b,9 The order of magnitude of nitrate binding of [Pd224]4+ can be seen as weak, as (in the same matrix) a comparable [Pd(pyridyl)4]2+ complex has been reported with Ka1:1 = 91960 M−1.9a A very similar titration involving [Pd214]4+ could also be modelled with this 1:3 stoichiometry (Fig. S46†), but in this instance with a Ka1:1 = 1864 M−1 (and smaller Ka1:2 = 537 M−1 and Ka1:3 = 316 M−1, with r2 = 0.957).
It thus appears that [Pd214]4+ readily hosts other molecules, but its CH → N analogue does not. To gain insight into the possible origin for this loss in binding capability, molecular modeling was conducted using density functional theory (DFT). The resulting models are shown in space filling mode in the left-hand side of Fig. 5 and are similar to previously reported crystal structures (see also Fig. S54 and S55†).4b,5b Also given in the figure are the inner dimensions (in Å) of the models that were obtained by measuring intramolecular distances and subtracting twice the van der Waals radius of Hydrogen (1.09 Å) or Nitrogen (1.55 Å). While the complex with ligand 2 is about 1.4 Å wider (N–N versus CH–HC distance), the complex is also 1.4 Å less high (2.5 versus 3.9 Å). Actually, the height of [Pd224]4+ of 2.5 Å is just large enough for host a nitrate anion (3.0 Å in height) when assuming van der Waals overlap in the order of 0.5 Å. However, the 2.5 Å height of [Pd224]4+ is much smaller than the height of a glucoside (4.9 Å) and as a result very unlikely to fit. The dimensions of [Pd214]4+ on the other hand are much more congruent with the dimensions of NO3− and a glucoside, thus rationalizing why this complex binds to both (and with much greater affinities). Moreover, due to the preorganization of the amides in ligand 2 (see Fig. 1) its complex is expected to be very rigid. As a result, [Pd224]4+ might well lack the conformational flexibility that could enable it to encapsulate a glycoside. Such a rational was also proposed previously for the comparison of isophthalamide versus dipicolinamide covalent cages in carbohydrate binding.10 Actually, inspection of the NOESY spectrum of [Pd224]4+ (Fig. S27†) reveals the complete absence of a close proximity of the amide hydrogens (s3NH) and the aromatic CH hydrogens (s2) of the dipicolinamide fragment. This indeed implies that rotation of the amides is locked into a position with the NH hydrogens pointing to the interior of the complex (bound by the dipicolinic N). In the NOESY spectrum of [Pd214]4+ (Fig. S20†) on the other hand there is a clear nuclear Overhauser effect cross peak between the amidic NH's and the inwards pointing s4, as well as the outwards pointing s2. This is consistent with the reported flexibility of these amides in the solid state structures of related cage complexes.4 Finally, modelling of interior bound BF4− (Fig. S55†) shows a much tighter and energetically more stable fit within the [Pd224]4+ binding pocket, suggesting that interior bound BF4− might hamper further binding. Such an adverse effect of BF4− on the binding potential of an M2L4 cage has been reported before.2b The increased flexibility of [Pd214]4+ compared to [Pd224]4+, the smaller size of [Pd224]4+, and the increased stability of the BF4− complex with [Pd224]4+ together rationalizes the stark contrast in binding properties observed for both complexes.
 | ||
| Fig. 5 Space filling representations of molecular models of both [Pd2(Ligand)4]4+ complexes as well as a nitrate anion and the sugar part of n-octyl-β-D-glucoside 5. The van der Waals corrected dimensions of the interior of the complexes and the exterior of NO3− and the glucoside are given in Å. Models generated with DFT/ωB97X-D/6-31G*. | ||
Summary and conclusion
In summary, ligands were synthesized that bear two dipyridyl donor groups linked by an isophthalamide (1) or a dipicolinamide moiety (2). Both ligands formed [Pd2(Ligand)4][BF4]4 complexes in CD2Cl2 containing 5% dmso-d6. Cage [Pd2(1)4][BF4]4 was shown to bind to n-octyl glycosides 3–6 with affinities of about 250–500 M−1 in the order 3 > 5 > 4 = 6, and to nitrate anions with a 1:1 affinity Ka = 1862 M−1. In sharp contrast, cage [Pd2(2)4][BF4]4 did not appear to bind to glycosides and bound to nitrate with a 1:1 affinity of merely 159 M−1. The difference in binding properties could be rationalized based on the reduced flexibility and size of the [Pd2(2)4]2+ cage, and its stronger complexation to a BF4− anion. It is thus concluded that preorganization of the amides in 2 by intramolecular NH⋯N hydrogen bonding has an adverse effect on the binding properties of [Pd2(2)4]2+ compared to its CH analogue [Pd2(1)4]2+, at least for n-octyl glycosides 3–6 and nitrate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Netherlands Organization for Scientific Research (NWO) with VIDI grant number 723.015.006.References
- (a) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (b) Z. Li, N. Kishi, K. Yoza, M. Akita and M. Yoshizawa, Chem. – Eur. J., 2012, 18, 8358–8365 CrossRef CAS PubMed; (c) A. Schmidt, A. Casini and F. E. Kuhn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS; (d) L. Xu, Y. X. Wang and H. B. Yang, Dalton Trans., 2015, 44, 867–890 RSC; (e) G. H. Clever and P. Punt, Acc. Chem. Res., 2017, 50, 2233–2243 CrossRef CAS PubMed; (f) D. Bardhan and D. K. Chand, Chem. – Eur. J., 2019, 25, 12241–12269 CrossRef CAS PubMed; (g) H. Y. Hou, K. Zhou, F. L. Jiang, Q. H. Chen and M. C. Hong, Isr. J. Chem., 2019, 59, 140–150 CrossRef CAS.
- (a) S. Freye, J. Hey, A. Torras-Galan, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS PubMed; (b) D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022–15026 CrossRef CAS PubMed.
- (a) M. Yamashina, M. Akita, T. Hasegawa, S. Hayashi and M. Yoshizawa, Sci. Adv., 2017, 3, 6 Search PubMed; (b) D. Yang, L. K. S. Krbek, L. Yu, T. K. Ronson, J. D. Thoburn, J. P. Carpenter, J. L. Greenfield, D. J. Howe, B. Wu and J. R. Nitschke, Angew. Chem., 2021, 60, 4485–4490 CrossRef CAS PubMed; (c) X. Schaapkens, E. O. Bobylev, J. N. H. Reek and T. J. Mooibroek, Org. Biomol. Chem., 2020, 18, 4734–4738 RSC; (d) X. Schaapkens, J. H. Holdener, J. Tolboom, E. O. Bobylev, J. N. H. Reek and T. J. Mooibroek, ChemPhysChem, 2021, 22(12), 1187–1192 CrossRef CAS PubMed.
- (a) N. Yue, Z. Q. Qin, M. C. Jennings, D. J. Eisler and R. J. Puddephatt, Inorg. Chem. Commun., 2003, 6, 1269–1271 CrossRef CAS; (b) N. L. S. Yue, D. J. Eisler, M. C. Jennings and R. J. Puddephatt, Inorg. Chem., 2004, 43, 7671–7681 CrossRef CAS PubMed; (c) N. L. S. Yue, M. C. Jennings and R. J. Puddephatt, Inorg. Chim. Acta, 2016, 445, 37–45 CrossRef CAS.
- (a) A. J. Baer, B. D. Koivisto, A. P. Cote, N. J. Taylor, G. S. Hanan, H. Nierengarten and A. Van Dorsselaer, Inorg. Chem., 2002, 41, 4987–4989 CrossRef CAS PubMed; (b) D. Tripathy, A. K. Pal, G. S. Hanan and D. K. Chand, Dalton Trans., 2012, 41, 11273–11275 RSC.
- (a) T. J. Mooibroek, B. J. J. Timmer, X. Schaapkens and A. Kooijman, Angew. Chem., Int. Ed., 2021, 60(29), 16178–16183 CrossRef PubMed; (b) O. Beyer, B. Hesseler and U. Luning, Synthesis, 2015, 2485–2495 CAS.
- T. Tateishi, S. Takahashi, A. Okazawa, V. Marti-Centelles, J. Z. Wang, T. Kojima, P. J. Lusby, H. Sato and S. Hiraoka, J. Am. Chem. Soc., 2019, 141, 19669–19676 CrossRef CAS PubMed.
- C. Frassineti, S. Ghelli, P. Gans, A. Sabatini, M. S. Moruzzi and A. Vacca, Anal. Biochem., 1995, 231, 374–382 CrossRef CAS PubMed.
- (a) B. J. J. Timmer and T. J. Mooibroek, Chem. Commun., 2021 10.1039/D1CC02663A; (b) D. K. Chand, K. Biradha and M. Fujita, Chem. Commun., 2001, 1652–1653 RSC; (c) L. P. Zhou and Q. F. Sun, Chem. Commun., 2015, 51, 16767–16770 RSC; (d) R. Sekiya, M. Fukuda and R. Kuroda, J. Am. Chem. Soc., 2012, 134, 10987–10997 CrossRef CAS PubMed; (e) J. Lee, S. Lim, D. Kim, O. S. Jung and Y. A. Lee, Dalton Trans., 2020, 49, 15002–15008 RSC; (f) U. Siriwardane and F. Fronczek, CSD Communication, 2017, CCDC deposition Nr. 1562063 Search PubMed; (g) T. Y. Kim, N. T. Lucas and J. D. Crowley, Supramol. Chem., 2015, 27, 734–745 CrossRef; (h) J. E. M. Lewis and J. D. Crowley, Supramol. Chem., 2014, 26, 173–181 CrossRef CAS; (i) E. Sone, M. Sato, K. Yamanishi, C. Kamio, H. Takemoto and M. Kondo, Dalton Trans., 2016, 45, 894–898 RSC; (j) S. Samantray, S. Krishnaswamy and D. K. Chand, Nat. Commun., 2020, 11, 11 CrossRef PubMed.
- T. Velasco, G. Lecollinet, T. Ryan and A. P. Davis, Org. Biomol. Chem., 2004, 2, 645–647 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01185e |
| This journal is © The Royal Society of Chemistry 2021 |