
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of size engineering-enabled electrocatalysts for Li–S chemistry
Xi
Zhang
 a,
Yaping
Zhang
a,
Xijun
Wei
a,
Chaohui
Wei
*b and
Yingze
Song
*a
a,
Yaping
Zhang
a,
Xijun
Wei
a,
Chaohui
Wei
*b and
Yingze
Song
*a
aState Key Laboratory of Environmental-Friendly Energy Materials, School of Materials Science and Engineering, Southwest University of Science and Technology, Mianyang, Sichuan 621010, P. R. China. E-mail: yzsong@swust.edu.cn
bCollege of Energy, Jiangsu Provincial Key Laboratory for Advanced Carbon Materials and Wearable Energy Technologies, Soochow University, Suzhou 215006, P. R. China. E-mail: chwei@suda.edu.cn
First published on 10th August 2021
Abstract
Li–S batteries (LSBs) have received extensive attention owing to their remarkable theoretical capacity (1672 mA h g−1) and high energy density (2600 W h kg−1), which are far beyond those of the state-of-the-art Li-ion batteries (LIBs). However, the retarded sulfur reaction kinetics and fatal shuttle effect have hindered the practical implementations of LSBs. In response, constructing electrocatalysts for Li–S systems has been considered an effective strategy to date. Particularly, size engineering-enabled electrocatalysts show high activity in the sulfur redox reaction, considerably contributing to the latest advances in Li–S system research. In this tutorial review, we provide a systematic summary of nano- to atomic-scale electrocatalysts employed in Li–S chemistry, aiming at figuring out the working mechanism of size engineering-enabled electrocatalysts in the sulfur redox reaction and guiding the rational construction of advanced LSBs toward practically viable applications.
 Xi Zhang | Xi Zhang received his B.S. degree from the Shandong University of Technology in 2020. At present, he is a master's candidate at the State Key Laboratory of Environment-friendly Energy Materials, Southwest University of Science and Technology under the supervision of Prof. Yingze Song. His current research interests focus on the activity design of electrocatalysts for Li–S chemistry. |
 Yaping Zhang | Yaping Zhang earned her PhD in Polymer Chemistry and Physics from the University of Science and Technology of China in 2006. Now, she is a full professor at the School of Materials Science and Engineering, Southwest University of Science and Technology. Her current research interests focus on polymer membrane design for energy storage systems and electrochemistry. |
 Xijun Wei | Xijun Wei obtained his PhD from Chongqing University in 2020 under the supervision of Prof. Yunhuai Zhang and Prof. Peng Xiao. Since July 2020, he became a distinguished associate professor at the School of Materials Science and Engineering, Southwest University of Science and Technology. His current research interest centres around advanced material design for energy storage and conversion systems. |
 Chaohui Wei | Chaohui Wei received her bachelor's degree from Xi'an Jiaotong University in 2014 and PhD from University of Nottingham in 2019. She is currently a postdoctoral research fellow at the College of Energy, Soochow University under the supervision of Prof. Jingyu Sun and Prof. Ruizhi Yang. Her current research interests include electrocatalyst design for lithium–sulfur and lithium–oxygen batteries, controllable preparation of MXene materials, and utilization of 3D printing technique enabling highly loaded electrodes for practical energy storage devices. |
 Yingze Song | Yingze Song obtained his PhD from Soochow University in 2019 under the supervision of Prof. Zhongfan Liu and Prof. Jingyu Sun. During 2013–2015, he worked at Ningbo Institute of Materials Technology & Engineering, CAS. Since July 2019, he became a distinguished professor and doctoral supervisor in the State Key Laboratory of Environment-Friendly Energy Materials, Southwest University of Science and Technology. His current research interests deal with advanced material design for energy storage systems. |
1. Introduction
The booming electrical vehicles and portable electronic equipment have urged the demand for advanced energy storage systems with high energy densities and low cost. Before the concept of rechargeable Li–S batteries (LSBs), Li-ion batteries (LIBs) were regarded as the key energy storage system to meet practical needs. Unfortunately, the energy density of LIBs is approaching the theoretical limit, probably because of failing to store large-scale energy for future applications.1 LSBs can serve as one of the most promising alternatives to LIBs due to their high theoretical specific capacity and outstanding energy density. Furthermore, the abundance and non-toxic nature of sulfur endow LSBs with low manufacturing cost and favorable environmental friendliness, respectively.2,3 However, several other obstacles hinder the commercialization of LSBs. The shuttle effect, which is caused by polysulfide (PS) migration between the cathode and the anode, leads to irreversible sulfur loss and anode corrosion. Other than that, the insulating nature of sulfur and discharge products (Li2S/Li2S2) give rise to the retarded electrochemical reaction redox kinetics, as well as the low utilization efficiency of active material. The combination of these issues results in the low discharge capacity and inferior cycling stability.4,5To address the above-mentioned issues, various mediators involving carbon-based materials2 and metal compounds6–8 have been extensively used as promotors in Li–S systems. Ideal promotors are expected to synergize the remarkable immobilization ability for PS shuttle and high electrocatalytic activity for better sulfur redox reaction kinetics. The current evidences corroborate that it is essential to build a smooth PS adsorption–diffusion–conversion process on the surface of mediators for LSBs. The PS adsorption ability of these promotors mainly lies in their polar surfaces, which has been widely investigated. Particularly, rationalizing the electrocatalysis of Li–S chemistry is still waiting to be throughout long-term endeavors from the activity design of electrocatalysts. In this regard, effective strategies such as interface,9 defect,10 and template engineering11 have emerged. These strategies have managed to enhance the activity of the electrocatalysts in Li–S chemistry to a certain extent. However, it is urgent to further enrich the design routes targeted at highly active electrocatalysts. Since the size of the electrocatalysts plays a pivotal role in determining the activity, size engineering on electrocatalysts by reducing their sizes has been proposed to rationalize the electrocatalysis of LSBs.
Downsizing the particles from the nanoscale12,13 to the cluster,14 molecule,15 and even atomic levels16 leads to an augmented number of active sites or optimized coordination configurations, which is favorable to boosting the sulfur redox reaction efficiency and thereby contributes to the high capacity and long lifespan of LSBs. This review focuses on the recent advances and prospects on electrocatalyst designs from the nano to atom scale toward high activity in Li–S chemistry, aiming at offering rational strategies and new insights for improving the electrocatalysis of Li–S systems.
2. Electrochemistry and challenges of LSBs
In a working LSB, solid sulfur molecules (S8) firstly dissolve to form high-order Li2S8 molecules and then are reduced to Li2S6 and Li2S4, contributing to the first typical discharge plateau at approximately 2.4 V (Fig. 1). The soluble Li2S4 intermediates continue to generate the low-order insoluble compounds Li2S2 and Li2S, giving rise to the second plateau at around 2.1 V, which contributes to three-quarters of the total capacity.17 The multi-step phase conversions from S8 to Li2S accompanied by multi-electron chemistry cause the notorious shuttle effect and slow the sulfur redox reaction kinetics, thus resulting in irreversible sulfur loss and continuous anode corrosions, as well as limited discharge depth and rate. Notably, the issue of retarded sulfur redox reaction kinetics mainly results from the insulating nature of sulfur and Li2S, viscosity change in the electrolyte, slow solid-state diffusion and Li2S precipitation on the lithium anode. Along this line, the problem of kinetics is complex and can be affected almost by the whole electrochemical reaction procedure. In particular, these issues become more formidable under the practical scenario of high sulfur loading. Therefore, the investigation of reaction kinetics can break the gap between fundamental exploration and real implementation for LSBs. Additionally, lithium dendrites also pose a grand threat to the electrochemical performance and safety of LSBs. Scientific and technological viewpoints have been proposed by applying size engineering-derived promotors, which are of utmost importance to probing the feasibility of accelerating Li–S chemistry and unravelling the underlying electrocatalytic mechanism. In this light, the focus will continue to be centred on the development of new routes for the synthesis of active electrocatalysts. | ||
| Fig. 1 Schematic of the construction of routine LSBs. | ||
3. Size engineering-enabled electrocatalysts for Li–S chemistry
The afore-mentioned issues limit the electrochemical performance of LSBs, leading to a sluggish commercialization process. The use of electrocatalysts in LSBs has been proposed as an effective strategy to address these challenges. Generally, the activity of electrocatalysts is largely restricted by their size. In this light, size engineering has been used as an emerging strategy to upgrade the activity of electrocatalysts by minimizing their sizes from the nanoscale to clusters, molecules and even the atom level. In this section, this review summarizes the progress of size engineering-enabled electrocatalysts, including nanoparticles, clusters, molecules and single metal atoms.A decrease in the size of electrocatalysts usually leads to larger surface areas or uniformly dispersed single atoms, implying an increased number of active sites or high atom utilization efficiency, respectively. In electrocatalysts, both the abundance of active sites and high atom utilization efficiency manifest superior chemical activity, mainly pertaining to outstanding PS-anchoring ability and remarkable electrocatalytic capability for improved sulfur redox reaction kinetics. Therefore, the active site number is usually evaluated effectively to guarantee the chemical activity of the electrocatalysts. Further, the focus must be on the balance between the large surface area and single atom utilization efficiency, PS adsorption ability, Li+-ion migration and electron conductivity to attain high electrocatalytic activity.
3.1 Milestones of electrocatalysts from nano to atomic scales
In recent years, electrocatalysts have been widely applied to improve the utilization and cyclic performance of LSBs.14,15,18 Since the concept of rechargeable LSBs was proposed in 2002,26 fruitful achievements have been made in size engineering-enabled electrocatalysts through constant explorations from fundamental to practical investigations (Fig. 2). A pioneering work by Nazar and co-workers in 2014 reported that Magneli-phase Ti4O7 offered an intrinsic polar surface to trigger interface-mediated redox reactions of PSs.19 Following this research, various nanocatalysts, including oxides,20 sulfides,21 nitrides,22 and phosphides,23 were used to improve the sulfur redox reaction kinetics. To further elevate their activity in Li–S chemistry, the sizes of the nanocatalysts were further decreased to cluster, molecule and single-atom levels. Zhang et al. proposed the application of atomic catalysts in heterogeneous catalysis in 2011.24 The model of single-atom catalysts (SACs) was conceived to make full use of the local metal atoms to realize fast electrochemical reactions in LSBs. For instance, SAFe was found to exhibit lower delithiation energy barriers, resulting in fast PS conversion and higher capacity.25 It is worth noting that SACs represent a new frontier in offering high-efficiency Li–S chemistry owing to their nearly 100% atom utilization efficiency and tunability.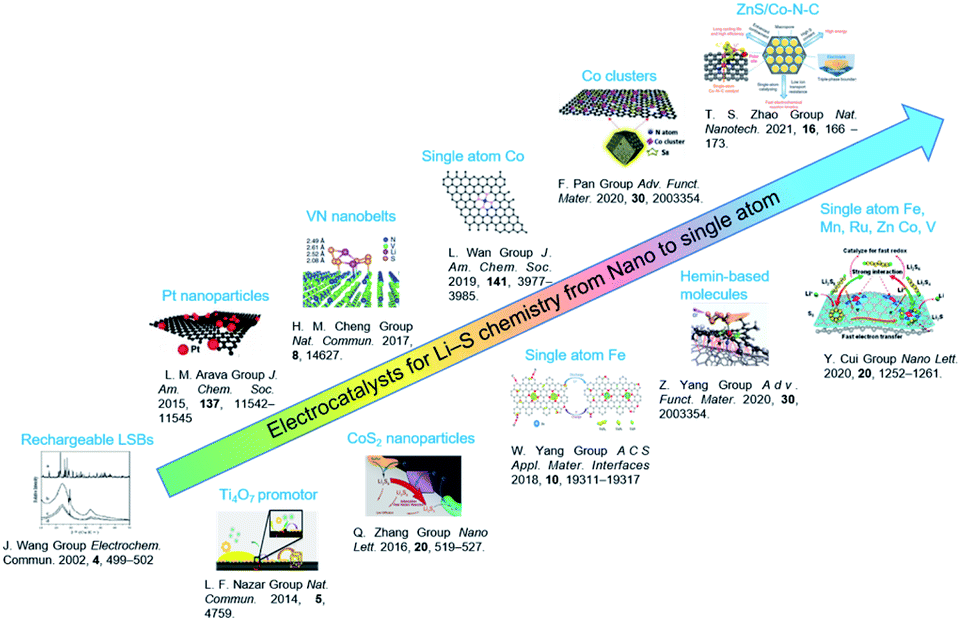 | ||
| Fig. 2 Electrocatalysts for LSBs from nano to single atom levels. Images reproduced with permission as follows: “Rechargeable LSBs”.26 Copyright 2002, Elsevier. “Ti4O7 promotor”.19 Copyright 2014, Nature Publishing Group. “Pt nanoparticles”.27 Copyright 2015, American Chemical Society. “VN nanobelts”.28 Copyright 2017, Nature Publishing Group. “CoS2 nanoparticles”.29 Copyright 2016, American Chemical Society. “Single atom Co”.30 Copyright 2019, American Chemical Society. “Single atom Fe”.31 Copyright 2019, American Chemical Society. “Co clusters”.32 Copyright 2020, Wiley-VCH. “Hemin-based molecule”.15 Copyright 2020, Wiley-VCH. “ZnS/Co–N–C”.33 Copyright 2020, Nature Publishing Group “Single atom Fe, Mn, Ru, Zn, Co, V”.34 Copyright 2020, American Chemical Society. | ||
3.2 Mechanism of electrocatalysts from nano to atomic levels
Electrocatalysts are extensively applied to deal with the stepwise reduction of S8 to soluble Li2S4 and further conversion to low-order insoluble Li2S. Particularly, the conversion of soluble Li2S4 to Li2S, which is called “Li2S nucleation and growth”, contributes to almost three-quarters of the total capacity, manifesting the critical electrochemical reaction steps.35,36 It is worth noting that this procedure refers to the formation of high-viscosity Li2S4 and the liquid–solid–solid conversion,37,38 implying that it is the speed-determining step of the electrocatalytic reaction. When the electrocatalyst is incorporated into the cathode of LSBs, the adsorption and electron transfer processes can be altered.39 The soluble PSs can be trapped by polar electrocatalysts followed by continuous and gradual conversion to solid Li2S during the reduction process.40 Therefore, the electrocatalytic mechanism can be explored by mainly investigating these two steps.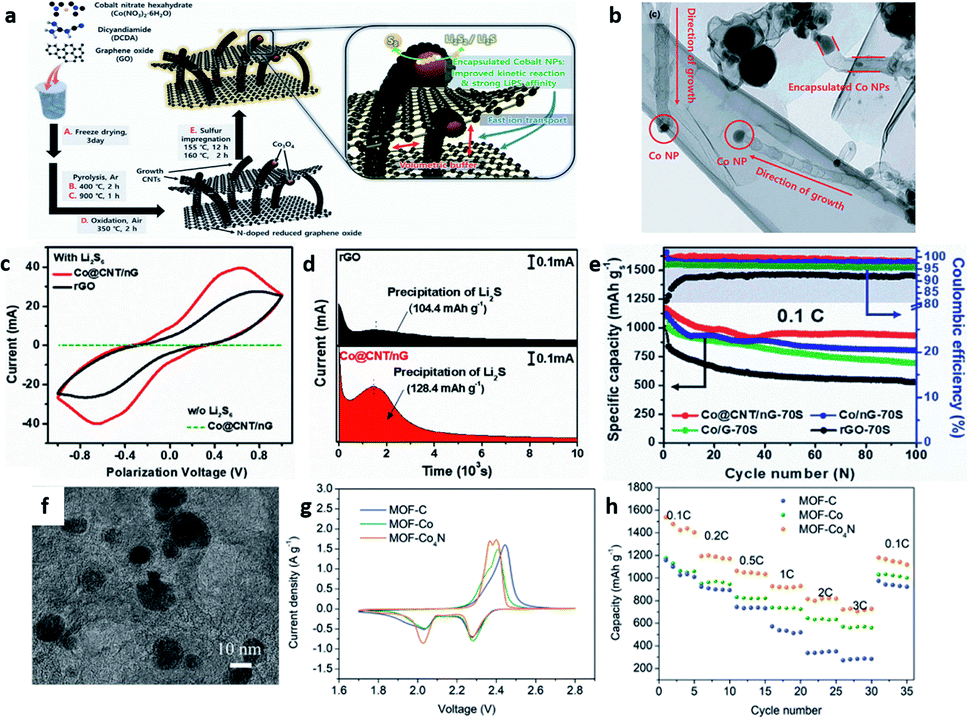 | ||
| Fig. 3 (a) The schematic illustration for the synthetic procedures and functional roles (zoomed image) of Co@CNT/nG-70S. (b) High-magnification TEM images of grown CNT of Co@CNT/nG. (c) CV curves of symmetric cells with Co@CNT/nG and rGO. (d) Potentiostatic discharge curves of Li2S8 solution at 2.06 V of Co@CNT/nG and rGO. (e) Cycling performance and coulombic efficiencies at 0.1C.42 Copyright 2021, Wiley-VCH. (f) TEM image of MOF-Co4N. (g) The second cycle of CV. (h) Rate performance of MOF-C, MOF-Co, and MOF-Co4N.43 Copyright 2019, Wiley-VCH. | ||
Conductive and nonconductive promotors usually present distinct mechanisms. In detail, the effect of nonconductive mediators, such as oxides, on Li–S chemistry relies on the adsorption and diffusion of PSs on their surface due to the limited electron conductivity. Conductive mediators show superior electrocatalytic activity on account of their complete catalysis throughout the whole electrochemical reaction process. Although size engineering can be applied to both types of electrocatalysts to form more active sites, it is very challenging to quantify the elevation of activity according to the scale change.
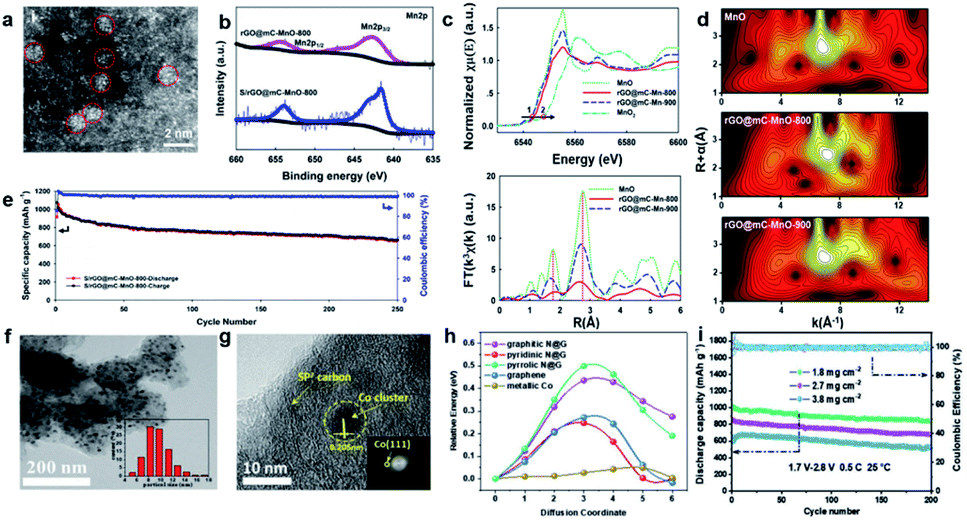 | ||
| Fig. 4 (a) HAADF-STEM images of MOC-embedded 2D rGO@mC nanosheets. (b) The Mn2p XPS spectra of rGO@mC-MnO-800 and S/rGO@mC-MnO-800. (c) XANES and k3-weighted FT-EXAFS in R space. (d) Wavelet transforms of rGO@mC-MnO-800 and the reference samples, including MnO, MnO2, and rGO@mC-MnO-900. (e) Rate capabilities of the cells assembled with S/rGO@mC-MnO-800 at various current densities.46 Copyright 2020, American Chemical Society. (f and g) SEM and HR-TEM of N-PC@uCo/S. (h) Calculated energy barriers for Li-ion diffusion. (i) Cycling capacity and coulombic efficiency of N-PC@uCo/S with the sulfur loadings of 1.8, 2.7, and 3.8 mg cm−2.32 Copyright 2020, Wiley-VCH. | ||
 ) during the discharge procedure in LSBs (Fig. 5b). Hence, an ultrahigh initial capacity of 1637.8 mA h g−1 at 0.2C and a low fading rate of 0.042% per cycle up to 1800 cycles at 1C were achieved (Fig. 5d). Lai et al. also developed a molecular catalyst, namely tris (4-fluorphenyl) phosphine (TFPP), as an interfacial mediator for longevous LSBs, which produced an impressive ultimate capacity of 545 mA h g−1 after 140 cycles at 5C with a high sulfur loading of 4.2 mg cm−2.9
) during the discharge procedure in LSBs (Fig. 5b). Hence, an ultrahigh initial capacity of 1637.8 mA h g−1 at 0.2C and a low fading rate of 0.042% per cycle up to 1800 cycles at 1C were achieved (Fig. 5d). Lai et al. also developed a molecular catalyst, namely tris (4-fluorphenyl) phosphine (TFPP), as an interfacial mediator for longevous LSBs, which produced an impressive ultimate capacity of 545 mA h g−1 after 140 cycles at 5C with a high sulfur loading of 4.2 mg cm−2.9
 | ||
| Fig. 5 (a) Schematic configuration of an LSB based on three CNTs-FG@hemin cathodes (FG = NH2, OH, COOH), and the mechanism of PS adsorption at the CNTs–COOH@hemin cathode. (b) The second cycle of the CV profiles of the CNTs–COOH, CNTs–NH2@hemin, CNTs–OH@hemin, and CNTs–COOH@hemin cathodes. (c) Energy diagram for the conversion of PSs on CNTs–COOH@hemin. (d) Cycling stability and coulombic efficiency of the CNTs–COOH, CNTs–NH2@hemin, CNTs–OH@hemin, and CNTs–COOH@hemin cathodes at 1C.15 Copyright 2020, Wiley-VCH. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −N bonds.51
−N bonds.51
Recently, Du et al. loaded SACo on N-doped graphene (Co–N/G) for a Li–S system (Fig. 6a).30 The Co–N–C coordination centres acted as dual-function electrocatalysts to accelerate the formation and decomposition of Li2S in the discharge and charge processes, respectively (Fig. 6b). The DFT calculations in Fig. 6e further indicate the lower Gibbs free energy of Li2S2 to Li2S conversion on Co–N/G than on N/G, indicating more favorable reaction thermodynamics. The in situ XANES in Fig. 6c and d further indicate that the introduction of SA on NG can facilitate the formation/decomposition of Li2S and Li2S2.30 Lu and co-workers devised a high-loading single metal atom material (SAFe@gC3N4) with an excellent catalytic activity that boosted the electrochemical conversion kinetics in LSBs (Fig. 6g–j).25 Owing to the strong coordination effect of the N sites, g-C3N4 was applied as a support for SAC with a high Fe atom content of 8.5 wt%. SAFe@gC3N4 could effectively suppress the shuttle effect of PSs and accelerate the conversion reaction, consequently reducing sulfur loss in the charge and discharge processes (Fig. 6k and l). These works offer new insights into the rational design of advanced SACs toward achieving high-performance LSBs. However, the single atom loading is low (<4.0 wt%), paving new development direction for SACs in Li–S chemistry.52
 | ||
| Fig. 6 (a) HAADF-STEM images of Co–N/G. (b) Evolution of S K-edge XANES during electrochemical cycling. (c) Evolution of the intensities of peak B and peak D during electrochemical cycling. (d) CVs of the symmetric cells with Co–N/G, N/G, Co/G, and rGO as electrodes. (e) Energy profiles for the reduction of PSs on the N/G and Co–N/G substrates. The optimized adsorption conformations of the intermediate species on the N/G and Co–N/G substrates are shown in the insets. Energy profiles of the decomposition of Li2S clusters on N/G. (f) Cycling performance of the S@Co–N/G electrode with the areal S loadings of 2.0, 3.4 and 6.0 mg cm−2 at 0.2C.30 Copyright 2019, American Chemical Society. (g) TEM image of b-Fe@g-C3N4 materials. (h) HAADF-STEM image of the SAFe@g-C3N4 material showing single iron atoms. (i) Fe K-edge XANES spectra of Fe foil and SAFe@g-C3N4 material. (j) Fourier transform curves of the Fe K-edge EXAFS spectra of Fe foil and SAFe@g-C3N4. Energy profiles for (k) Li2S g−1-C3N4, and (l) Li2S/SAFe@g-C3N4. (m) The long-term stability of the device at the current rates of 2 and 5C.25 Copyright 2020, American Chemical Society. | ||
The interactions of well-dispersed single metal atoms with carbon substrates, mainly pertaining to the direct coordination of SA-C and indirect coordination, such as SA-N-C and SA-O-C, represent the active centers for electrocatalytic reactions. The difference between the metal atoms in SACs and the heteroatoms in mediators favor the high activity of SACs. The heteroatoms on the mediators can also serve as active sites for Li–S chemistry. Particularly, lithiophilicity, which is formed by the interaction between the heteroatoms and Li+-ions, can guide the sulfur redox reaction kinetics.53 However, the electrocatalytic activity of these heteroatoms cannot be compared with single metal atoms. This is because the metal atoms in SACs generally display unique electronic structures, unsaturated coordination environment and exclusive selectivity, leading to superior electrocatalytic activity in LSBs.
4. Conclusions and perspectives
The retarded reaction kinetics and notorious PS shuttling severely reduce the working efficiency and electrochemical performance of LSBs. Appropriate electrocatalysts that possess a strong affinity for PSs and impressive catalytic capability towards their conversion reactions are needed to boost the electrochemical performance of LSBs (Table 1). The disadvantages of size reduction include limited synthetic routes, agglomeration and dissolution of electrocatalysts in the electrolyte during long-term cycling. Therefore, stabilizing the small-sized electrocatalysts on selected substrates, such as carbon and metal compounds, has been corroborated as a preferred strategy. Moreover, the substrates can be further tuned into various structures with favorable surface areas to mitigate agglomeration or dissolving into the electrolyte, which ensures the high utilization efficiency of electrocatalysts. For instance, loading single atoms on carbon substrates with large surface areas does not only result in near 100% atom utilization efficiency but also optimizes the coordination configurations, thus making it an effective strategy to tackle these disadvantages.| Types | Electrocatalysts | Areal loading of S (mg cm−2) | Capacities (mA h g−1) at various rate (C) | Cycle number | Capacity decay (per cycle) | Ref. |
|---|---|---|---|---|---|---|
| Nanoparticles | MOF-Co4N | 1 | 745 mA h g−1 at 1C | 400 | 0.043% | 43 |
| Co@NHCRs | 0.37 | 971 mA h g−1 at 0.5C | 100 | 0.27% | 54 | |
| N–CN-750@Co3Se4-0.1 | 1.5 & 3.1 | 1150.3 mA h g−1 at 0.2C | 800 | 0.067% | 55 | |
| Clusters | N-PC@uCo | 1.8–2.7 & 3.8 | 912 mA h g−1 at 1C | 500 | 0.028% | 32 |
| CE-NVO | 6 | 686 mA h g−1 at 1C | 200 | 0.1% | 56 | |
| rGO@mC-MnO-800 | 2.5 & 4.7 | 1535 mA h g−1 at 0.2C | 100 | 0.36% | 46 | |
| S@Co/PNC | 1.5 | 540 mA h g−1 at 1C | 300 | 0.064% | 57 | |
| rGO−CNT/PW12 | 3.1 & 4.1 | 1145 mA h g−1 at 1C | 150 | 0.084% | 58 | |
| Molecules | CNTs-COOH@hemin | 6.52 | 1637.8 mA h g−1 at 0.2C | 1800 | 0.042% | 15 |
| G@CB | 4.2 | 1032 mA h g−1 at 0.2C | 300 | 0.001% | 59 | |
| CNTs-S-TFPP | 4.2 | 517 mA h g−1 at 5C | 1000 | 0.042% | 9 | |
| SACs | Co–N/C | 2 & 6 | 926 mA h g−1 at 1C | 500 | 0.053% | 30 |
| Zn1–HNC | 4.6 & 7.8 | 1225 mA h g−1 at 0.5C | 100 | 0.009% | 60 | |
| SAFe@g-C3N4 | 3 | 1255 mA h g−1 at 0.2C | 200 | 0.05% | 25 | |
| S@SA-Zn-MXene | 1.7–5.3 | 802 mA h g−1 at 4C | 400 | 0.03% | 61 | |
| Fe-PNC | 1.3 | 1138.6 mA h g−1 at 0.1C | 300 | 0.2% | 31 |
The future applications of LSBs strongly depend on the progress of active electrocatalysts and the deep exploration of the underlying mechanisms. With the development of versatile size-engineering strategies, grand advances have been achieved in recent years.25,43 This tutorial review discusses the latest research achievements in terms of the electrocatalytic mechanisms in Li–S chemistry based on emerging electrocatalysts from the nano- to the atomic level and concludes with the key challenges and prospects in this field:
(i) Catalyst innovation: based on previous reports, an ideal electrocatalyst design involves an ample number of active sites, superb atom utilization efficiency, and high loading content, as well as remarkable conductivity. The activity of electrocatalysts in Li–S chemistry can be altered by delicate size engineering. However, newer electrocatalysts need to be further innovated by the continuous optimization of size engineering and the development of other new strategies.
(ii) Mechanism detection: an in-depth understanding of the mechanism of electrocatalysts is beneficial to further promote their activity. Emerging techniques, such as synchrotron radiation and neutron scattering, can record the real-time signals of PS intermediates in a working LSB, thus offering opportunities to understand the underlying electrocatalytic mechanism. Despite this, in view of their small sizes down to the atomic level, these electrocatalysts urgently call for advanced analytical techniques with higher resolution and wider detection range to achieve all-level surface and interface information for a clear interpretation of the electrocatalytic mechanisms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Project of State Key Laboratory of Environment-Friendly Energy Materials (SWUST, Grant No. 19FKSY16 and 18ZD320304) and National Natural Scientific Foundation of China (Grant No. U20A20125). We acknowledge the support from State Key Laboratory of Environment-Friendly Energy Materials (Mianyang, China).References
- T. D. Pham, A. Bin Faheem, S. Y. Chun, J. R. Rho, K. Kwak and K. K. Lee, Adv. Energy Mater., 2021, 11, 2003520 CrossRef CAS.
- S. Xin, L. Gu, N. H. Zhao, Y. X. Yin, L. J. Zhou, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef CAS PubMed.
- Y. Zhong, D. Chao, S. Deng, J. Zhan, R. Fang, Y. Xia, Y. Wang, X. Wang, X. Xia and J. Tu, Adv. Funct. Mater., 2018, 28, 1706391 CrossRef.
- J. H. Zuo and Y. J. Gong, Tungsten, 2020, 2, 134–146 CrossRef.
- Z. J. Cao, Y. Z. Zhang, Y. L. S. Cui, B. Li and S. B. Yang, Tungsten, 2020, 2, 162–175 CrossRef.
- H. Ye, Y. X. Yin, S. Xin and Y. G. Guo, J. Mater. Chem. A, 2013, 1, 6602–6608 RSC.
- Z. Zhang, Z. Li, F. Hao, X. Wang, Q. Li, Y. Qi, R. Fan and L. Yin, Adv. Funct. Mater., 2014, 24, 2500–2509 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- Y. Lai, H. Nie, X. Xu, G. Fang, X. Ding, D. Chan, S. Zhou, Y. Zhang, X. Chen and Z. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 29978–29984 CrossRef CAS.
- M. Li, Y. Yuan, Y. Zhao, S. Zhang, C. Zhong, J. Zhu, J. Lu and H. Zhang, ACS Nano, 2020, 14, 6673–6682 CrossRef PubMed.
- Y. Song, Z. Sun, Z. Fan, W. Cai, Y. Shao, G. Sheng, M. Wang, L. Song, Z. Liu, Q. Zhang and J. Sun, Nano Energy, 2020, 70, 104555 CrossRef CAS.
- Z. Li, C. Li, X. Ge, J. Ma, Z. Zhang, Q. Li, C. Wang and L. Yin, Nano Energy, 2016, 23, 15–26 CrossRef CAS.
- J. Pu, Z. Shen, J. Zheng, W. Wu, C. Zhu, Q. Zhou, H. Zhang and F. Pan, Nano Energy, 2017, 37, 7–14 CrossRef CAS.
- J. Ji, Y. Sha, Z. Li, X. Gao, T. Zhang, S. Zhou, T. Qiu, S. Zhou, L. Zhang, M. Ling, Y. Hou and C. Liang, Research, 2020, 2020, 5714349 CAS.
- X. Ding, S. Yang, S. Zhou, Y. Zhan, Y. Lai, X. Zhou, X. Xu, H. Nie, S. Huang and Z. Yang, Adv. Funct. Mater., 2020, 30, 2003354 CrossRef CAS.
- S. Wei, S. Xu, A. Agrawral, S. Choudhury, Y. Lu, Z. Tu, L. Ma and L. A. Archer, Nat. Commun., 2016, 7, 11722 CrossRef CAS PubMed.
- H. Yamin and E. Peled, J. Power Sources, 1983, 9, 281 CrossRef CAS.
- L. Ni, G. Zhao, G. Yang, G. Niu, M. Chen and G. Diao, ACS Appl. Mater. Interfaces, 2017, 9, 34793–34803 CrossRef CAS PubMed.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- K. Lu, H. Zhang, S. Gao, H. Ma, J. Chen and Y. Cheng, Adv. Funct. Mater., 2019, 29, 1807309 CrossRef.
- N. Zhou, W.-D. Dong, Y. J. Zhang, D. Wang, L. Wu, L. Wang, Z. Y. Hu, J. Liu, H. S. H. Mohamed, Y. Li, L. H. Chen and B. L. Su, Sci. China Mater., 2021 DOI:10.1007/s40843-021-1669-9.
- H. Zhang, D. Tian, Z. Zhao, X. Liu, Y. N. Hou, Y. Tang, J. Liang, Z. Zhang, X. Wang and J. Qiu, Energy Storage Mater., 2019, 21, 210–218 CrossRef.
- Y. Zhu, H. Zheng, J. Cen, A. Ali, X. Chen and P. K. Shen, J. Electro. Chem., 2021, 885, 114996 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- C. Lu, Y. Chen, Y. Yang and X. Chen, Nano Lett., 2020, 20, 5522–5530 CrossRef CAS PubMed.
- J. L. Wang, J. Yang, J. Y. Xie, N. X. Xu and Y. J. E. C. Li, Electrochem. Commun., 2002, 4, 499–502 CrossRef CAS.
- H. Al Salem, G. Babu, C. V. Rao and L. M. Arava, J. Am. Chem. Soc., 2015, 137, 11542–11545 CrossRef CAS.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef PubMed.
- Z. Yuan, H. J. Peng, T. Z. Hou, J. Q. Huang, C. M. Chen, D. W. Wang, X. B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu, H. Ji and L. J. Wan, J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed.
- Z. Liu, L. Zhou, Q. Ge, R. Chen, M. Ni, W. Utetiwabo, X. Zhang and W. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 19311–19317 CrossRef CAS.
- R. Wang, J. Yang, X. Chen, Y. Zhao, W. Zhao, G. Qian, S. Li, Y. Xiao, H. Chen, Y. Ye, G. Zhou and F. Pan, Adv. Energy Mater., 2020, 10, 1903550 CrossRef CAS.
- C. Zhao, G. L. Xu, Z. Yu, L. Zhang, I. Hwang, Y. X. Mo, Y. Ren, L. Cheng, C. J. Sun, Y. Ren, X. Zuo, J. T. Li, S. G. Sun, K. Amine and T. Zhao, Nat. Nanotechnol., 2021, 16, 166–173 CrossRef CAS.
- G. Zhou, S. Zhao, T. Wang, S. Z. Yang, B. Johannessen, H. Chen, C. Liu, Y. Ye, Y. Wu, Y. Peng, C. Liu, S. P. Jiang, Q. Zhang and Y. Cui, Nano Lett., 2020, 20, 1252–1261 CrossRef CAS.
- F. Y. Fan, W. C. Carter and Y. M. Chiang, Adv. Mater., 2015, 27, 5203–5209 CrossRef CAS PubMed.
- L. Kong, X. Chen, B. Q. Li, H. J. Peng, J. Q. Huang, J. Xie and Q. Zhang, Adv. Mater., 2018, 30, 1705219 CrossRef.
- N. Ding, X. D. Li, S. W. Chien, Z. L. Liu and Y. Zong, Chem. Commun., 2017, 53, 10152–10155 RSC.
- X. F. Yang, X. J. Gao, Q. Sun, S. P. Jand, Y. Yu, Y. Zhao, X. Li, K. Adair, L. Y. Kuo, J. Rohrer, J. N. Liang, X. T. Lin, M. N. Banis, Y. F. Hu, H. Z. Zhang, X. F. Li, R. Y. Li, H. M. Zhang, P. Kaghazchi, T. K. Sham and X. L. Sun, Adv. Mater., 2019, 31, 1901220 CrossRef PubMed.
- Q. Cheng, W. Xu, S. Qin, S. Das, T. Jin, A. Li, A. C. Li, B. Qie, P. Yao, H. Zhai, C. Shi, X. Yong and Y. Yang, Angew. Chem., Int. Ed., 2019, 58, 5557–5561 CrossRef CAS PubMed.
- Y. Song, W. Cai, L. Kong, J. Cai, Q. Zhang and J. Sun, Adv. Energy Mater., 2020, 10, 1901075 CrossRef CAS.
- J. He, L. Luo, Y. Chen and A. Manthiram, Adv. Mater., 2017, 29, 1702707 CrossRef PubMed.
- J. S. Yeon, Y. H. Ko, T. H. Park, H. Park, J. Kim and H. S. Park, Energy Environ. Mater., 2021, 1–10 Search PubMed.
- K. Xiao, J. Wang, Z. Chen, Y. Qian, Z. Liu, L. Zhang, X. Chen, J. Liu, X. Fan and Z. X. Shen, Small, 2019, 15, 1901454 CrossRef.
- F. Sun, J. Wang, D. Long, W. Qiao, L. Ling, C. Lv and R. Cai, J. Mater. Chem. A, 2013, 1, 13283–13289 RSC.
- C. Zhang, Y. Xie, M. Zhao, A. E. Pentecost, Z. Ling, J. Wang, D. Long, L. Ling and W. Qiao, ACS Appl. Mater. Interfaces, 2014, 6, 9751–9759 CrossRef CAS.
- P. Qiu, Y. Yao, W. Li, Y. Sun, Z. Jiang, B. Mei, L. Gu, Q. Zhang, T. Shang, X. Yu, J. Yang, Y. Fang, G. Zhu, Z. Zhang, X. Zhu, T. Zhao, W. Jiang, Y. Fan, L. Wang, B. Ma, L. Liu, Y. Yu and W. Luo, Nano Lett., 2021, 21, 700–708 CrossRef CAS PubMed.
- H. J. Peng, J. Q. Huang, X. B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1700260 CrossRef.
- J. G. Wang, K. Xie and B. Wei, Nano Energy, 2015, 15, 413–444 CrossRef CAS.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- X. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- U. Gulzar, T. Li, X. Bai, M. Colombo, A. Ansaldo, S. Marras, M. Prato, S. Goriparti, C. Capiglia and R. Proietti Zaccaria, ACS Appl. Mater. Interfaces, 2018, 10, 5551–5559 CrossRef CAS PubMed.
- Z. Zhang, Q. Kang, D. Wang and Y. Li, Nano Res., 2020, 13, 1856–1866 CrossRef.
- R. Zhang, X.-R. Chen, X. Chen, X.-B. Cheng, X.-Q. Zhang, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 7764–7768 CrossRef CAS PubMed.
- M. Zhang, C. Yu, C. Zhao, X. Song, X. Han, S. Liu, C. Hao and J. Qiu, Energy Storage Mater., 2016, 5, 223–229 CrossRef.
- D. Cai, B. Liu, D. Zhu, D. Chen, M. Lu, J. Cao, Y. Wang, W. Huang, Y. Shao, H. Tu and W. Han, Adv. Energy Mater., 2020, 10, 1904273 CrossRef CAS.
- Y. Yu, T. Li, H. Zhang, Y. Luo, H. Zhang, J. Zhang, J. Yan and X. Li, Nano Energy, 2020, 71, 2211–2855 Search PubMed.
- F. Zhang, S. Ji, H. Wang, H. Liang, X. Wang and R. Wang, Small Methods, 2021, 5, 2100066 CrossRef.
- L. Ni, G. Yang, Y. Liu, Z. Wu, Z. Ma, C. Shen, Z. Lv, Q. Wang, X. Gong, J. Xie, G. Diao and Y. Wei, ACS Nano, 2021, 15, 12222–12236 CrossRef CAS PubMed.
- J. Xie, H. J. Peng, J. Q. Huang, W. T. Xu, X. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 16223–16227 CrossRef CAS PubMed.
- H. Shi, X. Ren, J. Lu, C. Dong, J. Liu, Q. Yang, J. Chen and Z. S. Wu, Adv. Energy Mater., 2020, 10, 2002271 CrossRef CAS.
- D. Zhang, S. Wang, R. Hu, J. Gu, Y. Cui, B. Li, W. Chen, C. Liu, J. Shang and S. Yang, Adv. Funct. Mater., 2020, 30, 2002471 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |