
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe right tools for the job: the central role for next generation chemical probes and chemistry-based target deconvolution methods in phenotypic drug discovery
Manuela
Jörg
 *ab and
Katrina S.
Madden
*ab
*ab and
Katrina S.
Madden
*ab
aSchool of Natural and Environmental Sciences, Newcastle University, Bedson Building, Newcastle upon Tyne NE1 7RU, UK. E-mail: kate.madden@newcastle.ac.uk
bMedicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, Victoria 3052, Australia. E-mail: manuela.jorg@monash.edu
First published on 24th March 2021
Abstract
The reconnection of the scientific community with phenotypic drug discovery has created exciting new possibilities to develop therapies for diseases with highly complex biology. It promises to revolutionise fields such as neurodegenerative disease and regenerative medicine, where the development of new drugs has consistently proved elusive. Arguably, the greatest challenge in readopting the phenotypic drug discovery approach exists in establishing a crucial chain of translatability between phenotype and benefit to patients in the clinic. This remains a key stumbling block for the field which needs to be overcome in order to fully realise the potential of phenotypic drug discovery. Excellent quality chemical probes and chemistry-based target deconvolution techniques will be a crucial part of this process. In this review, we discuss the current capabilities of chemical probes and chemistry-based target deconvolution methods and evaluate the next advances necessary in order to fully support phenotypic screening approaches in drug discovery.
 Manuela Jörg | Dr. Manuela Jörg is a Monash and Newcastle (UK) University Research Fellow with an interest in developing small-molecular drugs and pharmacological tools to support different aspects of drug discovery. She obtained a PhD in Medicinal Chemistry from Monash University, in addition to a Bachelor and Masters in Chemistry at the University of Applied Sciences Northwestern Switzerland and the University of Basel, respectively. Prior to her academic career, she completed an apprenticeship as a chemical lab-technician and has experience working in industrial and government organisations. |
 Katrina Madden | Dr. Katrina Madden holds a Newcastle/Monash University Academic Track Fellowship in Drug Discovery at Newcastle University, where she performs research jointly between the School of Natural and Environmental Sciences and the Newcastle University Translational and Clinical Research Institute. She completed her PhD in natural product synthesis at Durham University supervised by Professor Andy Whiting, followed by postdoctoral appointments in phenotypic drug discovery and biocatalysis at the University of Oxford in the laboratories of Professor Angela Russell and Professor Kylie Vincent. Her research interests centre on phenotypic drug discovery and developing new chemical tools for precision control of the immune system. |
Introduction
Traditionally in drug discovery, compounds were evaluated empirically using in vitro and/or in vivo models that imitate a specific disease or a process linked to a disease, known as phenotypic drug discovery (PDD).1–6 Although this approach was never fully abandoned, progress in gene sequencing and molecular biology led to a shift in industry and academia towards target-based drug discovery (TBDD) efforts. Target-based approaches have some noteworthy benefits as they allow the efficient evaluation of many compounds via high-throughput screening (HTS). Furthermore, structure–activity relationship (SAR) studies are often well defined and, due to recent advances in structural biology, may be complemented with X-ray crystallography and cryo-EM data. This has led to the discovery of numerous life-saving therapies, particularly in the cancer field where specific molecular hallmarks have been targeted to provide better outcomes for these heterogeneous diseases.2,5,7–9 However, the overall success rate of TBDD programs in clinical trials has remained unsatisfactory for diseases with highly complex biology effected by multiple genetic and environmental factors e.g. autoimmune diseases, asthma and central nervous system (CNS) disorders. As a result, the scientific community is currently evaluating the gaps that need to be filled,4,10–26 leading to reinvigorated interest in PDD.3,6,10,11,19,27–32 This is partially attributed to the fact that the investigation of an isolated drug target is very distant from the complex biological mechanisms underlying human physiology, including disease although this is still a subject of some debate within the scientific community.19,33 Screening approaches in PDD investigate changes in biological systems more holistically, looking at entire biological pathways rather than isolated proteins, thereby generally providing superior correlation between model systems and disease states when assay cascades are constructed appropriately.34 Compounds are optimised based on their ability to cause the desired phenotype in cell-based assays naïve of the molecular target.4 The added complexity and increased technical requirements of primary PDD assays generally lead to reduced screening capacity of compounds and thus a slower, potentially more costly process. There is some dispute as to whether these costs outweigh those involved with testing multiple hypotheses early on in target-based approaches, and about the relative excellence of TBDD vs. PDD.4,10,19,30,32,33 The different strengths of PDD and TBDD are summarised in Table 1. It seems naïve to assert that one approach is ‘the correct approach’, and indeed the ability to tackle a specific challenge in drug discovery from either perspective, or a hybrid approach, will provide the most chance of success.| Phenotypic drug discovery |
|---|
| + Better correlation between phenotype and disease state |
| + Potential for discovering new biology & first-in-class drugs |
| + Potential to develop drugs for diseases with complex biology and pharmacology |
| Target-based drug discovery |
|---|
| + Simplicity of methods and data analysis |
| + Feasibility of high-throughput screening |
| + Defined structure–activity relationship |
| + Complementary use of structural biology methods, e.g. X-ray and cryo-EM |
The resurgence of PDD signals an exciting new chapter in medicine development, and one that arguably has the potential to impact patients' lives as much as the molecular biology and TBDD revolution.35 In order to realise this, we need to apply the considerable technological and theoretical advances made in the last few decades to elevate PDD to the level of sophistication required to tackle current unmet clinical needs. This review explores the central role that chemistry has to play in this process, particularly focussing on the next advances needed in chemical probe development to streamline the interrogation of complex biological phenomena. Herein, we provide an overview of the state-of-the-art methods used for phenotypic screening and chemistry-based target deconvolution. By highlighting the strengths and limitations of available approaches, we discuss new avenues for the design of chemical probes and chemistry-based target deconvolution methods that have the potential to improve PDD campaigns in the future.
Contemporary phenotypic screening
Historically drug discovery was based on the observation of phenotypic changes in specific test systems, e.g. the change of physiological, morphological and electrophysiological properties in animals, organ explants, tissue extracts as well as cells and bacteria in culture.5 Although advances in genomics and molecular biology have led to a shift towards TBDD, generally most programs have elements of both approaches, combining target-based research methods with functional output from in vitro and/or in vivo assays.19,33 To increase the translation of hits from phenotypic screening campaigns into meaningful outcomes for patients (often referred to as ‘the chain of translatability’), Moffat et al. recommend to use screening systems that have strong correlation with biology pathways in diseases.36 Commonly used model organisms in phenotypic screening include bacteria, fungi, worms, fish and rodents; generally higher organisms are more suitable to study complex biological systems but they are also more expensive. Ideally the systems have genetic similarity to humans and are relevant to the mechanism of action of a drug type. Vincent et al. has defined the “rule of three” emphasizing the key feature for a successful phenotypic screening campaign being a disease relevant model system, stimuli and endpoint.37 For instance, inhibition of bacterial growth has prevailed as a reliable indicator in the development of antibiotics due to its low cost and accessibility, and more importantly its predictive correlation between pharmacodynamic and therapeutic effects in animal models and humans.27,36Phenotypic screening often suffers from reduced throughput of samples when compared to TBDD, but progress in molecular and cellular biology are providing new opportunities to close the gap. Selection of the most appropriate cell system to perform screens in PDD is generally a balance between logistical considerations and disease relevance. The simplified, easy to culture and seed in multi-well plates, cellular model systems that have commonly been favoured in phenotypic screening are often not reflective of the complex human biology.38 Indeed, many cell types lose key characteristics ex vivo. For instance, cancer is made up of numerous molecularly and phenotypically distinct diseases. As a result, finding a disease-relevant model for phenotypic screening in a given application has been extremely challenging, and greater success has been shown with target-based approaches where patients can be selected by predictive genetic biomarkers.19 However, as cellular models become more precise in replicating cancer and the tumour environment (for example, through using co-culture experiments in which different cell types are present), phenotypic screening will likely be used more broadly in target and drug discovery for cancer.39,40 Microglia are another good example – these innate immune cells in the brain respond to signals in the local environment produced by other brain cells e.g. neurons, astrocytes and other stimuli such as pathogens or protein aggregates.41 Culturing them outside of this environment causes them to diverge from the phenotypes they might display in vivo. This can be in part mitigated through co-culturing different cell types, however, this still does not fully model the complexity of an organism and adds considerable extra challenges to the development and analysis of a phenotypic assay.42 Whilst such systems may be suitable for use in drug discovery with the understanding of the associated caveats and appropriate secondary assays, it is important to continually search for more phenotypically relevant model systems.43
There are a number of cell options available for in vitro phenotypic screening, with a general inverse correlation between disease relevance and throughput/ease of use, as illustrated in Fig. 1. Immortalised cell lines are cells which have either been deliberately altered e.g. by transformation using a viral gene such as the simian virus 40 (SV40) T antigen44 or have spontaneously transformed e.g. cancer cells that have overcome usual cell cycle processes to proliferate indefinitely.41 Examples include the Jurkat, BV-2 and SHY-5Y cell lines. These lines have the main advantage of being amenable to growing in large numbers, requiring minimal maintenance and being suitable for freezing and storing for long periods of time. As a result, these lines have become a key part of biomedical and drug discovery research, providing a cost-effective and scalable way to study disease, and are often the method of choice for high-throughput phenotypic screening. However, these cells come with a range of caveats that limit their effectiveness as disease models. In addition to the above mentioned limitations associated with being removed from their microenvironment, cells which have been deliberately transformed possess characteristics different to those they were derived from and even cancer cell lines, which have spontaneously developed show clear differences to in vivo tumours.19 It has also been shown that over-passaging of cell lines can cause spontaneous changes creating variation and potentially even more deviation from disease-relevance.45 There is also a considerable problem with cross contamination of cell lines, or a lack of verification data supporting cells donated between researchers.46 These changes do not necessarily prevent the cell line from being a suitable model, as demonstrated by their widespread usage, but care should be taken to consider whether they will provide an appropriate surrogate for the phenotype being targeted. Primary cells have been isolated directly from the species of study, including from human patients, and therefore better recapitulate the scenario in vivo.47 They can be obtained from patients suffering from the disease of interest, making them an invaluable tool for assessing drug candidates in a more disease-relevant setting. Primary cells cannot be expanded and passaged many times, unlike immortalised cell lines, meaning that obtaining large enough cell populations for screening is a huge barrier to their employment in PDD. They are subject to considerable variability (no two organisms are the same), making it harder for them to deliver on the metrics needed for a robust assay.48,49 They also are affected by being removed from their natural environment, as with all ex vivo models. Additionally, ensuring regular access to relevant patient-derived tissue samples is extremely challenging, and often a ‘no-go’ factor in their use for large screening efforts.
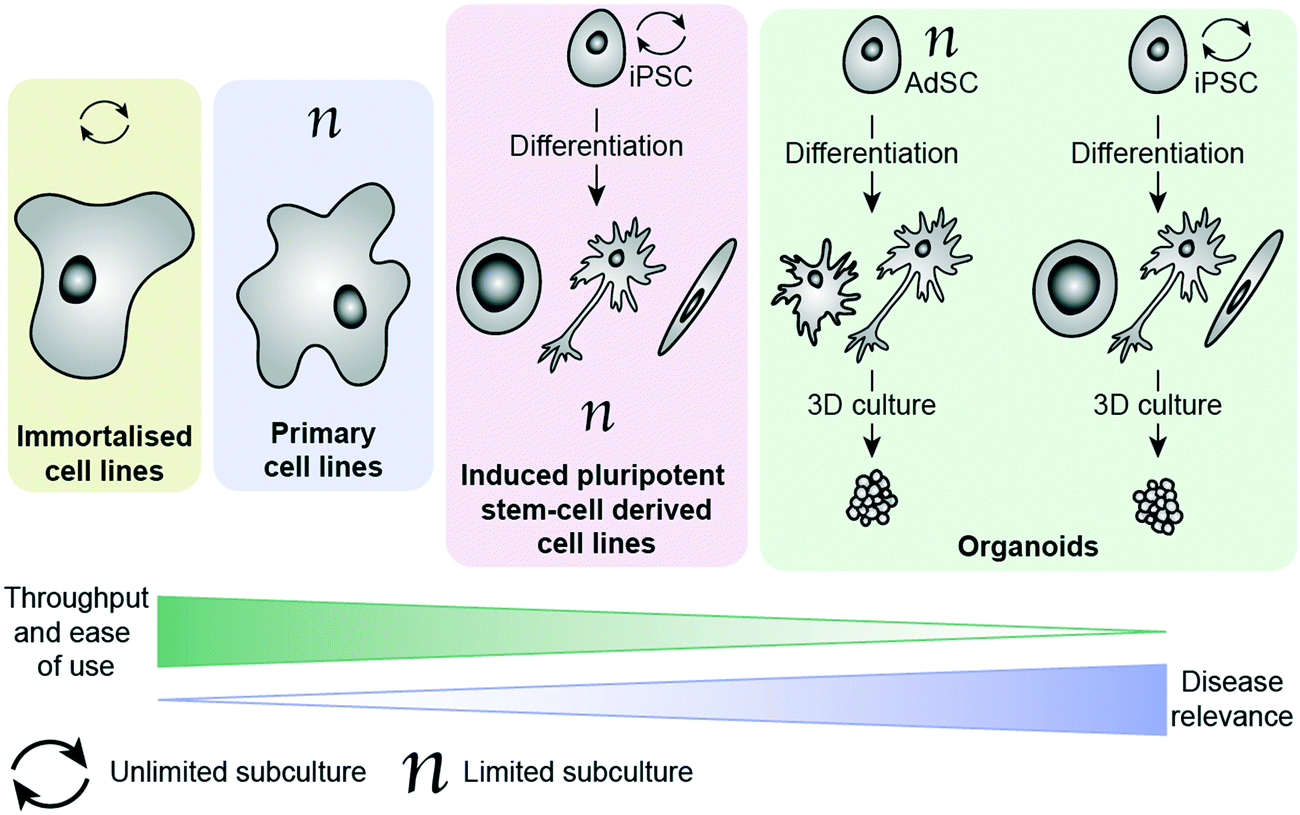 | ||
| Fig. 1 Illustration of different in vitro models available in phenotypic screening, (from left to right) in order of increasing adaptability to high-throughput screening, but decreased disease relevance. | ||
More recently, the progress in the area of stem cell research, specifically the development of human induced pluripotent cells (hiPSCs), has provided the ability study the unique genetic make-up of patients and their disorders. These cells are self-renewable and can be differentiated into numerous cell types, thereby generating relatively large quantities of cells (e.g. cardiomyocytes and neuronal cells) that are challenging to access otherwise. Such cells can then be used for the screening of compound libraries.50 Banks of hiPSCs are available to the scientific community, generated by international initiatives to collect and preserve as many different types as possible, making them much easier to access than primary patient-derived cells.51,52 By using genome editing technologies such as CRIPSR/Cas9 together in combination with human induced pluripotent cells has enabled researchers to gain insights into neurological disorders on a molecular level, by differentiating changes to the phenotype in the same cell type that is affected in patients.53 This method is particularly powerful for neurological disorders lacking disease-relevant in vivo models that accurately represent the human disease. The combination of patient-relevance and self-renewal is extremely exciting, having the potential not only to advance PDD, but also to enable personalised medicine. Excellent progress has been made in the development of protocols for culture and differentiation of hiPSC-derived cells, with some studies successfully employing them in screening efforts.54–57 Culture, maintenance and testing of hiPSC-derived cells, however, is both time-consuming and technically very challenging, making these systems largely inaccessible to those without highly specialised training.
The ability to grow 3D clusters of cells into organoids has represented a significant advance in available in vitro disease models.58 These systems can be grown either from iPSC-derived cells or from adult stem cells (AdSCs) and possess many of the same strengths and limitations as their 2D cultured counterparts.59,60 hiPSC-derived organoids can be established from patients, with the ability to create multiple tissue types from one hiPSC line. However, they require lengthy and complex protocols for reprogramming and differentiating the cells.59 AdSC-derived organoids are less challenging to culture and can also be taken from patients, but often suffer from the same limitations as primary cells (e.g. requirement for continued access to patient tissues).60 Additionally, they are more committed in their lineage than hiPSCs, meaning that cell types available for organoid culture are generally limited to the organ from which the cells were extracted.61 Organoids can be derived from tumour biopsies, and show indications of being more disease relevant than cell lines.62,63 Organoids are also amenable to genetic editing using methods such as CRIPSR/Cas9 and lentiviruses, providing further opportunities to diversify the cell models available.61 With improved recapitulation of the environment in vivo, this technology has enormous potential to improve translation in PDD. Organoids have already begun to be employed in HTS campaigns, demonstrating their potential to act as drug screening platforms.64–67 Biobanks of patient-derived tissues are providing greater choice in cell models and improving access to cells with specific molecular hallmarks, though this raises new ethical considerations around generating ‘organs’ from human donors.68 Although currently in its infancy, organoid testing could revolutionise drug discovery by abolishing or drastically reducing the need for animal testing in the future. Currently there are some limitations that will need to be overcome for organoids to be widely adopted in drug discovery, such as a lack of well-established protocols and variability in organoids produced from the same cells between groups.59 Culture of organoids currently relies heavily on extracellular matrix protein products such as Matrigel, which is obtained from Engelbreth–Holm–Swarm (EHS) mouse sarcoma cells. Variation in batches of Matrigel can be a cause of variability in organoid screening, which presents an imperative for synthetic, well-defined replacements that can support 3D culture instead.59,69 One way this is being addressed is by applying ‘organ-on-a-chip’ technology to create ‘organoids-on-a-chip’.70 Like hiPSC-derived cell culture in 2D, implementation of organoids for screening requires a high level of specialist technical skill, and does not yet possess reliable throughput and assay metrics to drive a drug discovery programme. Additionally, although organoids represent a considerable improvement towards phenotypic relevance, it must be kept in mind that these models do not fully replicate organ function.
Reporter-gene assays have increasingly found application in HTS platforms for PDD programs to address the problem of low compound throughput. The transfection of cells or organisms with a reporter gene (that ideally is not natively expressed in the selected studied system), allows monitoring of up- and down-regulation of specific proteins or pathways within a model system. Commonly used reporter proteins include green fluorescent protein (GFP), red fluorescent protein (dsRed), luciferase enzyme (luc), chloramphenicol acetyltransferase (CAT), β-glucuronidase (GUS) and β-galactosidase (lacZ). These generally have fluorescence or luminescence properties, which are used for the detection of changes to the cellular environment that are otherwise not easily measurable. Luciferase reporter assays are among the most widely employed tools in phenotypic screening due to their ease of use and the ability to generate a quantitative readout through measurement of fluorescence with minimal background-noise caused by autofluorescence. Additionally, this assay does not rely on post-translational modifications in mammalian cells, compared to green fluorescent protein (GFP) another broadly used reporter gene. Despite this, such assays have major drawbacks, namely that luciferase binding compounds can interfere with the signal, leading to higher rate of false positives or negatives, depending on the assay readout. A number of potent luciferase inhibitors have been discovered through their interference with a luciferase-based reporter cell assay.71–74 More generally, awareness of structural alerts of pan-assay interference compounds (PAINS) including luciferase inhibitors is important to flag potential non-specific binders to avoid disappointment further down in the drug discovery pipeline.75,76 More recently NanoLuc® luciferase systems, which are based on a 19.1 kDa luciferase enzyme that originates from deep sea shrimps, have provided researcher with new opportunities due to the enzyme's superior thermal and pH-stability, smaller size, and >150-fold increase in luminescence compared to traditionally technologies using firefly or Renilla luciferase.77,78 The improved brightness of the reporter permits measure proteins with a low-level of expression, including endogenous proteins. Furthermore, this has enabled the use luminescence microscopy more broadly as an alternative to confocal microscopy. NanoLuc has proven to be useful for a wide range in vitro and in vivo platforms including NanoBiT and NanoBRET. The main limitation of this technology is its blue-shifted emission maximum at 460 nm compared to traditional luciferases (480–485 nm), which is not optimal for in vivo investigations and requires further optimization.78
The power of phenotypic screening to investigate complex systems and diseases is also associated with the enormous challenge of analysing complex and large datasets.1,14–18,39,79–87 Advances in data analysis are already enabling the analysis of big and complex data from experiments and databases; such methods include neural-networks, machine-learning, and deep-learning methods.81,82,85,87–91 Additionally, advances in cellular imaging have ushered in the era of high content screening and have led to predictive toxicology procedures which draw attention to potentially problematic hits early in the discovery phase of PDD programs.92 Application of computational methods may expedite the identification of drug targets, toxicological structural alerts, and advance rational drug discovery approaches with regards to polypharmacology.93,94 Machine learning approaches may be able to reduce the need for in-depth knowledge of a drug target, by annotating and clustering complex protein networks based on multiple readouts from complex biological models and structural modifications from medicinal chemistry programs, which then can be used as the basis for rational drug optimization.
In summary, technical advances in phenotypic screening contributed to the spotlight on PDD into the 21st century, but there are still numerous challenges that limit widespread adoption of PDD in both academia and industry (Table 2).
| Advances in phenotypic screening |
|---|
| • High throughput screening |
| • Advanced data analysis |
| • Access to patient-derived samples |
| • Organoids |
| • Pluripotent stem cell technologies |
| • Gene-editing tools (CRISPR–Cas) |
| Challenges with phenotypic screening |
|---|
| • Low screening throughput |
| • Identification of false positives |
| • Ensuring chain of translatability |
| • Assay variability |
Target deconvolution from phenotypic screening
Whilst PDD allows for optimisation of compounds to induce a disease-relevant phenotype, doing so without knowledge of the targets involved can have major safety ramifications. Therefore, compounds discovered through PDD warrant at the very least meticulous investigation to rule out interactions with pathways deemed unsafe due to well-known drug-induced toxicities. Although the approval of drugs by authorities is not necessarily dependent on identifying the mechanism of action, knowing the drug target and disease biology generally eases the registration process due to increased confidence in the safety profile of a drug, whilst it also aids the drug development process.95 The identification of a target also permits stratification of patient groups via the use of predictive genetic biomarkers, an approach which has been particularly successful for the treatment of cancers.19 Consequently, the identification of drug targets from phenotypic screening, known as target deconvolution, is a central part of most phenotypic drug discovery programs.40,96–103 The synergistic effects of both PDD and TBDD is most significant when target identification is achieved early on in a drug discovery program (Table 1). As a result, advancement of both phenotypic screening methods and deconvolution strategies go hand-in-hand and play a critical role to improve the efficiency of the drug discovery process.Hit-to-lead optimization driven by phenotypic screening can be challenging due to the confounding effects of cell permeability, stability, solubility and uncertainty that analogues under comparison are engaging with the same target(s). Target deconvolution aims to overcome these limitations and, if performed successfully, aids the generation of well-defined SAR data in addition to potentially enabling structure-based design. Multiple excellent reviews have been published in recent years discussing target deconvolution and validation strategies, and we do not seek to replicate these.104–111 We will, however, discuss the aspects of target deconvolution involving small molecule engagement directly with protein targets, and the strengths and limitations of these techniques.
Target deconvolution based on genetic, chemical and biophysical methods is often time-consuming. Therefore, researchers routinely perform parallel screens of libraries consisting of a broad set of known biologically active compounds with well-established target activity and mode of action (approved and/or failed drugs, gold standards and chemical probes) to identify the drug targets of hits from high-throughput campaigns.83,84,112 Ideally these libraries of compounds cover a broad chemical and pharmacological space, while exhibiting respectable bioavailability and safety profiles. This approach allows for the direct comparisons of phenotypic changes between established compounds and novel hits from phenotypic screening campaigns, thereby assisting the elucidation of the drug target(s) while also identifying hits with novel mechanism of action. The possibility to identify polypharmacology of compounds with established in vivo efficacy is an important advantage of this method, allowing researchers to gain a better understanding of the complex underlying biology of many diseases and particularly CNS disorders, which remains a key challenge that needs to be addressed with modern drug discovery.22,23,25,113,114 Additionally, this approach might allow for repurposing of established drugs for new indications thereby fast-forwarding the optimization process of finding not only an efficacious, but also safe, drug with good physicochemical properties.115,116
Chemical probes in phenotypic screening and target deconvolution
Low success rates in clinical studies have highlighted the importance of high-quality chemical probes in drug discovery to ensure the robustness of drug targets and their clinical relevance.117,118 In the context of this review, chemical probes are defined as small-molecule tools that uniquely interact with a specific drug target and can be used to study protein function in a biological system. By designing a small molecule for this purpose, the behaviour of the protein target of interest can be studied in multiple biological systems where applicable, such as in different cell types or pathogens. High-quality probes become especially pivotal when complex screening assays are used, as is generally the case for PDD programs.119 There are many factors and properties that need to be considered when designing probes to guarantee the generation of robust data sets. These allow for informed and confident decision making regarding progressing a hit or terminating drug discovery projects at an early stage.120 Phenotypic screening relies on quality chemical probes to not only reliably establish the relationship between a target and its phenotype but also to assist with the deconvolution of drug targets and mechanism of action.121,122The development of target-selective probes to investigate drug targets has gained more attention in recent years, and has led to the establishment of dedicated libraries as well as of rules to guide the design of high-quality probes, which are collated in numerous recent reviews.120,121,123–125 A retrospective analysis performed by Pfizer determined the underlying factors that lead to the failure of their drug discovery programs, highlighted the importance of three key criteria (Table 3) to ensure the validity of results in cell-based assays for target validation using chemical probes, being 1) exposure at the site of action, 2) target engagement and 3) functional pharmacological activity.125 This concept was further complimented with a 4th pillar focusing on the relevance of the phenotype, essentially to assure that the observed change in the phenotype is relevant in the framework of human disease and is not caused by non-specific interactions.125 Certain properties are essential for all chemical probes, others might vary depending on type and location of a drug target, as well as model organism and the specific application of a probe (Table 3); hence, it is always essential to be aware of the limitations of a specific probe.
| Key criteria | Properties for consideration |
|---|---|
| 1) Exposure at the site of action | • On- and off-target affinity and selectivity |
| 2) Target engagement | • Solubility and toxicity profile |
| 3) Functional pharmacological activity | • Chemical and/or metabolic stability |
| Ensure requirements are fulfilled in all assays: biochemical, in-cell assays (in vitro) and animal models (in vivo). Confirm that the phenotype is relevant to human disease. | • Cell permeability/in vivo activity (including active efflux) |
• Lipophilicity (calculated or measured via clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) P) |
|
| • Assay interference and/or promiscuous binding | |
| • Structural integrity and purity (particularly important for purchased compounds from commercial vendors) |
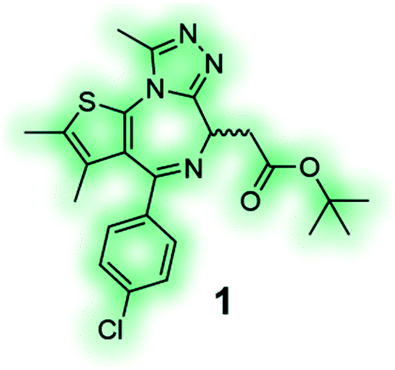
High affinity and good selectivity for the target of interest are key to ensure that the observed phenotypic effects are not due to another protein target. In some cases non-selective probes might still be suitable depending on the application, and off-target effects can be investigated via co-administration of selective inhibitors, however, this approach is undesirable as it adds significant complexity and uncertainty to the experiment.126 To understand the role of a protein in isolation, highly selective chemical probes for a specific drug target are desirable. High selectivity is not always readily achievable, especially for targets with highly conserved binding sites. It is, however, paramount that all probes well annotated to ensure they are appropriately used, and results interpreted correctly. A common source for false positive hits is the colour, auto fluorescent, cytotoxic or promiscuous binding nature of compounds. To confirm or disprove the validity of a hit it is always recommended to use at least one secondary orthogonal screening assay to eliminate false positives early in the drug discovery cascade. Furthermore, a probe's chemical composition and physicochemical properties play a crucial role for its cell-permeability, solubility, toxicity and stability. Without the careful consideration of these factors, exposure at the site of action, target engagement and, functional pharmacological activity cannot be guaranteed.120 BET bromodomain inhibitors, such as chemical probe JQ1 (1) (Table 4), are an example of excellence in the development and pharmacological evaluation of chemical probes.127,128 The (R)-enantiomer of JQ1 (1) is inactive and the ideal negative control when used in parallel to the active (S)-enantiomer of JQ1 (1).128 Their discovery showcases the step-by-step evaluation that should be sought after for the successful validation of hits from phenotypic screening and the associated target deconvolution.125,129
| Probe type | Example | Comments |
|---|---|---|
|
||
| Label-free chemical probes | ||

|
|
JQ1 (1) is a potent and selective BET bromodomain inhibitor; (S)-(+)-JQ1 is the active enantiomer and the (R)-(−)-JQ1 is the negative control.128 |
| Labelled chemical probes | ||
Reporter tag, no linker |
|
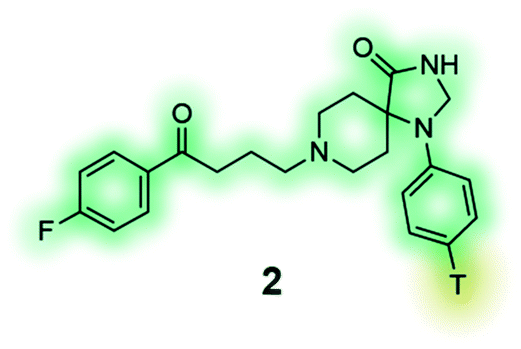
[3H] Spiperone (2) is a high-affinity radioligand for the dopamine receptors, commonly used in radioligand binding assays and compound screening.132,133 |
Reactive group and reporter tag, no linker |
|
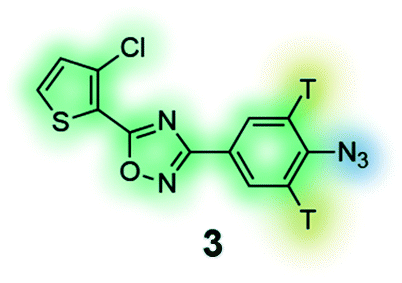
Photoaffinity probe with radioisotope reporter tag of the 3,5-diaryl-oxadiazole based lead MX-126374 to study selective apoptosis in malignant cells.134 |
Reporter tag with linker |
|
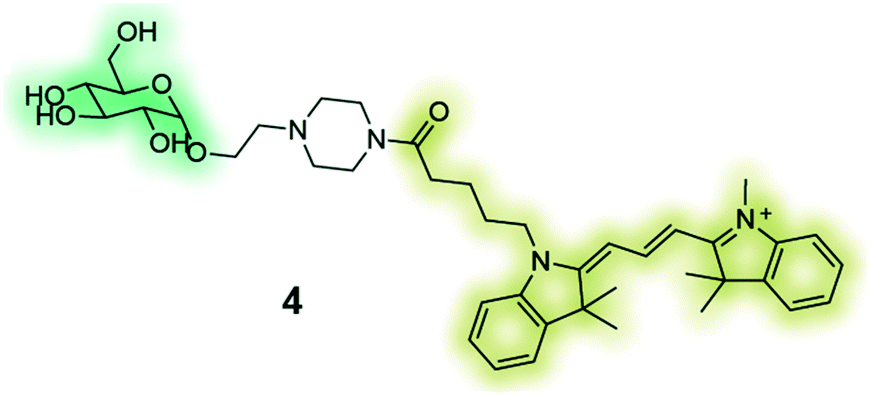
GB2-Cy3 (4) is a fluorescently-labelled glucose bioprobe for applicationsin high-throughput phenotypic screening to find therapeutic compounds for the treatment of cancer and diabetes.135 |
Reactive group and reporter tag with linker |
|
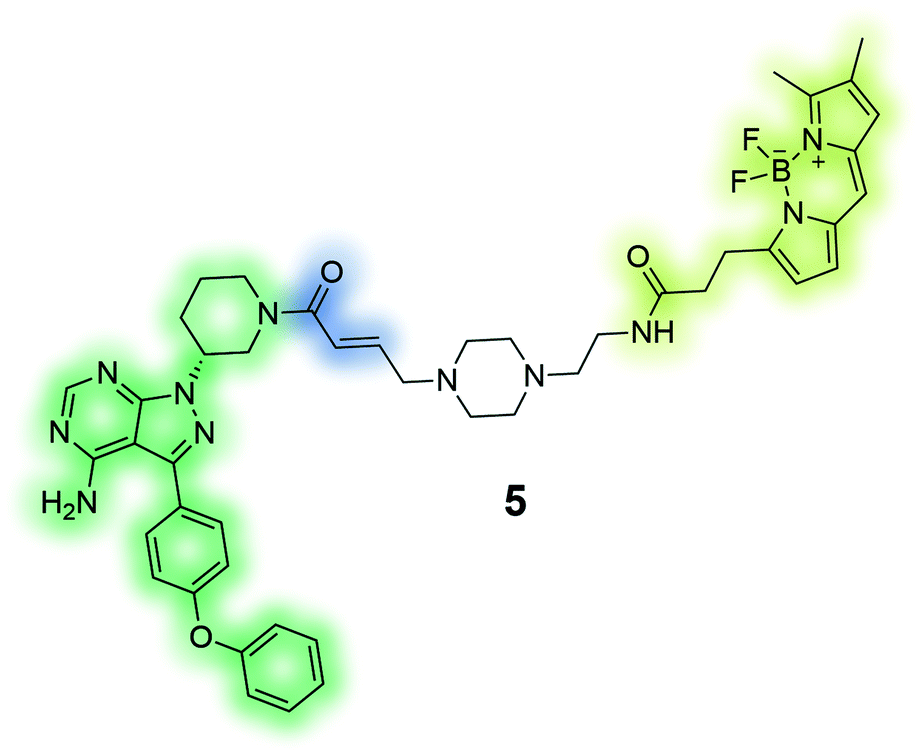
PCI-33380 (5) is an activity-based fluorescent probe of the anti-cancer drug ibrutinib. The depicted probe was used to measure occupancy levels in cells and tissue, and permitted the prediction of efficacious dosage of the drug for patients in clinical trials.136 |
Reactive group and reportertag with click handle |
|
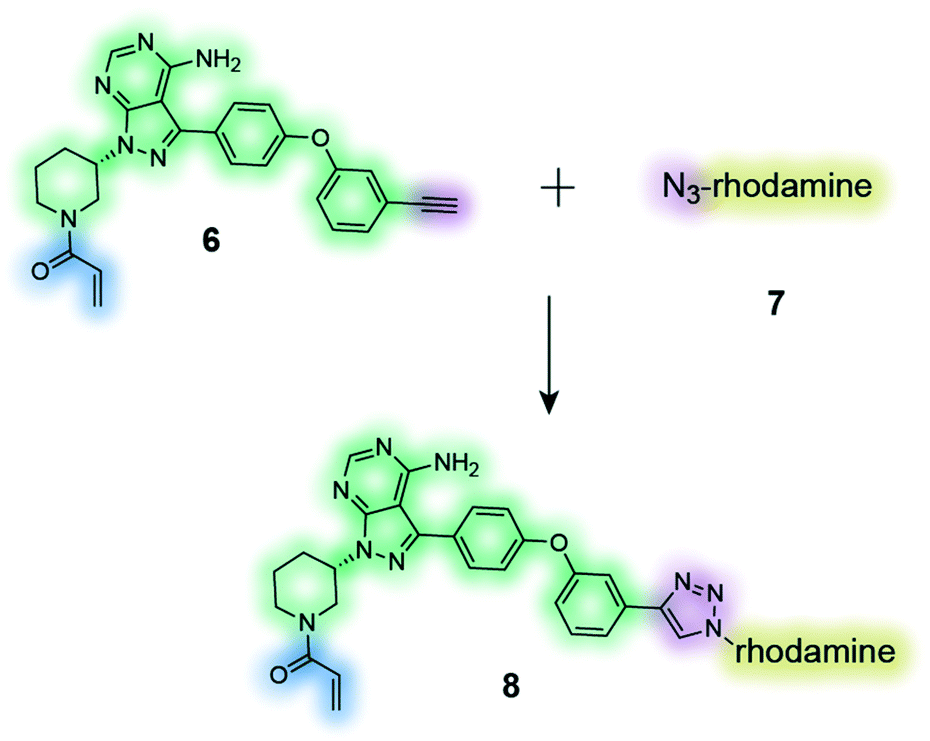
The clickable activity-based probe (6) of the anti-cancer drug ibrutinib was used to study in-cell proteome selectivity in human cancer cells. The probe was designed to have minimal effect on the reactivity of the Michael acceptor, which subsequently was optimized to improve the selectivity profile of next generation analogues.137 |
It is worth pointing out that the requirements for chemical probes are different to drug candidates, e.g. long-term therapeutic benefits and safety considerations are pivotal for the successful development of drugs, whereas the focus for chemical probes lies in gaining a robust understanding of the target and its mechanism of action.119,126 Nevertheless, the development and pharmacological evaluation of chemical probes is equally intricate as generally multiple factors, such as solubility, cell-permeability, metabolic stabilities as well as on- and off-target affinity and selectivity need to be optimized from an original screening hit. Strategies such as monitoring the lipophilicity of probes might be used to guide the development while reducing the risk of self-aggregation and off-target promiscuity of probes, but robust pharmacological evaluations are indispensable to ensure a probe is fit for purpose. It is also important to be on top of the latest progress in the field as superior chemical probes are likely to be developed over time, and the most commonly used and commercially available tools are not always the most suitable for use in a specific experiment.119 The value of systematic and detailed development and assessment of chemicals probes cannot be overstated for the robust validation of hits from phenotypic screening assays, and will be key to improving the odds of success of drug discovery programs.
In addition to the general qualities described above, tool compounds with distinctive properties, known as labelled probes, are strategically used to support phenotypic drug discovery programs in pursuit of hit identification as well as target deconvolution. There are some structural and functional features common to labelled probes, which we will discuss along with their applications in this section. Labelled probes consist of a biologically active moiety or pharmacophore that ensures affinity and selectivity for the target of interest, combined with a reporter group to enable their detection and/or a reactive group to facilitate target binding. Commonly used reporter groups include biotin, hemagglutinin, fluorophores or radioisotopes, which assist with the identification of the ligand–protein complex using a range of different experiments.130,131 In some cases, the installation of a linker is necessary to connect the pharmacophore with these functionalities to circumvent a detrimental loss of the parent compound's key pharmacological and physicochemical properties. Some representative examples of different labelled probes are discussed below and illustrated in Table 4. The time and effort required for the successful development of chemical probes should not be underestimated, as each part of a probe needs to be carefully optimized and often extensive SAR studies are necessary to be performed beforehand. Ultimately, chemical probes can play a key role by providing practical assay readouts, validating on-target action, target engagement and mechanism of action, and furthermore be valuable tools in assisting pull-down experiments.
Labelled chemical probes in phenotypic screening
Radioligands are chemical probes that contain a radioisotope, which allow for quantification of a molecule's ability to bind to a protein of interest. Tritium (3H) is the most used radioisotope for pharmacological experiments, mainly due to its long half-life (12.3 years), good safety profile (low beta energy emission), and the relative ease of its synthetic incorporation into molecules. Other radioisotopes are also used, such as 125I which has a half-life of 59.5 days and is commonly used to label peptides at tyrosine residues. In phenotypic screening radioligands can be used to establish if test molecules bind to the same binding site through competition experiments. Applications include the screening of compounds to discover structurally novel hits of the same drug target or assisting the identification of a drug target and mechanism of action from hits of a phenotypic screening campaign. Radiolabelled tool compounds also permit determination of the binding kinetics (association and dissociation rate constants) of hits without the need for labelling each compound individually. Apart from the insertion of a radioisotope, radioligands are often structurally identical or close analogues of the parent molecule, which increases the likelihood of retaining biological activity and physicochemical properties, the latter often permitting the use of these tools in live cell experiments.138 The radiolabelled [3H] spiperone (2) (Table 4), a dopamine receptor antagonist with extremely high affinity for the D2, D3 and D4 receptor subtypes e.g. has exhibited variances between results of binding studies performed in cells and membranes.132,133 To ensure specific binding for the target, radioligands ideally require rate constant (Kd) in the low nanomolar range and good selectivity, however, this often requires rigorous synthetic optimization and limits their application for types of ligands that commonly have low binding affinity, such as ligands that bind to allosteric sites of class A G protein-coupled receptors.Fluorescence has frequently been used in phenotypic screening for decades using reporter cell lines, which allows for measurement of the change in expression levels or subcellular location of a protein. As previously discussed, this approach has its limitations. The incorporation of a fluorophore into chemical molecules of interest, makes fluorescent probes suitable tools for a range of applications without the need for genetic manipulation.139–144 Fluorescent probes of a molecule with a known target can be used instead of radioligands. In addition to being superior in terms of safety, temporal and partial resolution, a fluorescent probe might also be used to study signalling processes, localisation and expression of a protein of interest. Similarly, the emerging research area of fluorescent bioprobes aims to investigate phenotypic changes in intact biological systems by measuring levels of fluorescently labelled biochemicals that have been linked to one or several diseases. A representative example is the development of fluorescently labelled-glucose bioprobes (Table 4, compound 4) that can be used to quantitatively measure cellular glucose uptake in real-time.135 Glucose levels are highly regulated in the human body and in the case of numerous diseases abnormal levels of glucose can be observed, e.g. in cancers, diabetes and Alzheimer's disease. As a result, fluorescent bioprobes for glucose have been used for diagnostic imaging and the successful identification of novel structural hits from medium-size phenotypic screens (>1000 compounds).135 Traditional image-based methods have lacked adaptability to high-throughput screening, but progress in imaging technology and computational methods is expected to even further broaden the scope of fluorescent-based phenotypic screening approaches.145–148
Labelled chemical probes in target deconvolution
The use of chemical probes to understand protein function, known as chemoproteomics, is a powerful strategy in target deconvolution.149 Like phenotypic screening, this field has made huge progress, largely driven by advances in mass spectrometry (MS), which is the main quantitative analytical method used to identify putative target proteins of small molecules.150 It has outstripped other analytic methods due to its speed, low sample loading requirements and flexibility, making it an essential tool in the target deconvolution process. Protein abundance can be calculated in a quantitative chemical proteomics approach through labelling methods such as stable isotope labelling by amino acids in cell culture (SILAC) and isobaric tags for relative and absolute quantitation (iTRAQ).151 These methods have made considerable progress in addressing the challenges of non-specific binding or low protein abundance that are often encountered in target deconvolution campaigns.152Fig. 2 illustrates various applications of labelled chemical probes in target deconvolution. Affinity chromatography (Fig. 2a) is one of the most well-known chemoproteomics techniques, which exploits the affinity of the small molecule to its protein target(s).102 The compound is derivatised to possess a chemical group (e.g. an amino group) suitable for immobilisation on a solid support, such as beads, if it doesn't have one already. A soluble protein fraction such as a cell lysate is then applied to the compound-immobilized surface, which is then repeatedly washed to remove unbound protein. Lastly, the bound proteins are eluted and identified by MS.107 An example of this is the discovery that (R)-roscovitine targets pyridoxal kinase in addition to cyclin-dependent kinases.153 This method is fairly disparate from the biological conditions of a phenotypic assay, which represents one of its major limitations. It also requires a high compound affinity for the protein to ensure that the complex survives the subsequent stringent washing steps, and to minimise contaminant interference or non-specific binding.104,107 Immobilisation of the compound can also present challenges by altering its conformation or reducing its ability to bind to its protein target(s), suggesting that validation of any targets using a method with free compound is key to developing confidence in any identified proteins.
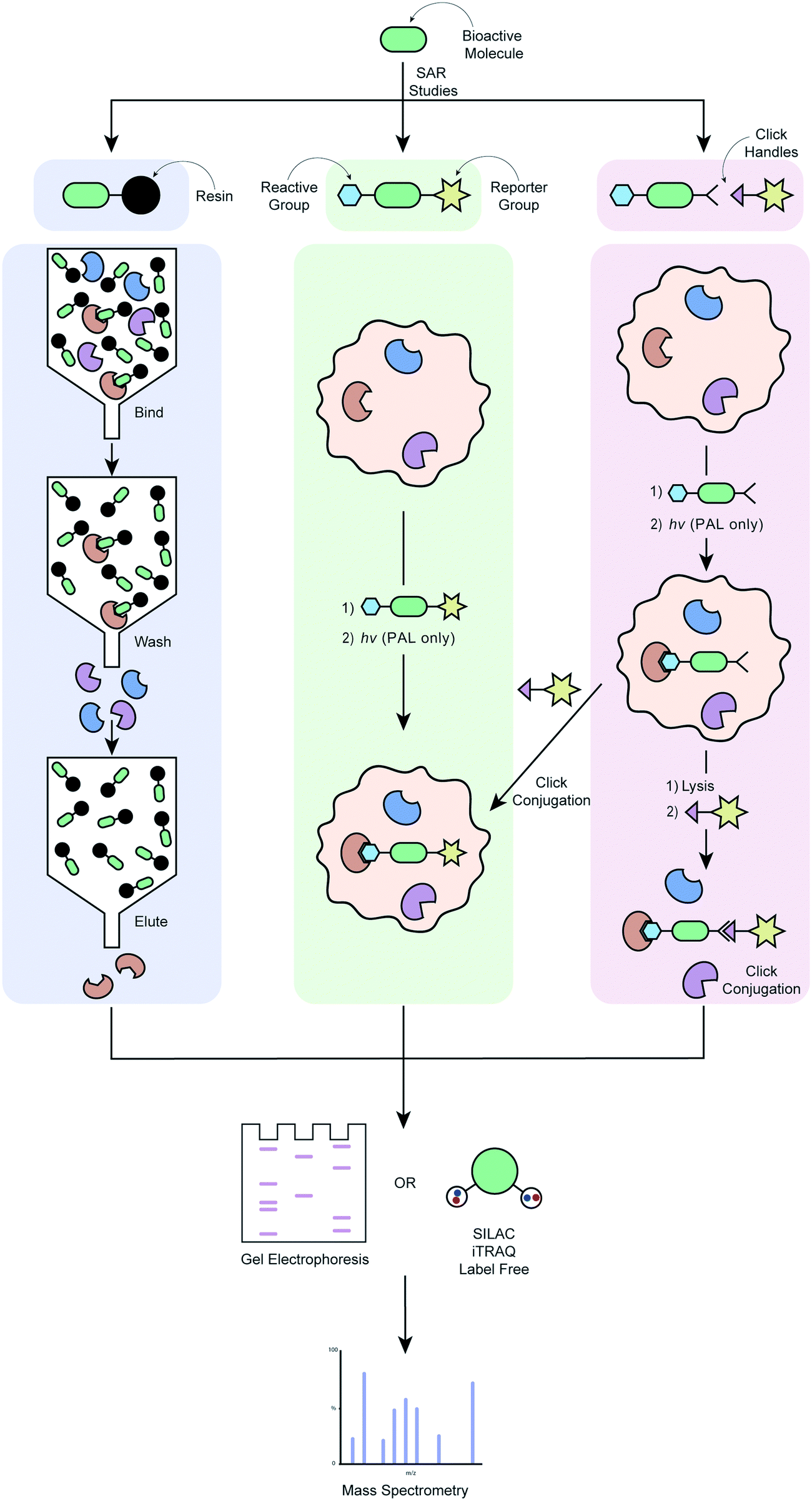 | ||
| Fig. 2 Workflow of different target deconvolution methods using labelled chemical probes, illustrating a) affinity chromatography, b) ABPP and AfBPP and, c) ABPP and AfBPP using click conjugation. | ||
In order to overcome the need for chemical probes with high target affinity, increasing campaigns are employing the use of modified chemical probes that allow direct engagement of this probe with targets in the proteome to be identified.34 Covalent probes possess a reactive group that is capable of forming irreversible covalent bond with an amino acid residue of a target, ideally within the proximity of a target binding site (Fig. 2b).154 The probe's affinity for the target(s) can therefore be much weaker, in the order of μM as opposed to the ideal nM required for pure affinity chromatography methods.107 Activity-based protein profiling (ABPP) involves a chemical probe that exploits nucleophilic amino acid residues in the binding site to form a covalent bond. If the drug is not already a covalent binder of its target protein, then it must be engineered to incorporate a chemoreactive group.151 This concept is demonstrated by the fluorescently tagged probe PCI-32765 (5) in Table 4, which is based on the marketed cancer drug ibrutinib, a covalent inhibitor blocking B-cell receptor signalling.136 Ibrutinib contains an α,β-unsaturated amide electrophile (Michael acceptors) that is known to form a covalent bond with an active site cysteine in the ATP-binding pocket of Burton's tyrosine kinase (BTK). Attachment of a Bodipy-FL fluorophore to the pharmacophore, PCI-32765 (5), was achieved via the introduction of a piperazine linker. The cell permeable probe was used to directly quantify the occupancy of the Btk active site by 5 in cells or target tissues, and determine minimum dosage levels in specific cancer patient cohorts.136 In affinity-based protein profiling (AfBPP)/photoaffinity labelling (PAL) a photoreactive group is incorporated into the probe which, on exposure to UV irradiation, can form a covalent bond with the target(s) it has an affinity for. The photoreactive group can either be incorporated directly into the pharmacophore or attached via a linker. There are pros and cons to each of these approaches, and both require optimisation to ensure that the biological activity of the parent compounds is recapitulated as well as possible. Installation of the photoreactive group via a linker can minimise interference with the pharmacophore's binding to the target, as can be a problem with a pharmacophore-incorporated photoreactive group. On the other hand, the addition of a linker results in a bigger difference structurally between the parent compound and the probe, and can have detrimental effects on the probe's cell permeability. AfBPP/PAL can also be referred to as capture compound mass spectrometry (CCMS).155–157 An example of this approach is the identification of the aryl hydrocarbon receptor as a molecular target for the Duchenne muscular dystrophy drug candidate, ezutromid.158 Groups have also been using targeted photoaffinity probes to take a snapshot of complex groups of proteins in living cells e.g. the Hsieh-Wilson group developed a photoaffinity probe which was used to identify >50 glycosaminoglycan binding proteins in cortical neuron cells, mapping a highly complex network of protein interactions.159 Cravatt and co-workers have been key drivers of innovation in the field of photoreactive probe development, using PAL not only as a method of target deconvolution, but also combining it with phenotypic screening to create powerful new platforms for drug development.160–166 Both types of probes have their unique strengths and weaknesses; chemoreactive probes rely on a nucleophilic amino acid residues in or near the active binding site, whereas photoreactive probes might be able to bind in more “natural” binding modes due to their ability to form a covalent bond in situ with any amino acid residue, but require UV radiation that might cause protein degradation.167 A major advantage of both of these techniques is that they do not require compound immobilisation before incubation with proteins. If a suitable probe can be developed, it can be incubated with live cells, the cells lysed and bound proteins purified using a suitable tag. This enables target engagement to be captured in a much more relevant biological context.
ABPP and AfBBP are extremely powerful approaches when successful, but the relative success rate of these versus affinity chromatography is low. Photo-crosslinking yields can be low, which is a considerable contributor to experiments with photoaffinity probes.107 Incorporation of the different groups required often represents a considerable chemical undertaking in terms of SAR development and synthesis (this is also true for more classical affinity chromatography studies), with the additional risk of loss or altered biological activity. Further considerations include the potential for metabolism of the probe if applied to live cells,34 as well as a detrimental effect on the probe's cell permeability.
Cell permeable probes can provide important information that is lost in biochemical assays, such as effects resulting from post-translational modification, autoinhibition, protein–protein interactions, substrate competition and the distinct subcellular distribution of proteins.168 This has led to the advancement of the field towards clickable probes (Fig. 2c), which are significantly smaller in size, often making these probes more suitable for experiments in live cells.168 Although various clickable groups are available, the most commonly used reaction is between an alkyne and azide group, which results into a 1,2,3-triazole linkage.169 This concept is again exemplified by the marketed cancer drug ibrutinib, which more recently was converted to a clickable activity-based probe (Table 4, entry 6) that was used to study in-cell proteome selectivity in human cancer cells.137 The probe was designed with the aim of not altering the reactivity of the Michael acceptors, while also having no or minimal effect on the compounds affinity of the target, which was achieved by introducing the alkyne functionality at the oxydibenzene group. Following cell penetration, the alkyne underwent a copper-catalysed Huisgen 1,3-dipolar cycloaddition with an azide-rhodamine (N3–Rh) reporter tag to facilitate isolation and identification of any bound proteins. This work revealed off-target interactions with a number of proteins, mainly ones that also contained an active site cysteine residue, and was the basis for the development of more selective analogues via chemical alteration of the reactivity of the electrophile (Michael acceptor).137
The principles discussed above are also applicable to the development of chemical probes for the yeast-three hybrid system. Detailed discussion of this chemical genetics approach is beyond the scope of this review, but incorporation of the probe into the system requires analogous linker chemistry.170,171
As the development of covalent probes is not without its challenges, some researchers have been advocating for the establishment of libraries of fully functionalized small-molecules containing both a photoreactive group for covalent cross-linking and an alkyne handle for the conjugation with a reporter tag.172 Although, this approach allows to expedite the target deconvolution process due to eliminating the need for further chemical optimization, extensive work is involved synthesizing such libraries, that have only limited structural diversity and lack of compounds with properties suitable as drug candidates. Although, some of the shortcomings of this method might be addressed with the development of novel photoreactive groups, there is a continuous need for more efficient and innovative ways to rationally design chemical probes. One might also foresee that the implementation of electrophile fragment screening methods into PDD could expedite the development of covalent probes for target deconvolution.173
Label-free chemical probes in target deconvolution
Many of the approaches for phenotypic screening and target deconvolution detailed in this review rely on small molecule chemical probes that exhibit affinity for their target(s). A small molecule under investigation often requires modification to allow for analysis of the proteins it binds to, although increasing options for label-free target deconvolution are available. As previously highlighted, the effect any modification might have on the activity of the chemical probe, especially the potential for the modification to alter the proteins targeted, is a real source of concern. As a result, label-free methods provide a level of reassurance as part of a package combining multiple approaches and readouts. The below methods work on the assumption that molecule binding to a protein target will alter its stability (Fig. 3). A major benefit of these techniques is that they do not require modification of a small molecule to generate a suitable chemical probe. This reduces the chemistry burden and allows visualisation of target engagement with the actual compound of interest. A key caveat with all these techniques is that not all ligand binding events will cause a change in the measured property, leading to the possibility of false negatives.174 The cellular thermal shift assay (CETSA) and thermal proteome profiling (TPP) exploit the thermal stabilisation of a protein on its complexation with a ligand to assess target engagement by a chemical probe.175 Incubation of the compound of interest with either protein lysates or cells, followed by lysis, and analysis at different temperatures allows profiling of the proteins' melting temperatures on compound treatment.176–180 Application of CETSA and TPP in a cellular context, instead of protein lysates, provides a more physiologically relevant snapshot of target engagement.178,181 Drug affinity-responsive target stability (DARTS) follows a similar premise to CETSA, this time assessing the change in stability of a protein to proteolysis on binding with the molecule of interest,103 and stability of proteins from rates of oxidation (SPROX) investigates the ligand-induced stability of a protein to oxidation.182 These techniques can be combined with MS proteomics to increase the data obtained.183 It is advisable to employ more than one unbiased target deconvolution approach to reduce the number of false positives, as exemplified by the identification of Plasmodium chaperonin TRiC/CCT as a molecular target for clemastine by Fitzgerald and Derbyshire.184 The researchers employed parallel TPP and SPROX chemoproteomic strategies in this work. | ||
| Fig. 3 Workflow of different label-free target deconvolution methods, illustrating a) DARTS, b) CETSA/TPP and, c) SPROX. | ||
Target deconvolution campaigns are being increasingly employed to uncover new biology and power drug discovery. The array of methods available, including non-chemistry led methods such as functional genetics, various-omics, knock-down studies and computational validation, has allowed scientists to uncover the biological targets of drug candidates with a high degree of confidence.110 These remain, however, challenging and time-consuming endeavours which require a great deal of effort and technical skills. The advances in chemistry-led target deconvolution and persisting challenges are summarised in Table 5.
| Advances in target deconvolution | Remaining challenges |
|---|---|
| • Annotated libraries of biologically active compounds | • Nonspecific binding events |
| • MS methods for chemoproteomics, including protein/peptide labelling | • Enrichment of target protein in low abundance samples |
| • Multiple activity and affinity-based approaches | • Identification of on- and off-targets |
| • Non-labelled approaches | • Rapid processing of large quantities of MS data |
| • Alternative analytical methods to MS |
What breakthroughs are needed next?
Phenotypic screening is at the forefront of drug discovery once more. The integration of new technologies and methods that have emerged over the past 50 years has already begun to transform PDD and improve the chances of discovering new medicines for complex indications. Despite this progress, further breakthroughs will be needed to fully realise the potential of PDD.One of the main strengths of PDD is its ability to capture polypharmacology i.e. interactions with multiple targets to produce both the desired biological effect, as well as other unwanted side effects. This is a key weakness of TBDD, as focussing on a single molecular target to drive compound design does not allow for a representative picture of the complex disease environment.32 As a result, a compound discovered through TBDD that has a high affinity for its target can fail to deliver the correct phenotypic effect linked to a desired therapeutic benefit. That does not mean that drugs produced through TBDD do not display polypharmacology – indeed many approved drugs have been shown to hit multiple targets regardless of their path of discovery.185 It should be noted that the presence of polypharmacology adds huge complexity to the challenge of target identification. Another considerable challenge that remains is the successful optimisation for multiple targets at once using structure-based design, however, there is considerable progress being made in this field.186 The ability to achieve this would harness the synergy between phenotypic drug discovery and phenotypic drug discovery, hopefully leading to greater translation.
Ensuring disease relevance in the assay cascade continues to represent a considerable challenge in PDD. Many factors influence the chain of translatability, therefore the data obtained from phenotypic screens must be systematically studied and validated. Development of phenotypic screening assays that better represent human disease and provide high confidence in translatability remain pivotal. Hence, there is a continuous need to further develop and optimize the previously mentioned technologies, such as iPSC-derived cells, high-content imaging, 3-dimensional organoid tissue models and machine learning methods, which have the potential to transformation the field, particularly when used to complement each other.
Validated, robust high-quality chemical probes for phenotypic screens are extremely crucial to ensure confidence in assay data. This is particularly relevant for novel more phenotypically relevant systems with high variation e.g. different batches of iPSC-derived or patient-derived cells.43 There are a number of information sources available to those seeking chemical probes but it is acknowledged that dissemination of our collective knowledge could be improved.187 An openly available and current collection of validated negative and positive controls, in addition of increased awareness of the limitations of existing probes, will help further direct and improve the effectiveness of drug discovery programs in academia and industry.
Another major limitation of current probes is the level of chemical modification required to induce fluorescence, covalent binding or attachment to solid support. The incorporation of moieties such as fluorophores or biotin generally results in a substantial change in chemical structure, molecular size and physiochemical properties in comparison to the parent compound. This is particularly profound for small non-peptide based hits, including those designed to pass the blood–brain barrier. As a result, they may not able to interact with the target or are poorly representative of how the parent compound interacts with the target, and additionally are often less active and drug-like. The ability to generate desirable characteristics, e.g. fluorescence, through minimal chemical and physiochemical modifications would represent a significant breakthrough in the field, allowing for more accurate modelling of the mechanism of action of the parent compound, and opening up the possibility of using such probes for in vivo applications.
Target deconvolution remains challenging with low to moderate affinity binders, resulting in often significant background noise.172 Methods are highly reliant on enrichment of proteins and their subsequent detection by MS. The development of methods based on fundamentally different principles of actions would increase the robustness of target deconvolution campaign. One might envisage the use of target degradation as a viable alternative to verify the results of current pull-down target deconvolution methods.188,189
Although our understanding of the underlying biological processes in the human body, and our ability to drug them, has evolved, approximately 85% of human proteins are considered “undruggable” proteins.190 This is due to a lack of sites that can be targeted using traditional drug discovery tactics; this includes transcription factors, non-enzymatic proteins and scaffolding proteins. The discovery of “small molecule” protein degraders, such as PROTACs and LYTACs, might be able to overcome some of the existing limitations, but continuous research is needed to find new avenues in our quest to target all therapeutically relevant proteins.190 This in turn means that current methods used for phenotypic screening, target deconvolution and the development of chemical probes require constant advancement to remain at the forefront of the field.
Conclusion
Unsatisfactory progress in drug discovery programs has resulted in a resurge of PDD, which has potential to identify better leads for complex diseases lacking robustly validated drug targets or well-defined biology. Despite the huge promise of this approach, phenotypic screening presents unique challenges that need to be addressed for it to reach its full potential. Researchers will have to integrate state-of-the-art methods, technology and data analysis to overcome remaining challenges and limitations associated with PDD. Additionally, progress in target deconvolution will further boost PDD programs by permitting the implementation of TBDD methods and allowing capitalisation of the synergy between these approaches.Chemical probes play an integral part in both phenotypic screening and target deconvolution, and the development of novel probes will be key to advance this research area and to attain its potential. There are different ways in which the development of chemical probes might be improved, including their design, synthesis and properties, supplemented by novel computational approaches to improve the analysis of pharmacological and biological results. Together, changes in this area will address key challenges that are limiting the success of these methods to date, such as the shortage of fundamentally different approaches for target deconvolution, the inaptness of probes for the performed studies as well as a limited understanding of key interactions between different proteins and signalling pathways. Methods such as CETSA and TPP are already beginning to overcome the latter issue, with these techniques able to provide information such as complex formation and binding partners in physiologically relevant environments.
By combining the latest methods in phenotypic screening, target deconvolution and the design of chemical probes, drug discovery programmes will be in a much-improved position to identify first-in-class drug candidates with well-defined mechanism-of-action. Due to the ageing population across the globe, it has never been more pressing to find therapeutic interventions for prevalent neurodegenerative disorders, such as Alzheimer's and Parkinson's Disease. Understanding and controlling the complex biology underlying disease states continues to be an enormous challenge but, considering the new possibilities that are available for researchers compared to only half a century ago, the future looks bright.
Author contributions
Both authors, M. J. and K. S. M., contributed equally to the conceptualization and writing of this review article.Conflicts of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.Acknowledgements
M. J. and K. S. M. both hold Newcastle/Monash University Academic Track (NUMAcT) Fellowships funded by Research England (ref.131911). The authors gratefully acknowledge Toni Pringle for generating the figures within this review.References
- J. C. Caicedo, S. Cooper, F. Heigwer, S. Warchal, P. Qiu, C. Molnar, A. S. Vasilevich, J. D. Barry, H. S. Bansal, O. Kraus, M. Wawer, L. Paavolainen, M. D. Herrmann, M. Rohban, J. Hung, H. Hennig, J. Concannon, I. Smith, P. A. Clemons, S. Singh, P. Rees, P. Horvath, R. G. Linington and A. E. Carpenter, Nat. Methods, 2017, 14, 849–863 CrossRef CAS PubMed.
- S. Rottenberg, J. E. Jaspers, A. Kersbergen, E. Van Der Burg, A. O. H. Nygren, S. A. L. Zander, P. W. B. Derksen, M. De Bruin, J. Zevenhoven, A. Lau, R. Boulter, A. Cranston, M. J. O'Connor, N. M. B. Martin, P. Borst and J. Jonkers, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17079–17084 CrossRef CAS PubMed.
- D. Haasen, U. Schopfer, C. Antczak, C. Guy, F. Fuchs and P. Selzer, Assay Drug Dev. Technol., 2017, 15, 239–246 CrossRef CAS PubMed.
- D. C. Swinney, Clin. Pharmacol. Ther., 2013, 93, 299–301 CrossRef CAS PubMed.
- D. C. Swinney, Phenotypic Drug Discovery, Royal Society of Chemistry, 2020, pp. 1–19 Search PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2015, 14, 807–809 CrossRef CAS PubMed.
- B. Evers, R. Drost, E. Schut, M. De Bruin, E. Der Van Burg, P. W. B. Derksen, H. Holstege, X. Liu, E. Van Drunen, H. B. Beverloo, G. C. M. Smith, N. M. B. Martin, A. Lau, M. J. O'Connor and J. Jonkers, Clin. Cancer Res., 2008, 14, 3916–3925 CrossRef CAS PubMed.
- G. Bollag, J. Tsai, J. Zhang, C. Zhang, P. Ibrahim, K. Nolop and P. Hirth, Nat. Rev. Drug Discovery, 2012, 11, 873–886 CrossRef CAS PubMed.
- G. Bollag, P. Hirth, J. Tsai, J. Zhang, P. N. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhang, G. Habets, E. A. Burton, B. Wong, G. Tsang, B. L. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K. Y. J. Zhang, D. R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. Dandrea, A. Koehler, M. Stumm, P. S. Lin, R. J. Lee, J. Grippo, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. B. Chapman, K. T. Flaherty, X. Xu, K. L. Nathanson and K. Nolop, Nature, 2010, 467, 596–599 CrossRef CAS PubMed.
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS PubMed.
- T. Dorval, B. Chanrion, M. E. Cattin and J. P. Stephan, Curr. Opin. Pharmacol., 2018, 42, 40–45 CrossRef CAS PubMed.
- A. Banik, R. E. Brown, J. Bamburg, D. K. Lahiri, D. Khurana, R. P. Friedland, W. Chen, Y. Ding, A. Mudher, A. L. Padjen, E. Mukaetova-Ladinska, M. Ihara, S. Srivastava, M. V. P. Srivastava, C. L. Masters, R. N. Kalaria and A. Anand, J. Alzheimer's Dis., 2015, 47, 815–843 Search PubMed.
- D. C. Swinney, Nat. Rev. Drug Discovery, 2004, 3, 801–808 CrossRef CAS PubMed.
- B. H. Munos and W. W. Chin, Sci. Transl. Med., 2011, 3(89), 1–3 Search PubMed.
- M. E. Bunnage, Nat. Chem. Biol., 2011, 7, 335–339 CrossRef CAS PubMed.
- F. Stegmeier, M. Warmuth, W. R. Sellers and M. Dorsch, Clin. Pharmacol. Ther., 2010, 87, 543–552 CrossRef CAS PubMed.
- S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, Nat. Rev. Drug Discovery, 2010, 9, 203–214 CrossRef CAS PubMed.
- K. I. Kaitin, Clin. Pharmacol. Ther., 2010, 87, 356–361 CrossRef CAS PubMed.
- J. G. Moffat, J. Rudolph and D. Bailey, Nat. Rev. Drug Discovery, 2014, 13, 588–602 CrossRef CAS PubMed.
- P. Horvath, N. Aulner, M. Bickle, A. M. Davies, E. Del Nery, D. Ebner, M. C. Montoya, P. Östling, V. Pietiäinen, L. S. Price, S. L. Shorte, G. Turcatti, C. Von Schantz and N. O. Carragher, Nat. Rev. Drug Discovery, 2016, 15, 751–769 CrossRef CAS PubMed.
- M. Gittelman, Resour. Policy, 2016, 45, 1570–1585 CrossRef.
- A. L. Hopkins, Nat. Chem. Biol., 2008, 4, 682–690 CrossRef CAS PubMed.
- M. Danhof, Eur. J. Pharm. Sci., 2016, 94, 4–14 CrossRef CAS PubMed.
- M. J. Waring, J. Arrowsmith, A. R. Leach, P. D. Leeson, S. Mandrell, R. M. Owen, G. Pairaudeau, W. D. Pennie, S. D. Pickett, J. Wang, O. Wallace and A. Weir, Nat. Rev. Drug Discovery, 2015, 14, 475–486 CrossRef CAS PubMed.
- J. Zhou, X. Jiang, S. He, H. Jiang, F. Feng, W. Liu, W. Qu and H. Sun, J. Med. Chem., 2019, 62, 8881–8914 CrossRef CAS PubMed.
- G. Maggiora and V. Gokhale, in ACS Symposium Series, American Chemical Society, 2016, vol. 1222, pp. 91–142 Search PubMed.
- W. E. Childers, K. M. Elokely and M. Abou-Gharbia, ACS Med. Chem. Lett., 2020, 11, 1820–1828 CrossRef CAS PubMed.
- J. A. Lee, M. T. Uhlik, C. M. Moxham, D. Tomandl and D. J. Sall, J. Med. Chem., 2012, 55, 4527–4538 CrossRef CAS PubMed.
- A. Friese, A. Ursu, A. Hochheimer, H. R. Schöler, H. Waldmann and J. M. Bruder, Cell Chem. Biol., 2019, 26, 1050–1066 CrossRef CAS PubMed.
- J. A. Lee and E. L. Berg, J. Biomol. Screening, 2013, 18, 1143–1155 CrossRef CAS PubMed.
- D. C. Swinney, J. Biomol. Screening, 2013, 18, 1186–1192 CrossRef PubMed.
- R. Ramabhadran, Modern Approaches in Drug Designing, 2018, 2, 1–3 CrossRef.
- J. Eder, R. Sedrani and C. Wiesmann, Nat. Rev. Drug Discovery, 2014, 13, 577–587 CrossRef CAS PubMed.
- S. Wang, Y. Tian, M. Wang, M. Wang, G. Sun and X. Sun, Front. Pharmacol., 2018, 9, 353 CrossRef PubMed.
- H. Al-Ali, MedChemComm, 2016, 7, 788–798 RSC.
- J. G. Moffat, F. Vincent, J. A. Lee, J. Eder and M. Prunotto, Nat. Rev. Drug Discovery, 2017, 16, 531–543 CrossRef CAS PubMed.
- F. Vincent, P. Loria, M. Pregel, R. Stanton, L. Kitching, K. Nocka, R. Doyonnas, C. Steppan, A. Gilbert, T. Schroeter and M. C. Peakman, Sci. Transl. Med., 2015, 7, 293ps15 CrossRef PubMed.
- P. Horvath, N. Aulner, M. Bickle, A. M. Davies, E. Del Nery, D. Ebner, M. C. Montoya, P. Östling, V. Pietiäinen, L. S. Price, S. L. Shorte, G. Turcatti, C. Von Schantz and N. O. Carragher, Nat. Rev. Drug Discovery, 2016, 15, 751–769 CrossRef CAS PubMed.
- H. A. Kenny, M. Lal-Nag, M. Shen, B. Kara, D. A. Nahotko, K. Wroblewski, S. Fazal, S. Chen, C. Y. Chiang, Y. J. Chen, K. R. Brimacombe, J. Marugan, M. Ferrer and E. Lengyel, Mol. Cancer Ther., 2020, 19, 52–62 CrossRef CAS PubMed.
- H. A. Kenny, M. Lal-Nag, E. A. White, M. Shen, C. Y. Chiang, A. K. Mitra, Y. Zhang, M. Curtis, E. M. Schryver, S. Bettis, A. Jadhav, M. B. Boxer, Z. Li, M. Ferrer and E. Lengyel, Nat. Commun., 2015, 6, 1–11 Search PubMed.
- M. J. Carson, J. Crane and A. X. Xie, Radio Sci., 2008, 5, 19–25 Search PubMed.
- E. L. Berg, Y. C. Hsu and J. A. Lee, Adv. Drug Delivery Rev., 2014, 69–70, 190–204 CrossRef PubMed.
- N. Thorne, N. Malik, S. Shah, J. Zhao, B. Class, F. Aguisanda, N. Southall, M. Xia, J. C. McKew, M. Rao and W. Zheng, Stem Cells Transl. Med., 2016, 5, 613–627 CrossRef CAS PubMed.
- J. M. Pipas, Virology, 2009, 384, 294–303 CrossRef CAS PubMed.
- M. J. Briske-Anderson, J. W. Finley and S. M. Newman, Exp. Biol. Med., 1997, 214, 248–257 CrossRef CAS PubMed.
- P. Hughes, D. Marshall, Y. Reid, H. Parkes and C. Gelber, BioTechniques, 2007, 43, 575–586 CrossRef CAS PubMed.
- E. Ferrari, A. Cardinale, B. Picconi and F. Gardoni, J. Neurosci. Methods, 2020, 340, 108741 CrossRef CAS PubMed.
- I. Coma, L. Clark, E. Diez, G. Harper, J. Herranz, G. Hofmann, M. Lennon, N. Richmond, M. Valmaseda and R. Macarron, J. Biomol. Screening, 2009, 14, 66–76 CrossRef CAS PubMed.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- M. C. Marchetto, K. J. Brennand, L. F. Boyer and F. H. Gage, Hum. Mol. Genet., 2011, 20, 109–115 CrossRef PubMed.
- M. Morrison, C. Klein, N. Clemann, D. A. Collier, J. Hardy, B. Heiβerer, M. Z. Cader, M. Graf and J. Kaye, Stem Cell Rev. Rep., 2015, 11, 681–687 CrossRef CAS PubMed.
- Y. Avior, I. Sagi and N. Benvenisty, Nat. Rev. Mol. Cell Biol., 2016, 17, 170–182 CrossRef CAS PubMed.
- B. P. Sridharan, C. Hubbs, N. Llamosas, M. Kilinc, F. U. Singhera, E. Willems, D. R. Piper, L. Scampavia, G. Rumbaugh and T. P. Spicer, Sci. Rep., 2019, 9, 1–14 CAS.
- B. van Wilgenburg, C. Browne, J. Vowles and S. A. Cowley, PLoS One, 2013, 8, e71098 CrossRef CAS PubMed.
- W. Haenseler, S. N. Sansom, J. Buchrieser, S. E. Newey, C. S. Moore, F. J. Nicholls, S. Chintawar, C. Schnell, J. P. Antel, N. D. Allen, M. Z. Cader, R. Wade-Martins, W. S. James and S. A. Cowley, Stem Cell Rep., 2017, 8, 1727–1742 CrossRef CAS PubMed.
- W. Haenseler, F. Zambon, H. Lee, J. Vowles, F. Rinaldi, G. Duggal, H. Houlden, K. Gwinn, S. Wray, K. C. Luk, R. Wade-Martins, W. S. James and S. A. Cowley, Sci. Rep., 2017, 7, 9003 CrossRef PubMed.
- T. S. Kapellos, L. Taylor, H. Lee, S. A. Cowley, W. S. James, A. J. Iqbal and D. R. Greaves, Biochem. Pharmacol., 2016, 116, 107–119 CrossRef CAS PubMed.
- M. Eisenstein, Nat. Methods, 2018, 15, 19–22 CrossRef CAS.
- J. Kim, B. K. Koo and J. A. Knoblich, Nat. Rev. Mol. Cell Biol., 2020, 21, 571–584 CrossRef CAS PubMed.
- M. Fujii and T. Sato, Nat. Mater., 2020, 1–14 Search PubMed.
- S. Bartfeld and H. Clevers, J. Mol. Med., 2017, 95, 729–738 CrossRef CAS PubMed.
- Z. Li, L. Yu, D. Chen, Z. Meng, W. Chen and W. Huang, Cell Press: STAR protocols, 2021, 2, 100239 Search PubMed.
- A. A. Duarte, E. Gogola, N. Sachs, M. Barazas, S. Annunziato, J. R. De Ruiter, A. Velds, S. Blatter, J. M. Houthuijzen, M. Van De Ven, H. Clevers, P. Borst, J. Jonkers and S. Rottenberg, Nat. Methods, 2018, 15, 134–140 CrossRef CAS PubMed.
- Y. Du, X. Li, Q. Niu, X. Mo, M. Qui, T. Ma, C. J. Kuo and H. Fu, J. Mol. Cell Biol., 2020, 12, 630–643 CrossRef PubMed.
- K. Boehnke, P. W. Iversen, D. Schumacher, M. J. Lallena, R. Haro, J. Amat, J. Haybaeck, S. Liebs, M. Lange, R. Schäfer, C. R. A. Regenbrecht, C. Reinhard and J. A. Velasco, J. Biomol. Screening, 2016, 21, 931–941 CrossRef CAS PubMed.
- H. Renner, M. Grabos, K. J. Becker, T. E. Kagermeier, J. Wu, M. Otto, S. Peischard, D. Zeuschner, Y. Tsytsyura, P. Disse, J. Klingauf, S. A. Leidel, G. Seebohm, H. R. Schöler and J. M. Bruder, eLife, 2020, 9, 1–39 CrossRef PubMed.
- B. Schuster, M. Junkin, S. S. Kashaf, I. Romero-Calvo, K. Kirby, J. Matthews, C. R. Weber, A. Rzhetsky, K. P. White and S. Tay, Nat. Commun., 2020, 11, 1–12 Search PubMed.
- S. N. Boers, J. J. Delden, H. Clevers and A. L. Bredenoord, EMBO Rep., 2016, 17, 938–941 CrossRef CAS PubMed.
- H. Xu, Y. Jiao, S. Qin, W. Zhao, Q. Chu and K. Wu, Exp. Hematol. Oncol., 2018, 7, 30 CrossRef CAS PubMed.
- S. E. Park, A. Georgescu and D. Huh, Science, 2019, 364, 960–965 CrossRef CAS PubMed.
- J. Günter, R. H. Wenger and C. C. Scholz, J. Photochem. Photobiol., B, 2020, 210, 111980–111986 CrossRef PubMed.
- I. V. L. Wilkinson, J. K. Reynolds, S. R. G. Galan, A. Vuorinen, A. J. Sills, E. Pires, G. M. Wynne, F. X. Wilson and A. J. Russell, Bioorg. Chem., 2020, 94, 103395–103403 CrossRef CAS PubMed.
- B. Somanadhan, C. Leong, S. R. Whitton, S. Ng, A. D. Buss and M. S. Butler, J. Nat. Prod., 2011, 74, 1500–1502 CrossRef CAS PubMed.
- L. H. Heitman, J. P. D. Van Veldhoven, A. M. Zweemer, K. Ye, J. Brussee and A. P. Ijzerrnan, J. Med. Chem., 2008, 51, 4724–4729 CrossRef CAS PubMed.
- Z. Y. Yang, J. H. He, A. P. Lu, T. J. Hou and D. S. Cao, Drug Discovery Today, 2020, 25, 657–667 CrossRef CAS PubMed.
- D. Ghosh, U. Koch, K. Hadian, M. Sattler and I. V. Tetko, J. Chem. Inf. Model., 2018, 58, 933–942 CrossRef CAS PubMed.
- N. Boute, P. Lowe, S. Berger, M. Malissard, A. Robert and M. Tesar, Front. Pharmacol., 2016, 7, 27 Search PubMed.
- C. G. England, E. B. Ehlerding and W. Cai, Bioconjugate Chem., 2016, 27, 1175–1187 CrossRef CAS PubMed.
- J. Henry and D. Wlodkowic, Mar. Drugs, 2019, 17(340), 1–20 Search PubMed.
- I. K. Moutsatsos and C. N. Parker, Expert Opin. Drug Discovery, 2016, 11, 415–423 CrossRef CAS PubMed.
- M. A. Bray, S. Singh, H. Han, C. T. Davis, B. Borgeson, C. Hartland, M. Kost-Alimova, S. M. Gustafsdottir, C. C. Gibson and A. E. Carpenter, Nat. Protoc., 2016, 11, 1757–1774 CrossRef CAS PubMed.
- L. Laraia, G. Garivet, D. J. Foley, N. Kaiser, S. Müller, S. Zinken, T. Pinkert, J. Wilke, D. Corkery, A. Pahl, S. Sievers, P. Janning, C. Arenz, Y. Wu, R. Rodriguez and H. Waldmann, Angew. Chem., Int. Ed., 2020, 59, 5721–5729 CrossRef CAS PubMed.
- S. Paricharak, A. P. Ijzerman, J. L. Jenkins, A. Bender and F. Nigsch, J. Chem. Inf. Model., 2016, 56, 1622–1630 CrossRef CAS PubMed.
- B. Seashore-Ludlow, M. G. Rees, J. H. Cheah, M. Cokol, E. V. Price, M. E. Coletti, V. Jones, N. E. Bodycombe, C. K. Soule, J. Gould, B. Alexander, A. Li, P. Montgomery, M. J. Wawer, N. Kuru, J. D. Kotz, C. S.-Y. Hon, B. Munoz, T. Liefeld, V. Dančík, J. A. Bittker, M. Palmer, J. E. Bradner, A. F. Shamji, P. A. Clemons and S. L. Schreiber, Cancer Discovery, 2015, 5, 1210–1223 CrossRef CAS PubMed.
- P. A. Clemons, Curr. Opin. Chem. Biol., 2004, 8, 334–338 CrossRef CAS PubMed.
- A. M. Stern, M. E. Schurdak, I. Bahar, J. M. Berg and D. L. Taylor, J. Biomol. Screening, 2016, 21, 521–534 CrossRef CAS PubMed.
- Y. Wang and J. L. Jenkins, in Methods in Molecular Biology, Humana Press Inc., 2018, vol. 1787, pp. 195–206 Search PubMed.
- S. Raschka and B. Kaufman, Methods, 2020, 180, 89–110 CrossRef CAS PubMed.
- T. Rodrigues and G. J. L. Bernardes, Curr. Opin. Chem. Biol., 2020, 56, 16–22 CrossRef CAS PubMed.
- Y. Fang, Expert Opin. Drug Discovery, 2016, 11, 269–280 CrossRef CAS PubMed.
- Y. Wang, A. Cornett, F. J. King, Y. Mao, F. Nigsch, C. G. Paris, G. McAllister and J. L. Jenkins, Cell Chem. Biol., 2016, 23, 862–874 CrossRef CAS PubMed.
- J. J. Xu, Front. Pharmacol., 2015, 6, 191 Search PubMed.
- H. Yang, C. Lou, W. Li, G. Liu and Y. Tang, Chem. Res. Toxicol., 2020, 33, 1312–1322 Search PubMed.
- X. Lin, X. Li and X. Lin, Molecules, 2020, 25, 1375 Search PubMed.
- E. L. Berg, Drug Discovery Today: Technol., 2017, 23, 53–60 Search PubMed.
- Q. T. L. Pasquer, I. A. Tsakoumagkos and S. Hoogendoorn, Molecules, 2020, 25(5702), 1–22 Search PubMed.
- M. Schenone, V. Dančík, B. K. Wagner and P. A. Clemons, Nat. Chem. Biol., 2013, 9, 232–240 CrossRef CAS PubMed.
- Y. Su, J. Ge, B. Zhu, Y. G. Zheng, Q. Zhu and S. Q. Yao, Curr. Opin. Chem. Biol., 2013, 17, 768–775 CrossRef CAS PubMed.
- F. Maqbool, A. Abid and I. Ahmed, Int. J. Pharmacol., 2017, 13, 773–784 CAS.
- J. R. Hill and A. A. B. Robertson, J. Med. Chem., 2018, 61, 6945–6963 CrossRef CAS PubMed.
- T. B. Durham and M. J. Blanco, Bioorg. Med. Chem. Lett., 2015, 25, 998–1008 CrossRef PubMed.
- A. Ursu and H. Waldmann, Bioorg. Med. Chem. Lett., 2015, 25, 3079–3086 CrossRef CAS PubMed.
- B. Lomenick, R. W. Olsen and J. Huang, ACS Chem. Biol., 2011, 6, 34–46 CrossRef CAS.
- G. C. Terstappen, C. Schlüpen, R. Raggiaschi and G. Gaviraghi, Nat. Rev. Drug Discovery, 2007, 6, 891–903 CrossRef CAS PubMed.
- M. J. Main and A. X. Zhang, SLAS Discovery, 2020, 25, 115–117 Search PubMed.
- M. Schirle and J. L. Jenkins, Drug Discovery Today, 2016, 21, 82–89 CrossRef CAS PubMed.
- K. Kubota, M. Funabashi and Y. Ogura, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 22–27 CrossRef CAS PubMed.
- J. Lee and M. Bogyo, Curr. Opin. Chem. Biol., 2013, 17, 118–126 CrossRef CAS PubMed.
- N. Panyain, C. R. Kennedy, R. T. Howard and E. W. Tate, in Target Discovery and Validation: Methods and Strategies for Drug Discovery, 2019, pp. 51–95 Search PubMed.
- I. V. L. Wilkinson, G. C. Terstappen and A. J. Russell, Drug Discovery Today, 2020, 25, 1998–2005 CrossRef CAS PubMed.
- G. C. Terstappen, Future Med. Chem., 2009, 1, 7–9 CrossRef CAS PubMed.
- J. P. Hughes, S. S. Rees, S. B. Kalindjian and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249 CrossRef CAS PubMed.
- L. Costantino and D. Barlocco, in Drug Selectivity: An Evolving Concept in Medicinal Chemistry, Wiley, 2017, pp. 161–205 Search PubMed.
- E. Proschak, H. Stark and D. Merk, J. Med. Chem., 2019, 62, 420–444 CrossRef CAS PubMed.
- A. G. Reaume, Drug Discovery Today: Ther. Strategies, 2011, 8, 85–88 CrossRef PubMed.
- W. Zheng, N. Thorne and J. C. McKew, Drug Discovery Today, 2013, 18, 1067–1073 CrossRef CAS PubMed.
- J. Arrowsmith and P. Miller, Nat. Rev. Drug Discovery, 2013, 12, 569–570 CrossRef CAS PubMed.
- J. Arrowsmith, Trial watch: Phase II failures: 2008–2010, Nature Publishing Group, 2011, vol. 23 Search PubMed.
- J. Blagg and P. Workman, Cancer Cell, 2017, 32, 9–25 CrossRef CAS PubMed.
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. M. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Müller, R. C. O'Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, R. Ross, B. Roth, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundström, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed.
- R. M. Garbaccio and E. R. Parmee, Cell Chem. Biol., 2016, 23, 10–17 CrossRef CAS.
- E. L. Berg, Drug Discovery Today: Technol., 2017, 23, 53–60 CrossRef PubMed.
- M. P. Castaldi, J. A. Hendricks and A. X. Zhang, Curr. Opin. Chem. Biol., 2020, 56, 91–97 CrossRef CAS PubMed.
- J. Quancard, B. Cox, D. Finsinger, S. M. Guéret, I. V. Hartung, H. F. Koolman, J. Messinger, G. Sbardella and S. Laufer, ChemMedChem, 2020, 15, 2388–2390 CrossRef CAS PubMed.
- M. E. Bunnage, E. L. P. Chekler and L. H. Jones, Nat. Chem. Biol., 2013, 9, 195–199 CrossRef CAS PubMed.
- H. Stark, Expert Opin. Drug Discovery, 2020, 15, 1365–1367 CrossRef PubMed.
- E. Nicodeme, K. L. Jeffrey, U. Schaefer, S. Beinke, S. Dewell, C. W. Chung, R. Chandwani, I. Marazzi, P. Wilson, H. Coste, J. White, J. Kirilovsky, C. M. Rice, J. M. Lora, R. K. Prinjha, K. Lee and A. Tarakhovsky, Nature, 2010, 468, 1119–1123 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- C. W. Chung, H. Coste, J. H. White, O. Mirguet, J. Wilde, R. L. Gosmini, C. Delves, S. M. Magny, R. Woodward, S. A. Hughes, E. V. Boursier, H. Flynn, A. M. Bouillot, P. Bamborough, J. M. G. Brusq, F. O. J. Gellibert, E. J. Jones, A. M. Riou, P. Homes, S. L. Martin, I. J. Uings, J. Toum, C. A. Clément, A. B. Boullay, R. L. Grimley, F. M. Blandel, R. K. Prinjha, K. Lee, J. Kirilovsky and E. Nicodeme, J. Med. Chem., 2011, 54, 3827–3838 CrossRef CAS.
- L. Zhang, Z. Yan, Y. Wang, C. Song and G. Miao, Molecules, 2019, 24, 2394 CrossRef PubMed.
- J. C. Green, Y. Jiang, L. He, Y. Xu, D. Sun, T. Keoprasert, C. Nelson, U. Oh, E. J. Lesnefsky, G. E. Kellogg, Q. Chen and S. Zhang, J. Med. Chem., 2020, 63, 11819–11830 CrossRef CAS PubMed.
- D. Armstrong and P. G. Strange, J. Biol. Chem., 2001, 276, 22621–22629 CrossRef CAS PubMed.
- A. Packeu, J. P. De Backer, I. Van Liefde, P. M. L. Vanderheyden and G. Vauquelin, Biochem. Pharmacol., 2008, 75, 2192–2203 CrossRef CAS.
- K. A. Jessen, N. M. English, J. Y. Wang, S. Maliartchouk, S. P. Archer, L. Qiu, R. Brand, J. Kuemmerle, H. Z. Zhang, K. Gehlsen, J. Drewe, B. Tseng, S. X. Cai and S. Kasibhatla, Mol. Cancer Ther., 2005, 4, 761–771 CrossRef CAS PubMed.
- A. Jo, J. Jung, E. Kim and S. B. Park, Chem. Commun., 2016, 52, 7433–7445 RSC.
- L. A. Honigberg, A. M. Smith, M. Sirisawad, E. Verner, D. Loury, B. Chang, S. Li, Z. Pan, D. H. Thamm, R. A. Miller and J. J. Buggy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13075–13080 CrossRef CAS PubMed.
- B. R. Lanning, L. R. Whitby, M. M. Dix, J. Douhan, A. M. Gilbert, E. C. Hett, T. O. Johnson, C. Joslyn, J. C. Kath, S. Niessen, L. R. Roberts, M. E. Schnute, C. Wang, J. J. Hulce, B. Wei, L. O. Whiteley, M. M. Hayward and B. F. Cravatt, Nat. Chem. Biol., 2014, 10, 760–767 CrossRef CAS.
- G. Vauquelin, I. Van Liefde and D. C. Swinney, Drug Discovery Today: Technol., 2015, 17, 28–34 CrossRef PubMed.
- J. V. Jun, D. M. Chenoweth and E. J. Petersson, Org. Biomol. Chem., 2020, 18, 5747–5763 RSC.
- M. Leopoldo, E. Lacivita, F. Berardi and R. Perrone, Drug Discovery Today, 2009, 14, 706–712 CrossRef CAS PubMed.
- A. J. Vernall, S. J. Hill and B. Kellam, Br. J. Pharmacol., 2014, 171, 1073–1084 CrossRef CAS PubMed.
- L. A. Stoddart, L. E. Kilpatrick, S. J. Briddon and S. J. Hill, Neuropharmacology, 2015, 98, 48–57 CrossRef CAS PubMed.
- E. A. Specht, E. Braselmann and A. E. Palmer, Annu. Rev. Physiol., 2017, 79, 93–117 CrossRef CAS PubMed.
- J. W. Conner, D. P. Poole, M. Jörg and N. A. Veldhuis, Future Med. Chem., 2021, 13, 63–90 CrossRef CAS PubMed.
- A. Jo, J. Park and S. B. Park, Chem. Commun., 2013, 49, 5138–5140 RSC.
- P. K. Sobhan, K. A. Mathew, J. Joseph, P. R. Pillai and T. R. Santhoshkumar, Apoptosis, 2014, 19, 269–284 CrossRef PubMed.
- F. Georgi, F. Kuttler, L. Murer, V. Andriasyan, R. Witte, A. Yakimovich, G. Turcatti and U. F. Greber, Sci. Data, 2020, 7, 1–12 CrossRef PubMed.
- S. Lin, K. Schorpp, I. Rothenaigner and K. Hadian, Drug Discovery Today, 2020, 25, 1348–1361 CrossRef CAS PubMed.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681–708 RSC.
- R. A. McClure and J. D. Williams, ACS Med. Chem. Lett., 2018, 9, 785–791 CrossRef CAS PubMed.
- H. Deng, Q. Lei, Y. Wu, Y. He and W. Li, Eur. J. Med. Chem., 2020, 191, 112151 CrossRef CAS PubMed.
- X. Chen, Y. K. Wong, J. Wang, J. Zhang, Y. M. Lee, H. M. Shen, Q. Lin and Z. C. Hua, Proteomics, 2017, 17, 1600212 CrossRef PubMed.
- S. Bach, M. Knockaert, J. Reinhardt, O. Lozach, S. Schmitt, B. Baratte, M. Koken, S. P. Coburn, L. Tang, T. Jiang, D. C. Liang, H. Galons, J. F. Dierick, L. A. Pinna, F. Meggio, F. Totzke, C. Schächtele, A. S. Lerman, A. Carnero, Y. Wan, N. Gray and L. Meijer, J. Biol. Chem., 2005, 280, 31208–31219 CrossRef CAS PubMed.
- M. Jörg and P. J. Scammells, ChemMedChem, 2016, 11, 1488–1498 CrossRef PubMed.
- J. J. Fischer, C. Dalhoff, A. K. Schrey, O. Y. Graebner, S. Michaelis, K. Andrich, M. Glinski, F. Kroll, M. Sefkow, M. Dreger and H. Koester, J. Proteomics, 2011, 75, 160–168 CrossRef CAS PubMed.
- C. Blex, S. Michaelis, A. K. Schrey, J. Furkert, J. Eichhorst, K. Bartho, F. Gyapon Quast, A. Marais, M. Hakelberg, U. Gruber, S. Niquet, O. Popp, F. Kroll, M. Sefkow, R. Schülein, M. Dreger and H. Köster, ChemBioChem, 2017, 18, 1639–1649 CrossRef CAS PubMed.
- J. J. Fischer, O. Graebner, M. Dreger, M. Glinski, S. Baumgart and H. Koester, J. Biomed. Biotechnol., 2011, 2011, 1–5 CrossRef PubMed.
- I. V. L. Wilkinson, K. J. Perkins, H. Dugdale, L. Moir, A. Vuorinen, M. Chatzopoulou, S. E. Squire, S. Monecke, A. Lomow, M. Geese, P. D. Charles, P. Burch, J. M. Tinsley, G. M. Wynne, S. G. Davies, F. X. Wilson, F. Rastinejad, S. Mohammed, K. E. Davies and A. J. Russell, Angew. Chem., Int. Ed., 2020, 59, 2420–2428 CrossRef CAS PubMed.
- A. M. Joffrin and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2020, 142, 13672–13676 CrossRef CAS PubMed.
- J. S. Cisar and B. F. Cravatt, J. Am. Chem. Soc., 2012, 134, 10385–10388 CrossRef CAS PubMed.
- A. T. Wright, J. D. Song and B. F. Cravatt, J. Am. Chem. Soc., 2009, 131, 10692–10700 CrossRef CAS PubMed.
- C. M. Salisbury and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1171–1176 CrossRef CAS.
- B. F. Cravatt and E. J. Sorensen, Curr. Opin. Chem. Biol., 2000, 4, 663–668 CrossRef CAS.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- G. M. Simon and B. F. Cravatt, J. Biol. Chem., 2010, 285, 11051–11055 CrossRef CAS PubMed.
- C. M. Salisbury and B. F. Cravatt, J. Am. Chem. Soc., 2008, 130, 2184–2194 CrossRef CAS PubMed.
- E. Smith and I. Collins, Future Med. Chem., 2015, 7, 159–183 CrossRef CAS PubMed.
- L. H. Jones, Future Med. Chem., 2015, 7, 2131–2141 CrossRef CAS PubMed.
- B. Ghosh and L. H. Jones, MedChemComm, 2014, 5, 247–254 RSC.
- F. Becker, K. Murthi, C. Smith, J. Come, N. Costa-Roldán, C. Kaufmann, U. Hanke, C. Degenhart, S. Baumann, W. Wallner, A. Huber, S. Dedier, S. Dill, D. Kinsman, M. Hediger, N. Bockovich, S. Meier-Ewert, A. F. Kluge and N. Kley, Chem. Biol., 2004, 11, 211–223 CrossRef CAS PubMed.
- S. Cottier, T. Mönig, Z. Wang, J. Svoboda, W. Boland, M. Kaiser and E. Kombrink, Front. Plant Sci., 2011, 2, 101 CAS.
- J. Oeljeklaus, F. Kaschani and M. Kaiser, Angew. Chem., Int. Ed., 2013, 52, 1368–1370 CrossRef CAS PubMed.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. Von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS PubMed.
- M. Luzarowski and A. Skirycz, J. Exp. Bot., 2019, 70, 4605–4617 CrossRef CAS PubMed.
- D. M. Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef CAS.
- I. Becher, T. Werner, C. Doce, E. A. Zaal, I. Tögel, C. A. Khan, A. Rueger, M. Muelbaier, E. Salzer, C. R. Berkers, P. F. Fitzpatrick, M. Bantscheff and M. M. Savitski, Nat. Chem. Biol., 2016, 12, 908–910 CrossRef CAS PubMed.
- T. Friman, Bioorg. Med. Chem., 2020, 28, 115174 CrossRef CAS PubMed.
- M. M. Savitski, F. B. M. Reinhard, H. Franken, T. Werner, M. F. Savitski, D. Eberhard, D. M. Molina, R. Jafari, R. B. Dovega, S. Klaeger, B. Kuster, P. Nordlund, M. Bantscheff and G. Drewes, Science, 2014, 346, 1255784 CrossRef PubMed.
- L. Dai, T. Zhao, X. Bisteau, W. Sun, N. Prabhu, Y. T. Lim, R. M. Sobota, P. Kaldis and P. Nordlund, Cell, 2018, 173, 1481–1494.e13 CrossRef CAS PubMed.
- W. Sun, L. Dai, H. Yu, B. Puspita, T. Zhao, F. Li, J. L. Tan, Y. T. Lim, M. W. Chen, R. M. Sobota, D. G. Tenen, N. Prabhu and P. Nordlund, Redox Biol., 2019, 24, 101168 CrossRef CAS.
- H. Franken, T. Mathieson, D. Childs, G. M. A. Sweetman, T. Werner, I. Tögel, C. Doce, S. Gade, M. Bantscheff, G. Drewes, F. B. M. Reinhard, W. Huber and M. M. Savitski, Nat. Protoc., 2015, 10, 1567–1593 CrossRef CAS PubMed.
- E. J. Walker, J. Q. Bettinger, K. A. Welle, J. R. Hryhorenko and S. Ghaemmaghami, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6081–6090 CrossRef CAS PubMed.
- A. Mateus, T. A. Määttä and M. M. Savitski, Proteome Sci., 2017, 15, 13 CrossRef PubMed.
- K. Y. Lu, B. Quan, K. Sylvester, T. Srivastava, M. C. Fitzgerald and E. R. Derbyshire, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5810–5817 CrossRef CAS PubMed.
- M. P. Gleeson, A. Hersey, D. Montanari and J. Overington, Nat. Rev. Drug Discovery, 2011, 10, 197–208 CrossRef CAS PubMed.
- J. Besnard, G. F. Ruda, V. Setola, K. Abecassis, R. M. Rodriguiz, X. P. Huang, S. Norval, M. F. Sassano, A. I. Shin, L. A. Webster, F. R. C. Simeons, L. Stojanovski, A. Prat, N. G. Seidah, D. B. Constam, G. R. Bickerton, K. D. Read, W. C. Wetsel, I. H. Gilbert, B. L. Roth and A. L. Hopkins, Nature, 2012, 492, 215–220 CrossRef CAS PubMed.
- A. A. Antolin, P. Workman and B. Al-Lazikani, Future Med. Chem., 2019, 1–18 Search PubMed.
- R. P. Nowak and L. H. Jones, SLAS Discovery, 2020, 1–10 Search PubMed.
- M. Kostic and L. H. Jones, Trends Pharmacol. Sci., 2020, 41, 305–317 CrossRef CAS.
- H. Pei, Y. Peng, Q. Zhao and Y. Chen, RSC Adv., 2019, 9, 16967–16976 RSC.
| This journal is © The Royal Society of Chemistry 2021 |