
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme entrapment, biocatalyst immobilization without covalent attachment
Hasan T.
Imam
 ,
Patricia C.
Marr
* and
Andrew C.
Marr
*
,
Patricia C.
Marr
* and
Andrew C.
Marr
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, UK. E-mail: p.marr@qub.ac.uk; a.marr@qub.ac.uk
First published on 24th June 2021
Abstract
In entrapment an active species, which is often a catalyst, is trapped within a material by a solid or gel forming event; thus, it becomes dispersed within the solid or semi-solid matrix. Entrapment methods can be used to immobilize isolated enzymes and render them more stable, and easier to separate and recycle. Entrapment immobilization methods are well established for whole cell biocatalysis. Despite this the applications of entrapment towards isolated enzyme immobilization have lagged the use of covalent attachment and crosslinking methods. In this review entrapment methods are contrasted with other methods of enzyme immobilization and literature methods of enzyme entrapment are reviewed. A key advantage of this approach is that no formal interaction with the protein is required, but this must be balanced against the threat of enzyme leaching, or introduction of mass transfer limitations. The main methods of entrapment are characterized, and some recent innovations are highlighted.
 Hasan Tanvir Imam | Dr Hasan Tanvir Imam is a post-doctoral research fellow in Queen's University Belfast, UK working with Dr A. C. Marr and Dr P. C. Marr. His interests include biocatalysis, bioinorganic chemistry and sustainable chemistry. He completed his PhD in Warwick University, UK on metal binding studies of cysteine rich metalloproteins. He previously worked as a PDRA in Texas A&M University, USA and with Prof. P. C. J. Kamer at the University of St-Andrews, UK. |
 Patricia C. Marr | Dr Patricia C. Marr is a senior faculty member in the School of Chemistry & Chemical Engineering, Queen's University Belfast, UK specialising in materials, green & sustainable chemistry and ionic liquids. Following a PhD in materials at the University of St-Andrews, UK, her interest in materials, colloids, neoteric solvents and green and sustainable chemistry intensified as a result of postdoctoral work at The University of Nottingham and the University of Bristol. |
 Andrew C. Marr | Dr Andrew C. Marr is a senior faculty member in the School of Chemistry & Chemical Engineering, Queen's University Belfast, UK specialising in green catalysis with biocatalysts, organometallic complexes and ionic liquids. After a PhD in the University of St-Andrews, UK, his fascination with biocatalysis was ignited during postdoctoral research on hydrogenases with Prof. Martin Schröder at The University of Nottingham. |
1. Introduction
When assessed against the guidelines of green chemistry and atom economy,1–4 isolated enzyme catalyzed reactions hold extreme promise. Enzymes catalyze a myriad of chemical transformations rapidly and cleanly in order to make the living cell functional. Location within the cell provides the enzyme with an optimized local environment in terms of pH, temperature, ionic strength and redox properties, and this supports high activity and exceptional selectivity.5–9 In order to harness the power of enzyme efficiency for green chemical synthesis, the enzyme must be optimized for use in a chemical reactor.10–14 Steps can be taken to mimic the cellular environment, for example, by using aqueous buffer, but overall the conditions will be alien to the natural enzyme; additionally many chemical reactions operate better in man-made media such as organic solvents or ionic liquids.12,15–19 Despite these challenges, many enzymes have been successfully applied as catalysts in industrial processes and isolated enzymes are becoming a major force in chemicals manufacture. This is in no small part due to rapid technical advancements in molecular biology, biochemistry and biotechnology that enable over expression of targeted enzymes in genetically modified organisms, e.g. E.coli, and isolation of enzymes in a partially or fully purified form. Industrial biocatalysis can be performed in two ways, whole cell and isolated enzyme biocatalysis.18,20,21 Whole cell biocatalysis retains the local environment around the enzyme, as the enzyme is accessed within a cell, such as a bacterium. Isolated enzyme methods require extra purification of the protein, and the loss of the cell environment around the protein, but this can be offset as the result of catalysis is often near total selectivity.18,22 The application of biocatalysts as isolated enzymes requires the operator to counter the inherent instability of biocatalysts under operational conditions. In isolated form enzymes have a wide range of stabilities. Lipase, a class of hydrolase tuned to operate on hydrophobic substrates, is an exceptional example, as this enzyme has proven stability in harsh reaction environments, leading to application in pharmaceuticals, fine chemicals, consumer chemicals, polymers and foods.23–28 However, for other classes of enzyme stability must be carefully optimized.16,29–34 A vital component of this is the application of immobilization methods (section 2).In applied industrial biocatalysis the choice of solvent(s) is a key consideration. The majority of enzymes will lend themselves naturally to aqueous enzymatic processes, however substrates may be insoluble or sparingly soluble in these media. Operating biocatalysis in organic solvents or in aqueous/organic biphasic media circumvents solubility challenges, but solvent toxicity can pose an environmental threat and/or damage the protein structure. Ionic liquids, famed for their low vapor pressure and tunable chemical and physical properties, have emerged as alternative solvents for biocatalysis (section 5).17,31,35–38
Established enzyme immobilization materials and methods have been reviewed.39,40,41–48,49 This review focuses on emerging enzyme immobilization methods that do not require covalent attachment of the protein to the support (section 3 & 4). These methods, termed ‘entrapment’ methods, are complementary with protein optimization, as they allow the protein to be supported as optimized with no further modification. In addition, entrapment methods offer the facile tuning and optimization of the support, so that the perfect environment can be created to stabilize and enhance the activity of the enzyme.
This review will mainly focus on enzyme immobilization via entrapment methods, highlighting pioneering work and recent developments. In the section 2, the suitability, and robustness of other enzyme immobilization methods, namely adsorption, covalent attachment and crosslinking, will be compared to entrapment.
2. Summary of enzyme immobilization methods
Biocatalysis technologies are rapidly developing and finding industrial applications. However, isolated enzymes can be short-lived and their inherent instability towards long term storage and reuse poses a threat to the sustainability of biocatalytic chemical processes. Technological advances, in particular genome mining (metagenomics), next generation gene sequencing and bioinformatics, assist the isolation of efficient natural enzyme catalysts.50,51 Introduction of directed evolution has greatly improved biocatalyst stability and activity.52–54 Computational aided, de novo design has further enhanced the understanding of biocatalysts leading to wider substrate scopes.55–61 However, the economic viability of such optimized biocatalytic systems commonly suffers from a lack of reusability, and reuse and recycling are vital components of a green and circular economy. Immobilization of the isolated enzyme, defined as the association of the biocatalyst with a support material, significantly lowers the environmental and economic impact of the enzyme catalyzed reaction. The support protects the enzymes from deactivation/degradation and facilitates separation and recycling; thus, biocatalyst immobilization has considerable industrial importance and has the potential to overcome challenges associated with widespread application. Immobilized enzymes can also be used in flow systems, thus offering operational flexibility.Immobilized enzymes have been applied in the fine chemical, food and pharmaceutical industries.28,62–66 Covalent attachment of the protein to a solid is a leading method of immobilization in industry. Supports are typically obtained off the shelf from third party manufacturers, with organic polymer supports featuring prominently. The efficiency of the immobilized enzyme depends on the complementarity of the support for the protein and the chemical process targeted.
In scientific research many methods of enzyme immobilization have been reported. These can be classified as depicted in Fig. 1. The main methods for rendering an enzyme insoluble and recyclable can be divided into physical and chemical methods. In the physical method, the enzyme physically (without forming any formal chemical bonds) interacts with the carrier material/support material. The protein can either be adsorbed on the surface of the material or entrapped within. In the chemical method, the enzyme is crosslinked or covalently bonded to the support material. As of 2021 the leading methods employed for enzyme immobilization in the chemical industry are chemical methods. Enzyme immobilization can be further classified into four categories-adsorption, entrapment, covalent linking and crosslinking (Fig. 1).18,44,67–70Table 1 summarizes and compares the different enzyme immobilization methods.
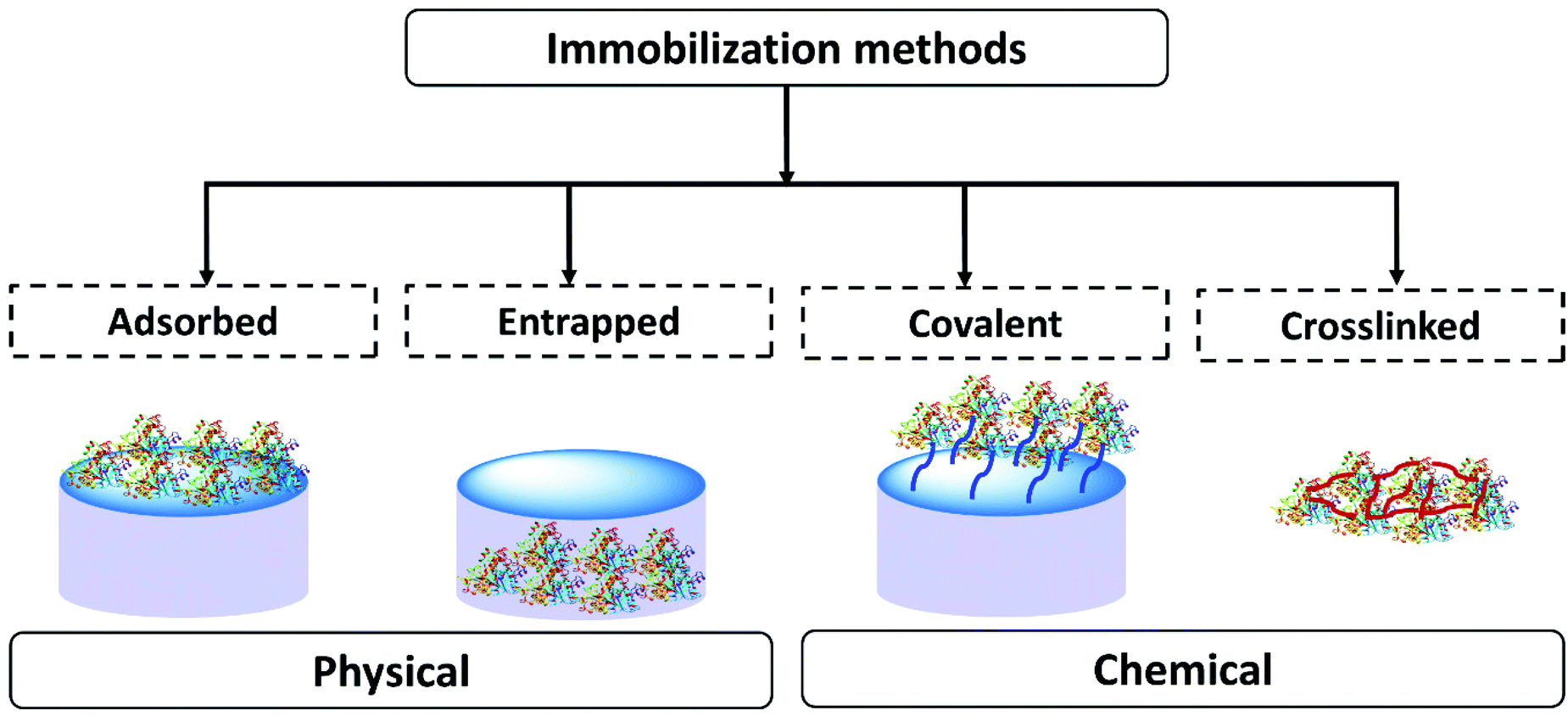 | ||
| Fig. 1 The classification of enzyme immobilization methods. | ||
| Observations | Adsorbed | Entrapped | Covalent | Crosslinked |
|---|---|---|---|---|
| Preparation | Enzyme binds to the support material by physical interaction | Support material forms in the presence of the enzyme trapping it | Enzyme binds to the support material via covalent bonds | Enzymes are cross-linked to enzyme/protein/support to form an enzyme aggregate or crystal |
| Process | Partial or incomplete adsorption on the surface of support | Support formed in the presence of the protein, protein fully entrapped | Number of attachment points to the support can vary | Extent of crosslinking can vary, can involve a support material |
| Requires an excess of enzyme to favor the equilibrium towards adsorption | Minimal loss of the support material and enzyme | Require an excess of the support to favor complete covalent attachment | Minimal loss of the support material and enzyme | |
| Protein and support material must be complimentary | Scope for tuning the support material | Custom support material | Scope for tuning crosslinking. Reproducibility can be challenging | |
| Enzyme structure | Enzyme interaction with the support is required | Minimal changes in enzyme structure. Some interaction with the matrix | Enzyme structure modified to form covalent bonds | Interaction with the crosslinkers |
| Operational stability | Binding is weak. Process stability depends on the reaction conditions | High operational stability. Leaching could be caused by soft material breakdown or poor enzyme matrix interaction | High operational stability. Strong binding to the support. Potential for some leaching depending on how well the protein is anchored | Good operational stability. Leaching could be caused by soft material breakdown or poor enzyme matrix interaction |
| Diffusion/mass transfer | Enzyme comes in close contact with the reaction medium. Easy diffusion is expected | Solid support protects the enzyme from direct contact with the reaction medium. Diffusion depends on the material design | Enzyme comes in close contact with the reaction medium. Possibility of reduction of mass transfer due to tethering | Structure can vary. Diffusion will depend on the size of the aggregate or crystal |
In the adsorption process, the enzyme is physically adsorbed onto a pre-prepared support material via hydrophilic–hydrophobic, van der Waals, H-bonding and/or ionic interactions.71 The process is easy and relatively cheap. As adsorption methods allow the enzymes close contact with the reaction media, the stability of this system is profoundly influenced by reaction conditions, like the solvent pH and ionic strength. The use of a pre-made support material provides little control over material properties. Interactions with the support material may change the enzyme surface charge distribution and activity. A couple of recent representative examples are given. Lipase from Aspergillus niger was adsorbed on a zirconium based metal organic frame work (UiO-66) and hydrophobic polydimethyl-siloxane (PDMS) modified UiO-66 via hydrophobic interactions, by mixing the enzyme with the support material in buffer.72 Increased activity was observed for PDMS modified UiO-66 in butyrin hydrolysis. Immobilized lipase on PDMS modified UiO-66 exhibited methanolysis of soybean oil for biodiesel production with 88% yield at 24 h and retained 83% activity after 10 runs. Lipase B from Candida Antarctica was immobilized in mesoporous silica (SBA-15) via electrostatic forces. The supported enzyme was active for ethyl hexanoate hydrolysis at pH 5, but at pH 7 the surface charge of the enzyme became negative, thus reducing the electrostatic interaction with the negatively charged support.260
Adsorption of an enzyme onto a support is relatively simple but the interaction between the support and protein is minimal, and these materials are vulnerable to leaching.
At the time of writing (2021) the covalent method was a common choice for enzyme immobilization. Enzymes are covalently attached to a support material, often a commercial polymer, by providing reactive points on the polymer that anchor onto the protein.73,74 For example, lipase A from Bacillus subtilis was immobilized via covalent modification of silica supported poly(sulfobetaine methacrylate) (PSBMA) polymer brushes.75,76 The enzyme was bound to the polymer brush by covalent modification of lysine with an NHS group (N-hydroxysuccinimide). The enzyme was immobilized on the polymer brush by mixing the enzyme with the polymer brush in buffer. At 20 °C, both immobilized and free enzymes exhibited similar activity in the hydrolysis of resorufin butyrate in buffer. At 50 °C the immobilized enzyme demonstrated a 100 fold increase in activity compared to the activity observed at 20 °C, whereas the free enzyme underwent a 2-fold increase in activity at its optimum temperature of 30 °C. Polymers that are ready to bind proteins are frequently provided by third party specialist manufacturers.
Covalently immobilized enzymes are expected to have good stability and recyclability under process conditions, and this has led to their commercial success. However, such strong attachment is a significant modification of the protein and can sometimes lead to changes in the protein hydrophilic/hydrophobic properties, which can significantly affect performance. There is some flexibility in material choice, with third parties providing a number of polymers with differing points of attachment and/or hydrophilicity/hydrophobicity, but there are few options for bespoke support design and polymers tend to be used as supplied. It may be necessary to modify the protein surface in order to improve the covalent attachment to the support, and it may be advantageous to optimize the protein and support together to ensure best performance.
In the crosslinking method, a material crosslinks enzymes with each other, with another protein, and/or with a support material.77–79 In general, enzyme crosslinking is a two-step process.80,81 In the first step the enzyme is precipitated to form aggregates or crystals, using different organic and inorganic precipitants. These precipitants can include glycol, dimethoxyethene, acetone, acetonitrile and ammonium sulphate. Often aqueous ammonium sulphate solution is used to make a crystal. The second step is the crosslinking of the aggregated or crystalized enzyme, to form a crosslinked enzyme aggregate (CLEA) or crosslinked enzyme crystal (CLEC), which is active and functional. The crosslinking material can target specific amino acids. Glutaraldehyde, benzoquinone and dextran-polyaldehyde are common crosslinking agents for lysine crosslinking whilst, polyethyleneimine and carboxylate activating carbodiimide are used for crosslinking glutamic or aspartic acid residues.78,82–85 CLEA is a leading method of enzyme immobilization. Sheldon et al. have highlighted the usefulness of CLEA for enzyme immobilization over the years and have recently updated the progress in their reviews.86,87 A representative example is given. The enzyme nitrile hydratase was precipitated with ammonium sulfate and crosslinked with glutaraldehyde to make CLEAs for the hydration of hexanenitrile.88 The nitrile hydratase CLEA showed 77% of the free enzyme activity at (8%v/v) substrate concentration. However, activity dropped significantly upon batch recycling.
Crosslinking is an established method of enzyme immobilization, and the materials can be relatively stable and recyclable under process conditions. Effort must be taken to ensure the mechanical stability of CLEAs and CLECs. Process optimization must concentrate on the reliability of the synthesis, ensuring that the structure formed is reproduceable.
In this review entrapment is defined as a method in which a solid or gel is formed or reformed in the presence of the enzyme, due to an event that occurs between molecules that end up as part of the support. This solid or gel forming event does not formally involve the enzyme, but the presence of the enzyme is expected to have a templating influence. The enzyme becomes trapped inside the material matrix as the support material grows, either due to size effects, supramolecular forces, or most likely, a combination of the two.49,70,89 The material formed is often a gel. A gel comprises a solid crosslinked matrix and a confined liquid. The liquid within provides an environment for the protein which can be engineered to optimize performance. Ideally the enzyme retains the native structure as no strong modifications happen within the gel. The matrix protects the enzyme from direct contact with the bulk reaction conditions, and renders the bulk material solid-like, yet allows the substrate and the product to diffuse, a schematic presentation of an ideal entrapped enzyme is given as Fig. 2.
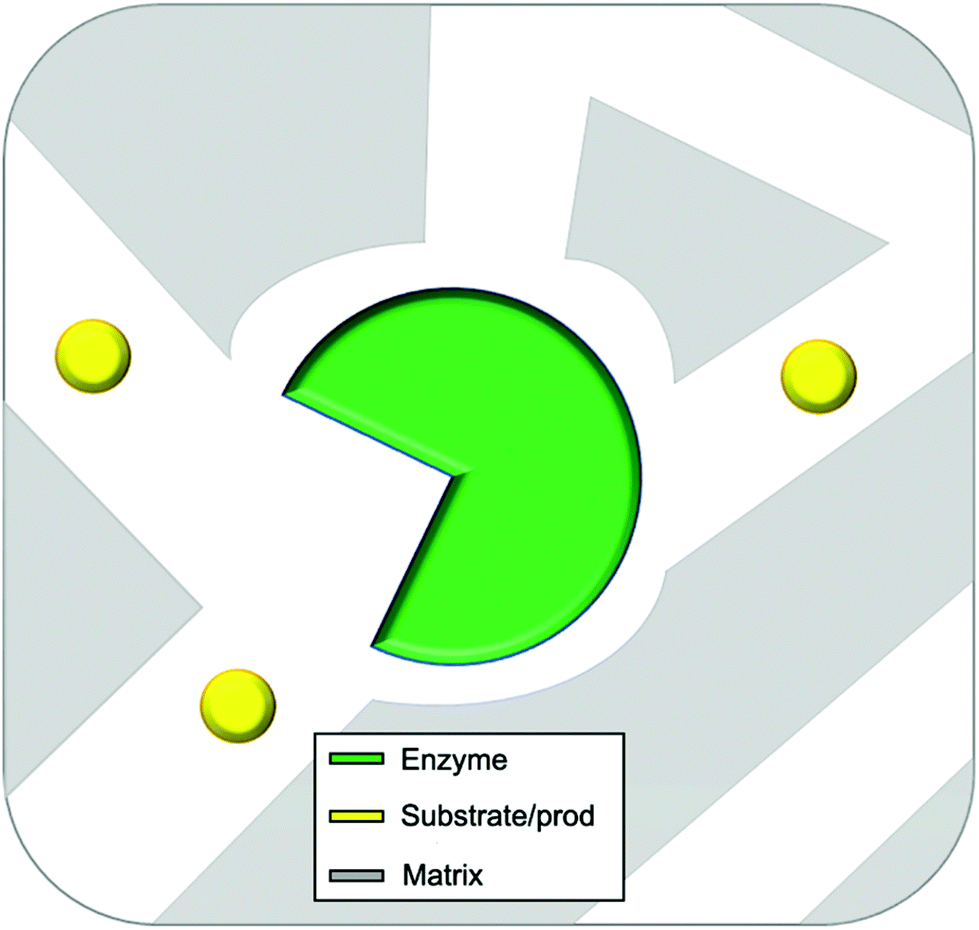 | ||
| Fig. 2 Idealized representation of an entrapped enzyme. The substrate/product and enzyme are of realistic proportional size. | ||
Mass transfer limitations can be a drawback of this method, and the material must be designed with appropriate pore size and matrix properties. However, a material that is too porous, or has pores that are too large will readily leach, and defects in the matrix may also lead to enzyme leaching. The key to successful entrapment is to provide the correct environment inside a porous material that allows for free diffusion of the substrate and product, but restricted movement of the protein. Differences in size and chemical nature of the enzyme protein and substrate molecule render this an achievable goal, but it must be considered as the primary design criterion. Many entrapment methods allow for control over the shape and thickness of the material, and this can help to control mass transport of the substrate and product.
The gel can be hard or soft. We define a hard gel as a gel in which the polymerization is essentially reversible. Hard gels tend to be the result of a covalent polymerization, such as the formation of an organic polymer, or an inorganic oxide. In a soft gel the gel can be disrupted and reformed, for example, by heating and cooling. Soft gels tend to be formed by supramolecular interactions.
This review is concerned with the development and possible future applications of entrapment methods that do not involve covalent linking to the protein. For more details on the research and industrial application of covalent bonding and crosslinking to enzyme immobilization an up to date review was recently published by Sheldon et al.87
3. Enzyme entrapment in gels
A gel comprises a liquid phase entrapped within a solid 3D network.90,91 To form a gel precursors to the solid matrix are self-assembled, dispersed or polymerized within the liquid phase. Depending on how the solid matrix is formed, gels can be classified as molecular (supra-molecular), colloidal and polymeric (macromolecular). However, gels are also classified based on the liquid phase, for example, if water is use as the liquid phase, the gel is known as a hydrogel.The entrapment of enzymes in gels is generally a mild process which allows the enzyme to retain its optimized structure. It can be achieved in four main ways: sol–gel methods, polymerisation to form insoluble polymers, crosslinking of biopolymers, and supramolecular assembly (Fig. 3). In general sol–gel forms a hard gel, supramolecular assembly forms a soft gel, and polymer and biopolymer gels can be hard or soft.
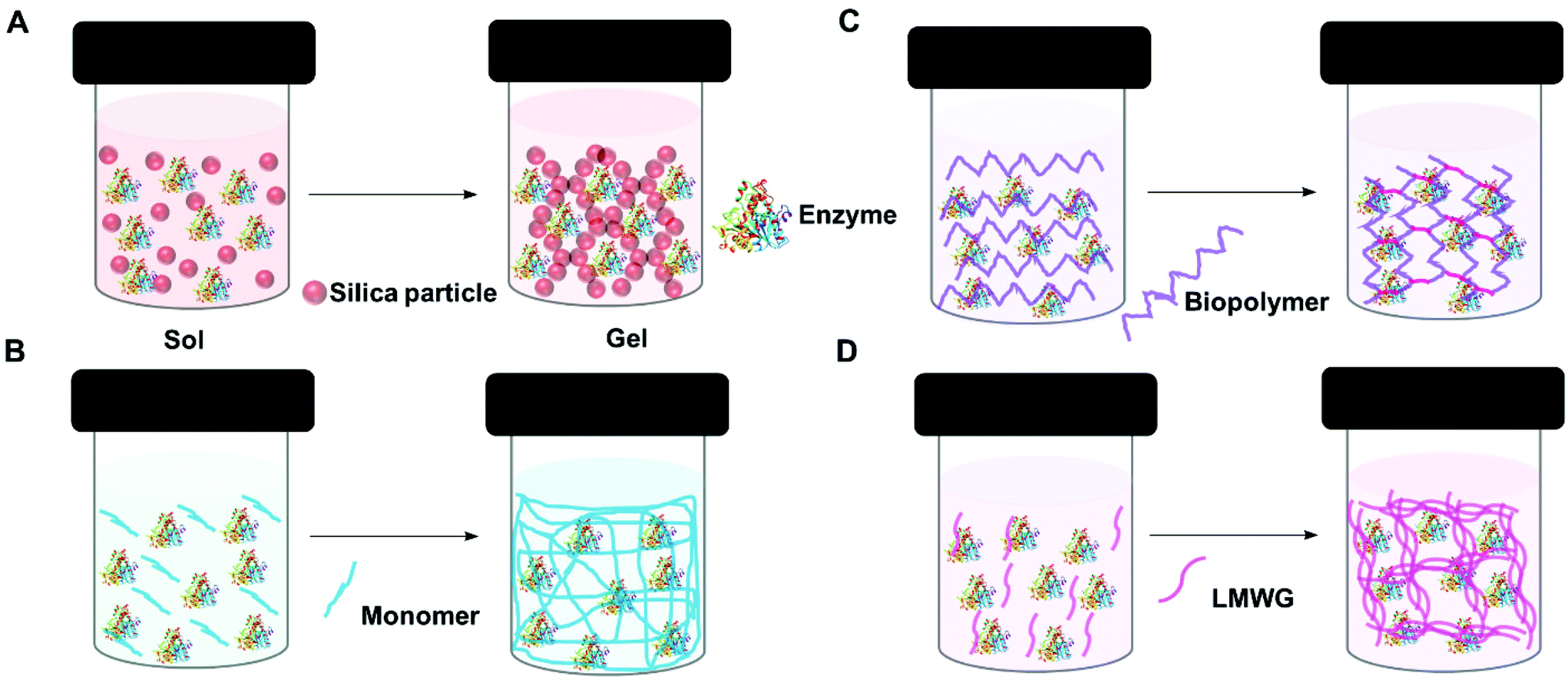 | ||
| Fig. 3 Schematic representation of enzyme entrapment in gels. (A) Sol–gel (silica gel) (B) polymerization (C) crosslinking biopolymer and (D) supra-molecular assembly (low molecular weight gel). | ||
3.1 Sol–gel methods
In the sol–gel process, soluble precursors to a solid oxide matrix, typically reactive oxides or alkoxides e.g., tetraethyl-orthosilicate (TEOS), are dissolved in a solvent, e.g., a mixture of alcohol and water. Upon initiation of the reaction, which may require the addition of and acidic or basic catalyst, a sol forms, (Fig. 4A). The suspended oxide particles of the sol react together until the gel point is reached (Fig. 4B). An enzyme can be added to the precursor solution resulting in an oxide gel containing an immobilized enzyme. During the gel formation the oxide precursor undergoes hydrolysis and condensation processes. Production of a stable gel requires ageing of the gel. During this time the reaction continues, but at a much slower rate. Drying plays a critical role in the gel morphology. Working up the gel in different ways enables the design of tailor-made materials with controlled pore and surface area. This affects diffusion rates through the final material. Sol–gel prepared oxides can be coated on a surface, or made into powders or spheres. Employing supercritical CO2 drying removes the solvent from the gel rapidly, yet retains the 3D gel network, and this produces a very high surface area material known as an aerogel (Fig. 4C). Normal drying at ambient conditions leads to greater pore collapse, and the production of a xerogel (Fig. 4D). Gel shrinking can be minimized by the inclusion of additives like synthetic or bio-polymers or ionic liquids (section 5).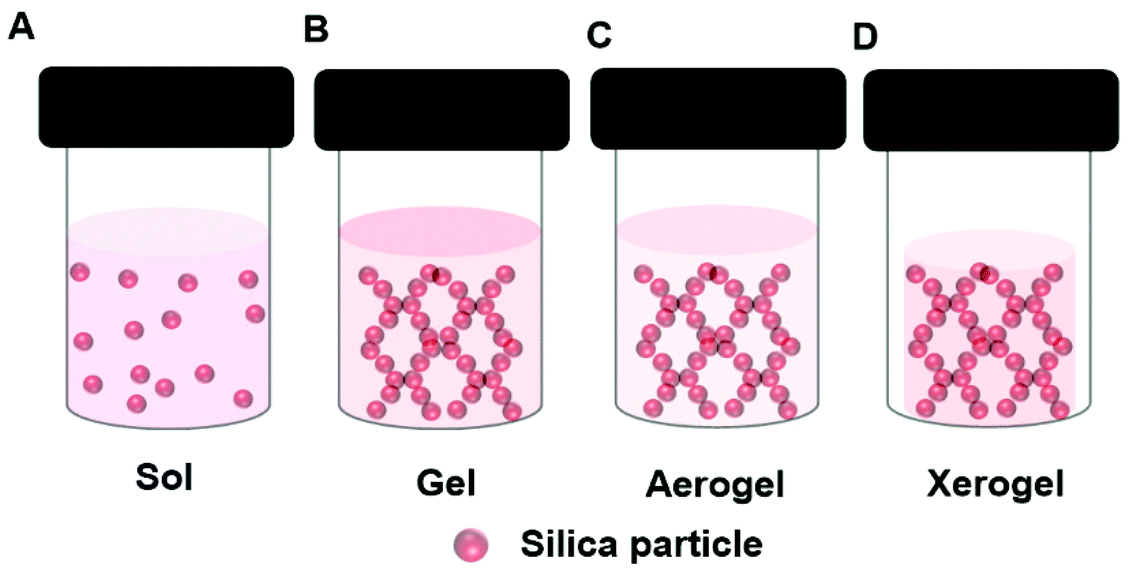 | ||
| Fig. 4 Schematic presentation of different structures formed during the sol–gel formation of a silica gel depicting a (A) Sol, (B) Gel, (C) Aerogel, (D) Xerogel. | ||
Some of the common reactions that are catalyzed by entrapped enzymes are summarized in Fig. 5.
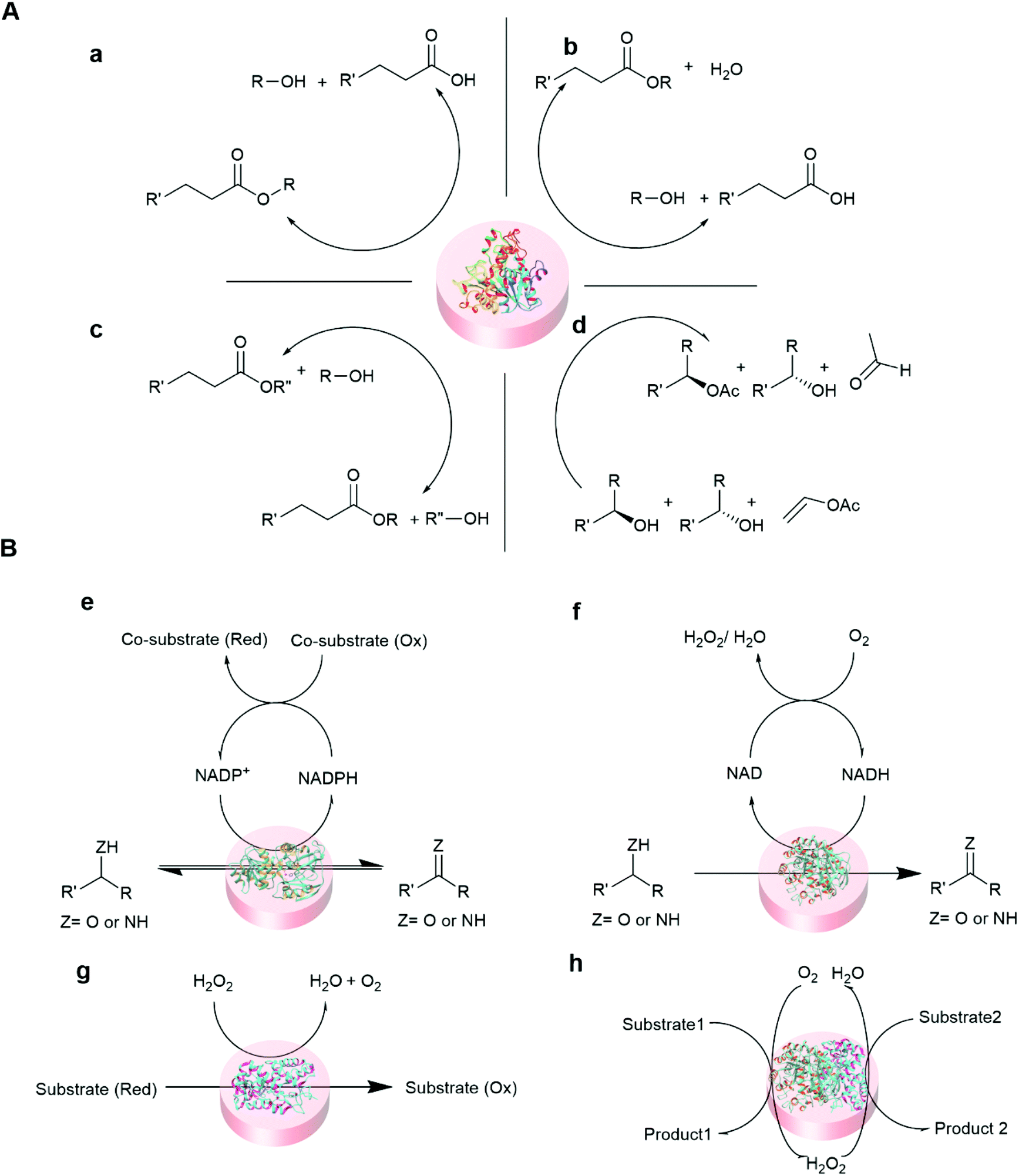 | ||
| Fig. 5 Representative reaction schemes of immobilized hydrolase and oxidoreductase biocatalysis. (A) Hydrolase (lipase) catalyzed reaction100,101 (a) hydrolysis (b) esterification (c) transesterification, and (d) kinetic resolution. (B) Oxidoreductase catalyzed reactions102,103 (e) dehydrogenase (f) oxidase (g) peroxidase and (h) cascade reaction of oxidase and peroxidase. | ||
Avnir and co-workers pioneered the entrapment of enzymes in silica using sol–gel processes. An aqueous solution of alkaline phosphatase was entrapped in silica gel, using a base catalyzed process and tetramethylorthosilicate (TMOS) as the silica precursor. The sol–gel process was performed in the presence of small amount of methanol by mixing an ice-cold enzyme solution with the TMOS solution, base and methanol at −20 °C, and the gel was dried and aged at 37 °C.92 The immobilized enzyme retained 30% of the activity of the free enzyme for the hydrolysis of p-nitrophenyl phosphate (pNPP) in glycine buffer. The immobilized enzyme exhibited enhanced thermal operation stability at 70 °C and retained activity for two months when stored in water at room temperature. Since then, an increasing number of enzymes have been entrapped in silicate gels using sol–gel procedures. Many of these materials have been applied as sensors.93,94 For example a tetraethylorthosilicate (TEOS) and ethanol sol–gel process was used to immobilized glucose oxidase and the material was applied to glucose sensing.95 Alcohol in these sol–gel processes can deactivate the immobilized enzyme and activity is typically lower than the free enzyme. Yamanaka and co-workers used aqueous buffer in an acid catalyzed TMOS sol–gel process, and entrapped single and multiple enzymes including glucose oxidase from Aspergillus niger and horseradish peroxidase, in a transparent gel.96 Sonication of TMOS, water and acid gave the sol, the sol was then buffered and finally the enzyme(s) in buffer was mixed with the buffered sol and left at 4 °C to dry. A cascade reaction was carried out using immobilized glucose oxidase and peroxidase. The immobilized enzymes retained 20% of the activity of the free enzymes. Aqueous solutions of glucose oxidase and peroxidase were also entrapped in silica gel using TMOS with 0.5% polydimethylsiloxane.97
The addition of additives to the sol–gel media further improved the sol–gel entrapment process. Addition of poly-ethylene glycol (PEG-6000) to a TMOS-derived gel of trypsin led to good retention of activity, as the trypsin stayed active for several months, and also increase the half-life of alkaline phosphatase.98
Metal oxide methods tend to be faster and harder to control than silica-based sol–gel methods. Hybrid materials of cellulose acetate with oxide of titanium isopropoxide (Ti(iOPr)4) and zirconium tetra-n-butoxide (Zr(OBu)4), respectively, were used to entrap urase from jack bean and glucose oxidase from Aspergillus niger using modified sol–gel methods.99 The hybrid Zr material was made by mixing cellulose acetate in acetone with the enzyme in buffer, followed by dropwise mixing of the cellulose-enzyme solution to (Zr(OBu)4) solution in acetone, the mixture was left for 30 min and the gel fibers were dried in the fridge. The immobilized glucose oxidase in cellulose-Zr gel fibers had 10 times lower activity than the free enzymes for glucose oxidation. However, increase in activity was observed with consequent runs, and it was speculated that the increase in activity with reuse originated from different orientations/configurations of the enzyme. A very similar result was observed for immobilized urase.
Using hydrophobic silica precursors for enzyme entrapment has proven a successful method for immobilizing biocatalysts for chemical synthesis, particularly for lipase.89,104–110 Reetz and co-workers added hydrophobic silica precursors to their sol–gel enzyme entrapment process. A lipase silica gel of Ps. cepaciu (Amano PS) derived from TMOS exhibited only 5% activity in lauric acid esterification. Using silica precursors TMOS/RSi(OCH3)3, or pure RSi(OCH3)3 (R = CnH2n+1, n = 1, 2, 3, 4, and 18), not only enhance the activity, but also increased the enzyme loading.105 In a sodium fluoride (NaF) catalyzed methyltrimethoxysilane MTMS (RSi(OCH3)3 (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3) derived silica gel of lipase, the esterification activity of lauric acid with n-octanol in isooctane was increased by 1300%, and >99% ee was obtained in the kinetic resolution of L-phenylethanol with acetic anhydride in benzene.104
CH3) derived silica gel of lipase, the esterification activity of lauric acid with n-octanol in isooctane was increased by 1300%, and >99% ee was obtained in the kinetic resolution of L-phenylethanol with acetic anhydride in benzene.104
The inclusion of additives into the sol–gel enzyme entrapment process can lead to significant improvements in enzyme stability. This has increased the scope of the chemistry that could be achieved with isolated enzymes. For example a TMOS derived gel of two highly pH sensitive enzymes alkaline phosphatase (optimal activity at pH 9.5) and acid phosphatase (optimal activity at pH 4.5), were doped with the surfactants sodium dioctylsulfosuccinate (AOT) and cetyltrimethlammoniumbromide (CTAB), and this enabled the application of the enzymes to the hydrolysis of p-nitrophenyl phosphate (pNPP) at low and high pH of 0.9 and 13.0 respectively.111 A TEOS derived silica gel of laccase from Trametes sp. retained 100% oxidation activity of 2,6-dimethoxyphenol (2,6-DMP) after 70 days of storage at 27 °C in the presence of polymer additives of polyvinyl alcohol (PVA), whereas the additive free immobilized enzyme retained only 30% activity.112 Isopropyl alcohol and polyvinyl alcohol were added to a silica gel of ω-Transaminase from Arthrobacter sp. derived from TMOS/RSi(OCH3)3 (R = CnH2n+1, n = 1, 2, 3, 4), enabling the kinetic resolution of 1-phenylethylamine with sodium pyruvate and pyridoxal-5-phosphate monohydrate using dimethylsulfoxide (DMSO) as co-solvent in buffer at relatively high substrate concentration of 100 mM.113 The conversion was 54% at 24 h with >99% ee obtained for the product 1-phenylethylamine. The immobilized enzyme was recycled 5 times with conversions of 51% at 24 h and >99% ee. Adding polyethylenimine to a silica gel of lipase from Thermomyces lanuginosus immobilized by a TEOS sol–gel process increased the enzyme loading, thermal stability and catalytic activity.114 Similarly adding poly-L-lysine to silica gels of lipase and glucose oxidase enhanced the stability, reusability and thermal stability.115,116 Adding polyethylene oxide to a TEOS derived silica gel of β-glucuronidase from Helix pomatia enabled the microfluidic (the reaction was operated in small quantities of liquids within narrow channels) hydrolysis of p-nitrophenyl-β-D-glucuronide (pNP-GluA).117
Enzymes can be entrapped in alumina (Boehmite) gels by sol–gel processes. Liu et al. claimed that hydrophilic alumina had advantages over silica, particularly, the retention of activity at high enzyme loading and the immobilization of low isoelectric point enzymes.118,119 Alumina entrapped enzymes are prepared from an alumina sol (Fig. 6). One approach is to make the sol from aluminium alkoxides at high temperature; this is followed by acid (such as HCl) catalyzed hydrolysis. Finally, the enzyme is added to the sol before gel formation (Fig. 6A). Aluminium alkoxides produce alcohols during the hydrolysis process, and this can denature the enzyme. If the enzyme is sensitive to alcohol, the alcohol can be evaporated prior to protein addition. Yang and co-workers used sodium aluminate in an acid catalyzed sol–gel process to entrap catalase from bovine liver in buffer.120 The gel morphology, studied by SEM and TEM, suggested boehmite (isoelectric point PI 9.1) formed positively charged nano-needles at pH 7, at that pH catalase (PI 5.4) remained negatively charged thus electrostatically attached to the boehmite, later, boehmite self-assembled into a nano strip and entrapped the enzyme. The catalase entrapped in alumina gel achieved 30% activity relative to the free enzyme in buffer at pH 7.0, 25 °C. However, the gel achieved 86% of the free enzyme's activity at pH 4. The gel exhibited poor recyclability due to enzyme leaching. Another approach was employed by Avnir and co-workers, who precipitated aluminium alkoxide in water, and ultrasonicated the precipitate to give a transparent sol (Fig. 6B). Human serum albumin was added to the sol at room temperature.121 Human serum albumin entrapped in alumina gel exhibited increased stability, with an increase in denature temperature of 54 °C. This was attributed to the ability of the alumina gel to retain water at elevated temperatures. The same group compared proteinase and xylanase activity in silica and alumina gels.122 Both the enzymes performed better in alumina gel than silica gel. In addition, a heat-treated alumina gel of xylanase at the elevated temperate of 200 °C for 3 min retained 73% activity in xylan degradation to xylose in buffer, whereas, the silica gel lost activity after heat treatment at 200 °C. The hard-crystalline alumina gel retained the enzyme's native structure and essential water within the confined environment of the gel. Plasminogen activator, a serine protease, entrapped in alumina gel was applied as an injectable material for thrombosis treatment.123 Alumina gels of three therapeutic enzymes, acid phosphatase (AcP), horseradish peroxidase (HRP), and asparaginase (ASP) all exhibited thermostability of 30–50 °C higher than the aqueous enzyme.124 The alumina sol–gel process was shown to refold an unfolded carbonic anhydrase and the entrapped enzyme exhibited activity of 180% higher than the native enzyme in hydrolysis of p-nitrophenyl acetate (pNPA) in buffer.125 Alumina gel also protected carbonic anhydrase, acid phosphatase and Horseradish peroxidase from photogedradation.126 Alumina gels of bovine serum albumin are used as a protein carrier for biomedical applications.127
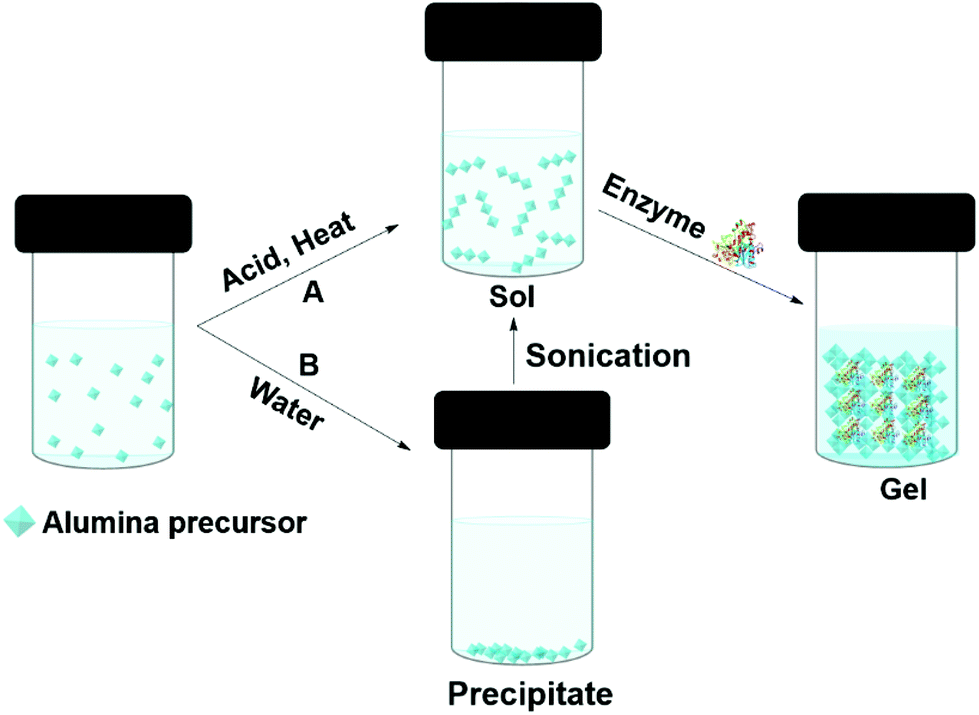 | ||
| Fig. 6 Different approaches (A and B) to entrapping enzymes in alumina (boehmite) gels by sol–gel methods. | ||
Sol–gel methods for enzyme entrapment have been demonstrated to form a wide range of active biocatalytic materials, particularly for hydrophobic enzymes such as lipase. In the presence of modifying groups, good activity and stability can be supported, and alumina gels appear to have significant advantages at higher temperatures. Historically the application of hard oxide gels suffered from the lack of structure–function understanding, with each entrapment method appearing as a ‘one off’, giving little opportunity for systematic tuning. The addition of ionic liquids could change this and lead to systematic design of the local enzyme environment. Ionic liquids are discussed in section 5.
3.2 Synthetic polymers
The immobilization of an enzyme using a synthetic polymer (Fig. 7A) is an old method, but it is still evolving and is therefore worthy of discussion.128,129 In general, entrapping an enzyme in a polymer matrix involves mixing the enzyme with monomers, and sometimes crosslinkers, and an initiator to mediate the polymerization process. Often polymerization is achieved via free radical formation. Different approaches are used to generate free radicals, some commonly used processes are photochemical, redox and enzyme mediation. Redox mediated free radical formation is a mild process and is commonly used for enzyme entrapment. Bernfeld and Wan pioneered methods for immobilizing enzymes and antigens in polymer gels, and these methods provide a good example to illustrate the scope of synthetic polymer entrapment.130 The monomers used in these and related methods (Fig. 7B) are known to form hydrogels in water. Bernfeld and Wan entrapped seven different enzymes and antigens in poly-acrylamide gels formed by the free radical polymerization of acrylamide 1 and N,N′-methylenebisacrylamide 7 in an aqueous buffer at 35 °C, the polymerization reaction was initiated using potassium persulphate (KP) 8a. The immobilized antigen exhibited antibody binding activity.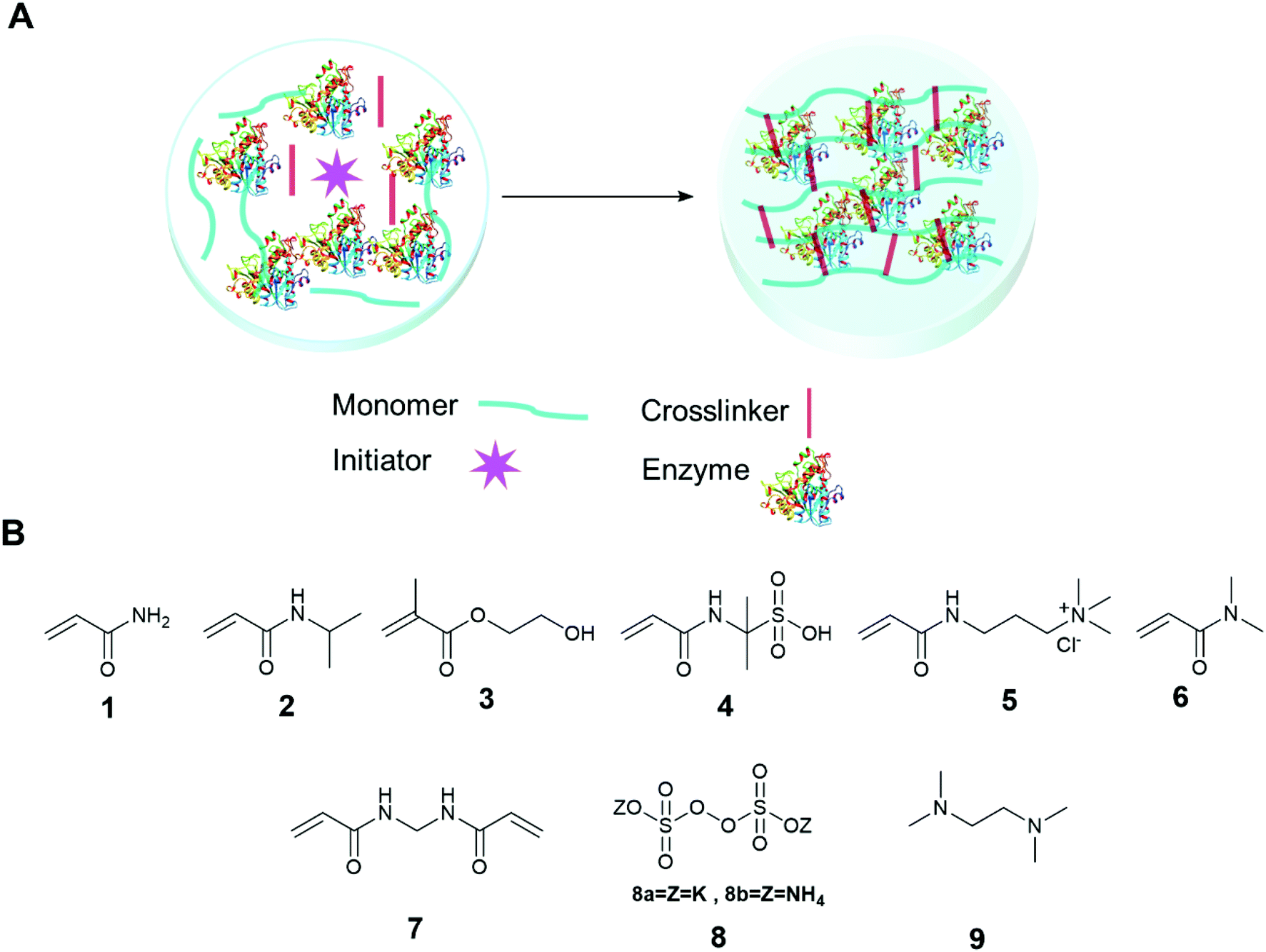 | ||
| Fig. 7 Enzyme entrapment in synthetic polymer gels. (A) Schematic presentation of entrapment in synthetic polymer gels; (B) chemical structure of some common monomers, crosslinker and initiators. | ||
Demirel and co-workers entrapped glucose isomerase in a poly(acrylamide) hydrogel using a free radical polymerization reaction of 1 and 7 in an aqueous solution using tetramethylethylenediamine (TEMED) 9 and ammonium persulfate (APS) 8b at room temperature.131 The immobilized enzyme exhibited optimal glucose isomerase activity at 60 °C, and retained 75% activity at the high temperature of 70 °C, at the same temperature the free enzyme retained only 40% activity. The immobilized enzyme retained 81% activity after 42 days storage at 4 °C and retained 98% activity after 25 recycles. Yamak et al. immobilized laccase in a poly(acrylamide-N-isopropylacrylamide) hydrogel by mixing the enzyme with 1, N-isopropylacrylamide 2, 7 (as a crosslinker), and 8b and 9 (as redox initiators) in buffer at room temperature.132 The immobilized laccase exhibited similar oxidation catalytic efficiency with substrate syringaldazine to free enzymes. The immobilized enzyme retained 91% activity after 56 days storage at 4 °C and retained 78% activity after 10 uses. Demirci et al. immobilized α-glucosidase and co-factor reduced L-glutathione in a poly(2-hydroxyethyl methacrylate) (p(HEMA)) gel.133 The entrapment process was carried out by mixing α-glucosidase and L-glutathione, with monomer 2-hydroxyethyl methacrylate, 3, and 7 and 9. The resulting solution was placed in a cryogenic (low temperature) environment of −18 °C and finally, 8a was added to the cold enzyme solution with mixing. The polymerized gel, a cryogel, formed at −18 °C. The hydrolytic activity of the immobilized enzyme was investigated using p-nitrophenyl-β-glucopyranoside (pNPG) as a substrate in buffer. The immobilized enzyme retained 92% of the activity of the free enzyme and 50% of its activity after 10 days storage at 25 °C. A drop in activity of 50% was observed after 10 reuses. The same research group further studied the entrapment of α-glucosidase and co-factor reduced L-glutathione in neutral, anionic and cationic polymers of 1, 2-acrylamido-2-methyl-1-propane sulfonic acid (AMPS) 4, 3-acrylamidopropyltrimethyl ammonium chloride 5 (APTMACl) cryogel and investigated the enzyme activity using β-glucopyranoside (p-NPG) as a substrate in buffer.134 The immobilized gels exhibited 81%, 62%, and 60% activity, respectively, compared to the free enzymes. The anionic (p(AMPS)) and cationic (p(APTMACl)) cryogels exhibited unusual pH stability and retained 100% activity at pH 8 and pH 5, respectively.
Wang and co-workers immobilized HRP in a self-catalyzed poly(N,N-dimethylacrylamide) gel in a water-in-oil (O/W) emulsion.135 The oil phase was prepared using the surfactants tween 20 and span 80 in octane and acetylacetone. An aqueous solution of HRP, N,N-dimethylacrylamide 6 and 7 were added slowly to the oil phase to form the water-in-oil emulsion. Addition of hydrogen peroxide to the emulsion initiated the HRP mediated radical formation of acetylacetone that mediated the polymerization reaction, and HRP was entrapped within the polymer hydrogel. The process was carried out under argon to suppress radical quenching. The catalytic activity of the immobilized HRP retained 75% activity compared to the free enzyme for the oxidation of o-phenylenediamine (OPD) in the presence of H2O2. The immobilized enzyme retained 98.5% activity after 3 months storage at 4 °C. The enzyme immobilized gel exhibited good thermal stability and retained 33% activity after heating at 70 °C for 30 min, higher than the free enzyme, which retained only 14% activity under the same conditions.
LentiKats® is a polymer entrapment system based on a polyvinyl alcohol (PVA) matrix which has demonstrated excellent performance with whole cell biocatalysts.136 LentiKats® technology has also been applied to enzyme immobilization. An enzyme can be entrapped in LentiKats® by dissolving PVA and polyethyleneglycol (PEG) in water at 90 °C, and while the solution is cooling at 40 °C, an enzyme solution is added. Beads are made by using a LentiPrinter device.137,138 Rebroš et al. immobilized the enzyme glucoamylase in LentiKats® for the hydrolysis of maltose to glucose.139 The immobilized enzyme exhibited 35% residual activity compared to the free enzyme, however, the immobilized enzyme maintained 80% activity after 100 recycles in a batch process at 30 °C. In addition, the immobilized enzyme retained the initial activity of 60% conversion for 1520 h in a continuous process at 60 °C. The initial activity loss of immobilized glucoamylae was high, however, the stability and reusability of the process circumvented the initial activity loss.
Polymerisation methods based on hydrogel formation have a wide applicability. Akin to sol–gel methods, the techniques involved can be quite specialized and would benefit from simple protocols that are widely applicable.
3.3 Biopolymers
The immobilization of enzymes and biocatalysts in biopolymers, such as proteins and carbohydrates is well established and has been the subject of recent reviews.46,140–142 Biomaterials are attractive due to their non-toxicity, biodegradability, flexibility and availability.142 This review will concentrate on methods in which the enzyme becomes entrapped. There is some overlap between entrapment and crosslinking methods, as biopolymers are frequently gelled by the addition of a crosslinking agent, such as Ca2+ for alginates. In this review we define entrapment as a solid or gel forming event which occurs in the presence of the enzyme and incorporates it within the structure, but does not directly involve the enzyme. The term ‘crosslinked’ is reserved for immobilization methods that formally involve the protein of the enzyme in the crosslinked structure. The biopolymer method depends on the initiation of additional bonds (supramolecular or covalent) to a macromolecule that is already formed (Fig. 3C). Biopolymer gels can be soft or hard gels depending on whether the method of crosslinking is reversible or not. Carbohydrate based biopolymers alginate 10, cellulose 11, chitosan 12, chitin 13 and agarose 14 (Fig. 8) are promising materials for enzyme immobilization. | ||
| Fig. 8 The chemical structure of some biopolymers used for enzyme entrapment. | ||
Carbohydrate-based biopolymers are highly biocompatible and contain multiple functional groups that make these molecules suitable for enzyme immobilization via entrapment methods. Bilal and Iqbal have recently reviewed and highlighted the potential of carbohydrate based natural polymer for enzyme immobilization.46 In this section progress on biopolymer-based materials for enzyme entrapment will be discussed, with representative examples. We refer the reader to published reviews for further details. Discussions will concentrate on alginate and silk fibroins for enzyme/biocatalyst entrapment.
Alginate 10 is a negatively charged polysaccharide, and in the presence of divalent metal ions, particularly calcium (Ca2+), alginate crosslinks to form a biopolymer gel. Arruda and Vitolo immobilized invertase in Ca-alginate for sucrose hydrolysis.143 The enzyme was immobilized by the dropwise addition of enzyme-alginate solution to CaCl2 solution and left for 24 h. The immobilized enzyme retained 73% activity compared with the free enzymes at pH 4.6 and 30 °C. Quiroga et al. immobilized cysteine protease araujiain in Ca-Alginate beads, and demonstrated high thermal stability at 70 °C and pH range 5–9.5.144 The enzyme retained 92% of its initial activity and 78% of its activity after 20 runs. The enzyme performance was optimized in biphasic 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 aqueous/ethyl acetate.
:1 aqueous/ethyl acetate.
The protein silk fibroin (SF) has a stable and flexible conformation that allows the design of immobilized materials with different shapes and sizes, like powders, scaffolds, membranes, films, and spheres.141 Fukui and co-workers first reported entrapment of β-glucosidase in a silk fibroin membrane.145 The enzyme immobilized silk membrane was made by mixing the aqueous solution of the silk fibroin and the enzyme, and the resultant mixture was air dried for 24 h at 20 °C. The membrane entrapped enzyme retained 47% of the activity of the soluble enzyme in the hydrolysis of p-nitrophenyl-β-D-glucopyranoside in acetate buffer (50 mM, pH 5.7) at 25 °C, and exhibited high thermal stability, retaining 90% activity at 55 °C, compare to 50% activity for the free enzyme. Activity dropped to 87% with 20 re-uses in 20 days with storage in the buffer containing the substrate. Glucose oxidase entrapped in a silk fibroin membrane exhibited 80% activity compared to the free enzyme in the oxidation of glucose in buffer at 25 °C, and was stable for 30 days.146 Immobilized enzyme activity was found to decrease with increased enzyme loading.
Silk fibroin entrapped horseradish peroxidase was used as a sensor for hydrogen peroxide detection using tetrathiafulvalene as the electron carrier.147 Murine anti-TGFb and IgG1 mono-clonal antibodies were entrapped in a silk fibroin hydrogel and lyophilized gel.148 The gels were made by sonicating silk fibroin to make a sol, then the antibody was added to the sol to form a silk fibroin hydrogel and lyophilization of the hydrogel produced the lyophilized gel, both the gels were used for controlled release of the antibody.
Carbonic anhydrase (CA) was entrapped in a metal mediated photochemically induced SF hydrogel.149 The hydrogel was made by mixing the enzyme and silk fibroin in buffer with a solution of APS 8b (Fig. 7B) and [Ru(bpy)3]Cl2. The resulting solution was mixed, and irradiated using white light (100 W fiber optics) for 10 min. The immobilized CA hydrogel exhibited >60% activity relative to the free enzyme in the hydrolysis of p-nitrophenyl acetate (pNPA) in buffer. Some increase in stability was reported at pH 3.
Biopolymer methods are often simple and cheap, and this makes them easily accessible. Development of these method must concentrate on achieving good reproducibility of structure and performance.
3.4 Supramolecular assembly using low molecular weight gelators
Low molecular weight gelators (LMWGs), are small to medium size (usually organic) molecules used to gel solvents by supramolecular assembly (Fig. 9). A LMWG that gels an organic solvent is referred to as an organogelator. LMWGs can also gel water to form a hydrogel, or an ionic liquid to form an ionic liquid gel. LMWGs form gels by supramolecular interactions such as H-bonds, hydrophobic interactions and π–π stacking to form supramolecular assemblies, thus LMW gels are frequently soft and reversible in nature.150,151 The self-assembly process requires the LMWG to dissolve in the solvent, and under influence of a stimulus, the gelator will self-organized to form the gel. The common stimuli are heating and cooling, pH change and enzyme activity.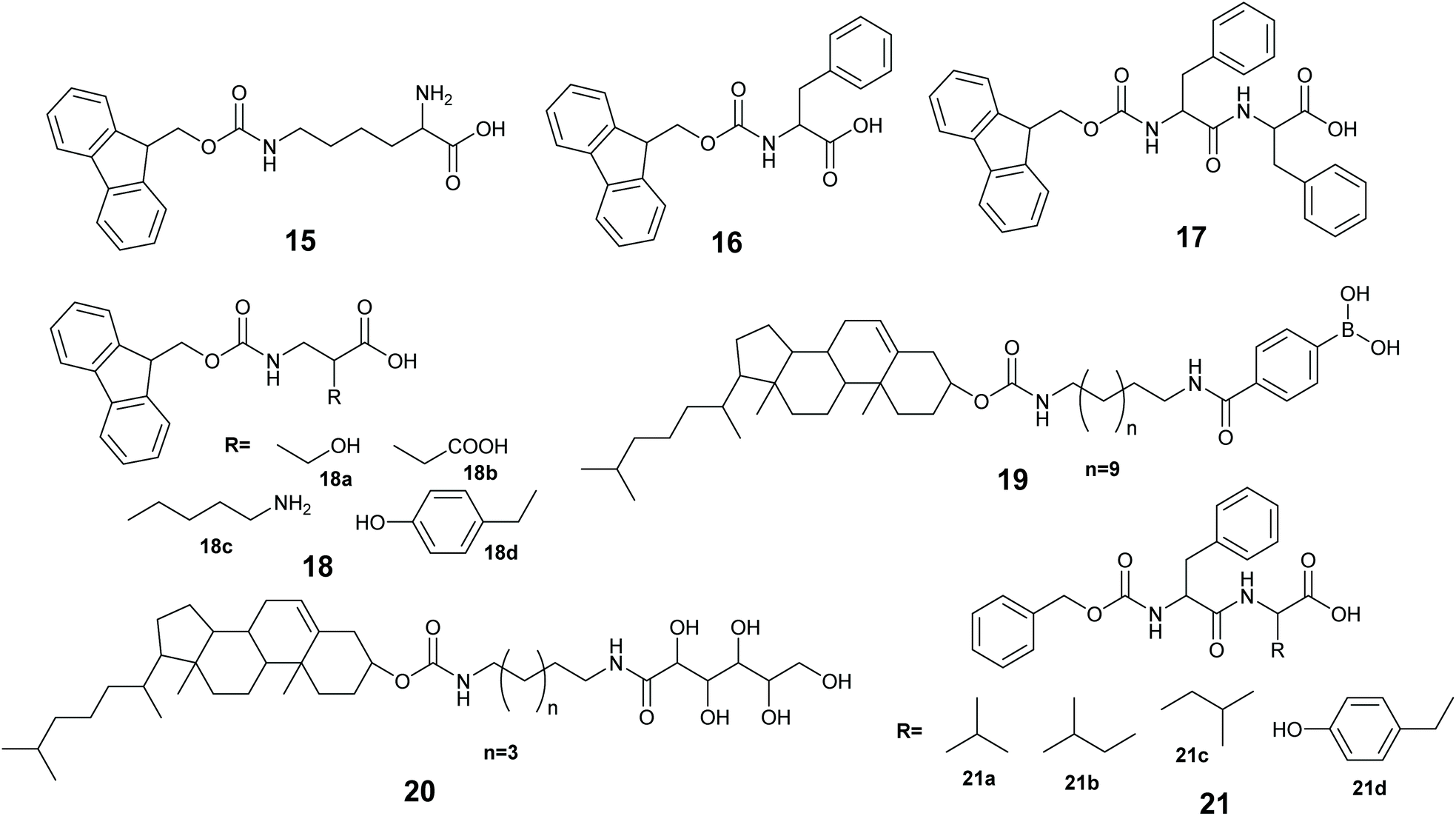 | ||
| Fig. 9 The chemical structures of LMWGs used to entrap enzymes in gels. | ||
Xu and co-workers immobilized methemoglobin, horseradish peroxidase, laccase and α-chymotrypsin in a 9-fluorenylmethoxycarbonyl-L-lysine (Fmoc-K) 15 and Fmoc-L-phenylalanine (Fmoc-F) 16 hydrogel.152 The enzyme immobilized hydrogel was made using a heating–cooling process by heating the aqueous suspension of 15, 16 and sodium carbonate at 60 °C to give a solution, when the solution was cooling at 35–40 °C, aqueous enzyme was added to the solution and cooling at room temperature gave the enzyme immobilized hydrogel. The immobilized enzyme gels exhibited better oxidation activity in toluene than in aqueous buffer. For example, immobilized methemoglobin and free methemoglobin showed very similar activity in pyrogallol oxidation to purpurgallin in buffer, however, the immobilized enzyme exhibited 8 times higher oxidation activity in toluene than the free enzyme oxidation in the aqueous buffer. UV-Vis and CD spectroscopic studies revealed that the immobilized enzymes in the hydrogel retained the structure as it was in water.
Ulijn and co-workers, immobilized lipase B in a peptide hydrogel of Fmoc-diphenylalanine (Fmoc-FF) 17 co-assembled with Fmoc-amino acid 18a–18d. The resultant gel was further stabilized using silica nano particles.153 The enzyme immobilized hydrogel was made using a pH variation method by dissolving 17 and 18 in basic aqueous media followed by sonication to give a solution, the pH was adjusted to 7.8 and addition of CaCl2 formed the gelator solution. The enzyme in buffer was added to the gelator solution, and the enzyme-gelator solution was dispersed into silica nanoparticles in heptane. After homogenization, the resultant mixture was left at 2–8 °C for silica stabilized peptide gel formation. The silica stabilized enzyme hydrogel made from 17 and 18a exhibited 4 times higher activity compared with the free enzyme in the transesterification of 1-octanol and octanoic acid in heptane.
Das and co-workers developed cholesterol based phenylboronic acid, and glucose based LMWGs 19 and 20 that gel DMF-water mixtures.154 A gel of 19 was used to entrap lipase and 20 was used to entrap the prodrug chloramphenicol ester or a model substrate p-nitrophenyl-n-octanoate. Mixing of the gels was found to destruct both due to the formation of a boronate-diol adduct that allowed the lipase to catalyze the prodrug/substrate and control the release of the active drug/product.
Roy and co-workers immobilized Lipase from Candida rugosa in a hydrogel via co-assembly of the enzyme with LMW dipeptides 21a–21d.155 The dipeptides 21a–21d were unable to form the hydrogel, however, addition of the enzyme induced gel formation.155 The gelation occurred via weak interactions of the peptide with lipase. The entrapped lipase hydrogel was active at catalyzing the hydrolysis of p-nitro phenylbutyrate (pNPB) in water.
The use of soft gels to support biocatalysts offers the advantages of ease of synthesis and recycle. An effective environment can be created swiftly to support enzyme activity at little cost, allowing enzymes to be operated in solvent environments they would not otherwise be stable in. The application of LMWGs to enzyme entrapment is less developed than sol–gel methods, but the area holds much promise, particularly if reliable methods to mechanically strengthen the gels can be found. The choice of LMWG should be informed by the principles of green chemistry and favour the use of molecules derived from nature which require little chemical modification to render them gel forming. Examples of sugar derived LMWGs are given in section 5.3.3.
4. Innovations in enzyme entrapment
In this section recent developments in the entrapment of enzymes, that do not fit exclusively into the traditional categories described in section 3, are discussed. Innovations that involve ionic liquids are discussed separately in section 5.4.1 Metal–organic frameworks
A metal organic framework (MOF) is a metallic cage in which metals are connected by organic ligands acting as linkers, that provide the cage with a high porosity and surface area. MOFs have been utilized to entrap many molecules, including enzymes.156–158 The enzyme can be adsorbed, crosslinked, covalent bonded, diffused and entrapped in a MOF.159 In entrapment by in situ processes, the MOF forms from the MOF precursor building blocks in the presence of the enzyme (Fig. 10). MOF enzyme entrapment can be achieved via biomineralization and co-precipitation methods.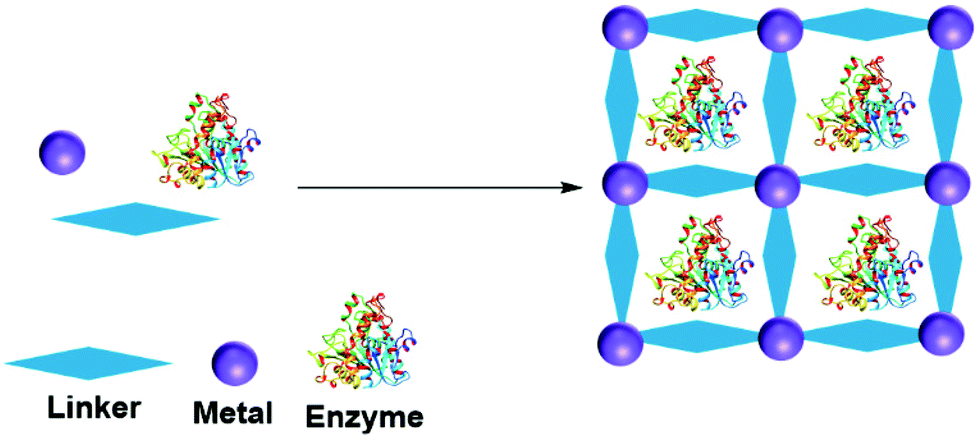 | ||
| Fig. 10 Schematic representation of in situ enzyme entrapment in a MOF. | ||
Liu and co-workers first reported in situ enzyme immobilization in a MOF.160 They immobilized Cytochrome C (Cyt C) in MOF ZIF-8 (a zeolitic imidazolate framework, constructed from Zn2+ and 2-methylimidazolate ions). Polyvinylpyrrolidone was added to stabilize the enzyme in methanol. The entrapped Cyt C exhibited 10 times higher peroxidase activity in the oxidation of 2,2′-azinobis(2-ethylbenzthiazoline-6-sulfonate) (ABTS) in the presence of H2O2 in buffer compared to the free Cyt C in solution. In parallel three other research groups developed mild aqueous processes for in situ non-covalent enzyme entrapment in MOFs.161–163 Tsung and co-workers immobilized catalase in ZIF-90 MOF crystals from ZIF-90 precursors of 2-imidazolecarboxaldehyde and zinc nitrate in an aqueous media. The enzyme was entrapped within pores that were smaller than the size of the enzyme.161 The MOF entrapped catalase exhibited hydrogen peroxide degradation activity and protected catalase from protease degradation. However, mass transfer limitations due to the structure resulted in low hydrogen peroxide degradation activity compared to the enzyme in solution.
Liang et al. immobilized bovine serum albumin in ZIF-8, using a biomineralization method.162 In the biomineralization method, the enzyme acts as the nucleation point and influences the MOF crystal growth and material porosity. This method allows the enzyme to be released from the MOF crystal by a pH change. Using the biomineralization method, they were able to immobilize 11 different proteins with 86–100% loading efficiency and with enhanced catalytic activity. Horseradish peroxidase (HRP) in ZIF-8 oxidized 88% of pyrogallol to purporogallin in the presence of trypsin, compare to 20% for the free enzyme.
Liu and co-workers entrapped glucose oxidase (GOx) and Horseradish peroxidase (HRP) for glucose detection in ZIF-8 in a one pot reaction employing the MOF building blocks in aqueous solution.163 Ouyang and co-workers immobilized glucose oxidase (GOx) and CytC in ZIF-8 MOF, from MOF precursors.164 They found that GOx in ZIF-8 retained a similar activity to the free enzyme, whereas Cyt C retained only 10% activity. The study revealed that the enzyme type and enzyme nucleation ability influenced the activity of the enzyme within the MOF. GOx initiated a rapid MOF nucleation and retained enzyme activity. In contrast to GOx, Cyt C does not take part in MOF nucleation leading to a slow nucleation. The slow nucleation process allowed Cyt C to interact with the MOF building blocks, resulting in deactivation. Thermophilic lipase QLM, immobilized in ZIF-8 MOF exhibited enhanced activity towards p-nitrophenyl caprylate (pNPC) hydrolysis and (R,S)-2-octanol kinetic resolution.165 lipase QLM in ZIF-8 retained 90% of hydrolysis activity compared to the free enzyme. When exposed to 1 mM of Zn2+ ions the free enzyme exhibited only 54% activity. The lipase QLM in ZIF-8 MOF retained activity for 5 days at 37°. 76% activity was retained after 5 days at 60 °C. In kinetic resolution of (R,S)-2-octanol with vinyl acetate in dichloromethane (DCM) the lipase QLM in ZIF-8 MOF gave 80% ee, lower enantioselectivity than the free lipase with 85% ee. One pot immobilization of lipase with ZIF-8 precursors increased the lipase heat stability, storage ability and recyclability.166 ZIF-8 nMOF (nano-MOF) enabled multiple enzyme entrapment for enzymatic cascade reactions.167 For example, enzymes GOx and HRP entrapped in ZIF-8 nMOF retained >90% free enzyme activity.
MOFs have exhibited some impressive qualities as matrices for enzyme entrapment. However, MOFs by nature are metal-rich and the life cycle, cost and recyclability of these systems will have be considered if they are to be employed in an industrial setting.
4.2 Smart gels: enzyme responsive entrapment
Enzyme assisted gel formation is a smart approach to the entrapment of enzymes in a non-covalent way, with applications in biomedical, drug delivery, sensing and biocatalysis (Fig. 11 & 12).168–170 The enzyme activates the gelator and initiates gelation via self-assembly, cross-linking, surface activation or polymerization.48 Low molecular weight gelators and polymeric materials are commonly used as the target for the enzyme.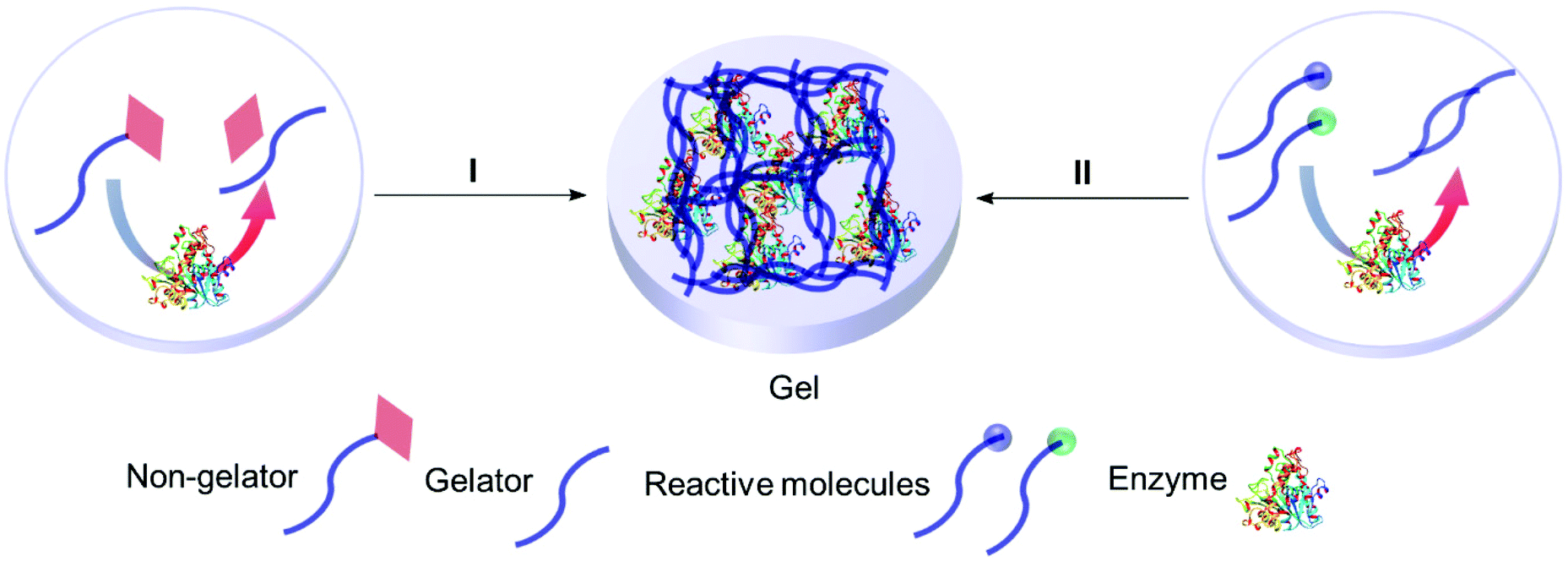 | ||
| Fig. 11 Schematic presentation of enzyme responsive gel formation. | ||
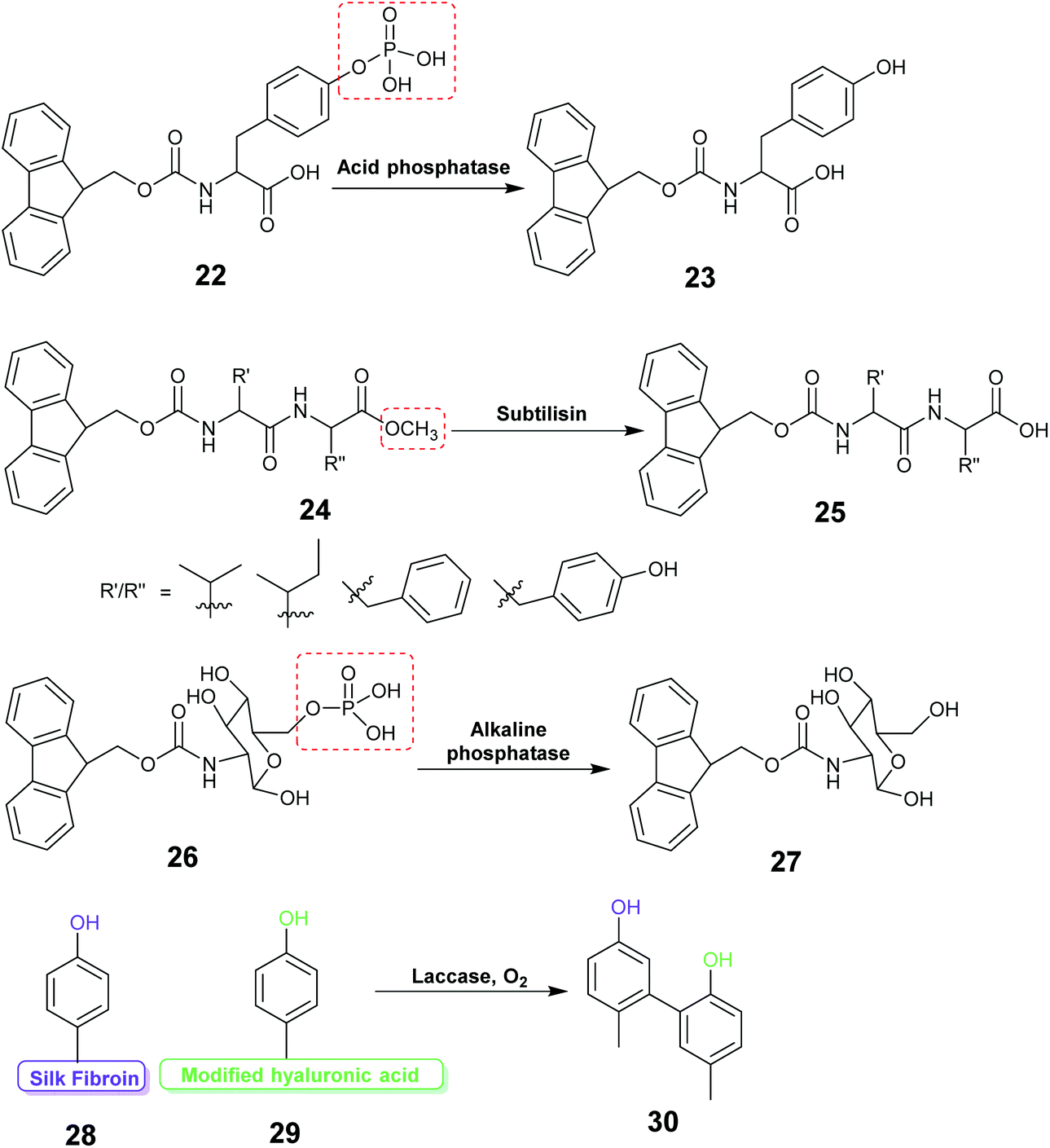 | ||
| Fig. 12 Examples of reaction schemes for enzyme assisted gel formation. | ||
Acid phosphatase (AP) was used to hydrolyze the phosphate group of the phosphate functionalized molecule 22 and liberate the gelator 23. This initiated hydrogelation, and the enzyme was entrapped within the hydrogel.171 The immobilized AP retained 75% activity compared with the free enzyme in the hydrolysis of p-nitrophenyl phosphate (pNPP) in water. The immobilized enzyme exhibited 100 times enhanced hydrolytic activity in chloroform compared to water. CD spectroscopy verified the structural integrity of the immobilized AP. Horseradish peroxidase (HRP) catalyzed the polymerization reaction of acetylacetone in the presence of hydrogen peroxide, silica nanoparticles, acrylolated Human Serum Albumin (HSA) and N,N-dimethylacrylamide (DMAA) resulting in a polymer hydrogel.172 The enzyme entrapped in the polymer hydrogel exhibited high thermal stability and recyclability in the oxidation reaction of o-phenylenediamine (OPD) in toluene and in water. Cascading enzymatic oxidations involving glucose oxidase (GOx) and horseradish peroxidase (HRP) led to co-immobilization of these enzymes into a self-assembled-polymerized hydrogel.173,174 In the cascade reaction, glucose and acetylacetone were oxidized in the presence of methacrylate PEGMA and NapFFK-acrylic acid as a LMWG. The gel showed activity for the oxidation of o-phenylenediamine (OPD) in the presence of H2O2.
Hydrolytic activity of the enzyme subtilisin was employed to convert the methyl ester protected Fmoc-dipeptide 24 to the parent acid 25, initiating the formation of a highly ordered gel structure via supramolecular assembly.175 Atomic force microscopy (AFM) revealed formation of self-assembled nano and micro structure near the enzyme active site forming a catalytic cluster within the gel. The enzyme content was found to affect the gel morphology.
Enzyme responsive gel formation has been demonstrated in vivo. Alkaline phosphatase induced dephosphorylation converted the carbohydrate based precursor 26 into the hydro-gelator 27.176 The process was investigated in vivo in the osteosarcoma cell line, SaOs2. The gelation depended on the alkaline phosphatase concentration thus allowing cell selective gelation. Recently the enzyme laccase was used to catalyze the coupling reaction between the tyrosine of silk fibroin (SF) protein and tyramine modified hyaluronic acid to form a highly elastic and mechanically stable SF hydrogel for drug delivery (28, 29, 30).177
Gels that form due to the activity of enzymes are an impressive development. These highly technical systems have specific applications, for example in sensors and medicine.
4.3 3D printing
Additive manufacturing, more well known as 3D printing, is a material fabrication process that enables the design of complicated bespoke 3D structures for various applications. Most often the material used as the ‘ink’ for printing is gel-like in nature. This provides opportunities for the 3D printing of enzyme-containing gels to form different shapes. This could find applications in, for example, flow biocatalysis.178β-Galactosidase (β-Gal) from Aspergillus oryzae in water-in-oil high internal phase emulsions (HIPEs) was used as a 3D printable bio-ink for enzyme entrapment in a cylinder shape hydrogel surrounded by a porous polymeric material.179 The water-in-oil emulsions were made by mixing an oil phase containing of 2-ethylhexyl acrylate (EHA), isobornyl acrylate (IBOA), trimethylolpropane triacrylate (TMPTA), surfactant Pluronic® L-121, and photo initiator (Darocur®TPO) with a water phase, made from acrylic acid (AA), poly(ethyleneglycol)diacrylate (PEG-DA700), lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) and β-galactosidase. The emulsion was molded in a 48 cylindrical well microplate and subjected to UV-LED for polymerization at 25mW cm−2 for 2 min to give a hydrogel surrounded by an outer porous polymeric material. Extensive washing was required to remove unbound material. Activity in the hydrolysis of ortho-nitrophenyl β-D-galactopyranoside (ONPG) in buffer depended on the content of the monomer and aqueous phase. The printing nozzle size influenced the activity, with a fourfold increase in specific activity observed for 110 μm nozzles, compare to 840 μm nozzles. The system suffered from mass transfer limitations and enzyme inactivation.
Laccase was immobilized within a cube shaped pore structure by 3D printing and the activity of the enzyme investigated for p-chloro phenol degradation.180 For 3D printing, the enzyme solution with or without hydroxyapatite (a calcium mineral) was added to a hydrogel solution of the biopolymer precursor sodium alginate 10 (Fig. 8) and CaCl2 with synthetic polymer (polyacrylamide) precursor 1 (Fig. 7B) and BIS 7, TEMED 9 and APS 8b. The enzyme hydrogel solution was molded into a cube shape using 3D printing. The immobilized laccase exhibited good stability, retaining over 80% of its initial activity after 72 h of storage. Conversion of the substrate dropped to 60% after 7 reuses.
3D printing holds a lot of promise, particularly for the application of industrial biocatalysis for flow reactions. This area is expected to expand rapidly.
4.4 Hybrid materials
In this review we use the term hybrid material to refer to substances that are a mixture of more than one material type. Such as an organic polymer modified with an inorganic oxide. Combining two or more different materials can afford better stability, mass transfer and reaction compatibility to the enzyme entrapped material. Many combinations can be envisaged of inorganic & organic materials, LMWGs, MOFs and biopolymers.Smith and co-workers entrapped alkaline phosphatase in a LMW gel of 31 strengthened with polymer 32 (Fig. 13). This hybrid enabled the formation of a ring shaped bioreactor.181 This was constructed by mixing the enzyme into a solution of 31 in DMSO/water at 80 °C, the hot enzyme-gelator solution was then poured into a glass mold to form the gel. The monomer for 32 and photo initiator (Irgacure 2959) were added and allowed to diffuse through the gel. Irradiation with long wavelength UV light resulted in alkaline phosphatase immobilized in the polymer reinforced LMW gel. The entrapped enzyme was active for the hydrolysis of p-nitrophenol phosphate (pNPP).
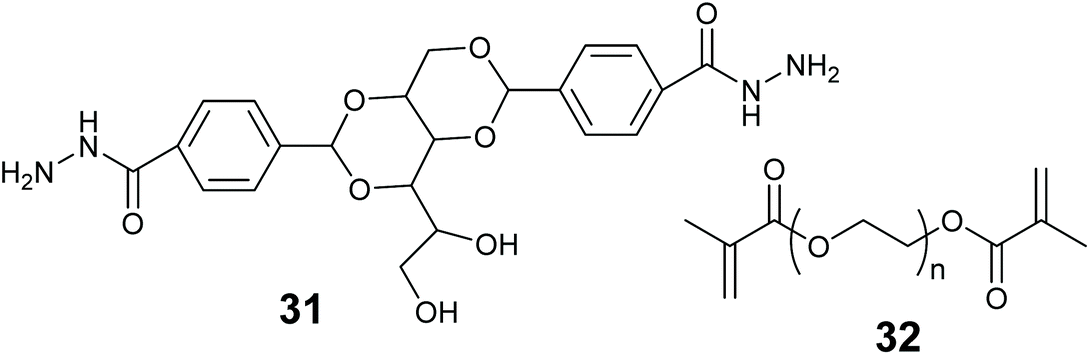 | ||
| Fig. 13 Chemical structure of the LMWG and polymer used to make a hybrid reactor for biocatalysis. | ||
Epoxide hydrolase SpEH and Bovine serum albumin (BSA) were entrapped into a hybrid inorganic -organic capsule made from Fmoc-FF 17 (Fig. 9), sodium silicate and polyethyleneimine in aqueous solution.182 The enzyme immobilized hybrid capsule was made by the dropwise addition of a homogeneous aqueous solution of enzyme, 17 and sodium silicate at pH 7–10 to an aqueous polyethyleneimine solution at pH 7.5. The enzyme capsule exhibited high enzyme loading, low enzyme leaching and high activity in the hydrolysis of cyclohexene oxide. The capsule was recycled 20 times and retained 87% of the initial activity.
The aluminium oxide boehmite was combined with alginate 10 (Fig. 8) to make hybrid beads which were used to immobilize yeast alcohol dehydrogenase (YADH).183 The enzyme was immobilized by mixing an enzyme solution in buffer into the boehmite sol, this was further mixed with an alginate solution. Finally, the homogeneous enzyme-boehmite-alginate solution was added dropwise to a CaCl2 solution to form beads. The immobilized YADH hybrid beads retained 7% of the activity of the free enzyme in the reduction of formaldehyde to methanol in the presence of nicotinamide adenine dinucleotide (NADH) in buffer. The low activity of the immobilized enzyme was attributed to mass transfer limitations due to the compact nature of the beads. After 12 cycles the immobilized enzyme retained 87% of its original activity without leaching.
Carbonic anhydrase (CA) was entrapped in magnetite sol–gel matrices by mixing the enzyme solution in buffer with a magnetite sol and drying for 24 h. The immobilized enzyme retained similar activity to the free enzyme for the hydrolysis of p-nitrophenyl acetate (pNPA) in buffer.184 The immobilized enzyme exhibited higher thermal stability, retaining 95% activity at 65 °C, at this temperature the free enzyme retained 55% activity. Incorporation of magnetic Fe3O4 nanoparticles during the sol–gel process facilitated the easy separation of the immobilized enzyme.109 The process enhanced the esterification activity of lauric acid with n-octanol by 200–300% and gave 97–99% ee in the kinetic resolution of 2-pentylamine with ethyl acetate in t-butylmethyl ether.
Lipase (Cal B) was immobilized in an agarose 14 (Fig. 8) hydrogel stabilized by magnetic a ZIF-8 MOF capsule.185 Cal B in the hybrid magnetic ZIF-8 MOF capsules exhibited 2.6 times higher activity compared to the Cal B in the hydrogel alone in the transesterification reaction of 1-butanol and vinyl acetate in acetone, however, with large substrates 3-(4-hydroxyphenyl)propan-1-ol and vinyl laurate, no detectable transesterification was observed for Cal B in hybrid magnetic ZIF-8 MOF capsules after 12 h, and only 7.5% conversion was observed at 48 h, whilst 100% conversion was obtained for the Cal B hydrogel at 12 h. Mass transfer limitations and capsule pore blockage were suggested as reasons for the low conversions.
Enzymes tyrosinase (Tyr) and β-glucosidase (β-glu) were co-immobilized in magnetic polydopamine-alginate beads using enzyme assisted polymerization and gelation processes.186 The enzyme immobilized hybrid beads were made by mixing a homogeneous solution of Fe3O4 nanoparticles, β-glucosidase and substrate D-(+)-gluconic acid δ-lactone into a homogeneous solution of tyrosinase, sodium alginate, substrate dopamine hydrochloride and hydroxylapatite (a calcium mineral). The polymerization and gelation process occurred as tyrosinase polymerized dopamine to poly-dopamine and β-glucosidase converted D-(+)-gluconic acid δ-lactone to gluconic acid. The gluconic acid subsequently released Ca2+ ions from the hydroxylapatite, initiating the gelation of the alginate. In order to form beads the homogeneous solution was dropped into a CaCl2 solution with excess D-(+)-gluconic acid δ-lactone. The immobilized enzyme beads were investigated for the inhibitory activity of tea polyphenol extracts using levodopa and p-nitrophenyl-β-D-glucosidase (pNPG) as substrates in buffer. The immobilized β-glucosidase and tyrosinase retained 79% and 78% activities compared to 54% and 46% for the free enzymes, respectively, after 35 days at 4 °C.
Methods that require relatively few synthetic steps and create hybrid materials with desirable properties promise to widen the potential applications of entrapped enzymes, by creating materials with physical properties that can be tuned and fitted to suit process parameters.
5 Ionic liquid materials for enzyme entrapment
5.1 A brief introduction to ionic liquids
Ionic liquids are a class of liquid comprised entirely of positive and negative ions held together by electrostatic interactions. The term ionic liquid (IL) is usually reserved for ionic materials that are liquid under mild to moderate conditions. ILs are a diverse class of materials, but research in the public domain has mainly concentrated on larger cations with significant organic groups (imidazolium, pyrrolidinium, ammonium, phosphonium) and relatively small anions (halide, trifate, sulfonate, acetate). The diversity of ILs provides opportunities to manipulate chemical and physical properties and create combinations that are significantly different from common salts and existing solvents.187,188 This tuning of the ionic liquid's properties makes ILs unique liquids for enzymes and biocatalysis, and the low volatility and flammability of ILs render them safer and less likely to damage biomaterials.35,37,38,189–195Enzymes tend to degrade when stored in conventional aqueous and organic solvents, potentially limiting their widespread industrial applications in chemical synthesis. The reuse and recovery of biocatalysts is particularly challenging. ILs can be tuned to support protein stability by manipulating properties such as hydrophobicity, electrostatic properties, H-bonding, and Brønsted acidity/basicity, thus offering a bespoke environment for a given enzyme, and supporting structural folding and catalytic activity.193 As the protein-stabilizing properties of ionic liquids have become apparent, the entrapment of enzymes within IL materials has begun. A green and sustainable enzyme entrapment method should retain the enzyme activity for a long time and render the enzyme re-usable without leaching of the enzyme nor the entrapment material to the reaction medium. The principle of entrapment in an ionic liquid material (Fig. 14) is that, by tuning the solubility of the IL, the passage of chemicals into and out of the gel can be controlled, in order to protect the catalyst from harsh reaction conditions.197 The ionic liquid keeps the enzyme in a liquid environment that promotes the right protein conformation for catalytic activity.63,161,162,196 Enzyme entrapment in an IL gel can thus act to encapsulate and store enzymes in their active form. Key to achieving these beneficial effects is the understanding of an enzyme's dissolution, stability and structure–activity relationships within ILs.
 | ||
| Fig. 14 Schematic presentation of an ionic liquid gel. The matrix exterior protects the enzyme and the ionic liquid supports enzyme activity. | ||
5.2 Enzymes in ionic liquids
A large variety of enzymes and ionic liquids exist, thus offering many opportunities to manipulate an enzyme-IL pair to maximize enzyme solubility and stability.Judicious choice of the ionic liquid can promote refolding of aggregated proteins. For example aggregated Concanavalin A (ConA) was shown to be resoluble in a hydrophobic phosphonium ionic liquid with a long alkyl chain (C16), and dihydrogen phosphate (dhp) anion at low water content.211 The intrinsically disordered protein Anti-Sigma factor (FlgM), folded in an aqueous solution of the hydrophobic IL 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)amide ([Bmpy][NTf2] Fig. 15, 39).212 The IL [Bmpy][NTf2] was shown to increase the alpha helix content of the protein.
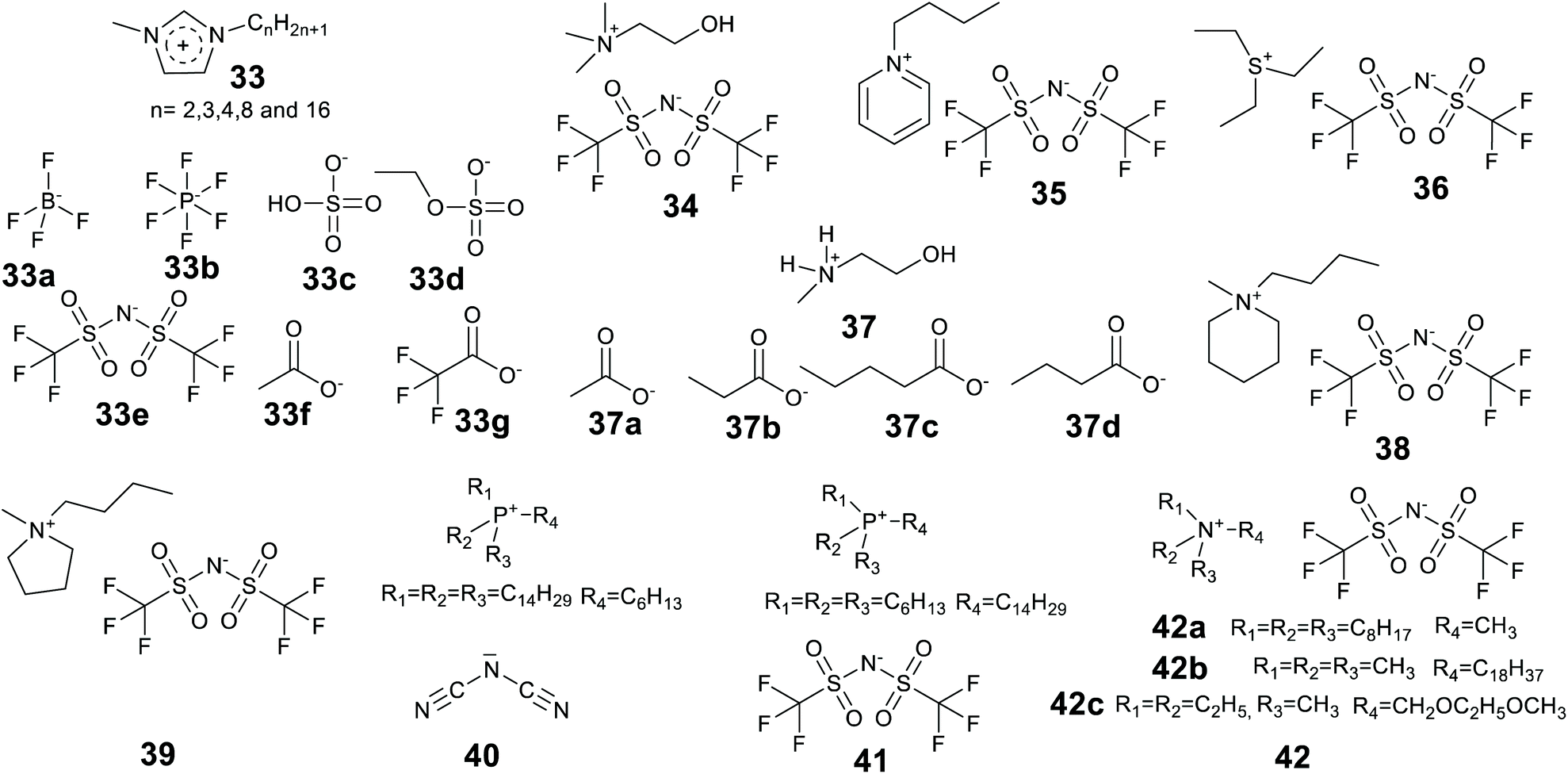 | ||
| Fig. 15 The chemical structure of some ionic liquids used in enzyme research. | ||
Recently ionic liquid research has shifted towards the design and application of biomass derived, less toxic, and environmentally begin ionic liquids. Many of these ILs will have good enzyme compatibility. Of major interest are IL cations derived from choline and lactam, and anions derived from amino acids and fatty acids. Protic ionic liquids are enjoying a come-back, as they can be derived from inexpensive, bulk chemicals, preferably from renewable resources, and are easier to make.213–215 Cholinium [Ch] (cation of Fig. 15, 34) ionic liquids can be biodegradable, non-toxic and aqueous water/buffer soluble.216,217 Cholinium ILs with different anions, for example amino acids, are emerging as a green and sustainable ionic liquid media for enzyme/protein stability and activity studies.204,217–219,220,221 Fatty acid222–225 and lactam226,227 based ionic liquids are also likely to significantly contribute towards the library of greener ILs for future industrial biocatalytic processes.
Proteins can be rendered more soluble in ionic liquids by manipulating the surface charges. The protein modification can be achieved by covalently bonding charged or neutral molecules onto the protein. For example polyether carboxylic acid and amine modifiers have been used to significantly enhance the activity of myoglobin dissolved in neat anhydrous ILs, thermal stability was also enhanced.228 In addition to covalent modification, genetic manipulation by site directed mutagenesis can also be used.
5.3 Co-entrapment of enzymes and ionic liquids
Ionic liquids have been found to assist the formation of gels for enzyme entrapment (Table 2). Methods were briefly reviewed in 2016,91 here we provide an updated and more comprehensive treatment of the progress of enzyme entrapment in ionic liquid gels.| Method | Matrix (catalyst) | Ionic liquid(s) | Enzyme | Test reaction | Ref. |
|---|---|---|---|---|---|
| Ionic liquids: Cations: [EMIM] 1-ethyl-3-methylimidazolium, [PMIM] 1-propyl-3-methyl-imidazolium, [BMIM] 1-butyl-3-methyl-imidazolium, [HMIM] 1-hexyl-3-methyl-imidazolium, [OMIM] 1-octyl-3-methylimidazolium, [HDMIM]1-hexadecyl-3-methylimidazolium, [N-MMEA] N-methylmonoethanol amine, [Ch] Cholinium, [N8,8,8,1] trioctylmethylammonium, [P6,6,6,14] trihexyltetradecylammonium, [P14,14,14,6] tritetradecylhexylammonium, [EPY] N-ethylpyridinium, [BMPI] 1-butyl-1-methylpiperidinium, [S2,2,2] triethylsulfonium. Anions: [BF4] tetrafluoroborate, [PF6] hexafluorophosphate, [NTf2] bis[(trifluoromethyl)sulfonyl]amide, [COOCH3] acetate, [COOC2H5] propionate, [COOC3H7] butyrate, [COOC4H9] pentanoate, [COOCF3] trifluoroacetate, [dca] dicyanamide, [HSO4] hydrogen sulphate, [EtSO4] ethyl sulfate, [Cl] chloride. | |||||
| Sol–gel | Silica (HCl) | [BMIM][BF4] | Peroxidase (Horseradish, HRP) | Oxidation | 236 and 237 |
| Sol–gel | Silica (HCl) | [EMIM][BF4], [EMIM][PF6], [OMIM][PF6], [EMIM][NTf2], [HDMIM][NTf2], [HDMIM][BF4], [HDMIM][Cl] | Lipase (Candida rugosa) | Hydrolysis /esterification | 243 |
| Sol–gel | Silica (HCl) | [EMIM][BF4], [OMIM][BF4], [BMIM][PF6], [OMIM][PF6], [EMIM] [NTf2], [OMIM][NTf2] | Lipase (Candida rugosa) | Hydrolysis /esterification | 238 |
| Sol–gel | Silica (NaF) | [EMIM][BF4], [EMIM][COOCH3], [EMIM][COOCF3], [PMIM][BF4], [BMIM][BF4], [HMIM][BF4], [BMIM][PF6], [BMIM][NTf2], [OMIM][BF4] | Lipase (Pseudomonas fluorescen) | Acylation | 239 |
| Sol–gel | Silica/glycerol (HCl) | Alkyl, hydroxy functionalized imidazolium ILs | Naringinase | Deglycosylation | 240 |
| Sol–gel | Silica (HCl) | [N-MMEA][COOCH3] | Lipase (Bacillus sp. ITP-001) (B. cepacia) | Hydrolysis | 244 and 245 |
| [N-MMEA][COOC2H5] | |||||
| [N-MMEA][COOC3H7] | |||||
| [N-MMEA][COOC4H9] | |||||
| Self-assembly silica coating | Silica (lipase) | [BMIM][PF6], [BMIM][BF4] | Lipase (Pseudomonas fuorescens) | Esterification | 241 and 242 |
| Entrapment polymer-IL gel | Polymer inclusion ionic liquid membranes/polyvinylchloride | [OMIM][NTf2], [OMIM][PF6], [EMIM][NTf2], [EPY][NTf2], [Ch][NTf2], [S2,2,2][NTf2], [BMIM][NTf2], [BMPI][NTf2], [N8,8,8,1][NTf2], [P6,6,6,14][NTf2], [P14,14,14,6][dca] | Laccases (Trametes versicolor) | oxidation | 246 |
| Entrapment polymer-IL gel | N-Iso-propylacrylamide/N,N0-methylene-bis(acrylamide)/[bis(n-5-cyclopentandienyl) iron]/dimethoxy-phenylacetophenone | [EMIM][EtSO4] | Lactate oxidase | oxidation | 247 |
| Microemulsion-based | Hydroxypropyl-methyl cellulose HPMC | [BMIM][PF6] | Lipase B (Candida Antarctica lipase B) | Esterification | 248 |
Lee and Kim introduced the technique of coating enzymes in ionic liquids. Lipase from Pseudomonas cepacia was coated in 1-(3′-phenylpropyl)-3-methylimidazolium hexafluorophosphate [PPMIM][PF6] ionic liquid for the preparation of acetate esters of secondary alcohols in toluene at 25 °C.230 This ionic liquid is solid at room temperature and becomes liquid at 53 °C. Enzyme powder was added to the liquid form of the ionic liquid and mixed to form a homogeneous solution and then the ionic liquid was allowed to solidify at room temperature. The ionic liquid coated enzymes were active and exhibited higher enantioselectivity than the unmodified enzymes. In addition, the ionic liquid enzymes were recycled 5 times maintaining 93% of activity of the native enzyme in the 5th recycle. The technique of coating has been significantly developed since, and the review of Itoh and the references therein provide a detailed story.37 Landmarks include the considerable enhancements of lipase performance, stability and recycling discovered by Itoh et al.231,232 Recently Itoh and co-workers demonstrated complete retention of lipase activity for 2 years when stored coated in the IL [N221MEM][NTf2] (Fig. 15, 42c).233
Lozano et al. developed a sponge-like hydrophobic ionic liquid with a long alkyl chain, namely N-octadecyl-N,N,N-trimethylammoniumbis(trifluoromethylsulfonyl)amide [C18tma][NTf2] (42b).234 This ionic liquid is solid and sponge-like at room temperature and forms a liquid at 60 °C. At 60 °C the liquid phase was used as the reaction medium for the production of biodiesel by lipase-catalyzed methanolysis of triolein with 100% yield at 8 h. The reaction mixture was then cooled and fractionated by centrifugation to separate three phases, the solid IL, the glycerol by-product and pure biodiesel. The enzyme could be fully recovered and reused. The system offered high enzyme stability with a half-life time of 1370 days at 60 °C.
These literature examples illustrate the potential of ionic liquids to facilitate the recycling and support the stable and enhanced activity of isolated enzyme biocatalysts.
Aqueous horseradish peroxidase (HRP) in buffer was immobilized in a 1-butyl-3-methylimidazolium tetrafluoroborate [BMIM][BF4] silica gel.236 The IL doped silica gel was 30 times more active for the oxidation of guaiacol in buffer in the presence of H2O2, at ambient temperature, compare to a silica gel without IL. It was noted that the IL doped silica gel also exhibited good thermally stability. It was suggested that the IL played a dual role as template for the silica matrix and protector of the enzyme structure. The same IL doped silica gel technology was later used in a biosensor.237 Lipase from Candida rugosa in buffer was entrapped in a TEOS-derived silica gel with various imidazolium ILs.238 The activities of the IL silica gels was investigated in the hydrolysis of p-nitrophenyl butyrate operated in buffer and the esterification of benzyl alcohol and benzyl acetate in water saturated hexane. The 1-methyl-3-octylimidazolium bis[trifluoromethylsulfonyl]amide ([OMIM][NTf2]) doped gels exhibited the best performance with 5 times higher hydrolytic activity and 16 times higher esterification activity compared to the silica gel without IL. The gel retained 80% of its initial activity after incubation at 50 °C for 5 days.
Lipase from Pseudomonas fluorescens was immobilized in a silica gel using different silane sol–gel precursors and various imidazolium based ILs as co-dopants.239 The gels were made two ways. In the first method silane precursors were added to a pre-mixed solution of lipase in buffer, IL, iPrOH and NaF. In the second approach silane precursors in EtOH were added to the lipase in buffer and IL solution. Gels made up from 1:1 octyl-tetramethoxysilane:tetramethoxysilane (OcTMOS:TMOS) silane precursors and [OMIM][BF4] exhibited the best performance in the acylation of 2-heptanol and vinyl acetate in hexane, however the specific activity was low. Naringinase from Penicilliun decumbens in buffer was immobilized in a silica gel (from TMOS) in the presence of different alkyl, and hydroxy functionalized alkyl imidazolium ILs with glycerol.240 The IL [OMIM][NTf2] and glycerol doped silica gel exhibited better recyclability in p-nitrophenyl α-L-rhamnopyranoside (pNRP) hydrolysis in buffer than a similar gel without IL. Bhargava and co-workers studied the self-assembly of the enzymes phytase and lipase in ionic liquids [BMIM][BF4] and [BMIM][PF6].241,242 In the ionic liquids the enzymes self-assembled to form enzyme capsules. The phytase capsule was added to TEOS and hydrolysis formed hollow and dense silica nano-structures. Two different orientations of the enzyme with the IL were observed by SEM and TEM analysis. In [BMIM][PF6] the enzyme active site was directed towards the hydrophilic part of the IL resulting in a hollow silica structure, whereas in [BMIM][BF4] the enzyme active site self-assembled inward to form a solid silica structure. The hollow silica structure retained enzyme activity and was reusable for four runs, whereas the solid silica structure was inactive.
Ionic liquid containing polymer gels are often termed ‘ionogels’. Eleven ionic liquids were separately co-entrapped with Laccase from Trametes versicolor in polymer gels.246 The gel was made by mixing an IL containing solution of the enzyme in buffer with a homogeneous solution of IL and polyvinyl chloride (PVC). [Ch][NTf2] was noted as the IL that supported the best performance in the oxidation of ABTS in buffer. This in an interesting example of the use of ionic liquids in the creation of immobilized enzymes, in this case the abilities of ILs to stabilize the protein and solubilize the polymer were exploited. A solid forming event did occur in the presence of ionic liquid and enzyme, but it is unclear whether it represents a true entrapment of the enzyme. Khodagholy et al., reported the entrapment of lactate oxidase (LOx) (for lactate sensing) in a polymer ionogel.247 The ionogel solution was made by mixing the ionic liquid 1-ethyl-3-methylimidazolium ethyl-sulfate ionic liquid, [EMIM][EtSO4], with monomer 2, crosslinker 7 (Fig. 7B), the metal complex [bis(n-5-cyclopentandienyl)iron] and photoinitiator dimethoxy-phenylacetophenone (DMPA). The resultant mixture was sonicated at 45 °C for 10 min to make a homogeneous solution. The enzyme in buffer was added to the ionogel solution, and a small amount of the enzyme-ionogel solution poured onto a dimethylsiloxane (PDMS) modified well of the sensor device. Photo-irradiation using UV-light at 365 nm with an intensity of approximately 330mW cm−2 for 1 min resulted in an enzyme ionogel. The device was able to detect lactate at the 10–100 mM concentration range.
The addition of ionic liquids to the sol–gel entrapment of enzymes has been shown to afford significant advantages compared to the entrapment in silica alone. The same protein stabilization is expected in the entrapment of enzymes in polymer ionogels, but the effects have been less explored. In this area the right balance must be struck between sufficient porosity to maximize substrate and product mass transfer, and good mechanical stability to promote ease of handling and minimize leaching.
 | ||
| Fig. 16 Chemical structure of some LMW ionic liquid gelators. 43,24944,250,25145,25246,25347,25448,25549,25650,25751,25852,25953.151 | ||
The best LMWGs are able to gel a wide range of ILs.151 Polymer poly(methyl methacrylate) (PMMA) coating of a LMW gel of 50 gel has been used to increase the mechanical strength.258
Supramolecular methods for co-entrapping enzymes and ionic liquids are comparatively rare, but the method shows considerable promise. An aqueous solution of Lipase B from Candida Antarctica and lipase from Chromobacterium viscosum in buffer was immobilized as a microemulsion with [BMIM][PF6] in a hydroxypropyl-methyl cellulose (HPMC) organogel.248 Immobilized lipase from Chromobacterium viscosum exhibited a >4 times higher activity than the enzyme operated in water emulsion for the esterification of lauric acid with 1-butanol in isooctane at 30 °C. The immobilized lipase retained 60% of its initial activity when stored in isooctane at 50 °C for 5 days, under the same storage conditions the free enzymes retained only 5% activity.
Even though a large number LWMGs have now been developed for ionic liquids, enzyme immobilization in LMW ionic liquid gels is largely unexplored, and this area is expected to expand significantly in the next few years. Key to the success of these materials will be the straightforward creation of robust materials with good mechanical stability and low leaching. This will require careful tuning of the supramolecular interactions, and the wide range of gelators and ILs available will be a significant advantage.
6. Conclusions and future directions
The use of isolated enzymes for the synthesis of chemicals is on the rapid increase. Isolated enzymes are relatively fragile, and in order to improve separation and ensure recycling enzyme immobilization should be encouraged. To be fit for purpose methods must complement the advances in biotechnology and be readily adapted in response to changes in the protein and/or bioprocess conditions.When adopting an immobilization method, a company must consider cost, reproducibility, and chemical regulations. As a result, long and complex methods of immobilization will not be adopted by biocatalysis producers nor end users. Simplicity and reliability are key to success. Such simplicity also lends itself to a better life cycle analysis (LCA) and represents greener chemistry.
The entrapment of an isolated enzyme in a support material has the potential to deliver adaptability, simplicity and reliability. Entrapment can be as simple as carrying out a polymerization in the presence of an enzyme or adding a low molecular weight gelator to an enzyme solution with heating and cooling. Entrapment does not depend on any direct interactions with the enzyme to achieve immobilization. The environment within the matrix can be changed to support enzyme activity, and changes in the protein within an enzyme library are unlikely to render an entrapment method obsolete.
As befits a successful immobilization strategy, and as demonstrated by the best chemical methods, entrapped enzymes have frequently demonstrated good enzyme activity, with enhanced thermal and pH stability, and there are examples in which the activity within the matrix is better than the activity of the free enzyme. Enzyme entrapment can be considered under-utilized in industry, and this can be attributed to doubts over simplicity and reliability. Moving forward researchers must consider diffusion, leaching and recycling as primary design criteria. Academia must work closely with industry to address the concerns of end users, and entrapment methods must be demonstrated under industrial conditions. Such collaboration will lead to greater adoption of enzyme entrapment for chemical manufacture and subsequent improvements in enzyme catalyzed bioprocesses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Professor Thomas Moody and Dr Stefan Mix of Almac Sciences and Dr Sebastian Cosgrove of Keele University, UK. This work is supported by Engineering and Physical Sciences Research Council (UK) grants EP/R026645/1, The UK Catalysis Hub ‘Science’: 2 Catalysis at the Water-Energy Nexus, and EP/M013219/1, Biocatalysis & Biotransformation: A 5th Theme for the National Catalysis Hub.References
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- P. T. Anastas, L. G. Heine and T. C. Williamson, Green Chemical Syntheses and Processes, ACS Symposium Series 767; American Chemical Society: Washington, DC, 2000, pp. 1—6 Search PubMed.
- R. A. Sheldon and J. P. M. Sanders, Catal. Today, 2015, 239, 3–6 CrossRef CAS.
- P. K. J. Kinnunen, Cell. Physiol. Biochem., 2000, 10, 243–250 CrossRef CAS PubMed.
- S. Martínez Cuesta, S. A. Rahman, N. Furnham and J. M. Thornton, Biophys. J., 2015, 109, 1082–1086 CrossRef PubMed.
- J. Boonstra and A. J. Verkleij, Adv. Enzyme Regul., 2004, 44, 61–73 CrossRef CAS PubMed.
- R. T. Schimke and D. Doyle, Annu. Rev. Biochem., 1970, 39, 929–976 CrossRef CAS PubMed.
- D. E. Atkinson, Annu. Rev. Microbiol., 1969, 23, 47–68 CrossRef CAS PubMed.
- E. Romero, B. S. Jones, B. N. Hogg, A. Rué Casamajo, M. A. Hayes, S. L. Flitsch, N. J. Turner and C. Schnepel, Angew. Chem., Int. Ed., 2021, 2–34 Search PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., 2021, 60, 88–119 CrossRef CAS PubMed.
- B. Hauer, ACS Catal., 2020, 10, 8418–8427 CrossRef CAS.
- R. A. Sheldon and D. Brady, Chem. Commun., 2018, 54, 6088–6104 RSC.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., 2021, 60, 88–119 CrossRef CAS PubMed.
- J. M. Woodley, Curr. Opin. Green Sustain. Chem., 2020, 21, 22–26 CrossRef.
- R. A. Sheldon, Curr. Opin. Green Sustain. Chem., 2019, 18, 13–19 CrossRef.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- J. M. Choi, S. S. Han and H. S. Kim, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef CAS PubMed.
- V. G. Yadav, M. De Mey, C. G. Lim, P. K. Ajikumar and G. Stephanopoulos, Metab. Eng., 2012, 14, 233–241 CrossRef CAS PubMed.
- T. Tanaka and A. Kondo, Biotechnol. Adv., 2015, 33, 1403–1411 CrossRef CAS PubMed.
- T. Hudlicky and J. W. Reed, Chem. Soc. Rev., 2009, 38, 3117–3132 RSC.
- B. R. Facin, M. S. Melchiors, A. Valério, J. V. Oliveira and D. De Oliveira, Ind. Eng. Chem. Res., 2019, 58, 5358–5378 CrossRef CAS.
- S. Naik, A. Basu, R. Saikia, B. Madan, P. Paul, R. Chaterjee, J. Brask and A. Svendsen, J. Mol. Catal. B: Enzym., 2010, 65, 18–23 CrossRef CAS.
- A. C. Carvalho, T. de S. Fonseca, M. C. de Mattos, M. da C. de Oliveira, T. L. de Lemos, F. Molinari, D. Romano and I. Serra, Int. J. Mol. Sci., 2015, 16, 29682–29716 CrossRef PubMed.
- N. Sarmah, D. Revathi, G. Sheelu, K. Yamuna Rani, S. Sridhar, V. Mehtab and C. Sumana, Biotechnol. Prog., 2018, 34, 5–28 CrossRef CAS PubMed.
- G. M. Borrelli and D. Trono, Int. J. Mol. Sci., 2015, 16, 20774–20840 CrossRef CAS PubMed.
- F. J. Contesini, M. G. Davanço, G. P. Borin, K. G. Vanegas, J. P. G. Cirino, R. R. de Melo, U. H. Mortensen, K. Hildén, D. R. Campos and P. de O. Carvalho, Catalysts, 2020, 10, 1–33 CrossRef.
- K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203–213 CrossRef CAS.
- B. A. Sandoval and T. K. Hyster, Curr. Opin. Chem. Biol., 2020, 55, 45–51 CrossRef CAS PubMed.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed.
- J. M. Woodley, Curr. Opin. Chem. Biol., 2013, 17, 310–316 CrossRef CAS PubMed.
- H. Kries, R. Blomberg and D. Hilvert, Curr. Opin. Chem. Biol., 2013, 17, 221–228 CrossRef CAS PubMed.
- N. J. Turner and M. D. Truppo, Curr. Opin. Chem. Biol., 2013, 17, 212–214 CrossRef CAS PubMed.
- A. A. M. Elgharbawy, M. Moniruzzaman and M. Goto, Biochem. Eng. J., 2020, 154, 107426 CrossRef CAS.
- M. Pätzold, S. Siebenhaller, S. Kara, A. Liese, C. Syldatk and D. Holtmann, Trends Biotechnol., 2019, 37, 943–959 CrossRef PubMed.
- T. Itoh, Chem. Rev., 2017, 117, 10567–10607 CrossRef CAS PubMed.
- P. Lozano, E. Alvarez, J. M. Bernal, S. Nieto, C. Gomez and G. Sanchez-Gomez, Curr. Green Chem., 2017, 4, 116–129 CAS.
- M. Romero-Fernández and F. Paradisi, Curr. Opin. Chem. Biol., 2020, 55, 1–8 CrossRef PubMed.
- R. C. Alnoch, L. A. Dos Santos, J. M. de Almeida, N. Krieger and C. Mateo, Catalysts, 2020, 10, 1–29 CrossRef.
- J. Liang and K. Liang, Adv. Funct. Mater., 2020, 30, 1–24 Search PubMed.
- J. Liu, R. T. Ma and Y. P. Shi, Anal. Chim. Acta, 2020, 1101, 9–22 CrossRef CAS PubMed.
- D. G. Filho, A. G. Silva and C. Z. Guidini, Appl. Microbiol. Biotechnol., 2019, 103, 7399–7423 CrossRef PubMed.
- B. Thangaraj and P. R. Solomon, ChemBioEng Rev., 2019, 6, 157–166 CrossRef CAS.
- G. Cottone, S. Giu, S. Bettati, S. Bruno, B. Campanini, M. Marchetti, S. Abbruzzetti, C. Viappiani, A. Cupane, A. Mozzarelli and L. Ronda, Catalysts, 2019, 9, 1–29 CrossRef.
- M. Bilal and H. M. N. Iqbal, Int. J. Biol. Macromol., 2019, 130, 462–482 CrossRef CAS PubMed.
- E. P. Cipolatti, A. Valério, R. O. Henriques, D. E. Moritz, J. L. Ninow, D. M. G. Freire, E. A. Manoel, R. Fernandez-Lafuente and D. De Oliveira, RSC Adv., 2016, 6, 104675–104692 RSC.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- D. Avnir, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013–1030 RSC.
- D. P. Gavin, F. J. Reen, J. Rocha-Martin, I. Abreu-Castilla, D. F. Woods, A. M. Foley, P. A. Sánchez-Murcia, M. Schwarz, P. O'Neill, A. R. Maguire and F. O'Gara, Sci. Rep., 2019, 9, 1–15 Search PubMed.
- M. Ferrer, F. Martínez-Abarca and P. N. Golyshin, Curr. Opin. Biotechnol., 2005, 16, 588–593 CrossRef CAS PubMed.
- J. C. Moore, A. Rodriguez-Granillo, A. Crespo, S. Govindarajan, M. Welch, K. Hiraga, K. Lexa, N. Marshall and M. D. Truppo, ACS Synth. Biol., 2018, 7, 1730–1741 CrossRef CAS PubMed.
- O. F. Brandenberg, R. Fasan and F. H. Arnold, Curr. Opin. Biotechnol., 2017, 47, 102–111 CrossRef CAS PubMed.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G. D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- W. Finnigan, L. J. Hepworth, S. L. Flitsch and N. J. Turner, Nat. Catal., 2021, 4, 98–104 CrossRef CAS PubMed.
- D. Rother and S. Malzacher, Nat. Catal., 2021, 4, 92–93 CrossRef CAS.
- J. Planas-Iglesias, S. M. Marques, G. P. Pinto, M. Musil, J. Stourac, J. Damborsky and D. Bednar, Biotechnol. Adv., 2021, 47, 107696–107718 CrossRef CAS PubMed.
- H. A. Bunzel, J. L. R. Anderson and A. J. Mulholland, Curr. Opin. Struct. Biol., 2021, 67, 212–218 CrossRef CAS PubMed.
- M. Ali, H. M. Ishqi and Q. Husain, Biotechnol. Bioeng., 2020, 117, 1877–1894 CrossRef CAS PubMed.
- S. Basler, S. Studer, Y. Zou, T. Mori, Y. Ota, A. Camus, H. A. Bunzel, R. C. Helgeson, K. N. Houk, G. Jiménez-Osés and D. Hilvert, Nat. Chem., 2021, 13, 231–235 CrossRef CAS PubMed.
- M. Kaushik, P. Sinha, P. Jaiswal, S. Mahendru, K. Roy and S. Kukreti, J. Mol. Recognit., 2016, 29, 499–503 CrossRef CAS PubMed.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- R. Di Cosimo, J. Mc Auliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- C. Ottone, O. Romero, C. Aburto, A. Illanes and L. Wilson, Compr. Rev. Food Sci. Food Saf., 2020, 19, 595–621 CrossRef CAS PubMed.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- J. Luo, S. Song, H. Zhang, H. Zhang, J. Zhang and Y. Wan, Eng. Life Sci., 2020, 20, 441–450 CrossRef CAS PubMed.
- X. Wang, P. C. Lan and S. Ma, ACS Cent. Sci., 2020, 6, 1497–1506 CrossRef PubMed.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS.
- A. C. Pierre, Biocatal. Biotransform., 2004, 22, 145–170 CrossRef CAS.
- T. Jesionowski, J. Zdarta and B. Krajewska, Adsorption, 2014, 20, 801–821 CrossRef CAS.
- Y. Hu, L. Dai, D. Liu and W. Du, Green Chem., 2018, 20, 4500–4506 RSC.
- L. S. Wong, F. Khan and J. Micklefield, Chem. Rev., 2009, 109, 4025–4053 CrossRef CAS PubMed.
- S. Smith, K. Goodge, M. Delaney, A. Struzyk, N. Tansey and M. Frey, Nanomaterials, 2020, 10, 1–39 CrossRef PubMed.
- J. S. Weltz, D. F. Kienle, D. K. Schwartz and J. L. Kaar, ACS Catal., 2019, 9, 4992–5001 CrossRef CAS.
- J. S. Weltz, D. F. Kienle, D. K. Schwartz and J. L. Kaar, J. Am. Chem. Soc., 2020, 142, 3463–3471 CrossRef CAS PubMed.
- O. Singh, P. Y. Lee, S. Matysiak and H. Bermudez, Phys. Chem. Chem. Phys., 2020, 22, 19779–19786 RSC.
- T. Akkas, A. Zakharyuta, A. Taralp and C. W. Ow-Yang, Enzyme Microb. Technol., 2020, 132, 109390 CrossRef CAS PubMed.
- R. A. Sheldon, Biochem. Soc. Trans., 2007, 35, 1583–1587 CrossRef CAS PubMed.
- H. Yamaguchi, Y. Kiyota and M. Miyazaki, Catalysts, 2018, 8, 4–6 CrossRef.
- R. A. Sheldon, Pharm. Process Chem., 2010, 159–181 Search PubMed.
- N. L. S. Clair and M. A. Navia, J. Am. Chem. Soc., 1992, 114, 7314–7316 CrossRef.
- L. Cao, F. Van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS PubMed.
- S. Wang, D. Zheng, L. Yin and F. Wang, Enzyme Microb. Technol., 2017, 107, 22–31 CrossRef CAS PubMed.
- M. Q. Xu, S. S. Wang, L. N. Li, J. Gao and Y. W. Zhang, Catalysts, 2018, 8, 1–20 Search PubMed.
- R. A. Sheldon, Catalysts, 2019, 9, 1–31 CrossRef.
- R. A. Sheldon, A. Basso and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC.
- S. van Pelt, S. Quignard, D. Kubáč, D. Y. Sorokin, F. van Rantwijk and R. A. Sheldon, Green Chem., 2008, 10, 395–340 RSC.
- P. Tielmann, H. Kierkels, A. Zonta, A. Ilie and M. T. Reetz, Nanoscale, 2014, 6, 6220–6228 RSC.
- A. C. Marr and P. C. Marr, Dalton Trans., 2011, 40, 20–26 RSC.
- P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC.
- S. Braun, S. Rappoport, R. Zusman, D. Avnir and M. Ottolenghi, Mater. Lett., 1990, 10, 2843–2846 CrossRef.
- D. Avnir, S. Braun, O. Lev and M. Ottolenghi, Chem. Mater., 1994, 6, 1–5 CrossRef.
- D. Avnir, S. Braun, O. Lev and M. Ottolenghi, Chem. Mater., 1994, 6, 1605–1614 CrossRef CAS.
- Y. Tatsu, K. Yamashita, M. Yamaguchi, S. Yamamura, H. Yamamoto and S. Yoshikawa, Chem. Lett., 1992, 1615–1618 CrossRef CAS.
- S. A. Yamanaka, L. M. Ellerbv, C. R. Nishida, J. I. Zink, F. Nishida, B. Dunn, S. J. Valentine and J. I. Zink, Chem. Mater., 1992, 4, 495–497 CrossRef CAS.
- S. Braun, S. Shtelzer, S. Rappoport, D. Avnir and M. Ottolenghi, J. Non-Cryst. Solids, 1992, 147–148, 739–743 CrossRef CAS.
- S. Shtelzer, S. Rappoport, D. Avnir, M. Ottolenghi and S. Braun, Biotechnol. Appl. Biochem., 1992, 15, 227–235 CAS.
- Y. Kurokawa, H. Ohta, M. Okubo and M. Takahashi, Carbohydr. Polym., 1994, 23, 1–4 CrossRef CAS.
- E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Soc. Rev., 2010, 39, 4504–4523 RSC.
- M. T. Reetz, Curr. Opin. Chem. Biol., 2002, 6, 145–150 CrossRef CAS.
- F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC.
- D. Romano, R. Villa and F. Molinari, ChemCatChem, 2012, 4, 739–749 CrossRef CAS.
- M. T. Reetz, A. Zonta and J. Simpelkamp, Angew. Chem., Int. Ed. Engl., 1995, 34, 301–303 CrossRef CAS.
- M. T. Reetz, A. Zonta and J. Simpelkamp, Biotechnol. Bioeng., 1996, 49, 527–534 CrossRef CAS PubMed.
- M. T. Reetz, P. Tielmann, W. Wiesenhöfer, W. Könen and A. Zonta, Adv. Synth. Catal., 2003, 345, 717–728 CrossRef CAS.
- M. T. Reetz, R. Wenkel and D. Avnir, Synthesis, 2000, 781–783 CrossRef CAS.
- M. T. Reetz, Adv. Mater., 1997, 9, 943–954 CrossRef CAS.
- M. T. Reetz, A. Zonta, V. Vijayakrishnan and K. Schimossek, J. Mol. Catal. A: Chem., 1998, 134, 251–258 CrossRef CAS.
- M. T. Reetz, A. Zonta, J. Simpelkamp and W. Konen, Chem. Commun., 1996, 1397–1398 RSC.
- H. Frenkel-Mullerad and D. Avnir, J. Am. Chem. Soc., 2005, 127, 8077–8081 CrossRef CAS PubMed.
- N. A. Mohidem and H. Bin Mat, J. Sol-Gel Sci. Technol., 2012, 61, 96–103 CrossRef CAS.
- M. Päiviö and L. T. Kanerva, Process Biochem., 2013, 48, 1488–1494 CrossRef.
- S. Escobar, C. Bernal, J. M. Bolivar, B. Nidetzky, F. López-Gallego and M. Mesa, Mol. Catal., 2018, 449, 106–113 CrossRef CAS.
- Y. Kawachi, S. I. Kugimiya, H. Nakamura and K. Kato, Appl. Surf. Sci., 2014, 314, 64–70 CrossRef CAS.
- H. Z. Ma, X. W. Yu, C. Song, Q. L. Xue and B. Jiang, J. Mol. Catal. B: Enzym., 2016, 127, 76–81 CrossRef CAS.
- C. Muderrisoglu, S. Sargin and O. Yesil-Celiktas, Biotechnol. Lett., 2018, 40, 773–780 CrossRef CAS PubMed.
- Z. Liu, B. Liu, M. Zhang, J. Kong and J. Deng, Anal. Chim. Acta, 1999, 392, 135–141 CrossRef CAS.
- Z. Liu, B. Liu, J. Kong and J. Deng, Anal. Chem., 2000, 72, 4707–4712 CrossRef CAS PubMed.
- Y. Zhu, Z. Jiang, L. Zhang, J. Shi and D. Yang, Ind. Eng. Chem. Res., 2012, 51, 255–261 CrossRef CAS.
- A. Rutenberg, V. V. Vinogradov and D. Avnir, Chem. Commun., 2013, 49, 5636–5638 RSC.
- V. V. Vinogradov and D. Avnir, RSC Adv., 2015, 5, 10862–10868 RSC.
- V. V. Vinogradov, A. V. Vinogradov, V. E. Sobolev, I. P. Dudanov and V. V. Vinogradov, J. Sol-Gel Sci. Technol., 2015, 73, 501–505 CrossRef CAS.
- V. V. Vinogradov and D. Avnir, J. Mater. Chem. B, 2014, 2, 2868–2873 RSC.
- V. V. Vinogradov and D. Avnir, Sci. Rep., 2015, 5, 1–7 Search PubMed.
- O. E. Shapovalova, D. Levy, D. Avnir and V. V. Vinogradov, Colloids Surf., B, 2016, 146, 731–736 CrossRef CAS PubMed.
- Y. V. Solovev, A. Y. Prilepskii, E. F. Krivoshapkina, A. F. Fakhardo, E. A. Bryushkova, P. A. Kalikina, E. I. Koshel and V. V. Vinogradov, Sci. Rep., 2019, 9, 1–14 CAS.
- K. F. O'Driscoll, Methods Enzymol., 1976, 44, 169–183 Search PubMed.
- M. Nöth, E. Gau, F. Jung, M. D. Davari, I. El-Awaad, A. Pich and U. Schwaneberg, Green Chem., 2020, 22, 8183–8209 RSC.
- P. Bernfeld and J. Wan, Science, 1963, 142, 678–679 CrossRef CAS PubMed.
- G. Demirel, G. Özçetin, F. Şahin, H. Tümtürk, S. Aksoy and N. Hasirci, React. Funct. Polym., 2006, 66, 389–394 CrossRef CAS.
- O. Yamak, N. A. Kalkan, S. Aksoy, H. Altinok and N. Hasirci, Process Biochem., 2009, 44, 440–445 CrossRef CAS.
- S. Demirci, M. Sahiner, S. Yilmaz, E. Karadag and N. Sahiner, Biotechnol. Rep., 2020, 28, e00534 CrossRef PubMed.
- S. Demirci and N. Sahiner, Chem. Eng. J., 2021, 409, 128233 CrossRef CAS.
- S. Bao, D. Wu, T. Su, Q. Wu and Q. Wang, RSC Adv., 2015, 5, 44342–44345 RSC.
- V. Krasňan, R. Stloukal, M. Rosenberg and M. Rebroš, Appl. Microbiol. Biotechnol., 2016, 100, 2535–2553 CrossRef PubMed.
- M. Rebroš, A. Pilniková, D. Šimčíková, L. Weignerová, R. Stloukal, V. Křen and M. Rosenberg, Biocatal. Biotransform., 2013, 31, 329–334 CrossRef.
- M. Rebroš, M. Rosenberg, Z. Mlichová and Ľ. Krištofíková, Food Chem., 2007, 102, 784–787 CrossRef.
- M. Rebroš, M. Rosenberg, Z. Mlichová, L. Krištofíková and M. Paluch, Enzyme Microb. Technol., 2006, 39, 800–804 CrossRef.
- S. Kaushik, P. D. Thungon and P. Goswami, ACS Biomater. Sci. Eng., 2020, 6, 4337–4355 CrossRef CAS PubMed.
- S. Lv, Molecules, 2020, 25, 4929–4954 CrossRef CAS PubMed.
- C. S. Bezerra, C. M. G. De Farias Lemos, M. De Sousa and L. R. B. Gonçalves, J. Appl. Polym. Sci., 2015, 132, 1–15 CrossRef.
- L. M. O. Arruda and M. Vitolo, Appl. Biochem. Biotechnol., Part A, 1999, 81, 23–33 CrossRef CAS.
- E. Quiroga, C. O. Illanes, N. A. Ochoa and S. Barberis, Process Biochem., 2011, 46, 1029–1034 CrossRef CAS.
- S. Miyairi, M. Sugiura and S. Fukui, Agric. Biol. Chem., 1978, 42, 1661–1667 CAS.
- M. Demura, T. Asakura and T. Kuroo, Biosensors, 1989, 4, 361–372 CrossRef CAS.
- J. Qian, Y. Liu, H. Liu, T. Yu and J. Deng, Biosens. Bioelectron., 1997, 12, 1213–1218 CrossRef CAS.
- N. Guziewicz, A. Best, B. Perez-Ramirez and D. L. Kaplan, Biomaterials, 2011, 32, 2642–2650 CrossRef CAS PubMed.
- Y. Han, S. Yu, L. Liu, S. Zhao, T. Yang, Y. Yang, Y. Fang and S. Lv, Mol. Catal., 2018, 457, 24–32 CrossRef CAS.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- P. McNeice, Y. Zhao, J. Wang, G. F. Donnelly and P. C. Marr, Green Chem., 2017, 19, 4690–4697 RSC.
- Q. Wang, Z. Yang, L. Wang, M. Ma and B. Xu, Chem. Commun., 2007, 1032–1034 RSC.
- G. Scott, S. Roy, Y. M. Abul-Haija, S. Fleming, S. Bai and R. V. Ulijn, Langmuir, 2013, 29, 14321–14327 CrossRef CAS PubMed.
- D. Mandal, P. Choudhury, D. Sarkar and P. K. Das, Chem. Commun., 2017, 53, 7844–7847 RSC.
- R. Jain, V. K. Pal and S. Roy, Biomacromolecules, 2020, 21, 4180–4193 CrossRef CAS PubMed.
- S. Huang, X. Kou, J. Shen, G. Chen and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 8786–8798 CrossRef CAS PubMed.
- J. Mehta, N. Bhardwaj, S. K. Bhardwaj, K. H. Kim and A. Deep, Coord. Chem. Rev., 2016, 322, 30–40 CrossRef CAS.
- S. S. Nadar, L. Vaidya and V. K. Rathod, Int. J. Biol. Macromol., 2020, 149, 861–876 CrossRef CAS PubMed.
- E. Gkaniatsou, C. Sicard, R. Ricoux, J. P. Mahy, N. Steunou and C. Serre, Mater. Horiz., 2017, 4, 55–63 RSC.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS.
- F. K. Shieh, S. C. Wang, C. I. Yen, C. C. Wu, S. Dutta, L. Y. Chou, J. V. Morabito, P. Hu, M. H. Hsu, K. C. W. Wu and C. K. Tsung, J. Am. Chem. Soc., 2015, 137, 4276–4279 CrossRef CAS PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 4–11 Search PubMed.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC.
- G. Chen, X. Kou, S. Huang, L. Tong, Y. Shen, W. Zhu, F. Zhu and G. Ouyang, Angew. Chem., 2020, 132, 2889–2896 CrossRef.
- H. He, H. Han, H. Shi, Y. Tian, F. Sun, Y. Song, Q. Li and G. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 24517–24524 CrossRef CAS PubMed.
- S. S. Nadar and V. K. Rathod, Enzyme Microb. Technol., 2018, 108, 11–20 CrossRef CAS PubMed.
- W. H. Chen, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Nat. Catal., 2018, 1, 689–695 CrossRef CAS.
- D. D. Díaz, D. Kühbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427–448 RSC.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- R. J. Williams, R. J. Mart and R. V. Ulijn, Biopolymers, 2010, 94, 107–117 CrossRef CAS PubMed.
- Q. Wang, Z. Yang, Y. Gao, W. Ge, L. Wang and B. Xu, Soft Matter, 2008, 4, 550–553 RSC.
- T. Su, D. Zhang, Z. Tang, Q. Wu and Q. Wang, Chem. Commun., 2013, 49, 8033–8035 RSC.
- Q. Wei, M. Xu, C. Liao, Q. Wu, M. Liu, Y. Zhang, C. Wu, L. Cheng and Q. Wang, Chem. Sci., 2016, 7, 2748–2752 RSC.
- Q. Wei, W. Xu, M. Liu, Q. Wu, L. Cheng and Q. Wang, J. Mater. Chem. B, 2016, 4, 6302–6306 RSC.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. Van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS PubMed.
- R. A. Pires, Y. M. Abul-Haija, D. S. Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn and I. Pashkuleva, J. Am. Chem. Soc., 2015, 137, 576–579 CrossRef CAS PubMed.
- L. Wang, F. Wang, B. Xu, M. Zhou, Y. Yu, P. Wang and Q. Wang, Langmuir, 2021, 37, 478–489 CrossRef CAS PubMed.
- V. Sans, Curr. Opin. Green Sustain. Chem., 2020, 25, 100367 CrossRef.
- L. Wenger, C. P. Radtke, J. Göpper, M. Wörner and J. Hubbuch, Front. Bioeng. Biotechnol., 2020, 8, 1–17 CrossRef PubMed.
- J. Liu, X. Shen, Z. Zheng, M. Li, X. Zhu, H. Cao and C. Cui, Int. J. Biol. Macromol., 2020, 164, 518–525 CrossRef CAS PubMed.
- P. R. A. Chivers, J. A. Kelly, M. J. S. Hill and D. K. Smith, React. Chem. Eng., 2020, 5, 1112–1117 RSC.
- R. Huang, M. Wu, M. J. Goldman and Z. Li, Biotechnol. Bioeng., 2015, 112, 1092–1101 CrossRef CAS PubMed.
- Q. Ai, D. Yang, Y. Zhu and Z. Jiang, Ind. Eng. Chem. Res., 2013, 52, 14898–14905 CrossRef CAS.
- A. S. Drozdov, O. E. Shapovalova, V. Ivanovski, D. Avnir and V. V. Vinogradov, Chem. Mater., 2016, 28, 2248–2253 CrossRef CAS.
- J. Huo, J. Aguilera-Sigalat, S. El-Hankari and D. Bradshaw, Chem. Sci., 2015, 6, 1938–1943 RSC.
- H. Zhang, M. Lu, H. Jiang, Z. Y. Wu, D. D. Zhou, D. Q. Li and F. Q. Yang, Colloids Surf., B, 2020, 188, 110800 CrossRef CAS PubMed.
- Z. Lei, B. Chen, Y. M. Koo and D. R. Macfarlane, Chem. Rev., 2017, 117, 6633–6635 CrossRef PubMed.
- T. Welton, Biophys. Rev., 2018, 10, 691–706 CrossRef CAS PubMed.
- R. A. Sheldon, RSC Catal. Ser., 2014, 2014-Janua, 20–43 Search PubMed.
- R. A. Sheldon, R. M. Lau, M. J. Sorgedrager, F. van Rantwijk and K. R. Seddon, Green Chem., 2002, 4, 147–151 RSC.
- A. Kumar, M. Bisht and P. Venkatesu, Int. J. Biol. Macromol., 2017, 96, 611–651 CrossRef CAS PubMed.
- L. E. Meyer, J. von Langermann and U. Kragl, Biophys. Rev., 2018, 10, 901–910 CrossRef CAS PubMed.
- A. Schindl, M. L. Hagen, S. Muzammal, H. A. D. Gunasekera and A. K. Croft, Front. Chem., 2019, 7, 1–31 CrossRef PubMed.
- H. Zhao, Biotechnol. Adv., 2020, 45, 107638 CrossRef CAS PubMed.
- A. A. M. Elgharbawy, M. Moniruzzaman and M. Goto, Curr. Opin. Green Sustain. Chem., 2021, 27, 100406 CrossRef.
- C. Rizzo, A. Mandoli, S. Marullo and F. D'Anna, J. Org. Chem., 2019, 84, 6356–6365 CrossRef CAS PubMed.
- P. McNeice, A. Reid, H. T. Imam, C. Mcdonagh, J. D. Walby, T. J. Collins, A. C. Marr and P. C. Marr, Environ. Sci. Technol., 2020, 54, 14026–14035 CrossRef PubMed.
- A. P. S. Brogan, C. J. Clarke, A. Charalambidou, C. N. Loynachan, S. E. Norman, J. Doutch and J. P. Hallett, Mater. Horiz., 2020, 7, 820–826 RSC.
- R. M. Lau, M. J. Sorgedrager, G. Carrea, F. Van Rantwijk, F. Secundo and R. A. Sheldon, Green Chem., 2004, 6, 483–487 RSC.
- M. Bihari, T. P. Russell and D. A. Hoagland, Biomacromolecules, 2010, 11, 2944–2948 CrossRef CAS PubMed.
- S. Strassburg, H. Bermudez and D. Hoagland, Biomacromolecules, 2016, 17, 2233–2239 CrossRef CAS PubMed.
- G. A. Baker and W. T. Heller, Chem. Eng. J., 2009, 147, 6–12 CrossRef CAS.
- J. A. Widegren, A. Laesecke and J. W. Magee, Chem. Commun., 2005, 1610–1612 RSC.
- K. Fujita and H. Ohno, Biopolymers, 2010, 93, 1093–1099 CrossRef CAS PubMed.
- Y. Kohno and H. Ohno, Chem. Commun., 2012, 48, 7119–7130 RSC.
- P. Lozano, T. de Diego, S. Gmouh, M. Vaultier and J. L. Iborra, Biocatal. Biotransform., 2005, 23, 169–176 CrossRef CAS.
- Y. Nikawa, S. Tsuzuki, H. Ohno and K. Fujita, Aust. J. Chem., 2019, 72, 392–399 CrossRef CAS.
- G. Chevrot, E. E. Fileti and V. V. Chaban, J. Mol. Model., 2016, 22, 258 CrossRef PubMed.
- K. Fujita, M. Kajiyama, Y. Liu, N. Nakamura and H. Ohno, Chem. Commun., 2016, 52, 13491–13494 RSC.
- K. Fujita, D. R. MacFarlene, M. Forsyth, M. Yoshizawa-Fujita, K. Murata, N. Nakamura and H. Ohno, Biomacromolecules, 2007, 8, 2080–2086 CrossRef CAS PubMed.
- K. Fujita, R. Nakano, R. Nakaba, N. Nakamura and H. Ohno, Chem. Commun., 2019, 55, 3578–3581 RSC.
- E. E. Carter, A. J. Heyert, M. De Souza, J. L. Baker and G. E. Lindberg, Phys. Chem. Chem. Phys., 2019, 21, 17950–17958 RSC.
- L. Gontrani, Biophys. Rev., 2018, 10, 873–880 CrossRef CAS PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2015, 115, 11379–11448 CrossRef CAS PubMed.
- H. Ohno and K. Fukumoto, Acc. Chem. Res., 2007, 40, 1122–1129 CrossRef CAS PubMed.
- M. Petkovic, J. L. Ferguson, H. Q. N. Gunaratne, R. Ferreira, M. C. Leitão, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Green Chem., 2010, 12, 643–664 RSC.
- A. Yazdani, M. Sivapragasam, J. M. Leveque and M. Moniruzzaman, J. Microb. Biochem. Technol., 2016, 8, 5 Search PubMed.
- M. Bisht, I. Jha and P. Venkatesu, ChemistrySelect, 2016, 1, 3510–3519 CrossRef CAS.
- Y. H. Bi, Z. Q. Duan, X. Q. Li, Z. Y. Wang and X. R. Zhao, J. Agric. Food Chem., 2015, 63, 1558–1561 CrossRef CAS PubMed.
- R. R. Mazid, R. Vijayaraghavan, D. R. MacFarlane, C. Cortez-Jugo and W. Cheng, Chem. Commun., 2015, 51, 8089–8092 RSC.
- D. R. MacFarlane, R. Vijayaraghavan, H. N. Ha, A. Izgorodin, K. D. Weaver and G. D. Elliott, Chem. Commun., 2010, 46, 7703–7705 RSC.
- R. Gusain, A. Khan and O. P. Khatri, J. Mol. Liq., 2020, 301, 112322 CrossRef CAS.
- T. Gundolf, N. Weyhing-Zerrer, J. Sommer, R. Kalb, D. Schoder, P. Rossmanith and P. Mester, ACS Sustainable Chem. Eng., 2019, 7, 15865–15873 CrossRef CAS.
- P. Oulego, J. Faes, R. González, J. L. Viesca, D. Blanco and A. H. Battez, J. Mol. Liq., 2019, 292, 111451–111459 CrossRef CAS.
- A. Mezzetta, L. Guazzelli, M. Seggiani, C. S. Pomelli, M. Puccini and C. Chiappe, Green Chem., 2017, 19, 3103–3111 RSC.
- A. M. M. Bessa, M. S. C. Venerando, F. X. Feitosa, L. M. Alexandre e Silva, F. A. N. Fernandes, R. S. de Santiago-Aguiar and H. B. de Sant'Ana, J. Mol. Liq., 2020, 313, 113586 CrossRef CAS.
- H. Yang, B. Jiang, Y. Sun, L. Hao, Z. Huang and L. Zhang, Chem. Eng. J., 2016, 306, 131–138 CrossRef CAS.
- A. P. S. Brogan and J. P. Hallett, J. Am. Chem. Soc., 2016, 138, 4494–4501 CrossRef CAS PubMed.
- T. De Diego, P. Lozano, S. Gmouh, M. Vaultier and J. L. Iborra, Biomacromolecules, 2005, 6, 1457–1464 CrossRef CAS.
- J. K. Lee and M. Kim, J. Org. Chem., 2002, 67, 6845–6847 CrossRef CAS PubMed.
- T. Itoh, S. Han, Y. Matsushita and S. Hayase, Green Chem., 2004, 6, 437–439 RSC.
- T. Itoh, Y. Matsushita, Y. Abe, S. H. Han, S. Wada, S. Hayase, M. Kawatsura, S. Takai, M. Morimoto and Y. Hirose, Chem. – Eur. J., 2006, 12, 9228–9237 CrossRef CAS PubMed.
- T. Nishihara, A. Shiomi, S. Kadotani, T. Nokami and T. Itoh, Green Chem., 2017, 19, 5250–5256 RSC.
- P. Lozano, J. M. Bernal, G. Sánchez-Gómez, G. López-López and M. Vaultier, Energy Environ. Sci., 2013, 6, 1328–1338 RSC.
- K. M. Bothwell and P. C. Marr, ACS Sustainable Chem. Eng., 2017, 5, 1260–1263 CrossRef CAS.
- Y. Liu, M. Wang, J. Li, Z. Li, P. He, H. Liu and J. Li, Chem. Commun., 2005, 1778–1780 RSC.
- Y. Liu, L. Shi, M. Wang, Z. Li, H. Liu and J. Li, Green Chem., 2005, 7, 655–658 RSC.
- S. H. Lee, T. T. N. Doan, S. H. Ha and Y. M. Koo, J. Mol. Catal. B: Enzym., 2007, 45, 57–61 CrossRef CAS.
- C. Zarcula, L. Corîci, R. Croitoru, A. Ursoiu and F. Peter, J. Mol. Catal. B: Enzym., 2010, 65, 79–86 CrossRef CAS.
- H. Vila-Real, A. J. Alfaia, J. N. Rosa, P. M. P. Gois, M. E. Rosa, A. R. T. Calado and M. H. Ribeiro, J. Biotechnol., 2011, 152, 147–158 CrossRef CAS PubMed.
- S. Sarkar, K. Mantri, D. Kumar, S. K. Bhargava and S. K. Soni, RSC Adv., 2015, 5, 105800–105809 RSC.
- S. K. Soni, R. Ramanathan, P. J. Coloe, V. Bansal and S. K. Bhargava, Langmuir, 2010, 26, 16020–16024 CrossRef CAS PubMed.
- S. H. Lee, T. T. N. Doan, S. H. Ha, W. J. Chang and Y. M. Koo, J. Mol. Catal. B: Enzym., 2007, 47, 129–134 CrossRef CAS.
- R. L. Souza, E. L. P. Faria, R. T. Figueiredo, S. Mettedi, O. A. A. Santos, A. S. Lima and C. M. F. Soares, J. Therm. Anal. Calorim., 2017, 128, 833–840 CrossRef CAS.
- R. L. de Souza, E. L. P. de Faria, R. T. Figueiredo, L. dos S. Freitas, M. Iglesias, S. Mattedi, G. M. Zanin, O. A. A. dos Santos, J. A. P. Coutinho, Á. S. Lima and C. M. F. Soares, Enzyme Microb. Technol., 2013, 52, 141–150 CrossRef CAS PubMed.
- S. Haj Kacem, S. Galai, A. Pérez de los Ríos, F. J. Hernández Fernández and I. Smaali, J. Chem. Technol. Biotechnol., 2018, 93, 174–183 CrossRef CAS.
- D. Khodagholy, V. F. Curto, K. J. Fraser, M. Gurfinkel, R. Byrne, D. Diamond, G. G. Malliaras, F. Benito-Lopez and R. M. Owens, J. Mater. Chem., 2012, 22, 4440–4443 RSC.
- I. V. Pavlidis, K. Tzafestas and H. Stamatis, Biotechnol. J., 2010, 5, 805–812 CrossRef CAS PubMed.
- N. Kimizuka and T. Nakashima, Langmuir, 2001, 17, 6759–6761 CrossRef CAS.
- W. Kubo, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, Chem. Commun., 2002, 4, 374–375 RSC.
- W. Kubo, S. Kambe, S. Nakade, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2003, 107, 4374–4381 CrossRef CAS.
- K. Hanabusa, H. Fukui, M. Suzuki and H. Shirai, Langmuir, 2005, 21, 10383–10390 CrossRef CAS PubMed.
- N. Mohmeyer, D. Kuang, P. Wang, H. W. Schmidt, S. M. Zakeeruddin and M. Grätzel, J. Mater. Chem., 2006, 16, 2978–2983 RSC.
- L. Tan, X. Dong, H. Wang and Y. Yang, Electrochem. Commun., 2009, 11, 933–936 CrossRef CAS.
- S. Dutta, D. Das, A. Dasgupta and P. K. Das, Chem. – Eur. J., 2010, 16, 1493–1505 CrossRef CAS PubMed.
- N. Minakuchi, K. Hoe, D. Yamaki, S. Ten-No, K. Nakashima, M. Goto, M. Mizuhata and T. Maruyama, Langmuir, 2012, 28, 9259–9266 CrossRef CAS.
- Y. Ishioka, N. Minakuchi, M. Mizuhata and T. Maruyama, Soft Matter, 2014, 10, 965–971 RSC.
- T. Kataoka, Y. Ishioka, M. Mizuhata, H. Minami and T. Maruyama, ACS Appl. Mater. Interfaces, 2015, 7, 23346–23352 CrossRef CAS PubMed.
- W. K. Restu, Y. Nishida, T. Kataoka, M. Morimoto, K. Ishida, M. Mizuhata and T. Maruyama, Colloid Polym. Sci., 2017, 295, 1109–1116 CrossRef CAS.
- N. S. Rios, M. P. Pinheiro, M. L. B. Lima, D. M. G. Freire, I. J. da Silva, E. Rodríguez-Castellón, H. B. de Sant’Ana, A. C. Macedo and L. R. B. Gonçalves, Chem. Eng. Res. Des., 2018, 129, 12–24 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |