
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of Sn(II) aminoalkoxide precursors for atomic layer deposition of SnO thin films†
James D.
Parish
*,
Michael W.
Snook
and
Andrew L.
Johnson
 *
*
Department of Chemistry, University of Bath. Claverton Down, Bath, BA2 7AY, UK. E-mail: A.L.Johnson@bath.ac.uk
First published on 8th September 2021
Abstract
We have successfully prepared and structurally characterized a family of eight tin(II) heteroleptic complexes, [Sn(NR2)(ON)]x (NR2 = NMe2 (1a–d) or N(SiMe3)2 (2a–d); x = 1 or 2) and four homoleptic systems, [Sn(κ2-ON)2] (3a–d) from a series of aminoalcohols and fluorinated aminoalcohols (H{ON}) having a different number of methyl/trifluoromethyl substituents at the α-carbon atom, [HOC(R1)(R2)CH2NMe2] (R1 = R2 = H (H{dmae}) (a); R1 = H, R2 = Me (H{dmap}) (b); R1 = R2 = Me (H{dmamp}) (c); R1 = R2 = CF3 (H{Fdmamp}) (d)). The synthetic route used reactions of either [Sn{N(SiMe3)2}2] or [Sn(NMe2)2] with one or two equivalents of the aminoalcohols (a–d) in dry aprotic solvents leading to elimination of amines and formation of the Sn(II) species 1a–d, 2a–d and 3a–d respectively. All complexes were thoroughly characterized by NMR spectroscopy (1H, 13C, 19F, and 119Sn) as well as single-crystal X-ray diffraction studies. In all case the solid state molecular structures of the complexes have been unambiguously established: the solid state structures 1a–b and 1c are dimeric with central {Sn2N2} cores resulting from bridging {μ2-NMe2} units, in which the Sn(II) atoms are four-coordinate. In contrast, the solid state structures of complexes 1c and 2a–c possess similarly dimeric structures, with four-coordinate Sn(II) atoms, in which the oxygen atoms of the {ON} ligand bridge two Sn(II) centres to form dimers with a central {Sn2O2} core. Uniquely in this study, 2d, [Sn(κ2-O,N-OCMe2CH2NMe2){N(SiMe3)2}] is monomeric with a three coordinate Sn(II) centre. The homoleptic complexes 3a–d are all isostructural with monomeric four-coordinate structures with disphenoidal geometries. Solution state NMR studies reveal complicated ligand exchange processes in the case of the heteroleptic complexes 1a–d and 2a–d. Contrastingly, the homoleptic systems 3a–d show no such behaviour. While complexes 1a–d and 2a–d displayed either poor thermal stability or multistep thermal decomposition processes, the thermal behaviour of the homoleptic complexes, 3a–d, was investigated in order to determine the effects, if any, of the degree of fluorination and asymmetry of the aminoalkoxide ligands on the suitability of these complexes as ALD precursors for the deposition of SnO thin films.
Introduction
Tin(II) oxide, which possesses a layered litharge structure, is one of a limited number of binary p-type oxide semiconductor materials, the exploration of which has increased markedly in recent years.1 Despite the intrinsic electrical limitations inherent within p-type oxide materials,2 a sustained drive for their development and application has seen SnO emerge as one of the most promising candidates for application in electronic devices,2,3 with reported hole mobilities of ∼18.71 cm2 (V s)−1 and field-effect mobilities of ∼6.8 cm2 V−1 s−1.4Aside from a range of interesting and desirable properties such as the ability to display superconducting behaviour under pressure at room temperature,5 and band gaps of 0.7 eV (indirect) and 2.5–3.0 eV (direct),6 SnO has attracted particular attention because of its potential utility within “true” complementary metal oxide semiconductor (CMOS) devices, i.e. the development of viable CMOS device structures comprising of both n- and p-type semiconducting oxides.7 Successful implementation of off-silicon CMOS devices is critical for low power, cost-effective, flexible and transparent electronics.
Currently, off-silicon based complementary devices rely on the combination of well-developed n-type oxide transistors and p-type organic or carbon-based thin film transistors. The incompatibility of processing makes the circuit design and integration more complicated and often unacceptable for practical applications.1a,b,2 It is imperative therefore to develop viable p-type, or bipolar, oxide semiconductor materials to allow fabrication of more compact CMOS devices.
Physical vapour deposition (PVD)4,8 and chemical vapour deposition (CVD)1c,9 have both been used to produce thin films of SnO with varying degrees of success. In the case of CVD, three molecular precursors capable of producing phase pure SnO have been reported to-date (Scheme 1, A–C).9b,10 Since modern devices are topographically diverse structures, a vapour phase technique capable of producing thin films with exceptional conformality is required. Atomic layer deposition (ALD) offers such a solution. Whilst a number of Sn-precursor/reactant combinations have been surveyed for the growth of SnO based materials,11 to date, only the Sn(II) aminoalkoxide complexes, Sn(dmamp)2 (R = Me),12 Sn(dmamb)2 (R = Et)13 have been found to produce phase pure SnO in an ALD process; for example Sn(dmamp)2 has been used in conjunction with H2O, between 90 °C and 210 °C, to produce crystalline SnO at temperatures of 150 °C.12b More recently the less effective bis-N-alkoxy carboxamide (Scheme 1, E) has been reported as a potential ALD precursor.14
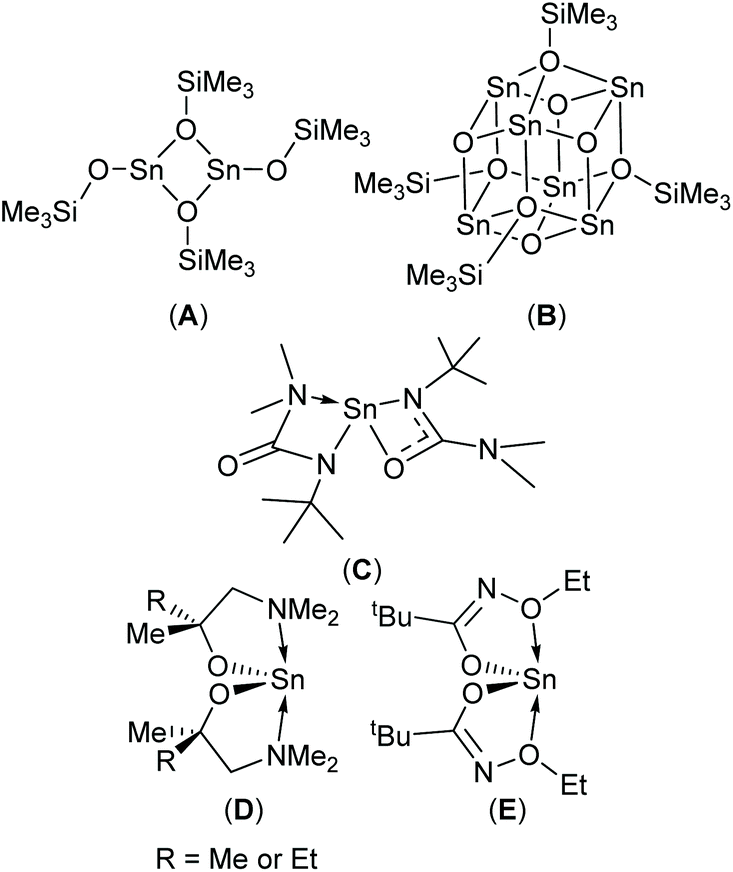 | ||
| Scheme 1 SnO CVD (A–C) and ALD (D, E) precursors. | ||
For divalent metals of group 14 elements (Ge(II), Sn(II) or Pb(II)), the metals possess both a lone pair of electrons and a vacant pz-orbital. One effective methodology for the stabilization of these reactive centres is the incorporation of intermolecular coordination (E → M(II) (E = N, O, P, S) by lariat donor groups, which occupy the vacant pz-orbital and stabilize the divalent metal centre with respect to oligomerisation.15 The complexes [M{OCH2CH2NMe2}2] (M = Ge, Sn) which contain the aminoalkoxide ligand {dmae}, were the first monomeric Sn(II) and Ge(II) compounds stabilized by intermolecular M → N interactions, negating the need for steric shielding by bulky substituents to achieve mono-nuclearity.16 Subsequently several of functionalised alkoxide ligands such as 1-methoxy-2-methyl-2-propanol (Hmmp)17 and 1-dimethylamino-2-methyl-2propanol (Hdmamp)18 have become important ligands in the development of MOCVD and ALD precursors.
The general dearth of suitable precursors for SnO production has prompted us, and others,14 to investigate new Sn(II)–ligand combinations. Aminoalcohols possess several attractive features as ligands in metal complexes. Recently, identical ligands have been exploited in the development of heteroleptic Sn(IV) alkoxide/aminoalkoxide complexes for SnO2 and F-doped-SnO2 by MOCVD.19 The incorporation of Lewis basic donor groups into alkoxide ligands, and their subsequent application as volatile MOCVD precursors, was pioneered by Herrmann with the development of a series of ether and amine functionalised alcohols.20 Sequential substitution of alkyl groups on the α-carbon atom in the functionalised alkoxide ligand has been shown to favour chelation over potential bridging modes. The presence of donor-functionalized groups. i.e. {OR} and {NR2}, has also been shown to enhance the volatility and reduce the hydrolytic susceptibility of metal alkoxide systems.21 Similarly, unsymmetrical substitution at both the α-carbon atom of the alkoxide ligand, and the alkyl section of the donor group has been used as a strategy to enhance precursor volatility. The introduction of fluorine into the ligand is also an established method by which volatility of a precursor can be increased. The substitution of H, CH3, or CR3 groups with fluorine moieties causes intermolecular repulsion due to the resulting negative “charge envelope”-and thus an increase in the volatility.20
In this work, we report an study into the synthesis and structural characterisation of the heteroleptic complexes 1a–d and 2a–d by reaction of the dimethylaminoethanol ligands Hdmae (a), Hdmap (b), Hdmamp (c) and HFdmamp (d) with [Sn{NMe2}2] and [Sn{N(SiMe3)2}2], respectively (Fig. 1), examining the effect of the incorporation of asymmetry, as well as fluorination on the solid state structures of these systems. We also report the synthesis and structural characterisation and thermal properties of the homoleptic Sn(II) complexes, 3a–d, (Fig. 1).
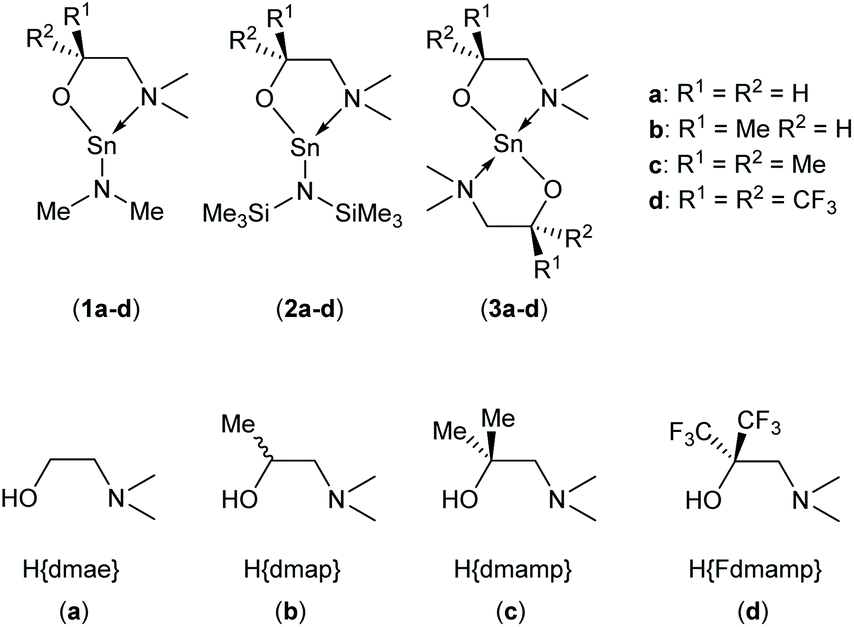 | ||
| Fig. 1 The target hetero- (1a–d and 2a–d) and homo-leptic Sn(II) systems (3a–d) derived from the aminoalcohol pro-ligands H{dmae} (a), H{dmap} (b), H{dmamp} (c) and H{Fdmamp} (d) respectively. | ||
Results and discussion
The development of a series of Sn(II) compounds was necessary prior to investigating their potential utility for the production of thin films of SnO via ALD. The synthesis and characterization of the various hetero- and homoleptic species isolated in this study are discussed below.The initial investigation focused on the synthesis and characterisation of the heteroleptic amide compounds “[Sn(L){NMe2}]” and “[Sn(L){N(SiMe3)2}]” (L = dmae, dmap, dmamp or Fdmamp). Complexes 1a–d and 2a–d were synthesized in near quantitative yields by the reaction of [Sn{NMe2}2] or [Sn{N(SiMe3)2}2] with one equivalent of the amino-alcohols (a–d) in hexane, as shown in Scheme 1.
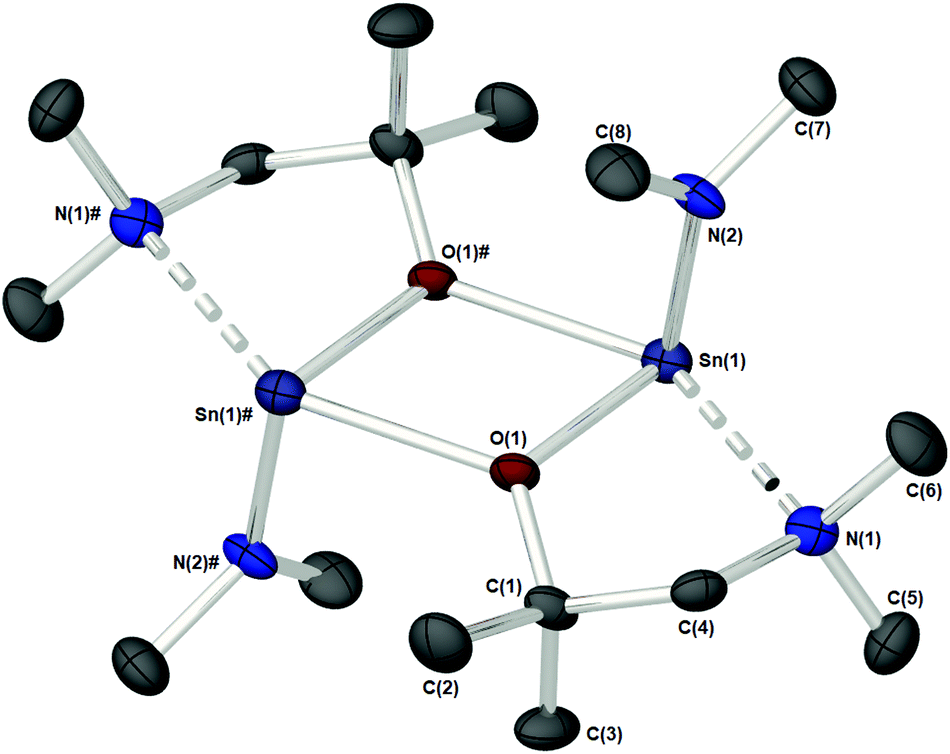
As part of our study, we undertook single crystal X-ray analysis to determine the absolute molecular structures of the complexes. Table S1† (in the ESI†) shows the structural data parameters for 1a–d, 2a–d and 3a–d. Compounds 1a–b and 1d are isostructural, each adopting a dinuclear arrangement in the solid state, with the two halves of the molecule related by a centre of inversion. Fig. 2 shows the molecular structures of complexes 1a and 1d, respectively. The molecular structure of 1b is included in the ESI.† The cores of 1a–b and 1d consist of planar, four-membered {Sn2N2} rings, in which the amide ligands bridge in an asymmetric fashion (Table 1). The Sn–O and Sn–N distances are within the range found in the CSD22 for tin(II) compounds with a central {Sn2N2} rings, and as with similar compounds, angles of ca. 90° are observed between the alkoxide bonds and the {Sn2N2} core [1a: 91.09(7), 1b: 89.27(11), 1d: 87.84(10)]. Appended to each Sn atom, in a mutually trans-configuration, are the aminoalkoxide ligands which chelate such that each metal centre possesses a 4-coordinate environment, with approximate trigonal bipyramidal geometry,11b,23 with a vacant equatorial coordination site. The fourth (axial) coordination position about the Sn centres is seen to be occupied by the lariat {NMe2} group of the aminoalkoxide ligands. It is noteworthy that these Sn⋯N interactions, while appreciably shorter than the sum of the respective van der Waals radii (2.17 (Sn) + 1.55 (N) = 3.72 Å), are longer than comparable Sn⋯N lariat interactions [Sn(1)⋯N(2), 1a: 2.795(2) Å, 1b: 2.720(3) Å, 1d: 2.717(6) Å]. We attribute the elongated nature of these interactions in part to electronic repulsive forces arising from the stereoactive lone pair of electrons on the Sn atoms, and it is plausible that these weak Sn⋯N interactions may result in the solution state fluxionality demonstrated in the 119Sn NMR (vide infra).
 | ||
| Fig. 2 (a) The solid state molecular structures of [Sn{μ-NMe2} {dmae}]2 (1a), and (b) [Sn{μ-NMe2}{Fdmamp}]2 (1d). Thermal ellipsoids are shown at 50% probability. H-Atoms are omitted for clarity. The symmetry equivalent atoms are generated by the operator: # −X, 1 − Y, 1 − Z (1a) and #1 − X, 2 − Y, 1 − Z (1d). | ||
| Bond lengths | |||
| 1a | 1b | 1d | |
| Sn–N(NMe2) | 2.795(2) | 2.720(3) | 2.717(6) |
| Sn–O | 2.045(2) | 2.048(2) | 2.067(5) |
| Sn–N(1)(μ-NMe2) | 2.218(2) | 2.217(3) | 2.212(7) |
| Sn(1)#–N(1)(μ-NMe2) | 2.320(2) | 2.350(3) | 2.323(7) |
| Sn–Sn | 3.4826(4) | 3.4825(4) | 3.4525(7) |
| O–C | 1.409(3) | 1.412(4) | 1.363(9) |
| ∑ Sn2N2 ring | 360 | 360 | 360 |
| Bond angles | |||
| N(NMe2)–Sn(1)–N(2)#(μ-NMe2) | 158.36(6) | 157.8(1) | 154.5(2) |
| Sn–N–Sn | 100.25(7) | 80.7(1) | 99.1(3) |
| O(1)–Sn–N(2) | 93.56(6) | 92.6(1) | 91.5(2) |
| Sn–O–C | 118.0(1) | 121.0(2) | 126.1(5) |
| C(2)–N(1)–Sn | 96.7(1) | 100.5(2) | 105.8(5) |
| C(2)–N(1)–C(3) | 111.6(2) | 111.9(3) | 111.7(8) |
| C(2)–N(1)–C(4) | 111.1(2) | 112.1(3) | 112.7(8) |
| C(3)–N(1)–C(4) | 108.3(2) | 109.0(3) | 107.9(9) |
| ∑ C–N(1)–C (sp3 ∼328.5°) | 331.0 | 333.0 | 332.3 |
Complex 1c (Fig. 3), which supports a {C(Me2)} unit in the α-position of the aminoalkoxide ligand, is also dimeric in the solid state. Interestingly however, the relative configuration of the ligands about the Sn atoms has changed significantly. Selected bond lengths and angles are presented in Table 2. At the core of this dimer is a planar asymmetric {Sn2O2} ring, in which a bridging {μ-OCH2CH2NMe2} group is bonded to each of the two Sn atoms in a chelating fashion. The Sn–O and Sn–N distances are within the range found in the Cambridge Structural Database for tin(II) compounds with central {Sn2O2} rings.22 The angle made between the {NMe2} groups on each Sn atom and the {Sn2O2} core is >90° [1c: 93.85(11)°], presumably because of the steric demands of the {NMe2} groups.
 | ||
| Fig. 3 The solid state molecular structure of [Sn[μ-dmamp]{NMe2}]2 (1c). Thermal ellipsoids are shown at 50% probability. H-Atoms are omitted for clarity. The symmetry equivalent atoms are generated by the operator: # −X, 1 − Y, 1 − Z. | ||
| Bond lengths | Bond angles | ||
|---|---|---|---|
| Sn–O(1) | 2.111(2) | N(1)–Sn–O(1)# | 142.3(1) |
| Sn–O(1)# | 2.338(2) | O(1)–Sn–N(2) | 94.7(1) |
| Sn–N(1) | 2.619(3) | Sn–O–C | 120.7(2) |
| Sn–N(2) | 2.058(3) | Sn–O–Sn | 110.0(1) |
| Sn–Sn | 3.6474(5) | O–Sn–O | 69.97(9) |
| O–C | 1.427(4) | C(2)–N(1)–Sn | 103.1(2) |
| C(2)–N(1)–C(3) | 110.1(3) | ||
| C(2)–N(1)–C(4) | 112.7(3) | ||
| C(3)–N(1)–C(4) | 108.7(3) | ||
| ∑ C–N–C (sp3 = ∼328.5°) | 331.5 | ||
Each Sn centre also bears a terminal, mutually-trans {NMe2} group, such that the geometry about each Sn(II) core can be described as trigonal–bipyramidal. In contrast to 1a–b and 1d the chelating interaction between the Sn atoms and the lariat {NMe2} group is approx. 4–7% shorter [Sn(1)–N(2), 2.619(3) Å], suggestive of a stronger Sn ← NMe2 interaction Cf.1a–b and 1d.
The specific reason for this variation of the coordination modes from {μ-NMe2}/{aminoalkoxide} in 1a–b and 1d to {μ-aminoalkoxide}/{NMe2 } in 1c, is unclear however we may postulate that a change in electron density on the O-atom of the aminoalkoxide ligand, or the NMe2 lariat, may be responsible.
Resonances of the 1H NMR spectra for 1a–d in C6D6 were in general found to be consistent with the respective aminoalkoxide and the amide ligands in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio; a feature also reflected in the 13C NMR spectra. For complex 1a the 1H and 13C NMR spectra contain a series of resonances attributed to the {OC
:1 ratio; a feature also reflected in the 13C NMR spectra. For complex 1a the 1H and 13C NMR spectra contain a series of resonances attributed to the {OC![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2}, {SnN
2}, {SnN![[M with combining low line]](https://www.rsc.org/images/entities/i_char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/i_char_0065_0332.gif) 2}, {NC2} and {CH2N2} groups respectively. Similarly, compound 1b displays resonances at δ = 4.17–4.19 ppm ascribed to the chiral CMe, with the remaining methylene group (i.e. {CH2}) giving rise to a broad multiplet at δ = 2.19–2.31 ppm and a doublet of doublets (J = 11.6, 2.5 Hz) at δ = 1.82 ppm.
2}, {NC2} and {CH2N2} groups respectively. Similarly, compound 1b displays resonances at δ = 4.17–4.19 ppm ascribed to the chiral CMe, with the remaining methylene group (i.e. {CH2}) giving rise to a broad multiplet at δ = 2.19–2.31 ppm and a doublet of doublets (J = 11.6, 2.5 Hz) at δ = 1.82 ppm.
The 1H NMR spectrum of 1c also shows the expected resonances attributed to the {Sn–NMe2}, {CH2}, {–NMe2} and {–CH3} groups. The resonances ascribed to the {![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H2} and {Me2} moieties appear to be coincidental in the 13C NMR spectrum, with the tertiary environment appearing more downfield (δ = 72.80 ppm) than the methylene (δ = 72.19 ppm), in contrast to compounds 1a and 1b, in which the alkoxide carbon environment is found further upfield of the methylene group.
H2} and {Me2} moieties appear to be coincidental in the 13C NMR spectrum, with the tertiary environment appearing more downfield (δ = 72.80 ppm) than the methylene (δ = 72.19 ppm), in contrast to compounds 1a and 1b, in which the alkoxide carbon environment is found further upfield of the methylene group.
The 1H, 13C, and 19F NMR spectra for 1d at room temperature display a pattern of resonances consistent with a highly fluxional system. At high temperature (i.e. 90 °C in d8-C7H8) resonances in the 1H NMR spectrum resolve into the expected {CH2}, {CH2NMe2} and {SnNMe2} signals. At the same temperature the 13C NMR spectrum displays signals for the {SnNMe2}, {NMe2}, {CH2} and {OC} moieties at δ = 41.68, 47.1, 57.50 and 82.48 ppm respectively. Peaks associated with the {CF3} moieties were obscured by resonances associated with solvent. At 90 °C the 19F NMR spectrum exhibits a major resonance at δ = −76.3 ppm, accompanied by a secondary, and substantially smaller resonance at δ = −76.7 ppm. This contrasts with the room temperature spectrum, which shows a redistribution of intensities across three resonances (δ = −76.3 (minor), −76.7 (major) and −77.5 (minor) ppm) indicative of multiple species present in equilibrium.
The solid-state structures of heteroleptic complexes 2a–2d, formed by stoichiometric reaction of pro-ligands a–d with [Sn{N(SiMe3)2}2], reveal dimeric systems in the case of 2a–c in which the bulkier {N(SiMe3)2} is terminally coordinated, whilst the fluorinated analogue 2d presents as a monomeric species. For the dimeric systems (2a–c) the solid-state structures consist of a central asymmetric {Sn2O2} core reminiscent of 1c, in which amide ligands are orientated about the centre in a transoidal fashion. Fig. 4 shows the solid-state molecular structures of complexes 2c and 2d. The molecular structures of complexes 2a and 2b are included in the ESI† and selected bond lengths and angles are shown in Table 3 (2a–c) and Table 4 (2d). Complex 2a has been previously described,24 but is included here as part of a wider narrative. Each Sn(II) centre possesses a trigonal–bipyramidal geometry, with Sn–O and Sn–N distances for 2a–2c within the range found in the Cambridge Structural Database22 for tin(II) compounds with a central {Sn2O2} core.16,24,25 In contrast to the terminal {NMe2} geometry in 1c, which is reflected in isostructural compounds 2a–c, the sterically encumbered {N(SiMe3)2} units result in more obtuse angles at the metal centres between the {N(SiMe3)2} ligands and the {Sn2O2} cores [2a: 99.47(19)°/103.45(19)°, 2b; 102.46(10)°/103.48(10)°, 2c: 104.78(7)°].
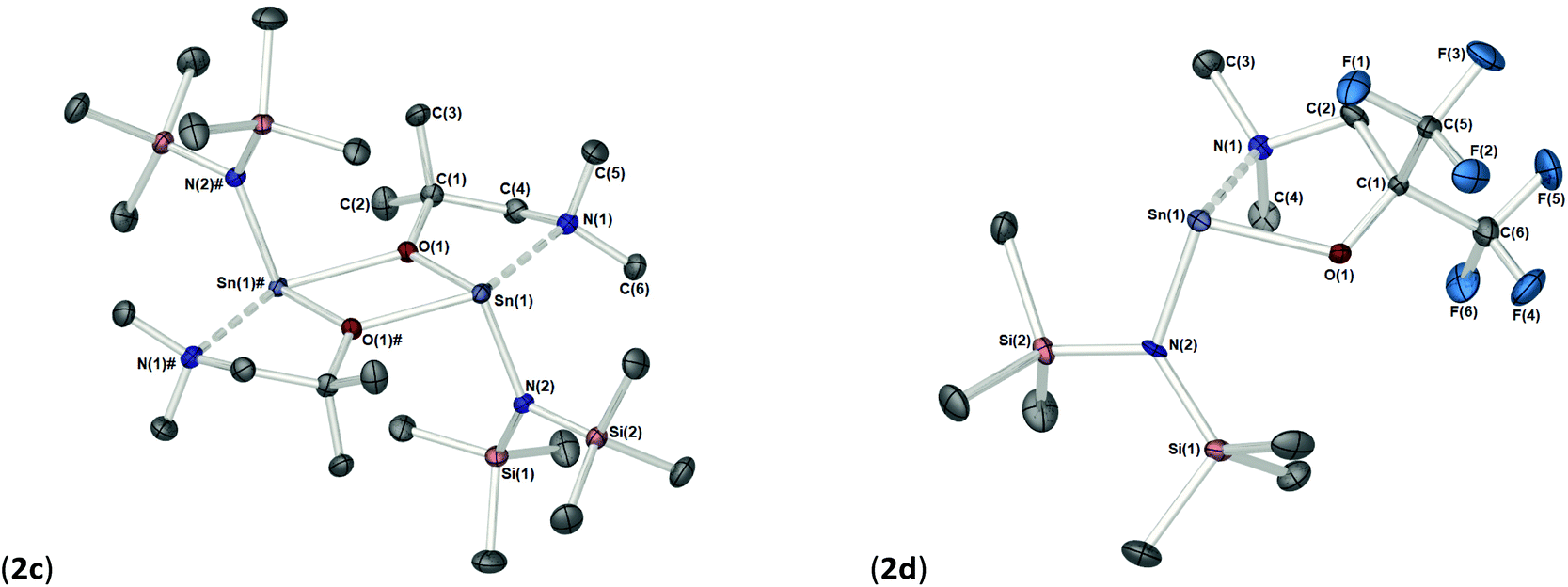 | ||
| Fig. 4 (a) The solid state molecular structure of [Sn[μ-dmamp}{N(SiMe3)2}]2 (2c). (b) The solid state molecular structure of [Sn[Fdmamp}{N(SiMe3)2}] (2d). Thermal ellipsoids are shown at 50% probability and H-atoms are omitted for clarity. The symmetry equivalent atoms in 2c are generated by the operator: # −X, 1 − Y, 1 − Z. | ||
| 2a | 2b | 2c | |
|---|---|---|---|
| Bond lengths | |||
| Sn(1)–N(NMe2) | 2.634(6), 2.613(6) | 2.596(8), 2.643(3) | 2.592(2) |
| Sn–μO | 2.126(5), 2.265(5), | 2.136(7), 2.341(6) | 2.130(1) |
| 2.303(5), 2.145(5) | 2.319(6) 2.120(6) | 2.381(1) | |
| Sn–N(HMDS) | 2.150(5), 2.155(5) | 2.147(7) | 2.183(2) |
| 2.158(7) | |||
| Sn–Sn | 3.6529(8) | 3.6679(8) | 3.7152(4) |
| O–C | 1.401(8), 1.399(7) | 1.43(1), 1.48(2) | 1.440(2) |
| ∑ Sn2O2 ring | 359.51(2) | 359.93(2) | 360 |
| Bond angles | |||
| μO–Sn–μO | 68.8(2), 67.8(2) | 69.0(2), 69.6(2) | 69.23(5) |
| Sn–μO–Sn | 111.0(2), 111.8(2) | 110.7(3), 110.6(3) | 110.77(6) |
| N(NMe2)–Sn–μO | 71.20(2), 71.02(2) | 72.56(10) | 73.50(4) |
| 139.5(2), 138.8(2) | 76.19(10) | 142.72(5) | |
| 141.52(6), 145.84(6) | |||
| μO–Sn–N(HMDS) | 105.8(2), 94.24(2) | 104.2(3) 97.84(3) | 105.64(6) |
| 103.2(2), 95.32(2) | 104.8(2) | 99.03(6) | |
| 96.75(2) | |||
| N(NMe2)–Sn–N | 90.15(2), 93.46(2) | 90.51(6) | 89.92(5) |
| 90.14(6) | |||
| ∑ N(NMe2) (sp3 = ∼328.5°) | 332.8(6) | 332.6(6) | 330.9(2) |
| Bond lengths | Bond angles | ||
|---|---|---|---|
| Sn(1)–O(1) | 2.087(2) | O–Sn(1)–N(1) | 72.1(1) |
| Sn(1)–N(NMe2) | 2.433(4) | O–Sn(1)–N(2) | 94.3(1) |
| Sn–N(HMDS) | 2.090(3) | N(1)–Sn(1)–N(2) | 102.3(1) |
| O(1)–C(1) | 1.364(5) | Sn(1)–O–C(1) | 117.4(2) |
| C(2)–N(1)–Sn(1) | 103.8(2) | ||
| C(2)–N(1)–C(3) | 111.6(3) | ||
| C(2)–N(1)–C(4) | 108.1(3) | ||
| C(3)–N(1)–C(4) | 109.8(3) | ||
| ∑ C–N–C (sp3 = ∼328.5°) | 329.5 | ||
| ∑ C–N–C (sp3 = ∼328.5°) | 331.5 | ||
Distinctively, the fluorinated compound 2d is monomeric in the solid state and is isostructural to the monomeric Ge(II) system [Ge{dmae}{N(SiMe3)2}],24 with a three-coordinate tin atom forming a single bond each to the {N(SiMe3)} and the oxygen atom of the {Fdmamp} ligand, whilst an additional dative Sn ← N interaction from the lariat {NMe2} group completes the coordination. Inspection of the Sn ← NMe2 distances across 2a–d clearly shows that a shorter interaction is present in 2d [Sn(1)–N(1), 2.433(4)] in comparison with interactions found in 1a–d and 2a–c (see Table 4). Concomitantly, both the Sn–O and Sn–N(SiMe3)2 bonds are also shortened in 2d compared with other complexes in this series.
Despite an acute bite angle [O(1)–Sn(1)–N(1): 72.1(1)°], the additional bond angles about the Sn(1) in 2d [N(1)–Sn(1)–N(2): 102.03(1)° and O(1)–Sn(1)–N(2): 94.3(1)°] suggest an absence of hybridisation at the Sn(II) centre. This would imply that the tin–ligand bonds in 2d almost exclusively involve the p-orbitals on Sn(II), resulting in a non-directional 5s2 configuration of the Sn lone pair.
As with 1a–c, the resonances of the 1H spectra for 2a–d in C6D6 were found to be consistent with the respective aminoalkoxide and amide ligands in a 1:1 ratio. For 2a the 1H and 13C NMR spectra were consistent with those previously reported.24
The homoleptic complexes 3a–d were synthesised in a similar fashion to 2a–d, through the addition of two equivalents of the desired aminoalcohol to a clear orange solution of [Sn{N(SiMe3)2}2] in hexane under an atmosphere of argon (Scheme 2). Independent of the aminoalcohol employed, the reaction mixture underwent an immediate transformation to a colourless solution and was stirred for 2 h before volatiles were removed in vacuo. The resultant solids were dissolved in a minimum volume of hexane, filtered, and stored at −28 °C to afford crystalline products. Complexes 3a and 3c have previously been reported by Zemlyansky et al.16 and Han et al.12b respectively. While 3c is, in our hands, a solid at room temperature, it has previously been described as colourless liquid.12b Our investigation suggests that small amounts of free ligand can act as a contaminant and have a significant effect on the physical form of 3c. Distillation of the neat product at 120 °C (10−2 mbar) into liquid nitrogen was found to yield a white-colourless crystalline material of high purity at room temperature, which when melted, often remained in liquid form at room temperature for some time. Overall, 3c exhibits an unusual solid–liquid behaviour; some crystalline samples remaining solid above 100 °C, whereas some samples remaining liquid at room temperature, crystallising in response to agitation, despite its high purity. A brief acknowledgement of these properties is made within the US patent,26 though no further discussion exists within the literature. The effect of the variability of these properties on the effectiveness of Sn(dmamp)2 (3c) as an atomic layer deposition precursor has yet to be studied.
 | ||
| Scheme 2 Synthetic scheme for complexes 1a–d and 2a–d. | ||
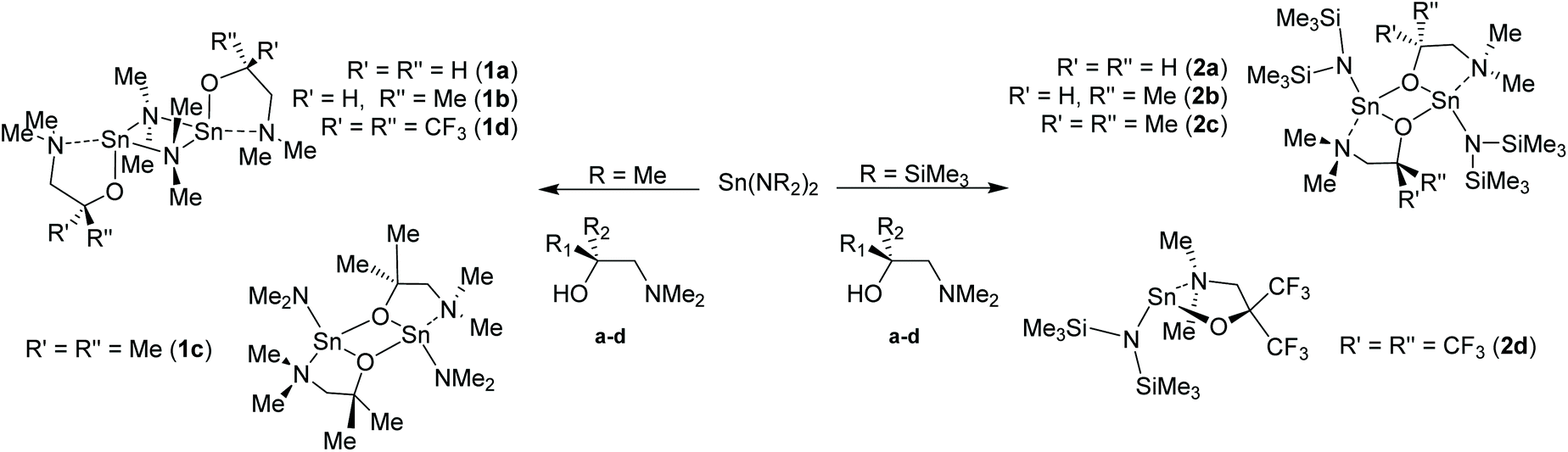
Fig. 5 shows the molecular structures of complexes 3b–d. The molecular structure of 3a is included in the ESI.† Selected bond lengths and angles are presented in Table 5. The homoleptic compounds 3a–d form an isostructural series of monomeric, four-coordinate complexes. Each possesses a pseudo-trigonal bipyramidal geometry (τ = 0.82 (3a), 0.78 (3b), 0.77 (3c) and 0.72 (3d))11b about the Sn(II) centre in which the two aminoalkoxide ligands are coordinated in the same κ2-fashion observed throughout this study, with the O-atoms of the ligands occupying two equatorial positions, and the pendent {NMe2} groups occupying the axial positions. The Sn–O [2.04–2.06 Å] and Sn ← NMe2 [2.46–2.58 Å] bond lengths in 3a–d are commensurate with comparable complexes and show no significant differences as this group of complexes is traversed.
 | ||
| Fig. 5 The solid-state molecular structures of (a) [Sn{dmap}2] (3b), (b) [Sn{dmamp}2] (3c) and (c) [Sn{Fdmamp}2] (3d). Thermal ellipsoids are shown at 50% probability and H-atoms are omitted for clarity. The symmetry equivalent atoms in 3b are generated by the operator: # 1 − X, Y, ½ − Z. | ||
| 3a | 3b | 3c | 3d | |
|---|---|---|---|---|
| a Smallest angle between N–Sn–N and O–Sn–O planes. | ||||
| Bond lengths | ||||
| O(1/2)–Sn | 2.052(4) | 2.0548(12) | 2.050(2), 2.038(2) | 2.050(2), 2.081(3) |
| N(1/2)–Sn | 2.464(4) | 2.4772(15) | 2.580(3), 2.436(3) | 2.449(3), 2.509(3) |
| C(1/11)–O | 1.412(7) | 1.409(2) | 1.412(4), 1.412(3) | — |
| Bond angles | ||||
| N–Sn–N | 145.86(19) | 143.24(7) | 144.25(9) | 141.9(1) |
| O–Sn–O | 96.4(2) | 96.39(7) | 98.30(9) | 98.3(1) |
| N–Sn–N/O–Sn–Oa | 87.2 | 87.0 | 84.26 | 84.55 |
| C(1/11)–O–Sn | 118.8(3) | 120.81(10) | 119.02(2), 122.1(2) | 126.4(2), 121.4(2) |
| C(2)–N–Sn | 101.0(3) | 100.49(10) | 101.7(2), 101.1(2) | 108.1(2), 105.7(4) |
| C(2/12)–N–C(3/13) | 111.8(4) | 110.6(1) | 110.6(2), 109.8(3) | 113.2(3), 113.2(6) |
| C(2/12)–N–C(4/14) | 110.6(4) | 112.0(1) | 112.2(3), 112.6(3) | 109.4(3), 111.8(6) |
| C(3/13)–N–C(4/14) | 109.9(4) | 110.0(1) | 109.0(3), 109.7(3) | 107.8(3), 108.7(6) |
| ∑ C–N–C (sp3 = 328.5°) | 332.3(4) | 332.6(1) | 331.8(3), 332.1(3) | 330.4(4), 333.7(4) |
A cursory analysis of the bond angles about the Sn(II) centre in 3a–d (see Table 5) suggests that the tin–ligand bonds almost exclusively involve the p-orbitals on Sn, and that the lone pair of electrons in 3a–d are therefore again largely 5s2 based.16
The 1H NMR spectra of 3a–d clearly show the absence of resonances associated with the {N(SiMe3)2} ligands and are consistent with the formation of the bis-aminoalkoxide complexes. For 3a and 3c the 1H and 13C NMR spectra for 3c match the reported data.
The 1H NMR spectrum for 3b is complicated by the chiral nature of the aminoalkoxide backbone, where chelation gives rise to a mixture of the RR, SS, RS and SR configurations. High temperature spectroscopy (90 °C in d8-C6H8) resolves the 1H and 13C NMR spectra into five resonances indicative of a highly fluxional system.
The 1H NMR spectrum of [Sn{Fdmamp}2] 3d, displays three environments, with a broad singlet at δ = 2.54 ppm associated with the {CH2} group, and two very broad signals of equal integration ascribed to the {Me} groups of the chelating {NMe2} unit (δ = 2.07 and 1.75 ppm), which exist in inequivalent environments. The 13C{1H} NMR spectrum shows a broad quartet at δ = 124.9 ppm, {CF3}, alongside four further resonances at δ = 82.8 (O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (CF3)2}, 58.07 (CH2), 47.8 (NMe) and 45.9 (NMe) ppm, signifying an inequivalence of the {NMe} groups. The presence of two resonances at δ = −76.40 and −77.55 ppm in the 19F NMR spectrum is also indicative of the two inequivalent {CF3} environments (cf. δ = −78.86 ppm for the free pro-ligand).
(CF3)2}, 58.07 (CH2), 47.8 (NMe) and 45.9 (NMe) ppm, signifying an inequivalence of the {NMe} groups. The presence of two resonances at δ = −76.40 and −77.55 ppm in the 19F NMR spectrum is also indicative of the two inequivalent {CF3} environments (cf. δ = −78.86 ppm for the free pro-ligand).
The purity of the bulk powders of all compounds 1a–3d was further investigated using elemental analysis. As is often noted for these types of compounds, obtaining useful elemental analyses is difficult due to the high volatility of the compounds, incomplete or premature decomposition of the precursor, or inclusion of solvents. Therefore, while the elemental analyses are within acceptable range for C and H for several complexes, elemental analysis for others were difficult to obtain.
119Sn NMR studies
As part of our investigation 119Sn{1H} NMR studies were performed on all complexes and are tabulated in Table 6. The NMR spectroscopic data for 1a–3d were recorded in benzene-d6 at 25 °C whenever possible, as 119Sn NMR shifts have previously been shown to be temperature sensitive. In the case of complex 1d NMR studies were also recorded at 90 °C in an attempt to resolve the spectra.For the homoleptic, 4-coordinate complexes 3a–d the 119Sn{1H} NMR spectra all display single well-defined resonances between δ = −279 and −322 ppm. Curiously, the 119Sn{1H} shift for 1a was recorded at −279 ppm, ∼30 ppm upfield of the literature value of δ = −310 (in d8-toluene). Such changes with solvent have been noted previously with upfield shifts being assignable to donor–acceptor interactions of variable strength. The 119Sn{1H} resonance for 3d is shifted upfield compared to that of 3c, presumably as a result of the proximity of electron withdrawing {CF3} groups. We note that complexes of the form [Sn{OC(CF3)2CH2L}2] (L = SeC6H4(CH2NMe2)27 or P(tBu)3}228) also possess upfield chemical shifts (119Sn{1H} δ = −392 and −292 ppm respectively).
119Sn chemical shifts for tin(II) complexes are known to be heavily influenced by coordination environment,29 and an increase in coordination number or electron density at the metal generally result a in upfield resonances. The observed 119Sn{1H} NMR shifts for 3a–d suggest an increase in electron density at the Sn(II) centre in the order 3c < 3b < 3a < 3d. The 119Sn{1H} chemical shifts for 3a–d (Table 6) fall in very narrow range (δ = 104 ppm) considering the fact that chemical shifts for tin(II) compounds have been reported over a 4500 ppm range. This suggests that 3a–d are all structurally comparable in solution and are likely to possess structures in solution akin to those established by X-ray diffraction.
For complexes 2a–d the observed 119Sn{1H} chemical shifts are spread over a wider ppm range and appear as single well-defined resonances. While complex 2a is already known to the literature16 its 119Sn{1H} NMR spectra has not previously been reported. In contrast to 3a–d the 119Sn{1H} chemical shifts of 2a–c, which are also 4-coordinate in the solid state, experience a downfield or de-shielding as the series is traversed. For 2d, which possesses a 3-coordinate Sn(II) centre in the solid state, a chemical shift of δ = 94 ppm is observed.
For the heteroleptic {NMe2} derivatives 1a–d the story is considerably more complicated, with the appearance of multiple Sn environments in the 119Sn{1H} NMR spectra of 1a–c, whilst 1a displays a single broad (ca. 40 ppm) 119Sn resonance at δ −77 ppm.
The complex solution-state activity is most clearly demonstrated in the 119Sn{1H} NMR of Sn(dmamp)NMe2 (1c), where four broad resonances are evident. At the two extremes of the range, a resonance attributed to [Sn{NMe2}2]2 (δ = +125 ppm) can be seen, alongside a resonance coincident with that of the homoleptic bis-substituted species Sn(dmamp)2 (3c) [δ = −218 ppm]. We hypothesis that these systems are in equilibria as shown in eqn (1), as similar equilibria have been previously noted in studies between Sn(II) amides and alcohols and phenols.30
A further two, upfield resonances at δ = −46 and −65 ppm, are attributed to two different isomers of the heteroleptic species: the peak observed at δ = −46 ppm is attributed to the {μ-O} bridged species observed in the solid state, whilst the resonance at δ = −66 ppm is assigned to the {μ-NMe2} isomer. Despite the lack of crystallographic evidence for the latter isomer, it is reasonable to suggest that the {μ-NMe2} form may be present given that this is the preferred method of bridging observed in the solid state for complexes 1a, 1b and 1d. To support this, analysis of the 119Sn{1H} NMR spectrum of 1b, which differs only by a single methyl substituent on the alkoxide, reveals a well-defined but broad resonance at δ = −65 ppm, attributed to the solid-state structure of 1b. A resonance of significantly lower intensity is also observed at δ = +125 ppm, suggestive of the previously described equilibrium depicted in eqn (1). However, a resonance at δ = −246 ppm associated with 3b was not observed.
The 119Sn NMR spectra for 1d displays resonances at δ = +80, +70, −117 and −132 ppm respectively. By comparison with other NMR data in this study, we speculate that the major peak at δ = +80 ppm corresponds to the monomeric form of 1d with a structure comparable to the 3-coordinate solid state structure of 2d. Whilst specific assignment is not possible, we venture that the remaining upfield peaks at δ = +70, +117 and −132 ppm may well represent dimeric species such as the (μ-NMe2) bridged dimer, with and without lariat {NMe2} coordination to the Sn(II) centre, as well as the corresponding (μ-Oalkoxide) bridged systems. Notably, none of these resonances correspond to the formation of the bis-amino-alkoxide complex 3d and [Sn{NMe2}2]2 as in the case of 1a–c. At 90 °C the spectra resolve into a single broad resonance at δ = +83 ppm, which we assume to be the 3-coordinate [(Me2N)Sn{κ2-OC(CF3)2CH2NMe2}].
Thermal profiles of homoleptic compounds (3a–d)
Two of the main precursor requirements for ALD applications are the need for volatility and thermal stability. As the primary goal of synthesising compounds 1a–d, 2a–d and 3a–d was driven by our interest in their application as precursors for the ALD of Sn(II) oxide films. The thermal profiles of complexes 1a–d and 2a–d where not investigated, as all displayed poor thermal stability: as well as displaying ligand exchange incompatible with application as ALD precursors, complexes 1a–d showed instability at room temperature, decomposing upon prolonged storage under inert atmosphere. In addition the complex ligand exchange processes observed for the heteroleptic systems 1a–d and 2a–d when in solution state call into question their suitability as stable ALD precursors. Complexes 2a–d were all found to possess multistep thermal decomposition processes, displaying mass losses over a wide temperature ranges. As such only the melting points and thermogravimetric analysis (TGA) and isothermal studies of the homoleptic complexes 3a–d are reported here. The melting points and analysis of these compounds were recorded with instruments housed in an argon filled glovebox in order to minimise reaction with atmospheric moisture/air. While complex 3a is known in the literature, its thermal properties, and their comparison to complexes 3b, 3c and 3d, have not previously been described.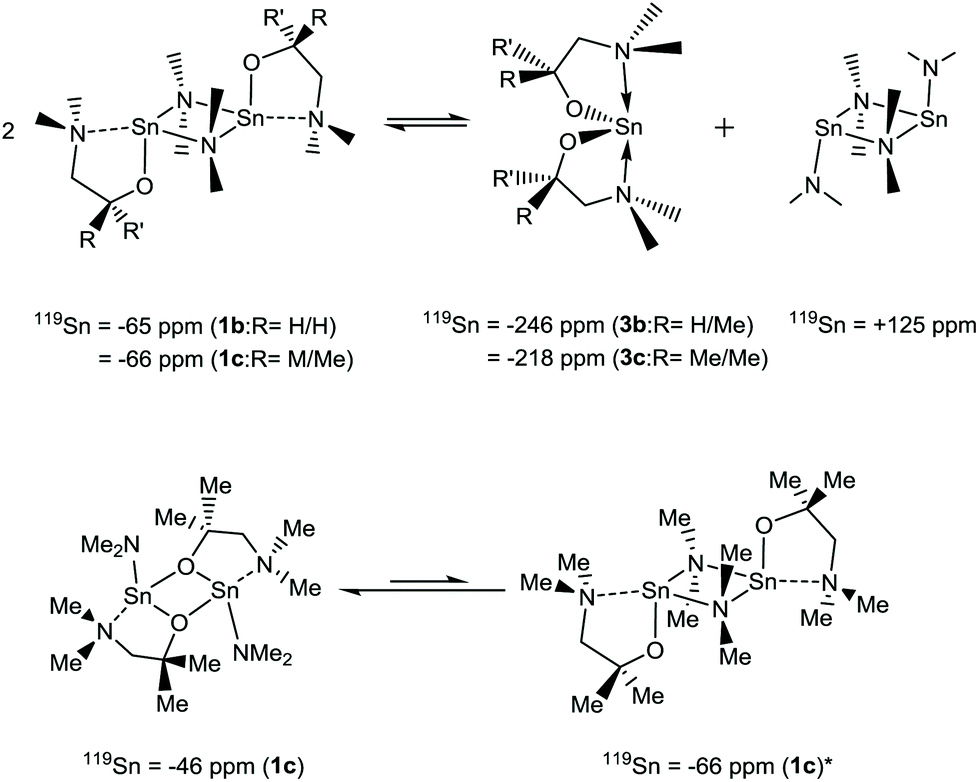 | (1) |
Fig. 6 shows the thermal profiles of [Sn{dmae}2] (3a), [Sn{dmap}2] (3b), [Sn{dmamp}2] (3c) and [Sn{Fdmamp}2] (3d), which were carried out under an inert atmosphere.
 | ||
| Fig. 6 Mass loss/temperature plots of [Sn{dmae}2] (3a), [Sn{dmap}2] (3b), [Sn{dmamp}2] (3c) and [Sn{Fdmamp}2] (3d). Ramp: 5 °C min−1, Ar flow 20 mL min−1. | ||
Apart from [Sn{dmae}2] (3a), all complexes display a single large mass loss event consistent with volatility or extensive one-step decomposition. The low residual masses (Table 7) indicate that the former is more likely, with the final masses for all four complexes presenting well below that which would be expected for metallic tin, indicating in all cases a of degree of volatility. While the TGA data provide an indication of the volatility of the complexes, decomposition characteristics are less easy to discern for complexes with significant volatility.
| Compound | M.p (°C) | Residual mass (%) | Expected residual mass (%) | ||
|---|---|---|---|---|---|
| SnO | SnO2 | Sn | |||
| (3a) | 45–46 | 13.1 | 45.7 | 51.1 | 40.2 |
| (3b) | 75–78 | 12.1 | 41.7 | 46.7 | 36.8 |
| (3c) | 56–60 | 11.2 | 38.4 | 42.9 | 33.8 |
| (3d) | 82–87 | 3.0 | 23.7 | 26.6 | 20.9 |
The thermal behaviour of complexes 3a–d were further investigated using isothermal TGA-studies (Fig. 7). At the fixed temperature of 70 °C, the mass loss for each compound was measured over a period of 120 min. In all measurements, an approximate linear weight loss was observed, which could be indicative of sublimation, with limited signs of decomposition.
 | ||
| Fig. 7 Isothermal plots at 70 °C of Sn(dmae)2 (3a), Sn(dmap)2 (3b), Sn(dmamp)2 (3c) and Sn(Fdmamp)2 (3d). Ar flow 20 mL min−1. | ||
As indicated by the initial TGA (Fig. 6), all compounds displayed degrees of volatility, with evaporation rates decreasing in the order (3c) ≫ (3b) > (3d) > (3a). This order is consistent with the observed onsets of volatility in the variable temperature TGA, and the observation that 3c and 3d can both be distilled under reduced pressure as part of the purification process.
The evaporation rates (Table 8 ESI†) were found to be in the range ∼33–119 μg min−1 cm−2. From the thermal studies, one can conclude that among the Sn(II) aminoalkoxides studied here, the dimethyl and monomethyl substituted complexes (3c) and (3b) show the greatest promise as ALD applications with evaporation rates of 118.7 μg min−1 cm−2 (3b) and 55 μg min−1 cm−2 (3c).
Conclusions
The development of precursors for ALD of electronically relevant materials such as SnO is essential to the future advancement of materials science. In this paper we present a family of homoleptic and heteroleptic tin(II) amino(fluoro)alkoxide complexes of the form [Sn(NR2)(ON)]x (NR2 = NMe2 (1a–d) or N(SiMe3)2 (2a–d); x = 1 or 2) and [Sn(κ2-ON)2] (3a–d), which have been synthesised and fully characterised. In the case of the heteroleptic systems, low thermal stabilities, and low volatilities preclude their application to ALD. However, the bis-aminoaloxide systems, of which 3c has previously been utilised as an ALD precursor for SnO deposition, all show stability, and some degree of volatility, especially 3b and 3c, as indicated by their rates of evaporation. Given the limited choice of precursors available for ALD of Sn(II) oxide thin films, complexes 3b and 3c, are clearly promising precursor candidates for vapour deposition processes.Indeed, while the work presented here primarily concerns precursor development and molecular characterisation, detailed studies on the ALD of Sn(II) oxides using these precursors, and subsequent thin film characterisation, will be published separately.
Experimental
General experimental
Full experimental and equipment detail can be found in the ESI.†Elemental analyses were performed using an Exeter Analytical CE 440 analyser. 1H, 13C and 119Sn NMR spectra were recorded on Bruker Advance 300 or 500 MHz FT-NMR spectrometers, as appropriate, as saturated solutions at room temperature. Chemical shifts are in ppm with respect to Me4Si (1H, 13C) neat SnCl4 (119Sn) and CFCl3 (19F). TGA and PXRD were performed using a PerkinElmer TGA7 and a Bruker D8 instrument (Cu-kα radiation), respectively.
Given the sensitivity of Sn(II) alkoxide and amide systems to both oxygen and moisture, and the propensity of such compounds to form oxo-clusters, inert atmosphere methodologies were employed throughout. Solvents were dried and degassed under an argon atmosphere over activated alumina columns using an Innovative Technology solvent purification system (SPS). The Sn(II) amides, [Sn{N(SiMe3)2}2]31 and [Sn{NMe2}2],32 were prepared by literature methods. The pro-ligands H{dmamp} and H{Fdmamp} (c and d) were made according to literature procedure,18c the pro-ligands H{dmae} and H{dmap} (a and b) were purchased from commercial sources.
[Sn(OCH2CH2NMe2)NMe2]2 – (1a)
A solution of dimethylaminoethanol (0.18 g, 2 mmol) in hexane (20 mL) was added to a cooled solution of Sn(NMe2)2 (0.41 g, 2 mmol) in hexane (20 mL) and left to stir for 2 h. After removal of the volatiles in vacuo, the solid residue was re-dissolved in hexane and filtered through Celite®. The volume was subsequently reduced, and colourless crystals obtained at −28 °C. (0.28 g, 55%) Elemental analysis: Found (calculated): 28.38 (28.72), 6.86 (6.43), 11.08 (11.16). 1H NMR (500 MHz, C6D6); δppm 4.01–4.03 (m, 2H, OC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 2.91 (br s, 6H, SnN
2), 2.91 (br s, 6H, SnN![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) 2), 2.20–2.22 (m, 2H, NC2), 1.98 (s, 6H, CH2N2). 13C NMR (125.7 MHz, C6D6); δppm 62.46 (1C, NH2), 61.22 (1C, OH2), 44.14 (2C, CH2N2), 42.42 (2C, SnN2). 119Sn NMR (186.3 MHz, C6D6); δppm −68 (br).
2), 2.20–2.22 (m, 2H, NC2), 1.98 (s, 6H, CH2N2). 13C NMR (125.7 MHz, C6D6); δppm 62.46 (1C, NH2), 61.22 (1C, OH2), 44.14 (2C, CH2N2), 42.42 (2C, SnN2). 119Sn NMR (186.3 MHz, C6D6); δppm −68 (br).
[Sn(OCH(CH3)CH2NMe2)NMe2]2 – (1b)
1b was prepared in a similar manner to 1a using 0.41 g of Sn(NMe2)2 (2 mmol) and 0.21 g of 1-dimethylamino-2-propanol (2 mmol). Yield = 0.37 g (70%). Elemental analysis: Found (calculated): 31.80 (31.72), 6.86 (6.85), 10.58 (10.57). 1H NMR (500 MHz, C6D6); δppm 4.17–4.19 (m, 1H, CMe), 2.82 (br s, 6H, SnN2), 2.19–2.31 (br m, 1H, C2NMe2), 2.01 (br s, 6H, CH2N2), 1.82 (dd, J = 11.6, 2.5 Hz, 1H, C2NMe2), 1.30 (d, J = 6.0 Hz, 3H, CH). 13C NMR (125.7 MHz, C6D6); δppm 68.84 (1C, H2) 66.64 (1C, HMe), 44.78 (2C, CH2N2), 42.57 (2C, SnN2), 24.18 (1C, CH). 119Sn NMR (186.3 MHz, C6D6); δppm −65.
[Sn(OC(CH3)2CH2NMe2)NMe2]2 – (1c)
1c was prepared in a similar manner to 1a using 0.41 g of Sn(NMe2)2 (2 mmol) and 0.24 g of 1-dimethylamino-2-methyl-2-propanol (2 mmol). Yield = 0.40 g (71%). Elemental analysis: Found (calculated): 34.80 (34.44), 7.22 (7.23), 10.04 (10.10). 1H NMR (500 MHz, C6D6); δppm 2.78 (s, 6H, SnN2), 2.19 (s, 2H, C2), 2.13 (s, 6H, CH2N2), 1.37 (s, 6H, C2). 13C NMR (125.7 MHz, C6D6); δppm 72.80 (1C, OMe2), 72.19 (1C, H2), 47.81 (2C, CH2N2), 42.96 (2C, SnN2), 33.88 (2C, C2). 119Sn NMR (186.3 MHz, C6D6); δppm 127 (Sn(NMe2)2), −46, −66, −217 (Sn(dmamp)2).
[Sn(OC(CF3)2CH2NMe2)NMe2] – (1d)
1d was prepared in a similar manner to 1a using HOC(CF3)2CH2NMe2 (0.45 g, 2 mmol) and 0.41 g (2 mmol) of Sn(NMe2)2. Yield = 0.22 g (28%). Elemental analysis: Found (calculated): 24.78 (24.83), 3.86 (3.65), 7.23 (7.24). 1H NMR (500 MHz, D8−tol, 90 °C); δppm 2.66 (br s, 2H, C2), 2.08 (br s, 6H, CH2N2), 2.52 and 2.26 (2:4, br s, 6H, SnN2). 13C NMR (125.7 MHz, D8−tol, 90 °C); δppm 41.68 (2C, SnN2), 57.50 (1C, H2). 19F NMR (470.6 MHz, D8−tol, 90 °C); δppm −76.29 (major), −76.75 (minor). 119Sn NMR (186.3 MHz, D8−tol, 90 °C); δppm 85 (br), −117, −132.
[Sn(OCH2CH2NMe2)HMDS]2 – (2a)
Compound 2a was synthesised according to an adapted literature procedure24 using 0.18 g (2 mmol) of dimethylaminoethanol and 0.89 g and (2 mmol), [Sn{N(SiMe3)2}2] Yield = 0.59 g (80%). 1H NMR (500 MHz, C6D6); δppm 3.73 (t, J = 5.3 Hz, 2H, OC2), 2.21 (br m, 2H, C2N), 1.99 (s, 6H, N2), 0.46 (s, 18H, Si3). 13C NMR (125.7 MHz, C6D6); δppm 61.15 (1C, OH2), 57.99 (1C, H2NMe2), 44.29 (2C, N2), 7.37 (d, 6C, 1JSiC = 54.6 Hz, ![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) Me3). 119Sn NMR (186.3 MHz, C6D6); δppm −168.
Me3). 119Sn NMR (186.3 MHz, C6D6); δppm −168.
[Sn(OCH(CH3)CH2NMe2)HMDS]2 – (2b)
A solution of 1-dimethylamino-2-propanol (0.21 g, 2 mmol) in hexane (20 mL) was added to a cooled solution of [Sn{N(SiMe3)2}2] (0.89 g, 2 mmol) in hexane (20 mL) and left to stir for 2 h. After removal of the volatiles in vacuo, the solid residue was re-dissolved in hexane and filtered through Celite®. The volume was subsequently reduced, and colourless crystals obtained at −28 °C. (0.57 g, 75%) Elemental analysis: Found (calculated): C 35.05 (34.65), H 7.66 (7.93), N 7.30 (7.35). 1H NMR (500 MHz, C6D6); δppm 4.11 (m, 1H, C()Me), 2.24 (t, J = 11.78 Hz, 1H, C2NMe2), 1.97 (s, 6H, N2), 1.71 (m, 2H, C2NMe2), 1.31 (d, J = 6.1 Hz, 3H, C(H)), 0.46 (s, 18H, Si3). 13C NMR (125.7 MHz, C6D6); δppm 68.87 (1C, O(H)Me), 67.25 (1C, H2), 45.25 (2C, N2), 22.44 (1C, OC(H)), 7.02 (6C, SiMe3). 119Sn NMR (186.3 MHz, C6D6); δppm −135.
[Sn(OC(CH3)2CH2NMe2)HMDS]2 – (2c)
2c was prepared in a similar manner to 2b using 0.89 g (2 mmol) of [Sn{N(SiMe3)2}2] and 0.24 g (2 mmol) of dimethylamino-2-methyl-2-propanol. Yield = 0.52 g (66%) Elemental analysis: Found (calculated): C 36.30 (36.46), H 8.20 (8.16), N 6.79 (7.09). 1H NMR (500 MHz, C6D6); δppm 2.01 (s, 6H, N2), 1.98 (s, 2H, C2), 1.23 (s, 6H, C2), 0.42 (s, 18H, Si3). 13C NMR (125.7 MHz, C6D6); δppm 75.61 (1C, OMe2), 71.56 (1C, H2), 47.90 (2C, N2), 32.42 (2C, C2), 6.40 (6C, 1JSiC = 55.0 Hz, SiMe3). 119Sn NMR (186.3 MHz, C6D6); δppm 123.
[Sn(OC(CF3)2CH2NMe2)HMDS] – (2d)
2d was prepared in a similar manner to 2b using 0.89 g (2 mmol) of [Sn{N(SiMe3)2}2] and 0.45 g (2 mmol) of HOC(CF3)2CH2NMe2. Yield = 0.35 g (35%). Elemental analysis: Found (calculated): 28.63 (28.64), 5.19 (5.21), 5.62 (5.57). 1H NMR (500 MHz, C6D6); δppm 2.34 (s, 2H, C2), 1.84 (s, 6H, N2), 0.29 (s, 18H, Si3). 13C NMR (125.7 MHz, C6D6); δppm 57.93 (1C, H2), 46.27 (2C, N2), 5.29 (6C, Si3). 19F NMR (470.6 MHz, C6D6); δppm = −76.88. 119Sn NMR (186.3 MHz, C6D6); δppm 94.
[Sn(OCH2CH2NMe2)2] – (3a)
Compound 3a was synthesised according to an adapted literature procedure16 using 0.36 g (4 mmol) of HOCH2CH2NMe2 and 0.89 g (2 mmol) of [Sn{N(SiMe3)2}2]. Yield = 0.45 g (76%). Elemental analysis: Found (calculated): 32.56 (32.58), 6.83 (6.83), 9.51 (9.50). 1H NMR (500 MHz, C6D6); δppm 4.24 (m, 2H, OC2), 2.31–2.39 (m, 4H, C2N), 2.10 (s, 12H, N2). 13C NMR (125.7 MHz, C6D6); δppm 63.43 (2C, OH2), 62.28 (2C, H2N), 43.46 (4C, N2). 119Sn NMR (186.3 MHz, C6D6); δppm −279.
[Sn(OCH(CH3)CH2NMe2)2] – (3b)
A solution of [Sn{N(SiMe3)2}2] (0.89 g, 2 mmol) in hexane (20 mL) was added to a cooled solution of HOC(H)(CH3)CH2NMe2 (0.41 g, 4 mmol) in hexane (20 mL) and left to stir for 2 h. After removal of the volatiles in vacuo, the solid residue was re-dissolved in hexane and filtered through Celite®. The volume was subsequently reduced, and colourless crystals obtained at −28 °C. (0.41 g, 64%) elemental analysis: Found (calculated): C 37.11 (37.18), H 7.47 (7.49), N 8.78 (8.67). 1H NMR (500 MHz, D8–tol, 90 °C); δppm 4.18 (m, 1H, CMe), 2.47 (m, 2H, C2), 2.16 (s, 12H, N2), 1.91 (m, 2H, C2), 1.21 (m, 6H, CH) 13C NMR (125.7 MHz, 90 °C); δppm 68.90 (2C, O(H)Me), 68.23 (2C, H2), 44.14 (4C, N2), 24.37 (2C, CH) 119Sn NMR (186.3 MHz, 90 °C); δppm −246.
[Sn(OC(CH3)2CH2NMe2)2] – (3c)
3c was prepared in a similar manner to 3b using 0.89 g (2 mmol) of [Sn{N(SiMe3)2}2] and 0.47 g (4 mmol) of HOC(CH3)2CH2NMe2. Yield = 0.51 g (72%). Elemental analysis: Found (calculated): 41.08 (41.05), 8.11 (8.04), 7.95 (7.98). 1H NMR (500 MHz, C6D6); δppm 2.34 (s, 4H, C2), 2.24 (s, 12H, N2), 1.39 (s, 12H, C2). 13C NMR (125.7 MHz, C6D6); δppm 74.28 (2C, OC()2), 71.02 (2C, H2), 46.78 (4C, N2), 34.45 (4C, C2). 119Sn NMR (186.3 MHz, C6D6); δppm −218.
[Sn(OC(CF3)2CH2NMe2)2] – (3d)
3d was prepared in a similar manner to 3b using 0.89 g (2 mmol) of [Sn{N(SiMe3)2}2] and 0.90 g (4 mmol) HOC(CF3)2CH2NMe2. Yield = 0.58 g, (51%). Elemental analysis: Found (calculated): 24.99 (25.42), H 2.97 (2.84), N 4.76 (4.94). 1H NMR (500 MHz, C6D6); δppm 2.54 (br s, 4H, C2), 2.07 (br s, 6H, N), 1.75 (br s, 6H, N). 13C NMR (125.7 MHz, C6D6); δppm 124.89 (q, 1JCF = 290 Hz, 4C, F3), 82.83 (m, 2C, O(CF3)2), 58.07 (H2), 47.78 (2C, N), 45.97 (2C, N). 19F NMR (470.6 MHz, C6D6); δppm −76.40 (6F, CF3), −77.55 (6F, CF3). 119Sn NMR (186.3 MHz, C6D6); δppm −322.
Single crystal X-ray diffraction
Experimental details relating to the single-crystal X-ray crystallographic studies for compounds 1a–d, 2a–d and 3a–d are summarised in Tables S1 (see ESI†). All crystallographic data was collected at 150(2) K on a SuperNova, Dual, EosS2 diffractometer using radiation Cu-Kα (λ = 1.54184 Å) or Mo-Kα (λ = 0.71073 Å). All structures were solved by direct methods followed by full-matrix least squares refinement on F2 using the WINGX-2014 suite of programs33 or OLEX2.34 All hydrogen atoms were included in idealised positions and refined using the riding model. Crystals were isolated from an argon filled Schlenk flask and immersed under oil before being mounted onto the diffractometer.Thermogravimetric analysis (TGA)
TGA was collected using a TGA 4000 PerkinElmer system, housed in an argon filled glovebox. Samples were prepared air sensitively, and TGAs were performed under a flow of Ar at 20 mL min−1 and heated from 30 °C to 400 °C at a ramp rate of 10 °C min−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the University of Bath (PhD studentships to J. D. P.), and the Department of Chemistry (Masters Studentship to M. W. S.). We also thank Dr Gabriele Kociok-Köhn for assistance with single crystal X-ray diffraction experiments.Notes and references
- (a) E. Fortunato, P. Barquinha and R. Martins, Adv. Mater., 2012, 24, 2945–2986 CrossRef CAS PubMed; (b) Z. Wang, P. K. Nayak, J. A. Caraveo-Frescas and H. N. Alshareef, Adv. Mater., 2016, 28, 3831–3892 CrossRef CAS PubMed; (c) I. Sayago, E. Hontañón and M. Aleixandre, Preparation of tin oxide nanostructures by chemical vapor deposition, Elsevier Inc., 2020 Search PubMed.
- K. H. L. Zhang, K. Xi, M. G. Blamire and R. G. Egdell, J. Phys.: Condens. Matter, 2016, 28, 383002 CrossRef PubMed.
- (a) K. J. Saji, K. Tian, M. Snure and A. Tiwari, Adv. Electron. Mater., 2016, 2, 1500453 CrossRef; (b) T. Daeneke, P. Atkin, R. Orrell-Trigg, A. Zavabeti, T. Ahmed, S. Walia, M. Liu, Y. Tachibana, M. Javaid, A. D. Greentree, S. P. Russo, R. B. Kaner and K. Kalantar-Zadeh, ACS Nano, 2017, 11, 10974–10983 CrossRef CAS PubMed; (c) K. Yim, Y. Youn, M. Lee, D. Yoo, J. Lee, S. H. Cho and S. Han, Npj Comput. Mater., 2018, 4, 17–24 CrossRef; (d) Y. R. Wang, S. Li and J. B. Yi, J. Phys. Chem. C, 2018, 122, 4651–4661 CrossRef CAS; (e) D. R. Kripalani, P.-P. Sun, P. Lin, M. Xue and K. Zhou, Phys. Rev. B, 2019, 100, 214112-1–214112-8 Search PubMed; (f) S. Yim, T. Kim, B. Yoo, H. Xu, Y. Youn, S. Han and J. K. Jeong, ACS Appl. Mater. Interfaces, 2019, 11, 47025–47036 CrossRef CAS PubMed; (g) M. Graužinytė, D. Tomerini, S. Goedecker and J. A. Flores-Livas, J. Phys. Chem. C, 2019, 123, 14909–14913 CrossRef.
- J. A. Caraveo-Frescas, P. K. Nayak, H. A. Al-Jawhari, D. B. Granato, U. Schwingenschlögl and H. N. Alshareef, ACS Nano, 2013, 7, 5160–5167 CrossRef CAS PubMed.
- (a) M. Oudah, J. N. Hausmann, S. Kitao, A. Ikeda, S. Yonezawa, M. Seto and Y. Maeno, Sci. Rep., 2019, 9, 1831 CrossRef PubMed; (b) M. K. Forthaus, K. Sengupta, O. Heyer, N. E. Christensen, A. Svane, K. Syassen, D. I. Khomskii, T. Lorenz and M. M. Abd-Elmeguid, Phys. Rev. Lett., 2010, 105, 157001 CrossRef CAS PubMed; (c) S. Xu, Y. Zou, J. Sun, Z. Liu, X. Yu, J. Gouchi, Y. Uwatoko, Z. G. Cheng, B. Wang and J. Cheng, Phys. Rev. B, 2020, 101, 104501–104508 CrossRef CAS.
- J. P. Allen, D. O. Scanlon, L. F. J. Piper and G. W. Watson, J. Mater. Chem. C, 2013, 1, 8194–8208 RSC.
- (a) I. C. Chiu, L. Yun-Shiuan, T. Min-Sheng and I. C. Cheng, IEEE Electron Device Lett., 2014, 35, 1263–1265 CAS; (b) Y. Li, J. Yang, Y. Wang, P. Ma, Y. Yuan, J. Zhang, Z. Lin, L. Zhou, Q. Xin and A. Song, IEEE Electron Device Lett., 2018, 39, 208–211 CAS; (c) H.-J. Joo, M.-G. Shin, H.-S. Jung, H.-S. Cha, D. Nam and H.-Y. Kwon, Materials, 2019, 12, 3815–3825 CrossRef CAS PubMed.
- (a) R. Dolbec, M. A. El Khakani, A. M. Serventi, M. Trudeau and R. G. Saint-Jacques, Thin Solid Films, 2002, 419, 230–236 CrossRef CAS; (b) K. Iizuka, M. Kambara and T. Yoshida, Sens. Actuators, B, 2011, 155, 551–556 CrossRef CAS; (c) S. H. Park, Y. C. Son, W. S. Willis, S. L. Suib and K. E. Creasy, Chem. Mater., 1998, 10, 2389–2398 CrossRef CAS; (d) T. Toyama, Y. Seo, T. Konishi, H. Okamoto and Y. Tsutsumi, Appl. Phys. Express, 2011, 4, 075503 Search PubMed.
- (a) T. Wildsmith, M. S. Hill, A. L. Johnson, A. J. Kingsley and K. C. Molloy, Chem. Commun., 2013, 49, 8773–8775 RSC; (b) M. S. Hill, A. L. Johnson, J. P. Lowe, K. C. Molloy, J. D. Parish, T. Wildsmith and A. L. Kingsley, Dalton Trans., 2016, 45, 18252–18258 RSC; (c) B. Kumar, D.-H. Lee, S.-H. Kim, B. Yang, S. Maeng and S.-W. Kim, J. Phys. Chem. C, 2010, 114, 11050–11055 CrossRef CAS.
- I. Barbul, A. L. Johnson, G. Kociok-Köhn, K. C. Molloy, C. Silvestru and A. L. Sudlow, ChemPlusChem, 2013, 78, 866–874 CrossRef CAS PubMed.
- (a) J.-H. Lee, M. Yoo, D. Kang, H.-M. Lee, W.-h. Choi, J. W. Park, Y. Yi, H. Y. Kim and J.-S. Park, ACS Appl. Mater. Interfaces, 2018, 10, 33335–33342 CrossRef CAS PubMed; (b) J. D. Parish, M. W. Snook, A. L. Johnson and G. Kociok-Köhn, Dalton Trans., 2018, 47, 7721–7729 RSC; (c) A. L. Johnson and J. D. Parish, in Organometallic Chemistry, RSC, 2018, pp. 1–53 Search PubMed.
- (a) S. H. Kim, I.-H. Baek, D. H. Kim, J. J. Pyeon, T.-M. Chung, S.-H. Baek, J.-S. Kim, J. H. Han and S. K. Kim, J. Mater. Chem. C, 2017, 5, 3139–3145 RSC; (b) J. H. Han, Y. J. Chung, B. K. Park, S. K. Kim, H.-S. Kim, C. G. Kim and T.-M. Chung, Chem. Mater., 2014, 26, 6088–6091 CrossRef CAS.
- M. G. Chae, S. H. Han, B. K. Park, T.-M. Chung and J. H. Han, Appl. Surf. Sci., 2021, 547, 148758 CrossRef CAS.
- S. M. George, J. H. Nam, G. Y. Lee, J. H. Han, B. K. Park, C. G. Kim, D. J. Jeon and T.-M. Chung, Eur. J. Inorg. Chem., 2016, 2016, 5539–5546 CrossRef CAS.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- N. N. Zemlyansky, I. V. Borisova, M. G. Kuznetsova, V. N. Khrustalev, Y. A. Ustynyuk, M. S. Nechaev, V. V. Lunin, J. Barrau and G. Rima, Organometallics, 2003, 22, 1675–1681 CrossRef CAS.
- (a) S. Ashraf, H. C. Aspinall, J. Bacsa, P. R. Chalker, H. O. Davies, A. C. Jones, P. O'Brien and J. S. Wrench, Polyhedron, 2015, 85, 761–769 CrossRef CAS; (b) H. C. Aspinall, J. Bacsa, A. C. Jones, J. S. Wrench, K. Black, P. R. Chalker, P. J. King, P. Marshall, M. Werner, H. O. Davies and R. Odedra, Inorg. Chem., 2011, 50, 11644–11652 CrossRef CAS PubMed; (c) H. C. Aspinall, J. F. Bickley, J. M. Gaskell, A. C. Jones, G. Labat, P. R. Chalker and P. A. Williams, Inorg. Chem., 2007, 46, 5852–5860 CrossRef CAS PubMed; (d) G. Carta, N. El Habra, G. Rossetto, L. Crociani, G. Torzo, P. Zanella, M. Casarin, G. Cavinato, G. Pace, S. Kaciulis and A. Mezzi, Thin Solid Films, 2008, 516, 7354–7360 CrossRef CAS; (e) L. Crociani, G. Carta, M. Natali, V. Rigato and G. Rossetto, Chem. Vap. Deposition, 2011, 17, 6–8 CrossRef CAS; (f) R. O'Kane, J. Gaskell, A. C. Jones, P. R. Chalker, K. Black, M. Werner, P. Taechakumput, S. Taylor, P. N. Heys and R. Odedra, Chem. Vap. Deposition, 2007, 13, 609–617 CrossRef; (g) R. J. Potter, P. A. Marshall, J. L. Roberts, A. C. Jones, P. R. Chalker, M. Vehkamaeki, M. Ritala, M. Leskelae, P. A. Williams, H. O. Davies, N. L. Tobin and L. M. Smith, Mater. Res. Soc. Symp. Proc., 2004, 784, 97–108 Search PubMed; (h) P. A. Williams, A. C. Jones, M. J. Crosbie, P. J. Wright, J. F. Bickley, A. Steiner, H. O. Davies, T. J. Leedham and G. W. Critchlow, Chem. Vap. Deposition, 2001, 7, 205–209 CrossRef CAS; (i) P. A. Williams, J. L. Roberts, A. C. Jones, P. R. Chalker, N. L. Tobin, J. F. Bickley, H. O. Davies, L. M. Smith and T. J. Leedham, Chem. Vap. Deposition, 2002, 8, 163–170 CrossRef CAS.
- (a) G. Carta, N. El Habra, G. Rossetto, G. Torzo, L. Crociani, M. Natali, P. Zanella, G. Cavinato, V. Matterello, V. Rigato, S. Kaciulis and A. Mezzi, Chem. Vap. Deposition, 2007, 13, 626–632 CrossRef CAS; (b) J. W. Park, H. S. Jang, M. Kim, K. Sung, S. S. Lee, T.-M. Chung, S. Koo, C. G. Kim and Y. Kim, Inorg. Chem. Commun., 2004, 7, 463–466 CrossRef CAS; (c) A. Verchere, S. Mishra, E. Jeanneau, H. Guillon, J.-M. Decams and S. Daniele, Inorg. Chem., 2020, 59, 7167–7180 CrossRef CAS PubMed; (d) S. H. Yoo, H. Choi, H.-S. Kim, B. K. Park, S. S. Lee, K.-S. An, Y. K. Lee, T.-M. Chung and C. G. Kim, Eur. J. Inorg. Chem., 2011, 1833–1839 CrossRef CAS.
- A. Verchère, S. Mishra, E. Jeanneau, H. Guillon, J.-M. Decams and S. Daniele, Inorg. Chem., 2020, 59, 7167–7180 CrossRef PubMed.
- W. A. Herrmann, N. W. Huber and O. Runte, Angew. Chem., Int. Ed. Engl., 1995, 34, 2187–2206 CrossRef CAS.
- L. McElwee-White, Dalton Trans., 2006, 5327–5333 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- V. N. Khrustalev, I. A. Portnyagin, N. N. Zemlyansky, I. V. Borisova, M. S. Nechaev, Y. A. Ustynyuk, M. Y. Antipin and V. Lunin, J. Organomet. Chem., 2005, 690, 1172–1177 CrossRef CAS.
- O. V. Chernov, A. Y. Smirnov, I. A. Portnyagin, V. N. Khrustalev and M. S. Nechaev, J. Organomet. Chem., 2009, 694, 3184–3189 CrossRef CAS.
- C. G. Kim, T.-M. Chung, Y. K. Lee, S. S. Lee, B. H. Ryu and S. J. Jang, 2009, Tinamino-alkoxide Complexes and Process for Preparing Thereof, Korean Research Institute of Chemical Technologyy, US8030507B2, 2009.
- A. Pop, L. Wang, V. Dorcet, T. Roisnel, J.-F. Carpentier, A. Silvestru and Y. Sarazin, Dalton Trans., 2014, 43, 16459–16474 RSC.
- A. S. Ionkin, W. J. Marshall and B. M. Fish, Organometallics, 2006, 25, 4170–4178 CrossRef CAS.
- (a) L. Wang, C. E. Kefalidis, T. Roisnel, S. Sinbandhit, L. Maron, J.-F. Carpentier and Y. Sarazin, Organometallics, 2014, 34, 2139–2150 CrossRef; (b) L. Broeckaert, P. Geerlings, A. Růžička, R. Willem and F. D. Proft, Organometallics, 2012, 31, 1605–1617 CrossRef; (c) L. Broeckaert, J. Turek, R. Olejník, A. Růžička, M. Biesemans, P. Geerlings, R. Willem and F. De Proft, Organometallics, 2013, 32, 2121–2134 CrossRef CAS.
- (a) L. Wang, V. Poirier, F. Ghiotto, M. Bochmann, R. D. Cannon, J.-F. Carpentier and Y. Sarazin, Macromolecules, 2014, 47, 2574–2584 CrossRef CAS; (b) M. J. McGeary, K. Folting and K. G. Caulton, Inorg. Chem., 1989, 28, 4051–4053 CrossRef CAS; (c) T. J. Boyle, T. Q. Doan, L. A. M. Steele, C. Apblett, S. M. Hoppe, K. Hawthorne, R. M. Kalinich and W. M. Sigmund, Dalton Trans., 2012, 41, 9349–9364 RSC.
- T. Fjeldberg, H. k. Hope, M. F. Lappert, P. P. Power and A. J. Thorne, J. Chem. Soc., Chem. Commun., 1983, 639–641 RSC.
- M. M. Olmstead and P. P. Power, Inorg. Chem., 1984, 23, 413–415 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2055535–2055545. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02480a |
| This journal is © The Royal Society of Chemistry 2021 |