
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination sphere hydrogen bonding as a structural element in metal–organic Frameworks
Chris S.
Hawes

School of Chemical and Physical Sciences, Keele University, Keele ST5 5BG, UK. E-mail: c.s.hawes@keele.ac.uk
First published on 1st April 2021
Abstract
In the design of new metal–organic frameworks, the constant challenges of framework stability and structural predictability continue to influence ligand choice in favour of well-studied dicarboxylates and similar ligands. However, a small subset of known MOF ligands contains suitable functionality for coordination sphere hydrogen bonding which can provide new opportunities in ligand design. Such interactions may serve to support and rigidity the coordination geometry of mononuclear coordination spheres, as well as providing extra thermodynamic and kinetic stabilisation to meet the challenge of hydrolytic stability in these materials. In this perspective, a collection of pyrazole, amine, amide and carboxylic acid containing species are examined through the lens of (primarily) inner-sphere hydrogen bonding. The influence of these interactions is then related to the overall structure, stability and function of these materials, to provide starting points for harnessing these interactions in future materials design.
Introduction
As the study of coordination polymers and metal–organic frameworks continues to progress, focus has begun to shift from understanding their preparation and fundamental properties towards their use in real-world applications.1 Many such applications have been suggested for such materials; although gas adsorption and separation is the most widely discussed,2 other applications including liquid-phase separations,3 nonlinear optics,4 anomalous thermal expansion,5 proton conduction,6 and molecular magnetism7 have been proposed and successfully demonstrated. For most proposed applications of coordination polymer materials, two fundamental challenges still remain to be overcome. Firstly and perhaps most importantly, the chemical stability of framework materials has been a source of difficulty for many years,8 particularly in the case of water stability in polycarboxylate cluster systems.9 Secondly, an element of design and predictability within coordination polymer structures is highly desirable, and can still prove elusive for some systems.10 Although substantial progress has been made to this end, especially in the case of carboxylate cluster systems and rigid linker molecules,11 further examples of structure prediction in non-traditional or highly flexible systems continue to prove challenging.Several approaches have been applied to the problem of hydrolytic stability in coordination polymer systems. Broad chemical stability can be introduced with the use of zirconium-carboxylate nodes or other hard cations,12 where the inert zirconium–oxygen bond can effectively prevent hydrolysis. Similarly, metal ions such as chromium(III) show excellent resistance to hydrolysis, which can be related to a combination of the chemical inertness of the ion itself and the high strength of the M–O bonds in the parent oxides.13 Where softer or more labile metal ions are desired, azolate-based ligands, particularly imidazolate, in combination with tetrahedral metal ions form notably stable frameworks with zeolitic topologies.14 In both cases, however, the high strength and corresponding low reversibility of the individual metal–ligand bonds can prove a hindrance to control in the material synthesis, which relies on a reversible self-assembly process. Mixed-ligand approaches, in which neutral and anionic bridging ligands are combined, have also been shown to improve the chemical stability of coordination polymer materials.15 Such systems tend to favour mononuclear metal nodes consisting of both charge-balancing anionic ligands and neutral co-ligands. This arrangement satisfies both the charge and coordination number requirements of transition metal ions without the need for hydrolytically vulnerable carboxylate bridges between metal ions, or reactive highly solvated metal nodes. Systems with mixed ligands, including the widely studied pillared-layer type MOFs, also provide multiple avenues for structural tuning across both ligand types to improve stability.16
Hydrogen bonding, particularly within the coordination sphere, is another strategy which may lead to enhancement in the chemical stability at the nodes, and therefor of the framework materials themselves. These interactions are well known to play key roles in controlling the geometry and reactivity of the coordination sphere within metalloenzymes,17 and have been widely and successfully employed in synthetic catalysts and biological models,18 not only as proton sources but as stabilising forces to favour specific active site geometries.19 In MOF chemistry, while the importance of hydrogen bonding at the coordination sphere has been demonstrated in terms of providing sites for preferential guest adsorption,20 and a key handle for proton conduction,21 the role of static hydrogen bonding as a structure directing element has received less attention.22 This is especially true given the typical nature of the most widely used MOF ligands; while hydrogen bond acceptor ligands such as carboxylates are ubiquitous,23 few common MOF ligands offer hydrogen bond donors proximal to the metal binding sites. As a consequence, inner-sphere hydrogen bonding is not the norm in coordination polymers or MOFs unless ligands capable of this function have been explicitly selected. Notable exceptions are the small-molecule O–H donor hydroxido, aqua or alcohol ligands. While these are often found bound to the coordination sphere, either originating from the reaction solvent or acting as a structural element within polynuclear cluster nodes, these groups are excluded from the present discussion. Firstly, while there are some examples of organic bridging ligands containing alcohol functional groups within MOFs,24 the majority of coordinated methanol, ethanol or water molecules tend to be removed during activation of the resulting frameworks. Secondly, these species do not tend to contribute to the overall topology of the frameworks, making them less useful to consider from a ligand design perspective.
In this perspective article, coordination sphere hydrogen bonding as a structure-directing force and a possible strategy for improving the stability of MOFs will be outlined. Key results in the structural chemistry of coordination polymers and MOFs derived from ligands containing hydrogen bond donors in close proximity to the metal binding functionality will be examined. The ligands of interest are presented in Fig. 1. These species are considered from the perspective of stable, reproducible and widely applicable building units for the design of new functional materials, and the role that incorporating hydrogen bonding as an element of ligand design may play in this task. The standard graph set nomenclature used to categorise hydrogen bonding in crystal structures will be used throughout,25 particularly those described by the ring designator Ryx(Z). This nomenclature conveniently classifies a motif by the number of hydrogen bond acceptors, donors and the ring size (x, y and z, respectively).
 | ||
| Fig. 1 Ligands of interest to this review. Naming schemes are derived from the fully protonated neutral forms of the ligands (based on the most acidic protons and typical coordination modes). | ||
Hydrogen bonding in MOFs: general considerations
When considering the total stabilisation energy associated with a coordinatively saturated d block ion, the additional stabilisation provided by a moderate strength intramolecular hydrogen bond (e.g. a neutral N–H donor and carboxylate acceptor) of ca. 10–20 kJ mol−1 is small.26 However, it has already been shown that such interactions can be synergic with other elements of the bonding involved, leading to outsized influences in rigidifying and stabilising the coordination sphere.27 The extra stabilisation energies are also very similar to the energetic tuning which can be achieved through other aspects of ligand design. Friščić and co-workers have recently shown that wholesale changes in ligand electronics, visualised as changes in the Hammett σ-parameter of substituted imidazoles from −0.3 to +0.7 in the formation of sodalite ZIFs, result in a total variation in enthalpy of formation of ca. 30 kJ mol−1.28 This is comparable to that accessible through installation of coordination sphere hydrogen bonding, with the demonstrated potential to influence reaction outcomes.Still, the addition of one or two hydrogen bonds within the coordination sphere is unlikely to convincingly shift the overall energy balance of hydrolysis and ligand dissociation in a hydrolytically labile MOF. Kinetic contributions in the stabilisation of the coordination sphere itself (both physically blocking access to the metal site and raising the energetic barrier to the first hydrolysis step), however, play an important role in stability.29 For MOFs formed from comparably labile metal ions such as zinc(II) and copper(II), in the few cases of high water stability this is frequently a kinetic influence.30 The stability of such systems in the presence of excess water is derived from the magnitude of the energy barrier towards ligand displacement, not necessarily the total energy of the intact coordination sphere. As such, even small stabilising influences such as hydrogen bonding may impact the lifetimes of these species in aqueous environments. Another aspect to consider when seeking to engineer these species is the complex energetic landscape on which MOFs exist. There are many structural permutations which may lie close together on complex potential energy surfaces in these species,31 hence allowing weak interactions and subtle directing forces to play a more substantial role.
Hydrogen bonding has recently been explicitly shown as a kinetic stabilisation strategy for restricting ligand rotations within MOFs. In 2020, She and co-workers reported significant stability enhancement in UiO-67 derivatives in the pH range 2–12 by installation of hydrogen-bonding amine groups arranged ortho to the coordinating carboxylate groups in L1.32 This was calculated to raise the rotational energy barrier by ca. 20 kJ mol−1 in the free ligand, and by as much as 60 kJ mol−1 in the MOF. Rather than purely altering the strength of the Zr–O bonding, this modification also acts to increase the rate of framework repair following ligand displacement by hydrolysis, by forcing the carboxylate group to remain in close proximity to the metal cluster node and thereby retaining the reverse hydrolysis reaction as a viable reaction pathway. Indeed, MOFs containing ortho-amino carboxylates have tended to show good stability in some systems,33 and generally ligand rigidification strategies have tended to improve stability in other materials. Zhou and co-workers have shown a strategy of ligand rigidification can be used to obtain improved water stability characteristics in zirconium MOFs.34 In that case, the authors employed covalent bonding or steric influences as the source of rigidification of the ligand core. Backbone hydrogen bonding influences on stability were also reported by Banerjee in a porphyrinic covalent-organic framework.35 Extending this hypothesis to the vulnerable metal ion itself, there is a plausible case to be made that hydrogen bonding within the coordination sphere may play a role in stabilising MOF architectures towards hydrolysis.
Owing to the donor–acceptor nature of hydrogen bonding, it also follows that such interactions are most favoured in heterotopic, mixed-ligand coordination spheres. The mixed-ligand approach to the design of MOFs and related materials has gathered popularity in recent years, being achieved either through the use of two different homoleptic ligands within a reaction mixture (commonly a bis-heterocycle and a dicarboxylate),36 or the use of bridging ligands containing more than one type of functional group. A significant drawback to the use of multiple homotopic ligands, however, is the increase in complexity of the reaction mixture. Treating the concentration or stoichiometry of each ligand as a variable, as well as the accompanying effects on pH, makes the full exploration of the reaction space an increasingly burdensome task. The potential also exists in these systems for the formation of mixtures with homoleptic side products, or co-crystals of the ligands or other reaction components.37 As such, the approach of incorporating both N-heterocycle and carboxylic acid groups within the same molecule (to achieve the benefits of a mixed coordination sphere with only a single organic component within the reaction mixture) can be beneficial.38
Solvent and/or pH choice can also play important roles in the successful crystallisation of such species. Many of the examples discussed below were crystallised from water or water-containing mixtures with acetonitrile or alcohols. The success of these protic solvents at the expense of the more commonly used (aprotic) amide solvents such as DMF or DMA is most likely a consequence of the pKa of the typical hydrogen bond donor ligands; for example, coordination to a metal ion decreases the pKa of imidazole by up to 5.39 As a result, reactions carried out at high pH or in the presence of excess base (such as that generated from the hydrolysis of amide solvents) may show a tendency to deprotonate N–H hydrogen bond donors and reduce the capacity for hydrogen bond formation.
Hydrogen bond donor ligands in MOF chemistry
Pyrazole
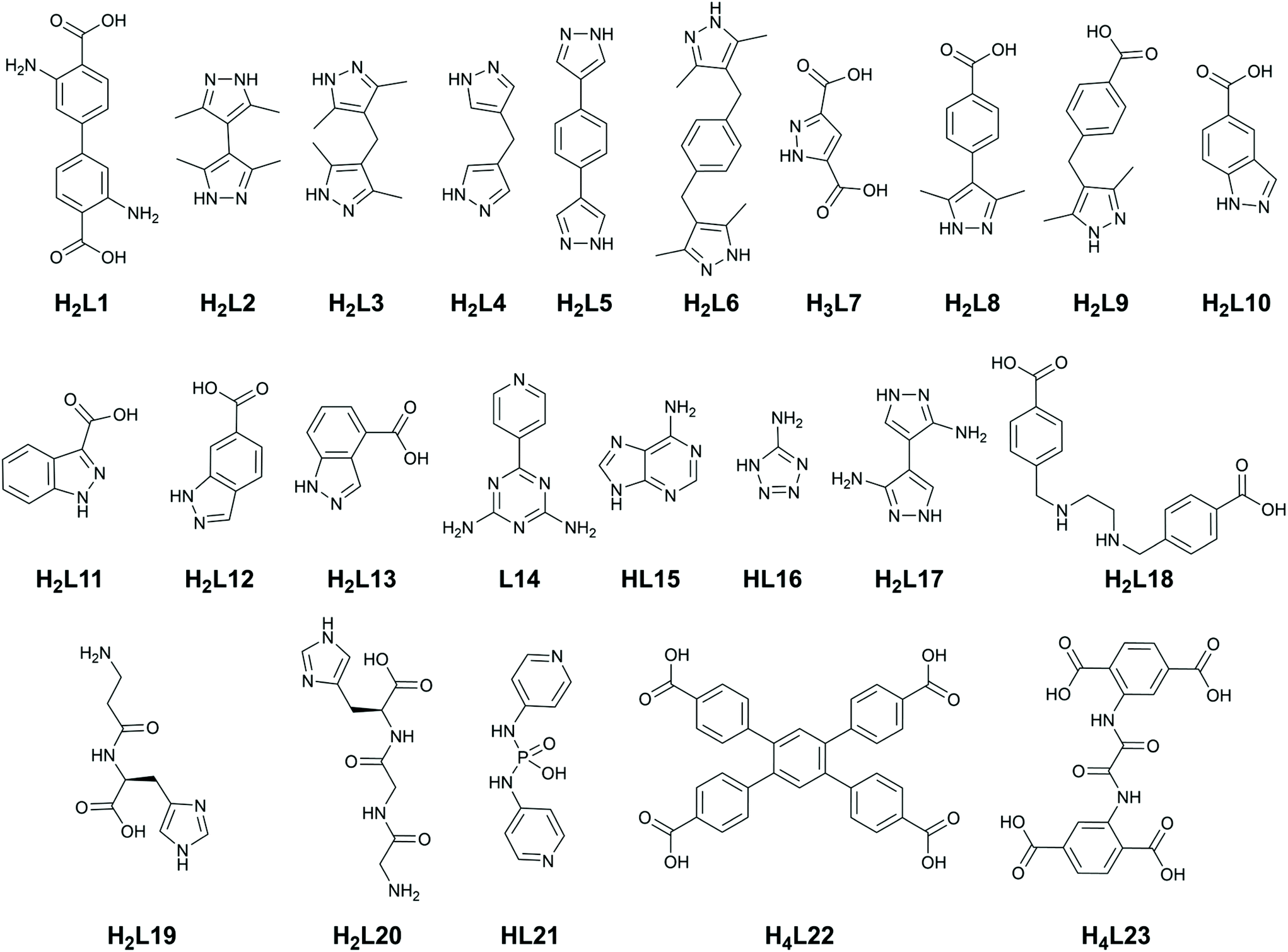
Pyrazole has gained significant popularity in MOF chemistry especially over the last 10 years,40 and bears the unique feature of a pyrrole-like N–H group directly adjacent to the coordinating pyridine-like nitrogen atom. While the neutral forms of the common 1,2,4-triazole and tetrazole rings can also conceivably include this functionality,41 those more acidic species are rarely encountered coordinating in their protonated forms. In pyrazoles, however, this functionality is ubiquitous. While pyrazoles can be deprotonated to form a highly stable bridging pyrazolate species, more relevant to the current discussion are the protonated forms which provide stabilising and structure-directing hydrogen bonding interactions in the vicinity of the metal site. The use of (poly)pyrazole ligands in homoleptic coordination polymer materials has been reviewed elsewhere,42 and will not be discussed in depth here. When used in combination with anionic carboxylate co-ligands, however, neutral 1H-pyrazole is a powerful, versatile and predictable structural tecton.The key hydrogen-bonding synthon of interest in pyrazole-carboxylate co-ligand complexes is the R11(7) intramolecular hydrogen bond between a neutral monodentate pyrazole ligand and an anionic monodentate carboxylate. Either one or two of such rings are regularly observed in tetrahedral metal ions such as cobalt(II) or zinc(II) with N2O2 coordination spheres, an example of which is shown in Fig. 2. The seven-membered ring motif is commonly observed in organic intramolecular hydrogen bonded systems with rings of this size expected to represent an optimum stability.43 A related but less commonly observed species (due to the energetically disfavoured 5-membered ring) is the R11(5) motif involving the coordinating oxygen atom, or when the carboxylate ligand is coordinated in a chelating κO,O′ coordination mode. These synthons have now been widely observed in several dozen studies involving mixed pyrazole-carboxylate systems which are further described below.
 | ||
| Fig. 2 Two commonly-encountered pyrazole-carboxylate coordination modes involving inter-ligand hydrogen bonding: (a) the R11(7) motif and (b) the R11(5) motif. In the former case, the typical D⋯A distances fall in the range 2.7–2.8 Å with D–H⋯A angles >150°, while in the latter case these values are more commonly 2.8–2.9 Å and 140–150°, respectively. | ||
Symmetric polypyrazoles
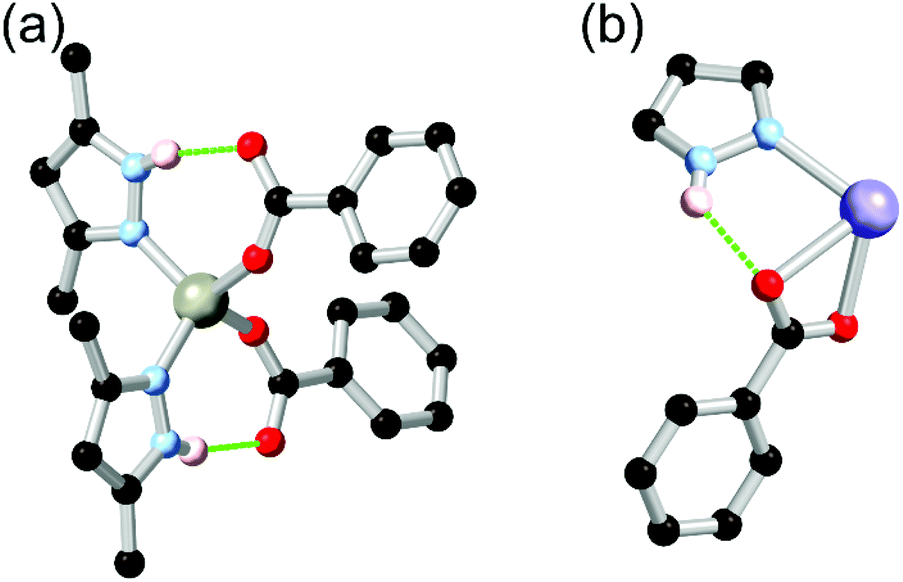
Many common polypyrazoles are prepared as 3,5-disubstituted species due to the easy access to the β-diketone precursors. As such, one of the most popular and earliest examples of a divergent polypyrazole linker in coordination polymer chemistry was 3,3′,5,5′-tetramethyl-4,4′-bipyrazole H2L2. While this species had seen prior use as a bridging ligand in discrete polynuclear species,44 its use as a linker in coordination polymers was first reported by Domasevitch and co-workers in two papers in 2001.45 In those works, the extensive coordination sphere hydrogen bonding which occurred in mixed pyrazole-carboxylate nodes was recognised, while hydrogen bonding with nitrate anions or solvent molecules as acceptors was also observed where carboxylate co-ligands were not present. While examples of the intramolecular R11(7) pyrazole-carboxylate synthon had been observed in discrete polynuclear systems or with monofunctional pyrazoles previously,46 the report by Boldog et al.45b was the first reported instance of this synthon in a polypyrazole-polycarboxylate mixed-ligand coordination polymer. As shown in Fig. 3, this example also included a chelating pair of two-donor one-acceptor R11(7) synthons around an octahedral cobalt(II) metal ion, giving four hydrogen bonds around a single metal ion.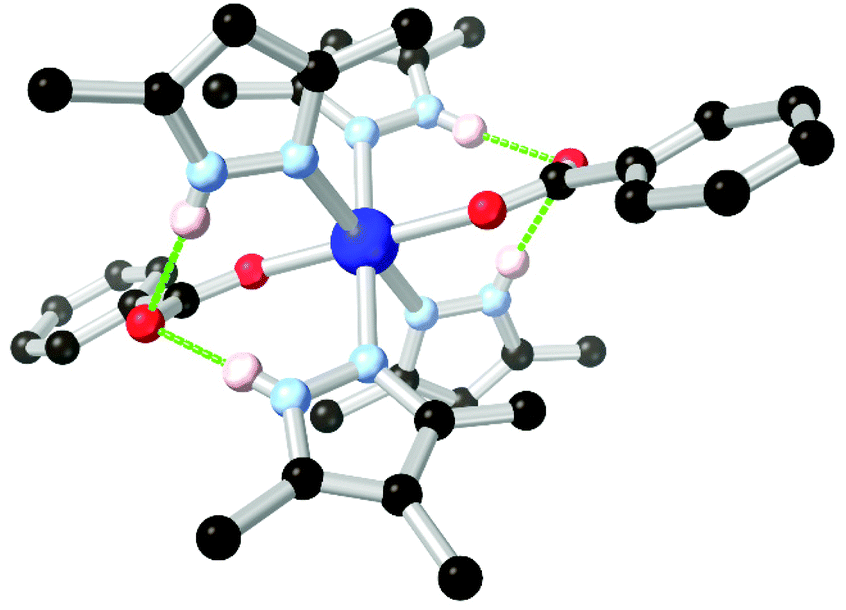 | ||
| Fig. 3 The chelating two-donor one-acceptor R11(7) rings observed by Boldog and co-workers in an N4O2 coordination sphere with H2L2.45b The D–A distances are 2.78 and 2.83 Å with D–H⋯A angles of 168 and 172°. | ||
Since then, the rigid linear H2L2 ligand has seen widespread use as a linker in both mixed-ligand and homoleptic coordination polymer systems, although often coordinating in the latter case as the deprotonated bis(pyrazolate) dianion L22−.47 While there are examples of permanently porous systems incorporating the dianionic form L22−,47a–d which tend to exhibit high stability as would be expected from the nature of the metal-azolate bonds, permanent porosity is less common in frameworks of the protonated form. One example is the chiral framework reported by Lu and co-workers in 2012,48 where a cadmium complex of H2L2 with an oxalyl dipeptide co-ligand formed a microporous framework in an ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixture. This material could be directly thermally activated without solvent exchange, and showed a moderate adsorption capacity for CO2 and H2. Another notable example is a recent report from Zaworotko and co-workers49 re-examining previously reported50 microporous copper(I) halide (Cl, Br, I) complexes of H2L2 for their potential in CO2/C2H2 separations. These structures exhibit diamondoid topologies and tetrahedral coordination spheres containing μ2-halido ligands and monodentate pyrazoles. Both show intramolecular R11(5) synthons with halide acceptors. Remarkably, despite the presence of coordinatively unsaturated metal ions and bridging halide ligands, after 2 months of accelerated stability testing at 75% relative humidity these materials showed no loss of crystallinity or adsorption performance.
:water mixture. This material could be directly thermally activated without solvent exchange, and showed a moderate adsorption capacity for CO2 and H2. Another notable example is a recent report from Zaworotko and co-workers49 re-examining previously reported50 microporous copper(I) halide (Cl, Br, I) complexes of H2L2 for their potential in CO2/C2H2 separations. These structures exhibit diamondoid topologies and tetrahedral coordination spheres containing μ2-halido ligands and monodentate pyrazoles. Both show intramolecular R11(5) synthons with halide acceptors. Remarkably, despite the presence of coordinatively unsaturated metal ions and bridging halide ligands, after 2 months of accelerated stability testing at 75% relative humidity these materials showed no loss of crystallinity or adsorption performance.
The related flexible ditopic linker 4,4′-methylenebis(3,5-dimethyl-1H-pyrazole) H2L3 has also become a common choice of ditopic bridging pyrazole ligand in the formation of network solids. At the time of writing, over 50 coordination compounds have been structurally characterised containing transition metal ions coordinated by H2L3 and carboxylate co-ligands. Additionally, many other instances are also known of homoleptic complexes of H2L3 and the singly or doubly deprotonated forms.51 In 2008, Mondal and co-workers reported the first instance of coordination polymers containing H2L3 with aromatic polycarboxylates, using zinc(II).52 Hydrogen bonding interactions were observed to varying extents in these structures depending on the nature of the carboxylate co-ligands, as exemplified in Fig. 4. The combination of H2L3 with 1,4-benzenedicarboxylic acid gave a 3-dimensional diamondoid network in which each zinc node bore two of the expected intramolecular R11(7) motifs (Fig. 2). Where 1,3-benzenedicarboxylate was employed, however, a one-dimensional chain structure resulted where the hydrogen bonding interactions were a combination of intra- and intermolecular hydrogen bonding, chelating to a single non-coordinated oxygen atom. In this case and as has been observed subsequently,53 steric hindrance from one or both ligands can lead to distortions in the local hydrogen bonding environment where the R11(7) motif is disfavoured compared with another interaction mode. This was also observed in a co-ligand complex with benzene-1,3,5-tricarboxylic acid,52 where a protonated carboxylic acid interrupted the coordination sphere hydrogen bonding. In subsequent reports,54 Mondal and others have shown the resilience of the hydrogen bonded [M(HPz)2(COO)2] tetrahedral coordination sphere for M = Co2+ or Zn2+ in the absence of strongly competing hydrogen bonding interactions or steric influences. The conformational flexibility of the V-shaped H2L3 ligand can give rise to intriguing structural features,54a,b and is often observed imparting a helical character onto the resulting networks. The propensity for H2L3 to form helical assemblies is also observed in the salts of the compound itself,55 and has also been exploited in the synthesis of discrete helical complexes from a 2-pyridyl appended L3 derivative.56 However, given the difficulties with preparing permanently porous frameworks containing flexible backbones, and the relatively large steric bulk of the H2L3 ligand, to date no permanently porous frameworks containing this ligand in its protonated form have been reported.
 | ||
| Fig. 4 Examples of hydrogen bonding modes of the H2L3 ligand with linear or angular dicarboxylates,52 showing (a) a combination of R11(7) modes and inter-framework bonding with the angular linker, and (b) saturation of the R11(7) motif with the linear linker. The D–A distances for both cases fall in the range 2.70–2.82 Å, and all D–H⋯A angles are in the range 148–167°. | ||
The 3,5-unsubstituted species 4,4′-methylenebis-1H-pyrazole H2L4 has also been explored in the synthesis of similar mixed-ligand frameworks.57 This ligand displays many properties similar to the tetramethyl analogue H2L3, including a propensity to undergo hydrogen bonding interactions with coordinating carboxylate oxygen atoms. Mondal and co-workers reported in 2013 the preparation of a series of H2L4-dicarboxylate coordination polymers containing a recurring {M2-H2L42} loop motif,58 which is presumably disfavoured on steric grounds in the equivalent H2L3 materials. In those structures, H2L4 also shows a much greater tendency to form species of coordination number 5 or 6, compared to the common tetrahedrally-coordinated Co2+ or Zn2+ species formed with H2L3. The donor–acceptor mismatch in these systems, which mostly adopt N2O4 or N4O2 coordination spheres, leads to a range of both intra- and inter-molecular hydrogen bonding modes being observed, although no permanently porous materials have yet been prepared from H2L4.
Larger polypyrazolyl ligands containing longer aromatic spacers have also been prepared and examined in similar coordination environments. As was the case with H2L2, most instances in which pyrazole groups have been linked with rigid spacers have been employed for coordination of the anionic pyrazolate species in a μ2-κN:κN′ coordination mode. A notable example is the 1,4-phenylenebispyrazole H2L5 employed by Long and co-workers in the synthesis of a remarkably robust porous coordination polymer material with Co2+ ions.59 The authors have rationalised this observation on the grounds of the high pKa of pyrazole systems compared with the notably unstable bridging carboxylate motifs more commonly seen in coordination polymer systems. The argument of higher pKa in depronated azolates lending greater stability also tends to hold true across pyrazolates, imidazolates and other azolate species.60 Xylylene-bridged bispyrazole systems have seen some use in coordination polymers,61 including in 2014 a report from Mondal detailing mixed pyrazole-carboxylate coordination polymers from p-xylylenebis(4-(3,5-dimethyl-1H-pyrazole)) H2L6.62 The same ligand was used by Tăbăcaru et al. as the dianion L62− to generate zinc and cobalt MOFs which showed gate-opening porosity based on their framework flexibility.63 Although a number of elegant discrete coordination complexes are known from the equivalent meta-substituted compound and the tripodal tris-pyrazole analogue,64 to date no carboxylate-containing coordination polymer species have been reported from these isomers.
Heterotopic pyrazoles and indazoles
In cases where heterocyclic carboxylates with N–H donor functionality are employed, coordination sphere hydrogen bonding tends to be observed mirroring that present in the two-component mixed ligand systems. The most widely used example in pyrazole ligands is the chelating ligand pyrazole-3,5-dicarboxylic acid H3L7 which has been widely used in the synthesis of a range of coordination polymers and MOFs.65 In this case, however, the thermodynamic drive to N,O-chelation tends to dominate the resulting coordination architectures from the L73− chelate, with N–H hydrogen bonding playing a smaller role.The quintessential example of a non-chelating carboxyphenyl-substituted pyrazole, 3,5-dimethyl-4-(4-carboxyphenyl)-1H-pyrazole H2L8, was first reported in the crystalline phase as the trifluoroacetate salt by Elguero and co-workers in 1996.66 Subsequently, this compound has seen use as a divergent ligand in several reports.67 In many of these cases, the use of cluster-based secondary building units has been crucial; the ability of pyrazolate groups to adopt similar μ2 bridging modes to carboxylates has been exploited, similarly to the approach adopted with the 4-carboxypyrazoles. Both Zhou and Janiak have observed mixed-valence Cu(I)/(II) frameworks containing two well-known homoleptic cluster SBUs, the tetracarboxylate ‘paddlewheel’ and the well-known triangular Cu3(pz)3 cluster, using the L82− dianion.67e,f This result mirrors the outcomes seen with pyrazole 4-carboxylic acid and its derivatives, as the coordination chemistry of the two species are closely related.68 However, coordination has also been observed from the monoanion HL81− in which the pyrazole group remains protonated, and can undergo additional supramolecular interactions. Several of these frameworks include the [M(Hpz)2(COO)2] (M = Co, Zn) node with associated intramolecular hydrogen bonding interactions, shown in Fig. 5, and equivalent to that observed in the mixed-ligand frameworks described above. A complex of HL81− was also presented containing both a copper paddlewheel cluster and a metal site containing a coordinating sulfate anion, complete with N–H⋯O–SO3 hydrogen bonding interactions on the periphery of the coordination sphere.67c
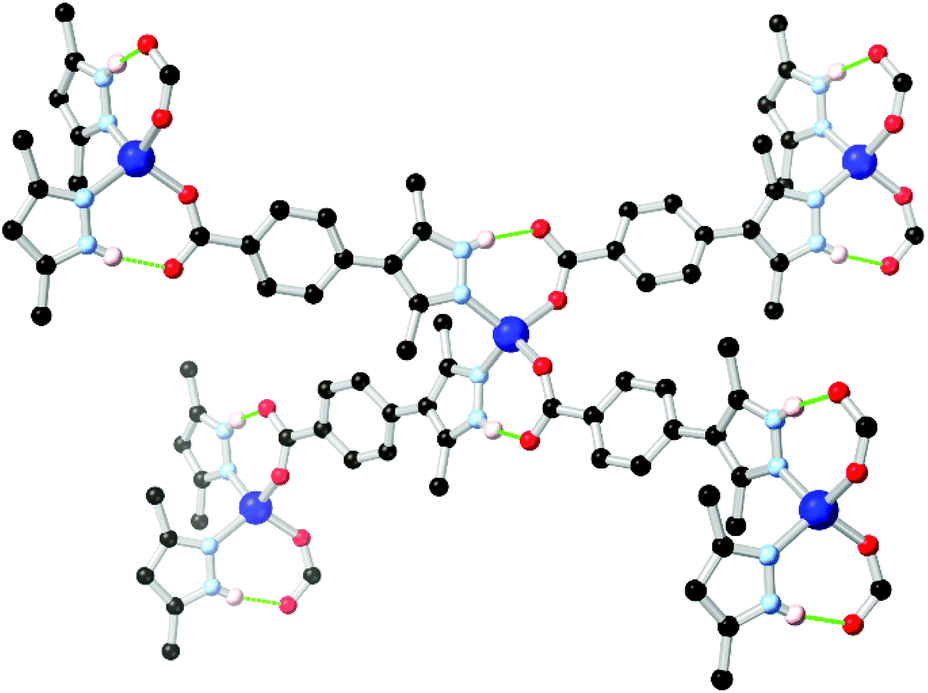 | ||
| Fig. 5 Example of the hydrogen bonding motif in a cobalt(II) coordination polymer of HL8 reported by Richardson,67c in which the R11(7) hydrogen bonding modes are equivalent to those seen in mixed-ligand pyrazole-carboxylate systems. The D–A distances are 2.73 and 2.67 Å, and the D–H⋯A angles are 165 and 166° respectively. | ||
More recently, Janiak and co-workers have presented several reports into the chemistry of H2L8, in which the N–H hydrogen bonding led to interesting structural consequences, shown in Fig. 6.67d,f Further examination of the copper coordination polymer with (3-dimensional) lvt topology reported by Richardson revealed that switching the synthesis solvent from methanol to acetonitrile or DMF gave a 2-dimensional sql square lattice network. In the sql case, intermolecular N–H⋯O hydrogen bonds to the carboxylate oxygen atoms of an adjacent coordination sphere were observed (Fig. 6A). Two such interactions support each pair of square planar Cu2+ ions into linear columns, leaving large and unobstructed rectangular solvent channels. In the lvt species, the pyrazole N–H group donates hydrogen bonds to lattice solvent molecules (Fig. 6B). The sql material showed greater uptake of both CO2 and N2 at low partial pressures. In H2O and EtOH vapour adsorption experiments, however, the sql material showed very low adsorption below P/P0 = 0.5, indicative of a hydrophobic pore environment. This is consistent with a structure in which all hydrogen bond donors were accounted for in intermolecular framework–framework interactions. This strongly contrasted to the lvt case, containing free hydrogen bond donors, which showed steep uptake of both vapours below P/P0 = 0.1 presumably by regeneration of the ligand-solvent hydrogen bonds.
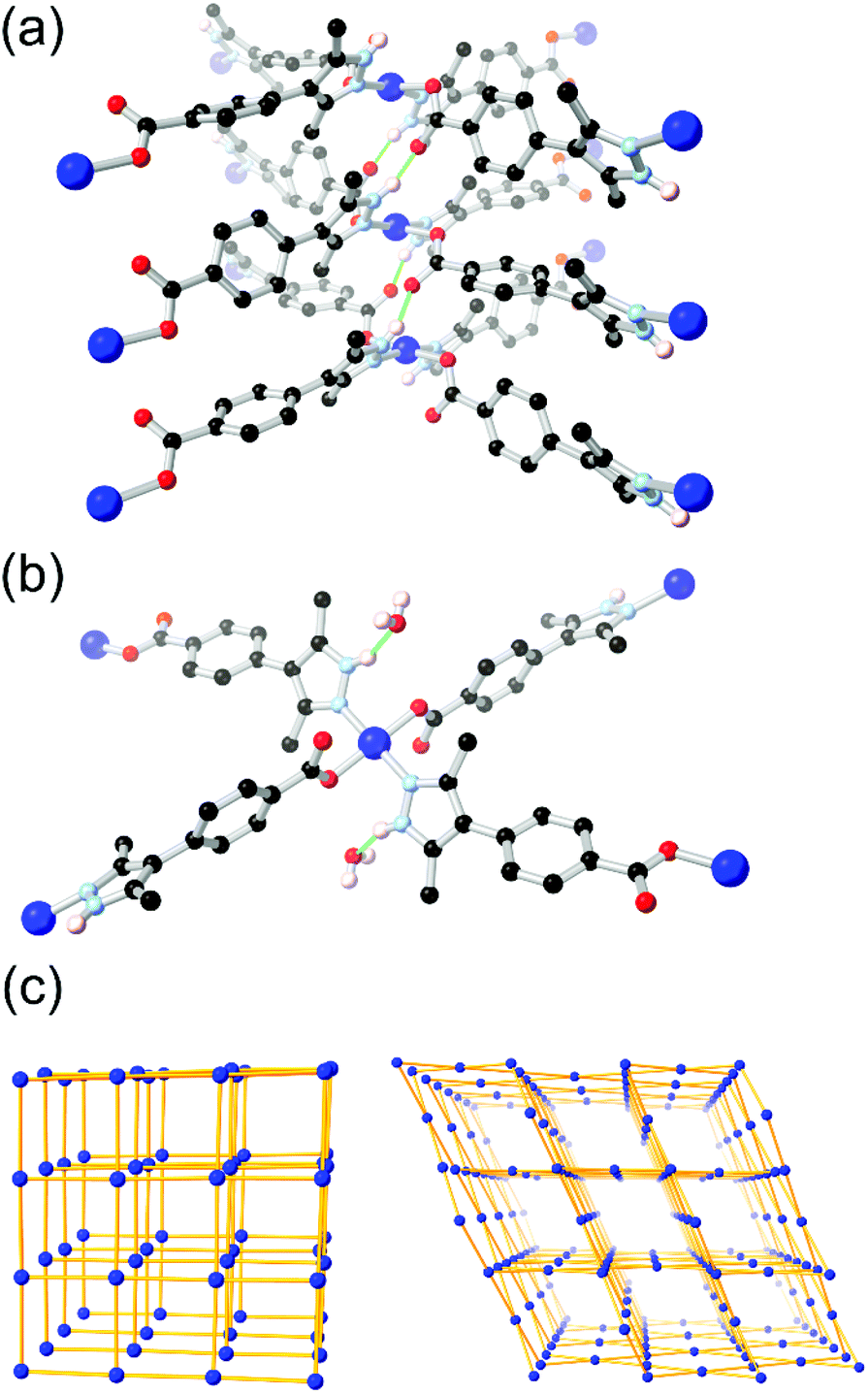 | ||
| Fig. 6 The two modes of hydrogen bonding observed in the isomeric sql (a) and lvt (b) MOFs reported by Janiak, and (c) representations of the sql (left) and lvt (right) topologies. In the sql case, the intermolecular hydrogen bonding is retained following evacuation, while the lvt case rapidly re-adsorbs lattice water molecules following evacuation to address the vacant N–H donor sites.67d The hydrogen bonds in the sql case are marginally shorter at 2.65 and 2.70 Å (D–A distance) compared to 2.81 Å for the lvt case. | ||
Analogous ligands can also be prepared incorporating flexible linkages, or by introducing further backbone functionalities. One example is the flexible ditopic pyrazole-carboxylate ligand 3,5-dimethyl-4-(4-carboxyphenylmethylene)-1H-pyrazole H2L9.69 On coordination with Co2+ or Cu2+, only one-dimensional polymeric materials could be generated, where the reproducible formation of M2L2 loops was thought to be favoured on thermodynamic grounds. In both cases, however, the hydrogen bonding behaviour of the pyrazole N–H group mirrored that observed by Mondal for the sterically constrained mixed-ligand assemblies of H2L3, forming only one intramolecular hydrogen bond while the other pyrazole N–H group participated in hydrogen bonding interactions with the solvent molecules or adjacent coordination spheres. A stable microporous cadmium(II) framework was subsequently prepared from this ligand by Li and co-workers,70 although coordinating in its L92− dianionic form.
Ring fusion of pyrazole into indazole gives further possibilities for derivatization into heterotopic ligands. The reaction of indazole-5-carboxylic acid H2L10 with Cu2+ under hydrothermal conditions gave a porous, 3-dimensional coordination polymer with twofold-interpenetrated nbo (niobium oxide) topology.71 This material proved stable to evacuation, exposure to air and immersion in hot water without loss of crystallinity despite the presence of a coordinatively unsaturated copper(II) node. The excellent water stability in this material was attributed to the inter-framework hydrogen bonding environment adjacent to the metal site, as shown in Fig. 7. These interactions both served to rigidify the square planar coordination sphere and shield the axial metal sites by locking both interpenetrating networks together at each metal node through inter-network N–H⋯O interactions. This behaviour also served to maximise the available pore space within the material, and is similar to that observed in the sql system reported by Janiak (Fig. 6b), although with a different extended topology.
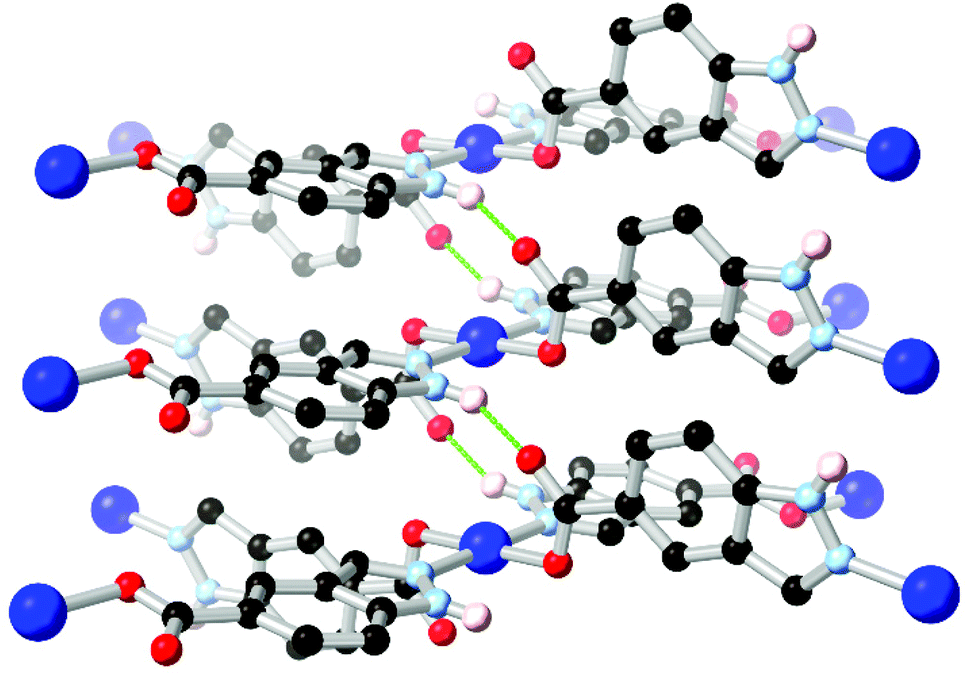 | ||
| Fig. 7 The inter-framework hydrogen bonds between interpenetrated nbo networks in the water-stable copper(II) MOF of HL10.71 The D–A distance for the N–H⋯O hydrogen bonds is 2.72 Å and the D–H⋯A angle is 143°. | ||
Subsequently, copper(II) coordination polymers were prepared from the isomeric indazole-3-carboxylic acid H2L11 and indazole-6-carboxylic acid H2L12.72 As with H3L7, the L112− dianion coordinates in a μ3-κN:κN′,O:κO′ mode, forming a 3-dimensional network identical to that observed with pyrazole-3-carboxylate. The divergent ligand H2L12, on the other hand, formed a two-dimensional polymeric framework as the HL121− anion, with copper(II) carboxylate paddlewheel and octahedral [Cu(Hin)4(ONO2)2] metal environments. Although the latter species contained a protonated N–H hydrogen bond donor adjacent to the coordinating nitrogen atom, hydrogen bonding from this site exclusively involves the lattice solvent molecules rather than the carboxylate co-ligands. In stark contrast to the 5-carboxylic acid isomer H2L10, these crystals showed poor stability to drying and exposure to atmospheric moisture and no permanent porosity was observed.
Further studies into the indazole-carboxylate system as a building block in coordination polymers and MOFs has been ongoing with several additional reports emerging within the last year. The group of Morzyk-Ociepa and co-workers reported the crystal structures of polymeric alkali metal complexes of HL11.73 In those structures, a uniform chelating μ2-κN,O:κO′ coordination mode was displayed by the ligand which coordinates in a monoanionic coordination mode, leaving the indazole N–H group free to participate in intramolecular hydrogen bonding to support the rod-shaped building units. Meanwhile, Garcıá-Valdivia et al. have recently explored H2L10 and H2L13 as linkers.74 While in those studies only the fully deprotonated L102− was observed, and therefore no hydrogen bonding was evident, HL131− coordinated exclusively as the monoanion in Ni, Co, Cu, Zn and Cd complexes. With the exception of the cadmium species, the other complexes each exhibited intermolecular R22(7) hydrogen bonded motifs involving the indazole and carboxylate groups of one coordination sphere and the aqua ligand of an adjacent metal site. While no porosity was observed in these systems, the authors credit this close association of metal ions to a spin-canted effect observed in magnetic susceptibility measurements of the copper species.
Aryl and heterocyclic amines
Amines represent another important class of coordination sphere hydrogen bond donors, with studies into hydrogen bond mediated assembly of metal-bound ammine ligands dating from the seminal work of Werner in the early 1900s.75 In terms of coordination polymer and MOF chemistry, amines are widely employed to enhance the enthalpy of adsorption of CO2, taking advantage of the well-studied affinity of carbon dioxide for basic nitrogen atoms and/or hydrogen bonding with N–H groups.76 The chemistry of these species, and their hydrogen bonding behaviour specifically, can be separated into two categories. While arylamines such as aminoterephthalic acid are readily available and widely used in amine-functionalised derivatives of common MOFs,77 the conjugated nature of these species leads to planarization of the substituent atoms and reduced basicity of the lone pair. Nonetheless, when positioned nearby to a coordinating atom, arylamines can engage in a range of stabilising hydrogen bonding interactions. This is exemplified by the 2,6-diaminotriazine derivative L14 employed by Zhang and co-workers in a zinc coordination polymer with a 4,4′-oxybis(benzoic acid) co-ligand.78 In this system, partially shown in Fig. 8, zinc carboxylate paddlewheel nodes are partially capped by a diaminotriazine ligand in the axial site, forming two N–H⋯O hydrogen bonds per cluster where the opposite site is capped by the pyridine group.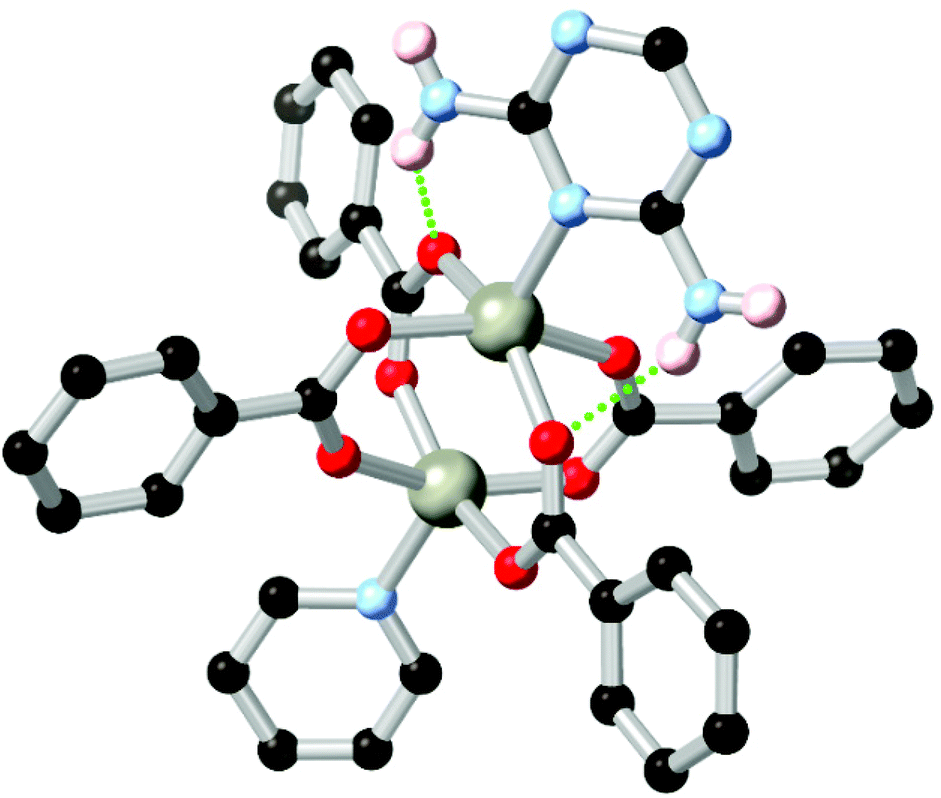 | ||
| Fig. 8 The heterocycle-capped copper paddlewheel nodes in the MOF of L14 reported by Zhang, showing the hydrogen bonding between the diaminotriazine group and two coordinating carboxylates.78 The D–A distances are 2.88 and 2.93 Å, and the D–H⋯A angles are 150 and 138° respectively. | ||
As arylamines tend to feature the N–H donor groups oriented in-plane with the parent aromatic ring, these functional groups are also known to form stabilising intramolecular hydrogen bonds within the ligand backbones. This influence has been explored by Xiang and co-workers,79 who found in a series of isoreticular zinc 1,2,4-triazole/terephthalate co-ligand MOFs that the aminoterephthalate derivative, FJU-40-NH2, was the only derivative of the series to show stability on exposure to air and immersion in water. This material was able to adsorb CO2 from humid air with sufficient enthalpy of adsorption and lattice ordering to be detected crystallographically, alongside adsorbed water molecules. The authors attribute the stability of this system to an intramolecular R11(6) motif which enhances the rotational barrier in this system, similar to that later observed by She in amine-functionalised UiO-67.32
Arylamines are also powerful hydrogen bonding tectons in combination with nitrogen heterocycles, with obvious parallels drawn to DNA bases and synthetic analogues in their hydrogen bond donor–acceptor capabilities.80 A well-known example is bio-MOF-1, a zinc-adeninate framework with a BPDC co-ligand reported by the Rosi group in 2009,81 and appearing in several subsequent studies.82 This species has shown excellent stability under aqueous conditions, despite the presence of coordinatively unsaturated zinc(II) ions within the structure. Perhaps most remarkably, bio-MOF-1 shows retention of crystallinity for a period of weeks even in the presence of strongly-coordinating aqueous PBS buffer, an important attribute for drug delivery studies and other applications requiring biological media. While part of the stability of bio-MOF-1 undoubtedly relates to the strongly coordinating L151− anion, two key R11(7) hydrogen bonding motifs are also present between the non-coordinating amine group and two coordinating oxygen atoms of the 4,4′-biphenyldicarboxylate co-ligand, as shown in Fig. 9. The geometry and rigidity of the heterocyclic ligand makes for favourable pre-organization of this site for hydrogen bonding, and both interactions exhibit N⋯O distances below 2.9 Å.
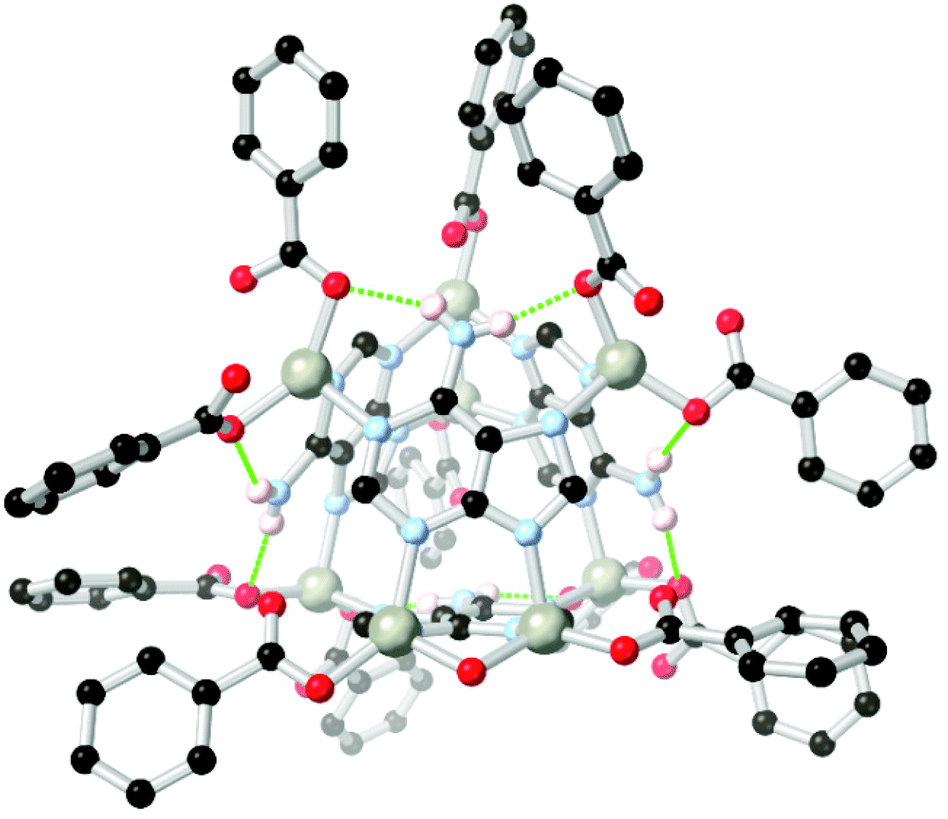 | ||
| Fig. 9 The hydrogen bonding between the amino group of adenine and coordinated carboxylate groups in the structure of BioMOF-1.82 The D–A distances are 2.84 and 2.87 Å and the D–H⋯A angles are 145 and 170° respectively. | ||
Nitrogen-rich azole and azolate species with amine substituents also widely exhibit coordination sphere hydrogen bonding in conjunction with carboxylate co-ligands. Two widely explored examples have been 3-amino-1,2,4-triazole, and 5-aminotetrazole HL16.83 Both ligands possess an amine group directly adjacent to two coordinating nitrogen atoms, and so are capable of contributing hydrogen bonds to two adjacent coordination spheres, as shown in Fig. 10. While edge-sharing R11(6) or R11(8) rings are common in such systems, the presence of amine functionality adjacent to the coordination sphere can also provide additional chemical functionality to these systems. Sun and co-workers recently implicated the amine group in L161− as a hydrogen bond donor in the metal-catalysed conversion of epoxides to cyclic carbonates.84 Intramolecular amine-amine hydrogen bonding involving the amine substituents of a diaminobipyrazole ligand H2L17 was also suggested as a plausible mechanism to explain the reactivity trends of a zinc(II) pyrazolate MOF in epihalohydrin conversion to cyclic carbonates by Galli and Rossin and co-workers.85
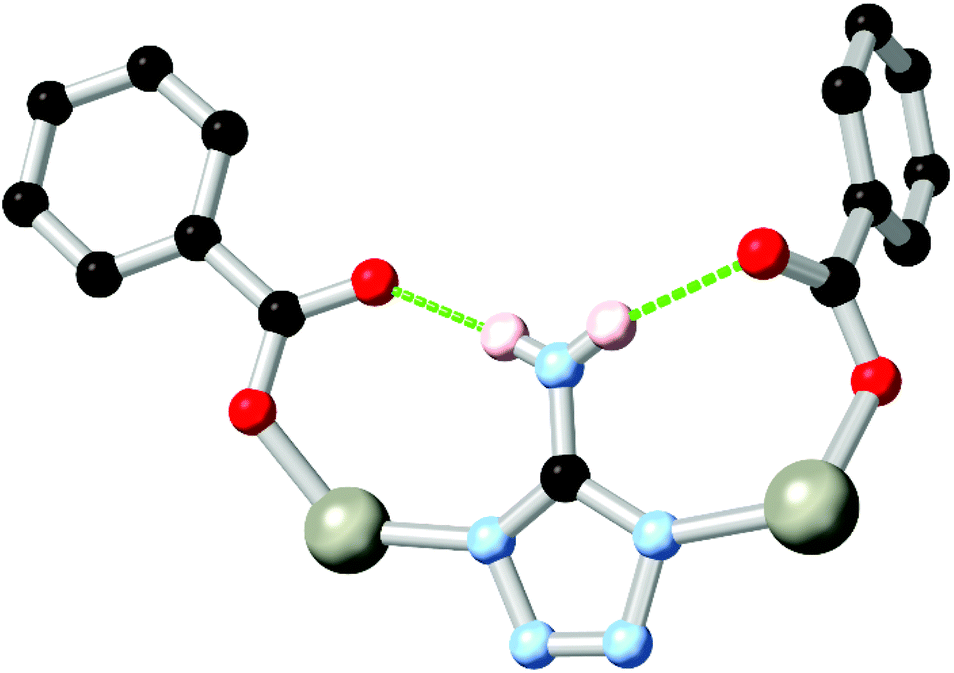 | ||
| Fig. 10 A representative example of the edge-sharing R11(8) rings formed in a 5-aminotetrazole carboxylate co-ligand MOF. Typical D–A distances are 2.8–3.0 Å with D–H⋯A angles >160°. | ||
Alkylamines
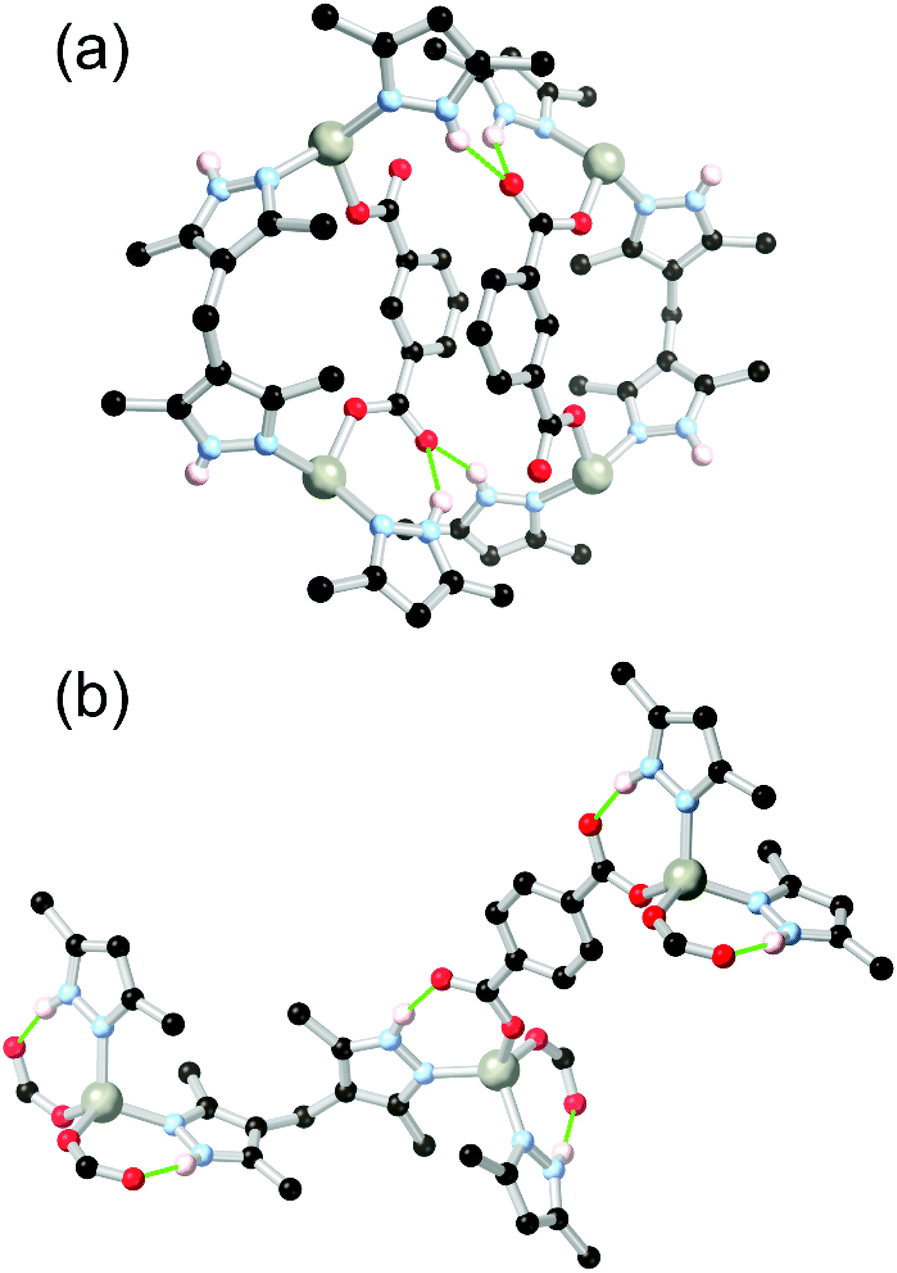
Unlike arylamines, substantially more Lewis basic non-conjugated alkylamines are typically found coordinated to the metal nodes in MOF syntheses.86 This can frustrate attempts to incorporate free alkylamines into open framework materials for CO2 capture unless larger steric bulk is employed.87 In the case of primary or secondary amines, further opportunities for coordination sphere hydrogen bonding are presented by the increased N–H acidity of metal-bound amines. Generally observed in mixed-ligand or heterotopic systems containing carboxylate groups and either linear or macrocyclic amines,88 the intramolecular R11(6) synthon between a coordinated secondary amine and the non-coordinating oxygen atom of a monodentate carboxylate is regularly observed as a stabilising force in these systems.Alkylamines such as divergent ethylenediamine-derived ligands also offer wide possibilities for structural engineering through hydrogen bonding, where the (R,R)/(S,S) and (R,S) conformers are related by a small energy barrier but with profound influence on their hydrogen bonding behaviour. Batten and co-workers reported a bis-(carboxybenzyl) derivative of ethylenediamine H2L18 which formed two closely related 2-dimensional coordination polymers with copper(II) ions, shown in Fig. 11.89 Depending on reaction conditions, either the trans-(R,R)/(S,S) or cis-(R,S) orientation of the ethylenediamine core could be selectively obtained. The permanently porous cis conformer, where both hydrogen bonding N–H groups were oriented in the same direction (Fig. 11B), formed a tight dimer between metal sites on adjacent sheets, with four hydrogen bonds enforcing a short distance of 4.213(2) Å between adjacent copper ions. In contrast the trans conformer contained both an intramolecular R11(6) synthon with the adjacent carboxylate group and a hydrogen bond to a lattice solvent molecule (Fig. 11A). Neither interaction contributing to the extended structure of the material, which was a non-porous twofold interpenetrated structure. The authors credit the hydrogen bonding in the cis conformer with rigidifying the material to allow evacuation and gas adsorption with retention of crystallinity, relatively unusual for a two-dimensional framework made from flexible ligands.
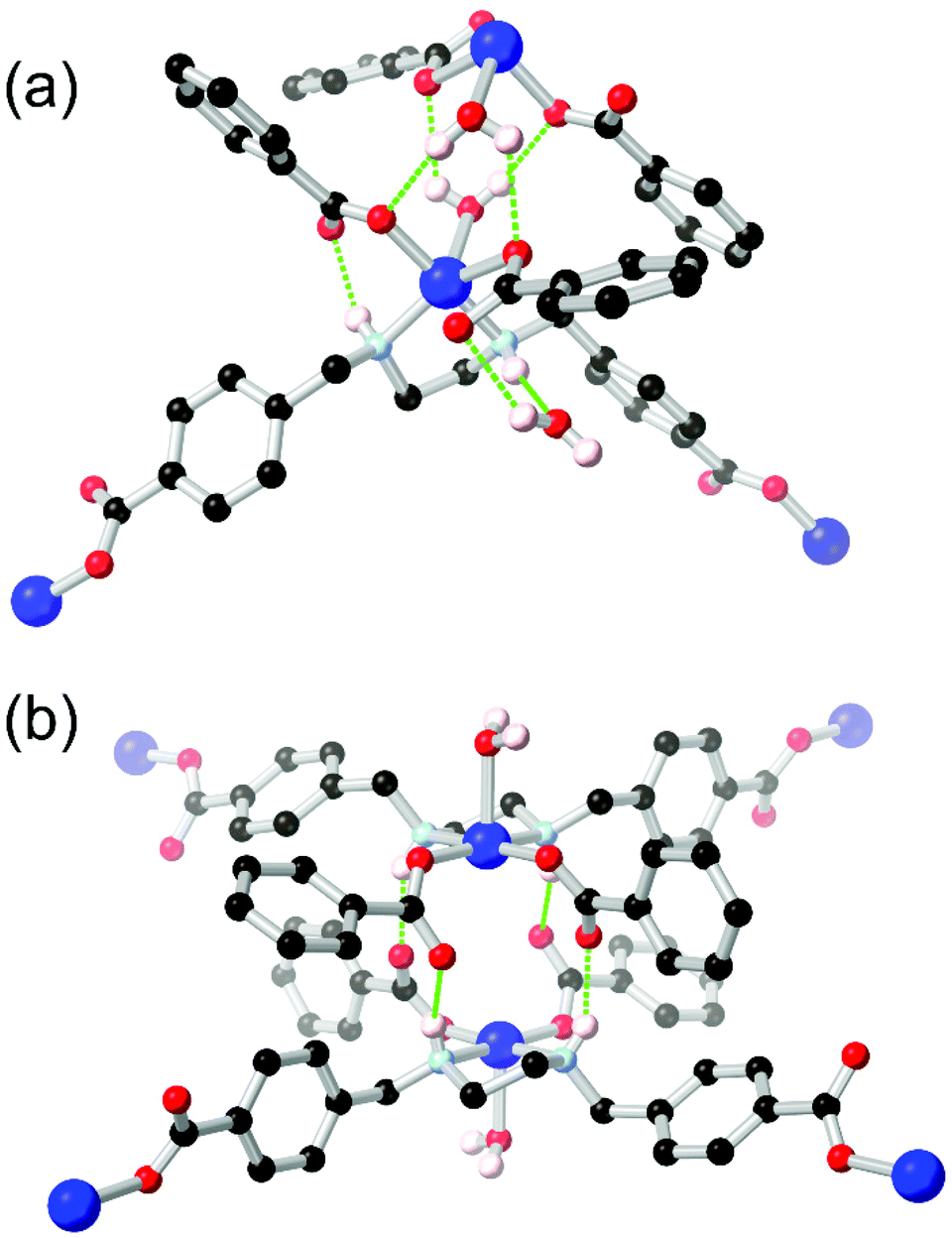 | ||
| Fig. 11 The coordination modes of the L182− ligand in the (a) trans, and (b) cis conformations, showing the disparity in hydrogen bonding modes between the two. Permanent porosity was observed in the cis case only.89 In the trans case, the D–A distance for the amine-carboxylate interaction was 2.83 Å with D–H⋯A angle 143°, while in the cis case the bonds were marginally longer at 2.85 and 3.03 Å, with D–H⋯A angles 149 and 147° respectively. | ||
Amides and related species
MOFs derived from ligands containing amide linkages, usually derived from amino acids, have been a widely used choice for the development of chiral framework materials for asymmetric catalysis and chemical sensing properties.90 In many such systems, amide linkages in the ligand backbone can make strong contributions to the overall structure of the material in ways that mirror the importance of these groups in defining the secondary structure of proteins.91 Protein-like secondary structures have been induced in MOFs containing synthetic peptide foldamers by the Gopi group recently.92 In that example, the structured peptide helix prevalent in the free peptide is reproduced in a silver(I) MOF, showing a clear dominance of the backbone hydrogen bonding as a structure-directing motif. The Rosseinsky group have reported several fascinating MOFs from di and tri-peptide ligands H2L19 and H2L20 where hydrogen bonding plays a key role in defining the extended structure as well as the overall stability of the materials. The L192−-based MOF was reported in 2013 which exhibits good stability to desolvation and exposure to water. A hydrated form ZnL19·H2O is related to the pristine material by structural rearrangement and extensive hydrogen bonding involving the peptide backbone.93 A flexible zinc MOF containing L202− was reported by the same group in 2019 where peptide hydrogen bonds also play a key role in defining nine unique structural minima, relating to the conformation of the flexible ligand backbone and accessible through guest uptake.94 Amide-amide hydrogen bonding can also feature as a structural element in coordination polymers derived from the popular 1,3,5-benzenetricarboxamide core, a widely used structural unit in soft materials chemistry due to its reliable formation of columnar superstructures.95Although not widely encountered in MOF chemistry, amide-like hydrogen bonding species bearing heavier main-group elements such as phosphoramides and thioureas also show great potential for coordination sphere stabilisation through hydrogen bonding, due to the ability of these species to enhance N–H hydrogen bond acidity.96 This is well-known from the anion binding literature, where the relatively higher acidity of the N–H donor groups of thiourea compared to urea allows for control of anion receptor capabilities between the two species.97
Boomishankar and co-workers reported a fascinating example of a diamidophosphate-based ligand HL21 which was derived from a partial in situ hydrolysis of the parent phosphoric tetramide in the presence of zinc nitrate in DMF.98 The resulting MOF, a 2-dimensional (6,3) network, contains tetrahedrally coordinated zinc ions bound by two pyridine groups, a monodentate formate ligand, and one oxygen atom of the diamidophosphate core. Two distinct modes of hydrogen bonding are observed in this structure, shown in Fig. 12. Firstly, each diamidophosphonate core participates in a dimeric R22(8) interaction with a ligand from an adjacent network, which serves to closely align adjacent networks without blocking pore space. The second N–H donor from each ligand core forms an (intramolecular) R11(8) hydrogen bonding motif with a terminal monodentate formate ligand from each zinc site. The resulting permanently porous network shows remarkable hydrolytic stability for a 2D MOF containing coordinatively unsaturated zinc(II) nodes, and is stable to desolvation and re-solvation with water or methanol without loss of crystallinity.
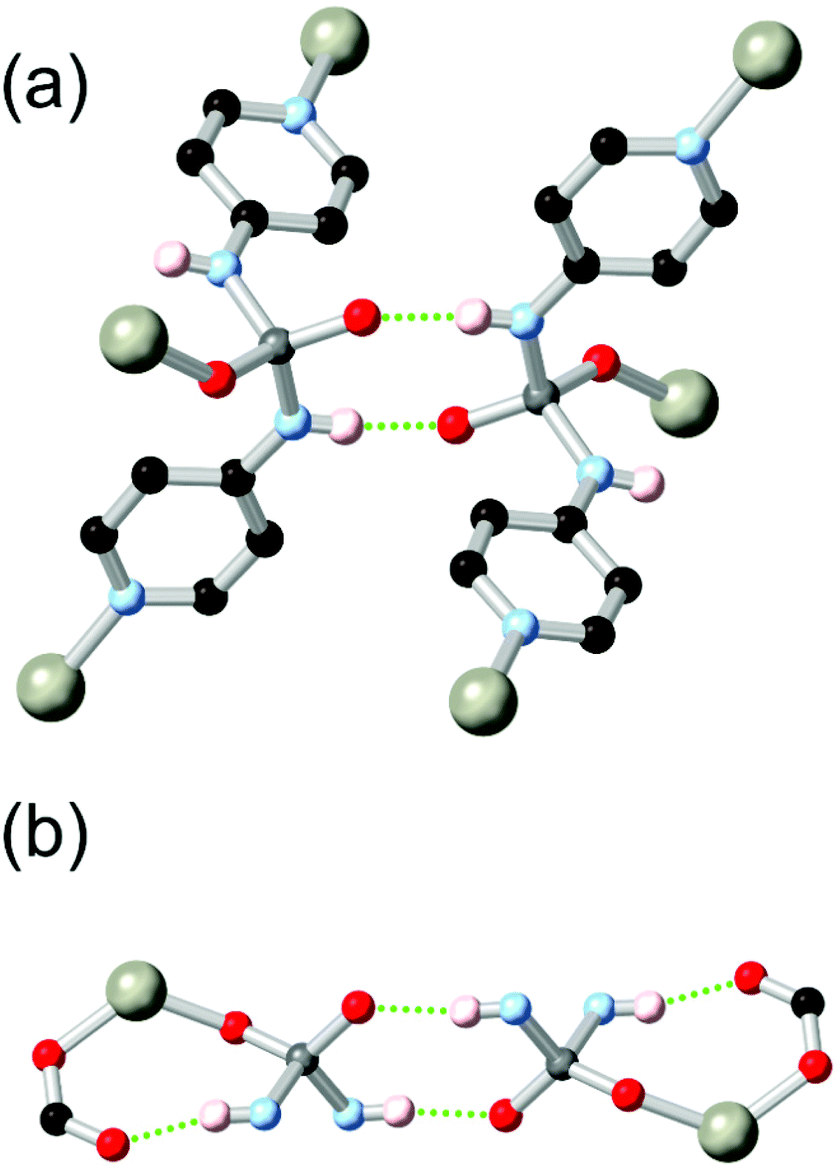 | ||
| Fig. 12 The hydrogen bonding modes exhibited by the diamidophosphate ligand L212− in a zinc(II) MOF reported by Boomishanker.98 Adjacent metal sites were linked by an R22(8) interaction (a), and further supported by an inner-sphere R11(8) motif (b). The D–A distances are 2.80 and 2.83 Å for formate and diamidophosphate acceptors, respectively, with D–H⋯A angles of 159 and 172°. | ||
Carboxylic acids
Given the ubiquitous use of carboxylates as ligands in MOF chemistry it is unsurprising that a vast array of coordination modes have been observed from polycarboxylates in MOFs and CPs. One coordination mode which is occasionally observed is coordination from protonated carboxylic acids through the ketonic oxygen atom, while the O–H proton is shared with an oxygen acceptor from an adjacent carboxylate group. This coordination mode is particularly prevalent in oxygen-rich coordination spheres of alkali and alkali earth metals and lanthanide elements.99 While neutral carboxylic acid coordination in isolation would be expected to show a relatively low stabilisation energy and be unlikely to prevail in isolation in the strongly competitive media of typical MOF syntheses, the coordination of the “[(RCOO)2H]−” unit as a chelate seems surprisingly resilient. The resulting R11(8) motif tends to enforce small angles between the terminal C–C bond vectors of each carboxylate group, typically either being parallel or folded about the M⋯H vector as represented in Fig. 13. | ||
| Fig. 13 Examples of the “[(RCOO)2H]−” chelating tecton in calcium(II) carboxylate MOFs, showing (a) the polymeric calcium-carboxylate/carboxylic acid chains and (b) the extended structure of the HL223− containing MOF reported by Parise,100 and (c) the structure of the highly water-stable calcium(II) carboxylate MOF reported by Margariti (coordinated solvent molecules are omitted for clarity).101 In both cases the D–A distances were particularly short (in the range 2.51–2.58 Å), with D–H⋯A angles in the range 156–171°. | ||
An interesting example of this was reported by Parise and co-workers,100 who reported a calcium(II) MOF with H4L22 which coordinates as the triply deprotonated species HL223−. In this structure, six-coordinate regular octahedral calcium sites are coordinated by four bridging carboxylates and two carboxylic acids, where hydrogen bonding helps to support a one-dimensional polymeric chain of calcium ions parallel to the axis of the solvent channels. This structure (Fig. 13a and b) was used for Xe/Kr separations, where the authors ascribe the low density of the framework to the presence of the carboxylate protons which provide a low molecular weight positive charge balance.
A similar mode of carboxylate coordination was observed in a calcium species of an oxalyldiamide ligand H4L23 reported by Margariti et al.,101 where isolated CaO6 octahedra are coordinated by two DMA solvent molecules, two deprotonated carboxylates and two protonated carboxylic acids. Two R11(8) rings are present supporting each coordination sphere, as shown in Fig. 13(b). Surprisingly, this species proved highly stable to water, exchanging only the axial solvent molecules. More remarkable, however, this material showed rapid metal exchange with copper(II) in water, exchanging almost all calcium sites within 24 hours, such that the material proved effective at removal of copper(II) from water samples.
Outlook
Coordination sphere hydrogen bonding, with a relatively small set of recurring motifs, is a consistent structural feature in MOFs and coordination polymers with ligand sets containing hydrogen bond donor functionality adjacent to metal binding sites. These interactions, if not always directly causal, certainly appear to favour specific outcomes where multiple energetically-similar structures are available. Less clear is the quantitative effect of these interactions on stability in these systems. While inferences can be drawn from the above examples that, anecdotally, there are instances of seemingly abnormally stable MOFs containing such interactions, hypothesis-driven studies on the subject are rare. This is especially the case given the role of serendipity in many studies involving new ligand design in MOFs.102Quantifying the impact of coordination sphere hydrogen bonding in the solid state, in the absence of the spectroscopic toolkit available to probe such equilibria in the solution state,103 is a challenging endeavour. Indeed, with the tendency of new MOF synthesis to show abundant sensitivity to slight changes in reaction conditions, control experiments of hydrogen-bonded versus non-hydrogen bonded versions of otherwise equivalent structures are conceptually challenging. Here post-synthetic ligand exchange may play a role,104 in generating directly related species in the presence and absence of hydrogen bonding for comparison. Deuteration studies have been successfully employed to probe similar interactions in solution measurements,105 and may also show similar usefulness here as a mechanism to address the strength of the hydrogen bonds themselves.
From the examples above, several key ligand design and synthesis strategies can be suggested to encourage the formation of coordination sphere hydrogen bonding in MOFs.
• For fully intramolecular hydrogen bonds, seven and eight-membered hydrogen bonded rings strike a good balance between avoiding ring strain and maintaining tight binding around the metal site. For carboxylate acceptors, this necessitates a hydrogen bond donor no more than two atoms removed from the coordinating atom.
• Complete saturation of all donors and acceptors in the coordination sphere is mostly readily achieved where the donor and acceptor numbers of the ligand systems are well matched, and where intramolecular hydrogen bonding is not disfavoured by backbone geometry.
• Where intramolecular ring motifs are disfavoured by geometry (such as in square planar coordination spheres or rigidly angled ligands), intermolecular hydrogen bonding may instead act to rigidify the extended structure of low-dimensional or interpenetrated nets.
• The choice of reaction solvent and synthesis conditions should take into account the need to avoid deprotonation of the hydrogen bond donor, with the expectation that multiple structurally distinct solvates may occupy similar positions on the energetic landscape.
Conclusions
The tendency of hydrogen bond donor-containing ligands to form supporting interactions around the coordination sphere in mixed-ligand MOFs is a potentially powerful structural tool for the design of these materials. Ligands including pyrazoles and indazoles, primary or secondary amines, amides and amidophosphates, and carboxylic acids have all shown coordination modes with persistent hydrogen bond donors proximal to metal binding groups. In the presence of hydrogen bond acceptors (typically carboxylate oxygens), cyclic hydrogen bonding synthons can be expected, with influences on the framework geometry, the pore chemistry, and potentially the overall stability of these systems. Such synthons may be considered as further handles alongside ligand and metal selection to influence the structural outcomes in MOF synthesis. With further quantitative study to understand the impact on stability in these systems, coordination sphere hydrogen boding may play a role in addressing the hydrolysis issues in many such materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author gratefully acknowledges the School of Chemical and Physical Sciences, Keele University, for funding support.Notes and references
-
(a) P. Silva, S. M. F. Vilela, J. P. C. Tomé and F. A. Almeida Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC
; (b) P. Kumar, K. Vellingiri, K.-H. Kim, R. J. C. Brown and M. J. Manos, Microporous Mesoporous Mater., 2017, 253, 251–265 CrossRef CAS
-
(a) B. M. Connolly, D. G. Madden, A. E. H. Wheatley and D. Fairen-Jimenez, J. Am. Chem. Soc., 2020, 142, 8541–8549 CrossRef CAS
-
(a) B. Van de Voorde, B. Bueken, J. Denayer and D. De Vos, Chem. Soc. Rev., 2014, 43, 5766–5788 RSC
-
(a) T. N. Nguyen, F. M. Ebrahim and K. C. Stylianou, Coord. Chem. Rev., 2018, 377, 259–306 CrossRef CAS
-
(a) J. E. Auckett, A. A. Barkhordarian, S. H. Ogilvie, S. G. Duyker, H. Chevreau, V. K. Peterson and C. J. Kepert, Nat. Commun., 2018, 9, 4873 CrossRef PubMed
-
(a) G. Shimizu, J. M. Taylor and S. Kim, Science, 2013, 341, 354–255 CrossRef CAS PubMed
-
(a) E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef
-
(a) B. S. Gelfand and G. K. H. Shimizu, Dalton Trans., 2016, 45, 3668–3678 RSC
-
(a) K. Tan, N. Nijem, Y. Gao, S. Zuluaga, J. Li, T. Thonhauser and Y. J. Chabal, CrystEngComm, 2015, 17, 247–260 RSC
- M. G. Goesten, F. Kapteijn and J. Gascon, CrystEngComm, 2013, 15, 9249–9257 RSC
-
(a) J. Keupp and R. Schmid, Faraday Discuss., 2018, 211, 79–101 RSC
- S. Yuan, J.-S. Qin, C. T. Lollar and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS PubMed
- I. J. Kang, N. A. Khan, E. Haque and S. H. Jhung, Chem. – Eur. J., 2011, 17, 6437–6442 CrossRef CAS PubMed
-
(a) K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed
-
(a) Z. Yin, Y.-L. Zhou, M.-H. Zeng and M. Kurmoo, Dalton Trans., 2015, 44, 5258–5275 RSC
-
(a) D.-S. Zhang, Y.-Z. Zhang, J. Gao, H.-L. Liu, H. Hu, L.-L. Geng, X. Zhang and Y.-W. Li, Dalton Trans., 2018, 47, 14025–14032 RSC
-
(a) J. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, T. Happe, E. Hofmann, U.-P. Apfel, M. Winkler, M. Senger, M. Haumann and S. T. Stripp, ACS Catal., 2019, 9, 9140–9149 CrossRef CAS
-
(a) E. W. Dahl and N. K. Szymczak, Angew. Chem., Int. Ed., 2016, 55, 3101–3105 CrossRef CAS
-
(a) A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller III and S. C. Marinescu, ACS Cent. Sci., 2018, 4, 397–404 CrossRef CAS PubMed
-
(a) I. Ahmed and S. H. Jhung, Chem. Eng. J., 2017, 310, 197–215 CrossRef CAS
- D.-W. Lim, M. Sadakiyo and H. Kitagawa, Chem. Sci., 2019, 10, 16–33 RSC
- R. S. Forgan, R. J. Marshall, M. Struckmann, A. B. Bleine, D.-L. Long, M. C. Bernini and D. Fairen-Jimenez, CrystEngComm, 2015, 17, 299–306 RSC
- B. J. Bucior, A. S. Rosen, M. Haranczyk, Z. Yao, M. E. Ziebel, O. K. Farha, J. T. Hupp, J. I. Siepmann, A. Aspuru-Guzik and R. Q. Snurr, Cryst. Growth Des., 2019, 19, 6682–6697 CrossRef CAS
-
(a) H. H-M. Yeung and A. K. Cheetham, Dalton Trans., 2014, 43, 95–102 RSC
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1990, 46, 256–262 CrossRef PubMed
-
(a) K. Wendler, J. Thar, S. Zahn and B. Kirchner, J. Phys. Chem. A, 2010, 114, 9529–9536 CrossRef CAS PubMed
-
(a) R. S. Forgan, B. D. Roach, P. A. Wood, F. J. White, J. Campbell, D. K. Henderson, E. Kamenetzky, F. E. McAllister, S. Parsons, E. Pidcock, P. Richardson, R. M. Swart and P. A. Tasker, Inorg. Chem., 2011, 50, 4515–4522 CrossRef CAS PubMed
- N. Novendra, J. M. Marrett, A. D. Katsenis, H. M. Titi, M. Arhangelskis, T. Friščić and A. Navrotsky, J. Am. Chem. Soc., 2020, 142, 21720–21729 CrossRef CAS PubMed
- H. Jasuja, N. C. Burtch, Y.-G. Huang, Y. Cai and K. S. Walton, Langmuir, 2013, 29, 633–642 CrossRef CAS PubMed
-
(a) P. Guo, D. Dutta, A. G. Wong-Foy, D. W. Gidley and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 2651–2657 CrossRef CAS PubMed
- C. L. Hobday and G. Kieslich, Dalton Trans., 2021, 50, 3759–3768 RSC
- K. Wang, Q. Wang, X. Wang, M. Wang, Q. Wang, H.-M. Shen, Y.-F. Yang and Y. She, Inorg. Chem. Front., 2020, 7, 3548–3554 RSC
-
(a) J. G. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 4560–1561 CrossRef CAS PubMed
- X.-L. Lv, S. Yuan, L.-H. Xie, H. F. Darke, Y. Chen, T. He, C. Dong, B. Wang, Z.-Z. Zhang, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2019, 141, 10283–10293 CrossRef CAS PubMed
- S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2013, 52, 13052–13056 CrossRef CAS PubMed
-
(a) P. Teo and T. S. A. Hor, Coord. Chem. Rev., 2011, 255, 273–289 CrossRef CAS
-
(a) R. Zhu, W. M. Bloch, J. J. Holstein, S. Mandal, L. V. Schäfer and G. H. Clever, Chem. – Eur. J., 2018, 24, 12976–12982 CrossRef CAS
- X.-L. Zhao and W.-Y. Sun, CrystEngComm, 2014, 16, 3247–3258 RSC
- M. F. Hoq and R. E. Shepherd, Inorg. Chem., 1984, 23, 1851–1858 CrossRef CAS
-
(a) S.-Q. Bai, D. J. Young and T. S. A. Hor, Chem. – Asian J., 2011, 6, 292–304 CrossRef CAS PubMed
-
(a) F. Ramondo, L. Bencivenni, G. Portalone and A. Domenicano, Struct. Chem., 1994, 5, 1–7 CrossRef CAS
- C. Pettinari, A. Tăbăcaru and S. Galli, Coord. Chem. Rev., 2016, 307, 1–31 CrossRef CAS
- R. A. Cormanich, M. P. Freitas, C. F. Tormena and R. Rittner, RSC Adv., 2012, 2, 4169–4174 RSC
-
(a) P. E. Kruger, G. D. Fallon, B. Moubaraki and K. S. Murray, J. Chem. Soc., Chem. Commun., 1992, 1726–1729 RSC
-
(a) I. Boldog, E. B. Rusanov, A. N. Chernega, J. Sieler and K. V. Domasevitch, Polyhedron, 2001, 20, 887–897 CrossRef CAS
-
(a) M. Döring, W. Ludwig and H. Görls, J. Therm. Anal., 1994, 42, 443–459 CrossRef
-
(a) L. Hou, L.-N. Jia, W.-J. Shi, L.-Y. Du, J. Li, Y.-Y. Wang and Q.-Z. Shi, Dalton Trans., 2013, 42, 6306–6309 RSC
- L. Ling, R. Yu, W. Tang, X.-Y. Wu and C.-Z. Lu, Cryst. Growth Des., 2012, 12, 3304–3311 CrossRef
- S. Mukherjee, Y. He, D. Franz, S.-Q. Wang, W.-R. Xian, A. A. Bezrukov, B. Space, Z. Xu, J. He and M. J. Zaworotko, Chem. – Eur. J., 2020, 26, 4923–4929 CrossRef CAS PubMed
-
(a) J. He, J. Duan, H. Shi, J. Huang, J. Huang, L. Yu, M. Zeller, A. D. Hunter and Z. Xu, Inorg. Chem., 2014, 53, 6837–6843 CrossRef CAS PubMed
-
(a) I. Timokhin, C. Pettinari, F. Marchetti, R. Pettinari, F. Condello, S. Galli, E. C. B. A. Alegria, L. M. D. R. S. Martins and A. J. L. Pombeiro, Cryst. Growth Des., 2015, 15, 2303–2317 CrossRef CAS
- R. Mondal, M. K. Bunia and K. Dhara, CrystEngComm, 2008, 10, 1167–1174 RSC
-
(a) X.-G. Guo, W.-B. Yang, X.-Y. Wu, Q.-K. Zhang and C.-Z. Lu, CrystEngComm, 2013, 15, 10107–10115 RSC
-
(a) A. Goswami, S. Sengupta and R. Mondal, CrystEngComm, 2012, 14, 561–572 RSC
- T. Basu and R. Mondal, CrystEngComm, 2010, 12, 366–369 RSC
- C. S. Hawes and P. E. Kruger, Polyhedron, 2013, 52, 255–260 CrossRef CAS
-
(a) S. Ganguly and R. Mondal, Cryst. Growth Des., 2015, 15, 2211–2222 CrossRef CAS
- S. Sengupta, S. Ganguly, A. Goswami, P. K. Sukul and R. Mondal, CrystEngComm, 2013, 15, 8353–8365 RSC
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311–1319 RSC
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC
- W. Ouellette, J. Gooch, S. Luquis and J. Zubieta, Inorg. Chim. Acta, 2015, 427, 188–197 CrossRef CAS
- S. Bhattacharya, A. Goswami, B. Gole, S. Ganguly, S. Bala, S. Sengupta, S. Khanra and R. Mondal, Cryst. Growth Des., 2014, 14, 2853–2865 CrossRef CAS
- A. Tăbăcaru, C. Pettinari, N. Masciocchi, S. Galli, F. Marchetti and M. Angjellari, Inorg. Chem., 2011, 50, 11506–11513 CrossRef PubMed
-
(a) S.-Y. Yu, Q. Jiao, J.-H. Li, H.-P. Huang, Y.-Z. Li, Y.-J. Pan, Y. Sei and K. Yamaguchi, Org. Lett., 2007, 9, 1379–1382 CrossRef CAS PubMed
-
(a) P. King, R. Clérac, C. E. Anson, C. Coulon and A. K. Powell, Inorg. Chem., 2003, 42, 3492–3500 CrossRef CAS PubMed
- C. Foces-Foces, C. Cativiela, M. M. Zurbano, I. Sobrados, N. Jagerovic and J. Elguero, J. Chem. Crystallogr., 1996, 26, 579–584 CrossRef CAS
-
(a) C.-T. He, J.-Y. Tian, S.-Y. Liu, G. Ouyang, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2013, 4, 351–356 RSC
- C. Heering, I. Boldog, V. Vasylyeva, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 9757–9768 RSC
- C. S. Hawes, B. Moubaraki, K. S. Murray, P. E. Kruger, D. R. Turner and S. R. Batten, Cryst. Growth Des., 2014, 14, 5749–5760 CrossRef CAS
- Q. Xiao, Y. Wu, M. Li, M. O'Keeffe and D. Li, Chem. Commun., 2016, 52, 11543–11546 RSC
- C. S. Hawes, R. Babarao, M. R. Hill, K. F. White, B. F. Abrahams and P. E. Kruger, Chem. Commun., 2012, 48, 11558–11560 RSC
-
(a) C. S. Hawes and P. E. Kruger, Dalton Trans., 2014, 43, 16450–16458 RSC
- K. Szmigiel-Bakalarz, M. Nentwig, D. Günther, O. Oeckler, M. Malik-Gajewska, D. Michalska and B. Morzyk-Ociepa, Polyhedron, 2020, 187, 114661 CrossRef CAS
-
(a) A. A. Garcıá-Valdivia, M. Peŕez-Mendoza, D. Choquesillo-Lazarte, J. Cepeda, B. Fernańdez, M. Souto, M. Gonzaĺez-Tejero, J. A. Garcıá, G. M. Espallargas and A. Rodrıǵuez-Dieǵuez, Cryst. Growth Des., 2020, 20, 4550–4560 CrossRef
- G. Schwarzenbach, Helv. Chim. Acta, 1967, 50, 38–63 CrossRef
- N. Planas, A. L. Dzubak, R. Poloni, L.-C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS PubMed
-
(a) T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Férey and N. Stock, Inorg. Chem., 2009, 48, 3057–3064 CrossRef CAS PubMed
- B.-W. Qin, X.-Y. Zhang and J.-P. Zhang, New J. Chem., 2019, 43, 13794–13801 RSC
- Z. Yao, Y. Chen, L. Liu, X. Wu, S. Xiong, Z. Zhang and S. Xiang, ChemPlusChem, 2016, 81, 850–856 CrossRef CAS PubMed
-
(a) C. F. Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, J. Am. Chem. Soc., 2000, 122, 4117–4128 CrossRef CAS
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed
-
(a) Y.-L. Huang, P.-L. Qiu, H. Zeng, H. Liu, D. Luo, Y. Y. Li, W. Lu and D. Li, Eur. J. Inorg. Chem., 2019, 4205–4210 CrossRef CAS
-
(a) H. Zhang, T. Sheng, S. Hu, C. Zhuo, H. Li, R. Fu, Y. Wen and X. Wu, Cryst. Growth Des., 2016, 16, 3154–3162 CrossRef CAS
- Y. Li, X. Zhang, J. Lan, P. Xu and J. Sun, Inorg. Chem., 2019, 58, 13917–13926 CrossRef CAS PubMed
- G. Mercuri, M. Moroni, K. V. Domasevitch, C. Di Nicola, P. Campitelli, C. Pettinari, G. Giambastiani, S. Galli and A. Rossin, Chem. – Eur. J., 2021, 27, 4746–4754 CrossRef CAS
-
(a) Y. Wen, Q. Liu, S. Su, Y. Yang, X. Li, Q.-L. Zhu and X. Wu, Nanoscale, 2020, 12, 12767–12772 RSC
- A. J. Emerson, A. Chahine, S. R. Batten and D. R. Turner, Coord. Chem. Rev., 2018, 365, 1–22 CrossRef CAS
-
(a) Y. Wen, T. Sheng, X. Zhu, C. Zhuo, S. Su, H. Li, S. Hu, Q.-L. Zhu and X. Wu, Adv. Mater., 2017, 29, 1700778 CrossRef PubMed
- C. S. Hawes, G. P. Knowles, A. L. Chaffee, D. R. Turner and S. R. Batten, Cryst. Growth Des., 2015, 15, 3417–3425 CrossRef CAS
-
(a) H. Tang, K. Yang, K.-Y. Wang, Q. Meng, F. Wu, Y. Fang, X. Wu, Y. Li, W. Zhang, Y. Luo, C. Zhu and H.-C. Zhou, Chem. Commun., 2020, 56, 9016–9019 RSC
-
(a) D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5216 CrossRef CAS
- R. Misra, A. Saseendran, S. Dey and H. N. Gopi, Angew. Chem., Int. Ed., 2019, 58, 2251–2255 CrossRef CAS PubMed
- A. P. Katsoulidis, K. S. Park, D. Antypov, C. Martí-Gastaldo, G. J. Miller, J. E. Warren, C. M. Robertson, F. Blac, G. R. Darling, N. G. Berry, J. A. Purton, D. J. Adams and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2014, 53, 193–198 CrossRef CAS PubMed
- A. P. Katsoulidis, D. Antypov, G. F. S. Whitehead, E. J. Carrington, D. J. Adams, N. G. Berry, G. R. Darling, M. S. Dyer and M. J. Rosseinsky, Nature, 2019, 565, 213–217 CrossRef CAS
-
(a) S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC
-
(a) D. A. Erzunov, G. V. Latyshev, A. D. Averin, I. P. Beletskaya and N. V. Lukashev, Eur. J. Org. Chem., 2015, 6289–6297 CrossRef CAS
- D. E. Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Org. Biomol. Chem., 2005, 3, 1495–1500 RSC
- A. K. Gupta, S. S. Nagarkar and R. Boomishankar, Dalton Trans., 2013, 42, 10964–10970 RSC
-
(a) L. K. Cadman, M. F. Mahon and A. D. Burrows, Dalton Trans., 2018, 47, 2360–2367 RSC
- X. Chen, A. M. Plonka, D. Banerjee, R. Krishna, H. T. Schaef, S. Ghose, P. K. Thallapally and J. B. Parise, J. Am. Chem. Soc., 2015, 137, 7007–7010 CrossRef CAS PubMed
- A. Margariti, S. Rapti, A. D. Katsenis, T. Friščić, Y. Georgiou, M. J. Manos and G. S. Papaefstathiou, Inorg. Chem. Front., 2017, 4, 773–781 RSC
-
(a) H. B. Alyappa, J. Masa, C. Andronescu, M. Muhler, R. A. Fisher and W. Schuhmann, Small Methods, 2019, 3, 1800415 CrossRef
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC
-
(a) M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed
-
(a) C. N. R. Rao, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 980–983 RSC
| This journal is © The Royal Society of Chemistry 2021 |