
Insights on the catalytic behaviour of sulfonic acid-functionalized ionic liquids (ILs) in transesterification reactions – voltammetric characterization of sulfonic task-specific ILs with bisulfate anions†
María B.
Martini
 a,
José L.
Fernández
*ab and
Claudia G.
Adam
*a
a,
José L.
Fernández
*ab and
Claudia G.
Adam
*a
aInstituto de Química Aplicada del Litoral (IQAL, UNL-CONICET) and Facultad de Ingeniería Química, Universidad Nacional del Litoral, Santiago del Estero 2829 (3000) Santa Fe, Argentina. E-mail: jlfernan@fiq.unl.edu.ar; cadam@fiq.unl.edu.ar
bPrograma de Electroquímica Aplicada e Ingeniería Electroquímica (PRELINE), Facultad de Ingeniería Química, Universidad Nacional del Litoral, Santiago del Estero 2829 (3000) Santa Fe, Argentina
First published on 15th January 2021
Abstract
This work shows for the first time the link between the amount of free sulfuric acid (as detected by cyclic voltammetry) and the activity of sulfonic-acid-functionalized ionic liquids (ILs) as acid catalysts for a transesterification reaction, and demonstrates that sulfonic acid groups, while are not directly involved in the catalysis, release the free acid during the reaction. Two imidazolic ILs with bisulfate as the counterion and their corresponding task-specific ILs (TSILs) that resulted from the addition of a sulfonic acid group inside the imidazolic-base structure were studied. The outstanding catalytic activity at room temperature of the TSILs 1-(4-sulfonic acid)-butyl-3-methylimidazolium bisulfate ([bsmim]HSO4) and 1-(4-sulfonic acid)-butyl-imidazolium bisulfate ([bsHim]HSO4) for the transesterification of p-nitrophenyl acetate with methanol was associated to the significant amounts of free sulfuric acid in equilibria with the ionic pairs. It was concluded that these TSILs function as reservoirs for releasing the free acid, which is the actual acid catalyst. In contrast, the corresponding non-sulfonic ILs supply very little amounts of free acid and consequently present low catalytic activities at room temperature, which in fact can be improved by increasing the reaction temperature up to 100 °C.
Introduction
The fascinating properties of ionic liquids (ILs) opened up a huge range of new research areas spanning different branches of Chemistry and Physics.1–3 In organic synthesis, ILs have been classified as “green solvents” due to their high physicochemical stability, low volatility, tunable solubility, and easy recyclability.4–6 These properties are being increasingly exploited for simplifying existing synthesis methods and/or for designing new environmentally benign processes.7,8 ILs also fit into the category of “designer solvents”, as their physicochemical properties can be modulated by the combination of a vast variety of cations and anions. Moreover, ILs can be specifically designed for accomplishing a particular task by introducing functional groups inside their chemical structures. In these ILs, known as “task-specific ionic liquids” (TSILs),9–12 the functional groups are incorporated into the cationic bases in order to modify a specific property that directs the IL behaviour for accomplishing a predefined specific task in a reacting system. In particular, the incorporation of sulfonic acid functional groups (SO3H) leads to TSILs with strong Brønsted acid properties, which are specially useful when using the counterion HSO4− for acid catalysis in esterification and transesterification reactions.13–16 These properties turned sulfonic-based TSILs into excellent alternative solvents and catalysts in processes for biodiesel synthesis.17–19 This dual solvent/catalyst role played by TSILs in these applications shows that they can be much more than just green solvents.For the design of TSILs, it is important to understand, at least in some extension, the mechanism of its performance. This is key information for proposing modifications of the main IL structure with functional groups toward a specific goal. However, an aspect that is at the same importance level and cannot be ignored is that the method used for the IL synthesis determines the species that may be naturally present within the IL. In many cases, the precursor reactants that were used to form the ionic pairs remain within the liquid.20,21 These are either spontaneously generated by natural equilibrium,22 or just cannot be removed during purification without reversing the IL synthesis reaction. This particular issue results critical because it may be the reason of some specific behaviours of the ILs that determine their performances. For example, it was recently reported that alkylammonium-derived ILs synthesized by acid–base addition contain the acid and base precursors in autoprotolysis equilibrium with the ionic pairs.23 The particular combination of ions and neutral species determines the properties of the synthesized IL, and then its performance in an application is influenced by all the interactions that are established within the reactive system.
In this context, our interest is to contribute to the design of imidazolic TSILs relating the structural modifications performed on the cationic bases of the ILs with their chemical compositions and behaviours as solvents and acid catalysts. The Brønsted acid properties of ILs are governed by the different functional groups within their structures that can function as proton donors. Thus, in principle one way to modify the acid properties should be through the incorporation of a sulfonic acid group in the cationic imidazolic structure. On this direction, four ILs with imidazolic bases and bisulfate counterion (two of them never reported) were synthesized and evaluated (see Table S1 in the ESI†). Two of them were sulfonic-type TSILs, where the imidazolic-base structures 1-methyl-3-buthylimidazolium ([bmim]) and 1-buthylimidazolium ([bHim]) were decorated with SO3H groups, leading to 1-(4-sulfonic acid)-butyl-3-methylimidazolium ([bsmim]) and 1-(4-sulfonic acid)-butyl-imidazolium ([bsHim]) cations, and to the resulting [bsmim]HSO4 and [bsHim]HSO4 TSILs. In addition, the corresponding ILs without the SO3H groups in the base structure were synthesized for comparison, leading to [bmim]HSO4 and [bHim]HSO4, respectively. In this way, we tuned the Brønsted acid properties of these TSILs, not only by incorporating a SO3H group (as explained previously), but also by adding other potentially labile proton sources in the imidazole base structure of [bHim] and [bsHim] cations. The goal of this analysis is to shed some light on the role of the SO3H group that is present in the TSIL structure in its global performance over a reactive system. As it is described in the literature, this performance might be governed by many structure-dependent variables such as the specific and non-specific interactions that may take place between catalyst, reactants and products, as well as the solubility, the stability, among others.1,24,25 However, as this performance also depends on the global composition of the IL, in this work this composition was monitored by cyclic voltammetry.
Electrochemical techniques are being intensely used for exploiting the outstanding properties of ILs as electrolytes,26 and particularly cyclic voltammetry has shown to be extremely useful as a detection technique for revealing the presence of free species other than the ionic pair.21,23 Thus, in this work the monitoring of ILs by cyclic voltammetry was carried out in order to perceive the effect of the IL synthesis conditions and parameters (synthesis reaction time, purification steps) on the amounts of these species. Our hypothesis is that a specific behaviour of a sulfonic TSIL in a reactive system cannot be attributed solely to the presence of the SO3H groups that functionalize the TSIL structure. There are many other variables (structural and compositional) that may affect the IL response, so it should be quite difficult to extend its behaviour to other similar ILs, even in the same family. This could be one of the reasons for the significant dispersion of results on the activity of these systems that is found in the literature. On the other hand, even though it is well known that the counterion also affects the catalytic properties of ILs, in this work the counterion HSO4− was kept constant in all the analyzed ILs because this is one of the most commonly used anions in these imidazolic combinations. The effect of changing the counterion will be analyzed in a following report. Finally, the catalytic performances of these four ILs were tested in a transesterification reaction by monitoring the reactant consumption and buildup of product concentration by UV-vis spectroscopy, which allowed the estimation of yields at different reaction times.
Experimental
Chemicals and materials
Imidazole (Him), methyl imidazole (mim), butyl bromide, 1,4-butanesultone, sodium ethoxide, p-nitrophenyl acetate, sulfuric acid (97–98%), and sodium bisulfate used as precursors or reactants were from Sigma-Aldrich. Methanol, sulfuric ether, ethyl acetate, hexane, acetone, butyl alcohol, and dichloromethane used as solvents or eluents were from Sigma-Aldrich. 1-Butyl-imidazole (bim) was synthesized by reaction of imidazole with butyl bromide through an aliphatic nucleophilic substitution, following a previously reported method that employs sodium ethoxide for deprotonating the imidazole,27,28 as it described in the ESI.†Instrumentation
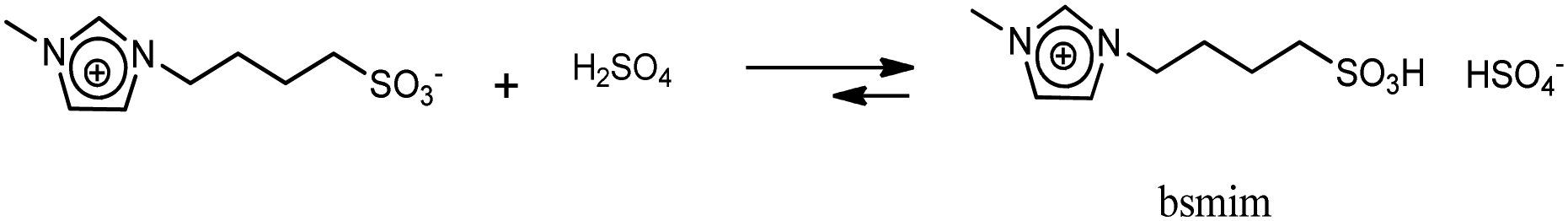
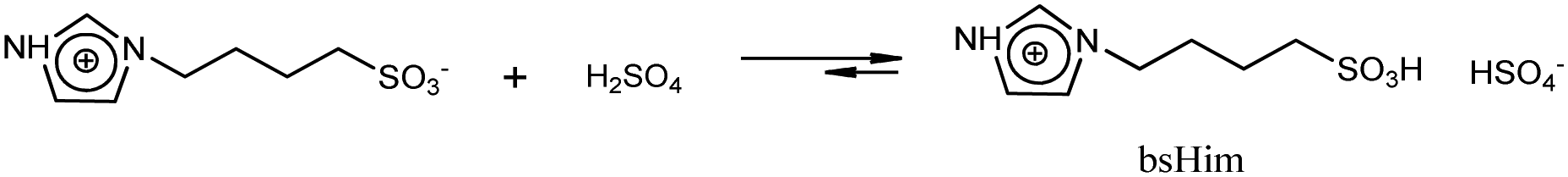
UV-vis spectra were taken using a Shimadzu UV-1800 UV/visible spectrophotometer. Voltammetric experiments were carried out using a CH Instruments CHI1140B potentiostat.Synthesis of sulfonic TSILs
The TSILs [bsmim]HSO4 and [bsHim]HSO4 were synthesized by reaction of sulfuric acid with the corresponding zwitterions, which are listed in Table S2 in the ESI.† Thus, the syntheses of these TSILs involved two steps: (i) the synthesis of the respective zwitterions; (ii) the protonation of the sulfonic groups at the zwitterions to obtain the cations of the final TSILs.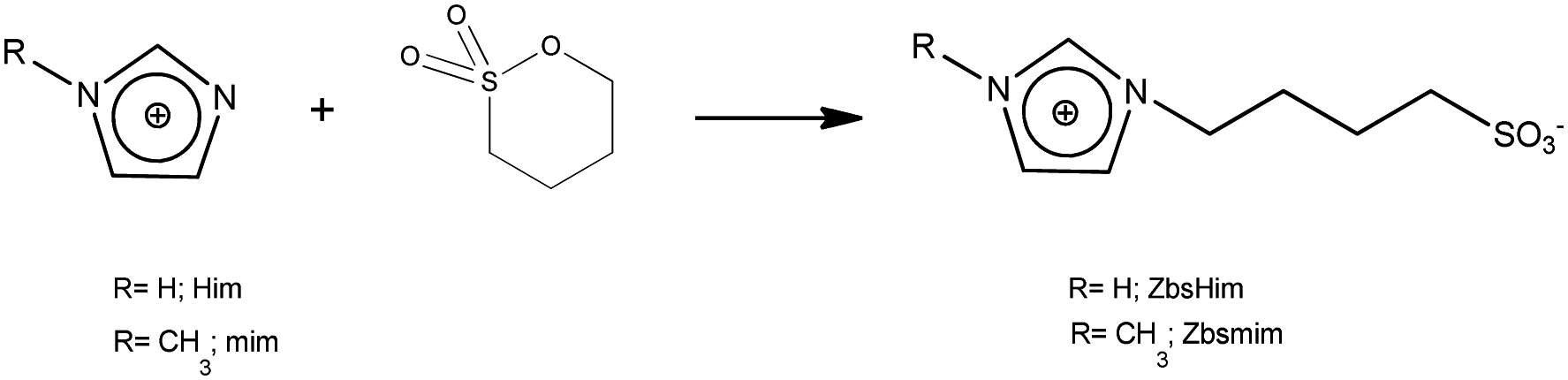 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent. Besides, the purification process was monitored by UV-vis spectroscopy.
:1) as eluent. Besides, the purification process was monitored by UV-vis spectroscopy.
 | (2) |
 | (3) |
:1) for [bsmim]HSO4 and hexane/ethyl acetate (1:2) for [bsHim]HSO4. In both cases the reaction product was a light yellow viscous liquid, which was placed in rotary evaporator for 1 h at 65 °C. The purification involved two extraction steps, first with butyl alcohol and then with sulfuric ether, a step under rotary evaporator for 2 h, and a final drying step at RT under ultra-high vacuum for 48 h.
Syntheses of the imidazolium-based ILs
 | (4) |
:1) as eluent. The solid NaBr that precipitated during the exchange reaction was separated by vacuum filtration. The supernatant viscous liquid was placed in rotary evaporator at 65 °C for 2 h to remove MeOH. It was finally dried under vacuum for 48 h, acquiring a light yellowish orange colour.
 | (5) |
:1) as eluent. The reaction product was a light yellow viscous liquid, which was placed in rotary evaporator at 80 °C for 2 h. Then it was purified by several extractions with sulfuric ether, brought to rotary evaporator for 2 h, and dried under ultra-high vacuum for 48 h.
Transesterification reaction
The model reaction that was used to evaluate the acid catalytic performance of the analyzed ILs was the transesterification reaction (or alcoholysis) between p-nitrophenyl acetate (PNPA) and MeOH giving 4-nitrophenol (PNP) and methyl acetate, according to reaction (6). This type of reaction, where an ester of a carboxilic acid is converted into a different one, can be displaced toward products with complete conversion by using an excess of alcohol (reactant) and an efficient acid or basic catalyst.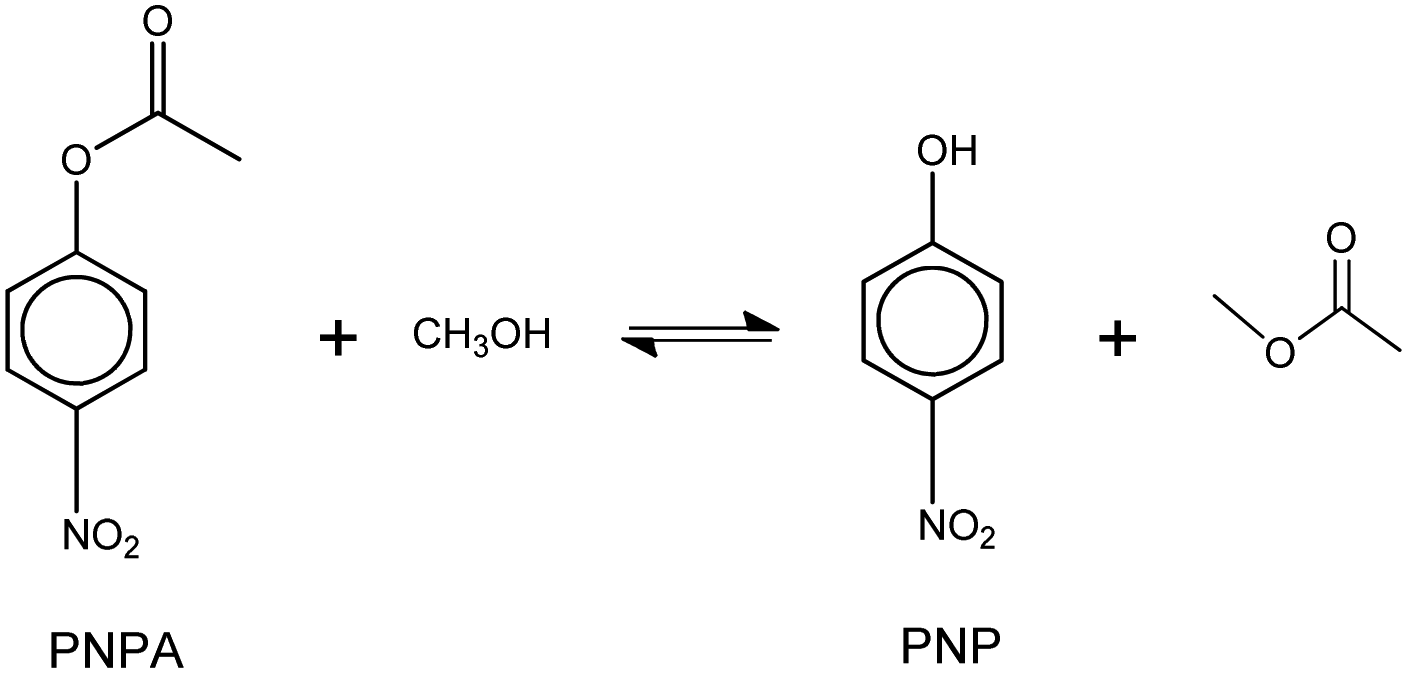 | (6) |
The reaction was carried out into a quartz cuvette (2 mL) thermostatted at the desired reaction temperature, starting from a PNPA solution in MeOH with a PNPA concentration 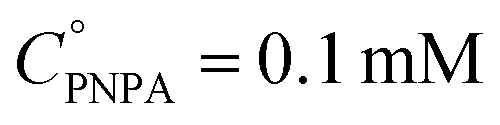 and an IL concentration of 0.1 M. For comparison of the performance against a well-known catalyst (as is sulfuric acid), this reaction was also carried out using 0.1 M H2SO4 instead of the IL. Most of the evaluations were done at 25.0 ± 0.1 °C, although some experiments were carried out at 60.0 ± 0.1 °C and at 100.0 ± 0.1 °C. The course of the reaction was monitored by UV-vis spectroscopy by acquiring spectra of the reaction media over the wavelength range between 220 and 500 nm every pre-defined time intervals. The PNP concentrations (CPNP) were estimated using these spectra taken at different reaction times (tR) from the absorbances measured at 310 nm, and using calibration curves measured on standard solutions of PNP in MeOH containing the respective ILs (see the ESI† for details on this procedure). This allowed the calculation of the reaction yields
and an IL concentration of 0.1 M. For comparison of the performance against a well-known catalyst (as is sulfuric acid), this reaction was also carried out using 0.1 M H2SO4 instead of the IL. Most of the evaluations were done at 25.0 ± 0.1 °C, although some experiments were carried out at 60.0 ± 0.1 °C and at 100.0 ± 0.1 °C. The course of the reaction was monitored by UV-vis spectroscopy by acquiring spectra of the reaction media over the wavelength range between 220 and 500 nm every pre-defined time intervals. The PNP concentrations (CPNP) were estimated using these spectra taken at different reaction times (tR) from the absorbances measured at 310 nm, and using calibration curves measured on standard solutions of PNP in MeOH containing the respective ILs (see the ESI† for details on this procedure). This allowed the calculation of the reaction yields 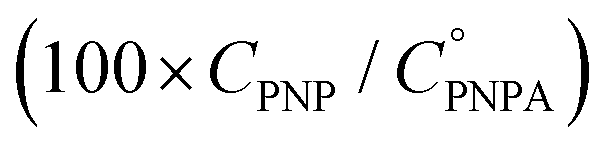 at different tR values.
at different tR values.
Voltammetric experiments
The synthesized ILs were characterized by cyclic voltammetry of Pt microelectrodes (MEs) for detecting free electroactive species (i.e. IL's parent species, or other species remaining from the synthesis). These experiments were carried out as described elsewhere.23 Briefly, a thermostatted low-volume (3 mL) three electrode cell was employed, with a disk-shaped 25 μm-diameter Pt ME as working electrode,35 and a Pt wire as counter-electrode. The reference electrode was a PdH wire36,37 placed inside a Luggin–Habber capillary that contained the same cell electrolyte. For operation, the cell was loaded at RT with the IL and closed with the Teflon cap that supported all the electrodes. Then, in order to melt those ILs that were solid, the cell temperature was raised to 80 °C. The resulting volume of the melted IL was ∼1.5 mL. The electrolyte was purged with inert gas (dry N2) by continuous bubbling. Cyclic voltammograms (CVs) were measured over a range of potentials (E) from a cathodic enough value (where reduction of the IL cation38 and/or hydrogen evolution23,37 was verified) to an anodic value where the oxidation of an IL component was detected, at a scan rate (v) of 0.1 V s−1. In order to confirm the assignment of voltammetric waves to the reduction or oxidation of a specific free species, when possible, pure precursors (i.e.[bmim]Br and zwitterions) were added during the experiments by using a canister placed at the cell cap. The absence of voltammetric peaks that could be associated to the reduction of platinum oxide formed from water traces allowed to confirm the inexistence of water at detectable levels.39,40Results and discussion
Voltammetric analysis of [bmim]Br and [bmim]HSO4
Imidazolic ILs with bmim as cation and with two different counterions (bromide and bisulfate) were analyzed first. As can be seen in the molecular structure of bmim (Table S1, ESI†), the imidazole has a H atom at the C atom in position 2 (which a priori should be the most labile H in bmim), and other less-labile H atoms at the C atoms in positions 4 and 5. Hydrogen atoms at the saturated carbon atoms in methyl and butyl substituent are hardly labile. The cathodic behaviours of ILs with this cation are very relevant for identifying the possible sources of protons at the imidazolic base that may be able to participate in processes with acid catalysis. The onset potentials of the observed cathodic voltammetric waves for H2 discharge from the different H atoms indicate how easy should be to draw on these proton sources. Moreover, as the bisulfate counterion is also a potential proton donor, the IL with bromide anion as counterion was analyzed first to identify the exclusive reduction of protons from the bmim cation.The CV of a Pt ME in [bmim]Br is shown in Fig. 1a. The anodic profile shows an exponential grow of the anodic current at E > 0.3 V, which never arrives to a limiting value (blue curve). This indicates that this current was generated by the oxidation of a large amount of reactant. Thus, it is most likely associated to the oxidation of bromide anions that form part of the IL ionic pair, generating bromine. In addition, an oxidation voltammetric peak is detected over the potential range −1 V < E < 0 V, whose charge slightly increased over the successive scans. Inspection of the Pt ME surface by SEM after being subjected to more than fifty cycles (inset in Fig. 1a) allowed to detect a faceting process of the Pt surface, which is typically caused by electro-dissolution using periodic signals.41 Thus, it is concluded that this peak was caused by the facile electro-oxidative dissolution of Pt in this highly concentrated bromide solution, presumably leading to dissolved platinum bromide complexes.
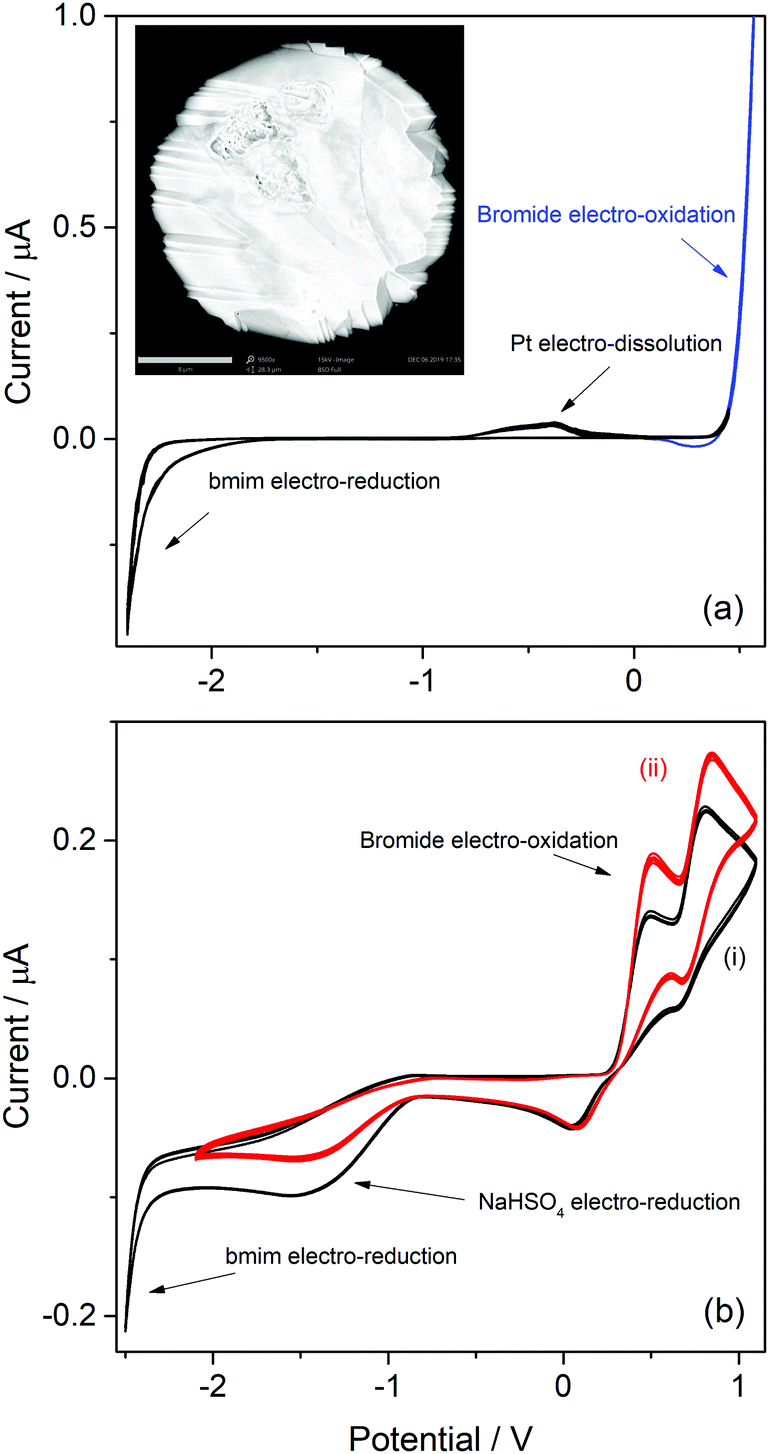 | ||
| Fig. 1 CVs of a Pt microelectrode in [bmim]Br (a) and in [bmim]HSO4 (b) before (i) and after (ii) addition of pure [bmim]Br. Inset in (a) shows a SEM picture of the Pt microelectrode after the voltammetric analysis (scale bar: 8 μm). T = 80 °C; v = 0.1 V s−1. | ||
Even though there are reports about this behaviour on Au,42 no similar antecedents were found for this response on Pt. Therefore, these results suggest a potential usefulness of [bmim]Br as an electrolyte for the environmentally friendly electrochemical etching of noble metal-based alloys for noble-metal recovering.43,44 Conversely, the scan toward cathodic direction only shows an exponential current increase at E < −2.3 V which, according to previous reports,38,45 is caused by the electroreduction of the imidazolium cation into the radical imidazol-2-yl, and its further decomposition. In conclusion, the potential window for analyzing possible cathodic discharges from additional proton sources that would be present at the next ILs that are studied in this work is extended down to −2.3 V (as below this limit the bmim cation is electro-reduced).
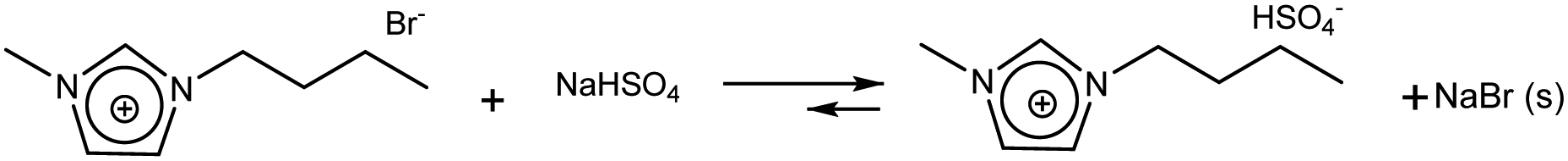
On the other hand, the voltammetric behaviour of a Pt ME in [bmim]HSO4 was analyzed, and the CV obtained at 80 °C is shown in Fig. 1b(i). In order to understand this CV, we should keep in mind that this IL has two important differences respect to its bromide analogue. First, the bisulfate counterion is an additional source of protons whose cathodic discharge could occur at potentials above the limit for bmim reduction. Second, as it was detailed in the Experimental section, the synthesis of [bmim]HSO4 involves the exchange reaction of [bmim]Br with NaHSO4, which leads to the formation of NaBr and the ionic pair through reaction (4). As NaBr has very low solubility, it precipitates and is separated by filtration, which allows displacing the exchange equilibrium toward the ionic pair. Taking into account these two aspects, the voltammetric waves in the CV (i) shown in Fig. 1b can be explained. Two oxidation peaks at 0.5 V and 0.8 V are detected, which could be associated to the electro-oxidation of bromide to bromine through a tribromide intermediate (which explains the presence of two peaks).46,47 These processes reach mass-transport limiting currents, which indicate that the concentration of bromide is low. The most probable source for these low (but detectable) amounts of bromide should be the free [bmim]Br that remains in equilibrium with the ionic pair through reaction (4). This was verified by the in situ addition of pure [bmim]Br to the analyzed IL, which caused an immediate increase of both anodic peaks, as can be seen in Fig. 1b(ii). It should be noted that, in the same way that there is an amount of free [bmim]Br, there also may remain some amount of NaHSO4 in equilibrium. Related to that, the cathodic scan shows a mass-transport controlled reduction wave at E < −0.8 V, which is tentatively caused by the reduction of this free amount of NaHSO4. This hypothesis was confirmed by the current decrease observed on this wave in Fig. 1b(ii) upon addition of pure [bmim]Br, which reacted with part of this free NaHSO4 through reaction (4) and decreased its concentration. At more cathodic potentials (E < −2.3 V) an exponential current increase is observed (as it was also detected in [bmim]Br), which is likely caused by the electro-reduction of the imidazolium cation.38,45 It should be noted that the bisulfate anion forming the ionic pair was not electro-reduced over the analyzed potential range, which indicates that the hydrogen atom of this group is strongly tightened into the highly compact structure of the IL, so it would be hardly available for acid catalysis. In summary, the IL [bmim]HSO4 conforms a compact arrangement of cations and anions with potential H sources that are deeply involved in the IL structure,48,49 so their availability is very limited. The only feasible source of protons in this IL is the free sodium bisulfate that remains from the synthesis (together with free [bmim]Br).
Voltammetric analysis of [bsmim]HSO4
As can be seen in the molecular structure of bsmim (Table S1, ESI†), the sulfonic acid group in this cation is a potential source of protons for acid catalysis. However, it is not clear how available these hydrogen atoms can be when these groups are involved in the compact IL structure. It was verified in the previously analyzed IL that H atoms from the anion are strongly compromised in building the IL network, so they have a completely different reactivity than the same H atoms in the free anion. If the sulfonic acid groups are usable sources of protons, it should be possible to detect by cyclic voltammetry the hydrogen evolution from these groups. This would constitute strong evidence in order to argue on how accessible these protons are in the IL. However, it should be kept in mind that H-containing free species can also be detected by this technique, as it was the case in the previous analysis of [bmim]HSO4 where hydrogen evolution from free NaHSO4 was detected. In this context, Fig. 2a(i) shows a CV measured on a Pt ME in [bsmim]HSO4 that was synthesized and purified as described in the Experimental section. This CV shows that in the cathodic scan, there is a significant current increase at E < −0.5 V (which is not very negative), indicating the discharge of hydrogen from a protonated species in quite high concentration. Electro-reduction of protons leads to dissolved hydrogen, whose concentration builds up at the electrode surface while it diffuses into the solution. The diffusion rate is affected by the diffusion coefficient of dissolved hydrogen, which in these highly viscous media should be much lower than in aqueous media.26 Thus, when high current densities are reached, oversaturation of dissolved hydrogen should occur,50 with the consequent nucleation of hydrogen bubbles at the electrode surface. The size and the dynamic behaviour of these bubbles are governed by the fluid viscosity, among other parameters.51 The formation and evacuation of bubbles at/from the ME surface generates random variations of the electrode current,52 which leads to a noisy signal in the CVs shown in Fig. 2. Taking into account that in [bmim]HSO4 a similar discharge (but with much smaller current) from free NaHSO4 was detected over this same potential range, it is likely that this current is caused by the discharge of the remaining free acid that was used to neutralize the zwitterions (H2SO4 in this case). This free sulfuric acid could be present in the IL not necessarily from remaining amounts that were not extracted, but also from the reverse displacement of the neutralization equilibrium given by reaction (2). In order to verify this hypothesis, the CVs of samples that were treated in different ways for deliberately changing the presumable amounts of free acid were analyzed. On the one hand, a first strategy was to modify the number of extraction steps used in the purification.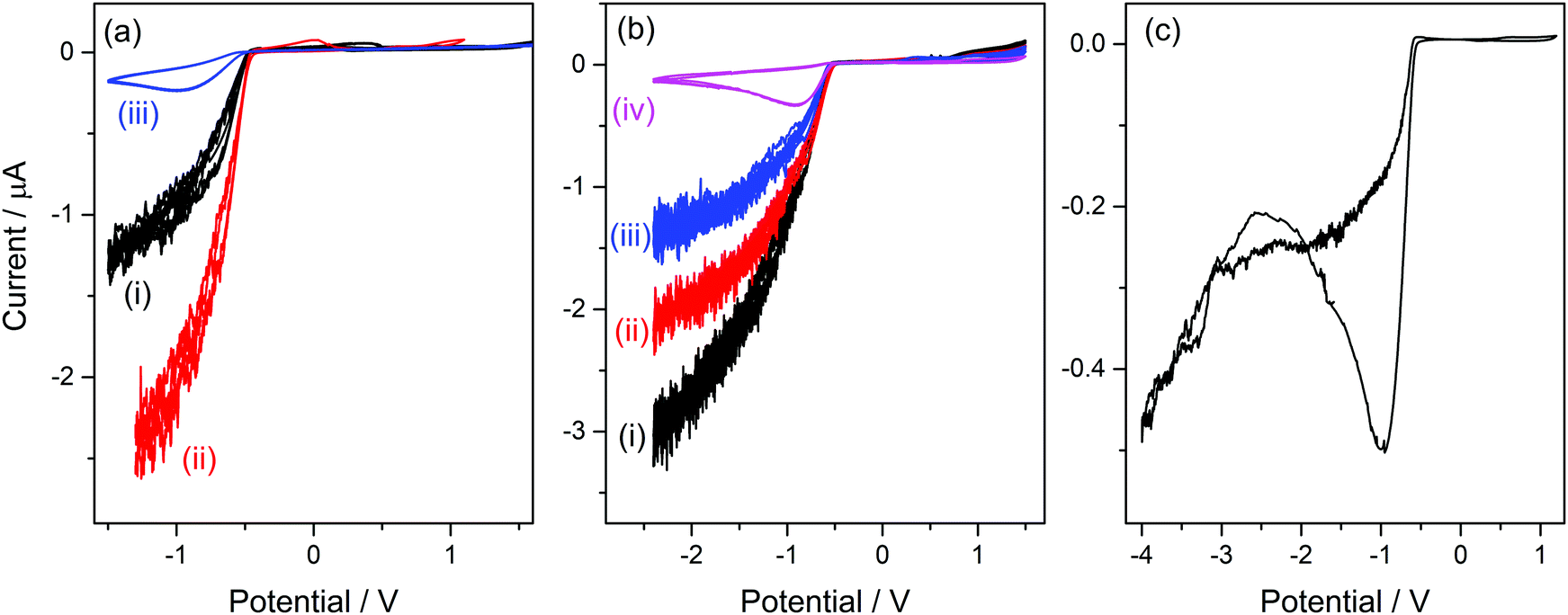 | ||
| Fig. 2 CVs of a Pt ME in [bsmim]HSO4 at 80 °C. (a) CVs in [bsmim]HSO4 purified with two extraction steps (i), in crude [bsmim]HSO4 (ii), and in [bsmim]HSO4 purified with six extraction steps (iii). (b) CVs in crude [bsmim]HSO4 (i), and after three successive additions (18 mg each) of pure Zbsmim (ii–iv). (c) Highly cathodic CV in [bsmim]HSO4 purified with six extraction steps. T = 80 °C; v = 0.1 V s−1. | ||
Thus, CVs were measured on three [bsmim]HSO4 samples with different number of extraction steps, one of them being the IL purified with two extraction steps whose CV was already shown in Fig. 2a(i). It should be noted that the extraction of free H2SO4 causes the back displacement of the neutralization equilibrium (reaction (2)), leading to an increase of the concentration of free Zbsmim. The CVs measured on an “as-prepared” (or crude) IL (without extraction steps) and on an IL with six extraction steps (where saturation and precipitation of Zbsmim was evident) are shown in Fig. 2a(ii) and (iii), respectively. The CV measured in the crude IL shows a significant H2-evolution current, indicating the presence of a very large amount of remaining free H2SO4. The CV taken in the IL with two extraction steps shows that this current is much smaller, being indicative of the good efficiency of the purification method for removing free H2SO4. The last CV shows that, even though the repetitive application of extractions (up to six times in this case) led to a decrease of the reduction current to very low levels, it was not possible to eliminate it completely. Moreover, saturation of the IL with Zbsmim was evident by detecting its precipitation, which is reasonable taking into account that a decrease of the free acid concentration leads to an increase of free Zbsmim concentration by displacement of reaction (2). In fact, once the saturation of the IL with Zbsmim was reached, the concentration of free H2SO4 became fixed by the equilibrium constant and could not be further decreased.
On the other hand, a second strategy to modify the free acid concentration was to add increasing amounts of pure Zbsmim to the crude IL. Thus, CVs measured on a crude [bsmim]HSO4 IL with different additions of Zbsmim are shown in Fig. 2b. A decrease of the current for H2 discharge is verified for increasing amounts of added Zbsmim, which reveals the displacement of reaction (2) generating the ionic pair with the consequent consumption of free H2SO4, in consonance with the results of the previous strategy. Results shown in Fig. 2a and b constitute very solid evidences that the IL [bsmim]HSO4 always contains free H2SO4 in concentrations that may vary from a small value in equilibrium with saturated Zbsmim to very large values (depending on the number of purification steps). This free H2SO4 is the most accessible source of protons in this IL.
The reduction of free H2SO4 in samples that contain large amounts of this species involves a significant current that impedes to detect any other parallel reduction process that may occur at E < −0.5 V. However, the CVs measured on the IL with the minimum possible amount of free H2SO4 shown in Fig. 2a(iii) and b(iv) allowed to confirm that no other species reduces over the analyzed potential range. By extending the cathodic potential limit to more negative values (down to −4 V), as it is shown in Fig. 2c, only one additional cathodic discharge is verified at E < −2.5 V. By comparing this response to those measured in [bmim]-based ILs that were described previously, it can be inferred that this very cathodic discharge should be associated to the reduction of the imidazolium group.38,45 Therefore, results shown in Fig. 2c indicate that the protons in the sulfonic acid groups constituting the cations in this TSIL are not electro-reduced over a very wide potential range, similarly to what was already detected for the protons in the bisulfate anions that conform the TSIL network. In summary, it is concluded that even though this TSIL has two potential sources of protons for acid catalysis (sulfonic acid groups at the cation and bisulfate groups at the anion), these protons are strongly bonded to the TSIL macrostructure, possibly playing important roles in keeping its integrity. Thus, the only hydrogen source that is easily available for chemical and electrochemical reactions is the free H2SO4 that is at equilibrium with [bsmim]HSO4 and dissolved Zbsmim.
Voltammetric analysis of [bHim]HSO4
The cation bHim, whose structure can be visualized in Table S1 (ESI†), contains a hydrogen atom in position 3 of the imidazole structure (instead of the methyl group in bmim), which may be labile enough for participating in acid catalysis. As it was detailed in the Experimental section, the imidazole group receives this hydrogen atom from a strong acid, which in this case is H2SO4, through reaction (5). The CV of a Pt ME in [bHim]HSO4 (Fig. S1(i) in the ESI†) evidences two cathodic processes. The first wave detected at E < −0.6 V corresponds to a mass-transport controlled process, which could be associated to the hydrogen discharge from a very low amount of free H2SO4. The addition of pure bim led to the vanishing of this wave (Fig. S1(ii) in the ESI†) due to its reaction with free H2SO4, confirming the previous hypothesis. The second discharge at E < −1.2 V is an exponential current increase from a species in excess. This is much less cathodic than the bmim discharge that occurred at E < −2.5 V, and is probably caused by hydrogen evolution from the imidazolic H atom in the bHim cation (H3). These results indicate that the IL [bHim]HSO4 contains very little amounts of free H2SO4 and an additional potential source of hydrogen that, even though is not very labile, could be available for acid catalysis.Voltammetric analysis of [bsHim]HSO4
The structure of the cation bsHim, which is visualized in Table S1 (ESI†), contains two potential sources of hydrogen, which are the imidazolic hydrogen (H3) and the proton at the sulfonic acid group. On the basis of the previous results obtained on [bsmim]HSO4 and on [bHim]HSO4, it seems like the imidazolic hydrogen is easier to reduce than the sulfonic proton, but this should also be verified in this TSIL that contains both sources simultaneously. As it was detailed in the Experimental section, the sulfonic group receives the proton from a strong acid, which in this case is H2SO4, through reaction (3), so it is very likely that some amount of free H2SO4 may be present. The CV of a Pt ME in [bsHim]HSO4 is shown in Fig. 3(i). A large cathodic current is verified at E < −0.5 V which, according to previous results, is most likely related to the hydrogen discharge from an excess of free H2SO4. This was verified by acquiring CVs after successive additions of increasing amounts of pure ZbsHim, causing the consumption of free H2SO4 by reaction (3) and the consequent drop of the cathodic currents, as it is observed in CVs (ii) and (iii) of Fig. 3. In fact the CV (iii) was measured in a solution saturated with ZbsHim, where an amount of solid zwitterion remained undissolved, so this shows the lowest amount of free H2SO4 that can be attained. Precisely in this last solution with such a low content of free H2SO4 it was possible to detect a second increase of the cathodic current at E < −1.3 V (as it is better observed in the inset graph of Fig. 3). This cathodic process shows up over the same potential range, not very cathodic, of a reduction current that was also observed in [bHim]HSO4 (Fig. S1(ii) in the ESI†) and assigned to the discharge of the imidazolic proton. No other cathodic discharges were detected, at least down to a potential of −3 V. Taking into account these evidences, it can be concluded that the protons at the sulfonic acid groups in [bsHim]HSO4 are not labile at all, similarly to what was verified in [bsmim]HSO4.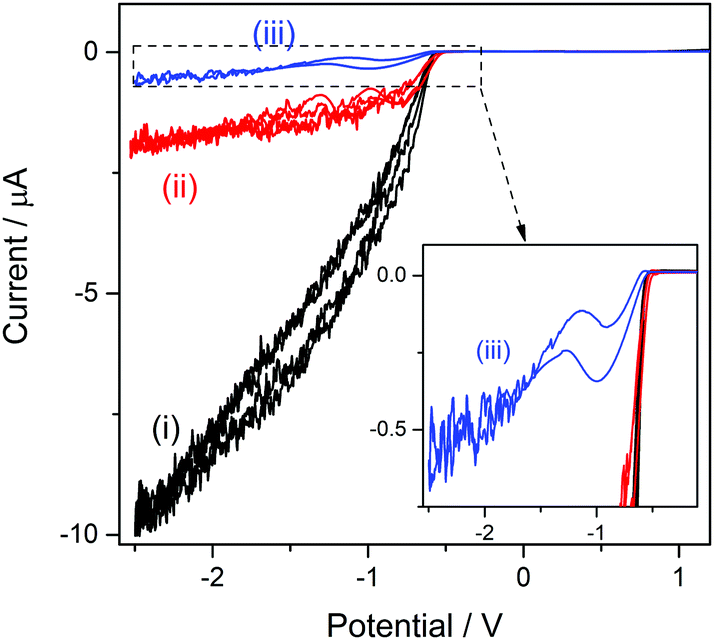 | ||
| Fig. 3 CVs of a Pt ME in [bsHim]HSO4 before (i) and after one (ii) and two (iii) additions of 15 mg of pure ZbsHim. Inset graph shows an expanded current scale over the potential interval −2.5 V ≤ E ≤ 0.0 V. T = 80 °C; v = 0.1 V s−1. | ||
Performance of ILs in a transesterification reaction monitored by UV-vis spectroscopy
Transesterification reactions are being widely studied in the last decades due to their technological application in manufacturing of biodiesel from vegetal oils.53–55 The recent interest on these processes is mostly focused on seeking alternatives for accomplishing the principles of Green Chemistry, which among other aspects, encourages the use of environmentally friendly and reusable catalysts. Thus, as these TSILs fit into this type of catalysts, our particular interest is to analyze and to understand their catalytic behaviour.Particularly in this work, we monitored by UV-vis spectroscopy the progress of the reaction of PNPA with MeOH leading to PNP and methyl acetate (MeAc) through reaction (6), using the four analyzed ILs as catalysts. As PNP is a pH indicator that exists at acidic pH values (<6), its detection in protonated form (and not as the phenolate) indicates the presence of an acid reaction medium. The UV-vis spectra of PNP and PNPA in methanolic solutions containing either of the studied ILs present absorption peaks at ∼269 nm (for PNPA)56 and ∼310 nm (for PNP).57 The hypsochromic shift of the PNPA absorption peak is caused by the lower delocalization of electrons at the oxygen atom in the ester respect to the OH in PNP. The absorption maxima of these peaks allows to follow the reaction conversion at different tR values. Thus, Fig. 4 shows the UV-vis spectra acquired on the reaction media at successive tR values when using the four studied ILs, as described in the Experimental section. An isosbestic point can be observed in all cases (in the wavelength range between 284 and 288 nm, depending on the used IL), which indicates that the conversion of reagents into products proceeds by a single and quantitative process. It is verified that much shorter tR values (less than 4 h) are needed to build a significant and invariant concentration of PNP (corresponding to almost complete conversion) when using both sulfonic TSILs, in contrast with the more than 20 h required when using [bmim]HSO4. Moreover, in [bHim]HSO4 the generation of PNP is clearly detected only after 48 h. In order to better visualize these differences, the PNP concentrations (CPNP) were estimated from these spectra as described in the Experimental section, and the reaction yields were calculated from these concentrations at different tR values, which are plotted in Fig. 5 for the four ILs. This shows the rapid increase of the reaction yields when using sulfonic TSILs, which tend to the maximum values at tR in the order of 20 h. These yields contrast with the low values detected when using the respective ILs, which only start to increase after 20 h and are still increasing after 100 h. Moreover, for a more quantitative comparison, apparent rate constants (kapp) were calculated from this data assuming a pseudo-first order kinetics respect to PNPA concentration (as described in the ESI†), which are shown in Table 1. It is verified that the kapp values measured on the ILs are more than two orders of magnitude smaller than those measured on the corresponding sulfonic TSILs. A first hypothesis for such a great catalytic difference between the TSILs and their corresponding ILs is that the catalytic performance would probably be associated to the large amounts of free sulfuric acid (as detected by voltammetry). It was shown that the other potential source of protons at the sulfonic acid group in TSILs is not easily accessible, and would hardly catalyze this reaction. In order to corroborate this hypothesis, the reaction was carried out using a TSIL ([bsmim]HSO4 in this case) with varying contents of free H2SO4. Thus, in addition to the purified [bsmim]HSO4 that was used in the previous catalytic test, the catalytic performance of other two [bsmim]HSO4 samples were evaluated. On the one hand, the crude [bsmim]HSO4 (without purification steps) containing a significant excess of free H2SO4 as noted by the CV in Fig. 2b(i), was used as a catalyst with a high content of free H2SO4. On the other hand, it was also used the [bsmim]HSO4 that received successive additions of Zbsmim for neutralizing the excess of free H2SO4 down to the minimum concentration in equilibrium with saturated Zbsmim, as verified by the CV in Fig. 2b(iv).
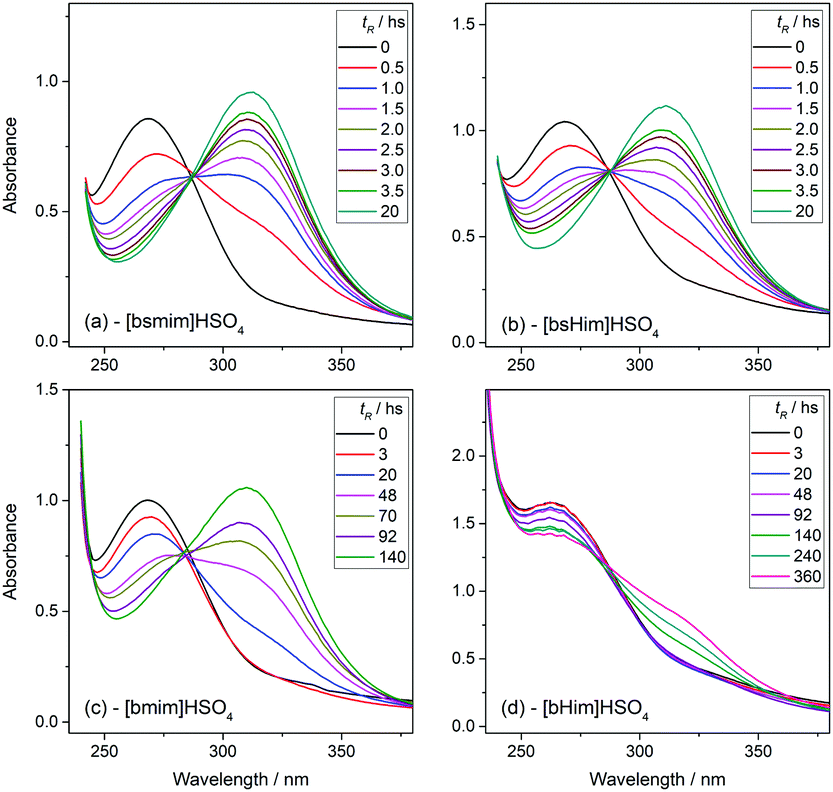 | ||
| Fig. 4 UV-vis spectra of the reaction media for the transesterification reaction of PNPA with MeOH at 25 °C in the presence of [bsmim]HSO4 (a), [bsHim]HSO4 (b), [bmim]HSO4 (c), and [bHim]HSO4 (d), acquired at different tR values, as indicated in each graph. | ||
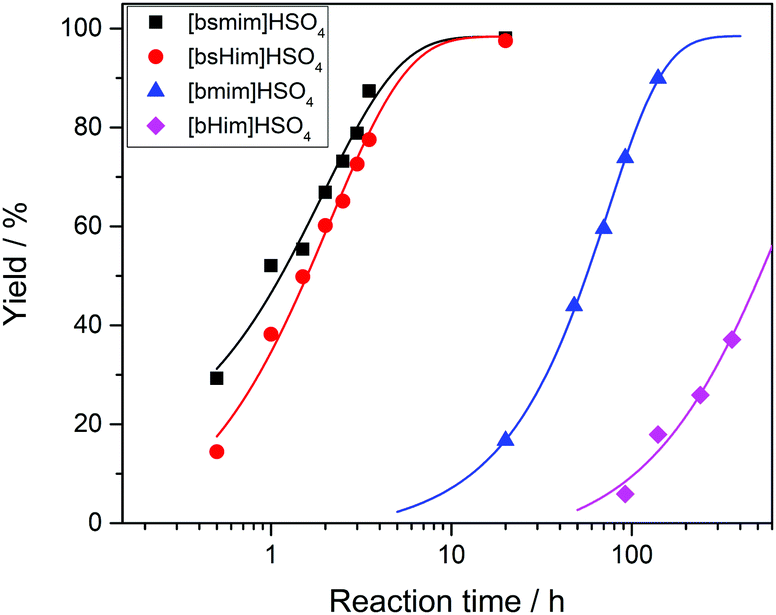 | ||
| Fig. 5 Yields for the transesterification reaction of PNPA with MeOH at 25 °C measured from the UV-vis spectra at different reaction times for the four studied ILs. Lines are drawn for a better trend perception. | ||
| IL | k app/s−1 |
|---|---|
| [bHim]HSO4 | (3.5 ± 0.2) × 10−7 |
| [bmim]HSO4 | (4.2 ± 0.2) × 10−6 |
| [bsHim]HSO4 | (1.21 ± 0.02) × 10−4 |
| [bsmim]HSO4 purified | (1.56 ± 0.06) × 10−4 |
| [bsmim]HSO4 crude | (1.25 ± 0.03) × 10−4 |
| [bsmim]HSO4 + Zbsmim | (3.1 ± 0.1) × 10−5 |
| 0.1 M H2SO4 | (2.14 ± 0.08) × 10−4 |
The reaction yields for each case as a function of tR are shown in Fig. 6 (complete spectra are provided in the ESI†), and the yields obtained when using pure H2SO4 are included as well for comparison. Besides, the kapp values calculated from this data are tabulated in Table 1. It is verified that the catalytic efficiencies of both TSILs with significant amounts of free H2SO4 are similar to that of pure H2SO4 (with kapp values in the same order of magnitude), where the reaction conversion is almost complete in around 10 h. Thus, it is concluded that the catalytic activity of sulfonic TSILs for this reaction is quite equivalent to that of pure sulfuric acid as long as it contains appreciable amounts of free acid coexisting in equilibrium with the ionic pair. Remarkably, the TSIL with minimum amounts of free H2SO4 presented a catalytic activity only slightly lower than the previous cases (with a kapp only one order of magnitude smaller), verifying almost complete conversion in around 40 h. These results demonstrate that even though the presence of large amounts of free H2SO4 accelerates the reaction to reach high yields in very short times, this is not critical and it takes just a few more hours of reaction for the TSIL to provide the protons that catalyze the reaction to attain complete conversion. The TSIL works as an efficient reservoir for the acid and is capable to dose it on demand to sustain an efficient acid catalysis. The advantages of using sulfonic TSILs instead of pure sulfuric acid are associated to its much safer handling, recycling, and disposal, properties that turn these catalysts very environmentally friendly.
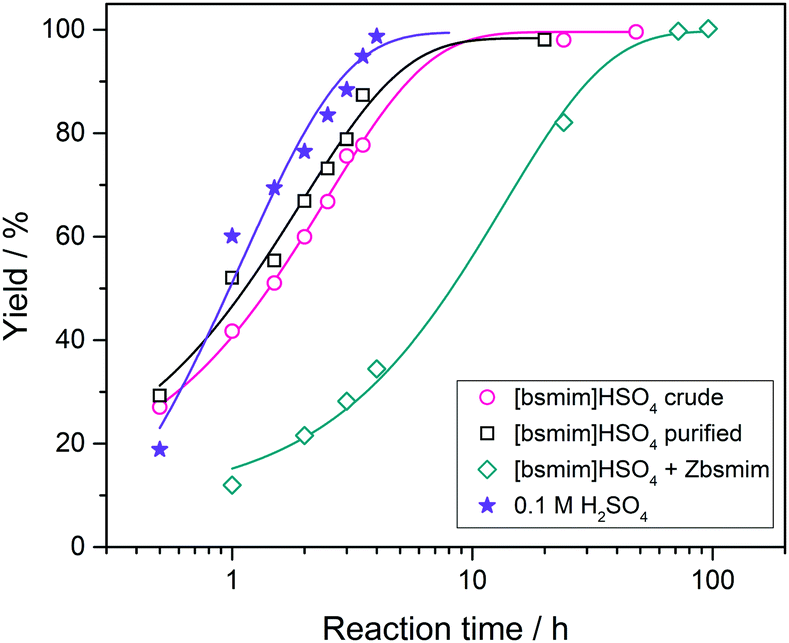 | ||
| Fig. 6 Yields for the transesterification reaction of PNPA with MeOH at 25 °C measured from the UV-vis spectra at different reaction times in the presence of [bsmim]HSO4 with three different contents of free H2SO4. Yields obtained when using 0.1 M H2SO4 are also included. Lines are drawn for a better trend perception. | ||
On the other hand, the respective non-sulfonic imidazolic ILs contain very little amounts of free acid, as it was verified by cyclic voltammetry, which leads to very low reaction rates for the catalyzed transesterification reaction at RT. It is evident that the reaction of PNPA with MeOH involves the active participation of the free acid (free bisulfate in [bmim]HSO4 or free sulfuric acid in [bHim]HSO4), which should be released by the ionic pairs through a back displacement of the neutralization/exchange reactions that were used to synthesize the ionic pairs (reactions (4) and (5), respectively). Although in [bmim]HSO4 the reversing of reaction (4) is impeded by the negligible availability of NaBr, the release of H2SO4 from [bHim]HSO4 from the back displacement of reaction (5) should be possible. However, the small amounts of H2SO4 verified even at 80 °C in the CVs (Fig. S1(i) in the ESI†) indicates that this reverse process seems to be thermodynamically and kinetically disfavoured, or in other words, the ion pair [bHim]HSO4 is much more stable than [bsHim]HSO4. In spite of that, these catalytic systems could still be useful when operating in high-temperature conditions. In order to analyze this effect, the catalytic performances of the imidazolic ILs were studied by measuring the yields at different reaction times at 60 °C and at 100 °C, verifying a notable increase in the rate to achieve complete conversion, as can be observed in Table 2.
| Temperature (°C) | IL | |
|---|---|---|
| [bmim]HSO4 | [bHim]HSO4 | |
| 25 | 20hs: <20% | 20hs: <10% |
| 92hs: 70–80% | 360hs: 30–40% | |
| 140hs: 90–95% | ||
| 60 | 20hs: 80% | 20hs: <30% |
| 48hs: 95–99% | 92hs: 80% | |
| 168hs: 95–99% | ||
| 100 | 20hs: 95–99% | — |
This evidence demonstrates the possibility of using imidazole-based ILs as efficient acid catalysts at temperatures only slightly above ambient conditions. As the synthesis and purification procedures of these ILs are simpler than those of the corresponding sulfonic TSILs (whose syntheses require specific reactants and involve several steps), their use as catalysts can still be a better choice than the respective TSILs in reactive systems where the reaction temperature can be raised.
Conclusions
The global compositions of imidazolic ILs and their derived sulfonic TSILs that use bisulfate as counterion, were analyzed by cyclic voltammetry. The goal was to detect the free species that coexist with the ionic pairs, and to explain on this basis their performances as acid catalysts. The transesterification of PNPA with MeOH to produce PNP was used as a catalyzed model reaction, as it proved to be a simple and thermodynamically efficient process. From these analyses, it was demonstrated for the first time that the outstanding catalytic activity of sulfonic TSILs is directly associated to the large amounts of free sulfuric acid that are contained within the ILs. However, in conditions of low free acid content, protons that are stored in the sulfonic acid groups constituting the functionalized IL network can be easily delivered through the reverse autoprotolysis equilibria, thus sustaining an acceptable catalytic activity. In addition, a number of other important conclusions about the IL's compositions, electrochemical performances, and reactivities were established, which are described below.Firstly, it was verified that neither the bisulfate anions conforming the ionic pairs of all the analyzed ILs nor the sulfonic acid groups of the studied TSILs can be electro-reduced. This evidence indicates that these protons may be strongly involved in building the IL's structures and are hardly available for participating in catalytic processes. In fact, the most accessible source of protons in all these ILs is the free acid precursor used for obtaining the bisulfate-based ILs, which remains in equilibrium with the ionic pair in variable amounts. It was verified that both [bsmim]HSO4 and [bsHim]HSO4 contained larger amounts of free H2SO4 when compared to their respective imidazolic ILs [bmim]HSO4 and [bHim]HSO4, which can explain the higher rates measured on these TSILs when catalyzing the transesterification reaction. The electro-reduction of these protons occurs at potentials that are not very cathodic, so the free acid can function as an effective source of hydrogen for acid catalysis. The amount of free H2SO4 may vary depending on the equilibrium constant of the autoprotolysis reaction, the number of extraction steps after synthesis, and the addition of pure base. Besides, the bim-based cations in [bHim]HSO4 and in [bsHim]HSO4 contain an additional hydrogen atom at position 3 of the imidazolic ring that can be electro-reduced at intermediate cathodic potentials, but it has no effect in the catalytic performance of these ILs.
In summary, the catalytic activity at RT of the imidazolic ILs was improved by designing TSILs that incorporate sulfonic acid groups into the imidazolic bases. In principle, the addition of this group was envisaged as an additional source of labile protons that would increase the acidity, but in fact in this work it was proved that these TSILs function as reservoirs for releasing the free acid, which is the actual acid catalyst. The respective imidazolic ILs without the sulfonic acid groups are poor acid catalysts at RT for the tested reaction, but their activity can be significantly improved by increasing the reaction temperature up to 100 °C. Thus, they still can be used as structurally simple efficient catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Universidad Nacional del Litoral (CAI+D 2016 234 and 018), Agencia Santafesina de Ciencia, Tecnología e Innovación (IO 2018 00161), Agencia Nacional de Promoción Científica y Tecnológica (PICT 2017 1340) and CONICET (PUE 229). M. B. M. thanks CONICET for a doctoral fellowship.Notes and references
- T. Welton, Proc. R. Soc. A, 2015, 471, 20150502 CrossRef.
- K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 16, 46430 Search PubMed.
- P. Nancarrow and H. Mohammed, ChemBioEng Rev., 2017, 4, 106 CrossRef.
- Ionic Liquids: Industrial Applications to Green Chemistry, ed. R. D. Rogers and K. R. Seddon, ACS Symposium Series, Washington, 2002 Search PubMed.
- Green Industrial Applications of Ionic Liquids, ed. R. D. Rogers, K. R. Seddon and S. V. Volkov, NATO Science Series: II. Mathematics, Physics and Chemistry, Springer, Dordrecht, 2003, vol. 92 Search PubMed.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. C. Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831 CrossRef CAS.
- M. Volland, V. Seitz, M. Maase, M. Flores, R. Papp, K. Massonne, V. Stegmann, K. Halbritter, R. Noe, M. Bartsch, W. Siegel, M. Becker and O. Huttenloch, Method for the separation of acids from chemical reaction mixtures by means of ionic fluids, World Pat., WO 03062251, 2003.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef CAS.
- A. Wierzbicki and J. H. Davis, Jr., Proceedings of the symposium on advances in solvent selection and substitution for extraction, AIChE, New York, 2000, p. 14F.
- A. Arfan and J. P. Bazureau, Org. Process Res. Dev., 2005, 9, 743 CrossRef CAS.
- R. Giernoth, Angew. Chem., Int. Ed., 2010, 49, 2834 CrossRef CAS.
- S. Lee, Chem. Commun., 2006, 1049 RSC.
- Y. Zhao, J. Long, F. Deng, X. Liu, Z. Li, C. Xia and J. Peng, Catal. Commun., 2009, 10, 732 CrossRef CAS.
- Y. Q. Cai, G. Q. Yu, C. D. Liu, Y. Y. Xu and W. Wang, Chin. Chem. Lett., 2012, 23, 1 CrossRef CAS.
- D.-J. Tao, J. Wu, Z.-Z. Wang, Z.-H. Lu, Z. Yang and X.-S. Chen, RSC Adv., 2014, 4, 1 RSC.
- H. Ding, W. Ye, Y. Wang, X. Wang, L. Li, D. Liu, J. Gui, C. Song and N. Ji, Energy, 2018, 144, 957 CrossRef CAS.
- P. Fan, S. Xing, J. Wang, J. Fu, L. Yang, G. Yang, C. Miao and P. Lv, Fuel, 2017, 188, 483 CrossRef CAS.
- X. Liang and J. Yang, Green Chem., 2010, 12, 201 RSC.
- Z. Ullah, A. S. Khan, N. Muhammad, R. Ullah, A. S. Alqahtani, S. N. Shah, O. B. Ghanem, M. A. Bustam and Z. Man, J. Mol. Liq., 2018, 266, 673 CrossRef CAS.
- P. Berton, S. P. Kelley, H. Wang and R. D. Rogers, J. Mol. Liq., 2018, 269, 126 CrossRef CAS.
- S. E. Goodwin, D. E. Smith, J. S. Gibson, R. G. Jones and D. A. Walsh, Langmuir, 2017, 33, 8436 CrossRef CAS.
- R. Kanzaki, H. Doi, X. Song, S. Hara, S. Ishiguro and Y. Umebayashi, J. Phys. Chem. B, 2012, 116, 14146 CrossRef CAS.
- M. V. Bravo, J. L. Fernández, C. G. Adam and C. D. Della Rosa, ChemPlusChem, 2019, 84, 919 CrossRef CAS.
- C. Reichardt and T. Welton, Solvent Effects in Organic Chemitry, Wiley-VCH, Weinheim, 4th edn, 2011 Search PubMed.
- Handbook of Solvents, ed. G. Wypych, ChemTech Publishing, Toronto, 2001 Search PubMed.
- Electrochemistry in Ionic Liquids, ed. A. A. J. Torriero, Springer, Cham, 2015, vol. 1 Search PubMed.
- C. G. Adam and G. Fortunato, J. Surfactants Deterg., 2019, 22, 501 CrossRef CAS.
- P. Bonhote, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Gratzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS.
- C. G. Adam, M. V. Bravo, P. M. E. Mancini and G. G. Fortunato, J. Phys. Org. Chem., 2014, 27, 841 CrossRef CAS.
- L. Farkas, O. Schachter and B. H. Vromen, J. Am. Chem. Soc., 1949, 71, 1991 CrossRef CAS.
- X. Cui, J. Cai, Y. Zhang, R. Li and T. Feng, Ind. Eng. Chem. Res., 2011, 50, 11521 CrossRef CAS.
- Y. Peng, X. Cui, Y. Zhang, T. Feng, Z. Tian and L. Xue, Appl. Catal., A, 2013, 466, 131 CrossRef CAS.
- Z. Yang, X. Cui, X. Yu, Y. Zhang, T. Feng and H. Liu, Catal. Lett., 2015, 145, 1281 CrossRef CAS.
- P. L. Silva, C. M. Silva, L. Guimarães and J. R. Pliego Jr., Theor. Chem. Acc., 2015, 134, 1591 Search PubMed.
- F. F. Fan, J. L. Fernández, B. Liu, J. Mauzeroll and C. G. Zoski, Ultramicroelectrodes, in Handbook of Electrochemistry, ed. C. G. Zoski, Elsevier, Amsterdam, 2007, ch. 6, pp. 189–197 Search PubMed.
- M. Fleischmann and J. N. Hiddleston, J. Phys. E: Sci. Instrum., 1968, 1, 667 CrossRef.
- J. A. Bautista-Martinez, L. Tang, J.-P. Belieres, R. Zeller, C. A. Angell and C. Friesen, J. Phys. Chem. C, 2009, 113, 12586 CrossRef CAS.
- R. Michez, T. Doneux, C. Buess-Herman and M. Luhmer, ChemPhysChem, 2017, 18, 2208 CrossRef CAS.
- A. García-Mendoza and J. C. Aguilar, Electrochim. Acta, 2015, 182, 238 CrossRef.
- C. L. Bentley, A. M. Bond, A. F. Hollenkamp, P. J. Mahon and J. Zhang, J. Phys. Chem. C, 2014, 118, 22439 CrossRef CAS.
- R. M. Cerviño, A. J. Arvía and W. Vielstich, Surf. Sci., 1985, 154, 623 CrossRef.
- P. H. Qi and J. B. Hiskey, Hydrometallurgy, 1993, 32, 161 CrossRef CAS.
- C. Deferm, J. Hulsegge, C. Möller and B. Thijs, J. Appl. Electrochem., 2013, 43, 789 CrossRef CAS.
- M. Balva, S. Legeai, N. Leclerc, E. Billy and E. Meux, ChemSusChem, 2017, 10, 2922 CrossRef CAS.
- R. Michez, J. Vander Steen, T. Doneux, M. Luhmer and C. Buess-Herman, Electrochim. Acta, 2018, 270, 434 CrossRef CAS.
- G. D. Allen, M. C. Buzzeo, C. Villagrán, C. Hardacre and R. G. Compton, J. Electroanal. Chem., 2005, 575, 311 CrossRef CAS.
- M. Tariq, Z. Phys. Chem., 2019, 234, 295 Search PubMed.
- J. Dupont and P. A. Z. Suarez, Phys. Chem. Chem. Phys., 2006, 8, 2441 RSC.
- J. Dupont, J. Braz. Chem. Soc., 2004, 15, 341 CrossRef CAS.
- S. Lubetkin, Electrochim. Acta, 2002, 48, 357 CrossRef CAS.
- Q. Chen, L. Luo, H. Faraji, S. W. Feldberg and H. S. White, J. Phys. Chem. Lett., 2014, 5, 3539 CrossRef CAS.
- J. Zhou, Y. Zu and A. J. Bard, J. Electroanal. Chem., 2000, 491, 22 CrossRef CAS.
- N. K. Patel and S. N. Shah, Biodiesel from plant oils, in Food, Energy, and Water, ed. S. Ahuja, Elsevier, Amsterdam, 2015, ch. 11, pp. 277–307 Search PubMed.
- D. A. Kotadia and S. S. Soni, Monatsh. Chem., 2013, 144, 1735 CrossRef CAS.
- M. Koberg and A. Gedanken, Using Microwave Radiation and SrO as a Catalyst for the Complete Conversion of Oils, Cooked Oils, and Microalgae to Biodiesel, in New and Future Developments in Catalysis, ed. L. SuibS, Elsevier, Amsterdam, 2013, ch. 9, pp. 209–227 Search PubMed.
- https://spectrabase.com/spectrum/LQ5aBGZth9Q .
- https://spectrabase.com/spectrum/FNHzrlrR4wS .
Footnote |
| † Electronic supplementary information (ESI) available: Structures of bases and zwitterions, additional cyclic voltammograms, additional UV-vis spectra, details on their processing, calibration curves, mechanistic details and kinetic calculations. See DOI: 10.1039/d0cp05674j |
| This journal is © the Owner Societies 2021 |