DOI:
10.1039/D0CE01316A
(Paper)
CrystEngComm, 2021,
23, 227-237
Enhanced solubility, permeability, and tabletability of nicorandil by salt and cocrystal formation†
Received
10th September 2020
, Accepted 2nd November 2020
First published on 3rd November 2020
Abstract
Cocrystallization is a rational selection crystal engineering approach for the development of novel solid forms with enhanced physicochemical and mechanical properties. Nicorandil (NCR) is a niacinamide vitamin derivative used to treat angina pectoris. A binary solid form screen of NCR with homologous dicarboxylic acids afforded NCR–oxalic acid (NCR–OA, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), NCR–fumaric acid (NCR–FA, 1:1), NCR–succinic acid (NCR–SA, 1:1), and NCR–suberic acid (NCR–SBA, 1:0.5). The binary solids were characterized by powder X-ray diffraction, IR and NMR spectroscopy, and DSC. NCR–FA and NCR–SBA were crystallized by slow evaporation from chloroform and toluene solvents, respectively. Single crystal X-ray diffraction confirmed that NCR–FA is a molecular salt, while NCR–SBA is a neutral cocrystal. NCR and the FA anion are connected via the robust carboxylate⋯pyridinium synthon, whereas in the NCR–SBA cocrystal, the components associate via the carboxylic acid⋯pyridine synthon. The phase stability, solubility, dissolution rate, diffusion rate and tabletability studies have demonstrated that the binary solids exhibit improved physical and mechanical properties compared to the NCR drug. Specifically, the NCR–FA salt and NCR–SBA cocrystal have higher solubility, dissolution rate, and hardness at lower pressures, making the formulation suitable for tablet compression.
:1), NCR–fumaric acid (NCR–FA, 1:1), NCR–succinic acid (NCR–SA, 1:1), and NCR–suberic acid (NCR–SBA, 1:0.5). The binary solids were characterized by powder X-ray diffraction, IR and NMR spectroscopy, and DSC. NCR–FA and NCR–SBA were crystallized by slow evaporation from chloroform and toluene solvents, respectively. Single crystal X-ray diffraction confirmed that NCR–FA is a molecular salt, while NCR–SBA is a neutral cocrystal. NCR and the FA anion are connected via the robust carboxylate⋯pyridinium synthon, whereas in the NCR–SBA cocrystal, the components associate via the carboxylic acid⋯pyridine synthon. The phase stability, solubility, dissolution rate, diffusion rate and tabletability studies have demonstrated that the binary solids exhibit improved physical and mechanical properties compared to the NCR drug. Specifically, the NCR–FA salt and NCR–SBA cocrystal have higher solubility, dissolution rate, and hardness at lower pressures, making the formulation suitable for tablet compression.
Introduction
Angina pectoris is the most common cardiovascular disease that occurs due to an imbalance in the myocardial oxygen supply, and requires constant monitoring.1 Potassium channel openers provide the vasodilatation of arterioles and large coronary arteries, which are important for the treatment of angina pectoris.2 Nicorandil (NCR) is a potassium channel activator that induces vasodilation of the coronary and systemic arteries, and has the ability to hyperpolarize smooth muscle cell membranes.3 NCR exhibits moderate solubility (1.19 mg mL−1 at 37 °C) and has a short half-life (<1 h) due to rapid metabolism and systematic elimination via oral administration.4,5 NCR is formulated in tablet and liquid dosage forms for oral and intravenous routes of drug delivery. Similarly, NCR transdermal patches are used for topical drug delivery.6 However, during the tablet making, NCR undergoes chemical degradation (NCR to denitrated NCR) due to the heat generated under the compressive pressure exerted by the punch machine. In another report, the co-formulation of NCR with fumaric acid, dimethicone, simethicone, and cyclodextrin compounds and their relative stabilities were studied.7 The sustained release formulation of NCR with dicarboxylic acids as excipients is described in a patent.7 The Cambridge Structural Database (CSD) contains a guest free form of NCR8 and four cocrystals with 1-hydroxy-2-naphthoic acid, salicylic acid, 3-hydroxybenzoic acid and 2,5-dihydroxybenzoic acid.9 However, there is no published report on the solid form screen of NCR for tabletability improvement.
Cocrystallization is an efficient crystal engineering approach targeted for small molecule pharmaceuticals to modify their crystal structures (without breaking/making covalent bonds), and to systematically alter physicochemical and mechanical properties.10,11 Cocrystals can be designed based on supramolecular heterosynthons, such as carboxylic acid⋯pyridine (COOH⋯Py), carboxylic acid⋯carboxamide (COOH⋯CONH2), sulfonamide⋯carboxamide (SO2NH2⋯CONH2), and amine⋯phenol (NH2⋯OH).12 Cocrystals are synthesized by liquid-assisted grinding, ball mill grinding, slurry technique, melt, and solution crystallization methods.13 In the case of NCR, liquid-assisted grinding followed by isothermal crystallization afforded crystals of binary solid forms. The homologous series of alkane/alkene dicaroboxylic acid coformers were selected to construct cocrystals for this study. NCR has a pyridyl functional group and can form the robust carboxylic acid⋯pyridine (COOH⋯N–Py) heterosynthon in crystal structures. We obtained four new binary products (two cocrystals and two salts), which were characterized through X-ray diffraction, spectroscopic, and thermal techniques. The solubility and dissolution rate, stability, permeability and diffusion rate, and tabletability of the novel crystalline binary products are compared with that of the parent drug NCR.
Experimental section
Materials
NCR was purchased from Yarrow Chemical Products (Mumbai, India). The purity of the compounds was confirmed by NMR, PXRD, and DSC. The coformers used in this study were purchased from Sigma-Aldrich, Hyderabad, India. All chemicals were of analytical grade, and solvents were of chromatographic grade.
X-ray crystallography
Bruker D8 QUEST, CCD diffractometer. Mo-Kα (λ = 0.71073 Å) radiation was used to collect X-ray reflections on all crystals. Bruker D8 Quest diffractometer equipped with a graphite monochromator and Mo-Kα fine-focus sealed tube (λ = 0.71073 Å). Data reduction was performed using CrysAlisPro 171.33.55 software (CrysAlisPro; 2010), and deduction was performed using APEX-II software.14 Intensities for absorption were corrected with SADABS. Structures were solved and refined using SHELXL-2018 (ref. 15) with anisotropic displacement parameters for non-H atoms. Hydrogen atoms on O and N were experimentally located in all crystal structures. Hydrogen atoms were experimentally located through the Fourier difference electron density maps in all crystal structures. All O–H and C–H atoms were fixed geometrically with HFIX command in the SHELX-TL program of Bruker-AXS. Crystal structure parameters are listed in Table 1, and the hydrogen bond distances shown in Table S1† are neutron normalized to fix the D–H distance to its accurate neutron value in the X-ray crystal structures (O–H 0.983 Å, N–H 1.009 Å, and C–H 1.083 Å). A check of the final CIF file was achieved using PLATON16,17 for any missed symmetry. X-Seed was used to prepare the packing diagrams.18
Table 1 Preparation of NCR solid forms
| Solid form |
NCR (mg, mmol) |
Coformer (mg, mmol) |
Method, solvent |
Solvent of crystallization |
| NCR–FA |
211.1, 0.1 |
116.01, 0.1 |
Mortar–pestle grinding, CH3CN (1 mL) |
CHCl3 (40 mg of cocrystal in 5 mL) |
| NCR–SBA |
211.1, 0.1 |
87.01, 0.05 |
Mortar–pestle grinding, CH3CN (1 mL) |
Toluene (40 mg of cocrystal in 5 mL) |
| NCR–OA |
211.1, 0.1 |
126.0, 0.01 |
Mortar–pestle grinding, CH3CN (1 mL) |
Unable to grow crystals |
| NCR–SA |
211.1, 0.1 |
118.0, 0.01 |
Mortar–pestle grinding, CH3CN (1 mL) |
Unable to grow crystals |
Powder X-ray diffraction
Powder X-ray diffraction (PXRD) was recorded on a Bruker D8 Advance diffractometer (Bruker-AXS, Karlsruhe, Germany) using Cu-Kα X-radiation (λ = 1.5406 Å) at an acceleration voltage of 40 kV and current of 30 mA power. The binary solids of the NCR diffraction patterns were scanned and collected over the 2θ range from 5–50° at a scan rate of 5° min−1. Powder Cell 2.455 (Federal Institute of Materials Research and Testing, Berlin, Germany) was used for the Rietveld refinement of experimental PXRD and calculated lines from the X-ray crystal structure.19
Thermal analysis
For all binary solids, differential scanning calorimetry (DSC) was performed on a Mettler-Toledo DSC 822e module, (Mettler-Toledo, Columbus, OH). Thermal gravimetry analysis (TGA) was carried out on a Mettler Toledo TGA/SDTA 851e module (Mettler-Toledo, Columbus, OH) to measure any solvent release from the crystal. The compound was placed in sealed pin-pricked aluminum pans for the DSC experiments. The characteristic sample size was 3–5 mg for DSC and 4–6 mg for TGA. The temperature range for the heating curves was 30–250 °C, and the sample was heated at a rate of 10 °C min−1. Samples were purged in a stream of dry nitrogen flowing at 80 mL min−1.
Vibrational spectroscopy
Infrared spectra of pure NCR and its binary solids were recorded on FT-IR spectrometer (Thermo-Nicolet 6700, Thermo Scientific, Waltham, MA) with the samples spread in KBr pellets. Omnic software (Thermo Scientific, Waltham, MA) was utilized to analyze the data. Each sample was scanned in the range of 400–4000 cm−1, and the spectrum was normalized with the background correction.
FESEM morphology
The morphology and size of the microstructures of NCR and its salts and cocrystals were examined by FESEM analysis on a Carl Zeiss field emission scanning electron microscope model 6027 with MERLIN compact using a beam voltage of 5 kV.
Equilibrium solubility and dissolution measurements
The solubility of NCR and their binary solid forms was measured using the Higuchi and Connors method20 in phosphate buffer saline (pH 7) media at 37 ± 1 °C. First, the absorbance of a known concentration of the NCR and its binary solids was measured at the given λmax (NCR 262 nm) in purified pH 7 phosphate buffer saline medium on a Thermo Scientific Evolution 300 UV–vis spectrometer (Thermo Scientific, Waltham, MA), respectively. These absorbance values were plotted against known concentrations to prepare the concentration versus intensity calibration curve. From the slope of the calibration curves, the molar extinction coefficients for NCR and its binary solids were calculated. The equilibrium solubility measurements of NCR were carried out in pH 7.0 phosphate buffer using the shake flask method.21 An excess amount of the sample (NCR/salts/cocrystals) was added to 3 mL of purified pH 7 phosphate buffer saline medium. The supersaturated solution was stirred at 800 rpm using a magnetic stirrer at 37 ± 1 °C. After 24 h, the suspension was filtered through a Whatman 0.45 μm syringe filter. Then, the equilibrium solubility was calculated as per procedure, and remaining residues of NCR/salts/cocrystals were characterized by PXRD.
The intrinsic dissolution rate study was carried out on a USP-certified Electrolab TDT-08L dissolution tester (Mumbai, India). Standard curves for all compounds were obtained spectrophotometrically at their respective λmax. The powder dissolution studies were performed with 350 mg of NCR, and the same equivalent weight of the salt/cocrystal active drug in 500 mL of pH 7 phosphate buffer saline medium. The pellet disk rotation was fixed at 150 rpm, and dissolution experiments were continued for up to 6 h at 37 ± 1 °C because the elimination half-life T½ of NCR is 1 h.22 At regular intervals, 5 mL of the dissolution medium was withdrawn and replaced by an equal volume of fresh medium to maintain a constant volume, and the concentration of the aliquots was determined with proper dilutions from the predetermined standard curves of the respective compounds.
Diffusion studies
Diffusion studies were carried out with glass Franz diffusion cells (model JFDC-07, Orchid Scientific, Maharashtra, India) with a 20 mL volume.23 The membranes used here were the dialysis membrane-135 (width 33.12 mm, and diameter 23.8 mm, capacity 4.45 mL cm−1), and purchased from HiMedia, India. The dialysis membrane was pretreated with 2% NaHCO3 at 80 °C for 30 min to remove traces of sulfides, followed by 10 mM of EDTA at 80 °C for 30 min to remove traces of heavy metal, and another 30 min of treatment with deionized water at 80 °C to remove glycerin. The treated dialysis membrane was placed between the two compartments fixed by a stainless-steel clamp with an effective mass transfer area of 3.14 cm2. The receptor compartment was filled with PBS solution, and air bubbles were removed. Next, the donor compartment was loaded with 20 mg pure crystalline NCR and its salts and cocrystals in equimolar ratio powders (20 mg drug active was maintained in all experiments), and 2 mL of PBS were added. The temperature of the diffusion medium was thermostatically maintained at 37 ± 1 °C throughout the experiment at 600 rpm, and permitted to diffuse through the membrane towards the receptor compartment. Aliquots of 1 mL were withdrawn from the receptor compartment at set time periods of up to 6 h, and fresh PBS was added to replenish the volume. The diffused concentration was determined by UV–vis spectrometer (Thermo Scientific EVOLUTION 300 UV–VIS). The cumulative concentration of drug that diffused into the receptor compartment at each interval was determined by taking into consideration the replacement of aliquots with PBS and the dilution derived from the addition of PBS buffer.
Compression properties
Well's method was adapted to assess the compression properties of NCR, NCR–FA, NCR–SA, NCR–SBA, and NCR–OA. About 150 mg of drug-coformer multicomponent form was prepared as per Well's protocol, and compacted using 13 mm die and punch at 50 MPa in an IR pellet press. The compressed compacts were kept in a desiccator for 24 h to achieve equilibration.24 After the equilibration time, these compacts were studied for hardness with the aid of the LABINDIA tablet hardness tester (TH 1050 M).
Compressibility
To evaluate the compressibility of the samples, 3 types of blends were used as per Well's protocol. These blends differ in their blending time and dwell time, as given in Table 2.
Table 2 Protocol for compressibility study
| Sample parameter |
Process parameter and time |
| Codes |
A |
B |
C |
| Blending time |
5 min |
5 min |
30 min |
| Dwell time |
2 s |
30 s |
2 s |
| Amount of sample |
150 mg of sample and 1% magnesium stearate |
| Compression |
13 mm die at 50 kg cm−2 |
Results and discussion
NCR (2-nicotinamidoethyl nitrate) is a niacinamide (vitamin) derivative with a weakly basic group (pKa 3.62 of pyridyl N). The pyridyl N of NCR has a pKa similar to nicotinamide (3.63), and is lower than that of an unsubstituted pyridine (pKa 5.2) due to the meta-amide group. The molecule has hydrogen bond acceptors (pyridine N and carboxamide O) and donor groups (carboxamide NH) that have a propensity to form cocrystals with carboxylic acid coformers. The cocrystallization of NCR with GRAS (generally regarded as safe additives in pharmaceuticals)25 dicarboxylic acid coformers using the liquid-assisted grinding method resulted in cocrystal and salt products, namely NCR–suberic acid (NCR–SBA, 1:0.5), NCR–oxalic acid (NCR–OA, 1:1), NCR–fumaric acid (NCR–FA, 1:1), and NCR–succinic acid (NCR–SA, 1:1). A summary of the experimental methods is described in the experimental section (Table 1), and the coformer molecular structures are represented in Scheme 1. The binary solid forms were characterized using powder X-ray diffraction, IR and NMR spectroscopy, and thermal DSC technique. NCR–FA and NCR–SBA crystal structures were established using the single crystal X-ray diffraction technique. The ORTEP (Oak Ridge Thermal Ellipsoid Plot) diagrams are presented in Fig. S1, ESI,† the unit cell and refinement parameters are summarized in Table 3, and the hydrogen bonds are listed in Table S1.† Furthermore, the binary solid form phase stability, solubility and dissolution, permeability, and tabletability properties were also examined and compared with those of pure NCR.
 |
| | Scheme 1 Molecular structures and acronyms of the NCR drug and coformers. | |
Table 3 Crystallographic parameters of NCR salt and cocrystal
| Compound |
NCR–FA |
NCR–SBA |
| Emp. form. |
C8H10N3O4, C4H3O4 |
C8H9N3O4, 0.5(C8H14O4) |
| Form. wt |
327.25 |
298.28 |
| Cryst. syst. |
Orthorhombic |
Monoclinic |
| Sp. gr. |
Pca21 |
P21/n |
|
T (K) |
298(2) |
298(2) |
|
a (Å) |
29.813(6) |
6.835(2) |
|
b (Å) |
4.787(2) |
23.752(5) |
|
c (Å) |
9.861(2) |
8.939(2) |
|
α (°) |
90 |
90 |
|
β (°) |
90 |
101.16 |
|
γ (°) |
90 |
90 |
|
Z
|
4 |
4 |
|
V (Å3) |
1407.2(7) |
1423.8(6) |
| Rflns. collect |
2478 |
2517 |
| Unique rflns. |
2353 |
2491 |
| Obsd. rflns. |
2245 |
1411 |
| Parameters |
220 |
190 |
|
R
1
|
0.0284 |
0.0606 |
| wR2 |
0.0706 |
0.1635 |
| GOF |
1.072 |
1.015 |
| Diffractometer |
BRUKER CCD detector |
BRUKER CCD detector |
Crystal structure analysis
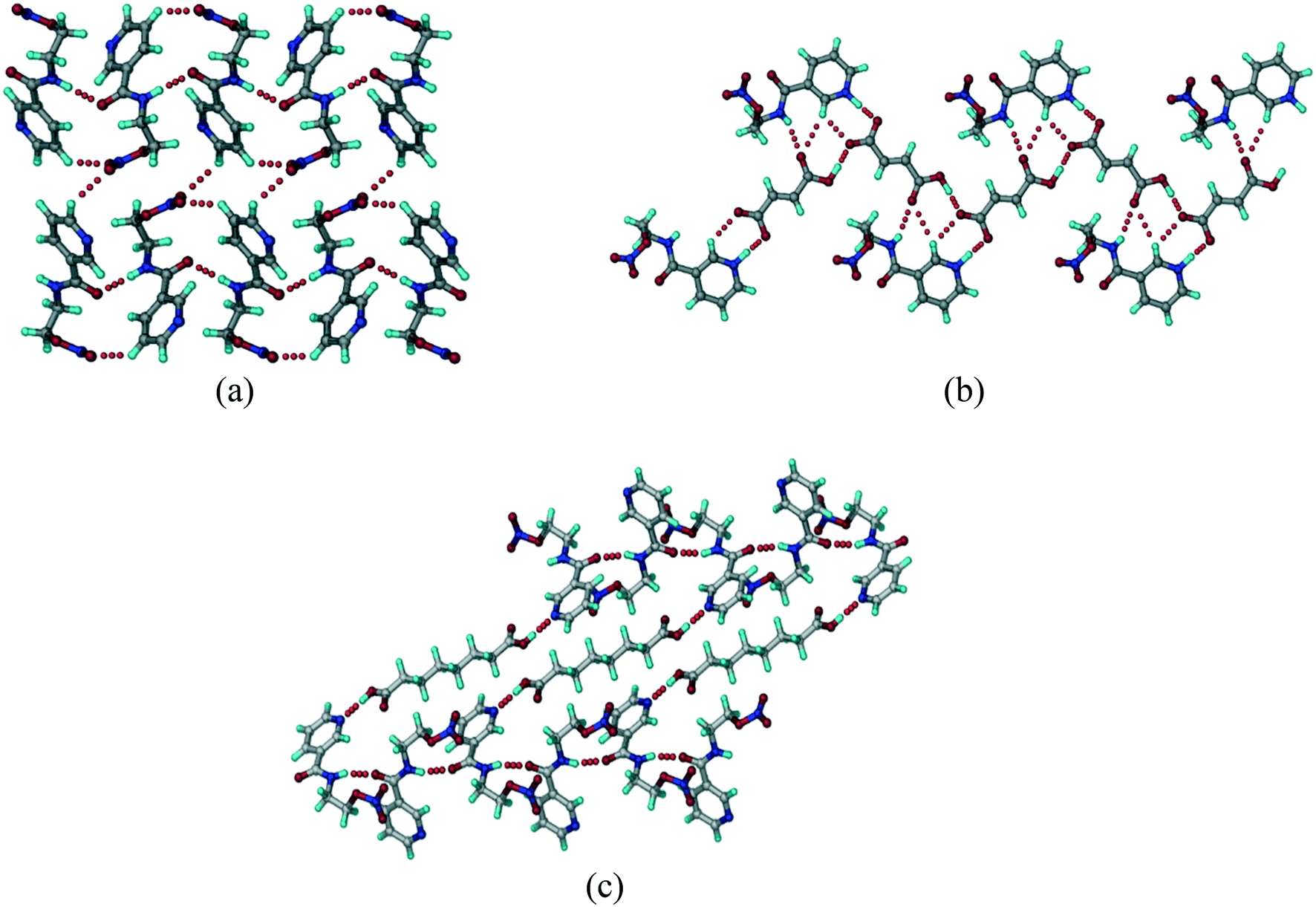
The crystal structure of the NCR drug (recode FUWNIC) shows adjacent molecules connected by a strong carboxamide catemer N–H⋯N chain (N2–H5⋯O1, 1.99 Å, 168°) and C–H⋯O (C3–H3⋯O1 2.35 Å, 147° and C2–H2⋯O3 2.46 Å, 132°) interactions, which extend in a 1D arrangement along the c-axis. Such 1D chains are connected via weak C–H⋯O (C3–H3⋯O1, 2.64 Å, 125°) interactions (Fig. 1a). In the NCR cocrystal and salt structures, these hydrogen bonds are replaced by the new and stronger acid⋯pyridine, and its ionic variant carboxylate⋯pyridinium salt heterosynthons. In addition, conformational changes were also observed. The secondary carboxamide (CONH) group is oriented in a different conformation in the salt/cocrystal structure. In pure NCR, pyridine N and carboxamide N are anti to each other, whereas in binary structures, they are syn to each other.
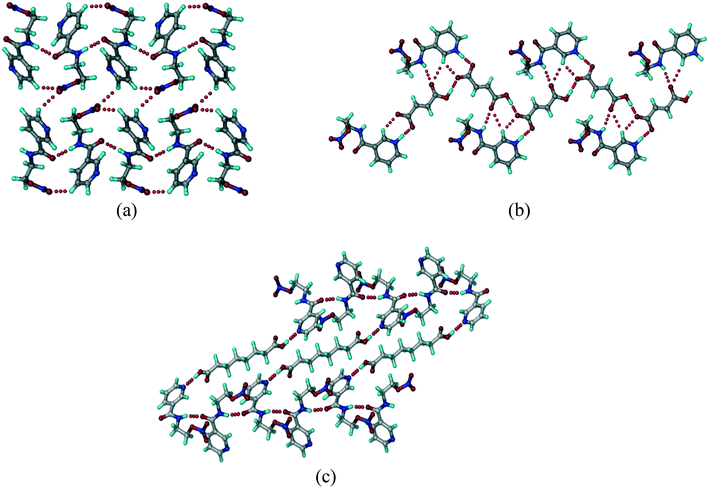 |
| | Fig. 1 (a) 1D chains of NCR molecules connected by the N–H⋯O catemer are bonded through auxiliary N–H⋯N and C–H⋯O interactions. (b) NCR–FA salt molecules with the pyridinium⋯carboxylate synthon are connected through N–H⋯O, O–H⋯O, and C–H⋯O interactions. (c) A zig-zag motif in the NCR–SBA cocrystal connected by an O–H⋯N hydrogen bond. | |
NCR–FA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) salt.
The NCR–FA 1:1 salt structure was solved and refined in the orthorhombic polar space group Pca21. The NCR and FA molecules are associated through strong N+–H⋯O− (N1–H1A⋯O51, 1.67 Å, 177°) and C–H⋯O (C1–H1⋯O6, 2.44 Å, 126°) hydrogen bonds in a dimeric R22(7) ring motif. The salt molecules extend in a sheet through N–H⋯O (N1–H1A⋯O6, 2.51 Å, 123° and N2–H2A⋯O8, 2.31 Å, 141°), O–H⋯O (O7–H7C⋯O6, 1.57 Å, 177°), and C–H⋯O (C7–H7B⋯O8, 2.55 Å, 162°) interactions (Fig. 1b). The sheets are connected by chalcogen⋯chalcogen, inter nitro group O⋯O (NO2⋯NO2, O5⋯O4, 2.98 Å, 129°) heteroatom bonding interaction (Fig. 1c).
:1) salt.
The NCR–FA 1:1 salt structure was solved and refined in the orthorhombic polar space group Pca21. The NCR and FA molecules are associated through strong N+–H⋯O− (N1–H1A⋯O51, 1.67 Å, 177°) and C–H⋯O (C1–H1⋯O6, 2.44 Å, 126°) hydrogen bonds in a dimeric R22(7) ring motif. The salt molecules extend in a sheet through N–H⋯O (N1–H1A⋯O6, 2.51 Å, 123° and N2–H2A⋯O8, 2.31 Å, 141°), O–H⋯O (O7–H7C⋯O6, 1.57 Å, 177°), and C–H⋯O (C7–H7B⋯O8, 2.55 Å, 162°) interactions (Fig. 1b). The sheets are connected by chalcogen⋯chalcogen, inter nitro group O⋯O (NO2⋯NO2, O5⋯O4, 2.98 Å, 129°) heteroatom bonding interaction (Fig. 1c).
NCR–SBA (1:0.5) cocrystal.
The crystal structure was solved and refined in the monoclinic space group P21/n, and contains one molecule of NCR and half molecule of SBA in the asymmetric unit. SBA interacts with NCR through the carboxylic acid⋯pyridine synthon. NCR molecules form a linear tape (Fig. 1c) through the carboxamide N–H⋯O hydrogen bond (O9–H9A⋯N1, 2.06 Å, 162°). Linear tapes of SBA molecules are bridged through the carboxylic acid⋯pyridine O–H⋯N (O9–H9A⋯N1, 1.86 Å, 170°) hydrogen bond.
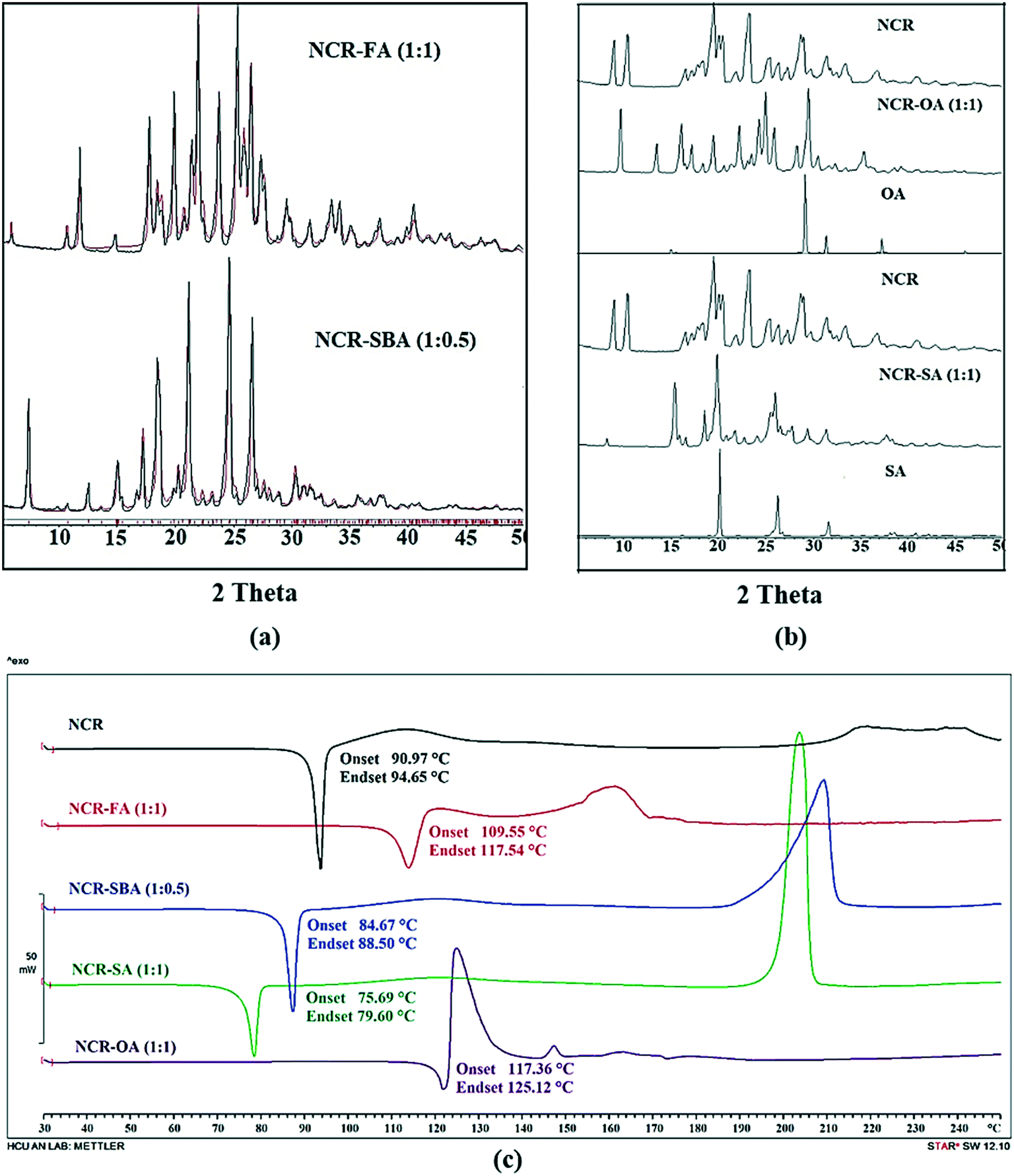
Powder X-ray diffraction (PXRD)
The NCR–FA (1:1) salt and NCR–SBA (1:0.5) cocrystal exhibit a unique diffraction line XRD pattern that matches the calculated profile from the X-ray crystal structure (Fig. 2a). NCR–OA and NCR–SA are different from the starting components and single-phase products (Fig. 2b). The stoichiometry of NCR–OA and NCR–SA was confirmed by proton NMR as a 1:1 molar ratio (Fig. S2 and S3, ESI†).
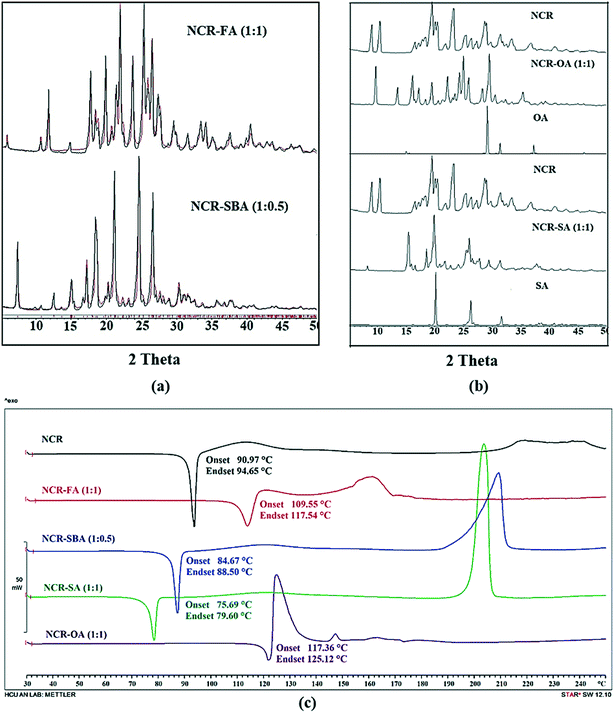 |
| | Fig. 2 (a) The experimental PXRD pattern of the NCR binary solids (NCR–FA and NCR–SBA, black) shows excellent match with the calculated line profile from the calculated X-ray crystal structure (red), indicating bulk purity and phase homogeneity. Rietveld refinement for NCR–FA gave Rp = 0.1382, Rwp = 0.1752, and Rexp = 0.5590; and Rp = 0.1324, Rwp = 0.2064, and Rexp = 0.8211 for NCR–SBA. (b) Representation of the experimental PXRD plot of NCR, NCR–OA, NCR–SA, and coformers (OA and SA). The diffraction pattern of these binary solids is different from the starting components, and indicates new crystalline phases. (c) DSC thermogram of NCR and binary solid forms NCR–FA and NCR–OA show a melting endotherm that is higher than the melting point of the pure NCR drug, whereas the NCR–SBA and NCR–SA cocrystal shows a melting endotherm that is lower than the melting point of pure NCR drug. | |
Thermal analysis
The thermal behavior of NCR and binary solids was tested using differential scanning calorimetry (DSC). The DSC heating curves and melting temperatures are represented in Fig. 2c. NCR exhibits a sharp melting endotherm followed by decomposition, and does not show any phase change/transformation on heating before melting. Similarly, the NCR–FA, NCR–SA, NCR–OA and NCR–SBA binary solids exhibit melting, followed by decomposition. There is no phase change before melting on heating at 10 °C min−1. The NCR–SA and NCR–SBA cocrystals displayed a lower melting point than the pure NCR and the coformer. In contrast, the melting point of the NCR–OA and NCR–FA salts lies between the melting temperatures of NCR and the coformer. Furthermore, TGA analysis showed that the NCR–OA salt and NCR–SA cocrystal for which the single crystal structure was not determined do not contain any unbound solvent in the crystal lattice, and are chemically stable (Fig. S4a and b, ESI†).
FT-IR spectroscopy
The carboxamide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and NH, and nitro (NO2) stretching bands appear at 1633 cm−1 and 3243 cm−1, and 1286 cm−1, respectively, in the IR spectrum of pure NCR. In salts and cocrystals with dicarboxylic acid coformers, these functional groups and coformer (CO) carbonyl group showed significant changes in the infrared spectra due to changes in the intermolecular interactions and molecular packing. Specifically, in the case of the NCR–OA and NCR–SA binary solids for which X-ray crystal structures could not be determined due to the unavailability of good quality single crystals, the signature peaks appeared at 1622 and 1643 cm−1 for the carboxylate (COO−) group, confirming the salt structures. The significant changes in the stretching frequency are listed in Table 4 and the IR spectra are shown in Fig. S5, ESI.† According to the ΔpKa rule,26 the difference in the pKa of NCR and the coformer binary solids is in the intermediate zone of about 0–3 (Table 5), which means that the product can be either a cocrystal or a salt. ΔpKa is negative for NCR–SA and NCR–SBA, and they are observed as cocrystals. Conversely, NCR–OA and NCR–FA are salts with small ΔpKa of <3, which is normally not indicative of proton transfer.
O and NH, and nitro (NO2) stretching bands appear at 1633 cm−1 and 3243 cm−1, and 1286 cm−1, respectively, in the IR spectrum of pure NCR. In salts and cocrystals with dicarboxylic acid coformers, these functional groups and coformer (CO) carbonyl group showed significant changes in the infrared spectra due to changes in the intermolecular interactions and molecular packing. Specifically, in the case of the NCR–OA and NCR–SA binary solids for which X-ray crystal structures could not be determined due to the unavailability of good quality single crystals, the signature peaks appeared at 1622 and 1643 cm−1 for the carboxylate (COO−) group, confirming the salt structures. The significant changes in the stretching frequency are listed in Table 4 and the IR spectra are shown in Fig. S5, ESI.† According to the ΔpKa rule,26 the difference in the pKa of NCR and the coformer binary solids is in the intermediate zone of about 0–3 (Table 5), which means that the product can be either a cocrystal or a salt. ΔpKa is negative for NCR–SA and NCR–SBA, and they are observed as cocrystals. Conversely, NCR–OA and NCR–FA are salts with small ΔpKa of <3, which is normally not indicative of proton transfer.
Table 4 FT-IR stretching frequency for binary solid forms and individual components
| Compound name |
Carbonyl νCO CONH and COO− (cm−1) |
Amide νNH CONH (cm−1) |
Nitro νNO NO3 (cm−1) |
Carbonyl νCO coformer (cm−1) |
| NCR |
1633 |
3243 |
1286 |
|
| OA |
— |
— |
— |
1666 |
| NCR–OA |
1622 |
3317 |
1282 |
1671 |
| SA |
— |
— |
— |
1693 |
| NCR–SA |
1643 |
3307 |
1276 |
1709 |
| FA |
— |
— |
— |
1674 |
| NCR–FA |
1626 |
3399 |
1280 |
1682 |
| SBA |
— |
— |
— |
1682 |
| NCR–SBA |
1645 |
3325 |
1281 |
1715 |
Table 5 ΔpKa valuesa of carboxylic acid coformers and NCR drug
| Compound name |
1st pKa of acid |
ΔpKa |
Molecular complex |
|
pKa values calculated using Marvin 5.10.1, 2012, ChemAxon, http://www.chemaxon.com. These values are closely matched with the pKa values compiled by R. Williams.
|
| NCR |
3.62 of pyridine N |
— |
— |
| OA |
1.23 |
2.39 |
1:1 salt |
| SA |
4.19 |
−0.57 |
1:1 cocrystal |
| SBA |
4.53 |
−0.91 |
1:0.5 cocrystal |
| FA |
3.03 |
0.59 |
1:1 salt |
FE-SEM morphology study
The morphology changes in the crystallites can significantly influence the physical and mechanical properties on the performance of oral dosage form.27 The reference compound NCR and its salts and cocrystal exhibited differences in the size and shape, which are visible by FE-SEM micrographs (Fig. 3). The reference sample NCR is composed of columnar morphology of approximately 15–30 μm length and 5–10 μm width (l × b) with a clear separation among the particles of curvaceous edges. The NCR–FA salt has sub-angular polygon morphology with a rough surface ranging from 2–20 μm, and small particles adhering on their surfaces. Among the other two complexes, the NCR–OA salt showed very fine adherent particles of thin flakes ranging 2–10 μm and 0.1–1 μm (l × b). The NCR–SA cocrystal is composed of fine acicular adherent clusters of thin needles ranging in 1–5 μm length and irregular particles. The NCR–SBA cocrystal is composed of sub-rounded, homogenous, smooth surface spheres of 2–10 μm. As a result, the aspect ratio of the two salts and two cocrystals of NCR are relatively small in size with no particular shape, typically isodimensional, when compared to the reference drug crystal NCR. Thus, the shape and crystallite size reduction of the salts and cocrystals of NCR are undoubtedly produced by the solvent-assisted grinding. In addition, the micron-sized crystals promote better flowability, tableting and dissolution behavior. Therefore, as the crystallites become smaller, the surface area-to-volume ratio increases. This allows for greater interaction with the solvent, resulting in higher solubility.28
 |
| | Fig. 3 Scanning electron microscopy of NCR and its binary solids. (a) The reference drug NCR with particle size 15–30 μm. (b) Orthorhombic salt form of NCR–FA with particle size of <20 μm. (c) Monoclinic form of the NCR–SBA cocrystal with particle size of <10 μm. (d) NCR–OA and (e) NCR–SA cocrystal composed of 1–10 μm particle size. The detailed view of the particle surface magnification for the salts and cocrystal of NCR and the reference drug NCR is 5.00 kX. | |
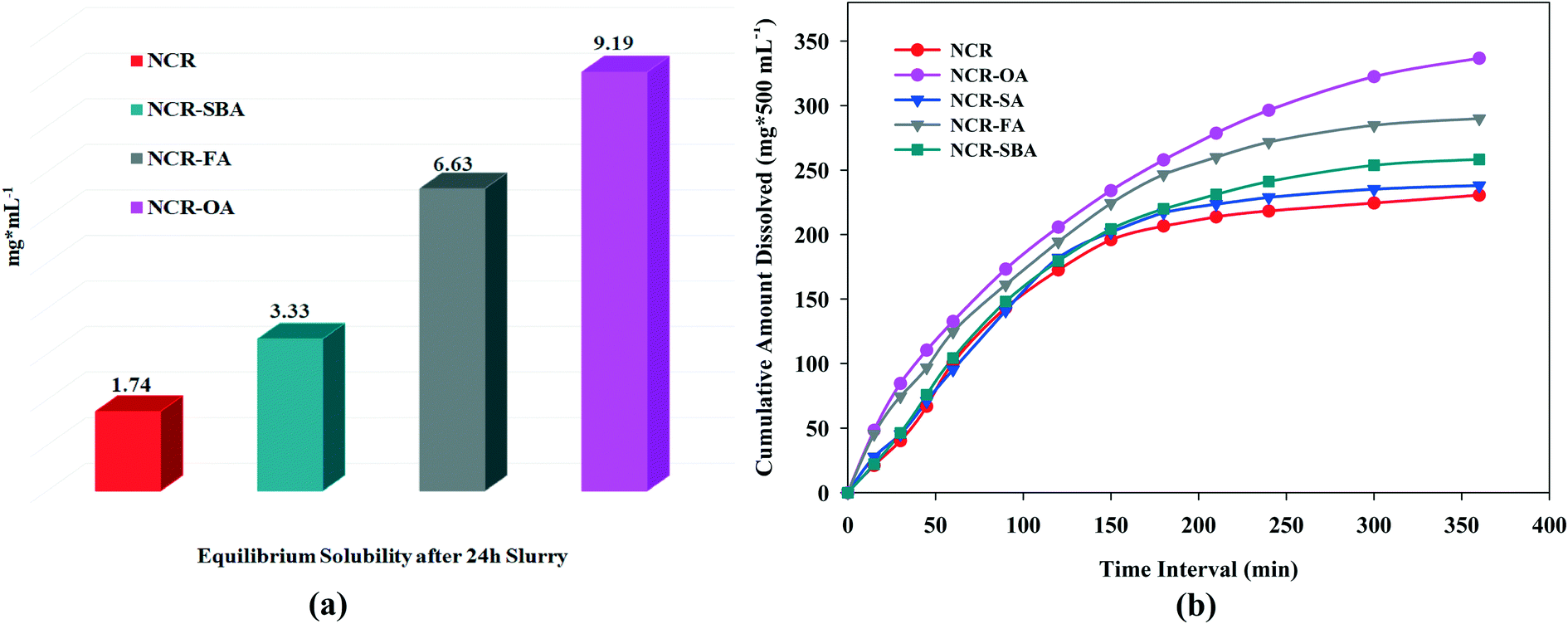
Solubility and dissolution
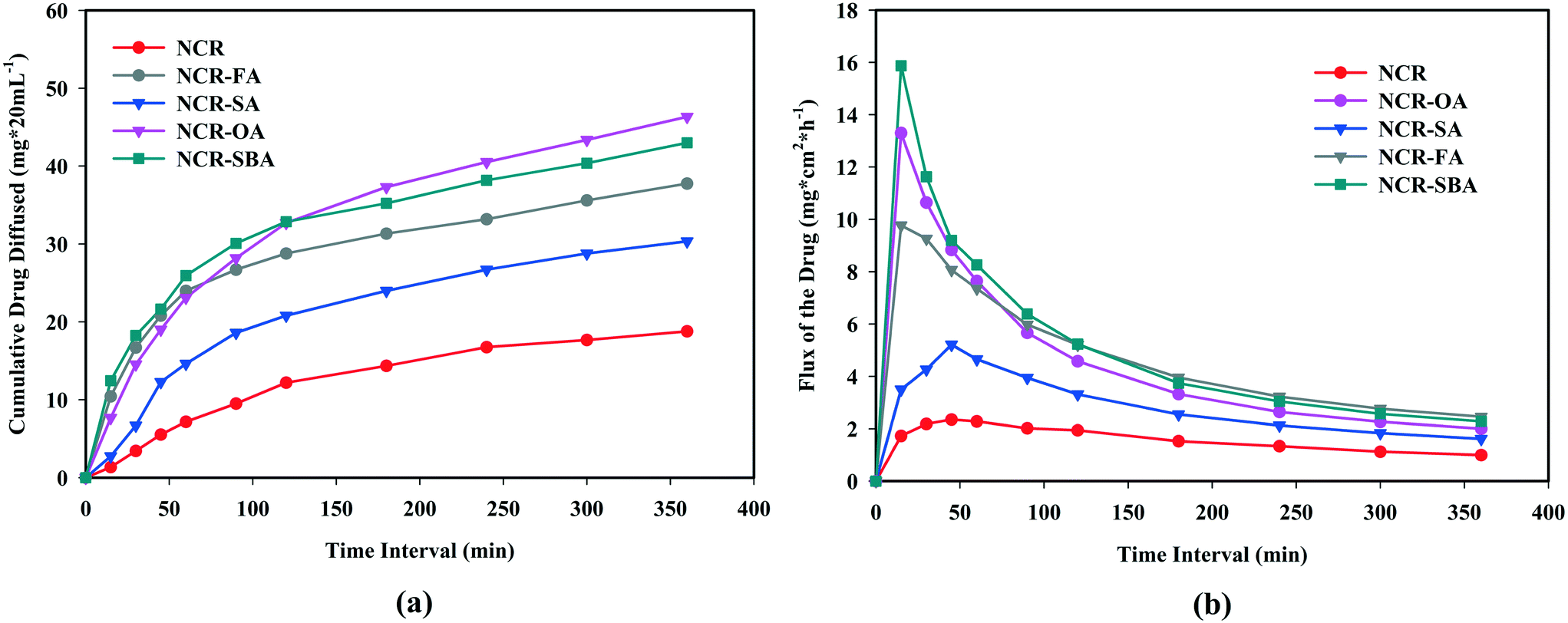
Drug solubility occurs under a dynamic equilibrium state, which determines the maximum concentration in a saturated solution when excess solid is present in the media. The dissolution rate is a kinetic process that measures the drug concentration, which passes into the media with respect to time. These two parameters are determined by the solvation of the molecular components and the strength of the crystal structure lattice.29,30 To improve the drug solubility, the solvent affinity must be increased and/or lower the lattice energy. These can be altered via cocrystal/salt formation, although it is also influenced by the coformer solubility. The equilibrium solubility and dissolution rate of the NCR solid forms were performed in phosphate buffer saline media (pH 7.0 ± 0.5) at 37 °C. The phase stability of the drug forms was analyzed by PXRD during the solubility/dissolution measurement period (Fig. S6a–g, ESI†). Under the equilibrium state (24 h), NCR–OA, NCR–FA, and NCR–SBA were stable due to strong ionic hydrogen bonding in the crystal structures of salts, and in the cocrystals via N–H⋯O and O–H⋯O supramolecular synthons. However, the NCR–SA cocrystal did not exhibit phase stability and showed disproportionation to the original components as a slurry in PBS media within 1 h (Fig. S6f, ESI†). The NCR–OA, NCR–FA, and NCR–SBA binary solids are stable and exhibited 5.2, 3.8, and 1.9 times higher solubility than the reference drug (Fig. 4a and Table 6). The IDR measurements for a shorter period of 6 h exhibited improved dissolution behavior compared to NCR for the cocrystals/salts (Fig. 4b). The profile of the NCR concentration vs. time indicates that rapid drug dissolution occurred, and half of the drug cocrystal/salt pellet dissolved in less than 3 h. Even though the data is on a limited number of salts and cocrystals, this study shows the difficulty in anticipating the stability of the binary complexes of pharmaceutical solids. The SA cocrystal is the least stable even though succinic acid is one of a preferred salt/cocrystal former in the drug formulation.
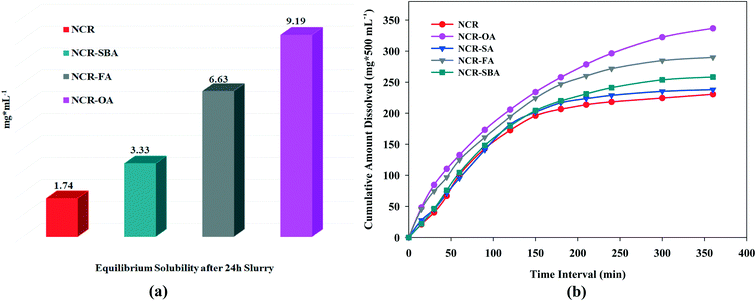 |
| | Fig. 4 (a) Equilibrium solubility and (b) intrinsic dissolution rate of the NCR binary solids in PBS media. | |
Table 6 Intrinsic dissolution of the NCR binary solids and n-fold enhancement compared to the drug is given in parentheses
| Compound |
Solubility of coformer in water (mg mL−1) |
Molar extinction coefficient (mM−1 cm−1) |
Intrinsic dissolution rate, IDR mg cm−2 min−1 |
Solubility of cocrystal/salt |
| NCR |
1.19 |
4.21 |
1.87 |
1.74 |
| NCR–OA (1:1) |
143 |
3.71 |
3.65 (×1.92) |
9.19 |
| NCR–FA (1:1) |
6 |
3.77 |
3.43 (×1.83) |
6.63 |
| NCR–SBA (1:0.5) |
12 |
3.64 |
2.64 (×1.41) |
3.33 |
| NCR–SA (1:1) |
211 |
4.59 |
2.60 (×1.39) |
— |
The above measurements show that the salts have higher solubility and dissolution rate than the cocrystal, and they are faster than the drug. Between NCR–OA and NCR–FA, the greater water solubility of the coformer OA results in higher solubility and dissolution rate of the product. Even though the SA coformer has high water solubility in its cocrystal, NCR–SA is not stable (rapid disproportionation) in PBS media, and so the dissolution behavior is similar to the reference drug. The improved solubility and dissolution rate of the novel solid forms are attributed to the ionic carboxylate⋯pyridinium synthon in the salts, which have higher solubility than the carboxylic acid⋯pyridine heterosynthon in the neutral cocrystal.31
Permeability studies
The proper selection of salt/cocrystal coformer does not necessarily equate to making the product with optimal aqueous solubility, but must be a balance between both high solubility and good permeability.32 Compared to the improved solubility and dissolution of cocrystals/salts, their diffusion behavior was measured in a Franz apparatus. The plot of flux and cumulative diffused drug amount is shown in Fig. 5. The NCR–OA, NCR–FA, and NCR–SBA flux plots display an initial sharp spike within 30 min, and then drop off to a steady state curve, corresponding to a near constant passage of drug until the end of the experiment up to 6 h. The rapid dissociation of the NCR–SA cocrystal in the solubility experiments means that its diffusion profile is difficult to measure, and the curve is slightly higher than NCR.
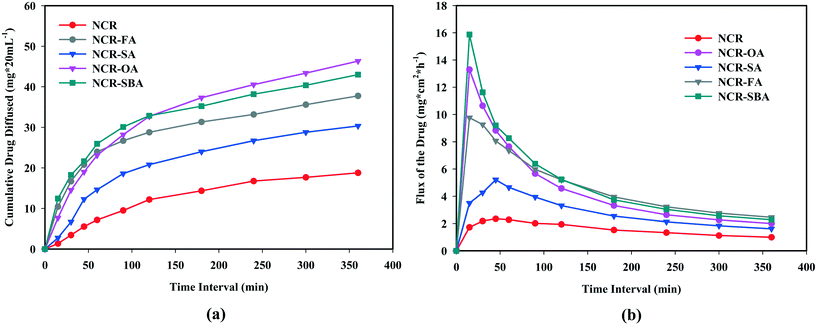 |
| | Fig. 5 (a) Membrane permeability and (b) flux rate vs. time of binary solids in PBS media. | |
The cumulative drug diffused (Fig. 5b) values for the salts NCR–OA (46.32 mg cm−2), NCR–FA (37.74 mg cm−2) and NCR–SA (30.33 mg cm−2) and the NCR–SBA cocrystal compared to the NCR (18.77 g cm−2) at 6 h (2.5, 2.0, 1.6 and 2.3 fold higher than the drug). The enhancement in the membrane diffusion of both salts and cocrystal is higher at the receiver compartment than that of the reference drug. The NCR–SBA cocrystal shows a sharp increase when compared to the NCR–OA and NCR–FA salts, while the dissolution profile is higher for the latter. The high hydrophobicity/lipophilicity of the suberic acid coformer compared to the smaller oxalic acid and fumaric acid means higher drug permeability. These observations are in line with the general principle of the drug hydrophilic (hydrogen bonding and ionic functional groups) nature, resulting in higher solubility/dissolution and higher hydrophobicity (lipophilic and carbon content), leading to higher permeability/diffusion. In balance, both need to be optimized simultaneously for BCS class I category drug classification.
Compressibility
The tabletability can be defined as the capacity of a powdered material to be transformed into a tablet of specified strength under the effect of the compaction pressure. The ideal goal in pharmaceutical development is to engineer a formulation that is suitable for direct compression with minimal addition of excipients as a stable compact tablet at low punch force, which does not stick to punch die, and has good flowability. A decrease in the crystallite size observed in the FE-SEM morphology images will enhance the compact hardness, which is due to the smaller crystallites having a larger number of contact points between crystals during compression.
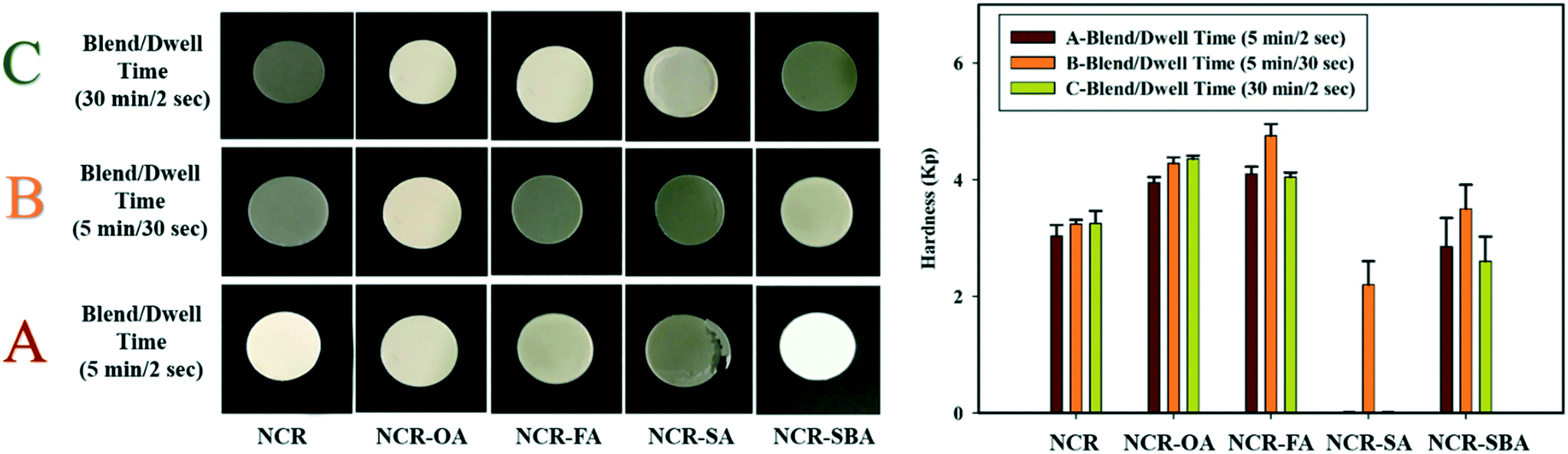
The tendency of the NCR powder to fragment during tableting was modified by forming multicomponent crystals NCR–OA, NCR–SA, NCR–FA, and NCR–SBA. A pictorial presentation of the compressed pellets is shown in Fig. 6, and the compaction properties are summarized in Table 7. The 3-dimensional crystal structure of NCR (Fig. 1a) has a fragmenting nature, and poses difficulty in tablet compression. Out of the coformers, OA and SA displayed the least impact on the alteration of the compression property of NCR, with SA showing a capping tendency and elastic behavior. This may have an adverse impact on the tableting behavior of NCR. NCR–OA showed anomalous behavior (blend A with lower hardness, and blend B and C exhibit similar hardness with a change in the dwell and blending time). The preparation of blends is detailed in Experimental section. NCR–FA and NCR–SBA were found to exhibit a plastic nature (plasticity of B > A > C), which is a desirable property for compression. NCR–FA has shown improved hardness in both A and B blends with different dwell times (2 s and 30 s), and blend C of low hardness, which can be attributed to poor binding due to the surface coating of magnesium stearate on increasing the blending time. To conclude, all of the examined salts/cocrystals of NCR demonstrated better tabletability than the reference drug NCR, except for the NCR–SA cocrystal. A quantitative correlation of compression and tabletability with crystal structure packing and intermolecular interactions is elusive because the single crystal X-ray structures are not available for the four crystalline solids.
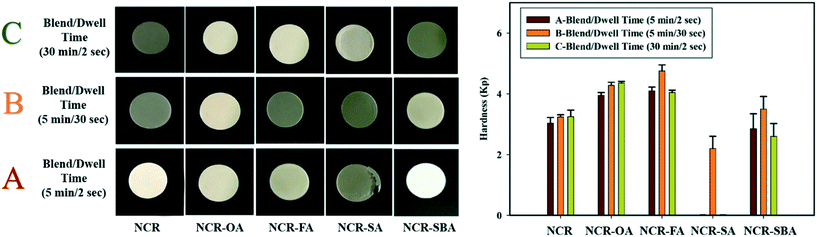 |
| | Fig. 6 The compression properties of the studied samples, where A–C are the process parameters with different dwell and blending times. | |
Table 7 Compressibility properties of the NCR cocrystals/salts
| Binary solid |
A |
B |
C |
Observation |
Comment |
|
Compression parameter: 13 mm die at 50 kg cm−2.
|
| NCR |
3.03 ± 0.19 |
3.24 ± 0.07 |
3.25 ± 0.21 |
No capping |
Fragmentation of A, B, and C are similar |
| NCR–OA |
3.95 ± 0.09 |
4.28 ± 0.1 |
4.35 ± 0.06 |
No capping |
C > B > A |
| NCR–FA |
4.1 ± 0.12 |
4.75 ± 0.2 |
4.04 ± 0.08 |
No capping |
Plastic nature B > A > C |
| NCR–SA |
Cap |
2.2 ± 0.4 |
Cap |
Capping |
Elastic nature |
| NCR–SBA |
2.85 ± 0.49 |
3.5 ± 0.41 |
2.6 ± 0.42 |
No capping |
Plastic nature B > A > C |
Conclusion
Cocrystallization of NCR with alkane/alkene dicarboxylic acids resulted in stable salts and cocrystal at ambient temperature and humidity conditions. The crystal structure analysis of the NCR–FA salt and NCR–SBA cocrystal showed that the former is connected through the ionic carboxylate⋯pyridinium synthon, and the latter has neutral carboxylic acid⋯pyridine hydrogen bonding. The NCR–carboxylic acid binary solids exhibit phase stability, and displayed enhanced solubility, dissolution rate and diffusion rate compared to the parent drug in PBS media (except NCR–SA). NCR–OA has the highest dissolution and diffusion behavior among the crystal forms examined. OA is a known counter ion in the marketed drug formulation (e.g., citalopram oxalate). NCR–OA is the optimal drug form for immediate release oral drug formulation with improved solubility, diffusion, and mechanical properties.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
Financial support by DST-SERB (EMR/2016/001226) to K. A. S. is gratefully acknowledged. A. K. N. thanks CSIR-NCL and DST-SERB (J. C. Bose fellowship, SR/S2/JCB06/2009 and Pharmaceutical materials and continuous process, CRG/2019/001388) for research funding, and the School of Chemistry, University of Hyderabad for providing infrastructure facilities. A. K. N. acknowledges financial and infrastructure support from the University Grants Commission (through UPE and CAS programs) and the Department of Science and Technology (through PURSE and FIST programs).
References
- B. Singh, T. Garg, A. K. Goyal and G. Rath, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 1498–1507 CrossRef CAS
 .
.
-
V. Fuster and B. Kelly, Promoting Cardiovascular Health in the Developing World, The National Academies Press, Washington, D.C., 2010 Search PubMed .
- A. M. Frydman, P. Chapell and H. Diekman, Am. J. Cardiol., 1989, 63, 25j–33j CrossRef CAS .
-
Drug Bank, https://www.drugbank.ca/drugs/DB09220, (accessed August 2020) .
- Gagandeep, T. Garg, B. Malik, G. Rath and A. K. Goyal, Eur. J. Pharm. Sci., 2014, 53, 10–16 CrossRef CAS .
- D. N. Tipre and P. R. Vavia, Pharm. Dev. Technol., 2002, 7, 325–332 CrossRef CAS .
-
Y. Lida and S. Sumida, EP0230932B1, 1991 .
- CSD v 2019, Feb 2020 update, https://www.ccdc.cam.ac.uk (accessed September 2020) .
- C. Guo, Q. Z. Bingqing, Z. Zhang, J. Bao, Q. Ding, G. Ren and X. Mei, Cryst. Growth Des., 2020, 20, 6995–7005 CrossRef CAS .
- G. R. Desiraju, J. Mol. Struct., 2003, 656, 5–15 CrossRef CAS .
- A. K. Nangia and G. R. Desiraju, Angew. Chem., Int. Ed., 2019, 58, 4100–4107 CrossRef CAS .
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS .
- T. Davin, L. Leigh and F. Tomislav, Chem. Commun., 2016, 52, 7760–7781 RSC .
-
Bruker SMART, Version 5.625, SHELXTL, Version 6.12, Bruker AXS Inc., Madison, Wisconsin, USA, 2000 Search PubMed .
-
G. M. Sheldrick, Program for Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS .
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed .
-
L. J. Barbour, X-Seed, Graphical Interface to SHELX-97 and POV-Ray, University of Missouri-Columbia, Columbus, MO, 1999 Search PubMed .
-
Powder Cell, A Program for Structure Visualization, Powder Pattern Calculation and Profile Fitting, http://www.ccp14.ac.uk/tutorial/powdcell/, (accessed August 2020) Search PubMed .
- T. Higuchi and K. A. Connors, Adv. Anal. Chem. Instrum., 1965, 4, 117–212 CAS .
- E. Baka, J. E. Comer and K. Takacs-Nova, J. Pharm. Biomed. Anal., 2008, 46, 335–341 CrossRef CAS .
- A. M. Frydman, P. Chapelle, H. Diekmann, R. Bruno, J. J. Thebault, J. Bouthier, H. Caplian, W. Ungethuem, C. Gailard, A. L. Liboux, A. Renard and J. Gaillot, Am. J. Cardiol., 1989, 63, J25–J33 CrossRef .
- M. K. C. Mannava, R. Dandela, S. Thothadi, K. A. Solomon and A. K. Nangia, Cryst. Growth Des., 2020, 20, 3064–3076 CrossRef CAS .
-
J. Wells and M. Aulton, The Science of Dosage Form Design, Pharmaceutics, Edinburgh, Churchill Livingstone, 2002 Search PubMed .
- Generally Regarded as Safe chemicals by the US-FDA, http://www.fda.gov/Food/IngredientsPackagingLabeling/FoodAdditivesIngredients/ucm091048.html, (accessed October 2019) .
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362–6365 RSC .
- J. Sun, F. Wang, Y. Sui, S. Zhennan, W. Zhai, C. Wang and Y. Deng, Int. J. Nanomed., 2014, 7, 5733–5744 Search PubMed .
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS .
- U. B. R. Khandavilli, S. Gangavaram, N. R. Goud, S. Cherukuvada, A. Raghavender, A. Nangia, S. Manjulatha, S. Nambiar and S. Pal, CrystEngComm, 2014, 16, 4842–4852 RSC .
- K. Suresh, V. S. Minkov, K. K. Namila, E. Derevyannikova, E. Losev, A. Nangia and E. V. Boldyreva, Cryst. Growth Des., 2015, 15, 3498–3510 CrossRef CAS .
- D. P. Elder, R. Holm and H. L. Diego, Int. J. Pharm., 2012, 453, 88–100 CrossRef .
- Y. Yan, J. M. Chen and T. B. Lu, CrystEngComm, 2013, 15, 6457–6460 RSC .
Footnote |
| † Electronic supplementary information (ESI) available: The ORTEP diagram of binary adducts of NCR (Fig. S1), hydrogen bond parameters in NCR salts and a cocrystal (Table S1), NMR of the NCR-SA cocrystal, NCR-OA salts (Fig. S2), to determine ratio of NCR-SA, NCR-OA by PXRD (Fig. S3), thermal analysis by both DSC and TGA for NCR-OA and NCR-SA for chemical stability (Fig. S4). FT-IR spectra of NCR salts and a cocrystal (Fig. S5), and powder XRD of NCR salts and cocrystals at the end of dissolution/phase stability experiment compared with the calculated line pattern from the X-ray crystal structure (Fig. S6). CCDC 2026185 and 2026186. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01316a |
|
| This journal is © The Royal Society of Chemistry 2021 |

 ab,
Anilkumar
Gunnam
ab,
Anilkumar
Gunnam